

Abstract
Notch signaling primarily determines T-cell fate. However, the molecular mechanisms underlying the maintenance of T-lineage potential in pre-thymic progenitors remain unclear. Here, we established two murine Ebf1-deficient pro-B cell lines, with and without T-lineage potential. The latter expressed lower levels of Lmo2; their potential was restored via ectopic expression of Lmo2. Conversely, the CRISPR/Cas9-mediated deletion of Lmo2 resulted in the loss of the T-lineage potential. Introduction of Bcl2 rescued massive cell death of Notch-stimulated pro-B cells without efficient LMO2-driven Bcl11a expression but was not sufficient to retain their T-lineage potential. Pro-B cells without T-lineage potential failed to activate Tcf7 due to DNA methylation; Tcf7 transduction restored this capacity. Moreover, direct binding of LMO2 to the Bcl11a and Tcf7 loci was observed. Altogether, our results highlight LMO2 as a crucial player in the survival and maintenance of T-lineage potential in T-cell progenitors via the regulation of the expression of Bcl11a and Tcf7.
Research organism: Mouse
Introduction
Hematopoietic stem cells (HSCs) have the ability to self-renew and differentiate into all blood cell types. HSCs begin to differentiate into various lineage cells, gradually losing their potential to become their descendants, hematopoietic progenitor cells (HPCs), and then differentiate into mature blood cells (Doulatov et al., 2012; Kosan and Godmann, 2016). This sequence of processes has long been imagined as a ball rolling down a valley track (Goldberg et al., 2007). Cell fate decisions in hematopoietic cells are controlled by continuous interactions between environmental influences and intrinsic cellular mechanisms, such as transcription factor networks and epigenetic regulation (Wilson et al., 2009).
In the process of specification from HPCs, only the T-cell lineage requires a specialized environment of the thymus, where the immigrant cells receive Notch signaling induced by the interaction of Notch1 on the immigrant cells and a Notch ligand, delta-like 4 (Dll4), on the thymic epithelium, which determines their fate to the T-cell lineage (Radtke et al., 1999; Hozumi et al., 2008; Koch et al., 2008). In addition, several transcription factors contribute to the acquisition of T-cell identity, including TCF1 (encoded by Tcf7), basic helix-loop-helix (bHLH) factors, E2A and HEB, PU.1, GATA3, Bcl11b, and Runx family members, all of which are essential for T-cell development in the thymus (Hosokawa and Rothenberg, 2021; Hosokawa et al., 2021a; Hosokawa et al., 2021b). Among them, one of the earliest Notch-activated genes, Tcf7, plays a critical role in Notch-mediated initiation of the T-lineage program (Weber et al., 2011; Johnson et al., 2018). The coordinated action of these environmental and intrinsic factors completes the commitment to T-cell lineage in double-negative (DN; CD4−CD8−) thymocytes at the transition from the DN2 to DN3 stages (Yui and Rothenberg, 2014; Hosokawa and Rothenberg, 2021). However, it remains unclear how the potential of the thymic immigrant cells is maintained to initiate their differentiation program toward the T-cell lineage.
LMO2 gene is located near the breakpoint of the chromosomal translocation t(11;14) (p13;q11) in human T-ALL (Boehm et al., 1991; Royer-Pokora et al., 1991), and LMO2 has been thought to function as a bridging factor in large transcriptional complexes with several DNA-binding and adaptor proteins (TAL1/SCL, E2A, GATA1, and Ldb1) (Wadman et al., 1997; Grütz et al., 1998; El Omari et al., 2013; Layer et al., 2016). Although deletion of Lmo2 in mice causes embryonic lethality due to embryonic erythropoiesis deficiency around 10 days post-fertilization (Yamada et al., 1998), conditional disruption of Lmo2 in T-lineage committed pro-T stages results in normal T-cell development in the thymus (McCormack et al., 2003), indicating that LMO2 is dispensable for T-cell development after T-lineage commitment. However, it remains unclear whether LMO2 plays an important role before T-lineage commitment, including the maintenance of T-lineage potential in pre-thymic progenitors.
It is well known that the recent gene therapy trials using retrovirus-mediated introduction of the common cytokine receptor gamma chain (γc; CD132) in X-linked severe combined immunodeficiency (X-SCID) patients resulted in the development of T-cell leukemia due to retroviral insertion at the LMO2 locus (Hacein-Bey-Abina et al., 2003; Hacein-Bey-Abina et al., 2008; Howe et al., 2008). Moreover, transgenic overexpression of Lmo2 in various murine tissues only results in T-cell leukemia, indicating its involvement exclusively in T-cell malignancies when abnormally expressed (Neale et al., 1995). On the other hand, Lmo2 is more highly expressed in HSCs and HPCs than in mature blood cells (Yoshida et al., 2019) and is known to induce reprogramming of HSCs from differentiated blood cells (Riddell et al., 2014) and fibroblasts (Batta et al., 2014; Vereide et al., 2014), suggesting that LMO2 actively contributes to the maintenance of the undifferentiated state of HSCs. Consistently, aberrant induction of Lmo2 in thymocytes generates self-renewing cells that retain the capacity for T-cell differentiation (McCormack et al., 2010; Cleveland et al., 2013). Thus, it is not clear why Lmo2 overexpression causes only T-cell malignancies and how LMO2 contributes to the maintenance of an undifferentiated state.
In attempts to establish HPC lines with T-cell differentiation potential, it has been found that pro-B cells without transcription factors essential for early B-cell development, including Pax5, Ebf1, and E2A (encoded by Tcf3), can be maintained in vitro with an HPC-like phenotype, retaining T-cell potential on OP9 stromal cells in the presence of IL-7 (Nutt et al., 1999; Rolink et al., 1999; Ikawa et al., 2004; Pongubala et al., 2008). In this study, based on their protocol, we established pro-B cell lines, called pro-B(+) cells, that retain T-cell potential on OP9 stromal cells. In contrast, proliferating pro-B cells, named pro-B(−) cells, were also obtained in our originally established thymic stromal cells, TD7, which die abruptly immediately following activation of Dll4-mediated Notch signaling in vitro. Thereafter, we identified Lmo2 as a gene whose expression is downregulated in pro-B(−) cells compared to pro-B(+) cells and found that the forced expression of Lmo2 was sufficient for the pro-B(−) cells to reacquire the ability to differentiate into T-cell lineage by Notch signaling. Furthermore, LMO2 ensures survival of pro-B cells through the activation of the Bcl11a/Bcl2 pathway and contributes to maintaining the accessibility of the Tcf7 locus, which is one of the earliest Notch downstream targets in thymic immigrant cells. These epigenetic alterations could be mediated by direct binding of the transcriptional complex, including LMO2, to the target loci. Together with loss-of-function experiments, we demonstrate that LMO2 is significant for maintaining T-cell progenitors for the progression of the T-cell differentiation program initiated by Notch signaling.
Results
LMO2 has a crucial role in the maintenance of T-cell differentiation capacity in Ebf1-deficient pro-B cells
Using different stromal cells, OP9 and TD7, with fetal liver progenitor cells from Ebf1-deficient mice, we established two types of pro-B cell lines, with and without the ability to differentiate into T-cell lineages designated pro-B(+) and pro-B(−) cells, respectively (Figure 1—figure supplement 1A,B). Both the Ebf1-deficient pro-B lines grew robustly on OP9 cells with IL-7, Flt3L, and SCF (Figure 1A, upper panels) and expressed intermediate levels of B220 (Figure 1—figure supplement 1C). Pro-B(+) cells were able to initiate differentiation into the T-cell lineage, DN2 (CD25+CD44+), DN3 (CD25+CD44lo), and double-positive (DP) stages on Dll4-expressing OP9 (OP9-Dll4) cells in vitro (Figure 1A, lower right, Figure 1—figure supplement 1D), and mature into CD4 and CD8 T cells in vivo (Figure 1—figure supplement 1E), as shown previously (Pongubala et al., 2008). In contrast, pro-B(−) cells remained in the pro-B cell phenotype (CD25−CD44+B220int) and died with Dll4-mediated Notch stimulation, although the expression levels of the Notch receptors were comparable (Figure 1A, Figure 1—figure supplement 1F). To explore their different characteristics at the molecular level, we carried out comparative microarray analysis and identified the differentially expressed genes between pro-B(+) and pro-B(−) cells (Figure 1B, Supplementary file 1). We identified approximately 400 differentially expressed genes (FC > 10) between the pro-B(+) and pro-B(−) cells, and these genes were enriched for genes related to the ‘Hematopoietic cell lineage’ pathway (Figure 1—figure supplement 1G). Among them, the functional importance of Meis1, Hmga2, and Bcl11a has been reported in undifferentiated HPCs (Wong et al., 2007; Ariki et al., 2014; Nishino et al., 2008; Copley et al., 2013; Yu et al., 2012). However, the introduction of Meis1 and Hmga2 failed to overcome the defective T-cell differentiation in the presence of Notch signaling, and that of Bcl11a was highly toxic in pro-B(−) cells (data not shown). Lmo2 has also been shown to be linked to the reprogramming of lineage-committed blood cells or mesenchymal cells to the induced HSCs (Riddell et al., 2014; Batta et al., 2014; Vereide et al., 2014). We found that the expression levels of Lmo2 mRNA and protein were approximately threefold higher in pro-B(+) than pro-B(−) cells (Figure 1B,C), and enforced expression of Lmo2 markedly provided their capacity to differentiate into T-cell lineage following Notch stimulation in vitro (Figure 1D). Therefore, these results suggest that LMO2 plays a pivotal role in the maintenance of the capacity to differentiate into T-cell lineage driven by Notch signaling in Ebf1-deficient pro-B cells.
Figure 1. LMO2 is critical for the maintenance of T-cell differentiation potential in Ebf1-deficient pro-B cells.
(A) Establishment of Ebf1-deficient pro-B cell lines with or without differentiation potential to the T-cell lineage. Lineage markers (CD19, Gr1, TER119, NK1.1)-negative, c-kit-positive cells in Ebf1−/− FL were cultured on TD7 or OP9 cells, and Ebf1-deficient pro-B cell lines were established. Stably growing pro-B cells with or without T-cell potential (pro-B(+) or pro-B(−)) were cultured on OP9-Mock (Mock) or OP9-Dll4 (Dll4) cells with Flt3L, SCF, and IL7 for 6 days and analyzed for the expression of CD44 and CD25 (right panels) in the lymphoid cell gate (FSC vs. SSC, left panels) by flow cytometry. The numbers in the profiles indicate the relative percentages in each corresponding quadrant or fraction. Numbers of CD25+ cells (fold expansion/input) are shown with standard deviation (SD) (right). Statistical analysis was performed using the two-tailed Student’s t-test. **p<0.01. Data are representative of three independent experiments with similar results. (B) Reverse transcription (RT)-quantitative PCR (qPCR) detection of Meis1, Hmga2, Bcl11a, or Lmo2 transcripts in pro-B(−) (closed columns) and pro-B(+) (open columns) cells. Data represent the mean values of three independent biological replicates, and all values are normalized to the expression of Actb. Error bars indicate SD. Three independent experiments were performed, and similar results were obtained. (C) Representative intracellular staining profiles of LMO2 and c-Myc in pro-B(−) (open blue line) and pro-B(+) (open red line) cells are shown. Closed lines (orange) represent staining with control rabbit mAb of pro-B(−) and pro-B(+), which were completely merged. The average mean fluorescent intensity (MFI) of LMO2 is shown with SD (right). A two-tailed Student’s t-test was used for statistical analysis. **p<0.01. Three independent experiments were performed with similar results. (D) Introduction of Lmo2 is sufficient to maintain the T-cell differentiation potential in pro-B cells. Empty vector- or Lmo2-transduced pro-B(−) cells (pro-B(−)/Mock or pro-B(−)/LMO2) were cultured on OP9-Dll4 for 6 days and analyzed for the expression of CD44 and CD25 (right panels) in lymphoid cell gate (left panels) and rat CD2+ (lentivirus-infected) CD45+ fraction. Numbers of CD25+ cells (relative expansion/input) are shown with SD (right). **p<0.01 by two-sided Student’s t-test. Six independent experiments were performed with similar results.

Figure 1—figure supplement 1. Characterization of pro-B(+) and pro-B(−) cells.

Disruption of Lmo2 in pro-B(+) cells leads to differentiation arrest following stimulation by Notch signaling
To confirm the necessity of LMO2 for the maintenance of the differentiation capacity into T-cell lineage, we tested the effect of Lmo2 disruption on the developmental potential of T-cell lineage in bone marrow (BM)-derived progenitor cells and pro-B(+) cells. BM progenitors from Cas9;Bcl2 Tg mice were infected with a bicistronic retroviral vector carrying an sgRNA against Lmo2 with the human nerve growth factor receptor (hNGFR) or control sgRNA against luciferase. Two days after sgRNA introduction, the cells were transferred onto an OP9-Dll1 monolayer to initiate T-lineage differentiation (Figure 2—figure supplement 1A). The ability sgLMO2-treated progenitors to differentiate into the T-lineage was comparable to that of control (Figure 2—figure supplement 1B). Next, Cas9-introduced pro-B(+) cells were transduced with sgRNA against LMO2, which caused specific loss of LMO2 protein 4 days after retroviral infection as detected by immunoblotting (Figure 2—figure supplement 1C). Five or ten days after sgLMO2 transduction, the cells were transferred onto a OP9-Dll4 monolayer to induce Notch-dependent T-lineage differentiation (Figure 2A). Lmo2-deficient pro-B(+) cells progressed to the DN2 stage (CD25+CD44+) as well as control cells on day 5, however, developmental arrest was observed ten days after sgLMO2 transduction (Figure 2B). These results indicate that LMO2 is necessary for pro-B(+) cells to maintain their ability to differentiate into T-cell lineage, and loss of their differentiation potential took several days (~10 days) after Lmo2 disruption. Thus, the process may require loss of LMO2-mediated slower time-scale transcriptional changes, including histone modifications, chromatin remodeling, and DNA methylation.
Figure 2. Loss of Lmo2 leads to the differentiation arrest in pro-B(+) cells.
(A) An experimental scheme for the deletion of Lmo2 using the CRISPR/Cas9 system in pro-B cell lines is shown. (B) Retroviral vectors encoding sgRNA against luciferase (sgCont.) or LMO2 (sgLMO2) were introduced into Cas9-expressing (GFP+) pro-B(+) cells. Five (cultured on OP9-Mock for 5 days, left panels) or 10 days (cultured on OP9-Mock for 10 days, right panels) after co-cultured on OP9-Mock cells following the infection, pro-B cells were cultured again on OP9-Mock (Mock) or OP9-Dll4 (Dll4) stromal cells for 3 days. GFP+hNGFR+ sgRNA-transduced cells were gated and analyzed for CD44 and CD25 expression (left). The percentages and numbers of CD25+ cells among GFP+hNGFR+ sgRNA-transduced cells, cultured on OP9-Dll4, are shown with SD (right). The data represent the mean values of three independent biological replicates. Each value is indicated by a closed circle. **p<0.01 by two-sided Student’s t-test.

Figure 2—figure supplement 1. CRISPR/Cas9-mediated deletion of Lmo2 in BM progenitors.

LMO2 regulates survival of Ebf1-deficient pro-B cells via Bcl11a/Bcl2 pathway
To identify the downstream targets of LMO2, we compared the gene expression profiles of pro-B(−), Lmo2-introduced pro-B(−) (pro-B(−)/LMO2), Meis1-introduced pro-B(−) (pro-B(−)/Meis1), and pro-B(+) cells (Figure 3A). We found that Lmo2-introduction restored the expression of Bcl11a in pro-B(−) cells, whereas Meis1 further downregulated Bcl11a expression. The expression of other potential target genes, including Meis1 and Hmga2, were also changed following Lmo2- and Meis1-transduction, which failed to restore T-lineage differentiation capacity in pro-B(−) cells (Figure 3A). Bcl11a is known to regulate the survival of lymphoid progenitors via induction of the anti-apoptotic gene, Bcl2 (Yu et al., 2012). Therefore, we examined the roles of the Bcl11a/Bcl2 pathway in the survival of pro-B(−) cells and their potential to differentiate into T-lineage cells. Transduction of Bcl2 significantly protected pro-B(−) cells from cell death, before and after Notch stimulation (Figure 3B, Figure 3—figure supplement 1). Conversly, overexpression of the oncogene, Bcl11a, was highly toxic in pro-B cells. Importantly, Bcl2-introduced pro-B(−) cells (pro-B(−)/Bcl2) still failed to progress into the CD25+ DN2 stage (Figure 3C). Thus, these results suggest that LMO2 would regulate survival of pro-B(−) cells via the Bcl11a/Bcl2 pathway, and other mechanisms contribute to the maintenance of the potential to differentiate into T-cell lineage.
Figure 3. LMO2 regulates survival of Ebf1-deficient pro-B cells via Bcl11a/Bcl2 pathway.
(A) RT-qPCR detection of Meis1, Hmga2, and Bcl11a transcripts in Lmo2- (red column), Meis1-transfected (green column) pro-B(−) cells, parent pro-B(−) (black column), and pro-B(+) (white column) cells, as shown in Figure 1B. Two independent experiments were performed with similar results. (B) Overexpression of Bcl2 improves cell survival of pro-B(−) cells. Pro-B(+) cells (open column) and empty vector (mock) or human BCL2 (Bcl2)-transduced pro-B(−) cells were cultured on OP9-Mock or OP9-Dll4 for 2 days. After culturing, the dead cells were detected by staining for Annexin V and 7-AAD in CD45+ and hNGFR+ (retrovirus-infected) cell populations (Figure 3—figure supplement 1). The cell death index was calculated as the difference in the percentage of dead cells in pro-B cells after co-culturing with OP9-Mock and OP9-Dll4. The data represent the mean values of three independent biological replicates with SD. **p<0.01 by two-sided Student’s t-test. (C) Bcl2 overexpression does not provide differentiation potential in pro-B(−) cells. Pro-B(−) or pro-B(+) cells with human BCL2 (pro-B(−)/Bcl2, pro-B(+)/Bcl2) were cultured on OP9-Dll4 as shown in Figure 1D. After culturing, the live cells were analyzed for the expression of CD44 and CD25 (left). The data represent the mean values of percentages of CD25+ cells in three independent biological replicates with SD (right). ***p<0.001 by two-sided Student’s t-test.

Figure 3—figure supplement 1. Overexpression of Bcl2 improves the cell survival in pro-B(−) cells.

LMO2 is required for the activation of Tcf7 after Notch signaling
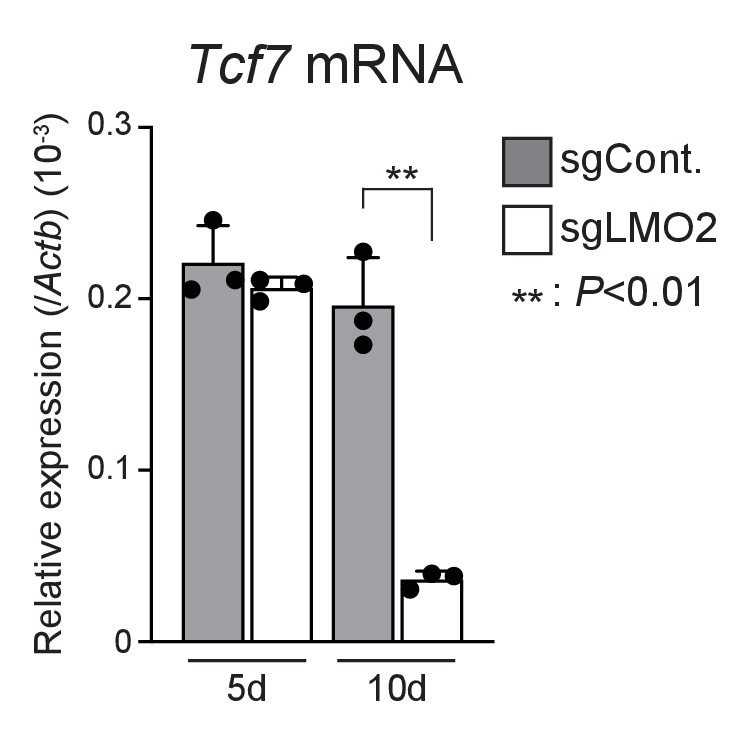
Our comparative microarray analysis of pro-B(−) and pro-B(+) cells showed that expression of Tcf7 (encoding TCF1), one of the most important and the earliest Notch target genes in thymic immigrant cells, was weakly detected in pro-B(+) cells; however, its level was approximately 100-fold lower in pro-B(−) cells. Therefore, we next examined the expression kinetics of Tcf7 in pro-B(+)/Bcl2 and pro-B(−)/Bcl2 cells that survive after Notch stimulation but fail to differentiate into T-cell lineages. The upregulation of Tcf7 expression observed in pro-B(+)/Bcl2 cells after Notch stimulation was significantly abrogated in pro-B(−)/Bcl2 cells (Figure 4A). In contrast, another Notch-regulated gene, Gata3, was upregulated by Notch stimulation but modestly lower in pro-B(−)/Bcl2 cells compared to that in pro-B(+)/Bcl2 cells (Figure 4A). These results were further confirmed at the protein level using pro-B(−)/Bcl2 and pro-B(+)/Bcl2 cells at 7 days post-Notch stimulation (Figure 4B). In addition, there was a decrease in Tcf7 expression in Lmo2-deficient pro-B(+) cells (Figure 2) after 10 days but not after 5 days of sgRNA transduction (Figure 4—figure supplement 1). We then tested the role of Tcf7 using a gain-of-function strategy and found that introduction of Tcf7 clearly rescued the defect in the differentiation capacity of pro-B(−)/Bcl2 into T-cell lineage (DN2 stage), after Notch signaling was provided (Figure 4C). These results indicate that LMO2 expression in T-cell progenitors plays a crucial role in the activation of Tcf7 once the progenitors migrate into the thymus and are stimulated by Notch signaling.
Figure 4. LMO2 is required for activation of Tcf7 after Notch signaling.
(A) Expression levels of Tcf7 and Gata3 in pro-B(−) and pro-B(+) cells with exogenous BCL2, at 0–5 days after the culture on OP9-Dll4, were analyzed by RT-qPCR. The relative expression (/Actb) is shown with SD. *p<0.05, **p<0.01 by two-sided Student’s t-test. (B) Intracellular staining of TCF1 or GATA3 in pro-B(+)/Bcl2 (upper panels) and pro-B(−)/Bcl2 (lower panels) was performed at day 7 after Notch stimulation; representative expression profiles of CD44 and CD25 are also shown (left). Results are representative of three independent experiments. (C) Introduction of Tcf7 provides differentiation potential for T-cell lineage to pro-B(−)/Bcl2. Pro-B(−)/Bcl2 cells were infected with either empty control or Tcf7-containing lentivirus, and the cells were co-cultured on OP9-Mock (upper panels) or OP9-Dll4 (lower panels) for 3 days. Lentivirus-infected cells were analyzed for the expression of CD44 and CD25. Three independent experiments were performed with similar results. The percentages of CD25+ cells are shown with SD (right). ***p<0.001 by two-sided Student’s t-test.

Figure 4—figure supplement 1. Down-regulation of Tcf7 in Lmo2-deficient pro-B(+) cells.
DNA methylation status of the Tcf7 locus is maintained by LMO2
Next, we examined the epigenetic status of the Tcf7 locus in pro-B cell lines. We observed that in pro-B(+) cells, the transcriptional start site (TSS) of the Tcf7 locus was highly enriched for the active histone mark H3K4-3Me. However, Lmo2 disruption caused a significant reduction in the H3K4-3Me marks 10 days post-sgRNA introduction (~30%), and a modest reduction in the H3K4-3Me levels after 5 days of Lmo2 deletion (~70%) (Figure 5—figure supplement 1). Moreover, we found a CpG island at the TSSs of Tcf7 (Figure 5A). Thus, we examined the DNA methylation levels of the CpG island at the TSSs of the Tcf7 locus in the pro-B cell lines. In pro-B(−) cells, the CpG island at the Tcf7 locus was highly methylated, in contrast to pro-B(+) cells that had a highly demethylated TSSs (77.6% vs. 29.8%) (Figure 5B). Moreover, enforced expression of Lmo2 in pro-B(−) cells induced demethylation of the CpG island at the TSSs (Figure 5B, pro-B(−)/LMO2). These results demonstrate that the epigenetic status of the Tcf7 locus in the progenitor cells is maintained by LMO2 in a transcriptionally poised chromatin state for quick responsiveness following Notch stimulation.
Figure 5. DNA methylation status of the Tcf7 locus is maintained by LMO2.
(A) A CpG island at the transcriptional start sites (TSSs) of the Tcf7 locus, which contains 25 potential CpG methylation sites. (B) The DNA methylation status of CpG island at the TSSs of the Tcf7 locus was determined by bisulfite sequencing in pro-B(−), pro-B(+), and LMO2-transduced pro-B(−) cells (pro-B(−)/LMO2). Bisulfite-converted genomic DNA around the TSSs of Tcf7 was amplified using PCR, and each PCR product was sequenced. The 25 horizontal circles each represent a CpG sequence derived from a single PCR product (17 clones from pro-B(−) cells, 9 clones from pro-B(+) cells, and 16 clones from pro-B(−)/LMO2 cells). Closed and open circles indicate methylated and demethylated CpG sites, respectively. The frequencies of the methylated CpGs are shown with SD. Data are based on two independent pooled experiments.

Figure 5—figure supplement 1. H3K4-3Me levels around the Tcf7 locus in Lmo2-deficient cells.

LMO2 directly binds to the Bcl11a and Tcf7 loci
Finally, we performed chromatin immunoprecipitation followed by deep-sequencing (ChIP-seq) analysis to identify genome-wide LMO2 occupancy sites in the pro-B(+) cells. We identified more than 1500 reproducible LMO2 binding peaks, and enrichment of Runx and ETS motifs that are frequently found in open chromatin regions of hematopoietic progenitors and pre-commitment pro-T cells (Yoshida et al., 2019; Ungerbäck et al., 2018; Figure 6A). The motif for a previously reported LMO2 interacting partner, the bHLH factors, was also coenriched (Figure 6A). Many of the bHLH-regulated genes, including Lyl1, Erg, and Hhex, were bound by LMO2 (Figure 6B). In addition, these motifs are highly relevant to the binding sites for major transcription factors, such as LMO2, in HPCs (Wilson et al., 2010). Moreover, two LMO2 binding peaks were detected in the downstream regions of the Bcl11a locus, one of the LMO2-sensitive genes (Figures 3A and 6B). Most importantly, a clear LMO2 peak was observed at the −35 kb upstream region of the Tcf7 locus. This binding site overlapped with RBPJ, a DNA-binding subunit of the Notch intracellular domain (NICD) transcriptional complex, binding sites at the Tcf7 locus, and co-occupied with Runx1 in pre-commitment DN1 cells (GSE148441, GSE110020) (Romero-Wolf et al., 2020; Hosokawa et al., 2018; Shin et al., 2021; Figure 6B, Figure 6—figure supplement 1). Taken together, LMO2 directly binds to the Bcl11a and Tcf7 loci and regulates their expression to maintain the ability of progenitors to differentiate into T-cell lineage.
Figure 6. LMO2 binds to the upstream region of the Tcf7 locus.
(A) LMO2 ChIP-seq analyses were performed using the pro-B(+) cell line. The top three enriched sequence motifs among the 1585 reproducible LMO2 peaks are shown. Data are based on ChIP-seq peaks scored as reproducible in two replicate samples. (B) ChIP-seq tracks showing two replicates of LMO2 binding profiles around the Lyl1, Erg, Hhex, Tcf7, and Bcl11a loci in pro-B(+) cell line with tracks for 1% input.

Figure 6—figure supplement 1. LMO2 binds to one of the RBPJ and Runx1 binding sites at the Tcf7 locus.

Discussion
Here, we demonstrate that LMO2 contributes to the differentiation capacity of T-cell lineage in Ebf1-deficient pro-B cells via activation of the Bcl11a/Bcl2 pathway, which is critical for cell survival, and maintenance of DNA methylation status of the Tcf7 locus. LMO2 directly binds to its target loci and enables the Tcf7 locus to achieve a state that is responsive to T-cell induction following Notch signaling.
We established our original thymic stromal cell line, TD7, in which HSCs differentiate into CD19+ B-lineage cells in the presence of IL-7 (Figure 1—figure supplement 1A) or into Thy1+CD25+ T-lineage cells upon receiving Notch signaling. To establish pro-B cell lines possessing pluripotency, we cultured HSCs from the fetal liver of Ebf1-deficient mice on TD7 or OP9 stromal cells, which have been reported to establish pro-B cell lines (Pongubala et al., 2008). The pro-B cells derived from cultures with TD7 grew robustly in the presence of IL-7, but abruptly died when Notch signaling is provided, in contrast to the cells from cultures with OP9, which differentiated into DN2/3 stages with Notch signaling. The characteristics observed in the former pro-B cells were found to be due to decreased expression of Lmo2. These results suggest that TD7 does not support the pluripotency of Ebf1-deficient pro-B cells, leading to loss of LMO2. Although the molecular machinery underlying LMO2 expression in HSCs has not been fully described, it was shown that several cis-regulatory elements around the Lmo2 locus cooperatively function and ensure the full expression pattern of Lmo2 (Landry et al., 2005; Landry et al., 2009), which appears to be driven in part by PU.1, TAL1, GATA factors, and LMO2 itself. The culture conditions provided by TD7 may be unable to sustain sufficient expression of these transcription factors. Alternatively, it is possible that TD7 could not support pro-B cells that retained their pluripotency because M-CSF, which is not produced by OP9 (Nakano et al., 1994), promoted the differentiation of HSCs into myeloid cells. In fact, the establishment of pro-B cells on TD7 was accompanied by a prolonged emergence of myeloid marker-positive cells compared to that on OP9, and the remaining pro-B cells lost their pluripotency (Figure 1—figure supplement 1B). Therefore, HSCs may be depleted by differentiation into the myeloid lineage, resulting in IL-7-dependent proliferation of pro-B cells without pluripotency that express reduced Lmo2. In either case, substantial expression of Lmo2 is critical for the maintenance of pluripotency in Ebf1-deficient pro-B cells, which is consistent with the finding that LMO2 is an important factor for the reprogramming of committed blood or mesenchymal cells to the induced HSCs (Riddell et al., 2014; Batta et al., 2014; Vereide et al., 2014).
A remarkable feature of LMO2 is that it only generates T-cell malignancies by abnormal expression (Neale et al., 1995; Curtis and McCormack, 2010; Matthews et al., 2013). A recent study indicated that LMO2 overexpression induces T-cell malignancies not only at the hematopoietic undifferentiated stages, but also after differentiation to B-lineage cells (García‐Ramírez et al., 2018). However, the strong association between LMO2 and T-cell malignancy is not well understood at the molecular level. We showed here that LMO2 plays an important role in maintaining the chromatin structure of the Tcf7 locus in an accessible state necessary for Notch signaling-mediated activation of Tcf7. The expression of Tcf7 is highly specific to the T-cell lineage among hematopoietic cells, and TCF1 is important for initiating the epigenetic identity of the T-cell lineage (Johnson et al., 2018). Therefore, it can be inferred that, as the Tcf7 locus remains in an accessible state in HPCs or lineage-committed cells with ectopic LMO2 expression, they efficiently differentiate into T-lineage cells and selectively develop T-cell malignancy. In addition, TCF1 forms a complex with β-catenin, which contributes to the development of T-cell malignancy (Bigas et al., 2020). Although the contribution of the β-catenin/TCF1 complex in normal T-cell development remains unclear, the complex, together with RAG molecules, induces various genetic instabilities including those in the Myc gene in immature T cells and causes Myc gene-targeted tumorigenesis (Dose et al., 2014; Gekas et al., 2016). As TCF1 is also involved in other tumorigenesis (Liu et al., 2019), aberrant expression of LMO2 may promote T-cell malignancy via TCF1.
Previous reports suggest that LMO2 forms complexes with the bHLH transcription factors, including E2A, Lyl1, and TAL1/SCL, and binds to their target genomic sites (Wadman et al., 1997; Grütz et al., 1998; El Omari et al., 2013; Layer et al., 2016). Consistently, in this study, we found that the binding motif for bHLH factors was enriched in LMO2 binding regions in pro-B(+) cells. Thus, LMO2 seems to function along with these bHLH factors in the lymphoid progenitors. In addition, we found that LMO2 directly binds to the Tcf7 locus. Initiation of Tcf7 expression requires Notch signaling and Runx factors in T-cell progenitors (Weber et al., 2011; Guo et al., 2008; Shin et al., 2021). Two NICD-RBPJ complex and Runx1 binding sites were found at the −31 and −35 kb upstream regions of the Tcf7 locus (Figure 6—figure supplement 1; Weber et al., 2011; Romero-Wolf et al., 2020; Hosokawa et al., 2018). Among these, the functional importance of the −31 kb region has been previously reported as a Notch-dependent enhancer of Tcf7 (Weber et al., 2011; Harly et al., 2020). The LMO2 binding site, identified in this study, overlapped with the other RBPJ binding site, at the −35 kb upstream region. While the physiological role of the −35 kb RBPJ binding site has not been clarified, our data suggest that direct binding of LMO2 to this region in progenitor cells plays an important role in maintaining the accessible chromatin configuration of the Tcf7 locus. In fact, Lmo2 expression levels were found to be associated with DNA methylation status around the Tcf7 locus and subsequent Notch-mediated activation of Tcf7 expression. Taken together, in the T-cell progenitor stage, LMO2 acts as a gatekeeper for maintaining the transcriptionally poised chromatin state of the Tcf7 locus via direct binding to the −35 kb region, and guarantees T-cell differentiation potential.
Materials and methods
Key resources table.
| Reagent type (species) or resource |
Designation | Source or reference |
Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (Mus musculus) | Ebf1+/- | Pongubala et al., 2008 | Provided by Dr. Grosschedl, Max Planck Institute of Immunobiology and Epigenetics |
|
| Genetic reagent (Mus musculus) | B6.Cg-Tg(BCL2)25Wehi/J | Jackson Laboratory | Stock# 002320 | |
| Genetic reagent (Mus musculus) | B6.Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J | Jackson Laboratory | Stock# 024858 | |
| Cell line (Mus musculus) | OP9 | Yokoyama et al., 2013 | Stromal cell line derived from fetal murine calvaria (B6 x C3H, op/op) | |
| Cell line (Mus musculus) | TD7 | This paper | Mouse fetal thymus (B6, E15.5)-derived mesenchymal cell line | |
| Cell line (Mus musculus) | Pro-B(+) | This paper | Ebf1-deficient fetal liver-derived hematopoietic progenitor cell line | |
| Cell line (Mus musculus) | Pro-B(−) | This paper | Ebf1-deficient fetal liver-derived hematopoietic progenitor cell line | |
| Cell line (Mus musculus) | OP9-Dll4 | Hirano et al., 2020 | ||
| Cell line (Homo sapiens) | HEK293T | Hirano et al., 2020 | RRID:CVCL_0063 | |
| Cell line (Homo sapiens) | PLAT-E | Hirano et al., 2020 | RRID:CVCL_B488 | |
| Antibody | FITC anti-mouse CD4 (Rat monoclonal) | BD Biosciences | Cat# 561835 RRID:AB_10894386 | FC (1:500) |
| Antibody | PE anti-mouse CD4 (Rat monoclonal) | BD Biosciences | Cat# 561829 RRID:AB_10926205 | FC (1:500) |
| Antibody | APC anti-mouse CD8a (Rat monoclonal) | BioLegend | Cat# 100711 RRID:AB_312750 | FC (1:500) |
| Antibody | APCCy7 anti-mouse CD8a (Rat monoclonal) | BioLegend | Cat# 100713 RRID:AB_312752 | FC (1:500) |
| Antibody | PerCpCy5.5 anti-mouse CD11b (Rat monoclonal) | BioLegend | Cat# 101227 RRID:AB_893233 | FC (1:500) |
| Antibody | PECy7 anti-mouse CD19 (Rat monoclonal) | Tonbo Biosciences | Cat# 60-0193 RRID:AB_2621840 | FC (1:250) |
| Antibody | PerCpCy5.5 anti-mouse CD25 (Rat monoclonal) | eBioscience | Cat# 45-0251-82 RRID:AB_914324 | FC (1:1000) |
| Antibody | APC-e780 anti-mouse CD25 (Rat monoclonal) | eBioscience | Cat# 47-0251-82 RRID:AB_1272179 | FC (1:200) |
| Antibody | FITC anti-mouse CD44 (Rat monoclonal) | BioLegend | Cat# 103005 RRID:AB_312956 | FC (1:500) |
| Antibody | APCCy7 anti-mouse CD44 (Rat monoclonal) | BioLegend | Cat# 103027 RRID:AB_830784 | FC (1:500) |
| Antibody | PECy7 anti-mouse CD45 (Rat monoclonal) | eBioscience | Cat# 25-0451-82 RRID:AB_2734986 | FC (1:400) |
| Antibody | APC anti-mouse CD45 (Rat monoclonal) | BioLegend | Cat# 103111 RRID:AB_312976 | FC (1:1000) |
| Antibody | PE anti-mouse CD45.1 (Mouse monoclonal) | BioLegend | Cat# 110727 RRID:AB_893348 | FC (1:250) |
| Antibody | PerCpCy5.5 anti-mouse CD45.2 (Mouse monoclonal) | BioLegend | Cat# 109807 RRID:AB_313444 | FC (1:250) |
| Antibody | PerCpCy5.5 anti-mouse B220 (Rat monoclonal) | BioLegend | Cat# 103235 RRID:AB_893356 | FC (1:200) |
| Antibody | APCCy7 anti-mouse Thy1.2 (Rat monoclonal) | BioLegend | Cat# 105327 RRID:AB_10613280 | FC (1:1000) |
| Antibody | FITC anti-mouse Gr-1 (Rat monoclonal) | BioLegend | Cat# 108405 RRID:AB_313370 |
FC (1:500) |
| Antibody | Biotin anti-mouse Gr-1 (Rat monoclonal) | eBioscience | Cat# 13-5931-86 RRID:AB_466802 | FC (1:300) |
| Antibody | Biotin anti-mouse TER119 (Rat monoclonal) | eBioscience | Cat# 13-5921-85 RRID:AB_466798 | FC (1:300) |
| Antibody | Biotin anti-mouse CD11b (Rat monoclonal) | eBioscience | Cat# 13-0112-86 RRID:AB_466361 | FC (1:300) |
| Antibody | Biotin anti-mouse CD11c (Armenian Hamster monoclonal) | eBioscience | Cat# 13-0114-85 RRID:AB_466364 | FC (1:300) |
| Antibody | Biotin anti-mouse CD19 (Rat monoclonal) | eBioscience | Cat# 13-0193-85 RRID:AB_657658 | FC (1:300) |
| Antibody | Biotin anti-mouse NK1.1 (Rat monoclonal) | eBioscience | Cat# 13-5941-85 RRID:AB_466805 | FC (1:300) |
| Antibody | Biotin anti-mouse CD3ε (Armenian Hamster monoclonal) | eBioscience | Cat# 13-0031-82 RRID:AB_466319 | FC (1:300) |
| Antibody | PE anti-rat CD2 (Mouse monoclonal) | BioLegend | Cat# 201305 RRID:AB_2073811 | FC (1:500) |
| Antibody | PE anti-human NGFR (Mouse monoclonal) | eBioscience | Cat# 12-9400-42 RRID:AB_2572710 | FC (1:500) |
| Antibody | PE Hamster IgG (Armenian Hamster monoclonal) |
BioLegend | Cat# 400907 RRID:AB_326593 | FC (1:200) |
| Antibody | PE anti-mouse Notch1 (Armenian Hamster monoclonal) |
BioLegend | Cat# 130607 RRID:AB_1227719 | FC (1:200) |
| Antibody | PE anti-mouse Notch2 (Armenian Hamster monoclonal) |
BioLegend | Cat# 130707 RRID:AB_1227725 | FC (1:200) |
| Antibody | PE anti-mouse Notch3 (Armenian Hamster monoclonal) |
BioLegend | Cat# 130507 RRID:AB_1227733 | FC (1:200) |
| Antibody | PE anti-mouse Notch4 (Armenian Hamster monoclonal) |
BioLegend | Cat# 128407 RRID:AB_1133997 | FC (1:200) |
| Antibody | Alexa Fluor 647 Mouse IgG1 (Mouse monoclonal) | BioLegend | Cat# 400130 RRID:AB_2800436 | FC (2 µl per test) |
| Antibody | Alexa Fluor 647 anti-GATA3 (Mouse monoclonal) | BD Biosciences | Cat# 560068 RRID:AB_1645316 | FC (8 µl per test) |
| Antibody | Rabbit-IgG (Rabbit monoclonal) | Cell Signaling Technology | Cat# 3900 RRID:AB_1550038 | FC (1:500) |
| Antibody | Anti-TCF1 (Rabbit monoclonal) | Cell Signaling Technology | Cat# 2203 RRID:AB_2199302 | FC (1:100) |
| Antibody | Anti-LMO2 (Rabbit monoclonal) | Abcam | Cat# ab91652 RRID:AB_2049879 | FC (1:200) ChIP (2ug/sample) |
| Antibody | Anti-c-Myc (Rabbit monoclonal) | Cell Signaling Technology | Cat# 5605 RRID:AB_1903938 | FC (1:100) |
| Antibody | DyLight 488 anti-rabbit IgG (Donkey polyclonal) |
BioLegend | Cat# 406404 RRID:AB_1575130 | FC (1:250) |
| Antibody | DyLight 649 anti-rabbit IgG (Donkey polyclonal) |
BioLegend | Cat# 406406 RRID:AB_1575135 | FC (1:500) |
| Antibody | Anti-Tubulinα (Mouse monoclonal) | Sigma | Cat# T6199 RRID:AB_477583 | WB (1:1000) |
| Antibody | Anti-LMO2 (Mouse monoclonal) | Novus | Cat# NB110-78626 RRID:AB_1084895 | WB (1:1000) ChIP (2 µg/sample) |
| Antibody | Anti-human LMO2 (Goat polyclonal) | R&D Systems | Cat# AF2726 RRID:AB_2249968 | ChIP (2 µg/sample) |
| Antibody | Anti-H3K4me3 (Rabbit polyclonal) | Millipore | Cat# 07-473 RRID:AB_1977252 | ChIP (5 µl/sample) |
| Recombinant DNA reagent (plasmid) | pCMV-VSV-G-RSV-Rev | RIKEN BRC | Cat# RDB04393 | Lentiviral packaging plasmid |
| Recombinant DNA reagent (plasmid) | pCAG-HIVgp | RIKEN BRC | Cat# RDB04394 | Lentiviral packaging plasmid |
| Recombinant DNA reagent (plasmid) | pLVS-EF-IR2 | This paper | Lentiviral vector with IRES-rat CD2 | |
| Recombinant DNA reagent (plasmid) | mLmo2/pLVS-EF-IR2 | This paper | pLVS-EF-IR2 Lentiviral vector encoding mLmo2 |
|
| Recombinant DNA reagent (plasmid) | mTcf7/pLVS-EF-IR2 | This paper | pLVS-EF-IR2 Lentiviral vector encoding mTcf7 |
|
| Recombinant DNA reagent (plasmid) | GCDN | Hirano et al., 2015 | Retroviral vector with IRES-human NGFR | |
| Recombinant DNA reagent (plasmid) | hBCL2/GCDN | This paper | GCDN Retroviral vector encoding hBCL2 |
|
| Recombinant DNA reagent (plasmid) | mMeis1/GCDN | This paper | GCDN Retroviral vector encoding mMeis1 |
|
| Recombinant DNA reagent (plasmid) | mHmga2/GCDN | This paper | GCDN Retroviral vector encoding mHmga2 |
|
| Recombinant DNA reagent (plasmid) | Cas9-GFP | Hosokawa et al., 2018 | Retroviral vector to express Cas9 and GFP |
|
| Recombinant DNA reagent (plasmid) | E42-dTet | Hosokawa et al., 2018 | Retroviral vector to express sgRNA and human NGFR |
|
| Recombinant DNA reagent (plasmid) | sgRNA against Luciferase (control) | Hosokawa et al., 2018 | 5’-ggcatttcgcag cctaccg-3’ |
|
| Recombinant DNA reagent (plasmid) | sgRNA against LMO2 #1 | This paper | 5’-tcgatggccgag gacattg-3’ |
|
| Recombinant DNA reagent (plasmid) | sgRNA against LMO2 #2 | This paper | 5’-aatgtcctcggc catcgaa-3’ |
|
| Recombinant DNA reagent (plasmid) | sgRNA against LMO2 #3 | This paper | 5’-gaaagccatcga ccagtac-3’ |
|
| Sequence-based reagent | ActB (Forward) | This paper | PCR primers | 5’-tacagcccgggg agcat-3’ |
| Sequence-based reagent | ActB (Reverse) | This paper | PCR primers | 5’-acacccgccac cagttc-3’ |
| Sequence-based reagent | Meis1 (Forward) | This paper | PCR primers | 5’-gacgctttaaag agagataaagatgc-3’ |
| Sequence-based reagent | Meis1 (Reverse) | This paper | PCR primers | 5’- catttctcaaa aatcagtgctaaga -3’ |
| Sequence-based reagent | Hmga2 (Forward) | This paper | PCR primers | 5’-aaggcagcaaaa acaagagc-3’ |
| Sequence-based reagent | Hmga2 (Reverse) | This paper | PCR primers | 5’-gccgtttttctc caatggt-3’ |
| Sequence-based reagent | Bcl11a (Forward) | This paper | PCR primers | 5’-ccaaacaggaac acacatagcaga-3’ |
| Sequence-based reagent | Bcl11a (Reverse) | This paper | PCR primers | 5’-ggggattagagc tccgtgt-3’ |
| Sequence-based reagent | Gata3 (Forward) | This paper | PCR primers | 5’-ttatcaagccca agcgaag-3’ |
| Sequence-based reagent | Gata3 (Reverse) | This paper | PCR primers | 5’-tggtggtggtct gacagttc-3’ |
| Sequence-based reagent | Lmo2 (Forward) | This paper | PCR primers | 5’-gaggcgcctcta ctacaa-3’ |
| Sequence-based reagent | Lmo2 (Reverse) | This paper | PCR primers | 5’-gatccgcttgtcacaggatg-3’ |
| Sequence-based reagent | Tcf7 (Forward) | This paper | PCR primers | 5’-cagctcccccatactgtgag-3’ |
| Sequence-based reagent | Tcf7 (Reverse) | This paper | PCR primers | 5’-tgctgtctatatccgcaggaa-3’ |
| Sequence-based reagent | Tcf7 promoter region (Forward) | This paper | PCR primers | 5’-ttaagtttttattggtgaatgagtt-3’ |
| Sequence-based reagent | Tcf7 promoter region (Reverse) | This paper | PCR primers | 5’-aaaaaactccaaaaataaaacccac-3’ |
| Sequence-based reagent | Tcf7 TSS (Forward) | This paper | PCR primers | 5’-gcagcaagggttgcattt-3’ |
| Sequence-based reagent | Tcf7 TSS (Reverse) | This paper | PCR primers | 5’-ttgtctgtactgggctgtttacat-3’ |
| Sequence-based reagent | Tcf7 -31kb (Forward) | This paper | PCR primers | 5’-ttccatccaccgttttgttt-3’ |
| Sequence-based reagent | Tcf7 -31kb (Reverse) | This paper | PCR primers | 5’-ggcgtgtggtgggaatacta-3’ |
| Sequence-based reagent | Tcf7 -35kb (Forward) | This paper | PCR primers | 5’-ctgcaagcagctggaagtc-3’ |
| Sequence-based reagent | Tcf7 -35kb (Reverse) | This paper | PCR primers | 5’-cactggaagctgtgagtgatg-3’ |
| Sequence-based reagent | Igk 3’UTR (Forward) | This paper | PCR primers | 5’-ggcacatctgttgctttcgc -3’ |
| Sequence-based reagent | Igk 3’UTR (Reverse) | This paper | PCR primers | 5’-ggggtaggga gcaggtgtat-3’ |
| Peptide, recombinant protein | PerCpCy5.5 streptavidin | BioLegend | Cat# 405214 | FC (1:200) |
| Peptide, recombinant protein | Recombinant Mouse SCF | PeproTech | Cat# 250-03 | |
| Peptide, recombinant protein | Recombinant Human FLT3L | PeproTech | Cat# 300-19 | |
| Peptide, recombinant protein | Recombinant Mouse IL-7 | PeproTech | Cat# 217-17 | |
| Commercial assay or kit | Foxp3 / Transcription Factor Staining Set | eBioscience | Cat# 00-5523-00 | Used to detect TCF1 and GATA3 |
| Commercial assay or kit | Fixation/Permeabilization Solution Kit with BD GolgiStop | BD Biosciences | Cat# 554715 | Used to detect Lmo2 and c-Myc |
| Commercial assay or kit | Permeabilization Buffer Plus | BD Biosciences | Cat# 561651 | Used to detect Lmo2 and c-Myc |
| Commercial assay or kit | High Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat# 4368814 | |
| Commercial assay or kit | NucleoSpin Tissue | TaKaRa Bio | Cat# 740952.50 | |
| Commercial assay or kit | MethylEasy Xceed | TaKaRa Bio | Cat# ME002 | |
| Commercial assay or kit | TaKaRa EpiTaq HS (for bisulfite-treated DNA) | TaKaRa Bio | Cat# R110A | |
| Commercial assay or kit | Mighty TA-cloning kit | TaKaRa Bio | Cat# 6028 | |
| Commercial assay or kit | Anti-Biotin MicroBeads | Miltenyi Biotec | Cat# 130-090-485 | |
| Commercial assay or kit | Dynabeads Protein A | Thermo Fisher Scientific | Cat# 10001D | |
| Commercial assay or kit | Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10003D | |
| Commercial assay or kit | Dynabeads M-280 Sheep Anti-Rabbit IgG | Thermo Fisher Scientific | Cat# 11203D | |
| Commercial assay or kit | NE-PER Nuclear and Cytoplasmic Extraction Reagents | Pierce | Cat# 78833 | |
| Commercial assay or kit | PCR purification Kit | Qiagen | Cat# 28004 | |
| Commercial assay or kit | Fast SYBR Green Master Mix | Thermo Fisher Scientific | Cat# 4385614 | |
| Commercial assay or kit | NEBNext Ultra II DNA Library Prep with Sample Purification Beads | NEB | Cat# E7103S | |
| Commercial assay or kit | NEBNext Multiplex Oligos for Illumina | NEB | Cat# E7500S | |
| Chemical compound, drug | Trizol reagent | Thermo Fisher Scientific | Cat# 15596026 | |
| Chemical compound, drug | 7-AAD | BioLegend | Cat# 420403 | FC (1:50) |
| Chemical compound, drug | Alexa Fluor 647 Annexin V | BioLegend | Cat# 640911 | FC (1:40) |
| Chemical compound, drug | DSG (disuccinimidyl glutarate) | Thermo Fisher Scientific | Cat# 20593 | 1 mg/ml |
| Software, algorithm | FlowJo | BD Biosciences | RRID:SCR_008520 |
Mice
Ebf1+/ mice were provided by R. Grosschedl (Max Planck Institute of Immunobiology and Epigenetics). Ebf1-deficient embryos were generated from Ebf1+/ intercrosses. https://www.jax.org/strain/002320 B6.Cg-Tg(BCL2)25Wehi/J (Bcl2-Tg), B6.Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (Cas9 knock-in), B6.Cg-Rag2tm1.1Cgn/J (RAG2 KO), and B6.129S4-Il2rgtm1Wjl/J (Cγ KO) mice were purchased from the Jackson Laboratory. All mice were maintained in specific pathogen free conditions, and animal experiments were approved by the Animal Experimentation Committee (Tokai University, Kanagawa, Japan).
Flow cytometry
For flow cytometric analysis, the following monoclonal antibodies (mAbs) and reagents were used: CD8a (53–6.7), CD11b (M1/70), CD44 (IM7), CD45 (30-F11), CD45.1 (A20), CD45.2 (104), B220 (RA3-6B2), Thy1.2 (30-H12), Gr-1 (RB6-8C5), rat-CD2 (OX34), Hamster IgG (HTK888), Notch1 (HMN1-12), Notch2 (HMN2-35), Notch3 (HMN3-133), Notch4 (HMN4-14), anti-Rabbit-IgG (Poly4064), mIgG1 (MOPC-21), AnnexinV and 7-AAD were purchased from BioLegend. CD25 (PC61.5), CD45 (30-F11), TER119 (TER-119), Gr-1 (RB6-8C5), CD11b (M1/70), CD11c (N418), CD19 (1D3), NK1.1 (PK136), CD3e (145–2 C11), hNGFR (ME20.4) were purchased from eBioscience. CD4 (GK1.5, RM4-5) and GATA3 (L50-823) were purchased from BD Biosciences. CD19 (1D3) was purchased from Tonbo Biosciences. TCF1 (C63D9), c-Myc (D84C12) and Rabbit-IgG (DA1E) were purchased from Cell Signaling Technology. LMO2 (EP3257) was purchased from Abcam. For intracellular staining, cells were fixed and permeabilized with Foxp3/Transcription Factor Staining Set (eBioscience) or Fixation/Permeabilization Solution Kit with BD GolgiStop (BD Biosciences) and Permeabilization Buffer Plus (BD Biosciences). Stained cells were measured with FACSCalibur (BD Biosciences) or FACSVerse (BD Biosciences). Data were analyzed using FlowJo (BD Biosciences).
Isolation of hematopoietic progenitors and establishment of pro-B(+) and pro-B(–) cell lines
For the establishment of HPC lines, Ebf1-deficient progenitor cells were isolated from Ebf1-deficient E14.5 fetal liver (both male and female). FL cells were incubated with biotin-conjugated mAbs against lineage markers (TER119 and Gr-1, Lin). The cells were then washed and incubated with anti-biotin Microbeads (Miltenyi Biotec), and Lin cells were enriched using autoMACS (Miltenyi Biotec). Lin− cells were cultured on OP9 or TD7 cells in the presence of mSCF, hFlt3L and mIL7. We established two pro-B cell lines, one that was maintained on OP9 cells was called as pro-B(+) cells, and the other that was maintained on TD7 cells was called as pro-B(–) cells.
Cell lines and cultures
TD7, thymic stromal cell line established from fetal thymus (B6, embryonic day 15.5) was cultured in RPMI 1640 (Nissui Pharmaceutical Co., Tokyo)with 10% FBS, sodium pyruvate (Sigma), l-glutamine (Wako), penicillin (Meiji Seika Pharma), streptomycin (Meiji Seika Pharma), 2-ME (Gibco). These cell line seems to be derived from thymic mesenchymal cells as they express PDGFRα. OP9 cell line was kindly provided from Dr. K. Yokoyama (University of Tokyo, Yokoyama et al., 2013) and cultured in α-MEM (Wako) with 20% FBS, penicillin, streptomycin, 2-ME. This cell line kept features in common with OP9-K (RRID:CVCL_KB57). These cell lines were confirmed to be mycoplasma-free status before experiments. Pro-B cell lines were cultured in IMDM (Wako) with 10% FBS, penicillin, streptomycin, 2-ME, 10 ng/ml mSCF (Peprotech), 10 ng/ml hFlt3L (Peprotech), 10 ng/ml mIL7 (Peprotech) on TD7 or mitomycin C (Kyowa-Kirin)-treated OP9. For T-cell induction, pro-B cells were co-cultured on OP9-Dll4 for 3–7 days under the same conditions as the maintenance of pro-B cell lines.
Viral vector transduction of pro-B cell lines
Retroviral vector GCDN (mock vector containing IRES-hNGFR) (Hirano et al., 2015), encoding hBCL2, mMeis1, and mHmga2 were generated by transient transfection into PLAT-E packaging cells. Lentiviral vector pLVS-EF-IR2 (mock vector containing IRES-rat CD2), encoding mLmo2 and mTcf7 were generated by transient transfection into 293T as described previously (Hirano et al., 2020). Pro-B cells were transduced with viral supernatants by spin infection as described previously (Hozumi et al., 2003). After the infection, cells were plated and maintained on stromal cells.
RNA preparation and RT-qPCR
Total RNA was isolated from 45105 cells using Trizol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. cDNA was synthesized with High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative PCR (qPCR) was performed with Fast SYBR Green Master Mix (Thermo Fisher Scientific) and LightCycler 480 System II (Roche). The following primer sets were used for qPCR are listed below.
ActB, 5’-tacagcccggggagcat-3’ and 5’-acacccgccaccagttc-3’
Meis1, 5’-gacgctttaaagagagataaagatgc-3’ and 5’- catttctcaaaaatcagtgctaaga −3’
Hmga2, 5’-aaggcagcaaaaacaagagc-3’ and 5’-gccgtttttctccaatggt-3’
Bcl11a, 5’-ccaaacaggaacacacatagcaga-3’ and 5’-ggggattagagctccgtgt-3’
Gata3, 5’-ttatcaagcccaagcgaag-3’ and 5’-tggtggtggtctgacagttc-3’
Lmo2, 5’-gaggcgcctctactacaa-3’ and 5’-gatccgcttgtcacaggatg-3’
Tcf7, 5’-cagctcccccatactgtgag-3’ and 5’-tgctgtctatatccgcaggaa-3’.
Microarray analysis
Total RNA was extracted from Ebf1-deficient pro-B(–) and pro-B(+) cells, and each 100 ng RNA was used for microarray analysis. Microarray experiments were performed with Whole Mouse Genome 4x44K ver. 2 Microarray Kit (Agilent) using the Agilent One-Color Microarray-Based Gene Expression Analysis protocol. Data from samples that passed the QC parameters were subjected to 75th percentile normalization and analyzed using Genespring GX (version 12, Agilent Technologies).
CRISPR/Cas9-mediated deletion of LMO2 in Pro-B cell lines sgRNA expression vector (E42-dTet) and Cas9-GFP expression vector were described previously (Hosokawa et al., 2018). 19-mer sgRNAs were designed using the Benchiling web tool (https://www.benchling.com) and inserted into the empty sgRNA-expression vector by PCR-based insertion. Three sgRNA-expression vectors were generated for one gene, and pooled retroviral plasmids were used to make retroviral supernatant. Sequences of sgRNAs used in this study are listed below.
Control (Luciferase); ggcatttcgcagcctaccg
LMO2 #1; tcgatggccgaggacattg
LMO2 #2; aatgtcctcggccatcgaa
LMO2 #3; gaaagccatcgaccagtac
Pro-B cell lines were transduced with retroviral vectors encoding Cas9-GFP, and 3 days after infection, GFP+ retrovirus-infected cells were sorted. Then, they were expanded for a week and subjected second retrovirus transduction with sgLMO2-hNGFR. They were transferred onto OP9-Dll4 on day5 or day10 after snd infection, then CD25 and CD44 profiles on GFP+ hNGFR+ retrovirus-infected cells were analyzed, 3 days later.
CRISPR/Cas9-mediated deletion of LMO2 in BM progenitors
BM was obtained from the femurs and tibiae of 2–3 monthold Cas9 and Bcl2 Tg mice. Suspensions of BM cells were prepared and stained for lineage (Lin) markers using biotin-conjugated lineage antibodies CD11b (eBioscience; 13-0112-86), CD11c (eBioscience; 13-0114-85), Gr-1 (eBioscience; 13-5931-86), TER-119 (eBioscience; 13-5921-85), NK1.1 (eBioscience; 13-5941-85), CD19 (eBioscience; 13-0193-85), and CD3ε (eBioscience; 13-0031-082); incubated with streptavidin-coated magnetic beads (Miltenyi Biotec); and passed through a magnetic column using AutoMACS with the ‘Depelete’ program (Miltenyi Biotec). Thereafter, the hematopoietic progenitors were transduced with retroviral vectors encoding sgRNA against LMO2 and cultured using the OP9 medium (α-MEM, 20% FBS, 50 μM β-mercaptoethanol, Pen-Strep-Glutamine) supplemented with 10 ng/ml of IL-7 (Pepro Tech Inc) and 10 ng/ml of SCF (Pepro Tech Inc). Two days after the sgLMO2 transduction, the cells were collected and cultured on OP9-Dll1 monolayers using OP9 medium supplemented with 10 ng/ml of IL-7 and 10 ng/ml of Flt3L (Pepro Tech Inc) for 4 days. The cultured cells were then disaggregated, filtered through a 40 μm nylon mesh, and subjected to flow cytometry analysis using surface antibodies against CD45 PECy7 (eBioscience; 25-0451-82), CD44 FITC (BioLegend; 103005), CD25 APC-e780 (eBioscience; 47-0251-82), human-NGFR PE (eBioscience; 12-9400-42), and a biotin-conjugated lineage cocktail (CD8α, CD11b, CD11c, Gr-1, TER-119, NK1.1, CD19, TCRβ, and TCRγδ) with streptavidin PerCPCy5.5.
Immunoblotting
Cytoplasmic and nuclear extracts, used to the detection of Tubulinα and LMO2, respectively, were prepared by using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce). Lysates were run on 12.5% polyacrylamide gel, followed by immunoblotting. The antibodies used for the immunoblot analysis were anti-Tubulinα (Sigma, T6199) and anti-LMO2 (Novus, NB110-78626).
DNA methylation analysis
Genomic DNA was isolated from pro-B(+), pro-B(–) and LMO2/pro-B(−) cells. The DNA was purified using NucleoSpin Tissue (TaKaRa Bio) and was treated with bisulfite using MethylEasy Xceed (TaKaRa Bio) according to the manufacturer’s instructions. For the evaluation of DNA methylation status of CpG island in Tcf7 promoter region, which contains 25 potential CpG methylation sites, bisulfite-converted DNA was amplified by PCR using TaKaRa EpiTaq HS (for bisulfite-treated DNA, TaKaRa Bio) and the following primer set: 5’-ttaagtttttattggtgaatgagtt-3’ and 5’-aaaaaactccaaaaataaaacccac-3’. Amplified PCR products were cloned into pMD20-T vector using Mighty TA-cloning kit (TaKaRa Bio) and then sequenced.
Chromatin immunoprecipitation (ChIP) and ChIP-sequencing
1 × 107 cells were fixed with 1 mg/ml DSG (Thermo Scientific) in PBS for 30 min at RT followed by an additional 10 min with addition of formaldehyde up to 1%. The reaction was quenched by addition of 1/10 vol of 0.125 M glycine and the cells were washed with HBSS (Gibco). Pelleted nuclei were dissolved in lysis buffer (0.5% SDS, 10 mM EDTA, 0.5 mM EGTA, 50 mM TrisHCl [pH 8] and PIC) and sonicated on SFX150 (Branson) for scycles of 20 s sonication followed by 1 min rest, with 30% amplitude. Six mof anti-LMO2 Abs (a mixture of 2 μg of NB110-78626 [Novus], 2 μg of ab91652 [Abcam] and 2 μg of AF2726 [R and D systems]) or anti-H3K4-3Me (07–043, Millipore) were pre-bound to Dynabeads Protein A/G or Dynabeads anti-Rabbit Ig (Invitrogen) and then added to the diluted chromatin complexes. The samples were incubated overnight at 4°C, then washed and eluted for 6 hr at 65°C in ChIP elution buffer (20 mM Tris–HCl, pH 7.5, 5 mM EDTA, 50 mM NaCl, 1% SDS, and 50 μg/ml proteinase K). Precipitated chromatin fragments were cleaned up using PCR purification Kit (Qiagen).
Quantitative PCR analysis was performed on QuantStudio3 (Applied Biosystems) using the Fast SYBR Green Master Mix. Data are shown as mean values (% input). The primers used are listed below:
Tcf7 TSS, 5ʹ-gcagcaagggttgcattt-3ʹ and 5ʹ-ttgtctgtactgggctgtttacat-3ʹ
Tcf7-31kb, 5ʹ-ttccatccaccgttttgttt-3ʹ and 5ʹ-ggcgtgtggtgggaatacta-3ʹ
Tcf7-35kb, 5ʹ-ctgcaagcagctggaagtc-3ʹ and 5ʹ-cactggaagctgtgagtgatg-3ʹ
Igk 3ʹ UTR, 5ʹ-ggcacatctgttgctttcgc −3ʹ and 5ʹ-ggggtagggagcaggtgtat-3ʹ
ChIP-seq libraries were constructed using NEBNext Ultra II DNA Library Prep with Sample Purification Beads (E7103S, NEB) and NEBNext Multiplex Oligos for Illumina (E7500S, NEB) and sequenced on Illumina NextSeq500 in single read mode with the read length of 75 nt. Base calls were performed with RTA 1.13.48.0 followed by conversion to FASTQ with bcl2fastq 1.8.4 and produced approximately 30 million reads per sample. ChIP-seq data were mapped to the mouse genome build NCBI37/mm10 using Bowtie (v1.1.1; http://bowtie-bio.sourceforge.net/index.shtml) with ‘-v 3 k 11 m 10 t --best –strata’ settings and HOMER tagdirectories were created with makeTagDirectory and visualized in the UCSC genome browser (http://genome.ucsc.edu). ChIP peaks were identified with findPeaks.pl against a matched control sample using the settings ‘-P. 1 -LP. 1 -poisson. 1 -style factor’. The identified peaks were annotated to genes with the annotatePeaks.pl command against the mm10 genomic build in the HOMER package. Peak reproducibility was determined by a HOMER adaptation of the IDR (Irreproducibility Discovery Rate) package according to ENCODE guidelines (https://sites.google.com/site/anshulkundaje/projects/idr). Only reproducible high-quality peaks, with a normalized peak score ≥ 15, were considered for further analysis. Motif enrichment analysis was performed with the findMotifsGenome.pl command in the HOMER package using a 200 bp window.
Acknowledgements
We thank Drs. R Goitsuka and K Nakashima for providing the expression vector for murine Meis1 and Hmga2, respectively; Dr. R Grosshedl for giving Ebf1+/ mice; Dr. K Yokoyama for providing OP9 cell line and critical advice for its culture conditions; Drs. J Takano and T Ikawa for technical advice and support; N Abe S Ochiai for experimental assistance and members of the Support Center for Medical Research and Education at Tokai University for their technical help. This work was partly supported by the Tokai University General Research Organization Research and Study Project. We would like to thank Editage for English editing.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Katsuto Hozumi, Email: hozumi@is.icc.u-tokai.ac.jp.
Juan Carlos Zúñiga-Pflücker, University of Toronto, Sunnybrook Research Institute, Canada.
Satyajit Rath, Indian Institute of Science Education and Research (IISER), India.
Funding Information
This paper was supported by the following grants:
Japan Society for the Promotion of Science 16K08848 to Katsuto Hozumi.
Japan Society for the Promotion of Science JP19H03692 to Hiroyuki Hosokawa.
Japan Society for the Promotion of Science 17H05802 to Katsuto Hozumi.
Japan Society for the Promotion of Science JP20K07730 to Katsuto Hozumi.
Uehara Memorial Foundation Research grant to Hiroyuki Hosokawa.
Naito Foundation Research grant to Hiroyuki Hosokawa.
Takeda Science Foundation Research grant to Hiroyuki Hosokawa.
Yasuda Medical Foundation Research grant to Hiroyuki Hosokawa.
SENSHIN Medical Research Foundation Research grant to Hiroyuki Hosokawa.
Daiichi Sankyo Foundation of Life Science Research grant to Hiroyuki Hosokawa.
Tokyo Biochemical Research Foundation Research grant to Hiroyuki Hosokawa.
Princess Takamatsu Cancer Research Fund Research grant to Hiroyuki Hosokawa.
Mitsubishi Foundation Research grant to Hiroyuki Hosokawa.
Tokai University School of Medicine Research Aid Research grant to Ken-ichi Hirano, Hiroyuki Hosokawa, Maria Koizumi.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Formal analysis, Investigation, Visualization, Writing - original draft, Writing - review and editing.
Conceptualization, Data curation, Formal analysis, Funding acquisition, Validation, Investigation, Visualization, Writing - original draft, Writing - review and editing.
Formal analysis, Investigation.
Formal analysis, Validation, Investigation, Methodology.
Formal analysis, Validation, Investigation, Methodology.
Supervision, Validation.
Conceptualization, Data curation, Formal analysis, Supervision, Funding acquisition, Validation, Investigation, Visualization, Methodology, Writing - original draft, Project administration, Writing - review and editing.
Ethics
Animal experimentation: This study was performed in strict accordance with the recommendations in the Guidelines for the Care and Use of Animals for Scientific Purposes at Tokai University, and approved by the Animal Experimentation Committee of Tokai University (Approval No.: 165015, 171002, 182026, 193040, 204028, 211006), which is further monitored by the Animal Experimentation Evaluation Committee of Tokai University with researcher for Humanities/Sociology and external expert.
Additional files
Data availability
Microarray expression data from Ebf1-deficient pro-B(-) and pro-B(+) cells, and the new deep-sequencing data reported in this paper are available via GEO (https://www.ncbi.nlm.nih.gov/geo/) (GSE162549 and GSE154472).
The following datasets were generated:
Hozumi K, Hirano Ki. 2020. Difference in gene expression signatures of Ebf1-deficient pro-B cell lines with and without the ability to differentiate into T cell lineage. NCBI Gene Expression Omnibus. GSE162549
Hirano Ki, Hosokawa H, Koizumi M, Endo Y, Hozumi K. 2020. LMO2 is essential to maintain the ability of progenitors to differentiate into T-cell lineage in mice. NCBI Gene Expression Omnibus. GSE154472
References
- Ariki R, Morikawa S, Mabuchi Y, Suzuki S, Nakatake M, Yoshioka K, Hidano S, Nakauchi H, Matsuzaki Y, Nakamura T, Goitsuka R. Homeodomain transcription factor Meis1 is a critical regulator of adult bone marrow hematopoiesis. PLOS ONE. 2014;9:e87646. doi: 10.1371/journal.pone.0087646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batta K, Florkowska M, Kouskoff V, Lacaud G. Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Reports. 2014;9:1871–1884. doi: 10.1016/j.celrep.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigas A, Guillén Y, Schoch L, Arambilet D. Revisiting β‐catenin signaling in t‐cell development and t‐cell acute lymphoblastic leukemia. BioEssays. 2020;42:1900099. doi: 10.1002/bies.201900099. [DOI] [PubMed] [Google Scholar]
- Boehm T, Foroni L, Kaneko Y, Perutz MF, Rabbitts TH. The rhombotin family of cysteine-rich LIM-domain oncogenes: distinct members are involved in T-cell translocations to human chromosomes 11p15 and 11p13. PNAS. 1991;88:4367–4371. doi: 10.1073/pnas.88.10.4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland SM, Smith S, Tripathi R, Mathias EM, Goodings C, Elliott N, Peng D, El-Rifai W, Yi D, Chen X, Li L, Mullighan C, Downing JR, Love P, Davé UP. Lmo2 induces hematopoietic stem cell-like features in T-cell progenitor cells prior to leukemia. Stem Cells. 2013;31:882–894. doi: 10.1002/stem.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley MR, Babovic S, Benz C, Knapp DJHF, Beer PA, Kent DG, Wohrer S, Treloar DQ, Day C, Rowe K, Mader H, Kuchenbauer F, Humphries RK, Eaves CJ. The Lin28b–let-7–Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nature Cell Biology. 2013;15:916–925. doi: 10.1038/ncb2783. [DOI] [PubMed] [Google Scholar]
- Curtis DJ, McCormack MP. The molecular basis of Lmo2-induced T-cell acute lymphoblastic leukemia. Clinical Cancer Research. 2010;16:5618–5623. doi: 10.1158/1078-0432.CCR-10-0440. [DOI] [PubMed] [Google Scholar]
- Dose M, Emmanuel AO, Chaumeil J, Zhang J, Sun T, Germar K, Aghajani K, Davis EM, Keerthivasan S, Bredemeyer AL, Sleckman BP, Rosen ST, Skok JA, Le Beau MM, Georgopoulos K, Gounari F. β-Catenin induces T-cell transformation by promoting genomic instability. PNAS. 2014;111:391–396. doi: 10.1073/pnas.1315752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- El Omari K, Hoosdally SJ, Tuladhar K, Karia D, Hall-Ponselé E, Platonova O, Vyas P, Patient R, Porcher C, Mancini EJ. Structural basis for LMO2-driven recruitment of the SCL:e47bhlh heterodimer to hematopoietic-specific transcriptional targets. Cell Reports. 2013;4:135–147. doi: 10.1016/j.celrep.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Ramírez I, Bhatia S, Rodríguez‐Hernández G, González‐Herrero I, Walter C, González de Tena‐Dávila S, Parvin S, Haas O, Woessmann W, Stanulla M, Schrappe M, Dugas M, Natkunam Y, Orfao A, Domínguez V, Pintado B, Blanco O, Alonso‐López D, De Las Rivas J, Martín‐Lorenzo A, Jiménez R, García Criado FJ, García Cenador MB, Lossos IS, Vicente‐Dueñas C, Borkhardt A, Hauer J, Sánchez‐García I. Lmo2 expression defines tumor cell identity during T‐cell leukemogenesis. The EMBO Journal. 2018;37:e98783. doi: 10.15252/embj.201798783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekas C, D’Altri T, Aligué R, González J, Espinosa L, Bigas A. β-Catenin is required for T-cell leukemia initiation and MYC transcription downstream of Notch1. Leukemia. 2016;30:2002–2010. doi: 10.1038/leu.2016.106. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Grütz GG, Bucher K, Lavenir I, Larson T, Larson R, Rabbitts TH. The oncogenic T cell LIM-protein Lmo2 forms part of a DNA-binding complex specifically in immature T cells. The EMBO Journal. 1998;17:4594–4605. doi: 10.1093/emboj/17.16.4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Maillard I, Chakraborti S, Rothenberg EV, Speck NA. Core binding factors are necessary for natural killer cell development and cooperate with Notch signaling during T-cell specification. Blood. 2008;112:480–492. doi: 10.1182/blood-2007-10-120261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. Journal of Clinical Investigation. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harly C, Kenney D, Wang Y, Ding Y, Zhao Y, Awasthi P, Bhandoola A. A shared regulatory element controls the initiation of Tcf7 Expression During Early T Cell and Innate Lymphoid Cell Developments. Frontiers in Immunology. 2020;11:470. doi: 10.3389/fimmu.2020.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano K, Negishi N, Yazawa M, Yagita H, Habu S, Hozumi K. Delta-like 4-mediated Notch signaling is required for early T-cell development in a three-dimensional thymic structure. European Journal of Immunology. 2015;45:2252–2262. doi: 10.1002/eji.201445123. [DOI] [PubMed] [Google Scholar]
- Hirano K, Suganami A, Tamura Y, Yagita H, Habu S, Kitagawa M, Sato T, Hozumi K. Delta-like 1 and Delta-like 4 differently require their extracellular domains for triggering Notch signaling in mice. eLife. 2020;9:e50979. doi: 10.7554/eLife.50979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa H, Ungerbäck J, Wang X, Matsumoto M, Nakayama KI, Cohen SM, Tanaka T, Rothenberg EV. Transcription factor PU.1 Represses and Activates Gene Expression in Early T Cells by Redirecting Partner Transcription Factor Binding. Immunity. 2018;48:1119–1134. doi: 10.1016/j.immuni.2018.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa H, Masuhara K, Koizumi M. Transcription factors regulate early T cell development via redeployment of other factors: Functional dynamics of constitutively required factors in cell fate decisions. BioEssays : News and Reviews in Molecular, Cellular and Developmental Biology. 2021a;43:e2000345. doi: 10.1002/bies.202000345. [DOI] [PubMed] [Google Scholar]
- Hosokawa H, Koizumi M, Masuhara K, Romero-Wolf M, Tanaka T, Nakayama T, Rothenberg EV. Stage-specific action of Runx1 and GATA3 controls silencing of PU.1 expression in mouse pro–T cells. Journal of Experimental Medicine. 2021b;218:e20202648. doi: 10.1084/jem.20202648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa H, Rothenberg EV. How transcription factors drive choice of the T cell fate. Nature Reviews Immunology. 2021;21:162–176. doi: 10.1038/s41577-020-00426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJT, Gale RE, Linch DC, Bayford J, Brown L, Quaye M, Kinnon C, Ancliff P, Webb DK, Schmidt M, von Kalle C, Gaspar HB, Thrasher AJ. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. Journal of Clinical Investigation. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hozumi K, Abe N, Chiba S, Hirai H, Habu S. Active form of notch members can enforce T lymphopoiesis on lymphoid progenitors in the monolayer culture specific for B cell development. The Journal of Immunology. 2003;170:4973–4979. doi: 10.4049/jimmunol.170.10.4973. [DOI] [PubMed] [Google Scholar]
- Hozumi K, Mailhos C, Negishi N, Hirano K, Yahata T, Ando K, Zuklys S, Holländer GA, Shima DT, Habu S. Delta-like 4 is indispensable in thymic environment specific for T cell development. Journal of Experimental Medicine. 2008;205:2507–2513. doi: 10.1084/jem.20080134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa T, Kawamoto H, Wright LY, Murre C. Long-term cultured E2A-deficient hematopoietic progenitor cells are pluripotent. Immunity. 2004;20:349–360. doi: 10.1016/S1074-7613(04)00049-4. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, Pear WS, Bhandoola A, Wherry EJ, Vahedi G. Lineage-Determining transcription factor TCF-1 initiates the epigenetic identity of T cells. Immunity. 2018;48:243–257. doi: 10.1016/j.immuni.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch U, Fiorini E, Benedito R, Besseyrias V, Schuster-Gossler K, Pierres M, Manley NR, Duarte A, MacDonald HR, Radtke F. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. Journal of Experimental Medicine. 2008;205:2515–2523. doi: 10.1084/jem.20080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosan C, Godmann M. Genetic and epigenetic mechanisms that maintain hematopoietic stem cell function. Stem Cells International. 2016;2016:1–14. doi: 10.1155/2016/5178965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry J-R, Kinston S, Knezevic K, Donaldson IJ, Green AR, Göttgens B. Fli1, Elf1, and Ets1 regulate the proximal promoter of the LMO2 gene in endothelial cells. Blood. 2005;106:2680–2687. doi: 10.1182/blood-2004-12-4755. [DOI] [PubMed] [Google Scholar]
- Landry J-R, Bonadies N, Kinston S, Knezevic K, Wilson NK, Oram SH, Janes M, Piltz S, Hammett M, Carter J, Hamilton T, Donaldson IJ, Lacaud G, Frampton J, Follows G, Kouskoff V, Göttgens B. Expression of the leukemia oncogene Lmo2 is controlled by an array of tissue-specific elements dispersed over 100 kb and bound by Tal1/Lmo2, Ets, and Gata factors. Blood. 2009;113:5783–5792. doi: 10.1182/blood-2008-11-187757. [DOI] [PubMed] [Google Scholar]
- Layer JH, Alford CE, McDonald WH, Davé UP. LMO2 oncoprotein stability in T-Cell leukemia requires direct LDB1 binding. Molecular and Cellular Biology. 2016;36:488–506. doi: 10.1128/MCB.00901-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Sun R, Zhang X, Qiu B, Chen T, Li Z, Xu Y, Zhang Z. Transcription factor 7 promotes the progression of perihilar cholangiocarcinoma by inducing the transcription of c-Myc and FOS-like antigen 1. EBioMedicine. 2019;45:181–191. doi: 10.1016/j.ebiom.2019.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews JM, Lester K, Joseph S, Curtis DJ. LIM-domain-only proteins in cancer. Nature Reviews Cancer. 2013;13:111–122. doi: 10.1038/nrc3418. [DOI] [PubMed] [Google Scholar]
- McCormack MP, Forster A, Drynan L, Pannell R, Rabbitts TH. The LMO2 T-cell oncogene is activated via chromosomal translocations or retroviral insertion during gene therapy but has no mandatory role in normal T-cell development. Molecular and Cellular Biology. 2003;23:9003–9013. doi: 10.1128/MCB.23.24.9003-9013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack MP, Young LF, Vasudevan S, de Graaf CA, Codrington R, Rabbitts TH, Jane SM, Curtis DJ. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science. 2010;327:879–883. doi: 10.1126/science.1182378. [DOI] [PubMed] [Google Scholar]
- Nakano T, Kodama H, Honjo T. Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science. 1994;265:1098–1101. doi: 10.1126/science.8066449. [DOI] [PubMed] [Google Scholar]
- Neale GA, Rehg JE, Goorha RM. Ectopic expression of rhombotin-2 causes selective expansion of CD4-CD8- lymphocytes in the thymus and T-cell tumors in transgenic mice. Blood. 1995;86:3060–3071. doi: 10.1182/blood.V86.8.3060.3060. [DOI] [PubMed] [Google Scholar]
- Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- Pongubala JMR, Northrup DL, Lancki DW, Medina KL, Treiber T, Bertolino E, Thomas M, Grosschedl R, Allman D, Singh H. Transcription factor EBF restricts alternative lineage options and promotes B cell fate commitment independently of Pax5. Nature Immunology. 2008;9:203–215. doi: 10.1038/ni1555. [DOI] [PubMed] [Google Scholar]
- Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, Aguet M. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10:547–558. doi: 10.1016/S1074-7613(00)80054-0. [DOI] [PubMed] [Google Scholar]
- Riddell J, Gazit R, Garrison BS, Guo G, Saadatpour A, Mandal PK, Ebina W, Volchkov P, Yuan GC, Orkin SH, Rossi DJ. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014;157:549–564. doi: 10.1016/j.cell.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolink AG, Nutt SL, Melchers F, Busslinger M. Long-term in vivo reconstitution of T-cell development by Pax5-deficient B-cell progenitors. Nature. 1999;401:603–606. doi: 10.1038/44164. [DOI] [PubMed] [Google Scholar]
- Romero-Wolf M, Shin B, Zhou W, Koizumi M, Rothenberg EV, Hosokawa H. Notch2 complements Notch1 to mediate inductive signaling that initiates early T cell development. Journal of Cell Biology. 2020;219:e202005093. doi: 10.1083/jcb.202005093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer-Pokora B, Loos U, Ludwig WD. TTG-2, a new gene encoding a cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia with the t(11;14)(p13;q11) Oncogene. 1991;6:1887–1893. [PubMed] [Google Scholar]
- Shin B, Hosokawa H, Romero-Wolf M, Zhou W, Masuhara K, Tobin VR, Levanon D, Groner Y, Rothenberg EV. Runx1 and Runx3 drive progenitor to T-lineage transcriptome conversion in mouse T cell commitment via dynamic genomic site switching. PNAS. 2021;118:e2019655118. doi: 10.1073/pnas.2019655118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerbäck J, Hosokawa H, Wang X, Strid T, Williams BA, Sigvardsson M, Rothenberg EV. Pioneering, chromatin remodeling, and epigenetic constraint in early T-cell gene regulation by SPI1 (PU.1) Genome Research. 2018;28:1508–1519. doi: 10.1101/gr.231423.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereide DT, Vickerman V, Swanson SA, Chu LF, McIntosh BE, Thomson JA. An expandable, inducible hemangioblast state regulated by fibroblast growth factor. Stem Cell Reports. 2014;3:1043–1057. doi: 10.1016/j.stemcr.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadman IA, Osada H, Grutz GG, Agulnick AD, Westphal H, Forster A, Rabbitts TH. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. The EMBO Journal. 1997;16:3145–3157. doi: 10.1093/emboj/16.11.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber BN, Chi AW-S, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, Bhandoola A. A critical role for TCF-1 in T-lineage specification and differentiation. Nature. 2011;476:63–68. doi: 10.1038/nature10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Trumpp A. Balancing dormant and self-renewing hematopoietic stem cells. Current Opinion in Genetics & Development. 2009;19:461–468. doi: 10.1016/j.gde.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Wilson NK, Foster SD, Wang X, Knezevic K, Schütte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, Pimanda JE, de Bruijn MF, Göttgens B. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7:532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Wong P, Iwasaki M, Somervaille TCP, So CWE, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes & Development. 2007;21:2762–2774. doi: 10.1101/gad.1602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Warren AJ, Dobson C, Forster A, Pannell R, Rabbitts TH. The T cell leukemia LIM protein Lmo2 is necessary for adult mouse hematopoiesis. PNAS. 1998;95:3890–3895. doi: 10.1073/pnas.95.7.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K, Yokoyama N, Izawa K, Kotani A, Harashima A, Hozumi K, Tojo A. In vivo leukemogenic potential of an interleukin 7 receptor α chain mutant in hematopoietic stem and progenitor cells. Blood. 2013;122:4259–4263. doi: 10.1182/blood-2012-08-451278. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, Desland F, Chudnovskiy A, Mortha A, Dominguez C, Tellier J, Kim E, Dwyer D, Shinton S, Nabekura T, Qi Y, Yu B, Robinette M, Kim K-W, Wagers A, Rhoads A, Nutt SL, Brown BD, Mostafavi S, Buenrostro JD, Benoist C. The cis-Regulatory Atlas of the Mouse Immune System. Cell. 2019;176:897–912. doi: 10.1016/j.cell.2018.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wang J, Khaled W, Burke S, Li P, Chen X, Yang W, Jenkins NA, Copeland NG, Zhang S, Liu P. Bcl11a is essential for lymphoid development and negatively regulates p53. Journal of Experimental Medicine. 2012;209:2467–2483. doi: 10.1084/jem.20121846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nature Reviews Immunology. 2014;14:529–545. doi: 10.1038/nri3702. [DOI] [PMC free article] [PubMed] [Google Scholar]