

Abstract
Adult neural plasticity is an important and intriguing phenomenon in the brain, and adult hippocampal neurogenesis is directly involved in modulating neural plasticity by mechanisms that are only partially understood. We have performed gain-of-function and loss-of-function experiments to study Smad2, a transcription factor selected from genes that are demethylated after exercise through the analysis of an array of physical activity-induced factors, and their corresponding gene expression, and an efficient inducer of plasticity. In these studies, changes in cell number and morphology were analyzed in the hippocampal dentate gyrus (cell proliferation and survival, including regional distribution, and structural maturation/differentiation, including arborization, dendritic spines, and neurotransmitter-specific vesicles) of sedentary male mice, after evaluation in a battery of behavioral tests. As a result, we reveal a role for Smad2 in the balance of proliferation versus maturation of differentiating immature cells (Smad2 silencing increases both the proliferation and survival of cycling cells in the dentate granule cell layer), and in the plasticity of both newborn and mature neurons in mice (by decreasing dendritic arborization and dendritic spine number). Moreover, Smad2 silencing specifically compromises spatial learning in mice (through impairments of spatial tasks acquisition both in long-term learning and working memory). These data suggest that Smad2 participates in adult neural plasticity by influencing the proliferation and maturation of dentate gyrus neurons.
SIGNIFICANCE STATEMENT Smad2 is one of the main components of the transforming growth factor-β (TGF-β) pathway. The commitment of cell fate in the nervous system is tightly coordinated by SMAD2 signaling, as are further differentiation steps (e.g., dendrite and axon growth, myelination, and synapse formation). However, there are no studies that have directly evaluated the role of Smad2 gene in hippocampus of adult animals. Modulation of these parameters in the adult hippocampus can affect hippocampal-dependent behaviors, which may shed light on the mechanisms that regulate adult neurogenesis and behavior. We demonstrate here a role for Smad2 in the maturation of differentiating immature cells and in the plasticity of mature neurons. Moreover, Smad2 silencing specifically compromises the spatial learning abilities of adult male mice.
Keywords: adult hippocampal neurogenesis, behavioral phenotype, neural plasticity, neuronal morphology, SMAD
Introduction
Smad2 is one of the main elements in the transforming growth factor-β (TGF-β) pathway, a cytokine signaling pathway that controls many processes involved in development and tissue homeostasis. A key feature of this pathway is its functional versatility, fulfilling different functions depending on the conditions. Cell proliferation, differentiation, migration, and apoptosis are events controlled by TGF-β signaling, and they are coordinated by SMAD proteins. SMAD2 acts as a transcription factor, and it represents a node for signal integrations (Massagué et al., 2005). The intracellular signaling driven by TGF-β involves the phosphorylation of a number of proteins that, in turn, activate multiple downstream pathways, including the SMAD2 protein with a Ser-X-Ser motif (Brown et al., 2007). This phosphorylation allows SMAD2 to bind to other proteins, forming strategic complexes that enter the nucleus, bind DNA, and act as a transcription factors (Brown et al., 2007).
The Smad family contains receptor-activated Smads (R-Smads), common mediators and inhibitory Smads. Vertebrate SMAD2 and three are R-Smads that are phosphorylated in response to TGF-β (Di Guglielmo et al., 2003), promoting their binding to SMAD4, their translocation to the nucleus, and binding to DNA. Efficient activation of downstream Smad-responsive genes depends on the existence of Smad binding elements in their promoter region, receptor-activated formation of SMAD2-SMAD4 transcriptional complexes, and different cofactors (Derynck and Zhang, 2003). Hence, although TGF-β can target several hundred genes in a given cell, each SMAD complex activates or represses the transcription of a few specific genes in each cell type. This cell-specific response to TGF-β is the rationale behind analyzing the role of Smad2 in specific cell types or distinct environmental situations, for example, cell proliferation or survival depending on the molecular milieu of a cell. Consequently, contrasting results have been reported regarding the role of Smad2 in neuronal precursor proliferation or differentiation, indicating that the effects of SMAD2 depend on local, temporal, and contextual features (Ueberham et al., 2009).
SMAD proteins are intricately involved in nervous system development, from the earliest stages of neuralization involving ectodermal cells to the onset of neurologic diseases. During neurulation, the suppression of Smad2 signaling is required to specify the neuroectoderm, while the stimulation of this pathway blocks neural induction and promotes non-neural cell fates (Chang and Harland, 2007). In the developing brain, primary neurogenesis is also dependent on SMAD proteins as the fate of neural stem cells is driven by specific signals. Thus, while TGF-β promotes the differentiation of radial glial cells toward astrocytes, activating mitogen-activated protein kinase (MAPK) signaling (Stipursky et al., 2012), SMAD2 activity is also required for neurogenesis. The commitment to cell fates in the nervous system is closely linked to the activity of the SMAD2 signaling cascade, as are further differentiation steps, such as dendrite and axon growth, myelination, and synapse formation.
Previous experiments with homozygous Smad2−/− mutant embryos indicated that this organism failed to form an organized egg because Smad2 plays a crucial role in establishing the anterior–posterior axis within the epiblast, and in the formation of endoderm, leading to a precocious death (Nomura and Li, 1998; Waldrip et al., 1998). Studies on mice that specifically lack Smad2 in the CNS (Smad2-CNS-KO) displayed behavioral abnormalities in motor coordination from an early postnatal age. Moreover, the cerebellar layers in these animals are aberrant, with increased apoptotic cell death, as well as delays in the migration and maturation of granule cells, and in the dendritic arborization of Purkinje cells (Wang et al., 2011).
To date, the role of the Smad2 gene in the adult hippocampus has not been directly evaluated, although there is indirect evidence of an influence of pathways downstream of TGF-β, particularly from studies focusing on membrane receptors (He et al., 2014) or Smad3 (Tapia-González et al., 2013; Munoz et al., 2016), or using in vitro models (Ishihara et al., 1994; Funaba et al., 2002). As the loss of Smad2 is lethal to embryos (Nomura and Li, 1998; Waldrip et al., 1998; Heyer et al., 1999; Tremblay et al., 2000; Liu et al., 2016), the most interesting data regarding the role of Smad2 in the adult brain has come from studying the Smad2-CNS-KO model (Wang et al., 2011). Hence, this study first aimed to test whether Smad2 participates in the neuroplasticity evident in the adult dentate gyrus (DG) given its possible involvement in both neurogenesis (specifically in the cell fate commitment and maturation of postmitotic cell populations) and the mature neuron structure. Modulating these events in the adult hippocampus may provide relevant information about the impact of Smad2 on hippocampal-dependent behaviors, enhancing our knowledge of the factors regulating adult neurogenesis and animal behavior.
Materials and Methods
Ethics approval.
All experiments were performed according to the European Community (Directive 2010/63/EU) and Spanish Guidelines (Royal Decree 53/2013), and they were approved by the Committee of Ethics and Animal Experimentation of the Cajal Institute (20/05/2016), the Ethics Committee (Subcommittee of Ethics) of the Spanish Research Council (07/27/2016), and the Animal Protection Area of the Ministry of Environment of the Community of Madrid (10/26/2016).
Animals.
C57BL/6J mice (male, 2 months of age) were housed at 22 ± 1°C on a 12 h light/dark cycle, and with ad libitum access to food and water. All the animals were handled in strict accordance with the Good Animal Practice guidelines defined by the national welfare bodies at the Cajal Institute and Consejo Superior de Investigaciones Científicas (CSIC). The work was previously approved by the Bioethics Committee of the Cajal Institute and CSIC, and was conducted in accordance with the European Commission directive 2010/63/EU. Ten animals (5 per experimental group, sedentary and runner) were used for the demethylation array and gene expression analysis, and 32 [8 per experimental group, 4 groups; Smad2 silencing (shRNA) and its control vector, and Smad2 overexpression (hSmad2) and its control vector)] were used for the gain-of-function and loss-of-function experiments.
Thymidine analogs.
Thymidine analogues were administered to the animals by intraperitoneal injection, as follows: bromodeoxyuridine (BrdU; Sigma-Aldrich) was injected at 50 mg/kg body weight (bw) prepared in 0.9% saline; 5-chloro-2'-deoxyuridine (CldU; Sigma-Aldrich) was injected at 42.75 mg/kg bw in 0.9% saline; 5-iodo-2'-deoxyuridine (IdU; Sigma-Aldrich) was injected at 56.75 mg/kg bw prepared in 0.1 m PBS with two drops of 5N NaOH added per 10–15 ml PBS. These doses were all based on equimolar doses of 50 mg/kg bw BrdU.
Moderate forced treadmill exercise.
Animals were habituated to the treadmill (Cibertec), and each group of animals, sedentary and exercising, was placed separately for 10 min in the inactive treadmill. The animals then ran for 5 min at a speed of 0.1 m/s, and finally for 10 min at 0.2 m/s. Both groups undertook habituation to avoid any effect because of the new environment. After 24 h, the exercising animals started their training, running for 40 min at a speed of 0.2 m/s, and 7 d/week over a period of 2 weeks, while the sedentary animals were placed in the treadmill for the same amount of time but without running.
Elevated plus maze.
The elevated plus maze (Cibertec) consists of a plus-shaped apparatus with two open and two enclosed arms, elevated 40 cm from the floor. The test involves a single 5 min trial for each animal, placing the mouse in the center of the maze, facing an open arm and away from the experimenter. During each trial, the mouse could move freely along the apparatus, and its movements were recorded with Tracking Interface software and analyzed using Video Maze software (Cibertec).
Morris water maze.
The apparatus to perform the hidden version of the Morris water maze (MWM) test consists of a 1-m-diameter pool with a 120l capacity, containing water at a temperature of 22 ± 1°C, with visual cues at the surface of the tank and an invisible methacrylate platform. The experiment was performed in three phases. (1) In the first session (habituation), the mouse was placed in the center of the tank, entering from the southern side, and after a 1 min session without a platform in the tank, the animal was returned to its cage. (2) After habituation, the animals began the acquisition sessions in which an invisible methacrylate platform was placed in the southeastern quadrant of the pool, submerged 1 cm below the water surface. Four daily trials were performed in which the site of entry of the mouse into the pool was changed. All the animals entered the pool at the same site in the same trial, and the entry sites were chosen in a random order each day. The mice swam in the pool for up to 1 min until they found the platform, on which they remained for 20 s before they were returned to their cage. If after 1 min they did not find the platform, the experimenter gently placed them on it. The number of acquisition days depended on the performance of each group, so the learning curve was monitored daily to avoid overtraining. Finally, (3) the third session consisted of the probe trial, performed just 1 h after the last acquisition session. There was no platform in the pool, and each mouse was introduced into the center of the tank from the south side. After 90 s of swimming, the mouse was removed from the water, and it returned to its cage.
The time the animals took to reach the platform (i.e., escape latency) was recorded with EthoVision XT 7 software. Daily mean escape latencies were measured to obtain the learning curve, and the searching paths of the animal were video recorded using the Ethovision XT 7 software to assess the time spent in every quadrant of the pool. The swim speed and path length were also recorded and compared between experimental groups in all the tests to rule out possible differences in locomotor capabilities, and to analyze searching strategies, respectively.
T-maze.
During the 10 min training, one of the shorter arms was closed and the animals were free to explore the maze. After 4 h, the mice were again put in the apparatus for 10 min, but with all of the arms available to explore. The number of entries and the time spent exploring the new arm were calculated, recording the trial with the Ethovision XT 7 software. An index of discrimination for the time spent in each arm was obtained by applying the following formula: (T_(New Arm) – T_(Old Arm))/T_tot (where T is time).
Killing.
Animals were killed by deep pentobarbital anesthesia (Euta-Lender). To obtain samples for histologic analysis, the mice were perfused transcardially with 0.9% saline followed by 4% paraformaldehyde in 0.1 m phosphate buffer (PB). The brain of the animal was removed and fixed again in the same fixative overnight at room temperature (RT). To obtain samples for molecular and biochemical analysis, mice were transcardially perfused with 0.9% saline alone; their brains were removed rapidly from the skull; and the hippocampal formation was isolated in an iced dish, preserved in dry ice, and stored at −80°C as quickly as possible.
Histology.
Approximately 100 serial coronal sections of the hippocampal formation (50 µm thick) were obtained on an automatic vibratome (model VT1000s, Leica), and they were collected individually in 96-multiwell culture plates. The sections were stored at 4°C in 0.1 m PB. For immunohistochemistry, the slices were initially preincubated for 5 min in 0.1 m PB with 0.5% Triton X-100 and 0.1% bovine serum albumin to blocking nonspecific binding. The primary antibodies were then diluted in the same following solution: a rat anti-BrdU (1:500; Abcam), a rat anti-CldU antibody (1:500; Accurate Chemicals), and a mouse anti-IdU antibody (1:500; BD Biosciences). To detect BrdU and IdU, slices were preincubated in HCl 2N for 30 min at RT. The antibodies were incubated with the sections for 1 h at RT and then at 4°C for 48 h. Primary antibody binding was detected with secondary Alexa Fluor-conjugated antibodies, again incubated for 1 h at RT and then at 4°C for 48 h with an Alexa Fluor 594-conjugated donkey anti-rat for BrdU (1:1000; Thermo Fisher Scientific), an Alexa Fluor 488-conjugated donkey anti-rat for the CldU antibody (1:1000; Thermo Fisher Scientific), and an Alexa Fluor 594-conjugated donkey anti-mouse for IdU (1:1000; Thermo Fisher Scientific). The primary anti-phospho-histone 3 (pH3) antibody used was a rabbit anti-pH3 antibody (1:500; Chemicon-Millipore) left over 2 d at 4°C and detected with an Alexa Fluor 594-conjugated donkey anti-rabbit (1:1000; Thermo Fisher Scientific) overnight at 4°C. A goat anti-doublecortin (DCX) antibody (1:500; Santa Cruz Biotechnology) was used for 1 h at RT and then at 4°C over 2 d. Doublecortin was recognized with an Alexa Fluor 594-conjugated donkey anti-goat (1:1000; Thermo Fisher Scientific) or a horse anti-goat biotinylated antibody (1:1000; Vector Laboratories) combined with a streptavidin-Alexa Fluor 633 antibody (1:1000; Thermo Fisher Scientific), incubated at 4°C overnight. The rabbit anti-calretinin antibody (1:3000; Swant) was left for 1 h at RT and then at 4°C over 2 d, and detected with an Alexa Fluor 488-conjugated donkey anti-rabbit antibody (1:1000; Thermo Fisher Scientific) incubated at 4°C overnight. To detect SMAD2, the slices were preincubated for 1 h at 80°C in citrate buffer, pH 6, for antigen retrieval. The slices were then incubated with the primary anti-SMAD2 antibody (1:500; Cell Signaling Technology) for 1 h at RT and overnight at 4°C. To amplify the signal, the slices were incubated with a goat anti-rabbit biotinylated antibody (1:1000; Sigma-Aldrich) and then with a streptavidin-Alexa Fluor 633 antibody (1:1000; Thermo Fisher Scientific), each for 1 h at RT and then overnight at 4°C. Slices were incubated with the primary rabbit anti-human SMAD2 antibody (1:500; Cell Signaling Technology) for 1 h at RT and then overnight at 4°C. The primary antibody was detected overnight at 4°C with an Alexa Fluor 594-conjugated donkey anti-rabbit antibody (1:1000; Thermo Fisher Scientific). To amplify the signal from the infected cells, the slices were incubated with a rat anti-green fluorescent protein (GFP; 1:1000; Nacalai Tesque) or a rabbit anti-GFP (1:1000; Thermo Fisher Scientific) antibody for 1 h at RT and then overnight at 4°C. The primary antibody was then detected with an Alexa Fluor 488-conjugated donkey anti-rat or anti-rabbit secondary antibody (1:1000; Thermo Fisher Scientific). To detect mature neurons, slices were incubated with the primary antibody mouse anti-NeuN (1:1000; Chemicon) for 1 h at RT and then for 48 h at 4°C. The primary antibody was detected overnight at 4°C with an Alexa Fluor 594-conjugated donkey anti-mouse antibody (1:1000; Thermo Fisher Scientific). The primary antibody used to detect the vesicular glutamate transporter (VGluT) was a guinea pig anti-VGluT antibody (1:2500; Chemicon) incubated for 1 h at RT and at 4°C over 2 d. The primary antibody was detected for 1 h at RT and then overnight at 4°C with a goat anti-guinea pig biotinylated antibody (1:1000; Vector Laboratories) combined with a streptavidin-Alexa Fluor 633 antibody (1:1000; Thermo Fisher Scientific). The primary antibody used to detect the glutamic acid decarboxylase (GAD) was a mouse anti-GAD antibody (1:500; Developmental Studies Hybridoma Bank) for 1 h at RT and at 4°C over the 2 d, and it was recognized with an Alexa Fluor 555-conjugated donkey anti-mouse antibody (1:1000; Thermo Fisher Scientific) incubated for 1 h at RT and then at 4°C overnight. The sections were finally counterstained for 12 min with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; 1:1000; Calbiochem) in 0.1 m PB, and the slices were mounted on gelatin slides and coverslipped in Gerbatol mounting medium (Sigma-Aldrich).
For each animal, one series of slices was chosen randomly, stained in Nissl solution, and measured. The area of the subgranular zone (SGZ) and the volume of the granule cell layer (GCL) were calculated considering the thickness of the slices (50 µm), and measuring the longitude of the SGZ and the area of the GCL under an optical microscope with the Neurolucida software.
Stereology.
Serial coronal sections (50 µm thick) were obtained on a Leica vibratome and collected in 96 multiwell culture plates for each hemisphere. One random series was chosen for immunohistochemistry with each antibody, constituting one slice every eight slices. To estimate the total number of BrdU+, IdU+, and pH3+ cells present in the DG, the physical multistage fractionator technique was applied. Briefly, the number of cells counted on an inverted fluorescent microscope (40× objective; model DMI6000B, Leica) in all the sections of a series was multiplied by 8 to obtain the total number of positive cells in the DG. The number of DCX+/GFP+ cells was also obtained applying the fractionator method, counting the cells under an optical microscope using a double cube filter that allowed double-stained cells to be detected. Again, the numbers of cells counted was multiplied by 8 to obtain the total number. The total number of doublecortin/calretinin (DCX/CLR) cells was analyzed by applying a physical dissector method developed for confocal microscopy (oil-immersion 63× objective; model TCS SP5 confocal microscope, Leica).
The 3D-U-disector (Llorens-Martín et al., 2006) is a modified version of the basic “unbiased brick” proposed by Howard and Reed (1998) in which optical sections were obtained in a continuous scan along the z-axis of the confocal microscope. Here, the “physical dissector” modification was used instead of the “optical dissector,” counting cells in successive series of confocal images with the support of a computer. For each animal, six stacks were analyzed (three stacks in the suprapyramidal blade of the DG and three stacks in the infrapyramidal blade), and each stack consisted of 11 images (100 × 100 µm) where z ∼ 17 µm. The images were obtained at a 512 resolution and a 2.46 zoom, and each stack represents the U-physical dissector. Cells in each pair of confocal sections were counted using the first as a reference section with the ImageJ software version 1.49, using the Cell Counter plugin. Accordingly, the cells were marked, and they were then identified in successive images of the dissector, ensuring that each cell was counted just once. In each stack, the following three different subpopulations of immature neurons were counted: DCX+/CLR–, DCX+/CLR+, and DCX–/CLR+. The number of immature neurons counted was divided by the area of the SGZ in the stack, thereby obtaining a reliable estimate of cell density by “unit of SGZ area.” The total number of immature cells was obtained by multiplying the cell density by the total extension of the SGZ. The total number of mature granule cells was counted using DAPI staining, but, because of the high density and large number, a physical dissector was used. For each animal, three stacks were analyzed, each consisting of six images (33 × 33 µm), where z ∼ 10 µm. The images were obtained on a confocal microscope (oil-immersion 63× objective; model TCS SP5, Leica) with a 7.45 zoom. The number of cells in each image was counted with the Cell Counter plugin of ImageJ, the mean density was calculated, and the total number of cells was obtained by multiplying by the GCL volume. The VGluT+ and GAD+ boutons on dendrites were detected, and, to quantify the relative area covered by each synapse, three confocal microscope images from the molecular layer were obtained from each sample (oil-immersion 63× objective; model TCS SP5 Confocal Microscope, Leica). Using the ROI Manager tool of ImageJ, a 100 × 100 circle was created and localized in the inner part of the molecular layer, near the upper limit of the granular layer. A threshold was assessed for each image and channel, and the total area occupied by the positive boutons was automatically calculated.
Morphometric analysis.
The morphology of the granule neurons was studied on the GFP+ cells. The cells chosen for the study were positive for the NeuN marker and negative for DCX to only analyze mature neurons. Cells were also chosen based on previous observations of the dendritic tree (no evident cutting of dendrites as a vibratome artifact) and based on the relative isolation from other GFP+ cells to simplify the reconstruction of single neurons. Images were obtained by scanning the entire cell every 0.8 µm with a confocal microscope (oil-immersion 40× objective; model TCS SP5, Leica), and the final projection was analyzed. The length of the dendrites was measured using the NeuronJ plugin of ImageJ, and a Sholl analysis was performed by applying concentric and equidistant circles centered on the cell body using the Concentric Circles plugin of ImageJ. For each experimental group, 20–30 cells were analyzed, quantifying the spine density on tertiary dendrites located in the molecular layer. The dendrites were scanned using a 63× oil-immersion confocal microscope objective and a 3.5 zoom, taking an image every 0.13 µm. The 16 bit images, with a 2048 × 512 resolution, were subjected to deconvolution to sharpen the image (LAS AF 2.6 software, Leica), and the spines were then counted and the dendrite length measured using the NeuronJ plugin of ImageJ to determine the linear spine density.
Western blot analysis.
Hippocampal tissue samples were homogenized in ice-cold RIPA buffer supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Sigma-Aldrich) by sonication. Protein concentration was calculated using Pierce BCA Protein Assay Kit (catalog #23225, Thermo Fisher Scientific). Protein (20 µg/well) was denatured in 1× SDS Laemmli buffer (2% SDS, 10% glycerol, 100 mm DTT, 0.5 mg/ml bromophenol blue, and 65 mm Tris-HCl, pH 6.8) and separated by SDS gel electrophoresis. Transferred nitrocellulose membranes were blocked with 5% nonfat dry milk in TTBS (150 mm NaCl, 0.05 Tween-20, and 20 mm Tris-HCl, pH 7.4), and then incubated with the following primary antibodies: anti-Smad2 (1:1000; catalog #5339, Cell Signaling Technology), anti-phospho-Smad2 (Ser465-467; 1:500; Cell Signaling Technology), and anti-b-III-tubulin (1:1000; catalog #MMS435P, Covance). After three washes in TTBS, blots were developed using secondary antibodies conjugated to HRP and ECL-Plus (Millipore) and scanned with the Versadoc Imaging System (Bio-Rad).
Epigenetic analysis.
DNA from the hippocampi stored at −80°C was extracted and purified using the DNeasy Blood and Tissue Kit (Qiagen). To prepare genomic DNA for the methylation analysis, the EpiTect Methyl DNA Restriction Kit (Qiagen) was used. DNA methylation profiles in the CpG islands of specific genes of interest were analyzed with the EpiTect Methyl qPCR Array (Qiagen), using an array for 24 mouse stem cell transcription factors (catalog #335211 MeAM-511A). The manufacturer instructions were followed in all cases.
Gene expression.
Total RNA was extracted from the hippocampus using the QuickGene RNA Tissue Kit S II, following the manufacturer instructions, and the RNA concentration and purity were measured in a spectrophotometer (ND-1000 NanoDrop, Thermo Fisher Scientific). The Applied Biosystems High Capacity RNA-to-cDNA Master Mix (Thermo Fisher Scientific) was used to synthesize cDNAs according to the manufacturer protocol and using the quantitative PCR (qPCR) primers designed with the Primer Express 3.0 software and obtained from Thermo Fisher Scientific. The expression of each gene was evaluated by qPCR, and, once the optimal conditions were established, quantitative PCR was conducted using SYBR Green (Thermo Fisher Scientific). The reactions were prepared in triplicate for each sample in 96-well plates, and the Applied Biosystems 7500 Real-Time PCR System (Thermo Fisher Scientific) was used under the following thermocycling conditions: 1 cycle at 60°C for 30 s, 1 cycle at 95°C for 10 min, and 40 cycles at 95°C for 15 s followed by 1 min at 60°C.
Cloning techniques.
The Mus Musculus Smad2 mRNA sequence was extracted from GenBank, and four shRNA sequences were designed with the help of the Shortcut OligoEngine software. The lyophilized primers were obtained from Biomers and dissolved in Milli-Q water to a final concentration of 100 nmol. To insert the shRNA in an expression vector, the oligos were annealed as DNA double strands by preparing a solution with the positive and negative sequences (25 µg of each), 10 µl of a 10× Medium Restriction Buffer, and ultrapure water up to a final volume of 100 µl (final concentration, 0.5 µg/µl). The thermal cycler conditions for annealing were as follows: 95°C for 3 min, and then 68°C for 10 min. The shRNA sequences were ligated into the linearized pLVTHM vector, a second-generation lentivector expressing the shRNA under an H1 promoter and with GFP [created by the Didier Trono laboratory, École polytechnique fédérale de Lausanne (EPFL), Lausanne, Switzerland; plasmid #12247; Addgene; Wiznerowicz and Trono, 2003].
The hSmad2 gene was obtained from the pCIG-Flag-hSmad2 expression vector. The linearized vector and the hSmad2 DNA fragment were separated and isolated by electrophoresis in a 1% agar gel, and the hSmad2 DNA was then inserted into pWPI, a second-generation lentiviral vector (created by the Didier Trono laboratory, EPFL, Lausanne, Switzerland; plasmid #12254, Addgene). This vector allows simultaneous expression of the transgene (in this case, the hSmad2 gene) and the GFP marker, under the control of the EF1-α (Elongation Factor1-α) promoter. The constructs were sequenced to guarantee the correct cloning of the inserts and the primers (Biomers) were designed using Oligo Explorer Software.
Lentivirus production.
Human embryonic kidney 293T cells (HEK293T) were used to produce lentiviral particles, and the cells were maintained in DMEM (high glucose, Na-pyruvate, no glutamine; Thermo Fisher Scientific), with fetal bovine serum (12%; Thermo Fisher Scientific), l-glutamine (4 mm; Thermo Fisher Scientific), and penicillin-streptomycin (1%; Thermo Fisher Scientific). A three-plasmid system was used to produce the lentiviral particles and the two helper plasmids were psPAX2 (second-generation lentiviral packaging plasmid) and pMD2.G (VSV-G envelope-expressing plasmid). The two helper plasmids were cotransfected with the engineered modified plasmid of interest using Lipofectamine 2000 (Thermo Fisher Scientific). The supernatant was collected 48 and 72 h post-transfection, filtered using a 0.45 µm filter to eliminate small debris, and stored at −80°C before ultracentrifugation. The supernatant containing the lentiviral particles was centrifuged in a BeckMAN centrifuge at 22,000 rpm and 4°C for 2 h, after which the supernatant was removed by aspiration without touching the viral pellet at the bottom of the tube. The pellet was resuspended in 50 µl of cold TBS-5 buffer, and the tubes were sealed with Parafilm and then stored at 4°C overnight. After soaking, the viral pellet was resuspended in the same buffer.
Stereotaxic injections.
Animals were deeply anesthetized with isoflurane (Isoflo, Esteve) and placed in the stereotactic device (David Kopf Instruments). To avoid any damage to the eardrums, jaw holder cuffs (Stoelting) were used to securely clamp the zygomatic processes of the skull. The lentiviral particles were injected bilaterally into the hippocampal DG of each animal with a 2 µl Hamilton microsyringe, performing two different injections in each hemisphere to guarantee infection the entire rostrocaudal axis. The 2 µl of lentivirus was injected at a rate of 0.2 µl/min using a microinjector, and when the four injections were completed, the animals were left in a cage placed over a heating pad until they recovered from the anesthesia. The condition of animals was monitored throughout the experiment.
Experimental design and statistical analysis.
Statistical analysis was performed using SPSS Statistics software package (version 23, IBM). Different statistical tests were applied depending on the groups and variables. To compare a mean value of the two groups, a Mann–Whitney U test was applied. A repeated-measures ANOVA was chosen to compare the repeated-measures factor within the same group (Morris water maze; if significantly different, a Mann–Whitney U test was used to compare experimental groups at specific days). A Wilcoxon signed-rank test was used to compare two sets of scores from the same group.
Data availability.
All materials, data and associated protocols used in this work are available to interested readers.
Results
We used moderate forced exercise on a treadmill as an intervention to induce plasticity and to increase the rate of adult hippocampal neurogenesis (AHN; van Praag et al., 1999). As little as 2 weeks of exercise can significantly increase the pH3+ (a marker of mitosis; Mann–Whitney U test, p = 0.029), DCX+ (a microtubule-associated protein, a marker of migrating neurons; Mann–Whitney U test, p = 0.022), and the fractin+ (caspase-specific cleavage product of actin, a marker of cell death; Mann–Whitney U test, p = 0.01) cell number, as well as the survival of 3-week-old newborn neurons (Mann–Whitney U test, p = 0.003; Fig. 1). This short period is sufficient to induce a moderate anxiolytic effect, as measured by the number of entries into the open arms of the elevated maze (Fig. 1f; Mann–Whitney U test, p = 0.079), although it is not adequate to significantly modify performance in a standard MWM task (Fig. 1). To investigate the epigenetic changes induced by exercise-induced plasticity, we used a DNA methylation array. We analyzed the methylation of 24 genes encoding stem cell transcription factors in sedentary and running mice, identifying significant changes in smad2 (control, 30.89 ± 2.74; runner, 5.73 ± 0.33; Mann–Whitney U test, p = 0.009), hdac1 (control, 37.28 ± 5.75; runner, 6.09 ± 1.68; Mann–Whitney U test, p = 0.009), and gata2 (control, 29.4 ± 7.67; runner, 9.77 ±3.71; Mann–Whitney U test, p = 0.047; Fig. 2a). We assessed whether these changes in methylation corresponded to significant alterations in mRNA expression (significant for Smad2: control, 1 ± 0.24; runner, 5.96 ± 1.29; Mann–Whitney U test, p = 0.02; a trend for Hdac1: control, 1 ± 0.04; runner, 2.48 ± 0.58; Mann–Whitney U test, p = 0.052; Fig. 2b) concomitant to an increase in the ratio of phosphorylated to total Smad2 protein (control, 1 ± 0.18; runner, 2.87 ± 0.84; Mann–Whitney U test, p = 0.043), and a trend toward a decrease in the Hdac1 protein (control, 100 ± 14.73; runner, 55.54 ± 14.37; Mann–Whitney U test, p = 0.057; Fig. 2c). These data suggest that Smad2 may be an active factor in these cells, playing a significant role in AHN-related plasticity.
Figure 1.

The influence of exercise on adult hippocampal neurogenesis and behavior in the experiment to analyze the epigenetic changes induced by moderate exercise (Fig. 2). AHN was characterized by immunohistochemically labeling cells with different markers. a–e, Changes were assessed in the total number of pH3+(a), IdU+ (b), DCX+/CLR– (c), DCX+/CLR+ (d), and fractin+ (e) cells. N = 5 animals/group. p < 0.05, p < 0.01. f–j, To analyze the epigenetic changes induced by moderate exercise, behavioral changes were assessed in the elevated plus maze (f, g) and the Morris water maze (h–j). The elevated plus maze represents both the number of entries in the open arms and the time spent in these areas. The water maze represents the escape latency along the acquisition trials (5 d) in seconds (h), the learning curve slope from the series of escape latencies of all days (i), and the time spent swimming during the probe trial (j) to compare the time spent in the platform quadrant to the mean time spent in the other three quadrants. N = 5 animals/group. #0.1 > p ≥ 0.05. *p < 0.05, **p < 0.01.
Figure 2.

Epigenetic control of gene expression in sedentary and exercising animals. a, DNA methylation array. The DNA methylation profile of 24 stem cell transcription factors was defined in the hippocampi of sedentary and exercising mice. The green line shows the threshold from which values up to 10% are considered as hypermethylation. A statistical difference in methylation was found for Smad2 (p = 0.009), Hdac1 (p = 0.009), and Gata2 (p = 0.047) genes. Values up to 10% were considered as hypermethylation. The majority of genes studied in this experiment were hypomethylated. It is worth noting that the Smad2 gene was found in a hypermethylated status in sedentary mice, while 2 weeks of physical exercise reduced the methylation level to under the threshold value (from 30.89 ± 2.74% to 5.73 ± 0.33%). Genes such as Hdac1 and Gata2 showed a methylation profile similar to those of Smad2 (Hdac1: from 37.28 ± 5.75% to 6.09 ± 1.68%; Gata2: from 29.40 ±7.67% to 9.77 ± 3.71%). b, Quantification by qPCR of mRNA expression with the most relevant changes in methylation. c, Smad2 protein quantification in Western blots, the pSmad2/total Smad2 ratio, and the HDAC1 protein. N = 5 animals/group. #0.1 > p ≥ 0.05. *p < 0.05, **p < 0.01
To investigate the role of Smad2 under basal conditions, we injected lentivirus carrying loss-of-function or gain-of-function vectors for Smad2 into the hippocampus of male mice. We followed the time course of infection to confirm they were adequate (Fig. 3a) and to define the best time to kill the animals after injection, indicating that infection was barely detected until 3 d had passed (Fig. 3b–d). We also tested whether the granule cell layer of the DG was targeted (Fig. 3e) by examining a rostrocaudal series of images representing the extension of infection in our animals 3 weeks after infection (Fig. 3f). Both loss-of-function (Smad2 shRNA) and gain-of-function (overexpressing hSmad2) constructs were tested (Fig. 3g–j; Fig. 3g,h shows a significant decrease of Smad2 protein: pLVTHM, 100 ± 2.1; pLVTHM-shRNA-Smad2, 38 ± 7.8; Mann–Whitney U test, p = 0.002; Fig. 3i,j shows a significant increase of Smad2 protein: pWPI, 100 ± 16; pWPI-hSmad2, 215 ± 12.5; Mann–Whitney U test, p = 0.005), and the infection detected corresponded to the Smad2 expression in the adult mouse hippocampus (Fig. 3k, representative pictures of Smad2 signal in the DG GCL; Fig. 3a, red box). Both immature and mature postmitotic neurons were infected across the whole width of the GCL, which indicates that our data are the sum of cell-autonomous and non-cell-autonomous effects.
Figure 3.

Altered Smad2 expression by injection of gain-of-function and loss-of-function lentiviral constructs. Green, GFP labeling; blue, DAPI staining. a, Experimental design: time course to evaluate lentivirus expression. The animals were killed at different times, and slices of their brain were observed under a fluorescent microscope. Drawing of a rostral brain section and a scheme of the hippocampal formation to identify where the images in Figure 2k were taken. b–d, The time course of lentiviral expression: 12 h (b), 3 d (c), and 4 d (d) after injection. GFP+ cells were not found before 3 d after injection. e, Experimental design for the results shown in g–k. f, Three weeks after the stereotaxic intervention, lentivirus expression was observed along the rostrocaudal axis of the DG with a high efficiency of infection: the dorsoventral (D and V) and mediolateral (M and L) orientations are indicated by white crosses (the first is valid for the top six pictures, the second for the four bottom pictures). g–j, Smad2 protein 3 weeks after stereotaxic intervention. The effect of different lentiviral constructs on the hippocampal levels of Smad2 protein quantified in Western blots: pLVTHM-shRNA-Smad2 to silence Smad2 (g, h); and pWPI-hSmad2 to overexpress Smad2 (i, j). k, Representative images of Smad2 immunohistochemistry in the DG GCL suprapyramidal blade (a, area of the red box) of an adult male mouse. Smad2 is expressed strongly in the DG, yet not all granule cells express Smad2 (white circles). The vast majority of positive cells show Smad2 in both the nucleus and the cytoplasm (pointing to a phosphorylated state; when the protein is in an unphosphorylated state, it is only found in the cytoplasm), although the cytoplasm of mature granule neurons is just a narrow rim because of the highly packed population (blue, DAPI staining; green, SMAD2 labeling). N = 3 animals/time point in a–d (total, 18); N = 8 animals/experimental group in e–j (total, 32). ML, Molecular layer. **p < 0.01.
In terms of cell proliferation and survival, silencing Smad2 in the DG GCL of male mice produced a significant increase in cell number, whereas hSmad2 overexpression caused a significant decrease in cell number (Fig. 4a). The effects of Smad2 silencing and hSmad2 overexpression were consistent (opposite) for proliferation/survival measured by BrdU+ uptake by 3-week-old cells (Fig. 4b–j). However, changes in cell proliferation at the end of the experimental time course, as measured by the number of pH3+ cells (Fig. 4k), were evident only following Smad2 silencing (Figs. 4l,n,p), while there was only a trend toward such a change following hSmad2 overexpression (Fig. 4m). The findings were valid for both the total cell numbers (Figs. 4b,c,l,m) and the ratio of labeled cells relative to the whole granule cell population (Figs. 4d,e,n,o) or the total cell number in the rostral half of the GCL (Fig. 4f,g,p). These differences were appreciated in the rostral part of the DG, both in the silencing and overexpression experiments when BrdU+ cells were assessed (Fig. 4h,i), yet only following silencing for pH3+ cells (Fig. 4r). In the latter case, a sparse difference only at 2.8 mm caudal from bregma was also found. We have found, specifically, the next results: for BrdU total cell number (pLVTHM, 1744 ± 148.74; pLVTHM-shRNA-Smad2, 2893.6 ± 148.64; Mann–Whitney U test, p = 0.007); for Smad2 silencing (Fig. 4b; pWPI, 1610 ± 147.8; pWPI-hSmad2, 470.67 ± 27.67; Mann–Whitney U test, p = 0.034); for hSmad2 overexpression (Fig. 4c); for the BrdU total cell number/granule cell number ratio (pLVTHM, 0.00132 ± 0.00015; pLVTHM-shRNA-Smad2, 0.0022 ± 0.00019; Mann–Whitney U test, p = 0.007); for Smad2 silencing (Fig. 4d; pWPI, 0.00089 ± 0.00,016; pWPI-hSmad2, 0.00031 ± 0.000039; Mann–Whitney U test, p = 0.036); for hSmad2 overexpression (Fig. 4e); for the BrdU+ cell number in the rostral half of the hippocampus (Mann–Whitney U test, p = 0.0065); for Smad2 silencing (Fig. 4f; Mann–Whitney U test, p = 0.043; for hSmad2 overexpression (Fig. 4g); for pH3 total cell number (pLVTHM, 792.8 ± 39.81; pLVTHM-shRNA-Smad2, 1428.8 ± 41.74; Mann–Whitney U test, p = 0.003); for Smad2 silencing (Fig. 4l; pWPI, 790.67 ± 61.71; pWPI-hSmad2, 453.3 ± 48.78; Mann–Whitney U test, p = 0.077); for hSmad2 overexpression (Fig. 4m); for the pH3 total cell number/granule cell number ratio (pLVTHM, 0.00048 ± 0.0000824; pLVTHM-shRNA-Smad2, 0.000914 ± 0.000185; Mann–Whitney U test, p = 0.003); for Smad2 silencing (Fig. 4n; pWPI, 0.000525 ± 0.0000884; pWPI-hSmad2, 0.000304 ± 0.0000821); no changes for hSmad2 overexpression; Fig. 4o); and for pH3+ cell number in the rostral half of the hippocampus (Mann–Whitney U test, p = 0.011 for Smad2 silencing).
Figure 4.

Proliferation/survival analysis of the hippocampal dentate gyrus in loss-of-function (shRNA-Smad2) and gain-of-function (hSmad2) experiments. The cell counts correspond to the GCL of the hippocampal DG. a, Scheme of the survival time for BrdU data. b–j, Proliferation/survival measured by BrdU immunohistochemistry and through fractionator cell counts: in the silencing experiment (b, d, f, h); in the overexpression experiment (c, e, g, i); total BrdU+ cell number (b, c); BrdU+ cell number/total granule cell number ratio (d, e); total BrdU+ cell number in the rostral half of the DG (f, g); and distribution of BrdU+ cell number at specific distances from bregma, in the entire DG (h) or in the rostral half of the DG (i), also represented in g. j, Representative images of the 3 week survival of BrdU+ cells in the DG. k–s, Proliferation measured by pH3 immunohistochemistry and fractionator cell count: representative images of the pH3 immunohistochemistry (k), in the silencing experiment (l, n, p, r), and in the overexpression experiment (m, o, q, s). l, m, Total pH3+ cell number. n, o, pH3+ cell/total granule cell ratio. p, q, Total pH3+ cell number in the rostral half of the DG. r, s, Distribution of pH3+ cell number at specific distances from bregma, in the entire DG (r) or in the rostral half of the DG (s), also represented in q. An increase in the total number of 3-week-old BrdU+ cells and the total number of pH3+ cells is shown in the silencing experiment, while a decrease in BrdU+ cells and a trend toward a decrease in pH3+ cells is shown in the overexpression experiment. N = 8 animals/experimental group (total, 32). *p < 0.05, **p < 0.01. #0.1 > p ≥ 0.05.
The regional, rostral–caudal distribution was significantly different for BrdU total cell number mostly at rostral regions of the hippocampus: at 1600 µm (Mann–Whitney U test, p = 0.018) and 2000 µm (Mann–Whitney U test, p = 0.01) from bregma after Smad2 silencing (Fig. 4h); and at 1200 µm (Mann–Whitney U test, p = 0.034) from bregma after hSmad2 overexpression (Fig. 4i). Similarly, for pH3 total cell number, the distribution was significantly different at 1600 µm (Mann–Whitney U test, p = 0.05), 2000 µm (Mann–Whitney U test, p = 0.017), and 2800 µm (Mann–Whitney U test, p = 0.048) from bregma after Smad2 silencing (Fig. 4r), while no changes were found after hSmad2 overexpression (Fig. 4s).
In terms of cell maturation, we found a significant increase in the number of immature neurons after silencing Smad2, as measured by DCX+/GFP+ labeling (Fig. 5a,b; pLVTHM, 100 ± 0.19; pLVTHM-shRNA-Smad2, 376 ±71.6; Mann–Whitney U test, p = 0.007), while there was a trend toward a decrease in this parameter after hSmad2 overexpression (Fig. 5c; pWPI, 100 ± 9.69; pWPI-hSmad2, 48.61 ±2.14; Mann–Whitney U test, p = 0.077). We also found an increase in the ratio of VGluT+/GAD+ terminals after Smad2 silencing (Fig. 5d–g; vGLUT/GAD ratio: pLVTHM, 1.51 ± 0.41; pLVTHM-shRNA-Smad2, 2.85 ± 0.91; Mann–Whitney U test, p = 0.027; Fig. 5d) because of a net decrease in GAD labeling (Fig. 5f; mean GAD area: pLVTHM, 17.32 ± 2.25; pLVTHM-shRNA-Smad2, 11.8 ± 1.8; Mann–Whitney U test, p = 0.05-), while no differences were found following overexpression (Fig. 6a–c). The complexity of the dendritic trees (Fig. 5o,p) was also used to measure the maturation of the GFP-labeled granule cell population. There was a decrease in the number of ramifications of granule dendrites in the range of 80–100 µm from the soma (Fig. 5h; Mann–Whitney U test: for 80 µm, p = 0.029; for 100 µm, p = 0.055), corresponding to tertiary dendrites (Fig. 5i; Mann–Whitney U test, p = 0.032), whereas no differences in dendrite number (Fig. 5j) or total branch points (Fig. 4k) were found. Similarly, the spine density on mature neurons (Fig. 5m,n) was significantly lower after Smad2 silencing (Fig. 5l; pLVTHM, 2.66 ± 0.1; pLVTHM-shRNA-Smad2, 2.04 ± 0.09; Mann–Whitney U test, p = 0.001). In terms of dendrite ramification, length, and number, and of spine density, hSmad2 overexpression did not produce any significant differences (Fig. 6d–h).
Figure 5.

Differentiation/maturation analysis of the hippocampal dentate gyrus in the loss-of-function (shRNA-Smad2) and gain-of-function (hSmad2) experiments. The cell counts correspond to the GCL of the hippocampal DG. The excitatory and inhibitory vesicles correspond to the inner third of the dentate molecular layer, suprapyramidal blade. Dendritic arborization and spine density in the DG GCL. a, DCX+/GFP+ cells. Representative immunohistochemistry in the DG GCL suprapyramidal blade of DCX–/GFP+ cells (green arrow), DCX+/GFP– cells (red arrows), and DCX+/GFP+ cells (orange arrow; with blue DAPI staining. b, c, The total number of DCX+/GFP+ cells (GFP+ cells coexpressing DCX in the DG GCL) increased following silencing (b), and there was a trend toward a decrease in the overexpressing experiments (c). d–g, VGluT/GAD immunohistochemistry in the inner third of the DG molecular layer: increased VGluT+/GAD+ ratio in shSmad2 animals relative to the controls (d); VGluT+ area (e); GAD+ area (f); and 8 bit images representing the density of vGLUT and GAD synaptic boutons in a control mouse (left) and a mouse injected with the silencing viral construct (right; g). A dotted red line marks the upper limit of the granule cell somas. h–p, Morphologic analysis of GFP+/NeuN+/DCX– mature neurons into the GCL. h–k, A Sholl analysis of the GFP+ cells to demonstrate the morphologic complexity; a decrease in the mean number of dendritic intersections per cell (80 μm, p = 0.029; 100 μm, p = 0.055; h); dendrite length (i), showing a decrease in tertiary dendrite number (j), and branch points (k). l–n, The spine density quantified on confocal stacks of tertiary dendrites of GFP+ cells showing a decrease in shSmad2 relative to the control animals (l), and examples of the different spine density in a tertiary dendrite of a control neuron and of a Smad2 silenced neuron (m, n). o, p, Schemes of DG granule cell dendritic trees from a representative control GFP+ neuron (o) and GFP+ Smad2 silenced neuron (p), from the brain region in h–k. N = 8 animals/experimental group (total, 32). For morphometric analysis, 20–30 cells were analyzed/experimental group. *p < 0.05, **p < 0.01, ***p < 0.001, #0.1 > p ≥ 0.05.
Figure 6.
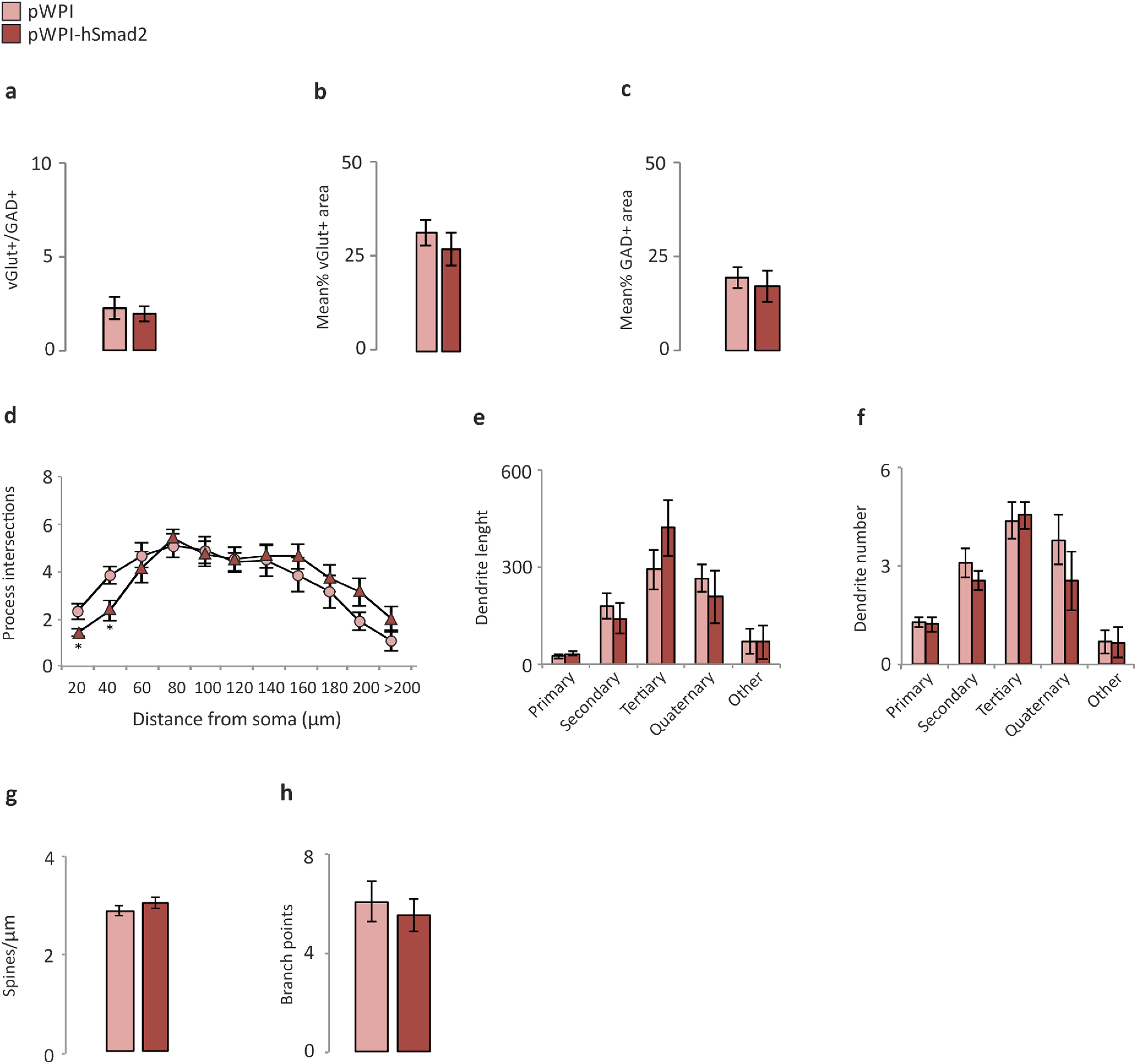
Maturation analysis of the hippocampal dentate gyrus in the gain-of-function experiments (hSmad2). a–c, VGluT/GAD immunohistochemistry in the inner third of the DG molecular layer: no changes in VGluT+/GAD+ ratio in hSmad2 animals relative to the controls (a); VGluT+ area (b); GAD+ area (c). d–f, h, A Sholl analysis of the GFP+ cells to demonstrate the morphologic complexity; no changes in the mean number of dendritic intersections per cell (d); no changes showing in dendrite length, (e), as well as in dendrite number (f) and branch points (h). g, The spine density quantified on confocal stacks of tertiary dendrites of GFP+ cells showing no changes in hSmad2 relative to the control animals. N = 8 animals/experimental group (total, 16). For morphometric analysis, 20–30 cells were analyzed/experimental group.
At the behavioral level, the anxiety-like status of the animals was analyzed, as well as their spatial learning and memory capabilities. Anxiety was evaluated in an elevated plus maze test to assess whether the animals spend more time in the closed arms, as expected, yet neither Smad2 silencing nor overexpression affected anxiety-like behavior (Fig. 7a; data not shown). In terms of learning and memory, we used an MWM test to analyze the capabilities of the mice to find a hidden platform based on internal and external cues (spatial memory), and a T-maze test to measure their working memory abilities. Silencing Smad2 had a detrimental effect on the spatial learning memory of the animals relative to the control animals (Fig. 7b; Mann–Whitney U test, p = 0.037), and on their discrimination of new/old arms in the T-maze test (Fig. 7c,d; Wilcoxon signed-rank test, p = 0.007 for time exploring the new or the old arms; Fig. 7c).
Figure 7.

Behavioral analysis of shSmad2 silencing. a, Elevated plus maze, with no differences in the total time spent in the open and closed arms. b, Morris water maze, significantly worse performance at the end of the task in the Smad2 silenced animals. c, d, T-maze, significantly longer time spent in the new arms during the probe trial in control animals, while the Smad silenced animals did not distinguish significantly between the arms (c); consequently, no positive discrimination index values were obtained for this group (d). N = 8 animals/ experimental group (total = 32). *p < 0.05, **p < 0.01.
Discussion
After analyzing the methylation of selected genes in the hippocampus following exercise, we selected Smad2 as a factor that potentially mediates adult neural plasticity in the hippocampal DG. The role of Smad2 in the DG GCL of adult mice was analyzed using the infection of lentiviruses that carry vectors to silence (shRNA) or overexpress (hSmad2) Smad2 under the control of constitutive promoters, given that Smad2 is expressed by both mature and immature neurons in the DG GCL (He et al., 2014). Most of transcription factors from a pool of stem cell-enriched transcription factors analyzed here were unmethylated, both in sedentary and exercising animals. However, Smad2 was hypermethylated in sedentary mice, and exercise induced demethylation of the promoter region of this gene, leading to an increase in Smad2 gene expression in exercising animals. Hdac1 demethylation and an increase in Hdac1 mRNA after exercise might account for the regulation of Smad2 methylation, although this issue requires further analysis.
The decrease in Smad2 expression in sedentary mice enhanced the proliferation of progenitor cells and the survival of immature neurons. In parallel, a deficit in differentiation from immature to mature neurons was evident, as suggested by the decrease in GAD+ terminals, while the mature neurons displayed defects in arborization and spine density. Hence, a decrease in Smad2 expression increases the proportion of excitatory versus inhibitory synapses. This effect could produce signals that indirectly induce hippocampal stem cell proliferation, which is directly related to a higher number of pH3+ cells. All these effects are coherent with the reported loss of TGF-βRI in progenitor cells infected with retroviral constructs expressing shRNA against ALK5, which alters the survival and maturation of newborn cells in the DG, dampening neurogenesis and affecting dendritic processes (He et al., 2014). Interestingly, these results are not specific for the brain region analyzed, the adult stage, or the species studied, as Smad2 knockdown also increased neurogenesis in chick embryo neural tube development (Míguez et al., 2013), pointing to a conserved role for Smad2 in regulating neurogenesis. According to the latter model, Smad2 and Smad3 show a dual cooperation/antagonism relationship in regulating neurogenesis. Further investigation is necessary to test whether this complex regulation of the TGF-β pathway is also present in mammals. By contrast, the overexpression of Smad2 in sedentary animals diminished the proliferation and survival of immature neurons, suggesting that progenitors exiting the cell cycle to initiate differentiation were affected but without inducing a variation in the VGluT/GAD ratios or in the dendritic maturation/arborization of mature neurons. These findings are useful to help understand the complex TGF-β pathway. While some authors have found that a chronic increase in TGF-β1 reduced the total number of immature hippocampal neurons and that TGF-β overexpression decreased hippocampal neurogenesis (Buckwalter et al., 2006), other authors have found a net promotion of neuronal survival and differentiation in the adult neurogenic niche after elevated TGF-β1 levels (Kandasamy et al., 2014). The use of different models can account for these discrepancies (aged vs adult animals), although it has been suggested the hypothesis that both a decrease in progenitor proliferation (through the control of progenitor cell quiescence) and an increase in postmitotic neuronal survival are possible and consistent with a role for TGF-β in the regulation of neural progenitor cell proliferation (Kandasamy et al., 2014). In the context of this hypothesis, the present findings after overexpressing Smad2 are similar in proliferation, but the opposite in survival. Smad2 is only one of the factors mediating the TGF-β-induced actions, and, as suggested above, a complex relationship exists between R-Smads. Together, these data and the gain-of-function and loss-of-function results presented here suggest that Smad2 is involved in AHN, indicating that it acts at several levels (promoting cell maturation and differentiation; and inhibiting proliferation and survival), while the cross talk with other Smads might account for the final effect of TGF-β signaling.
Our neuron morphology data (dendritic arborization measured by process intersections, dendrite length and number, branch points, and spine density) were analyzed in GFP+/NeuN+/DCX– cells, 3 weeks after viral injection. This approach guarantees that all neurons analyzed were mature cells, independent of their birth date. Because the virus infected equally both newborn and mature cells, a small proportion of infected cells is expected to be newborn cells potentially expressing DCX at the time of injection, but that switched off DCX expression during the 3 weeks from injection to kill, the common feature of all cells being that they are mature granule neurons in the GCL at the time of analysis.
In addition, the time from injection to kill is also relevant because it cannot be ruled out that some of the reported findings (changes in pH3+ cell number) take place in noninfected cells (in fact, it is highly probable considering the proportion of precursor and progenitor cells in the adult murine SGZ and GCL, and because a pH3+ cell is cycling at the time of kill, 3 weeks after the infection). This means that both cell autonomous and nonautonomous effects account for the present findings. Direct evidence that cell-nonautonomous effects may well be because of neurogenic niche-derived signals governing precursor and progenitor cell functioning have been long reported (Vicidomini et al., 2020; i.e., signals from neighboring infected, immature newborn cells). However, mature neuron population-derived signals have also been suggested to be influencing the neurogenic niche, as, for example, indirect evidence pointing to a strong relationship between the mature granule neuron number and the neurogenesis rate in the adult dentate gyrus (Llorens-Martín et al., 2006).
It is noteworthy that the significant changes found in cell proliferation/survival or cell proliferation alone are concentrated at the rostral-most portion of the DG, while tiny changes have been found in caudal portions. While these region-specific changes cannot be accounted for by a different regional induction of Smad2 silencing or overexpression (Fig. 3, viral vector expressions throughout the hippocampus) and considering that we have found here a net change in spatial learning/memory paradigms (both long-term and working memory); while no changes in anxiety behavior were observed, this result reinforces the idea of a different distribution of behaviors into hippocampal dorsal–ventral extension, but, more important, it raises a subregion-specific sensitivity to the role of Smad2 in hippocampal plasticity. This finding also supports the idea that different transcription factors and/or signal transduction pathways might be mediating this neural plasticity in different subregions of the hippocampus, as regional intrahippocampal and intra-dentate gyral specificity have been described for some receptor and neuropeptide signaling pathways in adult murine (Leonardo et al., 2006; Gresack et al., 2009; Christensen et al., 2010), adult human (Vogel et al., 2020), and postnatal hippocampus (Lee et al., 2017) settings. Similarly, other potent plasticity-mediator factors like IGF1 or BDNF have been reported displaying a specific rostral–caudal distribution (Poggini et al., 2019). Consistently, some activity-induced plasticity in the dentate gyrus has been reported recently showing dorsal–ventral specificity in the effects on growth factors and adult neurogenesis (Gualtieri et al., 2017).
Our results extend the developmental proneurogenic role of the TGF-β/Smad2/3 pathway to the adult brain, modulating associated learning and memory (Seth et al., 2017). They reveal its influence on proliferation/maturation in the adult DG, in direct relation to hippocampal-related learning and memory abilities. These findings might also explain the behavioral effects in mice lacking population-specific autocrine TGF-β signaling (Luo et al., 2016), implicating Smad2 in the control of excitatory/inhibitory synaptic balance, as indicated here by our VGluT/GAD data. Targeting the TGF-β/Smad2/3 pathway may be an interesting approach to alleviate the cognitive impairments induced by some interventions, as has proven useful in mice in a variety of hippocampal-related tasks accompanied by changes in neuron morphology (Tang et al., 2019).
It is worthy to consider that both the experimental silencing of Smad2 used here and physical activity induce increased proliferation and survival of progenitors and immature newborn neurons in the murine adult hippocampus. But while in the Smad2 silencing experiments we have found a net decrease in the maturation and dendritic arborization of mature granule neurons, the increased neurogenesis induced by exercise takes place in parallel with an increase in arborization and spines both in newborn and mature granule neurons (Glasper et al., 2010), mediated by IGF1 (Trejo et al., 2001), among other growth factors. This relevant difference may explain by itself the distinct effect of these interventions on behavior (Smad2 silencing induced an impairment in hippocampal-dependent cognition, while running induces precognitive, anxiolytic, and antidepressant effects (Llorens-Martín et al., 2008)). A second relevant consideration is that exercise induced Smad2 gene demethylation, and consequently increased Smad2 gene expression and activation as reported here, but this is not consistent with the Smad2 overexpression-induced effect described here on adult neurogenesis (decreased proliferation and survival), which is not found after physical exercise. Both considerations reinforce the idea that physical activity acts as a pleiotropic, multilevel intervention inducing plasticity mediated by a potent, well known growth factor cascade (Pérez-Domper et al., 2013) where the TGF-β/Smad2 signaling pathway may play a still unknown role, which is worthy to be investigated. It also substantiates the notion that adult hippocampal neurogenesis is a key process, but not the only process, in mediating the exercise-induced effects.
The data presented here demonstrate that Smad2 is a modulator of neuroplasticity in mature DG neurons, and that it regulates the late stages of AHN. Smad2 promotes the maturation and differentiation of granule neurons but not cell proliferation/survival, and it has a direct impact on hippocampal-related task performance. Nevertheless, exercise modulates the expression of Smad2 through epigenetic mechanisms like DNA methylation.
Footnotes
This work was supported by the Spanish Ministry of Economy and Competitiveness Project Grants BFU2013-48907-R and BFU2016-77162-R (to J.L.T.). We thank Laude Garmendia from the Animal facility at the Cajal Institute for invaluable help and advice, and the members of the Image Analysis Unit at the Cajal Institute.
The authors declare no competing financial interests.
References
- Brown KA, Pietenpol JA, Moses HL (2007) A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-beta signaling. J Cell Biochem 101:9–33. 10.1002/jcb.21255 [DOI] [PubMed] [Google Scholar]
- Buckwalter MS, Yamane M, Coleman BS, Ormerod BK, Chin JT, Palmer T, Wyss-Coray T (2006) Chronically increased transforming growth factor-beta1 strongly inhibits hippocampal neurogenesis in aged mice. Am J Pathol 169:154–164. 10.2353/ajpath.2006.051272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Harland RM (2007) Neural induction requires continued suppression of both Smad1 and Smad2 signals during gastrulation. Development 134:3861–3872. 10.1242/dev.007179 [DOI] [PubMed] [Google Scholar]
- Christensen T, Bisgaard CF, Nielsen HB, Wiborg O (2010) Transcriptome differentiation along the dorso-ventral axis in laser-captured microdissected rat hippocampal granular cell layer. Neuroscience 170:731–741. 10.1016/j.neuroscience.2010.07.016 [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425:577–584. 10.1038/nature02006 [DOI] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL (2003) Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol 5:410–421. 10.1038/ncb975 [DOI] [PubMed] [Google Scholar]
- Funaba M, Zimmerman CM, Mathews LS (2002) Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J Biol Chem 277:41361–41368. 10.1074/jbc.M204597200 [DOI] [PubMed] [Google Scholar]
- Glasper ER, Llorens-Martin MV, Leuner B, Gould E, Trejo JL (2010) Blockade of insulin-like growth factor-I has complex effects on structural plasticity in the hippocampus. Hippocampus 20:706–712. 10.1002/hipo.20672 [DOI] [PubMed] [Google Scholar]
- Gresack JE, Schafe GE, Orr PT, Frick KM (2009) Sex differences in contextual fear conditioning are associated with differential ventral hippocampal extracellular signal-regulated kinase activation. Neuroscience 159:451–467. 10.1016/j.neuroscience.2009.01.009 [DOI] [PubMed] [Google Scholar]
- Gualtieri F, Brégère C, Laws GC, Armstrong EA, Wylie NJ, Moxham TT, Guzman R, Boswell T, Smulders TV (2017) Effects of environmental enrichment on doublecortin and BDNF expression along the dorso-ventral axis of the dentate gyrus. Front Neurosci 11:488. 10.3389/fnins.2017.00488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zhang H, Yung A, Villeda SA, Jaeger PA, Olayiwola O, Fainberg N, Wyss-Coray T (2014) ALK5-dependent TGF-β signaling is a major determinant of late-stage adult neurogenesis. Nat Neurosci 17:943–952. 10.1038/nn.3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer J, Escalante-Alcalde D, Lia M, Boettinger E, Edelmann W, Stewart CL, Kucherlapati R (1999) Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proc Natl Acad Sci U S A 96:12595–12600. 10.1073/pnas.96.22.12595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard CV, Reed MG (1998) Unbiased Stereology. BIOS Scientific Publishers. Oxford. [Google Scholar]
- Ishihara A, Saito H, Abe K (1994) Transforming growth factor-β1 and -β2 promote neurite sprouting and elongation of cultured rat hippocampal neurons. Brain Res 639:21–25. 10.1016/0006-8993(94)91759-0 [DOI] [PubMed] [Google Scholar]
- Kandasamy M, Lehner B, Kraus S, Sander PR, Marschallinger J, Rivera FJ, Trümbach D, Ueberham U, Reitsamer HA, Strauss O, Bogdahn U, Couillard-Despres S, Aigner L (2014) TGF-beta signalling in the adult neurogenic niche promotes stem cell quiescence as well as generation of new neurons. J Cell Mol Med 18:1444–1459. 10.1111/jcmm.12298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AR, Kim JH, Cho E, Kim M, Park M (2017) Dorsal and ventral hippocampus differentiate in functional pathways and differentially associate with neurological disease-related genes during postnatal development. Front Mol Neurosci 10:331. 10.3389/fnmol.2017.00331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardo ED, Richardson-Jones JW, Sibille E, Kottman A, Hen R (2006) Molecular heterogeneity along the dorsal-ventral axis of the murine hippocampal CA1 field: a microarray analysis of gene expression. Neuroscience 137:177–186. 10.1016/j.neuroscience.2005.08.082 [DOI] [PubMed] [Google Scholar]
- Liu L, Liu X, Ren X, Tian Y, Chen Z, Xu X, Du Y, Jiang C, Fang Y, Liu Z, Fan B, Zhang Q, Jin G, Yang X, Zhang X (2016) Smad2 and Smad3 have differential sensitivity in relaying TGFbeta signaling and inversely regulate early lineage specification. Sci Rep 6:21602. 10.1038/srep21602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Martín M, Torres-Alemán I, Trejo JL (2006) Pronounced individual variation in the response to the stimulatory action of exercise on immature hippocampal neurons. Hippocampus 16:480–490. 10.1002/hipo.20175 [DOI] [PubMed] [Google Scholar]
- Llorens-Martín M, Torres-Alemán I, Trejo JL (2008) Growth factors as mediators of exercise actions on the brain. Neuromolecular Med 10:99–107. 10.1007/s12017-008-8026-1 [DOI] [PubMed] [Google Scholar]
- Luo SX, Timbang L, Kim JI, Shang Y, Sandoval K, Tang AA, Whistler JL, Ding JB, Huang EJ (2016) TGF-β signaling in dopaminergic neurons regulates dendritic growth, excitatory-inhibitory synaptic balance, and reversal learning. Cell Rep 17:3233–3245. 10.1016/j.celrep.2016.11.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19:2783–2810. 10.1101/gad.1350705 [DOI] [PubMed] [Google Scholar]
- Míguez DG, Gil-Guiñón E, Pons S, Martí E (2013) Smad2 and Smad3 cooperate and antagonize simultaneously in vertebrate neurogenesis. J Cell Sci 126:5335–5343. 10.1242/jcs.130435 [DOI] [PubMed] [Google Scholar]
- Munoz MD, Antolin-Vallespin M, Tapia-Gonzalez S, Sanchez-Capelo A (2016) Smad3 deficiency inhibits dentate gyrus LTP by enhancing GABAA neurotransmission. J Neurochem 137:190–199. [DOI] [PubMed] [Google Scholar]
- Nomura M, Li E (1998) Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 393:786–790. 10.1038/31693 [DOI] [PubMed] [Google Scholar]
- Pérez-Domper P, Gradari S, Trejo JL (2013) The growth factors cascade and the dendrito-/synapto-genesis versus cell survival in adult hippocampal neurogenesis: the chicken or the egg. Ageing Res Rev 12:777–785. 10.1016/j.arr.2013.06.001 [DOI] [PubMed] [Google Scholar]
- Poggini S, Golia MT, Alboni S, Milior G, Sciarria LP, Viglione A, Matte Bon G, Brunello N, Puglisi-Allegra S, Limatola C, Maggi L, Branchi I (2019) Combined fluoxetine and metformin treatment potentiates antidepressant efficacy increasing IGF2 expression in the dorsal hippocampus. Neural Plast 2019:4651031. 10.1155/2019/4651031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth B, Yadav A, Agarwal S, Tiwari SK, Chaturvedi RK (2017) Inhibition of the transforming growth factor-β/SMAD cascade mitigates the anti-neurogenic effects of the carbamate pesticide carbofuran. J Biol Chem 292:19423–19440. 10.1074/jbc.M117.798074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipursky J, Francis D, Gomes FC (2012) Activation of MAPK/PI3K/SMAD pathways by TGF-β(1) controls differentiation of radial glia into astrocytes in vitro. Dev Neurosci 34:68–81. 10.1159/000338108 [DOI] [PubMed] [Google Scholar]
- Tang CZ, Yang JT, Liu QH, Wang YR, Wang WS (2019) Up-regulated miR-192-5p expression rescues cognitive impairment and restores neural function in mice with depression via the Fbln2-mediated TGF-β1 signaling pathway. FASEB J 33:606–618. 10.1096/fj.201800210RR [DOI] [PubMed] [Google Scholar]
- Tapia-González S, Muñoz MD, Cuartero MI, Sánchez-Capelo A (2013) Smad3 is required for the survival of proliferative intermediate progenitor cells in the dentate gyrus of adult mice. Cell Commun Signal 11:93. 10.1186/1478-811X-11-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo JL, Carro E, Torres-Aleman I (2001) Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J Neurosci 21:1628–1634. 10.1523/JNEUROSCI.21-05-01628.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay KD, Hoodless PA, Bikoff EK, Robertson EJ (2000) Formation of the definitive endoderm in mouse is a Smad2-dependent process. Development 127:3079–3090. 10.1242/dev.127.14.3079 [DOI] [PubMed] [Google Scholar]
- Ueberham U, Lange P, Ueberham E, Brückner MK, Hartlage-Rübsamen M, Pannicke T, Rohn S, Cross M, Arendt T (2009) Smad2 isoforms are differentially expressed during mouse brain development and aging. Int J Dev Neurosci 27:501–510. 10.1016/j.ijdevneu.2009.04.001 [DOI] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH (1999) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 2:266–270. 10.1038/6368 [DOI] [PubMed] [Google Scholar]
- Vicidomini C, Guo N, Sahay A (2020) Communication, cross talk, and signal integration in the adult hippocampal neurogenic niche. Neuron 105:220–235. 10.1016/j.neuron.2019.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JW, La Joie R, Grothe MJ, Diaz-Papkovich A, Doyle A, Vachon-Presseau E, Lepage C, Vos de Wael R, Thomas RA, Iturria-Medina Y, Bernhardt B, Rabinovici GD, Evans AC (2020) A molecular gradient along the longitudinal axis of the human hippocampus informs large-scale behavioral systems. Nat Commun 11:960. 10.1038/s41467-020-14518-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ (1998) Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell 92:797–808. 10.1016/S0092-8674(00)81407-5 [DOI] [PubMed] [Google Scholar]
- Wang L, Nomura M, Goto Y, Tanaka K, Sakamoto R, Abe I, Sakamoto S, Shibata A, Enciso PLM, Adachi M, Ohnaka K, Kawate H, Takayanagi R (2011) Smad2 protein disruption in the central nervous system leads to aberrant cerebellar development and early postnatal ataxia in mice. J Biol Chem 286:18766–18774. 10.1074/jbc.M111.223271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiznerowicz M, Trono D (2003) Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol 77:8957–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All materials, data and associated protocols used in this work are available to interested readers.