

Abstract
Background
Mitochondria are key regulators in cell proliferation and apoptosis. Alterations in mitochondrial function are closely associated with inflammation and tumorigenesis. This study aimed to investigate whether mitochondrial transcription factor A (TFAM), a key regulator of mitochondrial DNA transcription and replication, is involved in the initiation and progression of colitis‐associated cancer (CAC).
Methods
TFAM expression was examined in tissue samples of inflammatory bowel diseases (IBD) and CAC by immunohistochemistry. Intestinal epithelial cell (IEC)‐specific TFAM‐knockout mice (TFAM △IEC) and colorectal cancer (CRC) cells with TFAM knockdown or overexpression were used to evaluate the role of TFAM in colitis and the initiation and progression of CAC. The underlying mechanisms of TFAM were also explored by analyzing mitochondrial respiration function and biogenesis.
Results
The expression of TFAM was downregulated in active IBD and negatively associated with the disease activity. The downregulation of TFAM in IECs was induced by interleukin‐6 in a signal transducer and activator of transcription 3 (STAT3)/miR‐23b‐dependent manner. In addition, TFAM knockout impaired IEC turnover to promote dextran sulfate sodium (DSS)‐induced colitis in mice. Of note, TFAM knockout increased the susceptibility of mice to azoxymethane/DSS‐induced CAC and TFAM overexpression protected mice from intestinal inflammation and colitis‐associated tumorigenesis. By contrast, TFAM expression was upregulated in CAC tissues and contributed to cell growth. Furthermore, it was demonstrated that β‐catenin induced the upregulation of TFAM through c‐Myc in CRC cells. Mechanistically, TFAM promoted the proliferation of both IECs and CRC cells by increasing mitochondrial biogenesis and activity.
Conclusions
TFAM plays a dual role in the initiation and progression of CAC, providing a novel understanding of CAC pathogenesis.
Keywords: colitis, colitis‐associated cancer, colorectal cancer, energy metabolism, inflammatory bowel diseases, intestinal homeostasis, mitochondrial transcription factor A (TFAM)
Our study uncover the opposite roles of TFAM in the initiation and progression of CAC, indicating that TFAM switches from tumor suppressor in the premalignant stages of tumorigenesis to oncogene at later stages of disease. Our data suggest that measuring TFAM status may serve as a clinically useful tool to identify patients at risk for colon dysplasia or cancer. In contrast, TFAM might also be a prognostic biomarker and pharmacological target for advanced CAC.
Abbreviations
- TFAM
Mitochondrial transcription factor A
- CAC
Colitis‐associated cancer
- IBD
Inflammatory bowel diseases
- IEC
Intestinal epithelial cell
- UC
Ulcerative colitis
- CD
Crohn's disease
- CRC
Colorectal cancer
- mtDNA
Mitochondrial DNA
- FITC
Fluorescein isothiocyanate
- AOM
Azoxymethane
- DSS
Dextran sulfate sodium
- qRT‐PCR
Quantitative real‐time reverse transcription PCR
- WB
Western blotting
- IHC
Immunohistochemistry
- OCR
Oxygen consumption rate
- EV
Empty vector
- CRP
C‐reactive protein
- IL‐6
Interleukin‐6
- TP53
Tumor protein p53
- KRAS
V‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog
- APC
Adenomatous polyposis coli
- MT‐ND1
Mitochondrial NADH dehydrogenase 1
- STAT3
Signal Transducer and Activator of Transcription 3
- mtDNA
Mitochondrial DNA
- GAPDH
Glyceraldehyde‐phosphate dehydrogenase
- ATCC
American Type Culture Collection
- EDTA
Ethylene Diamine Tetraacetic Acid
- HBSS
Hank's Balanced Salt Solution
- SD
Standard deviation
1. BACKGROUND
Inflammatory bowel diseases (IBD), which comprise ulcerative colitis (UC) and Crohn's disease (CD), are characterized by recurrent episodes of intestinal inflammation. Patients with IBD present an increased risk of colorectal cancer (CRC) [1], with a relative risk of 30 and 5.6 times higher in patients with UC [2] and CD [3], respectively, than that of the general population. Furthermore, compared with patients with sporadic CRC, patients with colitis‐associated cancer (CAC) were younger, had multiple cancerous lesions, and were diagnosed with advanced stage at presentation [4]. Although the severity and duration of inflammation appeared to be major factors associated with the development of CAC in patients with IBD, the pathogenesis of CAC remained poorly understood.
Within the intestine, maintaining the intestinal epithelium turnover is important for ensuring effective barrier function against the commensal microbiota and food digestion. However, a disruption of the intestinal epithelial barrier is commonly observed in IBD patients, which is usually associated with decreased intestinal epithelial cell proliferation and increased cell death, further promoting the inflammation‐to‐cancer transition [5, 6]. Thus, an interesting question is raised: what are the key factors that govern the transition from repressed cell proliferation under inflammatory conditions to rapid cell proliferation at the tumor stage.
Mitochondria are semi‐autonomous organelles that contain their own mitochondrial DNA (mtDNA), which encodes 13 essential proteins of the respiratory chain. During oncogenesis, mitochondria play a central role in the regulation of cell proliferation and apoptosis [7]. Recent scientific advances have further revealed that the intrinsic dynamicity of mitochondria played a crucial role in inflammatory signaling. Similarly, inflammatory mediators may also alter mitochondrial function [8]. For example, Haberman et al. [9] and Reifen et al. [10] have reported a marked decrease in mitochondrial and nuclear‐encoded mitochondrial genes across cohorts with active UC, which was also observed in rat colitis samples. Meanwhile, ultrastructural alterations in the mitochondria of intestinal epithelial cells (IECs), such as dissolved/irregular cristae, which are indicative of an impaired function, were evident in patients with CD as an early event of inflammation [11]. By contrast, our previous study [12] and other studies [13, 14] have demonstrated that the mitochondrial mass and mtDNA content in human sporadic CRC tissues were significantly higher than those in the normal colon mucosa. Furthermore, it was also reported that CRC cells relied on mitochondrial oxidative phosphorylation as their major source of energy [15, 16]. However, whether and how mitochondria are involved in the regulation of colitis and CAC remains unclear.
Mitochondrial transcription factor A (TFAM), which is encoded by a nuclear gene, plays a pivotal role in the transcription [17] and replication [18] of mtDNA coding for critical components of the electron transport chain, rendering TFAM a vital factor for energy production via oxidative phosphorylation [19]. The inactivation of TFAM function leaded to enlarged mitochondria with abnormal cristae, decreased energy production, and embryonic lethality [20]. The adipose‐specific deletion of TFAM increased mitochondrial oxidation and protected mice against obesity and insulin resistance [21]. In addition, the truncating mutation of TFAM induced mtDNA depletion and apoptotic resistance in microsatellite‐unstable CRC [22]. However, the genetic disruption of TFAM has not been investigated in the context of colitis and CAC.
Here, IEC‐specific TFAM knockout mice were used to determine the role of TFAM in the process of colitis and CAC. We also analyzed the expression of TFAM in human colitis and CAC tissues, and explored the underlying mechanism in vitro using normal human colorectal epithelial cell line and CRC cell lines.
2. MATERIALS AND METHODS
2.1. Reagents
Human tumor necrosis factor‐α (TNF‐α) (cat. no. 300‐01A) and interleukin‐6 (IL‐6; cat. no. 200‐06) were purchased from PeproTech EC Ltd (Rocky Hill, NJ, USA). The microRNA (miRNA/miR) mimics and inhibitors were synthesized by Shanghai GenePhama Co., Ltd. (Shanghai, China), and the sequences are listed in Supplementary Table S1. Oligomycin (cat. no. 495455), azoxymethane (AOM; cat. no. A5486) and fluorescein isothiocyanate (FITC)‐dextran (cat. no. 46944) were purchased from Merck KGaA (St. Louis, MO, USA). Dextran sulfate sodium (DSS; cat. no. 0216011080) was purchased from MP Biomedicals, LLC (Irvine, CA, USA). The signal transducer and activator of transcription 3 (STAT3) inhibitor Stattic (cat. no. S7024), the β‐catenin inhibitors IWR‐1 (cat. no. HY‐12238) and KYA1797K (cat. no. HY‐101090), and the β‐catenin agonists SKL2001 (cat. no. HY‐101085) and IM‐12 (cat. no. HY‐12292) were purchased from MedChemExpress (Monmouth Junction, NJ, USA). Penicillin‐Streptomycin solution (cat. no. P1410), agarose (cat. no. 9012‐36‐6) and ethylene diamine tetraacetic acid (EDTA, cat. no. E8040) were purchased from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China). Trizol (cat. no.15596‐026) was purchased from Invitrogen (Carlsbad, CA, USA). Puromycin was purchased from DIYIBio Co., Ltd. (Shanghai, China).
2.2. Cell culture and patient sample collection
The normal human colorectal epithelial cell line FHC and CRC cell lines SW1116, T84, RKO, SW480, SW620, LoVo, CaCo2 and HCT116 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Another normal human colorectal epithelial cell line NCM460 was purchased from Incell Corporation LLC (San Antonio, TX, USA). Cells were cultured in RPMI‐1640 medium (Gibco, Thermo Fisher Scientific, Inc., Waltham, MA, USA) or Dulbecco's Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin solution. Cells were routinely maintained in a humidified atmosphere containing 5% CO2 at 37°C. The STAT3 inhibitor Stattic (20 μmol/L) was added into cell culture medium of NCM460 and FHC for 24 h before Western blotting and reverse transcription‐quantitative PCR experiment.
A total of 10 paraffin‐embedded normal cadaveric colonic tissues from healthy controls, 88 fresh inflammatory tissues from patients with UC, 90 fresh inflammatory tissues from patients with CD, 30 fresh and 7 paraffin‐embedded tumor tissues from patients with CAC and paired venous blood samples from patients with IBD were collected from the Tangdu and Xijing Hospitals affiliated with Fourth Military Medical University (FMMU, Xi'an, Shaanxi, China). The clinical characteristics of patients with IBD are listed in Supplementary Table S2. All participants provided written informed consent, and this study was approved by the ethics committee of FMMU.
2.3. Experimental animals
TFAMflox/flox mice possessing LoxP sites flanking exons 6‐7 of TFAM were generously gifted by Professor Yongzhan Nie (Department of Digestive Diseases, Xijing Hospital, FMMU). Villin‐Cre mice were generously gifted by Professor Jian Zhang (Department of Biochemistry and Molecular Biology, Basic Medical Science Academy, FMMU). A mouse model lacking TFAM specifically in IECs (TFAM △IEC) was generated by crossing TFAM flox/flox mice with Villin‐Cre mice in a specific pathogen‐free (SPF) laboratory animal house (Laboratory Animal Center, FMMU). The genotypes of mice were evaluated by multiplex PCR and agarose gel electrophoresis. Agarose gels (2%) containing 0.5 μg/mL ethidium bromide were prepared in TAE buffer. The DNA samples were separated in agarose gel at 100 V for 30 min. A TFAM wild‐type allele produces a 396‐bp PCR product, whereas a TFAM deleted allele produces a 438‐bp PCR product as previously described [23]. All animal experimental procedures were approved by the Animal Care Committee of the FMMU.
2.4. Establishment of CAC and colitis mouse model
CAC was induced in mice as previously described [24]. Briefly, co‐housed TFAM △IEC and wild‐type (WT) littermate C57BL/6J mice aged 8‐10 weeks were intraperitoneally injected with 10 mg/kg AOM. Two days later, these mice were fed with 1.5% DSS for 5 consecutive days, followed by 14 days of feeding with normal water for recovery. The cycle from DSS to normal water was repeated three times. Mice without AOM/DSS treatment served as control. Mice were euthanized 1 or 3 months after AOM injection, and the colon was then cut open longitudinally. The tumor nodules were quantified under a stereomicroscope (M205, Leica Microsystems, Inc., Wetzlar, Germany).
To generate DSS‐induced colitis, mice were fed 2.5% DSS for 5 consecutive days, and then changed to normal water for another 5 days. Body weight, stool consistency (0, normal; 2, loose feces; 4, diarrhea) and hematochezia (0, occult blood negative; 2, occult blood positive; 4, gross bloody stool) were daily measured and recorded as previously described [25, 26]. Mice were euthanized by cervical dislocation on day 5 (end of DSS treatment) or day 10 (end of DSS and normal water treatment), and then the colon length was measured. Tissue samples were then frozen in liquid nitrogen or fixed in 10% formalin for further analysis.
2.5. Reverse transcription‐quantitative PCR (RT‐qPCR)
To determine the role of β‐catenin signaling in the regulation of TFAM expression in CRC cells, 20 μmol/L IWR‐1, 20 μmol/L KYA1797K, 30 μmol/L SKL2001, or 5 μmol/L IM‐12 was added into cell culture medium for 24 h. Total RNA was extracted from the mouse or human colon tissues, normal human colorectal epithelial cell lines, or CRC cell lines using Trizol and then reversely transcribed into cDNA using High Capacity cDNA Reverse Transcription Kits (cat. no. 4368814, Thermo Fisher Scientific, Inc., Waltham, MA, USA). RT‐qPCR was performed using TB Green® Premix Ex TaqTM II (cat. no. RR820A; Takara Bio, Inc, Tokyo, Japan) in a real‐time PCR instrument (Bio‐Rad, Hercules, CA, USA). The reaction conditions were 95°C for 2 min, then 40 cycles of 95°C for 10 s and 60°C for 30 s. The relative expression levels of the genes were calculated using the 2–∆∆Ct method. Glyceraldehyde‐phosphate dehydrogenase (GAPDH) and U6 snRNA (only for miRNAs) were used as reference genes. The primers used are listed in Supplementary Table S1.
2.6. Detection of mtDNA copy number
The relative mtDNA copy number was measured by RT‐qPCR as previously described [27]. Briefly, genomic DNA was extracted from FHC and RKO cells using QIAamp DNA Mini kits (cat. no. 51306; Qiagen, Inc, Hilden, Germany). The ratio of mitochondrial NADH dehydrogenase 1 (MT‐ND1) gene in mtDNA to the single copy nuclear gene human globulin (HGB) was determined from standard curves. The primers used are listed in Supplementary Table S1.
2.7. Western blotting (WB) and immunohistochemistry (IHC)
WB and IHC were performed as previously described [28]. WB were performed using proteins extracted from mouse colon tissues and normal human colorectal epithelial FHC and NCM460 cells and CRC cell lines, such as SW480, HCT116, RKO. Then the signals were detected using an ECL kit (Thermo Fisher Scientific, Inc.).
For IHC staining of human normal and inflammatory colonic tissues and CAC tissues, the intensity (0, none; 1, faint yellow; 2, yellow; 3, brown) and proportion of positive cells (0, 0‐9%; 1, 10‐25%; 2, 26‐50%; 3, 51‐75%; 4, 76‐100%) were determined within five microscopic visual fields per slide (magnification, × 200) by two independent pathologists blinded to the clinical data. IHC were then scored (ranging from 0 to 12) by multiplying the percentage of positive cells by the intensity. The expression levels of TFAM in the clinical samples were classified as low expression (score 0‐3), moderate expression (score 4‐7) and high expression (score 8‐12). The primary antibodies used in the study are listed in Supplementary Table S3.
2.8. Histological analysis
Hematoxylin and eosin (H&E) staining was performed as previously described [29]. The histological score of colitis in mice was evaluated using three independent parameters as previously described [30]: extent of injury (0, none; 1, mucosa only; 2, submucosa; 3, transmural), crypt damage (0, none; 1, basal 1/3 damaged; 2, basal 2/3 damaged; 3, almost entire epithelium damaged), and leukocyte infiltration severity (0, none; 1, mild; 2, moderate; 3, severe).
2.9. Detection of intestinal permeability
Intestinal permeability was evaluated by determining the amount of FITC‐dextran and D‐lactate in the blood, as previously reported [31, 32]. Briefly, mice were gavaged with FITC‐dextran (0.4 mg/g body weight) and then euthanized 4 h later. The serum FITC‐dextran concentration was measured using a fluorescent microplate reader with an excitation wavelength of 485 nm and emission wavelength of 535 nm (Thermo Fisher Scientific, Inc.). The serum D‐lactate concentration was detected using a D‐Lactate Colorimetric Assay Kit (cat. no. GMS70095.3, Shanghai Genmed Ltd., Shanghai, China) according to the manufacturer's instructions. The concentration of D‐lactate was calculated using the slope and intercept created from a standard curve.
2.10. Knockdown and forced expression of target genes
Lentiviral vectors for TFAM and its transcription factor c‐Myc knockdown or overexpression were constructed by Shanghai GenePharma Co., Ltd. (cat. no. D01001, D02001, Shanghai, China). Lentivirus was collected by co‐transfecting 297T cells with lentiviral expression construct and packaging plasmids. Normal human colorectal epithelial cells or CRC cells (5 × 104/well) were seeded into 6‐well plates and incubated at 37°C with 5% CO2 overnight. Lentivirus was then added into each well (10 multiplicity of infection) with serum‐free medium and incubated overnight. Successfully transfected cell clones were screened using puromycin (5 μg/mL). The expression levels of TFAM and c‐Myc in cells transfected with lentivirus were determined by WB.
2.11. Cell proliferation assay and cell cycle analysis
Cell proliferation was detected using the Cell‐Light 5‐ethynyl‐2’‐deoxyuridine (EdU) Apollo567 In Vitro Kit (cat. no. C10310, Ribobio Co., Ltd., Guangzhou, Guangdong, China) and a propidium iodide (PI)‐staining kit (cat. no. BB‐4104, Shanghai BestBio Co., Ltd.) as previously described [33]. A total of 2 μg/mL oligomycin was used to inhibit mitochondrial F0F1‐ATPase where appropriate. For the EdU assay, 2 × 105 cells were incubated with EdU for 2 h and fixed with 4% paraformaldehyde. Hoechst (cat. no. C1022, Beyotime Institute of Biotechnology; Shanghai, China) was used for nuclear staining. The images were visualized and photographed under a fluorescence microscope (Olympus Corporation, Tokyo, Japan). For cell cycle analysis, ∼1 × 106 cells were collected and incubated with PI staining solution at 4°C for 30 min. The cell cycle was immediately determined by flow cytometry (Beckman Coulter, Inc. Fullerton, CA, USA).
2.12. Detection of mitochondrial respiratory chain complex enzyme activity, oxygen consumption rate (OCR), and mitochondrial adenosine triphosphate (ATP)
Complex (I, III, IV, and V) enzyme activity in cell extracts was measured by the single‐wavelength spectrophotometric assay using detection kits (cat. no. BC0510, BC3240, BC0945 and BC1445, Beijing Solarbio Science & Technology) according to the manufacturers’ protocols. Briefly, 5 × 106 cells were collected and homogenized in lysis buffer. After two steps of centrifugation (600 × g for 10 min and then 11, 000 × g for 15 min) at 4˚C, the precipitate was collected for ultrasonic crushing and then added into the corresponding substrate to start the enzymatic reaction. The absorbance values at different wavelengths (I: 340 nm; III: 550 nm; IV: 550 nm; V: 660 nm) of the metabolic product was detected using a spectrophotometer (Thermo Fisher Scientific Inc.).
The OCR was measured using a 782 Oxygen Meter (Strathkelvin Instruments, Motherwell, UK) according to the manufacturer's instructions. The amplified signal was recorded at sampling intervals of 0.5 s.
Mitochondrial ATP was measured as previously described [34]. Briefly, the abundance of ATP was determined using the ATPlite assay system (PerkinElmer Inc., Waltham, MA, USA). Basal luminescence was recorded before the use of luciferin, and then cells were perfused with 20 μmol/L luciferin to record mitochondrial luminescence [35].
2.13. Intrarectal instillation of lentivirus
Intrarectal instillation of lentivirus was performed to increase the target gene expression in the colonic epithelium as previously described [36]. In brief, mice were anesthetized and received an intrarectal enema of 50% ethanol to increase the intestinal transduction with lentivirus. Mice then received intrarectal instillation of 100 μL solution containing 107 IU lentivirus expressing mouse TFAM (mTFAM) or empty vector (EV). Mice were then inverted for 30 s to prevent leakage. To ensure the efficiency of lentivirus transfection in IECs, lentivirus treatment was performed every 2 days.
2.14. Nude mouse xenograft model
BALB/c nude mice age 6 weeks were purchased from the Laboratory Animal Center of the FMMU and used to establish a xenograft model (6 mice per group). Human RKO cells with TFAM knockdown or overexpression were injected into the back of mice (5 × 106 cells/mouse). A time‐volume curve was plotted to investigate the growth of the xenograft. The mice were euthanized when the largest tumor reached a volume of ∼1000 mm3. Tumor volume (mm3) = [width2 (mm2) × length (mm)] / 2.
2.15. Isolation of IECs
IECs were isolated from the mouse colon as previously described [36]. Mice were anesthetized and then euthanized. The colons were dissected immediately and cut into pieces (1‐2 mm in diameter). Colon tissues were incubated in 5 mmol/L EDTA diluted by Hank's Balanced Salt Solution (HBSS) at 37˚C with shaking for 30 min. The supernatant was collected and centrifuged at 3000 × g for 10 min. The deposit was washed with ice‐cold phosphate buffer saline (PBS) and used for measuring TFAM expression.
2.16. Public dataset collection
To analyze the relationship between the expression of TFAM and c‐Myc, RNA‐seq data of colorectal cancer samples (n = 519) was downloaded from the Cancer Genome Atlas (TCGA) database using the Genomic Data Commons (GDC) Data Portal (https://gdc‐portal.nci.nih.gov). Trimmed mean of M‐values (TMM) algorithms were used for data normalization by using the calcNormFactors function in the edge R package (http://www.r‐project.org).
2.17. Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics 22.0 software (Armonk, NY, USA). A two‐sided P < 0.05 was considered significant. Data are expressed as the mean ± standard deviation (SD). Student's t test or Mann‐Whitney U test was used to examine differences between two groups after checking for normal distribution and equal variance of the data. Correlations between measured variables were analyzed using Spearman correlation analysis. Kaplan–Meier survival curves were plotted 15 days after DSS treatment or 3 months after AOM/DSS treatment. Survival data were compared by using a log‐rank test.
3. RESULTS
3.1. TFAM knockout increased susceptibility of mice to AOM/DSS‐induced CAC
To investigate the potential role of TFAM in CAC, the TFAM △IEC mouse model was generated, and the loss of TFAM expression in IECs was confirmed by agarose gel electrophoresis and WB (Supplementary Figure S1). Next, mouse CAC was induced by injection with the colonotropic mutagen AOM, followed by DSS‐supplemented water. When euthanized at 1 month after AOM/DSS treatment, TFAM △IEC mice developed intestinal polyps upon gross inspection, whereas intestinal polyps were rarely observed in WT mice. Three months after AOM/DSS treatment, both TFAM △IEC and WT mice developed intestinal polyps. However, TFAM △IEC mice exhibited significantly more and larger polyps when compared with WT mice. In the control groups, intestinal polyps were not clearly observed in both WT or TFAM △IEC mice at 1 and 3 months (Figure 1A‐C). Furthermore, histological analysis revealed that tumors in TFAM △IEC mice exhibited more advanced dysplasia when compared with those in WT mice (Figure 1D‐E). Of note, the conditional knockout of TFAM significantly reduced overall survival in the AOM/DSS‐induced CAC mouse model when compared with the WT control group (Figure 1F). These data demonstrate that the conditional knockout of TFAM increases the susceptibility of mice to AOM/DSS‐induced CAC.
FIGURE 1.
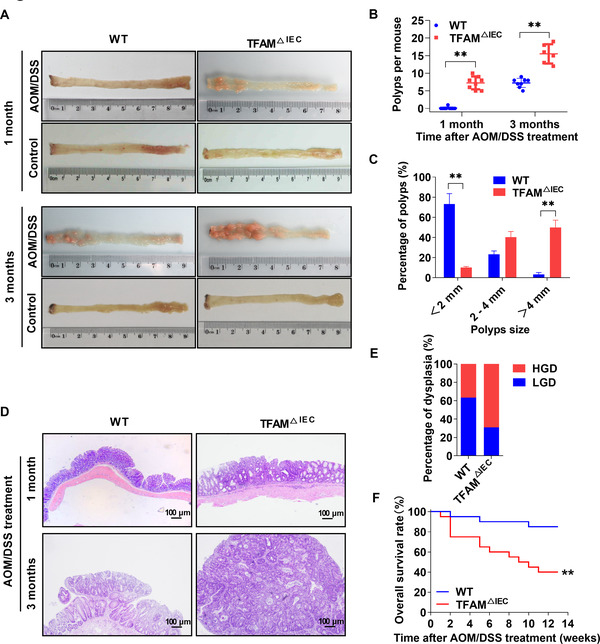
TFAM knockout increases the susceptibility of mice to AOM/DSS‐induced CAC. (A) Representative images of colon tissues from WT and TFAM △IEC mice treated with AOM/DSS as indicated. (B) Polyp numbers and (C) polyp size in WT and TFAM △IEC mice treated with AOM/DSS for 3 months. (D) Representative hematoxylin and eosin‐stained colon sections from WT and TFAM △IEC mice treated with AOM/DSS. (E) Percentage of low‐/high‐grade dysplasia of polyps in WT and TFAM △IEC mice treated with AOM/DSS for 3 months. (F) Kaplan‐Meier survival analysis of WT and TFAM △IEC mice treated with AOM/DSS for 3 months. Error bars represent mean ± SD. Data were analyzed using Mann‐Whitney U test (B and C). *P < 0.05, **P < 0.01. Abbreviations: TFAM, mitochondrial transcription factor; AOM, azoxymethane; DSS, dextran sulfate sodium; CAC, colitis‐associated cancer; WT, wild‐type; TFAM △IEC, knockdown of TFAM particularly in intestinal epithelial cells; LGD, low‐grade dysplasia; HGD, high‐grade dysplasia; SD, standard deviation
3.2. TFAM knockout promoted DSS‐induced intestinal inflammation and impaired IEC turnover in mice
Considering that inflammation plays a critical role in tumorigenesis, it was hypothesized that the increased susceptibility of CAC in TFAM △IEC mice may be caused by enhanced intestinal inflammation. To this end, TFAM △IEC and WT mice were treated with 2.5% DSS to establish an inflammation model. Intestinal inflammation activity was then assessed based on weight loss, stool consistency, gross rectal bleeding, and colon length. As shown in Figure 2A‐D, neither TFAM △IEC nor WT mice showed obvious intestinal inflammation without DSS treatment. However, TFAM △IEC mice developed considerably severer intestinal inflammation than WT mice following DSS treatment. Histological analysis of colon tissues revealed that both TFAM △IEC and WT mice had normal colonic mucosa prior to DSS treatment (Day 0), with the colonic crypts and intestinal epithelium integrity from both TFAM △IEC and WT mice completely destroyed at the end of DSS treatment (Day 5). In contrast, colonic mucosa of WT mice showed signs of re‐epithelialization on day 10, whereas that of TFAM △IEC mice still showed a high level of inflammation, as indicated by the greater extent of injury, inflammatory cell infiltration, decreased number of crypts, and higher histological scores (Figure 2E‐F). The recovery of intestinal epithelium integrity on day 10 was further evaluated by in vivo transepithelial permeability assays. The data showed a significant increase in FITC‐dextran and D‐lactate concentration in the blood of TFAM △IEC mice when compared with those in WT mice (Figure 2G‐H), indicating worse intestinal epithelium integrity and uncontrolled entry of solutes and pathogens. Consistently, the expression of pro‐inflammatory cytokines (IL‐1β, IL‐6, IL‐11, and TNF‐α) was much higher in the colonic mucosa of DSS‐treated TFAM △IEC mice than in that of WT mice (Figure 2I). Finally, the effect of TFAM on intestinal inflammation was further evaluated by continuous feeding with a lethal dose of DSS (3.5%). As shown in Figure 2J, TFAM △IEC mice exhibited a significantly shorter survival time than WT mice.
FIGURE 2.
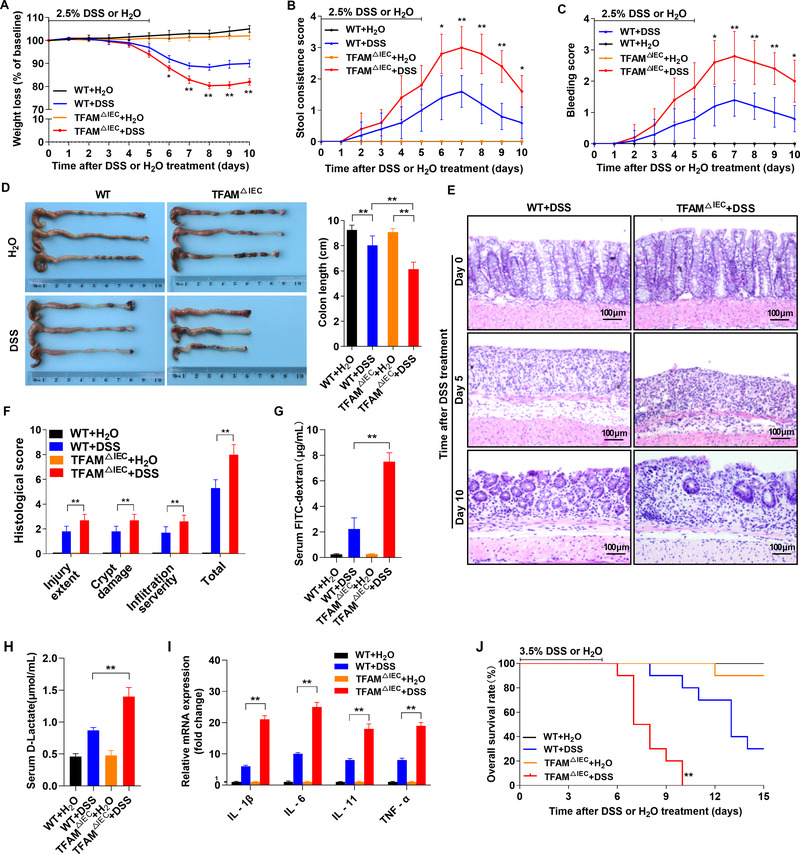
TFAM knockout promotes DSS‐induced intestinal inflammation and impairs IEC turnover in mice. (A‐C) Weight loss, stool consistency, and rectal bleeding score in WT and TFAM △IEC mice following 2.5% DSS or H2O treatment. (D) Gross morphology and length of the colons from WT and TFAM △IEC mice treated with 2.5% DSS or H2O (day 10). (E) Representative hematoxylin and eosin‐stained images of colon tissues from WT and TFAM △IEC mice treated with 2.5% DSS at different time points. (F) Histological score of colitis tissues from WT and TFAM △IEC mice treated with 2.5% DSS or H2O on day 10. (G and H) Serum FITC‐dextran and D‐lactate concentrations in WT and TFAM △IEC mice treated with 2.5% DSS or H2O on day 10. (I) Relative mRNA expression of pro‐inflammatory cytokines (IL‐1β, IL‐6, IL‐11, and TNF‐α) in colonic mucosa from WT and TFAM △IEC mice treated with 2.5% DSS or H2O treatment on day 10. (J) Kaplan‐Meier survival analysis of WT and TFAM △IEC mice treated with 3.5% DSS or H2O. Error bars represent mean ± SD. Data were analyzed using Mann‐Whitney U test. *P < 0.05, **P < 0.01. Abbreviations: TFAM, mitochondrial transcription factor; DSS, dextran sulfate sodium; IEC, intestinal epithelial cell; WT, wild‐type; FITC, fluorescein isothiocyanate; IL, interleukin; TNF, tumor necrosis factor; SD, standard deviation
3.3. The expression of TFAM was downregulated in active IBD and negatively associated with the disease activity
Next, the alteration of TFAM expression was examined by IHC analysis using colonic biopsies of IBD patients and healthy controls. As shown in Figure 3A‐B, the expression of TFAM was markedly downregulated in IECs from active CD and UC patients when compared with that in healthy controls, but not significantly changed in IECs from inactive UC and CD patients. Next, C‐reactive protein (CRP) and IL‐6, two major markers that reflect the activity and severity of inflammation, were detected for further analysis of the association between the disease activity of IBD and the expression of TFAM. The results showed that serum levels of CRP were much higher in IBD patients with low TFAM expression than in those with high TFAM expression (Figure 3C‐D). These results indicated that the expression of TFAM was inhibited in active IBD and negatively associated with disease activity.
FIGURE 3.
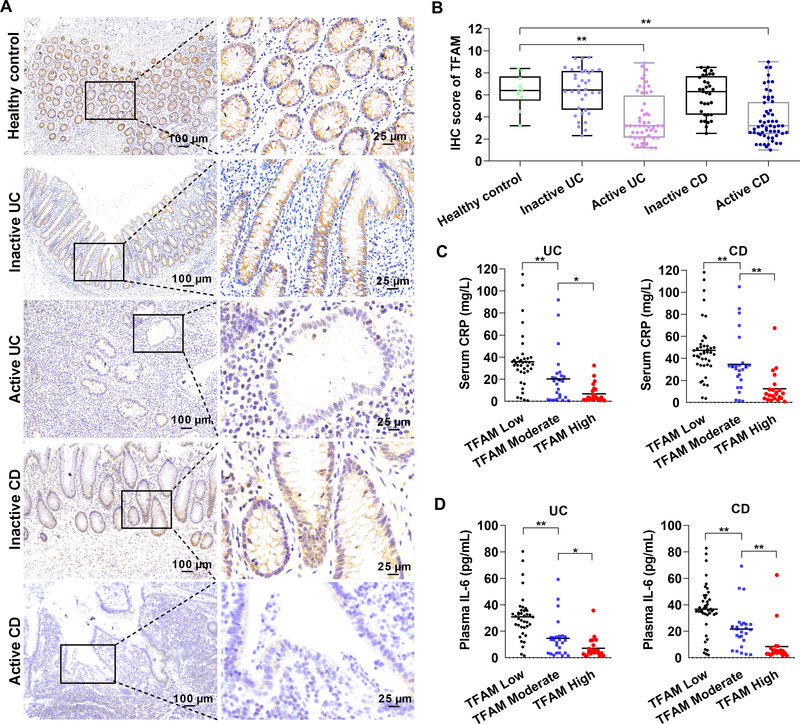
The expression of TFAM is decreased in active IBD and negatively associated with disease activity. (A) Representative IHC staining images of TFAM in colon tissues from healthy controls or patients with active/inactive UC or CD. (B) IHC score of TFAM in human colon tissues as indicated. (C) Serum levels of CRP in UC or CD patients with low, moderate, and high expression of TFAM. (D) Plasma levels of IL‐6 in UC or CD patients with low, moderate, and high expression of TFAM. Error bars represent mean ± SD. Data were analyzed using two‐tailed Student's t test (B) or Mann‐Whitney U test (C and D). *P < 0.05, **P < 0.01. Abbreviations: TFAM, mitochondrial transcription factor; UC, ulcerative colitis; CD, Crohn's disease; IBD, inflammatory bowel diseases; IHC, immunohistochemistry; CRP, C‐reactive protein; SD, standard deviation
3.4. IL‐6 induced TFAM downregulation in a STAT3/miR‐23b‐dependent manner in IECs
To explore the mechanism underlying the expression of TFAM in an inflammatory environment, normal colon epithelial NCM460 and FHC cells were stimulated by the major proinflammatory cytokines involved in colitis. The expression of TFAM was significantly downregulated upon exposure to IL‐6 (Figure 4A), but not to TNF‐α (Figure 4B). Considering that the janus kinase (JAK)/STAT3 signaling pathway, the central signaling hub in colitis, has a prominent role in mediating the effects of IL‐6, the phosphorylated STAT3 (p‐STAT3) was further evaluated. The results demonstrated that the level of p‐STAT3 was significantly increased by IL‐6. More importantly, the inhibitory effect of IL‐6 on TFAM expression was blocked by the STAT3 inhibitor Stattic (Figure 4A), implying that TFAM is a downstream target of IL‐6/STAT3 signaling. Since the expression of TFAM was downregulated, it was next determined whether miRNAs were involved in these processes. Among the seven miRNAs that were reported to repress TFAM expression in human cells, only miR‐23b was significantly upregulated by IL‐6 in both NCM460 and FHC cells (Figure 4C), which was markedly blocked by STAT3 inhibitor Stattic (Figure 4D). In addition, the influence of miR‐23b on TFAM expression was confirmed in normal colon epithelial cells using a miR‐23b mimic and inhibitor (Figure 4E). The association between the expression of TFAM and the regulatory IL‐6/p‐STAT3/miR‐23b axis was then further analyzed in intestinal tissue samples from patients with IBD. As shown in Figure 4F, the expression of TFAM was negatively associated with the colonic level of IL‐6. Consistently, the expression levels of p‐STAT3 and miR‐23b were also negatively associated with the level of TFAM (Figure 4G‐4H and Supplementary Table S4).
FIGURE 4.
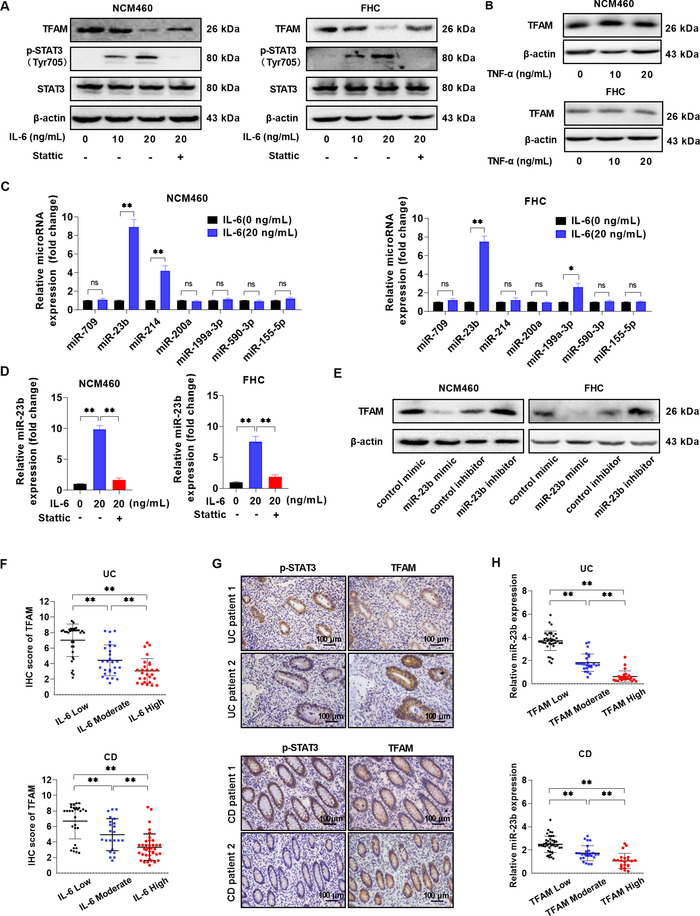
IL‐6 downregulates the expression of TFAM in a STAT3/miR‐23b‐dependent manner in IEC. (A) WB analysis of TFAM, STAT3, and p‐STAT3 expression in NCM460 and FHC cells treated with IL‐6 (0, 10, and 20 ng/mL) or IL‐6 (20 ng/mL) plus Stattic (STAT3 inhibitor, 20 μmol/L). (B) WB analysis for TFAM expression in NCM460 and FHC cells treated with TNF‐α(0, 10, and 20 ng/mL). (C) RT‐qPCR analysis of microRNA levels in NCM460 and FHC cells treated with or without IL‐6 (20 ng/mL). (D) RT‐qPCR analysis of miR‐23b level in NCM460 and FHC cells treated as indicated. (E) WB analysis of TFAM expression in NCM460 and FHC cells treated with miR‐23b mimic and inhibitor. (F) Colonic IL‐6 level in UC or CD patients with low, moderate, and high expression of TFAM. (G) Representative IHC staining images of p‐STAT3 and TFAM in inflammatory colonic tissues from UC or CD patients. (H) RT‐qPCR analysis of miR‐23b level in inflammatory colonic tissues from UC or CD patients with low, moderate, and high expression of TFAM. Error bars represent mean ± SD. Data were analyzed using Mann‐Whitney U test. *P < 0.05, **P < 0.01. Abbreviations: IL, interleukin; TFAM, mitochondrial transcription factor; RT‐qPCR, reverse transcription‐quantitative PCR; SD, standard deviation; WB, Western blotting; miR, microRNA; p‐, phosphorylated
3.5. Overexpression of TFAM protected mice from intestinal inflammation and colitis‐associated tumorigenesis
To further determine the effect of TFAM on intestinal inflammation and colitis‐associated tumorigenesis, TFAM expression was restored through the intrarectal instillation of lentivirus expressing TFAM (Supplementary Figure S2A). TFAM restoration in the colonic epithelia of both TFAM △IEC and WT mice markedly attenuated DSS‐induced colitis as evidenced by alleviation of colitis signs, including weight loss, loose or watery stool, gross rectal bleeding, and shortened colon length (Figure 5A‐D). Transepithelial permeability assays showed that TFAM restoration promoted the recovery of intestinal epithelium integrity following DSS treatment (Figure 5E). Consistently, mice with TFAM restoration showed a reduced level of inflammation, as indicated by increased number of crypts and improved histological scores (Figure 5F‐G). In addition, TFAM restoration reduced the susceptibility of mice to AOM/DSS‐induced CAC, as indicated by the decreases in the number and volume of polyps in both WT and TFAM △IEC mice (Figure 5H‐J). Histological analysis revealed that tumors exhibited a less advanced dysplasia in mice with TFAM restoration when compared with tumors in control groups (Supplementary Figure S2B‐C). Of note, the restoration of TFAM in the intestinal epithelium significantly prolonged overall survival in the AOM/DSS‐induced CAC mouse model (Supplementary Figure S2D).
FIGURE 5.
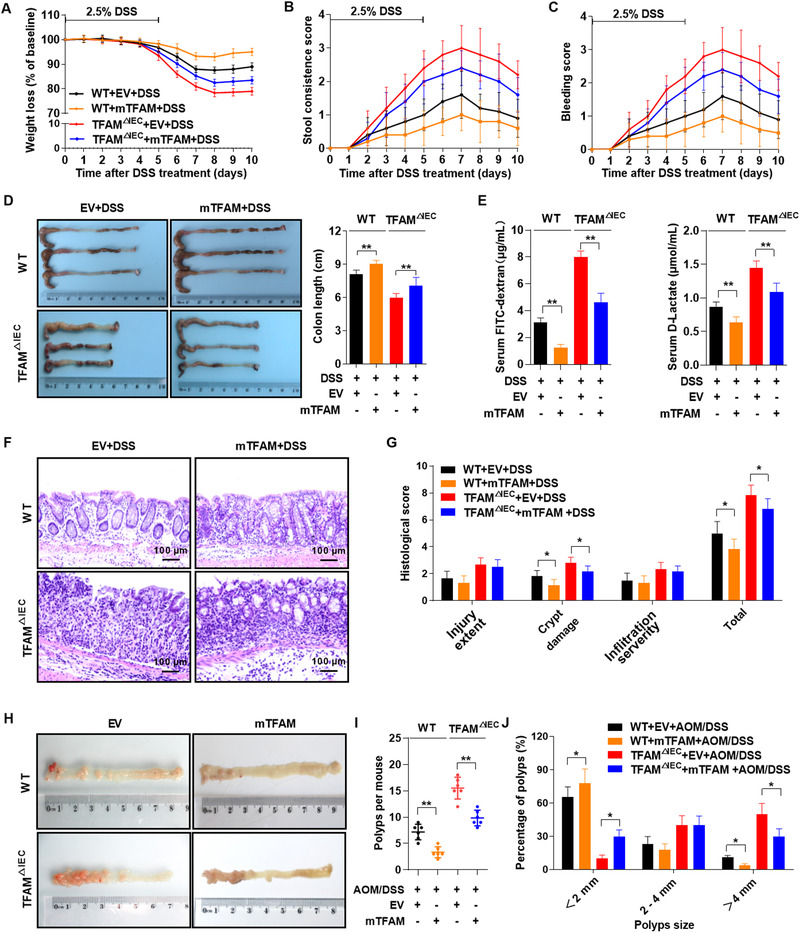
Overexpression of TFAM protects mice from intestinal inflammation and colitis‐associated tumorigenesis. (A‐C) Weight loss, stool consistency score, and rectal bleeding score in mice treated with 2.5% DSS and lentivirus expressing mouse TFAM (mTFAM). (D) Gross morphology and length of the colons from mice treated with lentivirus and DSS (day 10). (E) Serum FITC‐dextran and D‐lactate concentrations in WT and TFAM △IEC mice treated as indicated on day 10. (F) Representative hematoxylin and eosin‐stained images of colon tissues from mice treated as indicated on day 10. (G) Histological score of colitis tissues in mice treated as indicated on day 10. (H) Representative images of colon tissues from mice treated with lentivirus, followed by AOM/DSS treatment for 3 months. (I) Polyp numbers and (J) polyp size in mice treated as indicated for 3 months. Error bars represent mean ± SD. Data were analyzed using Mann‐Whitney U test. *P < 0.05, **P < 0.01. Abbreviations: TFAM, mitochondrial transcription factor; DSS, dextran sulfate sodium; FITC, fluorescein isothiocyanate; WT, wild‐type; AOM, azoxymethane; SD, standard deviation; EV, empty vector
3.6. TFAM was upregulated in CAC tissues and contributed to tumor growth in vivo
Next, the potential role of TFAM was assessed in human CAC. The protein expression of TFAM was much higher in CAC tissues than in peritumoral normal tissues (Figure 6A). This result was further confirmed by RT‐qPCR analysis (Figure 6B), indicating that TFAM was upregulated primarily at the transcriptional level. A xenograft model was established in nude mice to determine how TFAM regulates tumor growth in vivo. RKO cells were transfected with lentivirus containing TFAM short hairpin RNA (shTFAM) or control plasmid (shCtrl), and then transplanted into nude mice. As shown in Figure 6C‐E, TFAM knockdown markedly inhibited tumor growth when compared with controls. TFAM overexpression had the opposite effect (Figure 6F‐H). Consistently, the expression of the cell proliferation marker Ki‐67 was downregulated in tumors with TFAM knockdown and upregulated in tumors with TFAM overexpression (Figure 6I).
FIGURE 6.
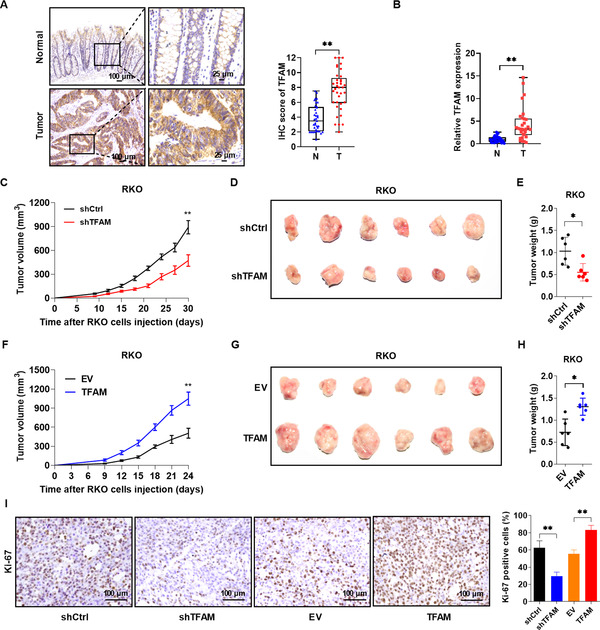
TFAM is upregulated in CAC tissues and contributes to tumor growth in vivo. (A) Representative IHC staining images of TFAM in paired CAC tissues (n = 37). (B) RT‐qPCR analysis for relative expression levels of TFAM in paired CAC tissues (n = 30). (C) Tumor growth curves of the subcutaneous xenograft tumor model developed from RKO cells stably expressing shRNA targeting TFAM (shTFAM) or control (shCtrl). (D‐E) Representative images (D) and weight (E) of harvested tumors in nude mice euthanized 4 weeks after a subcutaneous injection with RKO cells stably expressing shTFAM or shCtrl. (F) Tumor growth curves of subcutaneous xenograft tumor model developed from RKO cells stably overexpressing TFAM or empty vector (EV). (G and H) Representative images and weight of harvested tumors in nude mice euthanized 3 weeks after a subcutaneous injection with RKO cells stably overexpressing TFAM or empty vector. (I) Representative IHC staining images of Ki‐67 (left) and the percentage of Ki‐67‐positive cells (right) in different groups. Error bars represent mean ± SD. Data were analyzed using two‐tailed Student's t test (A) or Mann‐Whitney U test (B, C, E, F, H and I). *P < 0.05, **P < 0.01. Abbreviations: TFAM, mitochondrial transcription factor; T, tumor tissues; N, peritumoral normal tissues; SD, standard deviation
3.7. β‐catenin induced upregulation of TFAM through c‐Myc in CRC cells
CAC results from the accumulation of mutations in IECs. In the context of tumor, β‐catenin is a common downstream target of mutant genes such as tumor protein p53 (TP53), V‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS), and adenomatous polyposis coli (APC) [37]. Therefore, we hypothesized that β‐catenin signaling might govern the expression of TFAM in CAC cells. As shown in Figure 7A and Supplementary Table S5, the protein expression levels of nuclear β‐catenin and its target c‐Myc were positively associated with that of TFAM in human CAC tissue samples. These findings were further confirmed by the RNA‐seq dataset in TCGA database (Figure 7B), indicating a positive correlation between the c‐Myc and TFAM mRNA expression in human CAC samples. Moreover, colon tumor cells with higher β‐catenin activity exhibited an increased TFAM expression compared with those with lower β‐catenin activity (Figure 7C). Since c‐Myc was reported to transcriptionally activate TFAM expression in CRC cells [38], it was then examined whether the Wnt/β‐catenin signaling pathway could induce the expression of TFAM through c‐Myc in CRC cells. As shown in Figure 7D‐E, the inhibition of β‐catenin activity with IWR‐1 or KYA1797K significantly suppressed the expression of TFAM at both the mRNA and protein levels, whereas the activation of β‐catenin with SKL2001 and IM‐12 had the opposite effect. Consistently, c‐Myc overexpression significantly enhanced the expression of TFAM in CRC cells (Figure 7F), whereas the downregulation of c‐Myc inhibited the expression of TFAM (Figure 7G).
FIGURE 7.
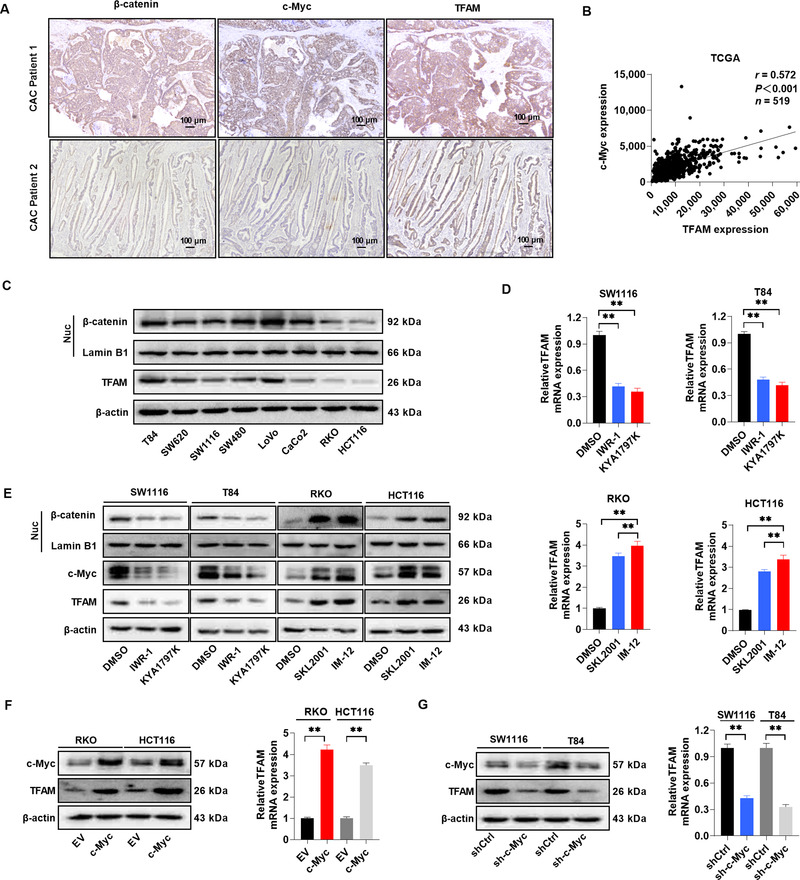
β‐catenin induces upregulation of TFAM through c‐Myc in CRC cells. (A) Representative IHC staining images of β‐catenin, c‐Myc, and TFAM in CAC tissues from different patients. Scale bar, 100 μm. (B) Correlation between c‐Myc and TFAM mRNA expression in TCGA database. (C) WB for the expression of nuclear β‐catenin and TFAM expression in different CRC cell lines. Lamin B1 (a nuclear envelope marker) and β‐actin were used as loading controls in the nuclear and cytoplasmic fractions, respectively. (D) RT‐qPCR analysis for TFAM expression in CRC cells treated with Wnt/β‐catenin signaling pathway inhibitors (IWR‐1 and KYA1797K) or agonists (SKL2001 and IM‐12) as indicated. (E) WB for the expression of nuclear β‐catenin, c‐Myc, and TFAM in CRC cells treated as indicated. (F) WB and RT‐qPCR analysis for the expression of c‐Myc and TFAM in RKO and HCT116 cells after lentiviral transfection with EV or c‐Myc expression vector. (G) WB and RT‐qPCR analysis for the expression of c‐Myc and TFAM in SW1116 and T84 cells expressing shCtrl or shRNA against c‐Myc (sh‐c‐Myc). Error bars represent mean ± SD. Data were analyzed using Mann‐Whitney U test. *P < 0.05, **P < 0.01. Abbreviations: TFAM, mitochondrial transcription factor; TCGA, The Cancer Genome Atlas; EV, empty vector; SD, standard deviation
3.8. TFAM promoted the proliferation of both IECs and CRC cells by increasing mitochondrial biogenesis and activity
Cell proliferation is crucial for tissue repair during intestinal inflammation and tumor progression. To determine the role of TFAM in cell proliferation, TFAM was overexpressed or knocked down in normal FHC cells and CRC RKO cells through lentiviral transfection (Supplementary Figure S3A). EdU assay results demonstrated that FHC and RKO cells with TFAM knockdown exhibited less EdU incorporation than the control cells (Figure 8A and Supplementary Figure S3B). Furthermore, cell cycle analysis indicated that the proportion of cells in the S phase was significantly decreased and that in G1 phase was markedly accumulated in FHC and RKO cells with TFAM knockdown (Figure 8B and Supplementary Figure S3C). TFAM overexpression demonstrated opposite effects (Figure 8A‐B, Supplementary Figure S3B‐C). The effect of TFAM on mitochondrial biogenesis and function was further determined. As shown in Figure 8C, TFAM knockdown significantly decreased and TFAM overexpression significantly increased the enzyme activity of mitochondrial complexes in FHC and RKO cells. Furthermore, the relative mtDNA copy number, OCR, and mitochondrial ATP level were also decreased in FHC and RKO cells with TFAM knockdown, but were increased in cells with TFAM overexpression (Figure 8D‐F). Finally, oligomycin, a mitochondrial F0F1‐ATPase inhibitor, was used to determine the association between TFAM‐mediated cell proliferation and mitochondrial function. The effects of TFAM on EdU incorporation and cell cycle transition were significantly attenuated by oligomycin treatment (Figure 8G‐H and Supplementary Figure S3D‐E).
FIGURE 8.
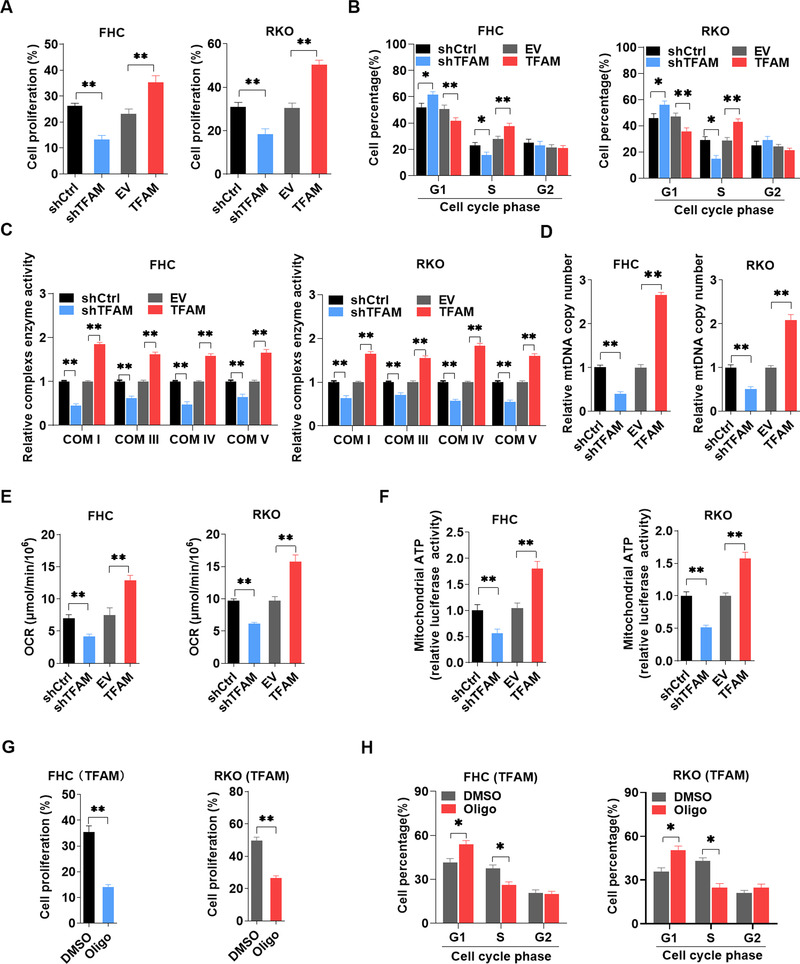
TFAM promotes the proliferation of both IECs and colorectal cancer cells by increasing mitochondrial biogenesis and activity. (A) EdU incorporation assay analysis for cell proliferation in FHC and RKO cells with TFAM knockdown or overexpression. (B) Flow cytometry for cell cycle analysis in FHC and RKO cells with TFAM knockdown or overexpression. (C) Relative mitochondrial respiratory chain complex enzyme (I, III, IV, and V) activity in FHC and RKO cells with TFAM knockdown or overexpression. (D‐F) Relative mtDNA copy number, OCR, and mitochondrial ATP levels in FHC and RKO cells with TFAM knockdown or overexpression. (G) EdU incorporation assay analysis for cell proliferation in FHC and RKO cells with TFAM overexpression and oligomycin treatment. (H) Flow cytometry for cell cycle analysis in FHC and RKO cells with TFAM overexpression and oligomycin treatment. Error bars represent mean ± SD. Data were analyzed using Mann‐Whitney U test. **P < 0.01, *P < 0.05. Abbreviations: TFAM, mitochondrial transcription factor; EdU, 5‐ethynyl‐2’‐deoxyuridine; OCR, oxygen consumption rate; SD, standard deviation; shCtrl, control shRNA; shTFAM, shRNA expression vector against TFAM; EV, empty vector; TFAM, expression vector encoding TFAM; shRNA, short hairpin RNA; Oligo: oligomycin
FIGURE 9.
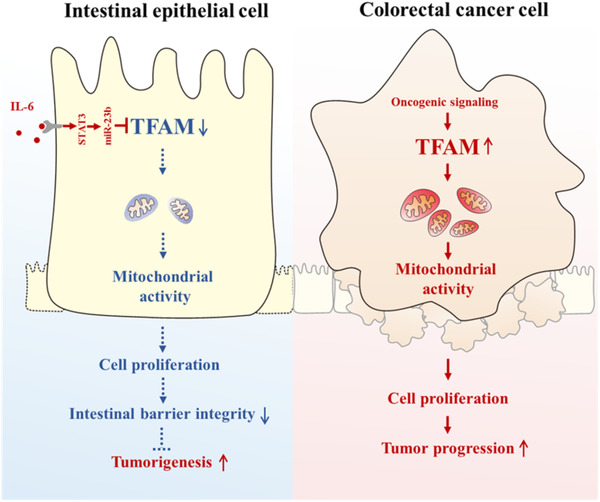
Schematic graph depicting the dual role of TFAM on the initiation and progression of CAC. Briefly, TFAM is downregulated by IL‐6 in active IBD tissues and plays a protective role in initiation of CAC. By contrast, TFAM expression is upregulated by oncogenic signaling in CAC tissues and contributed to tumor growth
4. DISCUSSION
Several key findings were obtained in the present study. First, TFAM expression was found to be downregulated in active IBD tissues but upregulated in CAC tissues. Second, TFAM played a protective role in the initiation of CAC, but a tumor‐promoting role in CAC progression. Third, two distinct pathways were found to regulate TFAM expression in IBD and CAC. The downregulation of TFAM in IECs was induced by IL‐6 in a STAT3/miR‐23b‐dependent manner. By contrast, β‐catenin induced the upregulation of TFAM through c‐Myc in CRC cells. Fourth, TFAM‐mediated mitochondrial activity played a key role in these processes. In IECs, TFAM promoted mitochondrial activity to protect the intestinal barrier and acted as a tumor suppressor. In CRC cells, TFAM acted as an oncogenic protein by accelerating tumor cell proliferation.
Recent researches have shed light on the effects of TFAM on inflammation. Mice with tubule‐specific deletion of TFAM developed severe mitochondrial loss, immune cell infiltration, and kidney fibrosis [39]. Similarly, TFAM +/− mouse embryonic fibroblasts exhibited a reduced oxidative mtDNA damage repair capacity, altered mtDNA packaging, and increased type I interferon responses [40]. Therefore, it may be important to further understand how TFAM broadly affects inflammation‐associated tumorigenesis for the establishment of therapies and screening strategies in patients with IBD. It was found in the present study that the expression of TFAM was downregulated in active IBD and negatively associated with disease activity. Considering the critical role of TFAM in mitochondrial biogenesis, this finding might partially explain the recent reports of mitochondrial gene expression and activity being downregulated in human and rat colitis samples [9, 10, 11].
The intestinal epithelium is in a constant state of turnover, which requires a considerable supply of energy. The present results demonstrated that TFAM knockout, specifically in IECs, aggravated DSS‐induced colitis and promoted AOM/DSS‐induced CAC in mice. It was also found that TFAM promoted the proliferation of IEC by increasing mitochondrial biogenesis and activity. Of note, Boone et al. [41] demonstrated that the altered mitochondrial oxidative phosphorylation activity influenced intestinal inflammation in models of experimental colitis using strains of conplastic mice, which have identical nuclear but diverse mitochondrial genomes. Similarly, Fratila et al. [42] reported that mucosal healing also resulted in an improved mitochondrial structure in the IECs of patients with UC. The present study suggested that the increased regeneration of the intestinal epithelia through increased mitochondrial function may be a key factor in combating intestinal inflammation.
Since mitochondrial dysfunction contributes to the metabolic reprogramming in tumor cells [43], TFAM has attracted considerable attention in the cancer research community. The initial experiments leading to the discovery of TFAM are based on its ability to enhance the malignant behavior of cancer cells [44, 45, 46, 47]. Consistently, these results showed that TFAM was upregulated in CAC tissues and contributed to CRC cell growth both in vitro and in vivo by increasing mitochondrial biogenesis and activity. Similarly, it has been reported that mitochondrial metabolism is essential for KRAS‐induced lung cancer cell proliferation and tumor formation [48]. However, the role of TFAM in the CRC mouse model using APC Min/+ mice is controversial. Wen et al. [49] found that TFAM deletion in IECs significantly inhibited the activation of Wnt signaling and tumorigenesis in APC‐driven mouse models of colon cancer. By contrast, Woo et al. [23] found that heterozygous TFAM whole‐body knockout increased the number and growth of tumors in the small intestine of APC Min/+ mice. A possible explanation for this is that the deletion of TFAM expression in other tissue types may contribute to the different phenotypes observed.
Previous studies have demonstrated that IL‐6 and activated STAT3 were increased in both CD and UC patients at diagnosis, and remains elevating to a very similar degree in treated CD patients with active disease [50, 51]. IL‐6‐deficient mice formed fewer and smaller adenomas following treatment with AOM/DSS compared with WT mice, and fewer myeloid cells were recruited to the IL‐6‐deficient colons following DSS administration [52]. In the present study, a novel target of this signaling was found to be involved in colitis, where the expression of TFAM was significantly downregulated by the IL‐6/STAT3/miR‐23b signaling pathway. Similarly, Wang et al. [53] have shown that the IL‐6/STAT3 signaling pathway activated miR‐23a to suppress gluconeogenesis in hepatocellular carcinoma. Furthermore, miR‐23b has also been found to directly regulate the expression of TFAM by binding to the 3’‐untranslated region in glioma cells [54].
Of note, the present study showed that the downregulation of TFAM in IECs under inflammatory conditions was reversed by the activation of the β‐catenin/c‐Myc axis in colon tumor cells. It was further confirmed that the expression of TFAM was considerably higher in CAC tissues and positively correlated with the protein expression levels of β‐catenin and c‐Myc. In fact, activated mutations in components of the Wnt/β‐catenin pathway are common in both CAC and sporadic CRC. A previous comprehensive genomic analysis showed that APC was one of the top three mutant genes in CAC and stimulated β‐catenin stabilization by inhibiting its proteasome‐dependent degradation [37]. In addition, β‐catenin may serve as a signaling hub in CAC progression. The other top mutant genes P53 and KRAS have also been reported to induce β‐catenin accumulation in human cancer cells [55, 56].
We acknowledge several limitations in the present study. First, since CAC cell model is lacking, the effects of TFAM on tumor cells were only measured in sporadic colon cancer cells. Second, small sample size of normal cadaveric colonic tissues from healthy controls were used. Third, we only investigated short‐term effects of TFAM knockout on intestinal inflammation in the animal model. Therefore, further investigations are necessary to confirm the association between TFAM and CAC.
5. CONCLUSIONS
In combination, these data revealed the opposite roles of TFAM in the initiation and progression of CAC, indicating that TFAM switches from a tumor suppressor in the premalignant stages of tumorigenesis to an oncogene at later stages of disease. The present data suggested that measuring TFAM status may serve as a clinically useful tool to identify patients at risk for colon dysplasia or cancer. In addition, TFAM may also be a prognostic biomarker and pharmacological target for advanced CAC.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the ethics committee of FMMU. Written informed consent was obtained by all participants. The animal study was carried out in compliance with the guidance suggestion of Animal Care Committee of the FMMU (permit number: IACUC‐20171207).
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
FUNDING
This work was supported by National Natural Science Foundation of China (82072722, 81830070, 81772935, and 81672340) and State Key Laboratory of Cancer Biology Project (CBSKL2019ZZ26).
AUTHOR CONTRIBUTIONS
SY and XH were involved in the acquisition, analysis, and interpretation of the data and drafting of the manuscript. SG was involved in bioinformatics analysis. JZ, DW, TG, YZ and GW were involved in the analysis and interpretation of data. CJ, ZY, YW and NW were involved in tissue sample collection. QH and JX were involved in involved in the conception and design of the study, securing research funding, acquiring, analyzing and interpreting the data, and critically revising the manuscript and overall study supervision. All authors read and approved the final manuscript.
Supporting information
SUPPORTING INFORMATION
SUPPORTING INFORMATION
ACKNOWLEDGMENTS
The authors thank Professor Yongzhan Nie (Xijing Hospital of Digestive Diseases, FMMU) and Professor Jian Zhang (Department of Biochemistry and Molecular Biology, FMMU) for donating the experimental mice.
Yang S, He X, Zhao J, Wang D, Guo S, Gao T, et al. Mitochondrial transcription factor A plays opposite roles in the initiation and progression of colitis‐associated cancer. Cancer Commun. 2021;41:695–714. 10.1002/cac2.12184
Contributor Information
Jinliang Xing, Email: xingjl@fmmu.edu.cn.
Qichao Huang, Email: huangqichao@fmmu.edu.cn.
DATA AVAILABILITY STATEMENT
All data in our study are available upon request.
REFERENCES
- 1.Rhodes JM, Campbell BJ. Inflammation and colorectal cancer: Ibd‐associated and sporadic cancer compared. Trends Mol Med. 2002;8(1):10–6. 10.1016/s1471-4914(01)02194-3. [DOI] [PubMed] [Google Scholar]
- 2.Viscido A, Bagnardi V, Sturniolo GC, Annese V, Frieri G, D'Arienzo A, et al. Survival and causes of death in italian patients with ulcerative colitis. A gisc nationwide study. Dig Liver Dis. 2001;33(8):686–92. 10.1016/s1590-8658(01)80046-3. [DOI] [PubMed] [Google Scholar]
- 3.Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in ibd: Potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol. 2009;6(5):297–305. 10.1038/nrgastro.2009.44. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe T, Konishi T, Kishimoto J, Kotake K, Muto T, Sugihara K, et al. Ulcerative colitis‐associated colorectal cancer shows a poorer survival than sporadic colorectal cancer: A nationwide japanese study. Inflamm Bowel Dis. 2011;17(3):802–8. 10.1002/ibd.21365. [DOI] [PubMed] [Google Scholar]
- 5.Asquith M, Powrie F. An innately dangerous balancing act: Intestinal homeostasis, inflammation, and colitis‐associated cancer. J Exp Med. 2010;207(8):1573–7. 10.1084/jem.20101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blander JM. On cell death in the intestinal epithelium and its impact on gut homeostasis. Curr Opin Gastroenterol. 2018;34(6):413–9. 10.1097/MOG.0000000000000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong L, Neuzil J. Targeting mitochondria as an anticancer strategy. Cancer Commun (Lond). 2019;39(1):63. 10.1186/s40880-019-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Missiroli S, Genovese I, Perrone M, Vezzani B, Vitto VAM, Giorgi C. The role of mitochondria in inflammation: From cancer to neurodegenerative disorders. J Clin Med. 2020;9(3): 10.3390/jcm9030740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haberman Y, Karns R, Dexheimer PJ, Schirmer M, Somekh J, Jurickova I, et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun. 2019;10(1):38. 10.1038/s41467-018-07841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reifen R, Levy E, Berkovich Z, Tirosh O. Vitamin a exerts its antiinflammatory activities in colitis through preservation of mitochondrial activity. Nutrition. 2015;31(11‐12):1402–7. 10.1016/j.nut.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Soderholm JD, Olaison G, Peterson KH, Franzen LE, Lindmark T, Wiren M, et al. Augmented increase in tight junction permeability by luminal stimuli in the non‐inflamed ileum of crohn's disease. Gut. 2002;50(3):307–13. 10.1136/gut.50.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang G, Wang Q, Huang Q, Chen Y, Sun X, He L, et al. Upregulation of mtssb by interleukin‐6 promotes cell growth through mitochondrial biogenesis‐mediated telomerase activation in colorectal cancer. Int J Cancer. 2019;144(10):2516–28. 10.1002/ijc.31978. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, He S, Zhu X, Qiao W, Zhang J. High copy number of mitochondrial DNA predicts poor prognosis in patients with advanced stage colon cancer. Int J Biol Markers. 2016;31(4):e382–8. 10.5301/jbm.5000211. [DOI] [PubMed] [Google Scholar]
- 14.Kaldma A, Klepinin A, Chekulayev V, Mado K, Shevchuk I, Timohhina N, et al. An in situ study of bioenergetic properties of human colorectal cancer: The regulation of mitochondrial respiration and distribution of flux control among the components of atp synthasome. Int J Biochem Cell Biol. 2014;55171–86. 10.1016/j.biocel.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Zheng J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (review). Oncol Lett. 2012;4(6):1151–7. 10.3892/ol.2012.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun X, Zhan L, Chen Y, Wang G, He L, Wang Q, et al. Increased mtdna copy number promotes cancer progression by enhancing mitochondrial oxidative phosphorylation in microsatellite‐stable colorectal cancer. Signal Transduct Target Ther. 2018;38. 10.1038/s41392-018-0011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramachandran A, Basu U, Sultana S, Nandakumar D, Patel SS. Human mitochondrial transcription factors tfam and tfb2m work synergistically in promoter melting during transcription initiation. Nucleic Acids Res. 2017;45(2):861–74. 10.1093/nar/gkw1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyonnais S, Tarres‐Sole A, Rubio‐Cosials A, Cuppari A, Brito R, Jaumot J, et al. The human mitochondrial transcription factor a is a versatile g‐quadruplex binding protein. Sci Rep. 2017;743992. 10.1038/srep43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, Dierckx A, Wanrooij PH, Wanrooij S, Larsson NG, Wilhelmsson LM, et al. Mammalian transcription factor a is a core component of the mitochondrial transcription machinery. Proc Natl Acad Sci U S A. 2012;109(41):16510–5. 10.1073/pnas.1119738109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor a is necessary for mtdna maintenance and embryogenesis in mice. Nat Genet. 1998;18(3):231–6. 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 21.Vernochet C, Mourier A, Bezy O, Macotela Y, Boucher J, Rardin MJ, et al. Adipose‐specific deletion of tfam increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab. 2012;16(6):765–76. 10.1016/j.cmet.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo J, Zheng L, Liu W, Wang X, Wang Z, Wang Z, et al. Frequent truncating mutation of tfam induces mitochondrial DNA depletion and apoptotic resistance in microsatellite‐unstable colorectal cancer. Cancer Res. 2011;71(8):2978–87. 10.1158/0008-5472.CAN-10-3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woo DK, Green PD, Santos JH, D'Souza AD, Walther Z, Martin WD, et al. Mitochondrial genome instability and ros enhance intestinal tumorigenesis in apc(min/+) mice. Am J Pathol. 2012;180(1):24–31. 10.1016/j.ajpath.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parang B, Barrett CW, Williams CS. Aom/dss model of colitis‐associated cancer. Methods Mol Biol. 2016;1422297–307. 10.1007/978-1-4939-3603-8_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang YJ, Kim WK, Han DH, Lee K, Ko G. Lactobacillus fermentum species ameliorate dextran sulfate sodium‐induced colitis by regulating the immune response and altering gut microbiota. Gut Microbes. 2019;10(6):696–711. 10.1080/19490976.2019.1589281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JJ, Shajib MS, Manocha MM, Khan WI. Investigating intestinal inflammation in dss‐induced model of ibd. J Vis Exp. 2012;(60): 10.3791/3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xing J, Chen M, Wood CG, Lin J, Spitz MR, Ma J, et al. Mitochondrial DNA content: Its genetic heritability and association with renal cell carcinoma. J Natl Cancer Inst. 2008;100(15):1104–12. 10.1093/jnci/djn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ros‐modulated coordinated regulation of the nfkb and tp53 pathways. Autophagy. 2016;12(6):999–1014. 10.1080/15548627.2016.1166318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song M, Chen FF, Li YH, Zhang L, Wang F, Qin RR, et al. Trimetazidine restores the positive adaptation to exercise training by mitigating statin‐induced skeletal muscle injury. J Cachexia Sarcopenia Muscle. 2018;9(1):106–18. 10.1002/jcsm.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez‐Canales M, Martinez‐Galero E, Nava‐Torres AD, Sanchez‐Torres LE, Garduno‐Siciliano L, Canales‐Martinez MM, et al. Anti‐inflammatory and antioxidant activities of the methanolic extract of cyrtocarpa procera bark reduces the severity of ulcerative colitis in a chemically induced colitis model. Mediators Inflamm. 2020;20205062506. 10.1155/2020/5062506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The nlrp3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32(3):379–91. 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu LL, Lyu YQ, Jiang HT, Shan T, Zhang JB, Li QR, et al. Effect of alemtuzumab on intestinal intraepithelial lymphocytes and intestinal barrier function in cynomolgus model. Chin Med J (Engl). 2015;128(5):680–6. 10.4103/0366-6999.151675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren T, Wang J, Zhang H, Yuan P, Zhu J, Wu Y, et al. Mcur1‐mediated mitochondrial calcium signaling facilitates cell survival of hepatocellular carcinoma via reactive oxygen species‐dependent p53 degradation. Antioxid Redox Signal. 2018;28(12):1120–36. 10.1089/ars.2017.6990. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Huang Q, Long X, Guo X, Sun X, Jin X, et al. Mitochondrial elongation‐mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene. 2017;36(34):4901–12. 10.1038/onc.2017.98. [DOI] [PubMed] [Google Scholar]
- 35.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13(5):589–98. 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsumoto H, Kimura T, Haga K, Kasahara N, Anton P, McGowan I. Effective in vivo and ex vivo gene transfer to intestinal mucosa by vsv‐g‐pseudotyped lentiviral vectors. BMC Gastroenterol. 2010;1044. 10.1186/1471-230X-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker AM, Cross W, Curtius K, Al Bakir I, Choi CR, Davis HL, et al. Evolutionary history of human colitis‐associated colorectal cancer. Gut. 2019;68(6):985–95. 10.1136/gutjnl-2018-316191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho K, Shin HW, Kim YI, Cho CH, Chun YS, Kim TY, et al. Mad1 mediates hypoxia‐induced doxorubicin resistance in colon cancer cells by inhibiting mitochondrial function. Free Radic Biol Med. 2013;60201–10. 10.1016/j.freeradbiomed.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 39.Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the sting pathway lead to renal inflammation and fibrosis. Cell Metab. 2019;30(4):784–99 e785. 10.1016/j.cmet.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.West AP, Khoury‐Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–7. 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boone DL, Teitell MA. The other genome: Mitochondrial DNA and protection from experimental colitis. Gastroenterology. 2013;145(5):933–5. 10.1053/j.gastro.2013.09.034. [DOI] [PubMed] [Google Scholar]
- 42.Fratila OC, Craciun C. Ultrastructural evidence of mucosal healing after infliximab in patients with ulcerative colitis. J Gastrointestin Liver Dis. 2010;19(2):147–53. [PubMed] [Google Scholar]
- 43.Tang Z, Xu Z, Zhu X, Zhang J. New insights into molecules and pathways of cancer metabolism and therapeutic implications. Cancer Commun (Lond). 2021;41(1):16–36. 10.1002/cac2.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Araujo LF, Siena ADD, Placa JR, Brotto DB, Barros, II, Muys BR, et al. Mitochondrial transcription factor a (tfam) shapes metabolic and invasion gene signatures in melanoma. Sci Rep. 2018;8(1):14190. 10.1038/s41598-018-31170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiao L, Ru G, Mao Z, Wang C, Nie Z, Li Q, et al. Mitochondrial DNA depletion, mitochondrial mutations and high tfam expression in hepatocellular carcinoma. Oncotarget. 2017;8(48):84373–83. 10.18632/oncotarget.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie D, Wu X, Lan L, Shangguan F, Lin X, Chen F, et al. Downregulation of tfam inhibits the tumorigenesis of non‐small cell lung cancer by activating ros‐mediated jnk/p38mapk signaling and reducing cellular bioenergetics. Oncotarget. 2016;7(10):11609–24. 10.18632/oncotarget.7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin CS, Liu LT, Ou LH, Pan SC, Lin CI, Wei YH. Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol Rep. 2018;39(1):316–30. 10.3892/or.2017.6087. [DOI] [PubMed] [Google Scholar]
- 48.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ros generation are essential for kras‐mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107(19):8788–93. 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wen YA, Xiong X, Scott T, Li AT, Wang C, Weiss HL, et al. The mitochondrial retrograde signaling regulates wnt signaling to promote tumorigenesis in colon cancer. Cell Death Differ. 2019;26(10):1955–69. 10.1038/s41418-018-0265-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carey R, Jurickova I, Ballard E, Bonkowski E, Han X, Xu H, et al. Activation of an il‐6:Stat3‐dependent transcriptome in pediatric‐onset inflammatory bowel disease. Inflamm Bowel Dis. 2008;14(4):446–57. 10.1002/ibd.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kusugami K, Fukatsu A, Tanimoto M, Shinoda M, Haruta J, Kuroiwa A, et al. Elevation of interleukin‐6 in inflammatory bowel disease is macrophage‐ and epithelial cell‐dependent. Dig Dis Sci. 1995;40(5):949–59. 10.1007/BF02064182. [DOI] [PubMed] [Google Scholar]
- 52.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. Il‐6 and stat3 are required for survival of intestinal epithelial cells and development of colitis‐associated cancer. Cancer Cell. 2009;15(2):103–13. 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang B, Hsu SH, Frankel W, Ghoshal K, Jacob ST. Stat3‐mediated activation of microrna‐23a suppresses gluconeogenesis in hepatocellular carcinoma by down‐regulating glucose‐6‐phosphatase and peroxisome proliferator‐activated receptor gamma, coactivator 1 alpha. Hepatology. 2012;56(1):186–97. 10.1002/hep.25632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang J, Yang J, Wang Z, Wu G, Liu F. Tfam is directly regulated by mir‐23b in glioma. Oncol Rep. 2013;30(5):2105–10. 10.3892/or.2013.2712. [DOI] [PubMed] [Google Scholar]
- 55.Cagatay T, Ozturk M. P53 mutation as a source of aberrant beta‐catenin accumulation in cancer cells. Oncogene. 2002;21(52):7971–80. 10.1038/sj.onc.1205919. [DOI] [PubMed] [Google Scholar]
- 56.Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, et al. Apc and oncogenic kras are synergistic in enhancing wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131(4):1096–1109. 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
SUPPORTING INFORMATION
Data Availability Statement
All data in our study are available upon request.