

Abstract
Sudden Unexpected Death in Epilepsy is a leading cause of epilepsy-related mortality, and the analysis of mouse Sudden Unexpected Death in Epilepsy models is steadily revealing a spectrum of inherited risk phenotypes based on distinct genetic mechanisms. Serotonin (5-HT) signalling enhances post-ictal cardiorespiratory drive and, when elevated in the brain, reduces death following evoked audiogenic brainstem seizures in inbred mouse models. However, no gene in this pathway has yet been linked to a spontaneous epilepsy phenotype, the defining criterion of Sudden Unexpected Death in Epilepsy. Most monogenic models of Sudden Unexpected Death in Epilepsy invoke a failure of inhibitory synaptic drive as a critical pathogenic step. Accordingly, the G protein-coupled, membrane serotonin receptor 5-HT2C inhibits forebrain and brainstem networks by exciting GABAergic interneurons, and deletion of this gene lowers the threshold for lethal evoked audiogenic seizures. Here, we characterize epileptogenesis throughout the lifespan of mice lacking X-linked, 5-HT2C receptors (loxTB Htr2c). We find that loss of Htr2c generates a complex, adult-onset spontaneous epileptic phenotype with a novel progressive hyperexcitability pattern of absences, non-convulsive, and convulsive behavioural seizures culminating in late onset sudden mortality predominantly in male mice. RNAscope localized Htr2c mRNA in subsets of Gad2+ GABAergic neurons in forebrain and brainstem regions. To evaluate the contribution of 5-HT2C receptor-mediated inhibitory drive, we selectively spared their deletion in GAD2+ GABAergic neurons of pan-deleted loxTB Htr2c mice, yet unexpectedly found no amelioration of survival or epileptic phenotype, indicating that expression of 5-HT2C receptors in GAD2+ inhibitory neurons was not sufficient to prevent hyperexcitability and lethal seizures. Analysis of human Sudden Unexpected Death in Epilepsy and epilepsy genetic databases identified an enrichment of HTR2C non-synonymous variants in Sudden Unexpected Death in Epilepsy cases. Interestingly, while early lethality is not reflected in the mouse model, we also identified variants mainly among male Sudden Infant Death Syndrome patients. Our findings validate HTR2C as a novel, sex-linked candidate gene modifying Sudden Unexpected Death in Epilepsy risk, and demonstrate that the complex epilepsy phenotype does not arise solely from 5-HT2C-mediated synaptic disinhibition. These results strengthen the evidence for the serotonin hypothesis of Sudden Unexpected Death in Epilepsy risk in humans, and advance current efforts to develop gene-guided interventions to mitigate premature mortality in epilepsy.
Keywords: SUDEP, HTR2C, 5-HT, sex differences, GABA
Massey et al. report that mice lacking serotonin 2C receptors are an adult-onset model of sudden unexpected death in epilepsy (SUDEP). Sparing this deletion in interneurons reveals that the complex epilepsy phenotype is not due solely to disinhibition. They also report 5-HT2C variants in cases of human SUDEP.
Graphical Abstract
Graphical Abstract.

Introduction
Sudden Unexpected Death in Epilepsy (SUDEP) is a leading cause of mortality in individuals with epilepsy, accounting for nearly 3000 deaths per year in the U.S.1–3 In patients with chronic refractory epilepsy, SUDEP is diagnosed as the cause of death in up to 50% of cases.4,5 The risk of sudden death is 20 times higher in patients with epilepsy compared to the general population and can be stratified according to clinical and genetic factors.6 Monogenic epilepsies, such as Dravet Syndrome (SCN1A), provide additional insight into the heterogeneity of SUDEP mechanisms in paediatric epilepsy populations, as well as an opportunity to more precisely identify and treat individuals at increased risk.7–9 Mouse models of these and related genes have been identified that significantly raise early SUDEP risk by impairing cardiorespiratory function following a tonic seizure,10–16 while few genes for adult-onset risk have been reported. Analysis of these early death models indicates the critical importance of defective synaptic inhibition. For example, in heterozygous Scn1a mutants, selective activation of inhibitory neurons by CRISPR-Cas9 engineering17 or a specific venom peptide toxin18 pinpoint the impairment of interneuron firing as a critical mechanism underlying seizures and SUDEP.
The neurotransmitter serotonin (5-hydroxytryptamine; 5-HT) is implicated in a number of neuropsychiatric disorders, including epilepsy, and the ‘serotonin hypothesis’ frames a clear mechanistic pathway contributing to SUDEP.19,20 5-HT signalling is anticonvulsant and decreases seizure susceptibility in different experimental models.21–23 Faingold et al.24,25 have shown that seizure-induced respiratory arrest and death following audiogenic seizures (AGSs) in inbred DBA/1 and DBA/2 mice can be prevented by pretreatment with the selective serotonin reuptake inhibitor, fluoxetine, or optogenetic stimulation of 5-HT neurons in the dorsal raphe. Moreover, expression of multiple 5-HT receptors, including 5-HT2C, is decreased in the brainstem of these mice compared to C57/Bl6 mice.26 5-HT2C (formerly known as 5-HT1C) is a 5-HT metabotropic receptor expressed by the X-linked gene HTR2C throughout the CNS, including retrosplenial and piriform cortex, amygdala, subthalamic nucleus, lateral habenula and brainstem.27–31 These receptors are predominantly coupled to Gq/11 proteins and activate phospholipase C to release intracellular Ca2+ stores, thereby altering excitability.32
5-HT2C receptors reside predominantly on GABAergic neurons, and their activation by 5-HT increases spontaneous activity of these interneurons.33,34 A 5-HT2C knockout model revealed that null male mice display AGS and increased mortality (38%) beginning at 2–4 months of age compared to wildtype littermates, presumably due to network disinhibition.35 A second knockout model in which a floxed transcriptional blocker was inserted between exons three and four of the Htr2c gene (loxTB Htr2c) also showed behavioural convulsions and reduced survival,36 however, neither of these models were analysed by EEG monitoring.
Here, we investigated the epileptic phenotype of the loxTB Htr2c mouse line and discovered that 5-HT2C mutant mice present a hitherto unreported syndrome of multiple adult-onset seizure types with a clear predominance of SUDEP in male mice. We then used a mouse line that expressed Cre recombinase under the control of Gad2 to excise the transcription block in loxTB Htr2c mice, preserving the expression of 5-HT2C receptors only in GAD2-positive cells. We studied this selective GAD2+ neuronal expression model to determine whether the loss of 5-HT2C receptors in a major population of GABAergic interneurons is the predominant driver of seizures and premature mortality in 5-HT2C mutant mice. In addition, we report here the first analysis of HTR2C variants found in human SUDEP and sudden infant death syndrome (SIDS) cohorts.
Methods
Animals
All experiments were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Mice were housed in groups of one to five animals in a temperature- and humidity-controlled room with a 14-h light and 10-h dark cycle, with access ad libitum to water and food. loxTB Htr2c mice were a gift of the Elmquist and Xu laboratories.36 These mice have a transcriptional blocker flanked by loxP sites (loxTB) inserted between exons three and four of the X-linked Htr2c gene, which prevents transcription of 5-HT2C receptors. These mice are functional knockouts, and we refer to male and female 5-HT2C-null mice as Htr2c−/Y and Htr2c−/−, respectively. We obtained male mice expressing Cre recombinase under the control of Gad2 (B6J.Cg-Gad2tm2(cre)Zjh/MwarJ; Stock #028867) from Jackson Labs and bred them to female Htr2c−/+ mice. This cross produced mice with the transcriptional blocker excised in only Gad2-positive neurons (Gad2Cre-Htr2c).
Survival analysis
We tracked mice up to 41–42 weeks old and counted deaths in the home cage during this period. Any animal requiring euthanasia per veterinary guidelines was excluded from the study. When possible, we documented the body position of deceased animals, and those that showed post-mortem hindlimb extension and a pronounced hunched back were considered probable SUDEP cases.
Video-EEG monitoring
Video-EEG recordings were performed as previously described.37 All surgical procedures were performed in a Baylor Center for Comparative Medicine surgery facility. Prior to surgery, mice were weighed and treated with meloxicam (5 mg/kg, s.c.) for analgesia. Mice were anesthetized with 2.5% isoflurane using a Patterson Scientific isoflurane vaporizer, and the scalp prepped using aseptic technique. Insulated silver EEG leads pre-wired to a nano strip connector (Omnetics Connector Corporation, Minneapolis, MN) were implanted in wildtype and mutant littermates through bilateral cranial burr over frontal and parietal cortex with a ground and reference electrode over the olfactory bulbs. Following surgery, mice were returned to their home cage and treated with a daily meloxicam (5 mg/kg, s.c.) injection for 3 days. Implanted mice were allowed 10–14 days post-operative recovery in their home cage before recordings began. EEG in freely behaving mice was recorded with a USBamp biosignal amplifier (g.tec USA, Albany, NY), digitized using a PowerLab 16/35 system (AD Instruments, Inc., Colorado Springs, CO), and analysed with LabChart 8 Pro software (AD Instruments, Inc.). A band-pass digital filter was applied during EEG analysis with a high cut-off frequency of 50 Hz and a low cut-off frequency of 0.3 Hz. Additional recording criteria can be found in Supplementary material.
AGS induction
A previous study reported that 5-HT2C-null mice are susceptible to AGS.38 To assess EEG during these sound-invoked convulsions, video-EEG recordings were performed for 12–48 h to obtain a baseline prior to AGS induction. Following the baseline period between the hours of 1–3 p.m. Central Time, animals were exposed to a 107 dB broadband acoustic tone generated by an analog twin bell alarm (La Crosse Technology, La Crosse, Wisconsin) placed in the recording cage until the appearance of convulsive movements or for a maximum of 60 s.
Htr2c expression
We determined regional expression levels of Htr2c mRNA in loxTB Htr2c and Gad2Cre-Htr2c mice by quantitative polymerase chain reaction (qPCR). Adult mice [postnatal day (PD) >45] were deeply anesthetized with isoflurane (>4%) and then decapitated. We harvested a 2 mm slab of coronal forebrain located 4 mm from the anterior pole that contained the majority of cortical and subcortical regions expressing Htr2c (dorsal hippocampus, habenula, thalamus, hypothalamus) which was flash frozen with dry ice. Next, we extracted RNA from this tissue using the RNAeasy Plus Mini Kit (Qiagen, Germantown, MD) and assayed RNA concentration using a NanoDrop One (Thermo Scientific, Waltham, MA). We used 200 ng of RNA material in the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) to generate cDNA for analysis. TaqMan probes targeting mouse Htr2c (Mm00434127_m1; Applied Biosystems) and Gapdh (Mm99999915_g1; Applied Biosystems) with TaqMan Fast Advanced Master Mix (Applied Biosystems) were used to assess gene expression. Samples were loaded on 96-well plates, and qPCR analysis was performed on an Applied Biosystems QuantStudio 3 system. Age-matched wildtype and mutant samples were assayed in triplicate, and male and female samples were assayed on separate plates. For a more detailed description of our qPCR analysis, see Supplementary material.
Cellular localization of Htr2c in mouse brain
Brains were extracted from wildtype mice and divided into forebrain and midbrain/hindbrain samples with a cut at the level of the superior colliculus. Each of these were hemisected by a midsagittal cut. Immediately following extraction, the right-sided samples were placed in a brain mold, covered in Optimal Cutting Temperature embedding medium, and placed on dry ice to preserve RNA quality. Tissue sections (of rostral and caudal blocks) were cryo-sectioned using a CM1950 cryostat (Leica Biosystems, Buffalo Gove, IL) into 15 µm thick hemi-coronal sections and mounted onto Superfrost Plus slides, then stored at −80°C until use.
RNAscope Multiplex Fluorescent V2 kit was used to perform RNAscope hybridization (Advanced Cell Diagnostics, Newark, CA). For our experiments, we used channel one probes targeting Gad2 (Mm-Gad2; Cat. no. 439371; Advanced Cell Diagnostics), channel two probes targeting Htr2c (Mm-Htr2c-C2; Cat. No. 401001-C2; Advanced Cell Diagnostics) and channel three probes targeting Slc32a1 (Mm-Slc32a1-C3; Cat. No. 319191-C3; Advanced Cell Diagnostics) mRNA species. Channel two and three probes were diluted into channel one probes at a 1:1:50 ratio. Probes were labelled using Opal fluorescent dyes (Akoya Biosciences, Marlborough, MA) at a 1:1500 dilution. We used Opal dye 520 for channel one probes (Cat. No. FP1487001KT), Opal dye 690 for channel two probes (Cat. No. FP1495001KT) and Opal dye 620 for channel three probes (Cat. No. FP1497001KT). 4′,6-Diamidino-2-phenylindole (DAPI; RNAscope Multiplex Fluorescent V2 kit) was used to label nuclei, and slides were mounted with coverslips using Prolong Gold Antifade Mountant (Cat. No. P36930, Thermo Fisher Scientific). For other details of RNAscope methods, see Supplementary material.
Fluorescent images were taken using a Nikon Eclipse TE2000-S epifluorescence microscope with three channels: DAPI (nuclei), green fluorescent protein (Gad2 channel one probe), Cyanine 5 (Htr2c channel two probe), TexasRed (Slc32a1 channel three probe). Full scan images were taken at ×40 magnification with appropriate filter settings and stitched at 10% overlap, to assess brain regions for positive probe signal using ImageJ and HALO Image Analysis software (Indica Labs, Albuquerque, NM).
Human HTR2C variant analysis
This study was approved by Institutional Review Boards at Baylor College of Medicine (H-32343) and Karolinska Institutet (2011/2146-31/3). Cases were enrolled in the STOP SUDEP Program at Baylor College of Medicine.10 We obtained a post-mortem venous blood sample or blood card from the proband and fresh venous blood samples from both parents. Genomic DNA was extracted using the Gentra Puregene Blood Kit (Qiagen) and stored at −80°C until use. DNA derived from blood cards was amplified as detailed.39 We performed a commercial next-generation exome sequencing on each sample (Prism Genomic Medicine, Inc., Houston, TX). The mean depth coverage was 155X, and the quality threshold was 97.6%. For the HTR2C gene, 100% of the coding region was covered at a minimum of 10X, in silico prediction of variant functional impact followed published principles40 built into our custom-built Mercury pipeline.41 Variant functional interpretation reported in ClinVar adhered to established guidelines.42
Statistical methods
Statistical analyses were performed using GraphPad Prism 8 version 8.4.3 (GraphPad Software, San Diego, CA) software. Unless otherwise noted, data are presented as mean ± standard deviation, and all error bars in figures represent standard deviation. The Shapiro–Wilk normality test was performed before any parametric statistical test that assumed Gaussian distribution. If data failed the Shapiro–Wilk test, we applied the appropriate non-parametric statistical test instead. In cases of multiple comparisons, appropriate post-hoc tests were used to correct P-values for multiple hypothesis testing. Statistical tests and post-hoc analyses are identified when reporting P-values. Survival analyses, surgeries, and EEG monitoring and analyses were all performed with the experimenter blinded to genotype.
Data availability
All supporting data in this study are available on request.
Results
loxTB Htr2c mutant mice show increased premature mortality
We first confirmed that loxTB Htr2c mutant mice did not express 5-HT2C receptors in the brain. Currently there are no validated antibodies to determine protein expression, therefore, we utilized qPCR to assay mRNA expression. We found that male Htr2c−/Y mice (n = 11) had undetectable (<0.02%) expression of Htr2c compared to wildtype (n = 16) littermates (P < 0.0001; Fig. 1A). Female Htr2c−/− (n = 10) also were devoid of Htr2c expression compared to wildtype (n = 10) mice (P < 0.0001, Dunn’s multiple comparisons test; Fig. 1B). Heterozygous female mice (Htr2c−/+; n = 11) had 43.68% of Htr2c expression compared to female wildtype (n = 10) mice (P = 0.0216, Dunn’s multiple comparisons test; Fig. 1B). To validate the loxTB Htr2c strain as a SUDEP model, we assessed mutants for decreased survival. We found that 37.89% (36/95) of Htr2c−/Y mice died prematurely (Fig. 1C, red line). This was significantly more than wildtype littermates, which had a premature mortality rate of 1.74% (2/115; P < 0.0001; Fig. 1C, black line) over the lifespan. The premature mortality rate was less than previously reported in male loxTB Htr2c mutant mice,36 but similar to that in the targeted deletion model of Htr2c.35 Since Htr2c is an X-linked gene and neither of these reports examined female mutant mice, we investigated the survival of female mutant loxTB Htr2c mice. We found that 16.47% (14/85; Fig. 1C, blue line) of female Htr2c−/− mice died prematurely compared with only 4.92% (6/122; Fig. 1, magenta line) of Htr2c−/+ and 1.72% (1/58; Fig. 1C, grey line) of female wildtype mice. There was no difference in premature mortality rates of male and female wildtype or Htr2c−/+ mice. Both Htr2c−/Y and Htr2c−/− mice had decreased survival, but Htr2c−/Y mice had a hazard ratio of 2.534, which corresponded to a mortality rate more than double that of females by PD 300 (P = 0.001, Log-rank test). Although male mutant mice showed substantial premature death, 62.11% of Htr2c−/Y mice (59/95) were still alive at PD 300, indicating incomplete penetrance of the SUDEP phenotype.
Figure 1.
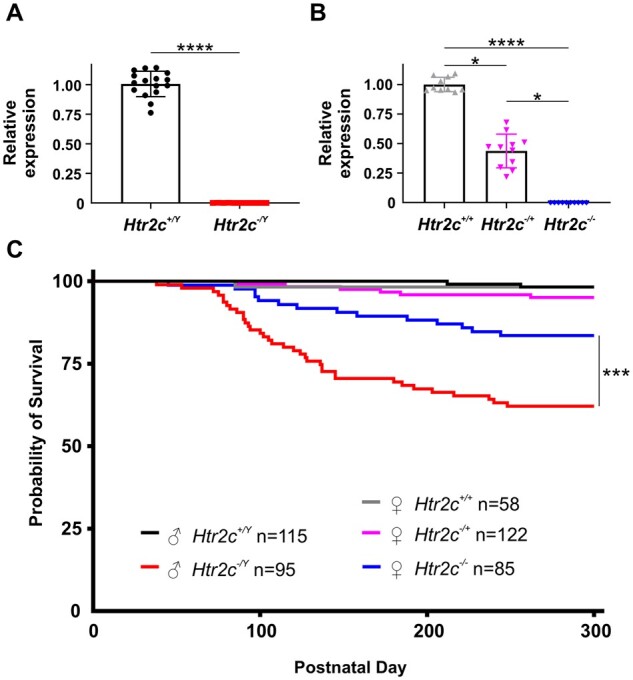
Mice lacking 5-HT2C receptors have increased premature death. (A) Quantification of forebrain Htr2c mRNA levels in male wildtype (n = 16) and loxTB Htr2c mutant (n = 11) mice. These mutant mice have a transcriptional blocker flanked by loxP sites (loxTB) inserted between exons three and four of the X-linked Htr2c gene, which prevents transcription of 5-HT2C receptors. These mice are functional knockouts, and we refer to male and female 5-HT2C-null mice as Htr2c−/Y and Htr2c−/−, respectively. Forebrain mRNA levels were normalized to the mean of wildtype expression. Htr2c−/Y mice had negligible (0.02 ± 0.04%) amplification of Htr2c compared to wildtype controls (P < 0.0001, Mann–Whitney test). (B) Quantification of Htr2c gene expression in female wildtype (n = 10), loxTB Htr2c Htr2c−/+ (n = 11) and Htr2c−/− (n = 10) mice. A one-way ANOVA test revealed a statistical difference in Htr2c gene expression in female loxTB Htr2c mice (P < 0.0001, Kruskal–Wallis test). Htr2c expression in heterozygous mice was 43.68 ± 14.29% of female wildtype mice (P = 0.0216, Dunn’s multiple comparisons test). Htr2c−/− mice had 0% Htr2c expression compared to wildtype animals (P < 0.0001, Dunn’s multiple comparisons test). Htr2c expression in these mice was also significantly lower than heterozygous mice (P = 0.0216, Dunn’s multiple comparisons test). (C) Survival rates of loxTB Htr2c mutant mice are decreased compared to wildtype mice (P = 0.0054, Log-rank test). Male Htr2c−/Y mice (n = 95, red line) had increased premature mortality with a 10.00 hazard ratio when compared to Htr2c+/Y (n = 115, black line) mice (P < 0.0001, Log-rank test). Female Htr2c−/− mice (n = 85, blue line) had increased death with a 4.288 hazard ration compare to Htr2c+/+ (n = 58, grey line) mice (P = 0.0054, Log-rank test). Male Htr2c−/Y mice had increased death compared to female Htr2c−/− mice (P = 0.001, Log-rank test). There was no difference in survival among Htr2c+/Y, Htr2c+/+ and Htr2c−/+ (n = 122; magenta line) mice (P = 0.2918, Log-rank test). * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001.
loxTB Htr2c deletion causes a complex epilepsy phenotype
After confirming premature mortality in loxTB Htr2c mutants, we investigated whether death was due to spontaneous seizures. Previous reports indicate that 5-HT2C mutants displayed spontaneous convulsive-like behaviours, but seizures were not confirmed electrographically, and their developmental onset was unspecified. Using simultaneous video-EEG recordings, we monitored brain activity in loxTB Htr2c mutants and wildtype littermates over prolonged periods from ages of PD 40–170. In young adult mutant mice (PD 40–70), the first EEG abnormality we observed in loxTB Htr2c mutants (of either sex) was frequent, bilateral spike-wave discharges (SWDs) (5–10 spikes/s, 0.5–2 s in duration, ranging from 0 to 25 per hour) similar to those seen in absence epilepsy models43 (Fig. 2A). These discharges appeared as early as PD 48 and were rarely seen in wildtype mice, occurring in only 17.65% (3/17; Fig. 2B) of Htr2c+/Y mice, whereas we observed SWDs in 74.07% (20/27; Fig. 2B) of Htr2c−/Y mice. Only one Htr2c−/Y died during this early stage (before PD70).
Figure 2.

loxTB Htr2c mutant mice have a complex epileptic phenotype with multiple seizure subtypes. (A) Representative trace from a P90 male Htr2c−/Y mouse demonstrating SWDs (6–9 Hz) observed in loxTB Htr2c mice. (B) Histogram quantifying the presence of SWDs during video-EEG recordings in Htr2c+/Y (n = 17), Htr2c−/Y (n = 27), Htr2c+/+ (n = 5), Htr2c−/+ (n = 11), Htr2c−/− (n = 18) mice. (C) Histogram quantifying premature mortality in mice monitored with video-EEG. Animals that died prematurely (before PD 300) are indicated in red. These include both animals which died during video-EEG recording and those found dead in their home cage with tonic hindlimb extension. (D) Representative trace of a generalized nonconvulsive seizure with behavioural arrest from a P89 male Htr2c−/Y mouse. (E) Representative GTCS in a PD 90 male Htr2c−/Y mouse. This trace is from the same mouse in D approximately 24 h following the nonconvulsive seizure. (F) Example of a SUDEP event following a GTCS in the same PD 90 male Htr2c−/Y mouse in D and E. (G) Representative trace of an induced AGS in a PD 79 male Htr2c−/Y mouse. Alarm sound began at point 1 and mouse exhibited wild running around the recording cage at point 2. Within seconds of wild running starting, the mouse fell on its side and had tonic hindlimb extension, which resulted in death. There was no observable cortical spiking activity across any of the leads and the aberrant EEG signal during wild running can be attributed to motion artefact.
One wildtype and 13 5-HT2C-null mice undergoing EEG monitoring died prematurely either during a recording period or between recordings (Fig. 2C). Mutants found dead in their cage typically displayed tonic hindlimb extension similar to those incidences of SUDEP we recorded with EEG monitoring. At this age we also observed generalized non-convulsive seizures with behavioural arrest (Fig. 2D). During these electrographic seizures, mice remained immobile without any convulsive movements and rapidly regained normal behavioural at the end of the abnormal EEG discharge. Within hours to days following the onset of non-convulsive seizures, these same mice developed generalized tonic-clonic seizures (GTCS). Typically, the seizures had the same electrographic profile of the preceding non-convulsive seizures, however, they displayed both a tonic and clonic motor phase, characterized by limb clonus and followed by loss of upright posture and coordination (Fig. 2E). In some cases, convulsive seizures progressed to wild running inside the recording cage and ended suddenly with generalized EEG flattening and tonic hindlimb extension. 5-HT2C-null mice died within 30 s of tonic hindlimb extension (Fig. 2F). We captured such spontaneous cortical seizure/SUDEP events in one Htr2c−/− and two Htr2c−/Y mice during video-EEG recordings.
Since this postictal pattern resembled that reported following evoked AGS in adult 5-HT2C-null mice,38 we induced AGSs in three mutant mice during simultaneous EEG recording. AGS in Htr2c−/Y mice resembled those in other rodent models (wild-running followed by tonic-clonic-tonic phase, hindlimb extension and sudden death), were much shorter in duration (approximately 7–10 s) than spontaneous events, which typically lasted 20–40 s, and were uniformly immediately fatal. As in other rodent models, there was no cortical electrographic seizure activity evident during evoked AGSs (Fig. 2G), consistent with evidence that the AGS is a subcortical event triggering a brainstem mechanism of cardiorespiratory arrest.44
Progressive hyperexcitability increase in adult loxTB Htr2c mutant brain
Our survival analysis indicated SUDEP first occurred after two months of age in both male and female mutants (Fig. 1), which is four to six weeks later than common monogenic developmental epileptic encephalopathy SUDEP models, such as Scn1a, Scn8a and Kcna1 mutants.45–48 This finding prompted us to investigate whether 5-HT2C mutant mice develop a slowly progressive increase in hyperexcitability and seizures during their lifespan. We recorded animals before the overt onset of convulsive seizures (<PD 75) and EEG activity was similar to wildtype littermates with only occasional interictal spikes or SWDs (Fig. 3A). However, after PD 75, we found increased abnormal hyperexcitability in 85% (23/27) of Htr2c−/Y and 78% (14/18) of Htr2c−/− mutant mice which coincided with the onset of spontaneous death in our colony (Fig. 1C). In one representative mouse (PD 98) we found SWDs with behavioural arrest, and a sharp increase in the number of interictal spikes (Fig. 3B). SWDs begins around PD 20 in most mouse models. In the same animal recorded approximately three weeks later (PD 122), we observed the first sustained generalized nonconvulsive seizure (Fig. 3C). Three days later (PD 125), we observed two short seizures that had minor motor manifestations, such as head nodding, but otherwise were nonconvulsive (Fig. 3D). Almost three weeks following this event at PD 142, additional seizures appeared that were longer in duration and progressed from nonconvulsive to GTCS (Fig. 3E). At PD 145, the mouse had a GTCS terminating in hindlimb extension and death (Fig. 3F). This representative animal exemplifies the progressive excitability increase we observed with EEG monitoring in cohorts of Htr2c−/Y mice that captured a recorded SUDEP event (n = 2) and those probable SUDEP cases that occurred outside of recording hours (n = 6). This pattern was also similar in Htr2c−/− mice undergoing EEG monitoring (n = 5), where we recorded one SUDEP event, and four probable SUDEP events occurred outside of recording hours.
Figure 3.

loxTB Htr2c mutant mice show a progressive increase in hyperexcitability over their lifetime. (A) Representative trace from a video-EEG recording of a PD 47 male Htr2c−/Y mouse. A few small SWDs occur periodically at this age. (B) Recording from the same animal at PD 98 shows more hyperactivity, including SWDs and spike discharges. (C) Three weeks later, at PD 122, the first monitored generalized nonconvulsive seizure appeared. (D) Three days later at PD 125, we observed short seizures with minor motor involvement, such as head nodding, but otherwise were nonconvulsive. (E) These seizures continued over the next three weeks and at PD 142 we recorded a convulsive GTCS. (F) Three days at PD 145, the mouse had another GTCS culminating in tonic hindlimb extension and death.
Restoring expression of 5-HT2C receptors in Gad2-positive neurons of null mutants does not improve survival
Previous studies indicate 5-HT2C receptors are expressed predominantly on GABAergic neurons.49–51 Moreover, when 5-HT2CR+ cells in the dorsal raphe nucleus are stimulated by serotonin, 5-HT2CR agonists, or optogenetic laser, they reduce the firing rate of principal outflow neurons, suggesting that 5-HT2C-expressing cells are inhibitory in this nucleus.33,34,49 Since available antibodies for the receptor lack specificity, we used RNAscope fluorescent in situ hybridization (FISH) to identify the distribution of Htr2c mRNA expression in mouse brain. We found that Htr2c showed sparse but widespread expression in both excitatory and inhibitory cells in the forebrain (Fig. 4A), midbrain (Fig. 4B) and medulla (Fig. 4C); two cortical areas showed dense expression in layers 2/3 of retrosplenial (Fig. 4D) and piriform (Fig. 4E) cortex. Htr2c expression was prominent in the lateral habenula (Fig. 4f) and thalamic reticular nucleus (TRN; Fig. 4G). Subthalamic structures such as zona incerta (Fig. 4H) and STN (Fig. 4I) expressed Htr2c mRNA at a high level. Htr2c signal was detected in both the CA3 region of the hippocampus (Fig. 4J) and amygdala (Fig. 4K).
Figure 4.

Htr2c mRNA is expressed throughout the forebrain and hindbrain. (A) Hemi coronal forebrain section taken from a PD 84 Htr2c+/Y mouse. Tissue was stained with DAPI (blue) and processed with RNAscope FISH using probes for Gad2 (green), Slc32a1 (red) and Htr2c (magenta). (B–C): Hemi section from the midbrain (B) and medulla (C) of a PD 84 Htr2c+/Y mouse. Tissue was stained with DAPI (blue) and processed with RNAscope FISH using probes for Gad2 (green), Slc32a1 (red) and Htr2c (magenta). (D–K): ×40 insets from section shown in panel (A) of the following regions: retrosplenial cortex (D), piriform cortex (E), zona incerta (F), STN (G), lateral habenula (H), TRN (I), CA3a region of the hippocampus (J) and lateral amygdala (K). Separate channel images for DAPI and each of the RNAscope FISH probes are available in Supplementary Figs. 1–4.
Based on earlier studies, we hypothesized that a loss of 5-HT2C receptors in GABAergic neurons would decrease inhibitory tone in the CNS and create a hyperexcitable state, and that allowing selective expression of 5-HT2C receptors only in inhibitory neurons of a 5-HT2C-null mouse would restore 5-HT2C-mediated excitation of interneurons, thereby preserving inhibition, reducing seizures, and preventing SUDEP. We chose the glutamate decarboxylase 2 (GAD2) Cre-promoter to restore the expression of 5-HT2C receptors exclusively in GABAergic neurons in the 5-HT2C-null mouse. GAD2 is one of two proteins utilized to synthesize GABA and is a pan-GABAergic cell marker with expression in multiple interneuron subtypes, including parvalbumin, cholecystokinin, neuropeptide Y and somatostatin.52
We bred female Htr2c−/+ mice with male mice homozygous for Gad2Cre which produced a line of mice with the loxTB element removed from the Htr2c gene only in Gad2+ cells (Gad2Cre-Htr2c). We verified expression of Htr2c by performing qPCR on forebrain tissue (Fig. 5A and B). We found that Htr2c−/Y; Gad2Cre mice (n = 12; Fig. 5A, red) express Htr2c mRNA at 35.25% of wildtype littermate (Htr2c+/Y; Gad2Cre) levels (n = 14; Fig. 5a, black). In female mice, Htr2c−/+; Gad2Cre mice (n = 14; Fig. 5B, magenta) and Htr2c−/−; Gad2Cre mice (n = 3; Fig. 5B, blue) expressed Htr2c mRNA at 62.89% and 29.30%, respectively, compared to wildtype mice (Htr2c+/+; Gad2Cre; n = 14; Fig. 5B, grey).
Figure 5.
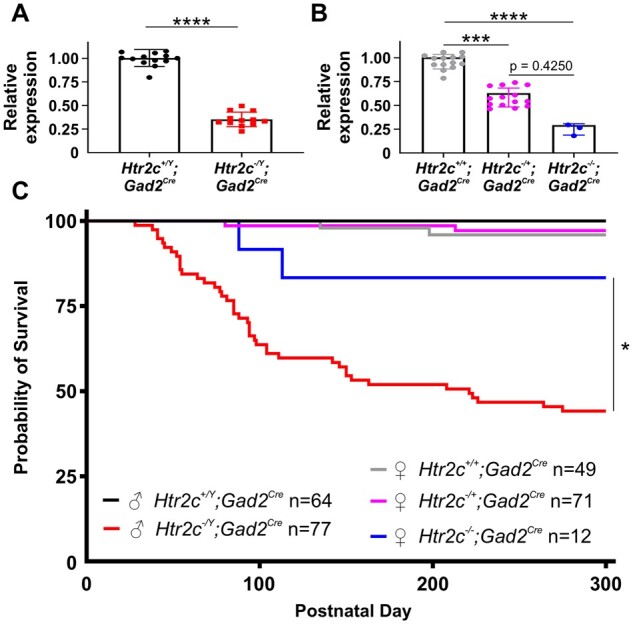
Expression of 5-HT2C receptors in Gad2-positive neurons does not prevent SUDEP. (A) We bred male mice expressing Cre recombinase under the control of Gad2 to female Htr2c−/+ mice. This cross produced mice with the transcriptional blocker excised in only Gad2-positive neurons. Mutant mice from this cross express 5-HT2C receptors in only GAD2+ neurons, and we refer to male and female mice as Htr2c−/Y; Gad2Cre and Htr2c−/−; Gad2Cre, respectively. Quantification of Htr2c gene expression in male Gad2Cre-Htr2c wildtype (n = 14) and mutant (n = 12) mice. Male Htr2c−/Y; Gad2Cre mice have 35.25 ± 7.71% of the Htr2c expression compared to wildtype littermates (P < 0.0001, Mann–Whitney test). (B) Quantification of Htr2c gene expression in female Gad2Cre-Htr2c mice. A one-way ANOVA test demonstrated there was a significant difference in gene expression among female Gad2Cre-Htr2c wildtype and mutant mice (P < 0.0001, Kruskal–Wallis test). Female Htr2c−/−; Gad2Cre (n = 3) mice have 29.30 ± 5.91% of the Htr2c expression of Htr2c+/+; Gad2Cre mice (n = 14; P = 0.0001, Dunn’s multiple comparisons test). Female Htr2c−/+; Gad2Cre (n = 14) had 62.89 ± 9.88% expression of Htr2c compared to female wildtype mice (P = 0.0003, Dunn’s multiple comparisons test). Statistical significance was not detected in expression between Htr2c−/+; Gad2Cre and Htr2c−/−; Gad2Cre (P = 0.4250, Dunn’s multiple comparisons test), which was most likely due to the low number of Htr2c−/−; Gad2Cre mice available for tissue collection. (C) Survival rates were different among wildtype and mutant Gad2Cre-Htr2c mice (P < 0.0001, Log-rank test). Male Htr2c−/Y; Gad2Cre mice (n = 77; red line) had increased death compared to male wildtypes (n = 64; black line) by P300 with a hazard ratio of 9.05 (P < 0.0001, Log-rank test). There was no difference in survival rates among male wildtype, female Htr2c−/+; Gad2Cre (n = 71; magenta line) and female Htr2c+/+; Gad2Cre (n = 49; grey line) mice (P = 0.3041, Log-rank test). We did not detect statistical differences in survival rates of female Gad2Cre-Htr2c mice, which was likely due to the low number of Htr2c−/−; Gad2Cre (n = 12, blue line) available to track in the colony (P = 0.0826, Log-rank test). * = P < 0.05, ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001.
We then tracked the survival of mice in this line to determine if spared expression of 5-HT2C receptor in Gad2+ neurons was sufficient to prevent SUDEP. Unexpectedly, we observed high mortality in Htr2c−/Y; Gad2Cre mice (55.84%; 43/77; Fig. 5C, red line) compared to wildtype littermates (0%; 0/64; P < 0.0001; Fig. 5C, black line). In females we recorded 16.67% mortality in Htr2c−/−; Gad2Cre mice (2/12; Fig. 5C, blue line) compared with only 1.49% in Htr2c−/+; Gad2Cre (1/67; Fig. 5C, magenta line) and 4.08% in Htr2c+/+; Gad2Cre (2/49; Fig. 5C, grey line) mice. These mortality rates were not lower than those observed in loxTB Htr2c male mutants (Fig. 1C). In fact, the Gad2Cre-Htr2c mutant mice showed death earlier than loxTB Htr2c mice. We observed only two deaths of Htr2c−/Y mice before PD 70, but in Htr2c−/Y; Gad2Cre mice we recorded 13 deaths by PD 70 (Figs. 1C and 5C). Moreover, the mortality rates of Htr2c−/Y; Gad2Cre mice (55.84%) were significantly higher than Htr2c−/Y mice (37.89%; P < 0.0001) (Figs. 1C and 5C). The larger gap in survival between Htr2c−/Y; Gad2Cre and Htr2c−/−; Gad2Cre mice could reflect a smaller number of Htr2c−/−; Gad2Cre mice in our colony, however, we consider it unlikely this disparity would shift survival enough to equal male Htr2c−/Y; Gad2Cre littermates.
Gad2Cre-Htr2c mice display the same complex epileptic profile as loxTB Htr2c mice
Since survival did not improve, we next determined whether restoration of expression of 5-HT2C receptors in GABAergic neurons had altered the epileptic phenotype of the mice. Using video-EEG recordings, we found that 75% (9/12) of Htr2c−/Y; Gad2Cre mice exhibited SWDs similar to those seen in loxTB Htr2c mice (Fig. 6A). In one representative Htr2c−/Y; Gad2Cre mouse, we recorded the same pattern of generalized nonconvulsive seizures (Fig. 6B) which then transitioned (over 7 days) into GTCS (Fig. 6C). At PD 93, the mouse had a GTCS that ended with hindlimb extension and death (Fig. 6D). Although these Gad2Cre-Htr2c mice spared the deletion of 5-HT2C receptors in GAD2+ inhibitory neurons, the mutant mice recapitulated the same spectrum and evolution of multiple seizure types observed in the loxTB Htr2c strain. Thus, expression of 5-HT2C in GAD2+ neurons was not sufficient to prevent the development of epilepsy, indicating that despite their powerful control over the epileptic phenotype, there is a minimal contribution of these receptors to 5-HT2C-mediated inhibitory signalling within the pathways underlying each of the various seizure subtypes.
Figure 6.

Gad2Cre-Htr2c mice display the same spontaneous seizure subtypes as loxTB Htr2c mice. (A) Representative trace demonstrating SWDs in a PD 80 male Htr2c−/Y; GadCre mouse. (B) Example of a generalized nonconvulsive seizure recorded in a PD 83 male Htr2c−/Y; Gad2Cre mouse. (C) In the same mouse as in B, we recorded a GTCS one week later at PD 90. (D) At PD 93, the same mouse died following a GTCS with tonic hindlimb extension.
HTR2C variants are enriched in cases of human sudden unexpected death
The role of 5-HT2C receptor gene variants in human premature mortality has not been explored. We reviewed our STOP SUDEP registry database of 534 patient exomes containing cases with epilepsy (n = 116), SUDEP (n = 137) and SIDS (n = 8). We identified 11 individuals with rare variants in HTR2C who died due to either definite SUDEP (n = 5, age range from 12 to 33 years, mean age 26.8 years), SIDS (n = 5, age range 2–5 months, mean age 3.5 months), or hypothermia (n = 1, age 34 years) (Table 1). Three of the five SUDEP cases were females (3/5; 67%), and each carried a non-synonymous (ns) single nucleotide variation in the HTR2C gene. In contrast, all but one of the SIDS cases were males (4/5; 80%), and carried either a ns variant (3/5), frameshift (1/5) or in-frame deletion (1/5). Review of HTR2C variation in gnomAD indicates that HTR2C is tolerant to loss of function variants (pLI = 0.04).53 Yet, the relative paucity of HTR2C variation among the 116 adult cases with presumed genetic epilepsies is intriguing and may warrant further screening in a more substantial patient sample.
Table 1.
Summary of SUDEP and SIDS cases with HTR2C variation
| Case | Pheno | Sex | AAD | Chr: Position | Variant† | AA change | Type | Zyg | Inh | Effect | Freq gnomAD | dbSNP | ClinVar |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2273 | SUDEP | M | 26 Y | X: 114082728 | c.512C>T | p.Ala171Val | Ns | hem | mat | likely benign | 0.054% ± 0.011% | rs139581423 | novel |
| 2353 | SUDEP | F | 32 Y | X: 113965907 | c.240G>A | p.Met80Ile | Ns | het | u | VUS | absent | novel | novel |
| 2347 | SUDEP | F | 33 Y | X: 113965926 | c.259G>A | p.Ala87Thr | Ns | het | u | VUS | absent | novel | novel |
| 2354 | SUDEP | F | 31 Y | X: 114141802 | c.1201G>A | p.Val401Ile | Ns | het | u | likely benign | absent | novel | novel |
| 2232 | SUDEP | M | 12 Y | X: 114141856 | c.1255A>G | p.Thr419Ala | Ns | hem | u | VUS | 0.078% ± 0.013% | rs76454046 | novel |
| 2475 | SIDS | M | 3.5 Mo | X: 113965847 | c.181_196delGTCATCATAATAATCA | p.Val61Ter | Fs | hem | u | VUS (P) | absent | novel | novel |
| 2099 | SIDS | M | 3 Mo | X: 114141797 | c.1196C>T | p.Pro399Leu | Ns | hem | u | VUS | absent | novel | novel |
| 2086 | SIDS | M | 5 Mo | X: 114141535 | c.934G>A | p.Val312Ile | ns | hem | u | VUS | absent | novel | novel |
| 2478 | SIDS | F | 4 Mo | X: 114141468 | c.874_876delAAG | p.Lys292del (in frame) | del | het | u | VUS | 0.143% ± 0.051% | novel | novel |
| 2474 | SIDS | M | 2 Mo | X: 113965886 | c.220delG | p.Val74Ter | fs | hem | u | VUS (P) | absent | novel | novel |
| 2346 | EPI* | M | 34 Y | X: 114141815 | c.1214C>T | p.Ala405Val | ns | hem | u | VUS | absent | novel | novel |
†—Reference transcript for variant calls: NM_001256760.1, GRCH38 build, AAD, age at death; Chr, chromosome; del, deletion; EPI*, patient with epilepsy that died due to hypothermia; fs, frameshift variant; het, heterozygous; Inh, inheritance; mat, maternal; Mo, months; ns, nonsynonymous; (P), possibly pathogenic; Pheno, phenotype; u, unknown inheritance; VUS, variant of uncertain clinical significance; Y, years; Zyg, zygosity.
We also reviewed the DECIPHER public copy number variation database54 (https://decipher.sanger.ac.uk/search?q=HTR2C#consented-patients/results, v.10.2, last accessed July 11, 2021) that houses 29 duplications and 35 deletions involving the HTR2C gene, ranging from 476 kb to 155.27 Mb in size, and often involving up to 60 other genes. Sex was reported in 43 cases (33 females and 10 males), and six duplications and 10 deletions were reported as either pathogenic or likely pathogenic in 12 females and four males. The most common reported phenotypes were intellectual disability, developmental delay and autism. Seizures were indicated in two females (cases 369118 and 288318), however, there were no reported cases of sudden death. The apparent absence of cases with copy number variations containing the HTR2C gene affected by sudden death may be due, in part, to the lack of clinical follow-up of patients referred for clinical genetic testing or due to the effect of genes co-duplicated or co-deleted within these large copy number variations.
Discussion
Pathophysiological mechanisms of SUDEP remain unclear, and each new genetic model represents a valuable opportunity to better define the distinct predispositions that lead to sudden death and develop effective interventions. The 5-HT signalling pathway has been implicated in both human and mouse models of SUDEP,55–57 and continues to attract interest to clinically intervene using 5-HT releasing and reuptake inhibiting pharmacology,58–63 however, few monogenic models in this pathway have been systemically explored for critical cellular targets. Here, we studied the loxTB Htr2c mouse model and found that both male and female 5-HT2C mutant mice display a complex epileptic phenotype with three different spontaneous seizure types and inducible AGSs. Unlike previous SUDEP mouse models, these mice experience a late-onset, progressive increase in hyperexcitability in adulthood, and a sex ratio imbalance in survival, with a mortality rate of male Htr2c−/Y mice double that of female Htr2c−/− mice. Although 5-HT2C receptors are expressed in GABAergic interneurons where various loss-of-function mutations are thought to cause epilepsy by disinhibition, selective expression of the receptors in Gad2+ interneurons was not sufficient to prevent epilepsy or SUDEP.
5-HT2C receptor knockout mice are an adult-onset SUDEP model
Two of the most studied SUDEP monogenic mouse models involve mutations of Kcna1 and Scn1a genes.45,46,48,64,65 Each has a high seizure burden and elevated mortality in the first few weeks of life. In contrast, the EEG in 5-HT2C mutant mice is unaffected before two months of age, convulsive seizures are not seen before PD 45 (Figs. 1C and 5C), and survival does not decrease until after 2 months of age. This post-pubescent onset of morbidity suggests that the contribution of 5-HT2C receptors in modulating CNS excitability builds during the mouse’s lifespan. While deterioration could be contingent upon progressive neurodegeneration, no anatomical pathology in the forebrain of 5-HT2C knockout mice has been reported.66
Another key difference between loxTB Htr2c mice and Kcna1 and Scn1a SUDEP models is seizure frequency. As in humans, mouse models with high SUDEP risk typically suffer from high seizure burden. For example, Kcna1−/− mice have very frequent daily seizures, and even those that survive until adulthood continue to have one to two seizures per hour throughout their life.48 In contrast, loxTB Htr2c mutant mice typically experience only infrequent (one to two per day) seizures. With intermittent EEG sampling, this complicated our ability to quantify absolute seizure burden. Furthermore, neither the epilepsy nor SUDEP phenotypes of loxTB Htr2c mice show full penetrance, which makes a full characterization of seizure severity and mortality risk more difficult, a feature of other SUDEP mouse models. Currently human studies have not yet determined if death is always related to the severity of the cortical seizure,67 or instead depends upon other intervening features, for example a lower threshold for brainstem spreading depolarization11 or other autonomic and cardiorespiratory factors.
Evidence linking 5-HT signalling and SUDEP
The links between 5-HT and SUDEP have been demonstrated pharmacologically in both animal models and humans, and treatments that increase 5-HT concentration also prevent seizure-induced mortality. Fluoxetine, a 5-HT reuptake inhibitor, reduces seizure-induced respiratory arrest in both DBA/1 and DBA/2 mice without altering AGS severity.58 Fenfluramine, a serotonin-releasing and reuptake inhibitor, prevents respiratory arrest and death in DBA/1 mice, and decreases seizure incidence and severity as well.58 Fenfluramine and fluoxetine reduced seizure frequency in patients with Dravet syndrome.60–63 Although fenfluramine is a weak 5-HT2C agonist, its metabolite, norfenfluramine, has high affinity for 5-HT2C receptors.68 Other 5-HT2C agonists, including lorcaserin and trazodone, have also shown effectiveness in reducing seizures in a Zebrafish model of Dravet syndrome.69 Interestingly, lorcaserin also suppresses SWDs in a rat genetic absence epilepsy model.70 Post-ictal generalized EEG suppression (PGES) is a possible biomarker for SUDEP risk in human patients.71 Although the mechanisms that underlie PGES remain unclear, recent evidence has demonstrated that increased 5-HT or 5-HT neuron activity reduces PGES in experimental models.55,72 Although our study was not designed to fully characterize PGES in 5-HT2C-null mice, we noted reduced EEG signal amplitude following non-fatal GTCS events (Figs. 2E and 4C). Future studies will investigate this potential biomarker for SUDEP risk in loxTB Htr2c mutant mice. Finally, in a post-mortem study of brainstem tissue, patients that died of SUDEP had decreased expression of tryptophan hydroxylase, the rate-limiting step in 5-HT synthesis, and the 5-HT transporter compared to epilepsy controls.73 Our findings, when paired with these supporting data, highlight 5-HT2C receptors as an important molecular target within the 5-HT signalling pathway capable of modulating seizures and premature death.
Sex ratio differences in SUDEP mortality
In human SUDEP, male sex confers a higher risk for mortality,6,74 but few SUDEP mouse models to date show a sexual dimorphism for survival. One recent study reported a sex difference in mortality of Scn1a+/− mice, however, in this study, female mutants had increased mortality.75 Our data indicate that mortality in male Htr2c−/Y mice is elevated compared to female Htr2c−/− mice (Fig. 1). The mechanisms contributing to the sex difference are obscure but could be hormonal. Ovarian sex hormones, including oestrogens and progesterones, play an important role in modulating CNS excitability.76 Interestingly, female rodents have increased concentrations of 5-HT and 5-hydroxyindolacetic acid, the primary metabolite of 5-HT.77–80 Ovarian hormones upregulate 5-HT synthesis and turnover, which are markers of 5-HT activity.77,78,81,82 In addition, hormone replacement therapy in ovariectomized rodents activates the 5-HT system.83–87 A Swedish population-based cohort study reported a higher SUDEP incidence in males, and the sex difference was most prominent in the youngest cohort,74 while the overwhelming majority of SUDEP cases in women occurred after the age of 50.74 These data suggest that SUDEP risk for women increases with age, which may be related to post-menopausal hormonal changes. Future studies are needed to explore mechanisms underlying SUDEP sex differences in 5-HT2C mutants and other SUDEP models.
Epilepsy in 5-HT2C mutant mice is not solely due to disinhibition mediated by GAD2+ interneurons
5-HT2C receptors are widely, but distinctly, distributed in the CNS.29–31 Liu et al.88 reported that 5-HT2C receptors were localized to GABAergic neurons in layer IV of the prelimbic prefrontal cortex in rat brain and are preferentially expressed on parvalbumin-positive cells compared to those expressing calbindin or calretinin. A study of 5-HT2C receptors in the anterior raphe nuclei showed that these receptors are located on GAD1-positive GABAergic interneurons.89 Our RNAscope data confirm overlapping interneuron co-expression in many of these areas, since an overwhelming number of cells co-expressed Htr2c and the GABAergic marker Gad2 and/or Slc32a1 (Fig. 4). These data suggest that 5-HT2C receptors play a role in a negative feedback loop by stimulating local GABAergic interneurons. Boothman et al.33 demonstrated that application of a 5-HT2C agonist, WAY 161503, decreased 5-HT neuron firing, and this effect could be blocked with 5-HT2C antagonists. This study also reported increased Fos expression in GABAergic neurons after WAY 161503 exposure suggesting 5-HT2C receptors activated these neurons.33 Another group used an optogenetic probe localized to 5-HT2C-expressing GAD2+ neurons to test the effect of 5-HT2C receptors on dorsal raphe activity and found that light stimulation activated these GABAergic neurons and inhibited 5-HT neurons.34 These data confirm a role for 5-HT2C receptors in inhibitory interneuron signalling.
We therefore hypothesized that loss of 5-HT2C receptors that activate GABAergic neurons provides a parsimonious explanation for epilepsy and SUDEP in loxTB Htr2c mice, however, in the Gad2Cre-Htr2c model with selective expression only in Gad2+ interneurons, we observed identical EEG abnormalities and spontaneous seizures, and premature mortality was increased rather than blunted (Fig. 5C). These data argue against a major role of 5-HT2C-linked disinhibition underlying the epilepsy/SUDEP phenotype and suggest that 5-HT2C receptors expressed in non-GABAergic neurons must play a more complex role in controlling CNS network excitability. Although most cells expressing 5-HT2C receptors are GABAergic, one study reported that 27% of 5-HT2C-expressing cells are non-GABAergic90 which may explain the complex seizure phenotype and sudden death in the Gad2Cre-Htr2c model. Future experiments that identify and selectively express 5-HT2C receptors in them may provide insight into critical pathways for each of these phenotypic elements.
HTR2C variants in SUDEP and SIDS populations
The sex-linked premature mortality in our model prompted us to perform the first variant analysis of HTR2C in a clinical exome sequencing cohort and in publicly available human SUDEP and SIDS databases. Although SIDS is a diagnosis of exclusion and by definition does not include cases with known epilepsy, there is neuropathological evidence of hippocampal malformations in many of these cases, suggesting increased risk of unrecognized seizure activity.91–93 A recent report indicates that up to 35% of SIDS cases may be due to genetic diseases including 9% of cases with likely pathological variants in ion channelopathy-associated genes.94 In addition, Brownstein et al.95 found likely pathogenic SCN1A variants in SIDS cases. Determining if there are HTR2C variants in these databases is important since disrupted serotonergic signalling is implicated in both SUDEP and SIDS.57,96,97 We found a sufficient enrichment of non-synonymous variants to warrant a larger scale human association study in the future. In our small cohort, non-synonymous variants dominate in male SIDS cases (4/5, 80%) whereas 67% (4/6) of SUDEP cases were older females. It is tempting to speculate that variants in SIDS were more detrimental and that early mortality precluded the appearance of seizures. Sex differences may also arise by imprinting of the variant allele. However, given the small sample size, and unclear functional effects of the variants, it is premature to draw human parallels, and collaborative large-scale studies of 5-HT receptor variation in human SUDEP and SIDS cohorts are needed.
In summary, our studies of loxTB Htr2c mice clearly reveal a SUDEP model with increased mortality beginning in adulthood and, to our knowledge, the first evidence for a gene-linked male sex predominance in SUDEP incidence in a monogenic animal model. We also demonstrate that whereas absence of this receptor causes epilepsy, selectively restoring 5-HT2C receptors only in interneurons is insufficient to prevent seizures and premature death in mice. This finding, consistent with the generally held view of serotonin as a central neuromodulator, implies a more complex network imbalance where dysmodulation of excitatory cells may play a more prominent role in epileptogenesis despite a relatively intact expression of 5-HT2C receptors on GABAergic inhibitory cells. Future studies using selective expression in excitatory neurons may confirm this mechanism, or possibly suggest that 5-HT2C receptors require precise rebalancing among different interneuron subtypes.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
We thank Drs. Joel Elmquist (UT Southwestern) and Yong Xu (Baylor College of Medicine) for providing loxTB Htr2c mice and genotyping protocols. We also thank Jill Mokry (Baylor College of Medicine) for her insightful comments. Graphical abstract was created with BioRender.com using an Academic License (Agreement #OQ22NAWIW). This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from contact@deciphergenomics.org. Those who carried out the original analysis and collection of the DECIPHER data bear no responsibility for the further analysis or interpretation of the data.
Funding
This work was supported by National Institute of Neurological Disorders and Stroke F32 NS105329 (C.A.M.), U01 NS090340 (J.L.N. and A.M.G.) and NS29709 (J.L.N.), a research grant from Citizens United for Research in Epilepsy (A.M.G.), and the Blue Bird Circle Foundation for Pediatric Research. Funding for the DECIPHER project was provided by Wellcome.
Competing interests
The authors report no competing interests.
Glossary
- AGS =
audiogenic seizure
- DAPI =
4′,6-diamidino-2-phenylindole
- FISH =
fluorescent in situ hybridization
- 5-HT =
serotonin
- GAD2 =
glutamate decarboxylase 2
- GTCS =
generalized tonic clonic seizure
- s.c. =
subcutaneous
- PD =
postnatal day
- PGES =
post-ictal generalized EEG suppression
- qPCR =
quantitative PCR
- SIDS =
sudden infant death syndrome
- SUDEP =
sudden unexpected death in epilepsy
- SWD =
spike-wave discharge
References
- 1. Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, Richerson G.. Sudden unexpected death in epilepsy: Epidemiology, mechanisms, and prevention. Lancet Neurol. 2016;15(10):1075–1088. [DOI] [PubMed] [Google Scholar]
- 2. Thurman DJ, Logroscino G, Beghi E, et al. ; the Epidemiology Commission of the International League Against Epilepsy. The burden of premature mortality of epilepsy in high-income countries: A systematic review from the Mortality Task Force of the International League Against Epilepsy. Epilepsia. 2017;58(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thurman DJ, Hesdorffer DC, French JA.. Sudden unexpected death in epilepsy: Assessing the public health burden. Epilepsia. 2014;55(10):1479–1485. [DOI] [PubMed] [Google Scholar]
- 4. Lhatoo SD, Sander JW.. Cause-specific mortality in epilepsy. Epilepsia. 2005;46 (Suppl 11):36–39. [DOI] [PubMed] [Google Scholar]
- 5. Tomson T, Walczak T, Sillanpaa M, Sander JW.. Sudden unexpected death in epilepsy: A review of incidence and risk factors. Epilepsia. 2005;46 (Suppl 11):54–61. [DOI] [PubMed] [Google Scholar]
- 6. Hesdorffer DC, Tomson T, Benn E, et al. ; for the ILAE Commission on Epidemiology; Subcommission on Mortality. Combined analysis of risk factors for SUDEP. Epilepsia. 2011;52(6):1150–1159. [DOI] [PubMed] [Google Scholar]
- 7. Cooper MS, Mcintosh A, Crompton DE, et al. Mortality in Dravet syndrome. Epilepsy Res. 2016;128:43–47. [DOI] [PubMed] [Google Scholar]
- 8. Johannesen KM, Gardella E, Scheffer I, et al. Early mortality in SCN8A-related epilepsies. Epilepsy Res. 2018;143:79–81. [DOI] [PubMed] [Google Scholar]
- 9. Kim Y, Bravo E, Thirnbeck CK, et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest. 2018;128(3):1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klassen TL, Bomben VC, Patel A, et al. High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia. 2014;55(2):e6–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aiba I, Noebels JL.. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med. 2015;7(282):282ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aiba I, Wehrens XH, Noebels JL.. Leaky RyR2 channels unleash a brainstem spreading depolarization mechanism of sudden cardiac death. Proc Natl Acad Sci U S A. 2016;113(33):E4895–E4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldman AM, Behr ER, Semsarian C, Bagnall RD, Sisodiya S, Cooper PN.. Sudden unexpected death in epilepsy genetics: Molecular diagnostics and prevention. Epilepsia. 2016;57 (Suppl 1):17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Loonen ICM, Jansen NA, Cain SM, et al. Brainstem spreading depolarization and cortical dynamics during fatal seizures in Cacna1a S218L mice. Brain. 2019;142(2):412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trosclair K, Dhaibar HA, Gautier NM, Mishra V, Glasscock E.. Neuron-specific Kv1.1 deficiency is sufficient to cause epilepsy, premature death, and cardiorespiratory dysregulation. Neurobiol Dis. 2020;137:104759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wenker IC, Teran FA, Wengert ER, et al. Postictal death is associated with tonic phase apnea in a mouse model of sudden unexpected death in epilepsy. Ann Neurol. 2021;89(5):1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamagata T, Raveau M, Kobayashi K, et al. CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol Dis. 2020;141:104954. [DOI] [PubMed] [Google Scholar]
- 18. Richards KL, Milligan CJ, Richardson RJ, et al. Selective NaV1.1 activation rescues Dravet syndrome mice from seizures and premature death. Proc Natl Acad Sci U S A. 2018;115(34):E8077–E8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bagdy G, Kecskemeti V, Riba P, Jakus R.. Serotonin and epilepsy. J Neurochem. 2007;100(4):857–873. [DOI] [PubMed] [Google Scholar]
- 20. Richerson GB, Buchanan GF.. The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia. 2011;52(Suppl 1):28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kilian M, Frey HH.. Central monoamines and convulsine thresholds in mice and rats. Neuropharmacology. 1973;12(7):681–692. [DOI] [PubMed] [Google Scholar]
- 22. Browning RA, Hoffmann WE, Simonton RL.. Changes in seizure susceptibility after intracerebral treatment with 5,7-dihydroxytryptamine: Role of serotonergic neurons. Ann N Y Acad Sci. 1978;305:437–456. [DOI] [PubMed] [Google Scholar]
- 23. Buchanan GF, Murray NM, Hajek MA, Richerson GB.. Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol. 2014;592(19):4395–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tupal S, Faingold CL.. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia. 2006;47(1):21–26. [DOI] [PubMed] [Google Scholar]
- 25. Zhang H, Zhao H, Zeng C, et al. Optogenetic activation of 5-HT neurons in the dorsal raphe suppresses seizure-induced respiratory arrest and produces anticonvulsant effect in the DBA/1 mouse SUDEP model. Neurobiol Dis. 2018;110:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uteshev VV, Tupal S, Mhaskar Y, Faingold CL.. Abnormal serotonin receptor expression in DBA/2 mice associated with susceptibility to sudden death due to respiratory arrest. Epilepsy Res. 2010;88(2-3):183–188. [DOI] [PubMed] [Google Scholar]
- 27. Yu L, Nguyen H, Le H, et al. The mouse 5-HT1C receptor contains eight hydrophobic domains and is X-linked. Brain Res Mol Brain Res. 1991;11(2):143–149. [DOI] [PubMed] [Google Scholar]
- 28. Humphrey PP, Hartig P, Hoyer D.. A proposed new nomenclature for 5-HT receptors. Trends Pharmacol Sci. 1993;14(6):233–236. [DOI] [PubMed] [Google Scholar]
- 29. Molineaux SM, Jessell TM, Axel R, Julius D.. 5-HT1c receptor is a prominent serotonin receptor subtype in the central nervous system. Proc Natl Acad Sci U S A. 1989;86(17):6793–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abramowski D, Rigo M, Duc D, Hoyer D, Staufenbiel M.. Localization of the 5-hydroxytryptamine2C receptor protein in human and rat brain using specific antisera. Neuropharmacology. 1995;34(12):1635–1645. [DOI] [PubMed] [Google Scholar]
- 31. Clemett DA, Punhani T, Duxon MS, Blackburn TP, Fone KC.. Immunohistochemical localisation of the 5-HT2C receptor protein in the rat CNS. Neuropharmacology. 2000;39(1):123–132. [DOI] [PubMed] [Google Scholar]
- 32. Cussac D, Newman-Tancredi A, Duqueyroix D, Pasteau V, Millan MJ.. Differential activation of Gq/11 and Gi(3) proteins at 5-hydroxytryptamine(2C) receptors revealed by antibody capture assays: Influence of receptor reserve and relationship to agonist-directed trafficking. Mol Pharmacol. 2002;62(3):578–589. [DOI] [PubMed] [Google Scholar]
- 33. Boothman L, Raley J, Denk F, Hirani E, Sharp T.. In vivo evidence that 5-HT(2C) receptors inhibit 5-HT neuronal activity via a GABAergic mechanism. Br J Pharmacol. 2006;149(7):861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spoida K, Masseck OA, Deneris ES, Herlitze S.. Gq/5-HT2c receptor signals activate a local GABAergic inhibitory feedback circuit to modulate serotonergic firing and anxiety in mice. Proc Natl Acad Sci U S A. 2014;111(17):6479–6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tecott LH, Sun LM, Akana SF, et al. Eating disorder and epilepsy in mice lacking 5 HT2C receptors. Nature. 1995;374(6522):542–546. [DOI] [PubMed] [Google Scholar]
- 36. Xu Y, Jones JE, Kohno D, et al. 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate energy homeostasis. Neuron. 2008;60(4):582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Olivetti PR, Maheshwari A, Noebels JL.. Neonatal estradiol stimulation prevents epilepsy in Arx model of X-linked infantile spasms syndrome. Sci Transl Med. 2014;6(220):220ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brennan TJ, Seeley WW, Kilgard M, Schreiner CE, Tecott LH.. Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet. 1997;16(4):387–390. [DOI] [PubMed] [Google Scholar]
- 39. Montier L, Haneef Z, Gavvala J, et al. A somatic mutation in MEN1 gene detected in periventricular nodular heterotopia tissue obtained from depth electrodes. Epilepsia. 2019;60(10):e104–e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu X, Wu C, Li C, Boerwinkle E.. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37(3):235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reid JG, Carroll A, Veeraraghavan N, et al. Launching genomics into the cloud: Deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics. 2014;15:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maheshwari A, Noebels JL.. Monogenic models of absence epilepsy: Windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. Prog Brain Res. 2014;213:223–252. [DOI] [PubMed] [Google Scholar]
- 44. Kommajosyula SP, Randall ME, Brozoski TJ, Odintsov BM, Faingold CL.. Specific subcortical structures are activated during seizure-induced death in a model of sudden unexpected death in epilepsy (SUDEP): A manganese-enhanced magnetic resonance imaging study. Epilepsy Res. 2017;135:87–94. [DOI] [PubMed] [Google Scholar]
- 45. Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL.. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30(15):5167–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Glasscock E, Qian J, Yoo JW, Noebels JL.. Masking epilepsy by combining two epilepsy genes. Nature Neurosci. 2007;10(12):1554–1558. [DOI] [PubMed] [Google Scholar]
- 47. Kalume F, Westenbroek RE, Cheah CS, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123(4):1798–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smart SL, Lopantsev V, Zhang CL, et al. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20(4):809–819. [DOI] [PubMed] [Google Scholar]
- 49. Liu R, Jolas T, Aghajanian G.. Serotonin 5-HT(2) receptors activate local GABA inhibitory inputs to serotonergic neurons of the dorsal raphe nucleus. Brain Res. 2000;873(1):34–45. [DOI] [PubMed] [Google Scholar]
- 50. Bubar MJ, Stutz SJ, Cunningham KA.. 5-HT(2C) receptors localize to dopamine and GABA neurons in the rat mesoaccumbens pathway. PLoS One. 2011;6(6):e20508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Craige CP, Lewandowski S, Kirby LG, Unterwald EM.. Dorsal raphe 5-HT(2C) receptor and GABA networks regulate anxiety produced by cocaine withdrawal. Neuropharmacology. 2015;93:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ledri M, Madsen MG, Nikitidou L, Kirik D, Kokaia M.. Global optogenetic activation of inhibitory interneurons during epileptiform activity. J Neurosci. 2014;34(9):3364–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karczewski KJ, Francioli LC, Tiao G, et al. Genome Aggregation Database Consortium. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. American Journal of Human Genetics. 2009;84(4):524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murugesan A, Rani MRS, Hampson J, et al. Serum serotonin levels in patients with epileptic seizures. Epilepsia. 2018;59(6):e91–e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Murugesan A, Rani MRS, Vilella L, et al. Postictal serotonin levels are associated with peri-ictal apnea. Neurology. 2019;93(15):e1485–e1494.[CVOCROSSCVO] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Petrucci AN, Joyal KG, Purnell BS, Buchanan GF.. Serotonin and sudden unexpected death in epilepsy. Exp Neurol. 2020;325:113145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tupal S, Faingold CL.. Fenfluramine, a serotonin-releasing drug, prevents seizure-induced respiratory arrest and is anticonvulsant in the DBA/1 mouse model of SUDEP. Epilepsia. 2019;60(3):485–494. [DOI] [PubMed] [Google Scholar]
- 59. Lacuey N, Martins R, Vilella L, et al. The association of serotonin reuptake inhibitors and benzodiazepines with ictal central apnea. Epilepsy Behav. 2019;98(Pt A):73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meador KJ. Seizure reduction with fluoxetine in Dravet syndrome. Epilepsy Behav Case Rep. 2014;2:54–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ceulemans B, Boel M, Leyssens K, et al. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia. 2012;53(7):1131–1139. [DOI] [PubMed] [Google Scholar]
- 62. Ceulemans B, Schoonjans AS, Marchau F, Paelinck BP, Lagae L.. Five-year extended follow-up status of 10 patients with Dravet syndrome treated with fenfluramine. Epilepsia. 2016;57(7):e129–e134. [DOI] [PubMed] [Google Scholar]
- 63. Schoonjans A, Paelinck BP, Marchau F, et al. Low-dose fenfluramine significantly reduces seizure frequency in Dravet syndrome: A prospective study of a new cohort of patients. Eur J Neurol. 2017;24(2):309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cheah CS, Yu FH, Westenbroek RE, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A. 2012;109(36):14646–14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kalume F. Sudden unexpected death in Dravet syndrome: Respiratory and other physiological dysfunctions. Respir Physiol Neurobiol. 2013;189(2):324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tecott LH, Logue SF, Wehner JM, Kauer JA.. Perturbed dentate gyrus function in serotonin 5-HT2C receptor mutant mice. Proc Natl Acad Sci U S A. 1998;95(25):15026–15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ochoa-Urrea M, Lacuey N, Vilella L, et al. Seizure clusters, seizure severity markers, and SUDEP risk. Front Neurol. 2021;12:643916.[CVOCROSSCVO] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Griffin A, Hamling KR, Knupp K, Hong S, Lee LP, Baraban SC.. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain. 2017;140(3):669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fitzgerald LW, Burn TC, Brown BS, et al. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol. 2000;57(1):75–81. [PubMed] [Google Scholar]
- 70. Venzi M, David F, Bellet J, et al. Role for serotonin2A (5-HT2A) and 2C (5-HT2C) receptors in experimental absence seizures. Neuropharmacology. 2016;108:292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lhatoo SD, Faulkner HJ, Dembny K, Trippick K, Johnson C, Bird JM.. An electroclinical case-control study of sudden unexpected death in epilepsy. Ann Neurol. 2010;68(6):787–796. [DOI] [PubMed] [Google Scholar]
- 72. Petrucci AN, Joyal KG, Chou JW, Li R, Vencer KM, Buchanan GF.. Post-ictal generalized EEG suppression is reduced by enhancing dorsal raphe serotonergic neurotransmission. Neuroscience. 2021;453:206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Patodia S, Somani A, O’Hare M, et al. The ventrolateral medulla and medullary raphe in sudden unexpected death in epilepsy. Brain. 2018;141(6):1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sveinsson O, Andersson T, Carlsson S, Tomson T.. The incidence of SUDEP: A nationwide population-based cohort study. Neurology. 2017;89(2):170–177. [DOI] [PubMed] [Google Scholar]
- 75. Niibori Y, Lee SJ, Minassian BA, Hampson DR.. Sexually divergent mortality and partial phenotypic rescue after gene therapy in a mouse model of Dravet syndrome. Hum Gene Ther. 2020;31(5-6):339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Scharfman HE, MacLusky NJ.. The influence of gonadal hormones on neuronal excitability, seizures, and epilepsy in the female. Epilepsia. 2006;47(9):1423–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rosecrans JA. Differences in brain area 5-hydroxytryptamine turnover and rearing behavior in rats and mice of both sexes. Eur J Pharmacol. 1970;9(3):379–382. [DOI] [PubMed] [Google Scholar]
- 78. Haleem DJ, Kennett GA, Curzon G.. Hippocampal 5-hydroxytryptamine synthesis is greater in female rats than in males and more decreased by the 5-HT1A agonist 8-OH-DPAT. J Neural Transm Gen Sect. 1990;79(1-2):93–101. [DOI] [PubMed] [Google Scholar]
- 79. Watts AG, Stanley HF.. Indoleamines in the hypothalamus and area of the midbrain raphe nuclei of male and female rats throughout postnatal development. Neuroendocrinology. 1984;38(6):461–466. [DOI] [PubMed] [Google Scholar]
- 80. Carlsson M, Carlsson A.. A regional study of sex differences in rat brain serotonin. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12(1):53–61. [DOI] [PubMed] [Google Scholar]
- 81. Johnson MD, Crowley WR.. Acute effects of estradiol on circulating luteinizing hormone and prolactin concentrations and on serotonin turnover in individual brain nuclei. Endocrinology. 1983;113(6):1935–1941. [DOI] [PubMed] [Google Scholar]
- 82. Carlsson M, Svensson K, Eriksson E, Carlsson A.. Rat brain serotonin: Biochemical and functional evidence for a sex difference. J Neural Transm. 1985;63(3-4):297–313. [DOI] [PubMed] [Google Scholar]
- 83. Ladisich W. Effect of progesterone on regional 5-hydroxytryptamine metabolism in the rat brain. Neuropharmacology. 1974;13(9):877–883. [DOI] [PubMed] [Google Scholar]
- 84. Crowley WR, O'Donohue TL, Muth EA, Jacobowitz DM.. Effects of ovarian hormones on levels of luteinizing hormone in plasma and on serotonin concentrations in discrete brain nuclei. Brain Res Bull. 1979;4(4):571–574. [DOI] [PubMed] [Google Scholar]
- 85. Cone RI, Davis GA, Goy RW.. Effects of ovarian steroids on serotonin metabolism within grossly dissected and microdissected brain regions of the ovariectomized rat. Brain Res Bull. 1981;7(6):639–644. [DOI] [PubMed] [Google Scholar]
- 86. Biegon A, Reches A, Snyder L, McEwen BS.. Serotonergic and noradrenergic receptors in the rat brain: Modulation by chronic exposure to ovarian hormones. Life Sci. 1983;32(17):2015–2021. [DOI] [PubMed] [Google Scholar]
- 87. Di Paolo T, Diagle M, Picard V, Barden N.. Effect of acute and chronic 17 beta-estradiol treatment on serotonin and 5-hydroxyindole acetic acid content of discrete brain nuclei of ovariectomized rat. Exp Brain Res. 1983;51(1):73–76. [DOI] [PubMed] [Google Scholar]
- 88. Liu S, Bubar MJ, Lanfranco MF, Hillman GR, Cunningham KA.. Serotonin2C receptor localization in GABA neurons of the rat medial prefrontal cortex: Implications for understanding the neurobiology of addiction. Neuroscience. 2007;146(4):1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Serrats J, Mengod G, Cortés R.. Expression of serotonin 5-HT2C receptors in GABAergic cells of the anterior raphe nuclei. J Chem Neuroanat. 2005;29(2):83–91. [DOI] [PubMed] [Google Scholar]
- 90. Nocjar C, Alex KD, Sonneborn A, Abbas AI, Roth BL, Pehek EA.. Serotonin-2C and -2a receptor co-expression on cells in the rat medial prefrontal cortex. Neuroscience. 2015;297:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Noebels J. Hippocampal abnormalities and sudden childhood death. Forensic Sci Med Pathol. 2016;12(2):198–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kinney HC, Poduri AH, Cryan JB, et al. Hippocampal formation maldevelopment and sudden unexpected death across the pediatric age spectrum. J Neuropathol Exp Neurol. 2016;75(10):981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kon FC, Vázquez RZ, Lang A, Cohen MC.. Hippocampal abnormalities and seizures: A 16-year single center review of sudden unexpected death in childhood, sudden unexpected death in epilepsy and SIDS. Forensic Sci Med Pathol. 2020;16(3):423–434. [DOI] [PubMed] [Google Scholar]
- 94. Neubauer J, Lecca MR, Russo G, et al. Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases. Eur J Hum Genet. 2017;25(4):404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Brownstein CA, Goldstein RD, Thompson CH, et al. SCN1A variants associated with sudden infant death syndrome. Epilepsia. 2018;59(4):e56–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kinney HC, Haynes RL.. The serotonin brainstem hypothesis for the sudden infant death syndrome. J Neuropathol Exp Neurol. 2019;78(9):765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cummings KJ, Hodges MR.. The serotonergic system and the control of breathing during development. Respir Physiol Neurobiol. 2019;270:103255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All supporting data in this study are available on request.