

Abstract
Sonic hedgehog (Shh) signaling from the primary cilium drives cerebellar granule cell precursor (GCP) proliferation. Mutations of hedgehog (Hh) pathway repressors commonly cause medulloblastoma, the most prevalent and malignant childhood brain tumor that arises from aberrant GCP proliferation. We demonstrate that Nestin Cre-driven conditional knock-out (CKO) of a Shh pathway repressor-Rab23 in the mouse brain of both genders caused mis-patterning of cerebellar folia and elevated GCP proliferation during early development, but with no prevalent occurrence of medulloblastoma at adult stage. Strikingly, Rab23-depleted GCPs exhibited upregulated basal level of Shh pathway activities despite showing an abnormal ciliogenesis of primary cilia. In line with the compromised ciliation, Rab23-depleted GCPs were desensitized against Hh pathway activity stimulations by Shh ligand and Smoothened (Smo) agonist-SAG, and exhibited attenuated stimulation of Smo-localization on the primary cilium in response to SAG. These results implicate multidimensional actions of Rab23 on Hh signaling cascade. Rab23 represses the basal level of Shh signaling, while facilitating primary cilium-dependent extrinsic Shh signaling activation. Collectively, our findings unravel instrumental roles of Rab23 in GCP proliferation and ciliogenesis. Furthermore, Rab23's potentiation of Shh signaling pathway through the primary cilium and Smo suggests a potential new therapeutic strategy for Smo/primary cilium-driven medulloblastoma.
SIGNIFICANCE STATEMENT Primary cilium and Sonic hedgehog (Shh) signaling are known to regulate granule cell precursor (GCP) proliferation. Aberrant overactivation of Shh signaling pathway ectopically increases GCP proliferation and causes malignant childhood tumor called medulloblastoma. However, the genetic and molecular regulatory cascade of GCP tumorigenesis remains incompletely understood. Our finding uncovers Rab23 as a novel regulator of hedgehog (Hh) signaling pathway activity and cell proliferation in GCP. Intriguingly, we demonstrated that Rab23 confers dual functions in regulating Shh signaling; it potentiates primary cilium and Shh/Smoothened (Smo)-dependent signaling activation, while antagonizes basal level Hh activity. Our data present a previously underappreciated aspect of Rab23 in mediating extrinsic Shh signaling upstream of Smo. This study sheds new light on the mechanistic insights underpinning Shh signaling-mediated GCP proliferation and tumorigenesis.
Keywords: ciliogenesis, granule cell precursor, medulloblastoma, primary cilium, Rab GTPase, sonic hedgehog
Introduction
Cerebellar development in mammals is highly dependent on sonic hedgehog (Shh) signaling. In particular, Shh signaling dictates the proliferation of granule cell precursors (GCP; Wallace, 1999; Wechsler-Reya and Scott, 1999). GCPs give rise to granule neurons, the most abundant neuronal type in the brain. In the developing cerebellum, GCPs receive mitotic signals from Shh ligands released from the neighboring Purkinje cells to sustain its proliferation (Wallace, 1999; Wechsler-Reya and Scott, 1999). Besides paracrine Shh signaling, GCPs were also capable of self-regulated autocrine-induced cell proliferation (Gao et al., 1991). Perturbation of Shh pathway activity during early embryonic or postnatal development results in cerebellar dysplasia, hypoplasia as well as malignant childhood brain tumor medulloblastoma (Dahmane and Ruiz i Altaba, 1999; Wechsler-Reya and Scott, 1999; Lewis et al., 2004; Roussel and Hatten, 2011; Basson and Wingate, 2013). For example, genetic mutations of Shh signaling components such as Patched (PTCH), Smoothened (SMO), Gpr161, or Suppressor of fused (SUFU) are known to lead to the formation of medulloblastoma (Raffel et al., 1997; Dong et al., 2000; Taylor et al., 2002; Dey et al., 2012; Begemann et al., 2020).
In the past decade, emergence of primary cilium as an indispensable organelle for Shh signal transduction has facilitated discoveries that highlighted the seminal roles of primary cilium in cerebellar development and medulloblastoma formation. The primary cilium is a non-motile cilium found on the surface of nearly every cell. It functions primarily as an “antenna” on the cell membrane to receive and transduce extracellular signals. In the Shh pathway, Shh ligand binds to the Ptch receptor to release its suppression of Smo on the cell membrane. This subsequently triggers Smo and cytosolic factors such as the Gli transcription factors and SuFu to interact within the primary cilium before translocating into the nucleus to activate Shh downstream target genes (Haycraft et al., 2005; Wong et al., 2009; Tukachinsky et al., 2010). Although the exact molecular mechanism and trafficking cargoes that mediate dynamic ciliary entry and exit of Shh signaling components remain incompletely understood, it has been well established that Shh signal transduction is inevitably deregulated in the absence of a functional primary cilium. For instance, knock-out (KO) of genes known to be required for primary cilium formation (i.e., Kif3a or Ift88) diminished Shh activities in the cerebellum and contributed to the manifestation of cerebellar hypoplasia and distorted foliation because of substantial shrinkage of GCPs pool (Chizhikov et al., 2007; Spassky et al., 2008).
Intriguingly, recent findings have revealed that the primary cilium could exert both inducing or suppressing forces on Shh pathway and cancer progression (Han et al., 2009; Wong et al., 2009; Bay et al., 2018). Depending on the pathogenic origin of the medulloblastoma, primary cilia could potentiate tumor growth driven by Smo, and on the other hand, inhibit tumor growth driven by Gli2 (Han et al., 2009). Adding to the complexity of the tumor biology, the same study also showed that there are ciliated and non-ciliated subcategories of medulloblastoma, with the ones bearing primary cilia often associated with increased Shh and Wnt pathway activities, whereas those without cilia do not exhibit Shh or Wnt pathway activation (Han et al., 2009). Given the opposing functions of primary cilium on Shh pathway-mediated tumor progression, as well as the heterogeneity in ciliation capacity among the tumor cells; the multifaceted functions of primary cilium might underlie variable patients' responses to Smo-specific drug, Vismodegib treatment in the clinical trials that targets Shh-subtype medulloblastoma (Gajjar et al., 2013). Therefore, further insights on the interaction between primary cilium and the Shh pathway, and their roles in GCP proliferation would lay critical foundation for future development of effective intervention for medulloblastoma.
Rab23 is a brain-enriched small GTPase (Guo et al., 2006) known to antagonize the Shh pathway in vivo, as evidenced by developmental mouse genetic studies. In humans, mutations of RAB23 cause Carpenter syndrome, an autosomal recessive disorder characterized by aberrant skull fusion, polydactyly and brachydactyly. Other variable developmental abnormalities include heart defect, genu valgum, cornea defect, umbilical hernia, obesity, developmental delay, as well as CNS-related conditions including cerebral and cerebellar malformations, hydrocephaly, intellectual disability and schizophrenia (Taravath and Tonsgard, 1993; Balci et al., 1997; Bersani et al., 2003; Tarhan et al., 2004; Jenkins et al., 2007; Ivaniutsin et al., 2009; Alessandri et al., 2010; Haye et al., 2014). In mouse, the Rab23-encoding open brain (opb) null allele mutant exhibited embryonic lethality at midgestation stage, exencephaly and ectopic neural tube ventralization (Eggenschwiler et al., 2001, 2006), which largely recapitulated the phenotypes of other Shh repressor mutants such as Patched1 (Ptch1) and Suppressor of fused (Sufu) KOs (Cooper et al., 2005; Jeong and McMahon, 2005; Svärd et al., 2006). Nonetheless, owing to the early embryonic lethality of Rab23-null mutant in mouse, true implications of Rab23 in Shh signaling-mediated CNS development beyond the midgestation stage are not known.
Genetic study revealed that Rab23 represses hedgehog (Hh) activities via Gli2 and promotes the proteolytic cleavage of Gli3 into its cleaved repressor form (Eggenschwiler et al., 2006). In addition, Rab23 also appeared to regulate Hh pathway activity through Smo. Concomitant deletion of Smo in the Rab23-null mutant has partially weakened Shh activation level in the neural tube as compared with that of Rab23 mutant (Eggenschwiler et al., 2006). Besides, a molecular study in mammalian cell line model reported that Rab23 mediates the protein turnover dynamics of Smo in the primary cilium, although it was not clear as to how this may influence Shh pathway activity (Boehlke et al., 2010). Another in vitro study further revealed that Rab23 antagonizes the nuclear translocation of Gli1 transcription activator to impede Shh pathway activation (Chi et al., 2012). Taken together, these findings suggest that Rab23 casts multiple actions in the modulation of Hh signaling cascade. However, how it orchestrates Shh pathway in the context of GCP proliferation and medulloblastoma formation remains to be determined.
Although independent studies have implicated the functions of Rab23 in primary cilium formation and ciliary trafficking, its role in ciliogenesis remains obscure because of inconsistent observations from different cell types. For instance, overexpression of the dominant-negative form, Rab23DN perturbed ciliation in the immortalized retinal pigmented epithelial cells (Yoshimura et al., 2007). Supporting this observation, a recent study has identified that the GDP-GTP exchange factors (GEFs) of Rab23 namely Inturned and Fuzzy, were localized to the primary cilium at proximal end, and played essential role in the primary cilia formation of human and mouse cells (Gerondopoulos et al., 2019). The same study demonstrated that depletion of GEF (i.e., Intu and Fuzz), or Rab23 perturbed primary cilium formation in culture IMCD3 cells. On the contrary, Rab23−/− mouse embryo showed unaltered node cilia during early development (Fuller et al., 2014). Taken together, these data suggest that Rab23's action in the primary cilium formation is possibly operating in a context-dependent manner. In the IMCD3 cells that have morphologically normal primary cilium, Rab23 forms protein complex with Kif17 and dopamine receptor 1 (D1R), and it was required for their ciliary localization (Leaf and Von Zastrow, 2015; Lim and Tang, 2015). These findings indicated that Rab23 plays crucial roles in ciliary protein targeting. Despite the known function of Rab23 in primary cilium formation and Hh signaling, as well as the long-perceived function of primary cilium-dependent Hh signaling in GCP proliferation, whether Rab23 is required for the primary cilium formation in the CNS and cerebellar GCP is not known. Moreover, how Rab23 may mediate primary cilium-dependent Shh signal transduction, and its impact on GCP proliferation and cerebellar development remain to be further characterized.
In this study, we demonstrate that conditional KO (CKO) of Rab23 in the developing mouse brain at E10.5 resulted in abnormal cerebellar foliation, as well as unexpected opposing changes in the cerebellar sizes and Shh activities during embryonic and postnatal cerebellar development. Interestingly, our data suggest that loss of Rab23 did not cause medulloblastoma despite an increase in the basal level of Shh pathway activities and GCP proliferation. We found that KO of Rab23 affected ciliation in GCP, and rendered the cells less responsive to pathway activation by Shh and Smo agonist. These results suggest that the Rab23-KO GCPs have an attenuated response to paracrine Shh stimuli from primary cilium. Taken together, we have uncovered novel functions of Rab23 in GCP proliferation, acting both positively and negatively via Shh signaling. Our results indicate that Rab23 represses basal level of Shh signaling pathway activities, while facilitating Smo-mediated Shh pathway activation in a primary cilium-dependent manner.
Materials and Methods
Animals
Rab23-floxed animal was generated by Ozgene Pty Ltd. Conditional Rab23-floxed allele was designed by flanking exon 4 for Rab23 gene with loxP sites. Nestin-Cre (The Jackson Laboratory catalog #003771) was a kind gift from Je H. S. Shawn form Duke-NUS Medical School. Animals were housed in animal facility, Department of Chemistry, HKBU, and specific pathogen-free (SPF) animal facility at Duke-NUS Medical School, Singapore. All animal related procedures were conducted in compliance to animal handling guidelines and protocol approved by IACUC Singhealth, Singapore, and Department of Health, Hong Kong. The animal experiments described in this project included unbiased data from both female and male mice unless otherwise specified.
Experimental design
Neural progenitor cell-specific conditional deletion of Rab23 [from approximately embryonic day (E)10.5] was achieved by cross-breeding homozygous Rab23f/f mouse with double allele Rab23f/f;NestinCre+/− mouse to obtain littermates consisting mixture of Rab23f/f and Rab23f/f;NestinCre+/− within the same litter. Throughout all experiments described, control animal refers to heterozygous or homozygous Rab23-flox male/female, whereas mutant or KO refers to homozygous Rab23-flox (Rab23f/f) with heterozygous NestinCre double allele mutant, Rab23f/f;NestinCre+/−.
Expression vectors
For in vitro viral transduction assay, Rab23 overexpression or cDNA were cloned into lentiviral pFUGW backbone. Wild-type (WT) Rab23 overexpression construct, previously described full-length Rab23 sequence (Guo et al., 2006) was subcloned into pFUGW vector driven by Ubc promoter. All plasmids were amplified according to the recommended protocol using Endofree plasmid purification kit (QIAGEN).
Viral transduction and culturing of mouse primary GCP
For viral transduction of primary GCP, self-inactivating murine lentiviruses were prepared according to previously described protocol (Hor and Goh, 2018). GCP culture method was modified from standard protocol. Briefly, P6 or P7 cerebellar tissues dissected were cut into small pieces and digested in digestion buffer [EBSS/Papain, 1000 times dilution factor (Worthington Biochemical Corporation catalog #3126)/0.1 mg/ml DNaseI (Roche catalog #11284932001)/5.5 mm cysteine-HCl] for 15 min at 37°C before dissociation into single cells. Digestion was terminated by resuspension in 10% fetal bovine serum/culture medium. Suspension culture was passed through 70-μm cell strainer (Corning catalog #352350) to remove undigested tissue clumps. Dissociated single-cell GCPs were preplated on 100 µg/ml poly-D-lysine (Sigma-Aldrich, catalog #P6407)-coated culture plates for 20 min at 37°C. After which cells were dislodge and immediately seeded on low concentration poly-D-lysine and matrigel precoated culture plates at the desired cell densities in Neurobasal (Invitrogen, Life Technologies) medium containing B27 supplement, 200uM GlutaMAX-I (Invitrogen, Life Technologies), sodium pyruvate (1 mm), penicilin/streptomycin, and KCl (250 μm). Half of the culture medium was refreshed every other day. Viral transduction was performed 2–3 h after culture while replacing fresh culture medium. The efficiencies of overexpression were validated by GFP signals and real-time quantitative (Q)PCR assay of DIV7 culture. For SAG stimulation, 0.2 μm SAG (Cayman Chemical, catalog #11914-1) was added to the DIV1 culture 24 h before total RNA extraction. Equal volume of dimethylsulfoxide (DMSO) was added as the untreated negative control group. For Shh stimulation, 2 μg/ml of Shh (Stem Cell Technologies, catalog #78065) was added to the DIV1 culture 24 h before total RNA extraction.
EdU-pulse labeling assays
EdU labeling assay was conducted according to the manufacturer's protocol. Click-iT EdU Alexa Fluor 647 Imaging kit (ThermoFisher Scientific, catalog #C10340). For GCP culture labeling, 10 μm Edu was added to the culture and incubated for 4 h before fixation. For E15.5 embryos labeling, 0.25 mg EdU was injected intraperitoneally into the pregnant mice 2 h before fixing the embryo. For postnatal animals, 25 µg of EdU was injected subcutaneously 2 h before brain fixation.
Gli activity reporter assay
Lentiviral carrying 7Gli:GFP reporter construct (Addgene #110494; Li et al., 2016) at preoptimized viral transduction titer were used to infect primary GCPs on 96-well plate 2 h after cell seeding. GCPs were seeded at equal cell density at 6.6 × 105 per well. Green fluorescence reporter signals were measured with fluorescence microplate reader on DIV3 where optimal signal intensity was detectable.
Cryosectioning, immunohistochemistry, and imaging
Mice were perfused with saline followed by fixative in 4% paraformaldehyde (Sigma-Aldrich, catalog #P6148)/HistoChoice (Amresco, catalog #H120) mixture of 1:1 ratio, and whole brains extracted were postfix at 4°C for 2 h, saturated in 30% sucrose in 0.12 m phosphate buffer and subjected to cryosection at 20-μm thickness. All cerebellar tissues were sectioned at sagittal angle and mounted on precoated glass slides (Superfrost Plus, Fisherbrand). Midsagittal sections were selected for immunostaining. Antibodies and the dilution factor used were: Pax6 (Covance, 1:1000), Nestin (Sigma, 1:800), NeuN (Milipore, 1:800), glial fibrillary acidic protein (GFAP; Milipore, 1:1000), and Arl13b (Proteintech, 1:1000). For histoimmunostaining, tissue sections were incubated at 100°C for 10 min in pH 6, 10 mm sodium citrate buffer with 0.05% Tween 20 (Sigma-Aldrich, catalog #P1379) for antigen retrieval, washed twice with PBS, blocked 1 h in 1% BSA (Sigma-Aldrich, catalog #A7906)/2% horse serum/0.3% Triton X-100 (Sigma-Aldrich, catalog #X100) and incubated 4°C overnight with primary antibodies diluted in blocking buffer. After three times of 5-min washes with PBS, tissue sections were incubated with secondary antibodies (Alexa Fluor, Life Technologies) for 1 h at room temperature. Tissue sections were mounted in mounting media after three times PBS washes. Fluorescence images were taken using Zeiss LSM710 confocal system.
Real-time QPCR
Total RNA was extracted using QIAGEN's RNeasy Mini kit. Equal amount of total RNAs from each sample were subjected to reverse transcription to produce cDNA. Equal volume of cDNA was used to perform QPCR assay using SYBR Select Master Mix (Applied Biosystems #4472908). Standard QPCR protocol was conducted according to manufacturer's instruction manual. Primers used were: mouse GAPDH: F-5′-TTCACCACCATGGAGAAGGC-3′, R-5′-GGCATGGACTGTGGTCATGA-3′; mouse Rab23: F-5′-AGGCCTACTATCGAGGAGCC-3′, R-5′-TTAGCCTTTTGGCCAGTCCC-3′; mouse Gli1: F-5′-CCCATAGGGTCTCGGGGTCTCAAAC-3′, R-5′-GGAGGACCTGCGGCTGACTGTGTAA-3′; mouse Gli2: F-5′-CATGGTATCCCTAGCTCCTC-3′, R-5′-GATGGCATCAAAGTCAATCT-3′; mouse Gli3: F-5′-CATGAACAGCCCTTTAAGAC-3′, R-5′-TGATATGTGAGGTAGCACCA-3′; mouse Ptch1: F-5′-TGCTGTGCCTGTGGTCATCCTGATT-3′, R-5′-CAGAGCGAGCATAGCCCTGTGGTTC-3′; mouse Atoh1: F-5′-AGTCAATGAAGTTGTTTCCC-3′, R-5′-ACAGATACTCTTATCTGCCC-3′; mouse Ki67: F-5′-CATTGACCGCTCCTTTAGGTATGAAG-3′, R-5′-TTGGTATCTTGACCTTCCCCATCAG-3′.
Statistical analysis
Unpaired Student's t test was used for comparison between two groups. For comparison of more that two groups, one-way AVONA, Bonferroni's multiple comparison test was used. For comparison between two or more groups with two variable factors, two-way ANOVA, Bonferroni post hoc tests was used; ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05, unless otherwise stated. Error bars depict ±SEM. For primary GCP culture data, all statistical results were collected from at least three to five independent primary cultures of GCPs isolated from multiple cerebellar tissues in which samples of the same genotype were grouped together in each experiment.
Results
Rab23 dictates proper cerebellar morphogenesis and development
In order to investigate the functions of Rab23 in CNS development, mouse bearing Nestin-cre (Nes) was crossed with Rab23-floxed (Hor and Goh, 2018) homozygous mutant to achieve CKO of Rab23 in the neural progenitor cells at ∼E10.5. Gross morphologic examination of the whole brain isolated from Nes-CKO mutant revealed noticeable cerebellar enlargement at earlier developmental stages [i.e., postnatal day (P)1 and P4] but appeared smaller at later adult stage as compared with the control (Rab23f/f) counterpart (Fig. 1A,B,E, yellow asterisk). Histologic examination of the midsagittal cerebellar sections by hematoxylin-eosin (H&E) staining revealed cerebellar dysplasia in Nes-CKO brains. This was consistently observed at P1, P4, and adult stages (Fig. 1C,D,F). Disrupted patterning of the cerebellar folia was more prominent at the caudal region. Moreover, the external granular layer (EGL) at the posterior lobules appeared thicker and disorganized as compared with the control group (Fig. 1C,D, red arrows). In the adult mutant, the posterior cerebellar folia were irregularly formed and lack of distinctive laminar layering of molecular layer (ML), Purkinje cell layer (PCL), and internal granule layer (IGL; Fig. 1F). Taken together, these data indicate that loss of Rab23 resulted in defective cerebellar folia patterning during postnatal CNS development.
Figure 1.

Nestin-Cre-driven KO of Rab23 causes expanded cerebellar size and abnormal foliation. A, Representative whole mount images of control and Nes-CKO mutant brains showing gross morphology of mouse cerebellum at P1, P4, and P15. B, P4 cerebellar sizes as determined by measuring 2D surface area of cerebellum on images captured at similar angle. Control, n = 6; CKO, n = 4. Statistical significance, unpaired Student's t test; **p ≤ 0.01. Error bars depict ±SEM. C, D, Representative images showing H&E staining of control and Nes-CKO cerebellar sagittal sections of P1 (C) and P4 (D) animals. Red arrows highlight morphologic changes in the external granule layer of Nes-CKO compared with the control. E, Representative image showing whole brain of two-month adult mice. Yellow asterisk shows smaller cerebellum of Nes-CKO mutant compared with the control. F, Representative images showing H&E staining of sagittal cerebellar sections of 10-month adult mice.
Depletion of Rab23 disrupted cerebellar radial glial scaffold and innervations of granule cells
The disorganized laminar layering, as well as the cerebellar folia anomaly prompted further examination of the Bergmann glial (Bg) scaffold, which acts as the cytoarchitectural scaffold to aid in neuronal migration and lamination (Sidman and Rakic, 1973; Yue et al., 2005; Xu et al., 2013). We used antibodies against Nestin and GFAP to immunolabel radial glia and the Bg scaffold at early postnatal and later adult stages, respectively. In the Rab23f/f (control) group, radial fibers of Bg at P1, P4, and P15 appeared perpendicularly aligned and extended from the cell bodies at the lower ML and PCL toward the pial surface of the cerebellum. In contrast, the processes of Nes-CKO mutant Bg in the disrupted lobules appeared tangled and misaligned, with some of them unable to extend processes to the pial surface, thus indicating an impairment of the Bg scaffold (Fig. 2A–C). Additionally, hyperplastic lesion-like ectopic nuclei accumulation were detectable at the pial surface in P4 (Fig. 2B, asterisk) and adult cerebellum (Fig. 2D, white arrows). In line with this defect, the NeuN-positive granule cells in the adult mutant were aberrantly localized to the pial surface and the ML instead of the deeper IGL. In addition, a subpopulation of the granule cells was randomly scattered at the posterior region, concomitant with a loss of laminar structure. The GFAP-positive astrocytes/Bg were also found to be ectopically misplaced at the pial surface and ML, indicating a misalignment of radial glial scaffold at the adult stage (Fig. 2D,D',D''). These data indicate that an abnormal glial scaffold in Nes-CKO mutants may hinder proper invagination and migration of granule cells to the deeper IGL during early postnatal cerebellar development.
Figure 2.

KO of Rab23 perturbs radial glial scaffold formation and causes partial loss of cerebellar laminar structure. A, B, Representative images showing immunostaining of Nestin (red) on P1 (A) and P4 (B) sagittal cerebellar tissue sections to illustrate radial glial scaffold. C, Representative images showing immunostaining of GFAP (red) on P15 sagittal cerebellar tissue sections to illustrate radial glial scaffold. D, Representative images showing co-immunostaining of NeuN and GFAP on 10-month adult cerebellum to illustrate cerebellar cytoarchitecture, laminar layers and glial cells. D', D'', Close up images showing NeuN-positive granule neurons and GFAP-positive glial cells at the IGL and pial surface.
Given the defective radial glial scaffold and ectopic accumulation of granule cells at the pial surface and ML, we asked whether the inward radial migration of granule cell at the earlier embryonic and postnatal stages was affected. Anti-Pax6 antibody was used to immunolabel both amplifying GCPs transiently residing in the EGL (source of granule cells), and the early inwardly migrated postmitotic granule cells in the granular layer at E15.5 (Chung et al., 2010). The Pax6-expressing granule cells in the control group were more well dispersed and scattered further into the deeper granule layer. Conversely, Rab23-depleted granule cells appeared less dispersed and are largely confined to the region adjacent to the EGL (Fig. 3A, yellow dotted lines). Since all granule cells arise from the EGL and innervate the inner granular layer as they undergo maturation, we quantified the innervation/migration rate by counting the number of Pax6-positive cells that have populated the inner granular layer (innervated) against total Pax6-positive cells. Indeed, the proportion of innervated granule cells in the Nes-CKO mutant appeared markedly reduced compared with the control counterpart (Fig. 3B), suggesting an impaired or delayed innervation. In addition, a 2-h EdU-pulse labeling assay was used to track all early innervated progenitor cells. The proportion of EdU-labeled cells that have innervated (from EGL, magenta arrows and VZ, yellow arrows) the granular layer was scored against all EdU-labeled cells. Similarly, Rab23-depleted cells showed a lower percentage of innervated cells in the granular layer (Fig. 3C).
Figure 3.
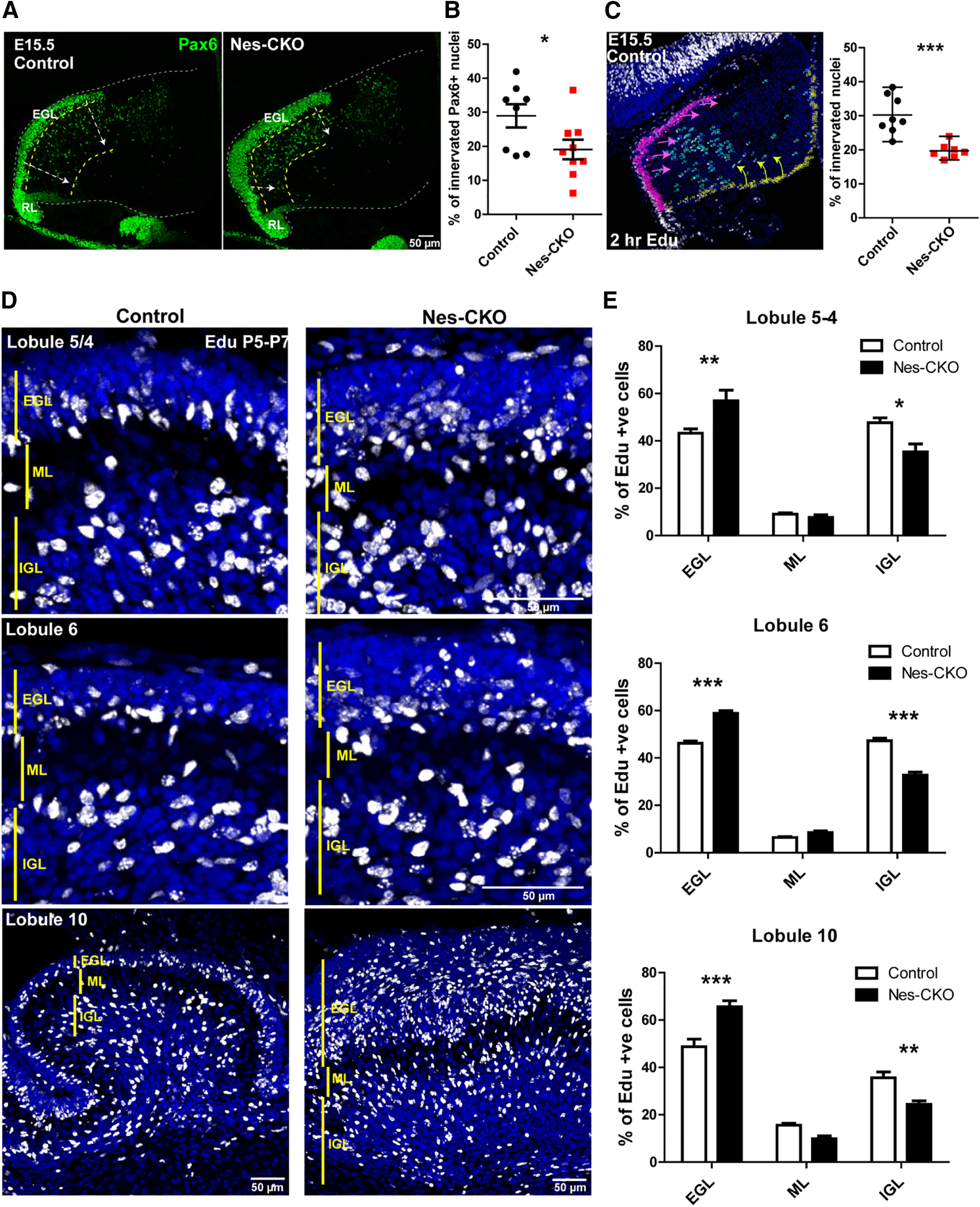
Depletion of Rab23 leads to GCP migration defect. A, Representative images showing immunostaining of Pax6 on E15.5 sagittal sections of cerebellar primordium to illustrate the GCPs residing in the EGL and early inward migrating GCPs. White arrows show inward migration paths. RL, rhombic lip. B, Quantification of the proportion of innervated Pax6+ GCPs against all Pax6-labeled GCPs. Two to three sections (∼50–100 μm apart) of the cerebellar primordium were counted for each animal. Control, n = 3; CKO, n = 3. Statistical significance, unpaired Student's t test; *p ≤ 0.05. Error bars depict ±SEM. C, Representative image and graph showing 2-h EdU-labeled progenitors in the cerebellar primordium. Magenta arrows show migration paths of progenitors from EGL, yellow arrows show migration paths of progenitors from VL. Control, n = 3; CKO, n = 3. Two to three sections (∼50–100 μm apart) were counted for each animal. Statistical significance, unpaired Student's t test; ***p ≤ 0.001. Error bars depict ±SEM. D, Representative images showing cerebellar lobules of 48-h Edu-labeled cells from P5 to P7 to illustrate proportions of cells innervated the IGL after 48 h of pulse-chase labeling. E, Quantification of the proportion of Edu-labeled cells in each laminar layer as indicated. Control, n = 3; CKO, n = 3. Statistical significance, two-way ANOVA, Bonferroni post hoc tests; ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05. Error bars depict ±SEM. EGL- External Granular Layer; ML-Molecular Layer; IGL- Internal Granule Layer.
For the postnatal stage, we examined the migration of granule cells by 48-h EdU-pulse labeling for P5–P7. The percentages of cell innervation in different lobules were then analyzed by quantifying the percentages of EdU-labeled cells residing in the EGL, ML, and IGL of each lobule. In agreement with the results from the embryonic stage (Fig. 3A–C), the proportions of mutant cells reaching IGL were greatly reduced, concomitant with an increase in the percentage of cells that were accumulated in the EGL (Fig. 3D,E). Taken together, these data suggest that deletion of Rab23 caused a misalignment of the radial glial scaffold, leading to perturbations in granule cells innervation and lamination. As a result, the cerebellar laminae and folia could not be properly formed during postnatal cerebellar development.
Rab23 ablation caused thickened EGL and enhanced GCP proliferation but no discernible tumorigenesis
In view of the thickened EGL observed in H&E staining, as well as the enlarged cerebellum of Nes-CKO at P1 and P4, a more detailed analysis of cell proliferation is warranted. We performed co-immunolabeling of Pax6 and Calbindin to visualize the organization of GCP and Purkinje cells, respectively. At P1, there was an overall increase in the number of Pax6-expressing GCPs in the Nes-CKO mutant as compared with the control. Besides, the Pax6-labeled EGL in Nes-CKO appeared greatly thickened, which was more profound near the posterior folia (Fig. 4A, white asterisks). On the other hand, the Calbindin-expressing Purkinje cells in the mutant PCL appeared to be lower in density and more sparsely distributed as compared with the more densely aligned PCL in the control counterpart (Fig. 4B), implying a perturbed PCL lamination.
Figure 4.
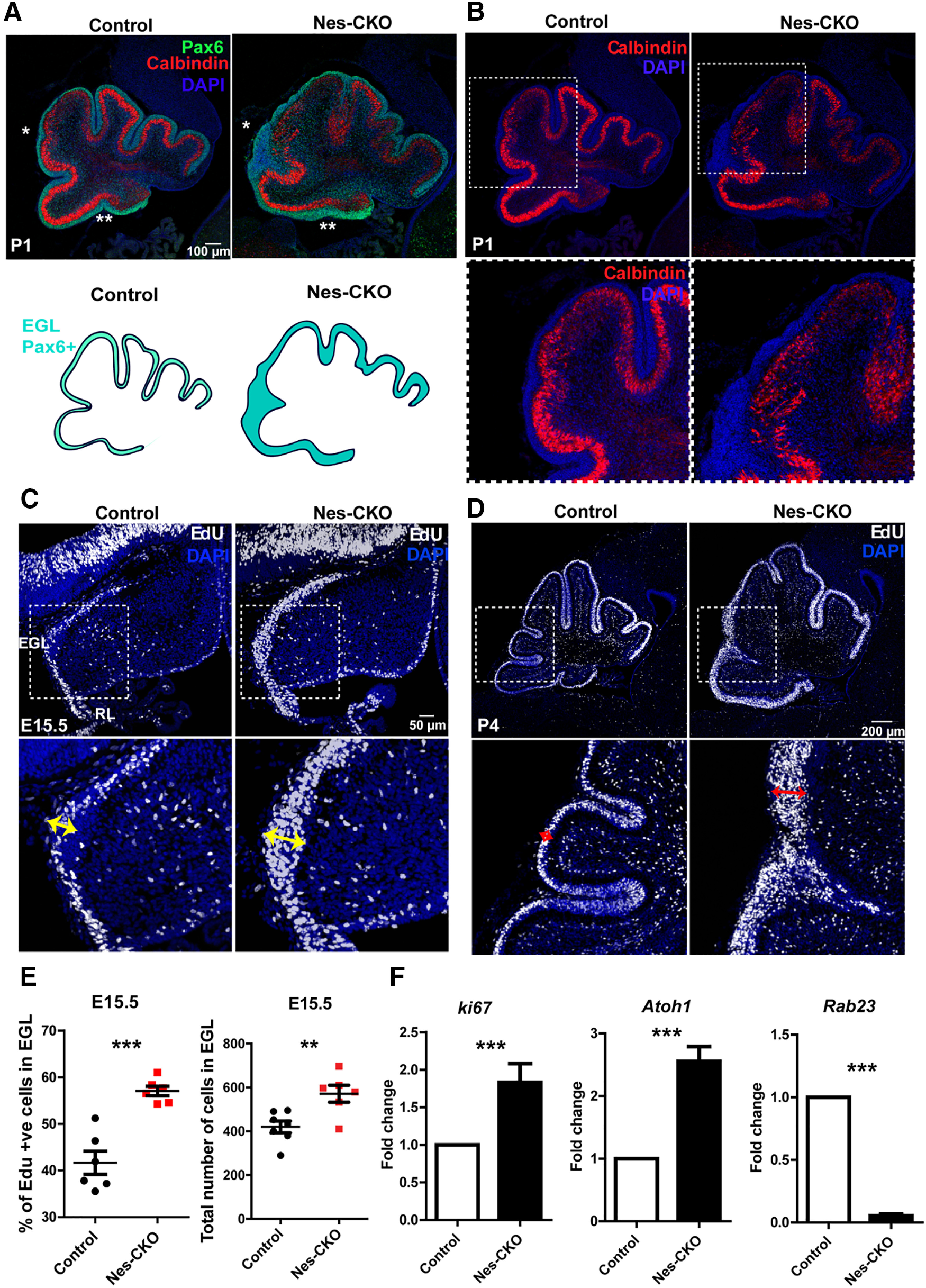
Rab23-deficient cerebellum exhibited thickened EGL and elevated GCP proliferation. A, B, Representative images showing co-immunostaining of Pax6 (green) and Calbindin (red) on P1 sagittal sections of cerebellum to illustrate the GCPs and Purkinje cells layers. Asterisks show a thickened EGL layer in Nes-CKO cerebellum compared with the control. C, D, Representative images showing 2-h EdU-labeled dividing progenitors in the E15.5 (C) and P4 (D) cerebellum. Double headed arrows highlight expanded pools of dividing cells in the Nes-CKO EGL as compared with the control counterparts. E, Graphs depict quantification of the percentages, and the total numebr of 2-h Edu-labeled proliferative cells in the EGL at E15.5. Two to three sections (∼100 μm apart) of the cerebellar primordium were counted for each animal. Control, n = 3; CKO, n = 3. Statistical significance, unpaired Student's t test; ***p ≤ 0.001, **p ≤ 0.01. Error bars depict ±SEM. F, Graphs illustrating the fold change of the gene expression levels of E15.5 cerebellar tissues quantified by real-time QPCR. Control, n = 4; CKO, n = 4. Nes-CKO values were normalized to its respective control group. Statistical significance, unpaired Student's t test; ***p ≤ 0.001. Error bars depict ±SEM.
Given the thickened EGL observed, 2-h EdU-pulse labeling was performed on E15.5 and P4 animals to probe GCP proliferation in further details. Compared with the control, the pools of EdU-positive proliferative cells are substantially expanded in the EGL of Nes-CKO mutant at both time points (Fig. 4C,D, red and yellow double heads arrows), indicating aberrantly enhanced GCP proliferation during both embryonic and postnatal cerebellar development. These phenotypes were further confirmed by a quantification of EdU-positive nuclei in the EGL at E15.5, which revealed a significant upregulation of proliferative cells in the mutant EGL as compared with the control group (Fig. 4E). Accordingly, another cell proliferation marker, Ki67, and a GCP-specific marker Atoh1, also showed markedly elevated expression levels in the cerebellar tissue of Nes-CKO mutants (Fig. 4F). Taken together, these data suggest that depletion of Rab23 potentiated GCP proliferation during early cerebellar development. Excessive GCP proliferation often give rise to medulloblastoma (Yoshioka, 2005; Roussel and Hatten, 2011). Given this, one would speculate the development of medulloblastoma at a later postnatal stage. However, we did not find detectable manifestation of medulloblastoma in adult mutant animals, despite the occurrence of hyperplastic lesions-like tissue clumps at P4 (Figs. 1F, 2B, asterisk, 2D, white arrows).
Shh signaling is differentially perturbed in the developing cerebellum
Previous genetic studies have reported that Rab23 negatively regulates Shh signaling (Eggenschwiler et al., 2001; Evans et al., 2005). As Shh signaling is the key signaling pathway that modulates GCP proliferation (Wallace, 1999; Wechsler-Reya and Scott, 1999; Corrales et al., 2004; Lewis et al., 2004), we reasoned that it is likely a main factor driving aberrant GCP proliferation in the Rab23-deficient cerebellum. To address this possibility, we examined the expressions of Gli transcription factors, which are downstream effectors of Shh signaling in CNS development. The Shh signaling activities in cerebellar tissue were examined in both embryonic E15.5 and late postnatal P15. In accordance with the increased CGP proliferation in Nes-CKO at E15.5 and P4, Shh signaling pathway activities were robustly upregulated at E15.5, as shown by an increase in Gli1 and Gli2 expressions compared with the control group (Fig. 5A). Intriguingly, at P15, the expression level of Gli1 transcripts was significantly downregulated in the Nes-CKO mutant cerebellum, despite upregulated levels of Gli2, Gli3, Ki67, and Atoh1 (Fig. 5B). Given that Gli1 activates Shh-regulated genes, and its expression is dependent on both Gli2 and Gli3, it could serve as the ultimate readout of Shh signaling pathway activity (Lee et al., 1997; Bai and Joyner, 2001; Bai et al., 2004). We also compared the expression profile of Gli1 transcripts at embryonic and postnatal stages. Compared with the control group which exhibited relatively unaltered Gli1 transcript level between E15.5 and P15, the Nes-CKO mutant showed a significant reduction in Gli1 transcript level at P15 compared with its embryonic stage (Fig. 5C). Given the perturbed Shh signaling pathway activities, we further examined whether the Shh transcripts were affected. Interestingly, Shh transcripts level in the Nes-CKO appeared largely unchanged at both developmental time points (Fig. 5A,B), implying that the mutant cerebellar tissues are not short of Shh stimulants despite the greatly mis-patterned cerebellum. Taken together, these data show that the Shh signaling activities in the Nes-CKO mutants were initially enhanced during embryonic cerebellar patterning, but became downregulated at later postnatal time point as compared with the control counterpart. Notably, this correlated well with the differential changes in the cerebellar size as aforementioned (Fig. 1A,B,E). Together, these results revealed that Shh signaling pathway activities were differentially perturbed as a result of Rab23 deficiency during embryonic and postnatal stages of cerebellar patterning.
Figure 5.

Shh activity is differentially perturbed in the embryonic and postnatal cerebellar tissues. A, Graphs illustrating the fold change of the gene expression levels of E15.5 cerebellar tissues quantified by real-time QPCR. Control, n = 4; CKO, n = 4. Nes-CKO values were normalized to its respective control group. Statistical significance, unpaired Student's t test; **p ≤ 0.01. Error bars depict ±SEM; n.s., not significant. B, Graphs illustrating the fold change of the gene expression levels of P15 cerebellar tissues quantified by real-time QPCR. Nes-CKO values were normalized to its respective control group. Control, n = 3; CKO, n = 4. Statistical significance, unpaired Student's t test; ***p ≤ 0.001. Error bars depict ±SEM; n.s., not significant. C, Graphs illustrating the basal level Gli1 expression profiles of E15.5 and P15 cerebellar tissues quantified by real-time QPCR. E15.5 control, n = 4; CKO, n = 4; P15 control, n = 3; CKO, n = 4. Statistical significance, one-way AVONA, Bonferroni's multiple comparison test; ***p ≤ 0.0001. Error bars depict ±SEM; n.s., not significant.
Rab23 could regulate shh signaling in the GCP at basal level as well as in a cilium-dependent manner
The alterations of Shh signaling pathway activities on GCP in the whole cerebellar tissues could be a secondary effect resulting from abnormal changes in the cellular/tissue composition. We ruled out this possibility by monitoring Shh signaling in primary GCP culture isolated from P6/7 cerebellar tissue. Primary culture data showed that Rab23-KO GCP indeed exhibited significantly elevated basal level expression of Gli1 mRNA as compared with that of the control group (Fig. 6A). Moreover, to measure the basal level Gli transcriptional activity, we expressed 7Gli:GFP reporter containing seven Gli responsive elements that drive the expression of GFP reporter in the control and KO-GCPs, respectively. In resonance with the upregulated Gli expression, Rab23-KO GCP exhibited higher intrinsic Gli transcription reporter activity, indicating a transcriptional upregulation of Shh signaling pathway in the absence of Rab23 (Fig. 7A). Accordingly, primary culture of mutant GCP also displayed potentiated cell proliferation, as illustrated by the upregulation of Atoh1 and Ki67, as well as an increase in the percentage of EdU-positive proliferative GCPs (Fig. 6A–C). Furthermore, Shh pathway overactivation and cell proliferation were significantly inhibited by co-expressing Rab23 WT cDNA, or its constitutive active form, Rab23QL, in the KO GCP (Fig. 6A–C), suggesting that the effects observed were indeed because of the loss-of Rab23 gene functions. Together, these results suggest a negative role for Rab23 in regulating Shh signaling-mediated GCP proliferation.
Figure 6.
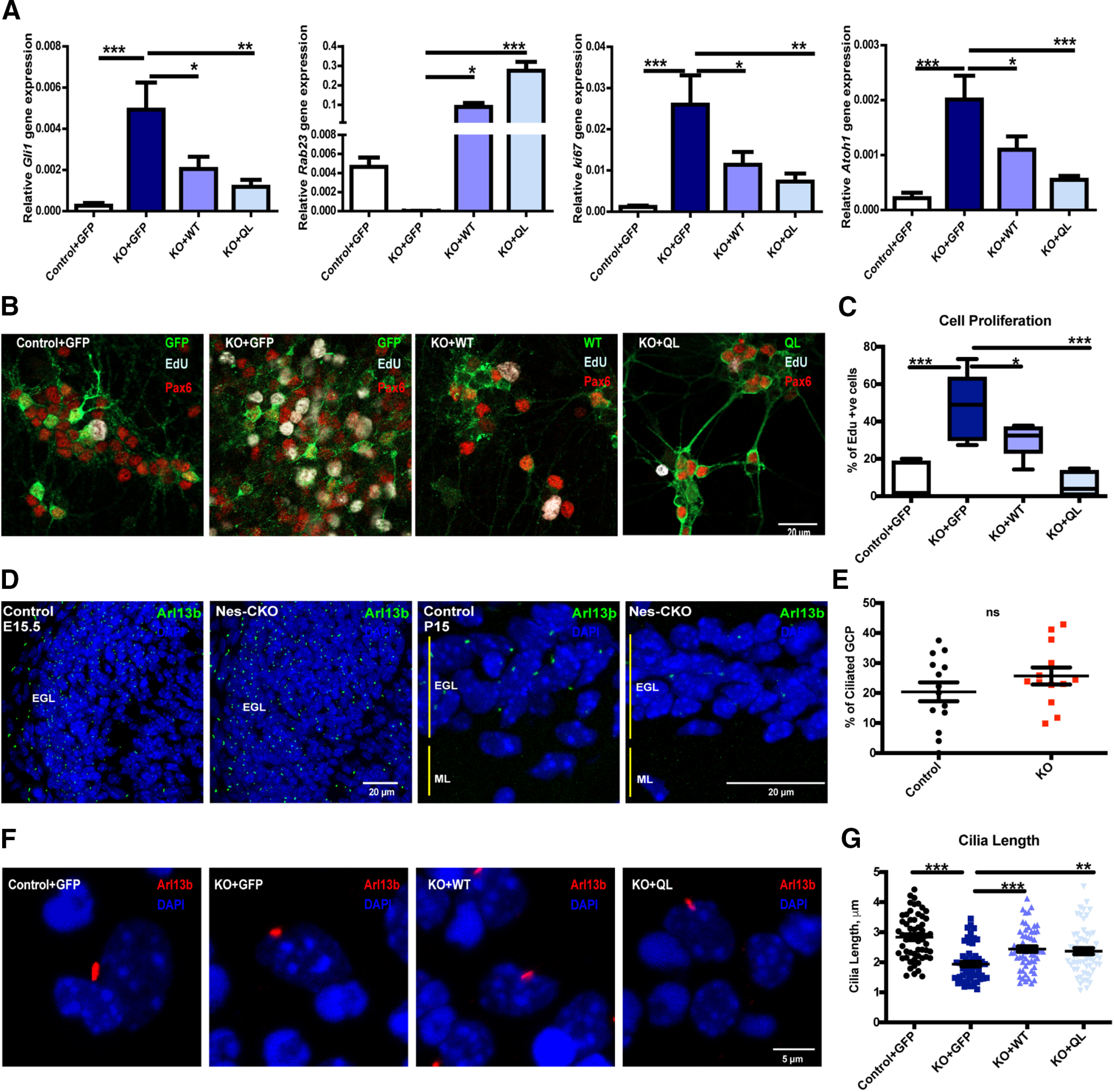
Rab23 regulates GCP proliferation and ciliogenesis. A, Graphs showing gene expression levels of P7 GCPs primary cultures harvested at DIV7. Lentiviral carrying the overexpression constructs as indicated were transduced into the primary cultures at day 0, 2 h after seeding cells. Quantifications depict four independent experiments. Statistical significance, one-way AVONA, Bonferroni's multiple comparison test; ***p ≤ 0.0001, **p ≤ 0.01, **p ≤ 0.05. Error bars depict ±SEM. B, C, Representative images (B) and graph (C) showing 4-h EdU-labeled (pseudocolored white; blue: DAPI; green: overexpressed GFP-positive construct as indicated; red: Pax6) dividing progenitors in P6 GCPs primary cultures of each indicated groups fixed at DIV6. Cell proliferation was determined by the percentages of Edu-labeled cells out of total number of GFP/Pax6-positive nuclei in each image taken. For quantification of each batch, two to three fluorescence images were randomly taken from each respective group as indicated. Quantifications depict three independent experiments. Statistical significance, one-way AVONA, Bonferroni's multiple comparison test; ***p ≤ 0.0001, *p ≤ 0.01. Error bars depict ±SEM. D, Representative images showing immunostaining of Arl13b on E15.5 (left) and P15 (right) sagittal sections to illustrate the primary cilia of GCPs residing in the EGL. E, Graph showing quantification of the percentages of ciliation in P6 primary cultures GCPs at DIV2 (Day In Vitro) determined by counting the number of Pax6-postive cells bearing Arl13b-labeled primary cilium against all Pax6-positive GCP nuclei in each image taken. For quantification of each independent experiment, four to five fluorescence images of 30–50 cells were randomly taken from each respective group as indicated. Quantifications depict three independent experiments. Statistical significance, unpaired Student's t test; n.s., not significant. Error bars depict ±SEM. F, G, Representative images (F) and graph (G) showing distribution of the primary cilia length in P6/7 GCPs primary cultures at DIV7. Each dot represents every Arl13b-labeled primary cilium length measurement on Pax6/GFP-positive (data not shown on representative images) viral-transduced cells. Quantification depicts three independent experiments of a total of 58 cells measured for each respective group as indicated. Statistical significance, one-way AVONA, Bonferroni's multiple comparison test; ***p ≤ 0.0001, **p ≤ 0.01. Error bars depict ±SEM.
Figure 7.
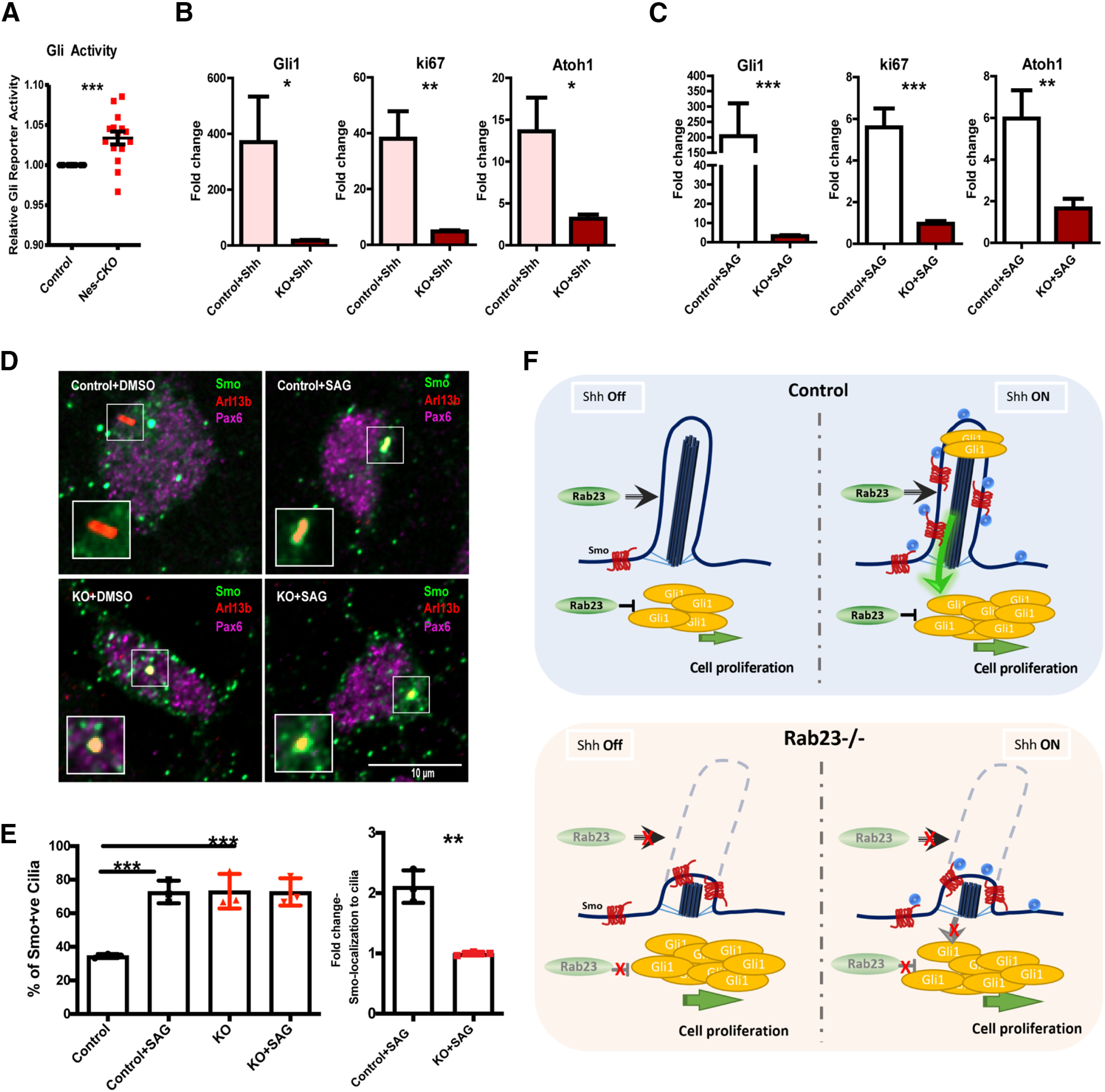
Rab23 regulates basal level and primary cilium-dependent Hh signaling activity. A, Graph depicts relative basal level 7Gli:GFP reporter signals on DIV3 (Day In Vitro) primary GCP cultures. Quantifications show quadruplicates of four independent experiments. Statistical significance, unpaired Student's t test; ***p ≤ 0.0001 B, C, Graphs showing gene expression levels of P7 GCPs primary cultures treated with Shh (B) and SAG (C) on DIV1, respectively. Total RNAs were extracted from DIV2 culture, 24 h after the respective treatments. Quantifications depict double δ Ct values of three independent experiments. δ Ct values of the treated groups were normalized to its respective untreated group, which gave double δ Ct values as plotted. Statistical significance, unpaired Student's t test; ***p ≤ 0.0001, **p ≤ 0.01. Error bars depict ±SEM. D, E, Representative images (D) and graphs (E) depict quantification of the percentage and the relative fold change of Smo-localization in the primary cilia of DIV3 Smo-EGFP-electorporated primary GCPs culture 24 h post-treated with DMSO and SAG, respectively. Quantifications show three independent experiments. Statistical significance, one-way AVONA, Bonferroni's multiple comparison test; ***p ≤ 0.0001, **p ≤ 0.01. Error bars depict ±SEM. F, Schematic illustration of proposed model. Top, Hh signaling in the presence of Rab23. Rab23 inhibits Gli1 expression intracellularly at basal level. Rab23 also promotes the maintenance of primary cilium on granule cell progenitor. Upon Shh ligand stimulation on the primary cilium, Smo translocates to cilia axoneme, Gli1 expression increased, and Hh signaling is elevated. Bottom, In the absence of Rab23, Smo is enriched in cilia axoneme, Gli1 expression is upregulated independent of Shh ligand stimulation. The primary cilium formation and/or maintenance are compromised, the GCP is not responsive to Shh ligand stimulation from the cell surface (paracrine signaling). Ligand-induced Smo-enrichment in cilia axoneme is not triggered.
Previous findings have demonstrated that primary cilium is required for Shh signaling-mediated CGP proliferation during cerebellar development (Chizhikov et al., 2007; Spassky et al., 2008). Although current evidence on the in vivo roles of Rab23 in ciliogenesis appear controversial, Rab23 has been reported to mediate ciliogenesis and ciliary signaling in some non-neuronal cell types in vitro (Yoshimura et al., 2007; Leaf and Von Zastrow, 2015; Lim and Tang, 2015). Therefore, it is conceivable that the impact of Rab23 on GCP proliferation was exerted through changes on the primary cilium. To address this hypothesis, we examined primary cilia morphology in the E15.5 and P15 cerebellar GCP in vivo by immunolabeling of Arl13b, a primary cilium-specific marker. Intriguingly, a temporal difference in the primary cilium morphology was observed. Our data showed that the Rab23-depleted GCPs in the external granule layer (EGL) exhibited similar frequency of ciliation as compared with that of control at E15.5. Conversely, an apparent reduction in the number of GCPs bearing Arl13b-positive primary cilium was observed in postnatal stage P15 mutant (Fig. 6D). Furthermore, in vitro analysis of primary cilium on primary GCP culture isolated from P6 cerebellar tissues revealed that Rab23-deleted GCPs showed comparable frequency of ciliation similar to that of the control counterpart (Fig. 6E). Nonetheless, Rab23-deleted GCPs in culture appreared to display a significantly shorter cilia length, which could be reversed by co-expressing Rab23 WT cDNA, or the constitutive active Rab23QL (Fig. 6F,G). Taken together, our data provide the first indication that Rab23 influences ciliogenesis in the cerebellar GCP in vivo. Importantly, these results also hinted at a novel primary cilium-dependent role of Rab23 in coordinating Shh pathway and GCP proliferation.
Given the perturbations in primary cilia morphology, we hypothesized that Rab23-KO GCP may have a compromised primary cilium-dependent Shh signal transduction. In order to address this hypothesis, primary GCP cultures were subjected to Shh ligand stimulation in vitro. Despite its higher basal level of Shh signaling activities as compared with the control cells (Figs. 6A, 7A), Rab23-KO GCP showed a weaker response to Shh ligand stimulation, as illustrated by a lower fold increase in the expression of Gli1 mRNA as compared with the control counterpart (Fig. 7B). This observation suggests that extrinsic Hh signaling transduction was impaired in the Rab23-KO GCPs. Besides, in response to Shh ligand stimulation, Rab23-KO GCPs also exhibited lower fold enhancement in cell division as compared with the control group, in which the expression of Ki67, as well as Atoh1 were both significantly lower than that of control group (Fig. 7B). This result indicates that Rab23 regulates GCP proliferation via Shh signaling pathway.
To further probe whether the Shh signaling activation occurs via Smo on the primary cilium, GCP cultures were treated with SAG, a Smo agonist that promotes Smo-localization to the cilium to activate Hh signaling pathway. Control GCPs exhibited robust elevation of Gli1 expression 24 h after SAG treatment (Fig. 7C). Conversely, Rab23-KO GCPs' response to SAG induction was significantly compromised, as shown by lower expression level of Gli1 (Fig. 7C), which implicates a desensitization to Shh signaling activation at the level of primary cilium and Smo. In order to further confirm that the Smo-translocation to the primary cilium was perturbed in the mutant GCPs, we examined the frequency of Smo-localization on the primary cilia in the KO GCP culture. Our results show that the frequency of Smo-localization to the primary cilium was significantly increased by about two-fold in the control GCPs on SAG stimulation. However, mutant GCPs exhibited unaltered frequency of Smo-localization to the primary cilium after the treatment of SAG (Fig. 7D,E), indicating that ligand-induced active translocation of Smo to the primary cilium was disrupted in the absence of Rab23, thereby desensitizing the cells to extrinsic Hh pathway stimuli.
Together, these data demonstrated that silencing Rab23 caused abnormal ciliation and perturbed ligand-induced localization of Smo at the primary cilium, leading to a weaken GCP's response to Shh ligand stimulations, thereby impeding Shh-mediated GCP proliferation (Fig. 7F). These unravel a novel positive role of Rab23 in modulating primary cilium-dependent Shh signaling and GCP expansion during early cerebellar development.
Discussion
Fine tuning Shh signaling during cerebellar development is essential to facilitate a temporally and spatially-defined transit amplification of GCPs to ensure proper patterning and growth of the cerebellar folia (Wallace, 1999; Wechsler-Reya and Scott, 1999; Miyazawa et al., 2000; Butts et al., 2014). We have demonstrated here that Rab23 has a role in the patterning and growth of cerebellar folia during early cerebellar development. Deletion of Rab23 resulted in foliation anomalies because of dramatically perturbed radial glial scaffold formation, granule cells lamination and GCP proliferation. This study presents Rab23 as a novel regulator of GCP proliferation, remarkably, acting both positively and negatively via the Shh signaling pathway. Excitingly, our data showed for the first time that Rab23 has a role in primary cilium-dependent Shh signal transduction during cerebellar development. This demonstration is made possible as the brain-specific KO of Rab23 in our genetic model did not result in midgestation lethality in mice as compared with a global loss of Rab23 in the open brain mutant.
Previous examination of primary cilia in the node of two to six somite stage Rab23-null embryo reported largely unaltered morphology and similar overall percentage of ciliation as compared with the control (Fuller et al., 2014). Interestingly, unlike the node cilia, our data revealed aberrant ciliation in the Rab23-null GCP during early postnatal cerebellar development, though not discernible at embryonic stage. siRNA-mediated knock-down studies performed on different cell lines have reported inconsistent conclusions with regards to the role of Rab23 in ciliogenesis (Yoshimura et al., 2007; Leaf and Von Zastrow, 2015; Lim and Tang, 2015; Hor et al., 2018). These discrepancies suggest that the functions of Rab23 in primary cilia could vary in a context or cell type-specific manner. Our data supported a GCP-specific role of Rab23 in ciliogenesis in vivo. In line with the in vivo data, primary culture of Rab23-KO GCPs also showed shortened primary cilia and compromised response to Shh ligand and SAG-mediated Smo activation, implicating a disrupted primary cilium-dependent Shh signaling.
Mutations of Shh pathway repressor genes, including Ptch1, Gpr161, and Sufu, commonly lead to the development of medulloblastoma (Dong et al., 2000; Zurawel et al., 2000; Taylor et al., 2002; Begemann et al., 2020) via Gli1 upregulation (Kimura et al., 2005). We showed that loss of Rab23, unlike other Shh repressors, did not promote the development of medulloblastoma despite the basal level upregulation of Shh pathway activity in the GCPs. We further showed that the overall amount of Shh ligands in Rab23-deleted cerebellar tissues remained relatively similar to that of control at both embryonic and postnatal stages, suggesting a sufficient source of Shh stimulants in the KO cerebellar tissue environment. Given the above, we deduce that one possible explanation for an absence of tumorigenicity is the defective primary cilium in Rab23-KO GCPs. The compromised response to primary cilium-dependent Shh activation may lead to insufficient paracrine pathway stimulations to drive tumor formation in the Rab23-KO cerebellum. This is in line with the indispensable role of primary cilium for medulloblastoma formation (Han et al., 2009; Barakat et al., 2013).
Harboring the primary cilium defect in GCP, Rab23-KO cerebellum partially phenocopied other ciliopathy mutants, which often exhibit severe cerebellar size shrinkage, abnormal foliation, and reduced GCP proliferation because of impaired Shh signaling (Chizhikov et al., 2007; Spassky et al., 2008; Han et al., 2009; Barakat et al., 2013). In this regard, the postnatal Nes-CKO displayed profoundly mis-patterned folia, and smaller cerebellum at later adult stage, similar to other ciliopathy mutants. Nevertheless, in contrast to most ciliopathy mutants, Shh signaling in the Rab23-KO mutant was not completely inhibited. Instead, there was a ligand-independent upregulation of Shh pathway at basal level, which underlies the increase in GCP proliferation and transiently enlarged cerebellum at earlier postnatal stages. Rab23 is known to influence Gli2 and Gli3 expression at the transcript level (Eggenschwiler et al., 2006), and it could also antagonize Gli1's nuclear translocation and transcriptional activation in cytosolic compartment in the absence of ligand stimulation (Chi et al., 2012). Given the ligand-independent function of Rab23 in Shh pathway, it is therefore plausible that the Shh pathway in GCP became overactivated because of a basal increase in Gli activations in the absence of Rab23 function. However, limited by the incompetency to respond to Smo activation from cell/cilium membrane (Fig. 7C–F), Rab23-deficient GCP could not reach or sustain the full capacity of ectopic Shh pathway activation, making them less susceptible to tumor formation as compared with other repressors such as Ptch1 and Sufu mutants that are not known to exhibit primary cilium defect.
Previous work has demonstrated that Rab23 maintains the overexpressed-SmoA1 protein turnover in the primary cilium of MDCK cells on Shh stimulation (Boehlke et al., 2010); however, the underlying mechanism, and how this regulation would affect Shh signaling output remain elusive. Our data show that Rab23-depleted GCPs were less responsive to a Smo agonist (SAG). Given that SAG activates Shh signaling pathway by facilitating Smo translocation to the cilium axoneme, the control cells showed more profound Smo-translocation at the primary cilium on SAG activation, in contrast, Rab23-deficient GCPs failed to stimulate nor sustain Smo-localization at the primary cilium in response to SAG. Notably, the percentage of Smo-positive primary cilium at basal level was profoundly higher in the Rab23-KO GCPs (Fig. 7E), indicating a basal level enhancement of Shh pathway activity, and that the active translocation Smo to the primary cilium is not totally impaired. Therefore, the compromised response to SAG as observed in mutant cells could possibly be caused by a lack of fully functional primary cilium for ligand-induced Smo-mediated signaling transduction, as well as an impaired maintenance of Smo turnover in the primary cilium of GCP. This finding is in line with previous implicated role of Rab23 on SmoA1 protein turnover in MDCK cells (Boehlke et al., 2010). Additionally, our results further proved that Rab23 is required for the ligand-induced Smo-localization and/or turnover in the primary cilium to promote extrinsic Shh signaling transduction.
Taken together, our findings suggest that Rab23 confers dual functions in regulating Shh signaling and GCP proliferation; it potentiates primary cilium and Shh/Smo-dependent signaling cascade, while intrinsically antagonizing basal level Gli transcriptional activation. Our data thus present a previously underappreciated aspect of Rab23 in mediating Shh signaling upstream of Smo. This study sheds new light into the genetic and mechanistic insights underpinning Shh signaling-mediated GCP proliferation and cerebellar development.
Footnotes
Acknowledgment: We thank C. C. Hui and B. L. Tang for sharing critical and constructive comments and suggestions for the successful completion of this project. This work was supported by the National Medical Research Council-Young Individual Research Grant NMRC/OFYIRG/0079/2018, the HKBU Tier 2 Start-up Grant RG-SGT2/18-19/SCI/009, and the Research Grant Council-Collaborative Research Fund CRF-C2103-20GF (to C.H.H.H.) and the National Medical Research Council-Collaborative Research Grant NMRC/CBRG/0094/2015 and Ministry of Education Tier 2 Grant MOE2015-T2-1-022 and Tier 3 Grant MOE2017-T3-1-002 (to E.L.K.G.).
The authors declare no competing financial interests.
References
- Alessandri JL, Dagoneau N, Laville JM, Baruteau J, Hébert JC, Cormier-Daire V (2010) RAB23 mutation in a large family from Comoros islands with carpenter syndrome. Am J Med Genet A 152A:982–986. 10.1002/ajmg.a.33327 [DOI] [PubMed] [Google Scholar]
- Bai CB, Joyner AL (2001) Gli1 can rescue the in vivo function of Gli2. Development 128:5161–5172. 10.1242/dev.128.24.5161 [DOI] [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL (2004) All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell 6:103–115. 10.1016/S1534-5807(03)00394-0 [DOI] [PubMed] [Google Scholar]
- Balci S, Onol B, Eryilmaz M, Haytoglu T (1997) A case of carpenter syndrome diagnosed in a 20-week-old fetus with postmortem examination. Clin Genet 51:412–416. 10.1111/j.1399-0004.1997.tb02501.x [DOI] [PubMed] [Google Scholar]
- Barakat MT, Humke EW, Scott MP (2013) Kif3a is necessary for initiation and maintenance of medulloblastoma. Carcinogenesis 34:1382–1392. 10.1093/carcin/bgt041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basson MA, Wingate RJ (2013) Congenital hypoplasia of the cerebellum: developmental causes and behavioral consequences. Front Neuroanat 7:29. 10.3389/fnana.2013.00029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay SN, Long AB, Caspary T (2018) Disruption of the ciliary GTPase Arl13b suppresses sonic hedgehog overactivation and inhibits medulloblastoma formation. Proc Natl Acad Sci USA 115:1570–1575. 10.1073/pnas.1706977115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann M, Waszak SM, Robinson GW, Jäger N, Sharma T, Knopp C, Kraft F, Moser O, Mynarek M, Guerrini-Rousseau L, Brugieres L, Varlet P, Pietsch T, Bowers DC, Chintagumpala M, Sahm F, Korbel JO, Rutkowski S, Eggermann T, Gajjar A, et al. (2020) Germline GPR161 mutations predispose to pediatric medulloblastoma. J Clin Oncol 38:43–50. 10.1200/JCO.19.00577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersani G, Maddalena F, Pasquini M, Orlandi V, Pancheri P (2003) Association of schizophrenia and carpenter syndrome. Acta Neuropsychiatr 15:304–305. 10.1034/j.1601-5215.2003.00046.x [DOI] [PubMed] [Google Scholar]
- Boehlke C, Bashkurov M, Buescher A, Krick T, John AK, Nitschke R, Walz G, Kuehn EW (2010) Differential role of Rab proteins in ciliary trafficking: rab23 regulates smoothened levels. J Cell Sci 123:1460–1467. 10.1242/jcs.058883 [DOI] [PubMed] [Google Scholar]
- Butts T, Green MJ, Wingate RJT (2014) Development of the cerebellum: simple steps to make a 'little brain.' Development 141:4031–4041. 10.1242/dev.106559 [DOI] [PubMed] [Google Scholar]
- Chi S, Xie G, Liu H, Chen K, Zhang X, Li C, Xie J (2012) Rab23 negatively regulates Gli1 transcriptional factor in a Su(Fu)-dependent manner. Cell Signal 24:1222–1228. 10.1016/j.cellsig.2012.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizhikov VV, Davenport J, Zhang Q, Shih EK, Cabello OA, Fuchs JL, Yoder BK, Millen KJ (2007) Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci 27:9780–9789. 10.1523/JNEUROSCI.5586-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SH, Kim CT, Jung YH, Lee NS, Jeong YG (2010) Early cerebellar granule cell migration in the mouse embryonic development. Anat Cell Biol 434:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AF, Yu KP, Brueckner M, Brailey LL, Johnson L, McGrath JM, Bale AE (2005) Cardiac and CNS defects in a mouse with targeted disruption of suppressor of fused. Development 132:4407–4417. 10.1242/dev.02021 [DOI] [PubMed] [Google Scholar]
- Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL (2004) Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development 131:5581–5590. 10.1242/dev.01438 [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz i Altaba A (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126:3089–3100. 10.1242/dev.126.14.3089 [DOI] [PubMed] [Google Scholar]
- Dey J, Ditzler S, Knoblaugh SE, Hatton BA, Schelter JM, Cleary MA, Mecham B, Rorke-Adams LB, Olson JM (2012) A distinct smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol Cell Biol 32:4104–4115. 10.1128/MCB.00862-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Gailani MR, Pomeroy SL, Reardon D, Bale AE (2000) Identification of PATCHED mutations in medulloblastomas by direct sequencing. Hum Mutat 16:89–90. 10.1002/1098-1004(200007)16:1<89::AID-HUMU18>3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- Eggenschwiler JT, Espinoza E, Anderson KV (2001) Rab23 is an essential negative regulator of the mouse sonic hedgehog signalling pathway. Nature 412:194–198. 10.1038/35084089 [DOI] [PubMed] [Google Scholar]
- Eggenschwiler JT, Bulgakov OV, Qin J, Li T, Anderson KV (2006) Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Dev Biol 290:1–12. 10.1016/j.ydbio.2005.09.022 [DOI] [PubMed] [Google Scholar]
- Evans TM, Simpson F, Parton RG, Wicking C (2005) Characterization of Rab23, a negative regulator of sonic hedgehog signaling. Methods Enzymol 403:759–777 [DOI] [PubMed] [Google Scholar]
- Fuller K, O'Connell JT, Gordon J, Mauti O, Eggenschwiler J (2014) Rab23 regulates nodal signaling in vertebrate left-right patterning independently of the hedgehog pathway. Dev Biol 391:182–195. 10.1016/j.ydbio.2014.04.012 [DOI] [PubMed] [Google Scholar]
- Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ, Goldman S, Chintagumpala M, Wallace D, Takebe N, Boyett JM, Gilbertson RJ, Curran T (2013) Phase I study of Vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res 19:6305–6312. 10.1158/1078-0432.CCR-13-1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WQ, Heintz N, Hatten ME (1991) Cerebellar granule cell neurogenesis is regulated by cell-cell interactions in vitro. Neuron 6:705–715. 10.1016/0896-6273(91)90168-Y [DOI] [PubMed] [Google Scholar]
- Gerondopoulos A, Strutt H, Stevenson NL, Sobajima T, Levine TP, Stephens DJ, Strutt D, Barr FA (2019) Planar cell polarity effector proteins inturned and fuzzy form a Rab23 GEF complex. Curr Biol 29:3323–3330.e8. 10.1016/j.cub.2019.07.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Wang T, Ng EL, Aulia S, Chong KH, Teng FYH, Wang Y, Tang BL (2006) Open brain gene product Rab23: expression pattern in the adult mouse brain and functional characterization. J Neurosci Res 83:1118–1127. 10.1002/jnr.20788 [DOI] [PubMed] [Google Scholar]
- Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A (2009) Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 15:1062–1065. 10.1038/nm.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK (2005) Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 1:e53. 10.1371/journal.pgen.0010053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haye D, Collet C, Sembely-Taveau C, Haddad G, Denis C, Soulé N, Suc AL, Listrat A, Toutain A (2014) Prenatal findings in carpenter syndrome and a novel mutation in RAB23. Am J Med Genet A 164A:2926–2930. [DOI] [PubMed] [Google Scholar]
- Hor CHH, Goh ELK (2018) Rab23 regulates radial migration of projection neurons via N-cadherin. Cereb Cortex 28:1516–1531. 10.1093/cercor/bhy018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hor CHH, Tang BL, Goh ELK (2018) Rab23 and developmental disorders. Rev Neurosci 29:849–860. 10.1515/revneuro-2017-0110 [DOI] [PubMed] [Google Scholar]
- Ivaniutsin U, Chen Y, Mason JO, Price DJ, Pratt T (2009) Adenomatous polyposis coli is required for early events in the normal growth and differentiation of the developing cerebral cortex. Neural Dev 4:3. 10.1186/1749-8104-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins D, Seelow D, Jehee FS, Perlyn CA, Alonso LG, Bueno DF, Donnai D, Josifova D, Josifiova D, Mathijssen IMJ, Morton JEV, Orstavik KH, Sweeney E, Wall SA, Marsh JL, Nurnberg P, Passos-Bueno MR, Wilkie AOM (2007) RAB23 mutations in carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am J Hum Genet 80:1162–1170. 10.1086/518047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, McMahon AP (2005) Growth and pattern of the mammalian neural tube are governed by partially overlapping feedback activities of the hedgehog antagonists patched 1 and Hhip1. Development 132:143–154. 10.1242/dev.01566 [DOI] [PubMed] [Google Scholar]
- Kimura H, Stephen D, Joyner A, Curran T (2005) Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene 24:4026–4036. 10.1038/sj.onc.1208567 [DOI] [PubMed] [Google Scholar]
- Leaf A, Von Zastrow M (2015) Dopamine receptors reveal an essential role of IFT-B, KIF17, and Rab23 in delivering specific receptors to primary cilia. Elife 4:e06996. 10.7554/eLife.06996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Platt KA, Censullo P, Ruiz i Altaba A (1997) Gli1 is a target of sonic hedgehog that induces ventral neural tube development. Development 124:2537–2552. 10.1242/dev.124.13.2537 [DOI] [PubMed] [Google Scholar]
- Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP (2004) Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol 270:393–410. 10.1016/j.ydbio.2004.03.007 [DOI] [PubMed] [Google Scholar]
- Li T, Fan J, Blanco-Sánchez B, Giagtzoglou N, Lin G, Yamamoto S, Jaiswal M, Chen K, Zhang J, Wei W, Lewis MT, Groves AK, Westerfield M, Jia J, Bellen HJ (2016) Ubr3, a novel modulator of Hh signaling affects the degradation of Costal-2 and Kif7 through poly-ubiquitination. PLoS Genet 12:e1006054–30. 10.1371/journal.pgen.1006054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YS, Tang BL (2015) A role for Rab23 in the trafficking of Kif17 to the primary cilium. J Cell Sci 128:2996–3008. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Himi T, Garcia V, Yamagishi H, Sato S, Ishizaki Y (2000) A role for P27/Kip1 in the control of cerebellar granule cell precursor proliferation. J Neurosci 20:5756–5763. 10.1523/JNEUROSCI.20-15-05756.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, James CD (1997) Sporadic medulloblastomas contain PTCH mutations. Cancer Res 57:842–845. [PubMed] [Google Scholar]
- Roussel MF, Hatten ME (2011) Cerebellum development and medulloblastoma. Curr Top Dev Biol 94:235–282. 10.1016/B978-0-12-380916-2.00008-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidman RL, Rakic P (1973) Neuronal migration, with special reference to developing human brain: a review. Brain Res 62:1–35. 10.1016/0006-8993(73)90617-3 [DOI] [PubMed] [Google Scholar]
- Spassky N, Han YG, Aguilar A, Strehl L, Besse L, Laclef C, Ros MR, Garcia-Verdugo JM, Alvarez-Buylla A (2008) Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol 317:246–259. 10.1016/j.ydbio.2008.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svärd J, Henricson KH, Persson-Lek M, Rozell B, Lauth M, Bergström Å, Ericson J, Toftgård R, Teglund S (2006) Genetic elimination of suppressor of fused reveals an essential repressor function in the mammalian hedgehog signaling pathway. Dev Cell 10:187–197. 10.1016/j.devcel.2005.12.013 [DOI] [PubMed] [Google Scholar]
- Taravath S, Tonsgard JH (1993) Cerebral malformations in carpenter syndrome. Pediatr Neurol 9:230–234. 10.1016/0887-8994(93)90092-Q [DOI] [PubMed] [Google Scholar]
- Tarhan E, Oğuz H, Safak MA, Samim E (2004) The carpenter syndrome phenotype. Int J Pediatr Otorhinolaryngol 68:353–357. 10.1016/j.ijporl.2003.10.009 [DOI] [PubMed] [Google Scholar]
- Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, Squire JA, Rutka JT, Hogg D (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31:306–310. 10.1038/ng916 [DOI] [PubMed] [Google Scholar]
- Tukachinsky H, Lopez LV, Salic A (2010) A mechanism for vertebrate hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol 191:415–428. 10.1083/jcb.201004108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace VA (1999) Purkinje-cell-derived sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 9:445–448. 10.1016/S0960-9822(99)80195-X [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron 22:103–114. 10.1016/S0896-6273(00)80682-0 [DOI] [PubMed] [Google Scholar]
- Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH, Dlugosz AA, Reiter JF (2009) Primary cilia can both mediate and suppress hedgehog pathway-dependent tumorigenesis. Nat Med 15:1055–1061. 10.1038/nm.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yang Y, Tang X, Zhao M, Liang F, Xu P, Hou B, Xing Y, Bao X, Fan X (2013) Bergmann glia function in granule cell migration during cerebellum development. Mol Neurobiol 47:833–844. 10.1007/s12035-013-8405-y [DOI] [PubMed] [Google Scholar]
- Yoshimura SI, Egerer J, Fuchs E, Haas AK, Barr FA (2007) Functional dissection of Rab GTPases involved in primary cilium formation. J Cell Biol 178:363–369. 10.1083/jcb.200703047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K (2005) Control of granule cell precursor proliferation in the developing cerebellum and in medulloblastoma. Biomed Rev 16:35. 10.14748/bmr.v16.92 [DOI] [Google Scholar]
- Yue Q, Groszer M, Gil JS, Berk AJ, Messing A, Wu H, Liu X (2005) PTEN deletion in Bergmann glia leads to premature differentiation and affects laminar organization. Development 132:3281–3291. 10.1242/dev.01891 [DOI] [PubMed] [Google Scholar]
- Zurawel RH, Allen C, Wechsler-Reya R, Scott MP, Raffel C (2000) Evidence that haploinsufficiency of Ptch leads to medulloblastoma in mice. Genes Chromosom Cancer 28:77–81. 10.1002/(SICI)1098-2264(200005)28:1<77::AID-GCC9>3.0.CO;2-Y [DOI] [PubMed] [Google Scholar]