

Abstract
Research conducted on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pathogenesis and coronavirus disease 2019 (COVID-19) generally focuses on the systemic host response, especially that generated by severely ill patients, with few studies investigating the impact of acute SARS-CoV-2 at the site of infection. We show that the nasal microbiome of SARS-CoV-2-positive patients (CoV+, n = 68) at the time of diagnosis is unique when compared to CoV− healthcare workers (n = 45) and CoV− outpatients (n = 21). This shift is marked by an increased abundance of bacterial pathogens, including Pseudomonas aeruginosa, which is also positively associated with viral RNA load. Additionally, we observe a robust host transcriptional response in the nasal epithelia of CoV+ patients, indicative of an antiviral innate immune response and neuronal damage. These data suggest that the inflammatory response caused by SARS-CoV-2 infection is associated with an increased abundance of bacterial pathogens in the nasal cavity that could contribute to increased incidence of secondary bacterial infections.
Keywords: SARS-CoV-2, nasal microbiome, inflammation, COVID-19, viral RNA load, coinfection, RNA-seq, Pseudomonas aeruginosa
Graphical abstract
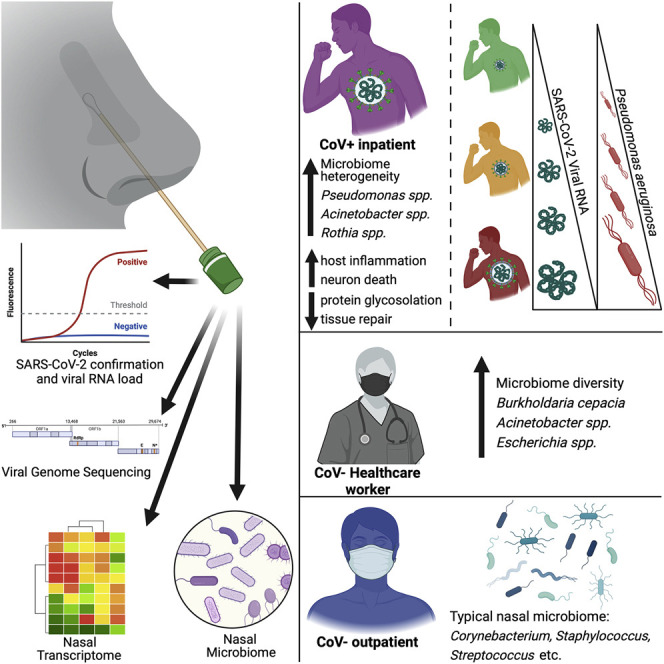
Rhoades et al. show that patients acutely infected with SARS-CoV-2 have a distinct nasal microbiome marked by an increase in bacterial pathogens such as Pseudomonas aeruginosa and accompanied by a robust inflammatory host transcriptional response. This provides a potential explanation for the high rate of bacterial co-infections in COVID-19 patients.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of coronavirus disease 2019 (COVID-19) (Zhou et al., 2020b; Zhu et al., 2020), a respiratory disease ranging in clinical presentation from an asymptomatic (∼80% of cases) to a fatal infection (∼1%–2%) (Guan et al., 2020; Hu et al., 2021). Long-term consequences of COVID-19 range from anosmia and ageusia (Guan et al., 2020; Harrison et al., 2020; Hornuss et al., 2020) to respiratory (e.g., dyspnea) and nervous system (e.g., headache, anosmia) complications (Lopez-Leon et al., 2021). Numerous studies have identified advanced age, obesity, diabetes, and male sex as risk factors for severe COVID-19 (Callender et al., 2020; Ejaz et al., 2020; Harrison et al., 2020; Hu et al., 2021). Other factors, including mucosal microbial defenses and viral burden at the site of initial infection, have been underexplored.
SARS-CoV-2 is primarily spread through the inhalation of virus-laden respiratory droplets (Harrison et al., 2020). Initial entry and subsequent replication occur in the upper respiratory tract (URT) following the interaction of the viral spike protein and host ACE2 (Hoffmann et al., 2020; Murgolo et al., 2021; Shang et al., 2020). The nasal cavity is thought to be the initial site of viral replication rather than the oral cavity due to higher expression of ACE2 (Chen et al., 2020; Sungnak et al., 2020; Zou et al., 2020b) and greater levels of viral RNA (vRNA) at this site (Zou et al., 2020a). Rapid and uncontrolled replication can lead to infection of the lower respiratory tract and severe disease. However, a lack of correlation between disease severity and vRNA load in nasal cavities suggests that other factors contribute to disease outcomes (Lui et al., 2020; Yilmaz et al., 2021; Zou et al., 2020a).
The mucosal microbiome plays a critical role in modulating viral infection (Gallo et al., 2021; He et al., 2020; Khatiwada and Subedi, 2020; Wilks and Golovkina, 2012). Disruption of microbial communities by viral infection can exacerbate inflammation, facilitate coinfection, and deregulate the adaptive immune response (Gallo et al., 2021; He et al., 2020; Khatiwada and Subedi, 2020; Wilks and Golovkina, 2012). Furthermore, severe inflammation in nasal epithelium can lead to short- and long-term symptoms such as anosmia and ageusia (Melo et al., 2021; Rawson and Huang, 2009). However, a limited number of studies have examined the interplay between respiratory microbiome and acute SARS-CoV-2 infection. Studies of nasopharyngeal samples from small cohorts of mild and severe COVID-19 patients show variable differences in diversity and increased abundance of select phyla without accounting for the potential impact of viral load (Butler et al., 2021; De Maio et al., 2020; Nardelli et al., 2021; Rueca et al., 2021). Other studies of bronchoalveolar lavage and pharyngeal samples in severe patients reported a general dysbiosis of the lower respiratory microbiome similar to that of pneumonia patients (Budding et al., 2020; Shen et al., 2020).
Studies aimed at understanding the impact of COVID-19 and viral burden on nasal microbial communities are urgently needed to gain insight into acute and long-term consequences of COVID-19. In this study, we determined the impact of acute SARS-CoV-2 infection with a spectrum of vRNA loads on the nasal microbiome, local host transcriptional responses, and potential association with viral genome sequence diversity. Our cross-sectional study of CoV+ patients, CoV− outpatients, and CoV− healthcare workers from the same hospital indicated a distinct shift in the nasal microbiome of patients at the time of COVID-19 diagnosis, featuring the increased abundance of pathobionts such as Rothia, Acinetobacter, and Pseudomonas. This was accompanied by the upregulation of host antiviral genes and the downregulation of genes with roles in mucosal and neuronal cell homeostasis. Overall, our data suggest that SARS-CoV-2 infection is associated with an increased abundance of bacterial pathogens in the nasal cavity and virus-induced host dysregulation at the site of infection.
Results
SARS-CoV-2 infection is associated with increased abundance of pathobionts in the nasal microbiome
We utilized 16S rRNA gene amplicon sequencing to profile nasal microbiomes from SARS-CoV-2 positive (CoV+) patients at the time of diagnosis (n = 68), CoV− outpatients seeking elective procedures (n = 21), and CoV− healthcare workers (HCWs; n = 45) (Figure 1 A). These three groups did not differ by age or sex (Figure 1A; Table S1). We first determined whether microbial DNA co-eluted in vRNA extractions was reflective of a typical nasal microbiome. To that end, we compared our dataset to a previously published healthy nasal microbiome dataset obtained using conventionally DNA extraction methods (De Boeck et al., 2017) (Figure S1B). This analysis revealed a high degree of overlap between the two studies (Figure S1B). Additionally, we found that age and sex explain a significant amount of variation in unweighted and weighted UniFrac distance, respectively (Figures S1C–S1H). However, host CoV status exerted a greater impact on the nasal microbiome composition than did age and sex, accounting for 9.3% of the total variation of unweighted UniFrac distance (Figures 1B, S1G, and S1H).
Figure 1.

The nasal microbiome of SARS-CoV-2-infected patients is distinct
(A) Study design.
(B) Principal coordinate analysis of nasal microbial communities unweighted UniFrac distance colored by host status. The contribution of host status to the total variance in the unweighted UniFrac dissimilarity was measured using permutational multivariate analysis of variance (PERMANOVA).
(C–E) Violin plot of (C) average unweighted UniFrac distances, (D) number of observed ASVs, and (E) Shannon diversity. Significance for (C)–(E) was determined using a Kruskal-Wallis non-parametric ANOVA (p values inset at the bottom of each panel) with Dunn’s multiple comparison. ∗∗p < 0.01, ∗∗∗∗ = p < 0.0001.
(F) Bubble plots of bacterial genera with >1% average abundance across the entire study population. The size of each circle indicates the average relative abundance for each taxa, and the color of each circle denotes bacterial phyla.
Additionally, nasal microbial communities in CoV+ patients showed the highest intra-group variability followed by communities from CoV− HCWs and CoV− outpatients (Figure 1C). These same trends held for abundance-based weighted UniFrac distance (Figures S2A and S2B). We also observed an increased richness in nasal community from COV+ patients and HCWs as indicated by a higher number of observed amplicon sequencing variants (ASVs) compared to CoV− outpatients (Figure 1D). As expected, all three groups had comparable low community Shannon diversity values, indicative of a low-complexity community dominated by a few microbes (Figure 1E). We next probed the taxonomic landscape of the nasal microbiome to identify microbial taxa driving the shift in the overall community. At the phyla level, samples from all groups were dominated by Actinobacteria, Firmicutes, and Proteobacteria (Figures 1F and S2C). Common nasal genera such as Corynebacterium, Staphylococcus, Streptococcus, Dolosigranulum, and Neisseria were highly abundant in all three groups (Figures 1F and S2C).
Comparison of the three groups using linear discriminant analysis effect size (LEfSe) revealed enrichment of specific genera in each group (Figure 2 A; Table S2). Specifically, Anaerococcus was enriched in CoV− outpatients, while Lawsonella and Burkholderia were most abundant in CoV− HCWs (Figure 2A; Table S2). Finally, several pathogenic bacteria such as Rothia, Acinetobacter, and Pseudomonas were most abundant in CoV+ patients (Figure 2A; Table S1). To further explore the distribution of specific taxonomic groups, we plotted the abundance of selected taxa. The two dominant genera, Corynebacterium and Staphylococcus, were equally abundant in the groups (Figures 2B and 2C). Interestingly, abundance of Anoxybacillus was higher in both CoV− outpatients and CoV+ inpatients compared to HCWs (Figure 2D). Conversely, Burkholderia cepacia was significantly more abundant in CoV− HCWs compared to the two patient groups (Figures 1F and 2E). Alternatively, the genera Acinetobacter and Pseudomonas were most abundant in the CoV+ patients (Figures 2F and 2G). When probed at the species level, Pseudomonas aeruginosa was highly enriched in CoV+ patients (Figure 2H). These data suggest that SARS-CoV-2 infection is associated with a shift in the nasal microbiome and acquisition of pathobionts.
Figure 2.

The nasal microbiome of SARS-CoV-2-infected patients and healthcare workers are enriched in opportunistic bacterial pathogens
(A) Selected differentially abundant genera in the nasal microbiome between CoV−, HCWs, and CoV+ individuals. Differential abundance was determined using LEfSe (log10 linear discriminant analysis [LDA] score >2).
(B–H) Scatterplots of bacterial genera and species of interest identified by LEfSe analysis plotted as log10 relative abundance + 0.01. Horizontal black lines represent the mean and whiskers represent the SEM. Significance for (B)–(H) was determined using a Kruskal-Wallis non-parametric ANOVA, with a Dunn’s multiple comparison test. ∗p < 0.05, ∗∗∗p < 0.001.
Associations between SARS-CoV-2 vRNA load and the nasal microbiome
We next determined whether vRNA load is associated with changes in the nasal microbiome. We divided CoV+ inpatients into three groups based on cycle threshold (Ct) values at admission: high Ct value (<40–34.6; n = 23), middle Ct value (32.9–25.0; n = 23), and low Ct value (23.5–15.8; n = 22) (Figure S2D). We compared the number of observed ASVs and community composition based on unweighted and weighted UniFrac distance, as well as intra-group variability across the three Ct groups (Figures S2E–S2I). vRNA load did not impact the diversity of the nasal microbial community (Figures S2E–S2G). However, when the relative abundance of taxa was considered, viral loads explained 10% of the overall community composition (Figure S2H). Moreover, the nasal communities of low and middle Ct groups were comparable to each other and distinct from the high Ct group (Figure S2I), suggesting that shifts in the nasal microbiome are modulated by vRNA load.
LEfSe analysis between the three Ct groups showed higher abundance of Streptococcus in nasal microbial communities of patients with high Ct, higher abundance of Corynebacterium in patients with middle Ct, and high abundance of Cutibacterium, Neisseria, and Pseudomonas in patients with low Ct (Figure 3 A). The relative abundance of the genus Streptococcus was highest in the high Ct group (Figure 3B), while that of Corynebacterium was more abundant in the middle and low Ct groups (Figure 3C). Finally, Neisseria and Pseudomonas were more abundant in the low Ct group (Figures 3D and 3E), and P. aeruginosa was positively associated with viral loads (Figure 3F).
Figure 3.

The nasal microbiome of SARS-CoV-2-infected patients is associated with vRNA load
(A) Differentially abundant genera in the nasal microbiome between CoV+ individuals stratified by vRNA load into high, middle, and low Ct. Differential abundance was determined using LEfSe (log10 LDA score >2).
(B–F) Scatterplots of bacterial genera and species of interest identified by LEfSe analysis plotted as log10 relative abundance + 0.01. Horizontal black bars represent the mean and whiskers represent the SEM. Significance for (B)–(H) was determined using a Kruskal-Wallis non-parametric ANOVA, with Dunn’s multiple comparison test. ∗p < 0.05, ∗∗∗p < 0.001.
See also Figure S3.
SAR-CoV-2 infection remodels the nasal epithelium transcriptome
We compared the nasal transcriptomes of a subset of CoV− HCWs (n = 4) and CoV+ inpatients (n = 4) using RNA sequencing (RNA-seq) (Figure 4 ). We identified 692 differentially expressed genes (DEGs) in the CoV+ group relative to HCWs (Figure 4A). Upregulated DEGs (n = 377) enriched to multiple Gene Ontology (GO) terms associated with innate and adaptive host defense pathways such as “response to virus,” “inflammatory response,” “response to bacterium,” and “lymphocyte activation” (Figure 4B). A large number of interferon-stimulated (ISGs; e.g., IFITM3, ISG15) and type I interferon signaling genes (e.g., IRF7, STAT1) mapped to the GO term “response to virus” (Figure 4C). Genes with roles in nuclear factor κB (NF-κB) and JUN-ATP-1 signaling, such as BCL3 and MYD88, mapped to GO terms “inflammatory response.” Expression of genes involved in leukocyte chemotaxis (e.g., CCL2, CCR1), hematopoiesis (e.g., CD53, IKZF1), and B cell activation (e.g., IGHA, LCK) were upregulated in CoV+ patients. Interestingly, genes that encode inhibitory receptors (e.g., CD300E, LAGLS9) as well as genes involved in cell death (e.g., BCL2, NUPR1) were also highly upregulated. Additionally, upregulated DEGs (e.g., SNCA, MDK) belonged to “neuron death” (Figure 4B).
Figure 4.

Transcriptional profiling of the nasal passages reveals robust immune activation
(A) Volcano plot of gene expression changes in CoV+ patients relative to CoV− HCWs. Upregulated differentially expressed genes (DEGs) are indicated in red; downregulated genes are indicated in blue.
(B) Functional enrichment of upregulated DEGs. Horizontal bars represent the number of genes enriching to each GO term, with color intensity representing the negative log of the false discovery rate (FDR)-adjusted p value (−log[q value]).
(C) Heatmap of upregulated DEGs. Columns of all heatmaps represent the RPKM (reads per kilobase transcript per million mapped reads) of one individual. Range of colors per each heatmap is based on scaled and centered RPKM values of the represented DEGs (red indicates upregulated; blue indicates downregulated).
(D) Functional enrichment of downregulated DEGs as described in (C).
(E) Heatmap of downregulated DEGs. See (D) for additional details.
Downregulated DEGs (n = 315) mapped to GO terms related to tissue homeostasis (e.g., “response to wounding”), cellular organization (e.g., “microtubule-based movement”), and neuronal processes (e.g., “sensory organ development”) (Figure 4C). Notable downregulated DEGs encoded integrins (e.g., ITGA2/3/V, ITGB1/6), laminins (e.g., LAMA2/B2/B3/C2), and microtubules (e.g., DNAH3/7/10/11, DYNC2H1) comprising intracellular and extracellular structures such as cilia and cell-cell adhesion junctions. Genes associated with mucin production in nasal passages (e.g., MUC20, MUC5AC) were downregulated. Finally, genes encoding ion channels important for chloride ion balance and neuron homeostasis (e.g., ATP1A1, SLC39A, CLCN3) were downregulated (Figures 4A and 4E).
Viral genome recovery, but not genetics, correlates with viral loads
To identify associations between vRNA load, genome recovery, and viral evolution, we assembled SARS-CoV-2 genomes from CoV+ patients. As expected, the Ct value was negatively associated with viral genome coverage (Pearson correlation, r = −0.509, p = 0.0001), with 81.6%, 54.58%, and 8.3% of genomes from low, middle, and high Ct groups having greater than 90% genome coverage, respectively. All genomes with adequate coverage harbored the spike D614G and nsp12 P4715L mutations, a noncoding mutation in the 5′ UTR, and a synonymous mutation in nsp3 (F924) (Figure S3A). The nucleoprotein T205I mutation associated with the 501.V2 variant was found in two low and middle Ct samples. Genomes belonged to three main clades, that is, 20A, 20B, and 20C, with very few sequences belonging to the 20H clade, which emerged in October and no distinct relationship to current variants of concern (P.1, B.1.1.7, B.1.351, B.1.427, B.1.429) or partitioning according to Ct value (Figure S3B).
Discussion
In this study, we explored connections between acute SARS-CoV-2 infection, the nasal microbiome, and the local host transcriptional response. While a wealth of studies have focused on the systemic host response to SARS-CoV-2, few studies have investigated the impact of acute SARS-CoV-2 on the nasal epithelium. SARS-CoV-2 infection occurs primarily via respiratory droplets, with the initial viral replication taking place in the nasal epithelia (Murgolo et al., 2021; Sungnak et al., 2020). Most individuals who contract SARS-CoV-2 clear infection within the URT, resulting in asymptomatic to mild disease (Guan et al., 2020). When this initial response is insufficient, SARS-CoV-2 migrates into the lower respiratory tract, leading to moderate/severe COVID-19 characterized by acute respiratory distress syndrome, pneumonias, and cytokine storm (Hu et al., 2021; Kuri-Cervantes et al., 2020; Grasselli et al., 2021; Guan et al., 2020). Thus, there is a clear need to understand the host and microbial factors in the nasal cavity during acute infection.
The healthy nasal microbiome is dominated by Corynebacterium, Staphylococcus, Streptococcus, Dolosigranulum, and Moraxella (Whelan et al., 2014; Bassis et al., 2014; Stearns et al., 2015). This community is influenced by a multitude of external factors such as age, sex, antibiotic use, and pollutants (Wos-Oxley et al., 2010; Charlson et al., 2010; Ramakrishnan et al., 2018). Sex and age exerted a limited effect on the overall composition of the nasal microbiome compared to SARS-CoV-2 infection. However, we were unable to control for antibiotic use, exposure to pollutants, socioeconomic status, and race, all of which could differ considerably between the groups given the high prevalence of COVID-19 in Hispanic patients with lower socioeconomic status in Orange County (Khanijahani, 2021; Hatef et al., 2020). The composition of the nasal microbiome can impact host susceptibility and disease course following respiratory infection (Teo et al., 2015; Lee et al., 2019). In turn, acute viral infections can modulate bacterial communities potentially favoring the expansion of opportunistic pathogens (Edouard et al., 2018; Wolter et al., 2014). In this study, we show that the nasal microbiome of CoV+ patients was enriched in pathogenic bacteria such Acinetobacter, Rothia, Moraxella, and P. aeruginosa. Similarly, an increased abundance of Pseudomonas was also observed following influenza A infection and in throat samples collected from COVID-19 patients (Xu et al., 2021; Kaul et al., 2020). However, the cross-sectional nature of our study makes it impossible to determine whether viral infection alters the nasal microbiome or whether the nasal microbiome community state type pre-disposes an individual to viral infection. A recent longitudinal study found that rhinovirus infection did not significantly alter the nasal microbiome, but individuals with a Pseudomonas-dominated community prior to infection experienced more severe symptoms (Lehtinen et al., 2018). Therefore, high abundance of Pseudomonas in the nasal microbiome may predispose the host to severe respiratory viral infection. Our findings are in line with increased risk of secondary bacterial infections in COVID-19 patients, especially those who are severely ill (Feng et al., 2020; Vaillancourt and Jorth, 2020; Zhou et al., 2020a).
Hospital acquired infections (HAIs) are a major public health concern. Indeed, up to 46% of patients hospitalized with COVID-19 suffered from a HAI (Grasselli et al., 2021). One of the major reservoirs for opportunistic pathogens is the nasal cavity, which can act as the entry point and contribute to the spread of bacterial infections to the rest of the respiratory tract (Dimitri-Pinheiro et al., 2020). HCWs are often carriers of pathogens associated with nosocomial infection such as methicillin-resistant Staphylococcus aureus (MRSA) given their extended exposure to the hospital environment (El Aila et al., 2017). Our analysis found an enrichment of pathobionts such as B. cepacia, often detected in patients with underlying conditions such as cystic fibrosis (Coutinho et al., 2011; Kalish et al., 2006) and the elderly (El Chakhtoura et al., 2017), in the nasal communities of HCWs. The abundance of Acinetobacter was significantly higher in HCWs and CoV+ patients compared to CoV− outpatients. Since CoV+ samples were collected at hospital admission, it is possible that patients with respiratory infections are contributing to the high prevalence of bacterial pathogens in the hospital environment, which is being reflected in the nasal microbiome of HCWs. Additionally, Rothia was more abundant in the nasal microbiome of CoV+ patients. Interestingly, a recent study found that Rothia was the best predictor of detectable SARS-CoV-2 vRNA in patient samples and on the surfaces of COVID-19 patient rooms (Marotz et al., 2021).
We also profiled the host transcriptional profile of the nasal epithelium in a subset of patients. As recently described (Butler et al., 2021; Islam et al., 2021; Lieberman et al., 2020; Ng et al., 2021), we observed an upregulation of genes associated with innate immune cell activation, antiviral defense, inflammation, and cell death. Furthermore, the upregulation of genes associated with neuronal death and the downregulation of genes influencing epithelial integrity and sensory organ development support a mechanism for infection-induced anosmia and ageusia (de Melo et al., 2021; Melo et al., 2021; Rawson and Huang, 2009).
Additionally, we sought to determine the relationship between vRNA load, viral phylogeny, and viral genome recovery. Consistent with previous studies, genome recovery was negatively correlated with Ct value (La Scola et al., 2020; Bullard et al., 2020; Singanayagam et al., 2020). We detected the D614G amino acid change in all samples, which is expected given the global dominance of this genotype (Korber et al., 2020; Isabel et al., 2020). Mutations in the RDRP subunits nsp12 and nsp3, which could impact viral replication and immune evasion and immune antagonist, were also present in all samples (Brosey et al., 2021; Hillen et al., 2020). Given the limited viral genetic diversity we observed within our study population, we were unable to determine the impact of viral genetic variation on the composition of the nasal microbiome or host transcriptional response.
In summary, data presented herein show that SARS-CoV-2 infection is associated with a distinct shift in the composition of the nasal microbiome, including an increased abundance of bacterial pathogens such as P. aeruginosa. Additionally, HCW nasal microbiomes are enriched in pathogens known to cause nosocomial infections such as B. cepacia. Transcriptional profiling indicated a robust local immune response to SARS-CoV-2 infection and provided support for neuronal damage in the URT leading to anosmia. This study had some limitations. The samples used in this study were initially collected for SARS-CoV-2 diagnosis and were therefore not processed in the typical fashion for either host transcriptomics or microbiome analysis. Additionally, our study consisted of only one time point. A longitudinal study should be performed to gain much needed insight into the dynamic changes in microbial communities and host responses within the nasal cavity. Finally, analysis of samples from individuals with asymptomatic infection would provide additional valuable insight into the determinants of disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| QIAseq FX DNA Library Kit | QIAGEN | 180473 |
| Quick-RNA Viral 96 Kit | Zymo Research | R1041 |
| QIAseq SARS-CoV-2 Primer Panel | QIAGEN | 333895 |
| NEB Next Ultra II Directional RNA Library kit | New England Biolabs | E7760S |
| Deposited Data | ||
| Raw data | This Study | Sequence Read Archive- BioProject SRA: PRJNA745169 (https://www.ncbi.nlm.nih.gov) |
| Healthy Nasal Microbiome | http://journal.frontiersin.org/article/10.3389/fmicb.2017.02372/full | European Nucleotide Archive - study number ENA: PRJEB23057 (https://www.ebi.ac.uk/ena/browser) |
| Oligonucleotides | ||
| 16S rRNA gene 515F forward primer 5′-GTGYCAGCMGCCGCGGTAA-3′ | https://doi.org/10.1111/1462-2920.13023 | N/A |
| 16S rRNA gene 806R reverse primer 5′-GGACTACNVGGGTWTCTAAT-3′ | https://doi.org/10.3354/ame01753 | N/A |
| SARS-CoV-2 nucleoprotein qPCR forward primer 5′-GGGGAACTTCTCC TGCTAGAAT-3′ |
https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf | N/A |
| SARS-CoV-2 nucleoprotein qPCR reverse primer 5′-CAGACATTTTG CTCTCAAGCTG-3′ |
https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf | N/A |
| SARS-CoV-2 nucleoprotein qPCR probe 5′-FAMTTGCTGCTGCTTG ACAGATT-BHQ1-3′ |
https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf | N/A |
| Human RNaseP qPCR forward primer 5′-AGATTTGGACCTGCGAGCG-3′ | https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf | N/A |
| Human RNaseP qPCR reverse primer 5′-GAGCGGCTGTCTCCACAAGT-3′ | https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf | N/A |
| Human RNaseP qPCR probe 5′-FAM-TTCTGACCTGAAGGC TCTGCGCG–BHQ1-3′ |
https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf | N/A |
|
Software and algorithms | ||
| Quantitative Insights Into Microbial Ecology 2 (QIIME2)(2019.10) Built in tools: Dada2, mafft, sklearn, FastTree 2 | http://www.nature.com/articles/s41587-019-0209-9 | https://qiime2.org |
| GraphPad Prism 8 | GraphPad Software Inc | http://www.graphpad.com:443 |
| Metascape | 10.1038/s41467-019-09234-6 | https://metascape.org/ |
| R software (v4.0.2) R packages: vegan (v2.5-7), SystemPipeR (v1.26.3), edgeR (v3.34.0), ggplot2 (v3.3.3) | R Foundation | https://www.r-project.org |
| GitHub of commands used in data analysis | This Study | https://github.com/NickRhoades/COVID-19_nasal_microboime |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ilhem Messaoudi (imessaou@uci.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human subjects
In this study we utilized excess material from samples originally collected for diagnostic purposes that would have otherwise been discarded. These samples were de-identified prior to being used for research purposes. As such the UC Irvine IRB determined that the data included in this manuscript did not qualify as human subject research and did not require IRB approval. All samples for this study were collected at University of California Irvine Medical center. Nasal swabs were collected from 68 SARS-CoV-2 positive patients (CoV+), 45 SARS-CoV-2 negative healthcare workers (HCW), and 21 SARS-CoV-2 negative patients (CoV-). Samples from CoV+ were collected at hospital admission. Samples from HCW were collected from otherwise healthy individuals as part of regular asymptomatic screenings. Samples from CoV- were collected from otherwise healthy individuals for asymptomatic screening prior to elective outpatient procedures. A full breakdown of subjects age and gender identity can be found in Table S1. Samples were collected between August and November 2020.
Method details
Sample collection, extraction, and SARS-CoV-2 RNA quantification
RNA was extracted from remnant de-identified nasal swab samples collected in viral transport medium (VTM) using the Quick-RNA™ Viral 96 Kit (Zymo, Cat#R1041). SARS-CoV-2 viral loads were determined using qPCR with primers specific to the nucleoprotein (N). A one-step reaction was prepared using 5ul of extracted RNA or standard, 500nM of forward (5′-GGGGAACTTCTCCTGCTAGAAT-3′) and reverse (5′-CAGACATTTTGCTCTCAAGCTG-3′) SARS-CoV-2-nucleocapsid primers, 125 nM of SARS2-nucleocapsid probe (5′-FAM-TTGCTGCTGCTTGACAGATT-BHQ1-3′), and TaqPath 1-Step RT-qPCR Master Mix (Applied Biosystems, Foster City, CA, USA). PCR cycler conditions were 2 min at 95 °C, 15 min at 50 °C, denaturation for 2 min at 95 °C followed by 45 cycles of 3 s at 95 °C and 30 s 60 °C on the StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). An additional qPCR reaction was run on each sample using a primer/probe set to detect the human RNase P gene (RP) to control for extraction and sample quality. Blank extraction served as negative controls, while RNA extracted from SARS-CoV-2 cultures was used as Positive Template Control. All qPCR reactions were run in duplicates. Positivity was determined as a cycle threshold (Ct) value ≤ 40 for the N gene. Samples with a coefficient of variance > 20% were flagged to be re-run (none of the samples used in this study met this criteria). Samples were classified as: High Ct value (< 40- > 34.6-; n = 23); Mid Ct value (32.9-25.0; n = 23); and Low Ct values (< 23.5-15.8; n = 22).
16S amplicon libraries construction and data analysis
DNA that co-eluted with extracted RNA was used as the template to amplify the hypervariable V4 region of the 16S rRNA gene using PCR primers (515F/806R with the forward primer containing a 12-bp barcode) in duplicate reactions containing: 12.5 ul GoTaq master mix, 9.5 ul nuclease-free H20, 1 ul template DNA, and 1 ul 10uM primer mix. Thermal cycling parameters were 94°C for 3 minutes; 35 cycles of 94°C for 45 s, 50°C for 1 minute, and 72°C for 1 minute and 30 s; followed by 72°C for 10 minutes. PCR products were purified using a MinElute 96 UF PCR Purification Kit (QIAGEN, Valencia, CA, USA). Libraries were sequenced (2 × 300 bases) using Illumina MiSeq. Additional control samples were concurrently sequenced including extraction negative control, PCR blank negative control, and a microbial community standard positive control.
Raw FASTQ 16S rRNA gene amplicon sequences were uploaded and processed using the QIIME 2 version 2019.10 (Bolyen et al., 2019) analysis pipeline as we have previously described (Rhoades et al., 2019). Briefly, sequences were demultiplexed and quality filtered using the DADA2 plugin for QIIME 2 (Callahan et al., 2016), which filters chimeric and low-quality sequences. The generated sequence variants were then aligned using MAFFT (Katoh and Standley, 2013), and a phylogenetic tree was constructed using FastTree 2 (Price et al., 2010). Taxonomy was assigned to sequence variants using q2-feature-classifier against the SILVA Database (release: 138) (Quast et al., 2013). After taxonomic classification all sequences not assigned to a known phyla were removed along with any sequence assigned to mitochondria or chloroplast. Any Amplicon Sequencing Variant (ASV) that was found in > 1% abundance in either the extraction or PCR negative control and > 1% average abundance in samples was also removed. These ASVs were primarily assigned to two taxa which were highly abundant in the extraction controls (Thermoanaerobacterium saccharolyticum and Myxococcales 0319-6G20). We also found that Streptococcus was shared between the extraction control and samples, but these sequences were not shared at the ASV level and therefore included in the dataset (Figure S1A). Finally, we confirmed a lack of PCR or sequencing bias using duplicate community standard samples (Figure S1A).
After clean up taxonomy was reassigned to sequence variants using q2-feature-classifier against the expanded Human Oral Microbiome Database (eHOMD: release version 15.21) (Escapa et al., 2018). This curated and site-specific database was used to improve species-level resolution. To prevent sequencing depth bias, samples were rarified to 8,000 sequences per sample before α and β diversity analysis. QIIME 2 was also used to generate the following α diversity metrics: richness (as observed taxonomic units), Shannon evenness, and phylogenetic diversity. β diversity was determined in QIIME 2 using weighted and unweighted UniFrac distances (Lozupone et al., 2011).
To confirm that microbial DNA co-eluted in our was reflective of a typical nasal microbiome community, we compared our data to a previously published nasal microbiome dataset (De Boeck et al., 2017). This study as it sampled a large number of healthy subjects and used the same amplicon primer set as our current study. Nasal microbiome data from this study was downloaded from (BioProject: PRJEB23057), merged with our dataset and analyzed using the exact parameters described above. Due to low sequencing depth of some samples in BioProject: PRJEB23057 after applying our analysis parameters, this combined dataset was rarified to 3000 sequences per sample prior to calculating α and β diversity metrics. A full analysis pipeline including parameters used for all commands can be accessed at https://github.com/NickRhoades/COVID-19_nasal_microboime.
RNA-seq library preparation and analysis
Quantity and quality of RNA extracted from 8 nasal swabs of (4 CoV+ and 4 HCW chosen at random, Table S1) was determined using an Agilent 2100 BioAnalyzer. cDNA libraries were constructed using the NEB Next Ultra II Directional RNA Library kit (Thermo Fischer). Briefly, RNA was treated with RNaseH and DNase I after depletion of ribosomal rRNA. Adapters were ligated to cDNA products. The ∼300 bp amplicons were PCR-amplified and indexed with a unique molecular identifier. cDNA libraries were assessed for quality and quantity on the BioAnalyzer prior to single-end sequencing (x100bp) using the Illumina NovaSeq platform.
Bioinformatic analysis was performed using the systemPipeR RNA-Seq workflow (Backman and Girke, 2016). RNA-Seq reads were demultiplexed, quality-filtered and trimmed for quality and adaptor removal using Trim Galore (average Phred Score cut-off = 30, minimum length of 50 basepairs). Tophat was used to align filtered reads to the reference genome Homo sapiens (GRCh38). The file was used for annotation. Raw expression values (gene-level read counts) were generated using the summarizeOverlaps function and normalized (read per kilobase of transcript per million mapped reads, rpkm) using the edgeR package.
SARS-CoV-2 genome library construction and analysis
Enrichment for vRNA was performed using the QIASeq SARS-CoV-2 Primer Panel V2 (QIAGEN) Panel V2) followed by library construction with the QIAGEN FX DNA library preparation kit. Samples were indexed, pooled and validated with 2100 Agilent BioAn. Prior to Illumina sequencing (2x100bp, 1 M reads).
Adapters and primers were removed from demultiplexed sequencing readings using TrimGalore and MaskPrimers.py (pRESTO). Merged reads were aligned to SARS-CoV-2 Wuhan isolate NC_045512.2 with BWA-mem software version 0.7.17. Genomes with greater than 90% coverage and 10X depth were retained for phylogenetic analysis with Nextstrain. Amino acid changes and clade assignments were identified using Nextclade. A set of comparator sequences representing the original Wuhan isolate (NC_045512.2), variants of concern (B.1.351, EPI_ISL_960123; P.1, EPI_ISL_833167; B.1.1.7, EPI_ISL_659057), isolates from January 2020 New York (EPI_ISL_1293138) and Washington (EPI_ISL_404895), and representative isolates from California January-November 2020 (EPI_ISL_406036, EPI_ISL_411954, EPI_ISL_429875, EPI_ISL_436642, EPI_ISL_444023, EPI_ISL_569672, EPI_ISL_548382, EPI_ISL_548612, EPI_ISL_582897, EPI_ISL_625601, EPI_ISL_753324) were used for phylogenetic analysis. All sequences from Orange County, California from January 2020 to December 2020 were also retrieved from GISAID.
Quantification and statistical analysis
PERMANOVAs were performed using the Vegan (Dixon, 2003) function ADONIS. 1-way, non-parametric Kruskal-Wallis ANOVA were implemented using PRISM (V8) to generate p values and utilizing the Dunns post hoc-test when the initial ANOVA was significant. The LEfSe algorithm was used to identify differentially abundant taxa and pathways between groups with a logarithmic Linear discriminant analysis (LDA) score cutoff of 2 (Segata et al., 2011). For host transcriptional data edgeR was used to determine differentially expressed genes (DEGs) meeting the following criteria: genes with median rpkm of ≥ 1, a false discovery rate (FDR) corrected p value ≤ 0.05 and a log2fold change ≥ 1 compared to CoV- samples. Functional enrichment of DEGs was performed using Metascape to identify relevant Gene Ontology (GO) biological process terms (Zhou et al., 2019). Heatmaps, bar graphs and volcano plots were generated using R package ggplot2.
Acknowledgments
This study was supported by NIH grants UL1 TR001414, 1R01AI152258-02, and 3R01AA028735-01S1. N.S.R. is supported by NIH T32 AI007319. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Experimental design and graphical abstract figures were generated using graphics from BioRender.com. We thank the Chao Family Comprehensive Cancer Center (P30CA062203) for providing the de-identified biospecimens used in this study.
Author contributions
N.S.R. and I.M. conceived and designed the experiments. N.S.R., A.N.P., A.N.M., B.M.D., A.J., and I.R.C. performed the experiments. N.S.R, A.N.P., and A.N.M. analyzed the data. N.S.R., A.N.P., and I.M. interpreted the results and wrote the paper. All authors have read and approved the final draft of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: August 13, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109637.
Supplemental information
Data and code availability
16S rRNA gene amplicon, host transcriptional and viral genome sequencing data have been deposited at the NCBI Sequence Read Archive (SRA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. However, a GitHub has been generated for the analysis of all sequencing data in this manuscript and can be accessed here (https://github.com/NickRhoades/COVID-19_nasal_microboime). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Bassis C.M., Tang A.L., Young V.B., Pynnonen M.A. The nasal cavity microbiota of healthy adults. Microbiome. 2014;2:27. doi: 10.1186/2049-2618-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosey C.A., Houl J.H., Katsonis P., Balapiti-Modarage L.P.F., Bommagani S., Arvai A., Moiani D., Bacolla A., Link T., Warden L.S. Targeting SARS-CoV-2 Nsp3 macrodomain structure with insights from human poly(ADP-ribose) glycohydrolase (PARG) structures with inhibitors. Prog. Biophys. Mol. Biol. 2021;163:171–186. doi: 10.1016/j.pbiomolbio.2021.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budding A., Sieswerda E., Wintermans B., Bos M. 2020. An age dependent pharyngeal microbiota signature associated with Sars-Cov-2 infection.https://ssrn.com/abstract=3582780 [Google Scholar]
- Bullard J., Dust K., Funk D., Strong J.E., Alexander D., Garnett L., Boodman C., Bello A., Hedley A., Schiffman Z. predicting infectious severe acute respiratory syndrome coronavirus 2 from diagnostic samples. Clin. Infect. Dis. 2020;71:2663–2666. doi: 10.1093/cid/ciaa638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler D., Mozsary C., Meydan C., Foox J., Rosiene J., Shaiber A., Danko D., Afshinnekoo E., MacKay M., Sedlazeck F.J. Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat. Commun. 2021;12:1660. doi: 10.1038/s41467-021-21361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J., Holmes S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callender L.A., Curran M., Bates S.M., Mairesse M., Weigandt J., Betts C.J. The impact of pre-existing comorbidities and therapeutic interventions on COVID-19. Front. Immunol. 2020;11:1991. doi: 10.3389/fimmu.2020.01991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlson E.S., Chen J., Custers-Allen R., Bittinger K., Li H., Sinha R., Hwang J., Bushman F.D., Collman R.G. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS ONE. 2010;5:e15216. doi: 10.1371/journal.pone.0015216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Shen W., Rowan N.R., Kulaga H., Hillel A., Ramanathan M., Jr., Lane A.P. Elevated ACE2 expression in the olfactory neuroepithelium: Implications for anosmia and upper respiratory SARS-CoV-2 entry and replication. bioRxiv. 2020 doi: 10.1101/2020.05.08.084996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho C.P., Dos Santos S.C., Madeira A., Mira N.P., Moreira A.S., Sá-Correia I. Long-term colonization of the cystic fibrosis lung by Burkholderia cepacia complex bacteria: Epidemiology, clonal variation, and genome-wide expression alterations. Front. Cell. Infect. Microbiol. 2011;1:12. doi: 10.3389/fcimb.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boeck I., Wittouck S., Wuyts S., Oerlemans E.F.M., van den Broek M.F.L., Vandenheuvel D., Vanderveken O., Lebeer S. Comparing the healthy nose and nasopharynx microbiota reveals continuity as well as niche-specificity. Front. Microbiol. 2017;8:2372. doi: 10.3389/fmicb.2017.02372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maio F., Posteraro B., Ponziani F.R., Cattani P., Gasbarrini A., Sanguinetti M. Nasopharyngeal microbiota profiling of SARS-CoV-2 infected patients. Biol. Proced. Online. 2020;22:18. doi: 10.1186/s12575-020-00131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo G.D., Lazarini F., Levallois S., Hautefort C., Michel V., Larrous F., Verillaud B., Aparicio C., Wagner S., Gheusi G. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021;13:eabf8396. doi: 10.1126/scitranslmed.abf8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitri-Pinheiro S., Soares R., Barata P. The microbiome of the nose—Friend or foe? Allergy Rhinol. (Providence) 2020;11 doi: 10.1177/2152656720911605. 2152656720911605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon P. VEGAN, a package of R functions for community ecology. J. Vegetation Sci. 2003;14:927–930. [Google Scholar]
- Edouard S., Million M., Bachar D., Dubourg G., Michelle C., Ninove L., Charrel R., Raoult D. The nasopharyngeal microbiota in patients with viral respiratory tract infections is enriched in bacterial pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 2018;37:1725–1733. doi: 10.1007/s10096-018-3305-8. [DOI] [PubMed] [Google Scholar]
- Ejaz H., Alsrhani A., Zafar A., Javed H., Junaid K., Abdalla A.E., Abosalif K.O.A., Ahmed Z., Younas S. COVID-19 and comorbidities: Deleterious impact on infected patients. J. Infect. Public Health. 2020;13:1833–1839. doi: 10.1016/j.jiph.2020.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Aila N.A., Al Laham N.A., Ayesh B.M. Nasal carriage of methicillin resistant Staphylococcus aureus among health care workers at Al Shifa hospital in Gaza Strip. BMC Infect. Dis. 2017;17:28. doi: 10.1186/s12879-016-2139-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Chakhtoura N.G., Saade E., Wilson B.M., Perez F., Papp-Wallace K.M., Bonomo R.A. A 17-year nationwide study of Burkholderia cepacia complex bloodstream infections among patients in the united states veterans health administration. Clin. Infect. Dis. 2017;65:1253–1259. doi: 10.1093/cid/cix559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escapa I.F., Chen T., Huang Y., Gajare P., Dewhirst F.E., Lemon K.P. New insights into human nostril microbiome from the expanded Human Oral Microbiome Database (eHOMD): A resource for the microbiome of the human aerodigestive tract. mSystems. 2018;3:e00187-18. doi: 10.1128/mSystems.00187-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Ling Y., Bai T., Xie Y., Huang J., Li J., Xiong W., Yang D., Chen R., Lu F. COVID-19 with different severities: A multicenter study of clinical features. Am. J. Respir. Crit. Care Med. 2020;201:1380–1388. doi: 10.1164/rccm.202002-0445OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo O., Locatello L.G., Mazzoni A., Novelli L., Annunziato F. The central role of the nasal microenvironment in the transmission, modulation, and clinical progression of SARS-CoV-2 infection. Mucosal Immunol. 2021;14:305–316. doi: 10.1038/s41385-020-00359-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasselli G., Scaravilli V., Mangioni D., Scudeller L., Alagna L., Bartoletti M., Bellani G., Biagioni E., Bonfanti P., Bottino N. Hospital-acquired infections in critically ill patients with COVID-19. Chest. 2021;160:454–465. doi: 10.1016/j.chest.2021.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.-J., Ni Z.-Y., Hu Y., Liang W.-H., Ou C.-Q., He J.-X., Liu L., Shan H., Lei C.-L., Hui D.S.C., China Medical Treatment Expert Group for Covid-19 Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman T.W., Girke T. systemPipeR: NGS workflow and report generation environment. BMC Bioinformatics. 2016;17:388. doi: 10.1186/s12859-016-1241-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison A.G., Lin T., Wang P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. 2020;41:1100–1115. doi: 10.1016/j.it.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatef E., Chang H.Y., Kitchen C., Weiner J.P., Kharrazi H. Assessing the impact of neighborhood socioeconomic characteristics on COVID-19 prevalence across seven states in the United States. Front. Public Health. 2020;8:571808. doi: 10.3389/fpubh.2020.571808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Wang J., Li F., Shi Y. Main clinical features of COVID-19 and potential prognostic and therapeutic value of the microbiota in SARS-CoV-2 infections. Front. Microbiol. 2020;11:1302. doi: 10.3389/fmicb.2020.01302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen H.S., Kokic G., Farnung L., Dienemann C., Tegunov D., Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020;584:154–156. doi: 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornuss D., Lange B., Schröter N., Rieg S., Kern W.V., Wagner D. Anosmia in COVID-19 patients. Clin. Microbiol. Infect. 2020;26:1426–1427. doi: 10.1016/j.cmi.2020.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B., Guo H., Zhou P., Shi Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabel S., Graña-Miraglia L., Gutierrez J.M., Bundalovic-Torma C., Groves H.E., Isabel M.R., Eshaghi A., Patel S.N., Gubbay J.B., Poutanen T. Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation now documented worldwide. Sci. Rep. 2020;10:14031. doi: 10.1038/s41598-020-70827-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam A.B.M.M.K., Khan M.A., Ahmed R., Hossain M.S., Kabir S.M.T., Islam M.S., Siddiki A.M.A.M.Z. Transcriptome of nasopharyngeal samples from COVID-19 patients and a comparative analysis with other SARS-CoV-2 infection models reveal disparate host responses against SARS-CoV-2. J. Transl. Med. 2021;19:32. doi: 10.1186/s12967-020-02695-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish L.A., Waltz D.A., Dovey M., Potter-Bynoe G., McAdam A.J., Lipuma J.J., Gerard C., Goldmann D. Impact of Burkholderia dolosa on lung function and survival in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2006;173:421–425. doi: 10.1164/rccm.200503-344OC. [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul D., Rathnasinghe R., Ferres M., Tan G.S., Barrera A., Pickett B.E., Methe B.A., Das S.R., Budnik I., Halpin R.A. Microbiome disturbance and resilience dynamics of the upper respiratory tract during influenza A virus infection. Nat. Commun. 2020;11:2537. doi: 10.1038/s41467-020-16429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanijahani A. Racial, ethnic, and socioeconomic disparities in confirmed COVID-19 cases and deaths in the United States: A county-level analysis as of November 2020. Ethn. Health. 2021;26:22–35. doi: 10.1080/13557858.2020.1853067. [DOI] [PubMed] [Google Scholar]
- Khatiwada S., Subedi A. Lung microbiome and coronavirus disease 2019 (COVID-19): Possible link and implications. Hum. Microb. J. 2020;17:100073. doi: 10.1016/j.humic.2020.100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., Sheffield COVID-19 Genomics Group Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R.S., Mathew D., Baxter A.E., Vella L.A. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020;5:eabd7114. doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Scola B., Le Bideau M., Andreani J., Hoang V.T., Grimaldier C., Colson P., Gautret P., Raoult D. Viral RNA load as determined by cell culture as a management tool for discharge of SARS-CoV-2 patients from infectious disease wards. Eur. J. Clin. Microbiol. Infect. Dis. 2020;39:1059–1061. doi: 10.1007/s10096-020-03913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.H., Gordon A., Shedden K., Kuan G., Ng S., Balmaseda A., Foxman B. The respiratory microbiome and susceptibility to influenza virus infection. PLoS ONE. 2019;14:e0207898. doi: 10.1371/journal.pone.0207898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen M.J., Hibberd A.A., Männikkö S., Yeung N., Kauko T., Forssten S., Lehtoranta L., Lahtinen S.J., Stahl B., Lyra A., Turner R.B. Nasal microbiota clusters associate with inflammatory response, viral load, and symptom severity in experimental rhinovirus challenge. Sci. Rep. 2018;8:11411. doi: 10.1038/s41598-018-29793-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman N.A.P., Peddu V., Xie H., Shrestha L., Huang M.L., Mears M.C., Cajimat M.N., Bente D.A., Shi P.Y., Bovier F. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020;18:e3000849. doi: 10.1371/journal.pbio.3000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Leon S., Wegman-Ostrosky T., Perelman C., Sepulveda R., Rebolledo P., Cuapio A., Villapol S. More than 50 long-term effects of COVID-19: A systematic review and meta-analysis. Sci. Rep. 2021;11:16144. doi: 10.1038/s41598-021-95565-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C., Lladser M.E., Knights D., Stombaugh J., Knight R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui G., Ling L., Lai C.K.C., Tso E.Y.K., Fung K.S.C., Chan V., Ho T.H.Y., Luk F., Chen Z., Ng J.K.C. Viral dynamics of SARS-CoV-2 across a spectrum of disease severity in COVID-19. J. Infect. 2020;81:318–356. doi: 10.1016/j.jinf.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotz C., Belda-Ferre P., Ali F., Das P., Huang S., Cantrell K., Jiang L., Martino C., Diner R.E., Rahman G. SARS-CoV-2 detection status associates with bacterial community composition in patients and the hospital environment. Microbiome. 2021;9:132. doi: 10.1186/s40168-021-01083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo G.D.D., Lazarini F., Levallois S., Hautefort C., Michel V., Larrous F., Verillaud B., Aparicio C., Wagner S., Gheusi G. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021;13:eabf8396. doi: 10.1126/scitranslmed.abf8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgolo N., Therien A.G., Howell B., Klein D., Koeplinger K., Lieberman L.A., Adam G.C., Flynn J., McKenna P., Swaminathan G. SARS-CoV-2 tropism, entry, replication, and propagation: Considerations for drug discovery and development. PLoS Pathog. 2021;17:e1009225. doi: 10.1371/journal.ppat.1009225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardelli C., Gentile I., Setaro M., Di Domenico C., Pinchera B., Buonomo A.R., Zappulo E., Scotto R., Scaglione G.L., Castaldo G., Capoluongo E. Nasopharyngeal microbiome signature in COVID-19 positive patients: Can we definitively get a role to Fusobacterium periodonticum? Front. Cell. Infect. Microbiol. 2021;11:625581. doi: 10.3389/fcimb.2021.625581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D.L., Granados A.C., Santos Y.A., Servellita V., Goldgof G.M., Meydan C., Sotomayor-Gonzalez A., Levine A.G., Balcerek J., Han L.M. A diagnostic host response biosignature for COVID-19 from RNA profiling of nasal swabs and blood. Sci. Adv. 2021;7:eabe5984. doi: 10.1126/sciadv.abe5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M.N., Dehal P.S., Arkin A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan V.R., Holt J., Nelson L.F., Ir D., Robertson C.E., Frank D.N. Determinants of the nasal microbiome: Pilot study of effects of intranasal medication use. Allergy Rhinol. (Providence) 2018;9 doi: 10.1177/2152656718789519. 2152656718789519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson N.E., Huang L. Impact of oronasal inflammation on taste and smell: An introduction. Ann. N Y Acad. Sci. 2009;1170:581–584. doi: 10.1111/j.1749-6632.2009.04489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades N., Barr T., Hendrickson S., Prongay K., Haertel A., Gill L., Garzel L., Whiteson K., Slifka M., Messaoudi I. Maturation of the infant rhesus macaque gut microbiome and its role in the development of diarrheal disease. Genome Biol. 2019;20:173. doi: 10.1186/s13059-019-1789-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueca M., Fontana A., Bartolini B., Piselli P., Mazzarelli A., Copetti M., Binda E., Perri F., Gruber C.E.M., Nicastri E. Investigation of nasal/oropharyngeal microbial community of COVID-19 patients by 16S rDNA sequencing. Int. J. Environ. Res. Public Health. 2021;18:2174. doi: 10.3390/ijerph18042174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA. 2020;117:11727–11734. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z., Xiao Y., Kang L., Ma W., Shi L., Zhang L., Zhou Z., Yang J., Zhong J., Yang D. Genomic diversity of severe acute respiratory syndrome-coronavirus 2 in patients with coronavirus disease 2019. Clin. Infect. Dis. 2020;71:713–720. doi: 10.1093/cid/ciaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singanayagam A., Patel M., Charlett A., Lopez Bernal J., Saliba V., Ellis J., Ladhani S., Zambon M., Gopal R. Duration of infectiousness and correlation with RT-PCR cycle threshold values in cases of COVID-19, England, January to May 2020. Euro Surveill. 2020;25:2001483. doi: 10.2807/1560-7917.ES.2020.25.32.2001483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns J.C., Davidson C.J., McKeon S., Whelan F.J., Fontes M.E., Schryvers A.B., Bowdish D.M., Kellner J.D., Surette M.G. Culture and molecular-based profiles show shifts in bacterial communities of the upper respiratory tract that occur with age. ISME J. 2015;9:1246–1259. doi: 10.1038/ismej.2014.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sungnak W., Huang N., Bécavin C., Berg M., Queen R., Litvinukova M., Talavera-López C., Maatz H., Reichart D., Sampaziotis F., HCA Lung Biological Network SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26:681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo S.M., Mok D., Pham K., Kusel M., Serralha M., Troy N., Holt B.J., Hales B.J., Walker M.L., Hollams E. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. 2015;17:704–715. doi: 10.1016/j.chom.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillancourt M., Jorth P. The unrecognized threat of secondary bacterial infections with COVID-19. MBio. 2020;11:e01806-20. doi: 10.1128/mBio.01806-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan F.J., Verschoor C.P., Stearns J.C., Rossi L., Luinstra K., Loeb M., Smieja M., Johnstone J., Surette M.G., Bowdish D.M. The loss of topography in the microbial communities of the upper respiratory tract in the elderly. Ann. Am. Thorac. Soc. 2014;11:513–521. doi: 10.1513/AnnalsATS.201310-351OC. [DOI] [PubMed] [Google Scholar]
- Wilks J., Golovkina T. Influence of microbiota on viral infections. PLoS Pathog. 2012;8:e1002681. doi: 10.1371/journal.ppat.1002681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter N., Tempia S., Cohen C., Madhi S.A., Venter M., Moyes J., Walaza S., Malope-Kgokong B., Groome M., du Plessis M. High nasopharyngeal pneumococcal density, increased by viral coinfection, is associated with invasive pneumococcal pneumonia. J. Infect. Dis. 2014;210:1649–1657. doi: 10.1093/infdis/jiu326. [DOI] [PubMed] [Google Scholar]
- Wos-Oxley M.L., Plumeier I., von Eiff C., Taudien S., Platzer M., Vilchez-Vargas R., Becker K., Pieper D.H. A poke into the diversity and associations within human anterior nare microbial communities. ISME J. 2010;4:839–851. doi: 10.1038/ismej.2010.15. [DOI] [PubMed] [Google Scholar]
- Xu R., Lu R., Zhang T., Wu Q., Cai W., Han X., Wan Z., Jin X., Zhang Z., Zhang C. Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Commun. Biol. 2021;4:240. doi: 10.1038/s42003-021-01796-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz A., Marklund E., Andersson M., Nilsson S., Andersson L.-M., Lindh M., Gisslén M. Upper respiratory tract levels of severe acute respiratory syndrome coronavirus 2 RNA and duration of viral RNA shedding do not differ between patients with mild and severe/critical coronavirus disease 2019. J. Infect. Dis. 2021;223:15–18. doi: 10.1093/infdis/jiaa632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., China Novel Coronavirus Investigating and Research Team A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L., Ruan F., Huang M., Liang L., Huang H., Hong Z., Yu J., Kang M., Song Y., Xia J. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 2020;382:1177–1179. doi: 10.1056/NEJMc2001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X., Chen K., Zou J., Han P., Hao J., Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020;14:185–192. doi: 10.1007/s11684-020-0754-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16S rRNA gene amplicon, host transcriptional and viral genome sequencing data have been deposited at the NCBI Sequence Read Archive (SRA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. However, a GitHub has been generated for the analysis of all sequencing data in this manuscript and can be accessed here (https://github.com/NickRhoades/COVID-19_nasal_microboime). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.