

Abstract

We report herein a modular class of organic catalysts that, acting as donors, can readily form photoactive electron donor–acceptor (EDA) complexes with a variety of radical precursors. Excitation with visible light generates open-shell intermediates under mild conditions, including nonstabilized carbon radicals and nitrogen-centered radicals. The modular nature of the commercially available xanthogenate and dithiocarbamate anion organocatalysts offers a versatile EDA complex catalytic platform for developing mechanistically distinct radical reactions, encompassing redox-neutral and net-reductive processes. Mechanistic investigations, by means of quantum yield determination, established that a closed catalytic cycle is operational for all of the developed radical processes, highlighting the ability of the organic catalysts to turn over and iteratively drive every catalytic cycle. We also demonstrate how the catalysts’ stability and the method’s high functional group tolerance could be advantageous for the direct radical functionalization of abundant functional groups, including aliphatic carboxylic acids and amines, and for applications in the late-stage elaboration of biorelevant compounds and enantioselective radical catalysis.
Introduction
The photochemistry of electron donor–acceptor (EDA) complexes1 is a powerful approach to generating radicals under mild conditions, providing fresh opportunities in synthetic chemistry.2 The strategy exploits the association of an electron acceptor substrate A and a donor molecule D to form a new molecular aggregate in the ground state (Figure 1a). Although the two components A and D may not absorb visible light themselves, the resulting EDA complex generally does. Visible-light excitation then triggers an intramolecular single-electron transfer (SET), leading to a radical ion pair ([D+•, A–•]). When a suitable leaving group (LG) is present, an irreversible fragmentation productively renders two reactive open-shell intermediates, which can engage in synthetically useful radical processes.2
Figure 1.

(a) Photochemistry of stoichiometric EDA complexes for radical generation. (b) Moving the EDA complex activation strategy into a catalytic regime: previous examples of catalytic donors. (c) A new modular class of donor organocatalysts for catalytic EDA complex photochemistry and their use in radical processes.
EDA complex photochemistry has attracted the interest of chemists because of the ease of operation, the unique ability to use visible light to activate colorless substances, and the possibility of generating radicals without exogenous photoredox catalysts.3 There are, however, aspects that lower the generality and versatility of this radical generation strategy. For example, the most straightforward synthetic application of EDA complex activation is based on the light-driven coupling of two stoichiometric donor and acceptor substrates.4 The moieties of the substrates will eventually end up in the core of the products, thus restricting their structural diversity. Implementing the EDA complex activation strategy within a catalytic regime would significantly expand its efficiency and synthetic applicability.
Moving away from stoichiometric reactivity would require the use of an electron-rich catalyst that could trigger the EDA complex formation upon aggregation with an electron-poor substrate (Figure 1b). A photoinduced SET would then lead to radicals, which could be intercepted by an external trap to form a product. The most problematic yet essential step of this catalytic plan is the effective turnover of the catalyst, which requires SET reduction of the catalyst radical cation, arising from the photoactivity of the progenitor EDA complex. So far, few reported protocols could address this requirement and develop a catalytic EDA complex strategy.5 Our group demonstrated that some chiral organocatalytic intermediates, including enamines,6a−6c iminium ions,6d,6e and enolates,7 could serve as catalytic donors in EDA complex formation to trigger photochemical radical formation while stereoselectively trapping the ensuing open-shell intermediates. One limitation of this approach was the inability to turn over the catalyst: in fact, the EDA complex photoactivity served as an initiation step to feed radicals in a self-propagating chain process.6b−6d Bosque and Bach8 and Stephenson and co-workers9 recently reported different strategies that could effectively turn over catalytic electron-donors. These catalytic platforms were unfortunately limited to the activation of specific radical precursors, which intrinsically narrowed their synthetic applicability and generality. A more flexible EDA complex catalytic approach was disclosed by Shang and Fu and their co-workers, who described how a combination of triphenylphosphine (Ph3P) and sodium iodide (NaI) could catalyze synthetically useful radical reactions.10 This catalytic platform could trigger the formation of photoactive three-component EDA complexes with a variety of radical precursors. However, all these previous strategies were limited to the development of redox-neutral radical processes only.
Herein, we report a general and modular class of electron-donor organocatalysts that, although they cannot absorb visible light themselves, can readily form photoactive EDA complexes with different radical precursors (Figure 1c). Specifically, commercially available dithiocarbamate anion and xanthogenate catalysts A and B can generate a variety of radicals under blue-light excitation, including nonstabilized primary carbon radicals and nitrogen-centered radicals. The modular nature of these organic catalysts allowed us to tune their properties, including stability toward acidic conditions, thus offering a versatile and robust EDA catalytic strategy. This flexibility secured the development of both redox neutral and net-reductive photoinduced radical processes. Mechanistic investigations, by means of quantum yield determination, established that a closed catalytic cycle is operational for all the developed reactions, thus highlighting the ability of the catalysts to turn over and iteratively drive every catalytic cycle. Overall, this organocatalytic system offers a simple, general way to promote a variety of synthetically useful and mechanistically distinct radical processes.11
Results and Discussion
Background and Design Plan
The present study was motivated by our interest in developing photochemical catalytic methods for generating radicals under mild conditions. Recently, we reported that commercially available nucleophilic organic catalysts, including the dithiocarbamate anion catalyst A, adorned with an indole chromophoric unit,12 and potassium ethyl xanthate catalyst B,13 can activate alkyl and acyl electrophiles, including chlorides, via an SN2 pathway (Figure 2a). Visible-light excitation of the resulting photon-absorbing intermediates I afforded radicals upon homolytic cleavage of the weak C–S bond. A merit of this SN2-based catalytic platform is that, by relying solely on the electrophilic properties of the precursors, it could grant access to open-shell intermediates from substrates that would be inert to classical radical-generating strategies. Its underlying mechanism, however, also limited the approach, since only substrates amenable to an SN2 displacement could be used. In addition, only stabilized radicals (including benzyl, allyl, and radicals bearing either a heteroatom or an electron-withdrawing moiety at the α-position) could be effectively generated and used in a variety of C–C12−14 and C–B bond-forming processes.14c
Figure 2.

(a) Our recently developed SN2-based radical generation method using nucleophilic organocatalysts A and B and the key turnover event, based on the reduction of the persistent radical II via either SET or HAT. (b) Translating the potential of catalysts A and B into an EDA complex photoactivation strategy: their use as catalytic donors to generate nonstabilized radicals. RA: redox auxiliary, which drives EDA complex formation and acts as a fragmenting group.
Extensive mechanistic studies13 allowed us to elucidate a crucial aspect of this system, namely, the mechanism of catalyst turnover. Specifically, we found that the sulfur radical II, which emerges from the photolytic cleavage of intermediate I, can dimerize to form III.13,14a Dimer III, which can absorb in the visible region, is in a light-regulated equilibrium with the progenitor sulfur-centered radical II. This dimerization manifold, by conferring a longer lifetime to radical II,15 enables an effective catalyst turnover. We demonstrated that the sulfur-centered radical II, which has a persistent character, can be effectively reduced and turned over by an SET event or by a hydrogen atom transfer (HAT) process.13
The versatile mechanism underpinning catalyst turnover, along with the electron-rich nature of A and B, made us wonder if these organic catalysts could be successfully used as catalytic donors for EDA complex photoactivation (Figure 2b). We were motivated by the following considerations: (i) it is synthetically appealing to develop a general EDA complex catalytic strategy based on commercially available organic catalysts and use it to generate a variety of radicals. (ii) Our understanding of the sulfur-centered radical II behavior, which would be generated upon EDA complex formation and photoinduced SET, may help in the design of photoinduced radical processes. By ensuring that different paths are available for turning over the catalyst, the relative kinetic stability of II could be used to develop mechanistically distinct radical transformations, including net-reductive processes that are not accessible via previously reported EDA complex catalytic platforms.8−10 (iii) Using A and B as catalytic EDA donors would significantly expand the synthetic potential and applicability of this family of organocatalysts beyond the SN2-based catalytic platform.12−14 This is because radical precursors not prone to an SN2 displacement could also become competent substrates. For example, using reaction partners decorated with a purposely installed electron-poor activating group, which serves as both a redox-auxiliary (RA, blue circle in Figure 2b, which triggers EDA complex formation) and leaving group, would allow the generation of previously inaccessible nonstabilized carbon radicals, including primary ones, and nitrogen-centered radicals.
Developing a Net-Reductive Process
To test the feasibility of our EDA complex catalytic strategy, we investigated the reaction of cyclohexyl N-(acyloxy)phthalimide161a and vinyl sulfone 2a catalyzed by the organic catalysts A and B. We selected this process as a testbed because it would require the photochemical formation of a nonstabilized cyclohexyl radical IV, which could not be generated using our previous SN2-based catalytic strategy.12−14 Mechanistically, the resulting Giese-type addition17 of the cyclohexyl radical IV to the electron-poor olefin 2a would require a net-reductive pathway in order to proceed. Figure 3 details the proposed mechanism of the overall process. The ground-state association between the electron-rich donor catalyst (A or B) and the electron-poor substrate 1a would lead to a visible-light-absorbing EDA complex. The formation of the EDA complex is feasible considering the tendency of stoichiometric thiolates and dithiocarbonyl anions to serve as donor partners for EDA complexes.18 A photoinduced SET would then generate the cyclohexyl radical IV along with the sulfur-centered radical II.
Figure 3.

Mechanistic plan for a net-reductive Giese-type addition manifold catalyzed by the excitation of a catalytic EDA complex. NPhth: phthalimide.
Upon interception of radical IV by 2a to forge a new C–C bond, the emerging electrophilic radical V would abstract a hydrogen atom from γ-terpinene (a H donor). This reductive step leads to product 3a and to the cyclohexadienyl radical VI. Overall, this sequence, which requires reduction of both the radical precursor 1a (via an SET) and intermediate V (via HAT), characterizes a net-reductive process. Crucial for catalyst turnover would be the reduction of the dithiocarbonyl radical II (Eox = 0.45–0.75 V vs SCE), which our previous studies established could proceed via an SET event from the cyclohexadienyl radical VI (Ered = −0.1 V vs SCE)12 or via an HAT pathway from γ-terpinene.13 Both reductive steps would eventually close the catalytic cycle by returning the organic catalysts. Importantly, the fact that catalyst turnover can be realized by simply using an external reductant (e.g., γ-terpinene), thus avoiding any specific interaction with a radical intermediate that is a progenitor to the reaction product, increases the versatility of this EDA catalytic system.
We conducted initial experiments reacting substrates 1a and 2a at 40 °C in dimethylacetamide (DMA) using a blue light-emitting diode (LED) strip emitting at 465 nm, γ-terpinene as the H donor (4 equiv), and 10 mol % of the donor catalyst (Table 1). The commercially available indole-containing dithiocarbonyl anion catalyst A and potassium ethyl xanthate catalyst B both provided the target Giese addition product 3a with high chemical yield (entries 1 and 2). Sodium diethyldithiocarbamate C was also a suitable catalyst for this transformation (entry 3). These results established that catalysts with different properties can be used as suitable EDA donors; for example, the dithiocarbonyl catalyst A possesses a higher electron-donor ability than B, as inferred by their redox properties,19 and it is more stable under acidic conditions (see section D1.4 in Supporting Information for details). The modular nature of these catalysts can therefore offer a versatile EDA complex catalytic platform. Further investigations were conducted using the inexpensive catalyst B. Interestingly, the reaction was also promoted by green light (λmax = 520 nm, entry 4), while the presence of air was deleterious for reactivity (entries 5). Control experiments showed the need for light and for the donor catalyst (entries 6 and 7). In addition, the reactivity was completely inhibited (entry 8) in the presence of a radical scavenger (TEMPO; interception of the cyclohexyl radical was observed and results are detailed in section D1.3 of the Supporting Information).
Table 1. Optimization Studiesa.

| entry | catalyst | deviation | yield (%)b |
|---|---|---|---|
| 1 | A | none | 81 |
| 2 | B | none | 95 (86)c |
| 3 | C | none | 85 |
| 4 | B | green LED (520 nm) | 95 |
| 5 | B | under air | 0 |
| 6 | B | no light | 0 |
| 7 | none | none | 0 |
| 8 | B | TEMPO (1.5 equiv) | 0 |
Reactions were performed under inert atmosphere on a 0.1 mmol scale at 40 °C for 16 h under illumination by a blue LED strip (λmax = 465 nm, 14 W) using 1.5 equiv of 2a and 4 equiv of γ-terpinene. Redox potentials of the catalysts were measured in CH3CN vs Ag/AgCl; see section D4 in the SI for details. Cy: cyclohexyl. NPhth: phthalimide.
Yield determined by 1H NMR analysis of the crude mixture using trimethoxybenzene as the internal standard.
Yield of the isolated product 3a.
We then performed additional mechanistically diagnostic investigations. The formation of an EDA complex under the reaction conditions was confirmed through UV/vis spectroscopic analysis (Figure 4). Immediately after mixing catalyst B with the phthalimide ester substrate 1a, the solution developed a marked yellow color, while its optical absorption spectrum showed a bathochromic displacement in the visible spectral region, diagnostic of a new EDA molecular aggregation in the ground state.
Figure 4.

Optical absorption spectra, recorded in DMA in 1 mm path length quartz cuvettes using a Shimadzu 2401PC UV/vis spectrophotometer, and visual appearance of the separate reaction components and of the colored EDA complex between catalyst B and 1a. [1a] = 0.10 M, [B] = 0.01 M.
In addition, we detected the formation of the xanthyl radical IIa by means of laser flash photolysis (Figure 5). Accordingly, when a 1:1 mixture of 1a and catalyst B was excited with a laser beam centered at 355 nm, we observed the formation of a transient species absorbing at 620 nm (half lifetime = 0.1 ± 0.01 ms), consistent with the characteristic line shape of xanthyl radical IIa.13 This observation corroborates the idea that the key event for radical generation is the photoinduced intracomplex SET within the EDA complex, formed between 1a and B.
Figure 5.

Absorption at 620 nm of the transient xanthyl radical IIa generated upon 355 nm laser excitation of a 1:1 mixture of 1a and catalyst B (30 mM) in DMA.
To gain further insight into the mechanism, we measure the quantum yield (Φ) of the overall model reaction of 1a and 2a catalyzed by B, which was as low as 0.01 (λ = 460 nm, using potassium ferrioxalate as the actinometer; see section D.5 in Supporting Information for details). This information, which is not consonant with a radical chain propagation manifold, corroborates the mechanistic scenario depicted in Figure 3: it supports our original plan that the donor B serves as an actual EDA catalyst, since it can effectively turn over while iteratively driving the formation of radicals in every catalytic cycle.
To increase the synthetic utility of our approach, we sought to implement a two-step telescoped sequence to form the redox-active radical precursor 1a in situ and use it without further purification (Scheme 1a). This one-pot procedure granted access to product 3a from readily available cyclohexanecarboxylic acid upon simple treatment with N-(hydroxy)phthalimide (NHPI). In addition, catalyst B proved compatible with a direct domino protocol, where all the reagents were added together at the onset of the reaction (Scheme 1b). These methods add a synthetically useful dimension to the EDA complex catalytic platform, since abundant aliphatic carboxylic acids can be directly functionalized and used as radical precursors without the need to isolate complex phthalimide esters.
Scheme 1. (a) One-Pot Two-Step Telescoped Procedure to Functionalize the Carboxylic Acid and (b) Domino Procedure, Where All Reagents Were Added at the Same Time,

The solvent was evaporated between the two steps.
Yields refer to the isolated product 3a.
Abbreviations: DIC, N,N′-diisopropylcarbodiimide; NHPI, N-(hydroxy)phthalimide; DCM, dichloromethane.
We then used the telescoped procedure to evaluate the scope of the decarboxylative Giese-type addition protocol catalyzed by the EDA donor B (Figure 6). A variety of carboxylic acids could be directly functionalized. Primary (products 3b and 3e), secondary (3c), and tertiary (3d) radicals were generated efficiently and trapped with different electron-poor olefins in good to excellent yields. The protocol tolerated a variety of functional groups. For example, amino acids could be used as radical precursors (adducts 3f–i), while a precursor bearing a chloride substituent selectively reacted at the carboxylic moiety under the optimized conditions (3e). We also demonstrated that this method is suitable for the direct functionalization of biorelevant compounds bearing unprotected polar functional groups. For example, oleanolic acid (3j), which contains a free hydroxyl group, dehydrocholic acid (3k), and biotin (3l) could all be efficiently functionalized. We finally demonstrated that the one-pot domino procedure detailed in Scheme 1b could be applied to directly functionalize complex carboxylic acid substrates (adducts 3g, 3j, and 3l).
Figure 6.
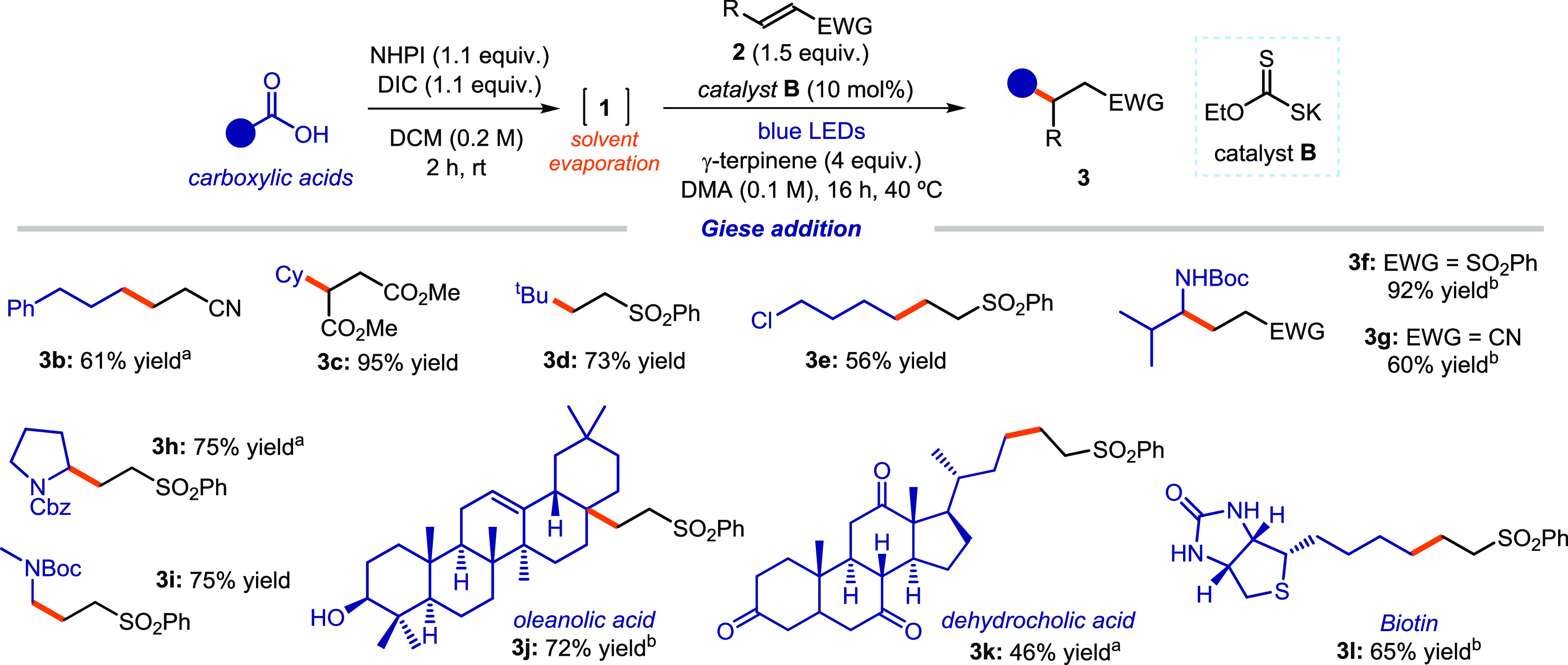
EDA complex catalytic strategy for the generation of alkyl radicals from carboxylic acids and their use in decarboxylative Giese addition processes. Reactions were performed on a 0.2 mmol scale using 1 equiv of acid 1. Yields of products refer to isolated products 3 after purification. The bold orange bond denotes the newly formed C–C bonds. Unless otherwise indicated, all entries were performed using a telescoped sequence without isolation of the phthalimide ester 1 by simply evaporating the solvent (DCM) after completion of the first step. Notes: aUsing the preformed phthalimide ester 1 as the radical precursor. bOne-pot domino procedure according to the conditions in Scheme 1b. Abbreviations: NHPI, N-hydroxyphthalimide; DIC, N,N′-diisopropylcarbodiimide; Cy, cyclohexyl; Pr, propyl; Boc, tert-butyloxycarbonyl; Cbz, carboxybenzyl; Bn, benzyl; Ts, tosyl; EWG, electron-withdrawing group.
To further explore the potential of our photochemical catalytic radical generation method, we investigated the activation of pyridinium salts 4. Substrates 4 are prone to EDA complex formation, acting as acceptors, and can provide alkyl radicals upon SET activation.19 The indole-based dithiocarbamate catalyst A proved more effective than catalyst B for the EDA complex activation of pyridinium salts 4 (see sections D1.2 and D1.4 in Supporting Information for details). This result highlights how the modular nature of these donor catalysts can be leveraged to optimize the activation of electronically different radical precursors.20
We therefore used catalyst A (20 mol %) to trigger the formation of nonstabilized secondary carbon radicals from 4, which were readily intercepted by electron-poor olefins (Figure 7a). This deamination strategy offers a complementary approach to the decarboxylative Giese-type addition protocol, since radical precursors 4 can be readily synthesized from amines. The applicability of the method was also showcased by developing a one-pot telescoped procedure where the primary amine 5 could be directly converted to product 3n through the in situ formation of the corresponding pyridinium salt 4 (Figure 7b). This telescoped procedure did not require any evaporation of the solvent and could be performed by simply adding the reagents sequentially.
Figure 7.

(a) EDA complex catalytic strategy for the deaminative Giese-type addition processes. Note: aProduct 3m was formed in a 3.8:1 ratio with the regioisomeric five-member ring adduct; see the Supporting Information for details. (b) One-pot telescoped procedure for functionalized amines. Reactions were performed on a 0.2 mmol scale.
We then envisaged that the same catalytic protocol used for the Giese-type addition could be successfully translated to perform a Barton decarboxylation process.21 When conducted in the absence of an olefin trap 2, the EDA-complex-based catalytic system would provide an alkyl radical prone to hydrogen atom abstraction (via HAT) from γ-terpinene, delivering the decarboxylation reductive product 6. We demonstrated the feasibility of this idea by applying a one-pot domino process, which allowed the direct reduction of carboxylic acids (Figure 8).
Figure 8.

EDA complex catalysis for the Barton decarboxylation. Reactions were performed on a 0.2 mmol scale using a one-pot domino process. Yields refer to isolated products 6 after purification. The bold orange bond denotes the newly formed bonds. Abbreviations: NHPI, N-hydroxyphthalimide; DIC, N,N′-diisopropylcarbodiimide; Boc, tert-butyloxycarbonyl; Bn, benzyl.
The xanthogenate catalyst B (10 mol %) secured an effective activation of the phthalimide ester substrate generated in situ.20 Primary (adduct 6a), secondary (6b), and tertiary (6c) acids were all competent substrates in this experimentally simple protocol. The Barton decarboxylation has found broad application in total synthesis.22 We therefore tested our methodology in the reduction of complex biologically relevant carboxylic acid-containing molecules, including gibberellic acid (6f) and a baclofen derivative (product 6g). The functionalization of dehydrocholic acid, leading to product 6d, was efficiently performed on a 4 mmol scale, demonstrating that this method is amenable to synthetically useful applications.
Finally, this catalytic process could be extended to include a deaminative reduction path, since pyridinium salts 4 were used as radical precursors, leading to products 6 (Scheme 2). Here, too, the indole-based catalyst A was the donor catalyst of choice for the activation of 4.
Scheme 2. EDA Complex Catalysis for Deaminative Reduction.

Mechanistically, we propose that this process proceeds via a net-reductive manifold resembling the general catalytic cycle depicted in Figure 3. The radical of type IV emerging from the EDA complex photoactivity is quenched by γ-terpinene to afford the reduced product 6, while the dithiocarbonyl radical II can be reduced by a SET (from the cyclohexadienyl radical VI) or HAT manifold (from γ-terpinene). To corroborate this scenario, we measured the quantum yield of the Barton decarboxylation leading to product 6b using the corresponding preformed phthalimide ester radical precursor. The quantum yield Φ was found to be 0.01 (λ = 460 nm, using potassium ferrioxalate as the actinometer). This indicates that a radical-chain process is highly unlikely, confirming the ability of the EDA catalytic donor to turn over and repeatedly trigger radical formation.
Developing a Redox-Neutral Process
The Giese addition and the Barton decarboxylation processes developed so far proceed via a net-reductive mechanism. One of our targets was to identify a versatile EDA complex catalytic platform suitable for the design of radical processes based on mechanistically divergent mechanisms. We considered the radical α-alkylation of silyl enol ethers 7 as a suitable test reaction to implement a redox-neutral process. We envisaged a catalytic cycle (Figure 9) where the excitation of the catalytic EDA complex, formed upon association of the donor catalyst with a radical precursor, such as the pyridinium salts 4, forms the target radical IV. The silyl enol ether 7 would then intercept IV, leading to the α-oxo radical VII. SET between VII and the sulfur-centered radical II, which is stabilized by a dimerization mechanism, would regenerate the EDA catalytic donor and form the oxocarbenium ion VIII, which can hydrolyze to afford the final α-alkylation ketone product 8. The overall sequence requires reduction of the radical precursor 4 and oxidation of intermediate VII, therefore constituting a redox-neutral process. In contrast to previous processes, an effective catalyst turnover would here require an SET between the sulfur radical II and a radical intermediate progenitor of the reaction product VII.
Figure 9.

Mechanistic plan for a redox-neutral transformation catalyzed by the excitation of a catalytic EDA complex.
Our catalytic platform was flexible enough to accommodate this mechanistic requirement. Both EDA catalysts A and B could effectively trigger the photochemical radical alkylation of silyl enol ethers (see section D1.2 in the Supporting Information for a comparison of the catalysts’ performance). Since the indole-based dithiocarbamate catalyst A offered better and more consistent results, it was selected to evaluate the generality of the process (Figure 10). A broad range of pyridinium salts 4 could be used as precursors of electrophilic radicals, which were trapped by 7. Both primary radicals (product 8a), including benzylic ones (8b), and secondary carbon radicals (adducts 8c,d) could be generated and intercepted. The approach displayed a good tolerance toward heterocycles containing nitrogen atoms (adducts 8e,f). We then demonstrated that silyl enol ethers 7 derived from both aliphatic (product 8g) and aromatic ketones could intercept an electrophilic α-ester radical. A variety of substitution patterns on the aryl ring, with different electronic or steric profiles (8h–k), could be easily accommodated. An easily oxidizable heterocyclic substrate (8i) was tolerated. In addition, a substrate derived from azaperone, containing an aminopyridine and a piperazine moiety, could be alkylated in moderate yield (8m). Last, nonstabilized secondary and primary radicals could be effectively generated from phthalimide esters 1 and successfully intercepted, providing products 8n and 8o in moderate yield.23
Figure 10.

Redox-neutral addition of alkyl radicals to silyl enol ethers under EDA complex catalysis. Reactions were performed on a 0.2 mmol scale using 2.0 mL of DMSO. Yields refer to isolated products 8 after purification. The bold orange bond denotes the newly formed C–C bond. Unless otherwise indicated, all entries were performed at 25 °C. Notes: a40 °C; b60 °C; c1:1 mixture of DMSO/DCE used as solvent; ein the absence of water; fusing alkyl N-(acyloxy)phthalimides 1 as radical precursors. TBS: tert-butyldimethylsilyl.
Given the different underlying mechanism of this redox-neutral process with respect to previously studied net-reductive transformations, we considered it pertinent to determine the quantum yield of the reaction leading to product 8h. The very low quantum yield (Φ = 0.02, λ = 460 nm) supports the mechanism depicted in Figure 9, where the EDA donor catalyst A is responsible for the formation of every radical and can effectively turn over by engaging in both a reduction and an oxidation process.
We then set out to develop a three-component reaction under EDA complex catalysis that combined a Giese addition with the radical trap by silyl enol ethers 7 (Figure 11). Specifically, we used phthalimide ester substrates 1 to generate nucleophilic radicals. Capitalizing upon the mismatched polarity between the photogenerated radical and the electron-rich silyl enol ether 7, we first favored the selective radical trap by an electron-poor olefin 2. The electron-deficient secondary radical emerging from this Giese-addition manifold had the right polarity to rapidly react with 7, affording the complex cascade products 9. This cascade sequence, which is reported here for the first time, was efficiently catalyzed by the xanthate catalyst B,20 providing rapid access to structurally complex products 9 from readily available substrates and using an experimentally simple protocol.
Figure 11.

Three-component process under EDA complex catalysis. NPhth: phthalimide.
We then wondered if this EDA complex catalytic platform could be applied to develop another redox-neutral process, namely, the radical C-alkylation of heteroarenes. The Minisci reaction involves the addition of a nucleophilic carbon-centered radical onto a protonated heteroaromatic compound. It is widely used in organic synthesis since it offers a direct way to functionalize heterocycles.24 Different variants of this transformation have been reported using a variety of alkyl radical precursors.25 These methods require a rearomatization of the radical cation IX, generated upon radical addition to the protonated heteroarene, which proceeds via an SET oxidation using either a stoichiometric oxidant or photoredox catalysis (Figure 12). We surmised that our EDA catalysts can first generate the carbon radicals and then oxidize intermediate IX (Ered = −1.01 V vs SCE),26 capitalizing on the kinetic stability of the sulfur-centered radical IIa (Eox = 0.45 V vs SCE, Table 1). The last step would provide the Minisci product while returning the EDA donor catalyst.20
Figure 12.

Common mechanistic pathway in Minisci-type reactions and its integration with our EDA complex catalytic strategy.
This idea was successfully realized using the indole-based catalyst A (10 mol %), which offered a better stability than catalyst B under the acidic conditions20,27 required for the Minisci process (Figure 13). Using preformed substrates 1 as radical precursors, we first explored the scope of the heterocycles amenable to this Minisci-type catalytic protocol. Quinolines (products 11a–c), isoquinolines (11d), and pyridine (11e) derivatives were all competent substrates. Remarkable functional group tolerance was observed, since the reaction conditions tolerate protected amines (11g) and unprotected alcohols (11h–j). To demonstrate the synthetic utility of the method, we successfully performed the alkylation of an intermediate used in the synthesis of the HIF protyl-hydroxylase inhibitor roxadustat (products 11h–i), the anticancer agent camptothecin (11j), and the neuroleptic drug azaperone (11k). Importantly, this catalytic method could be applied for the direct methylation of heterocycles (adducts 11c, 11g, and 11i), a useful process given the unique pharmacokinetic properties inferred by the methyl substituent to medicinally relevant azine derivatives.28 Finally, we demonstrated that a variety of primary, secondary, and tertiary carbon-centered radicals could be generated from phthalimide ester precursors 1 and installed within 2-methylquinoline (products 11l–p). A list of unsuccessful substrates for all the reactions discussed in this study is reported in section C9 of the Supporting Information.
Figure 13.

Photochemical catalytic generation of alkyl radicals and their addition to heterocycles. Reactions were performed on a 0.2 mmol scale using 2.0 mL of DMSO. Yields refer to isolated products 11 after purification. The bold orange bond denotes the newly formed C–C bond. Notes: aperformed in NMP as solvent; b3 equiv of TfOH; cperformed at 60 °C. Abbreviations: Cy, cyclohexyl; Ts, tosyl; NPhth, phthalimide.
We measured the quantum yield of the Minisci reaction leading to product 11b, which was <0.01 (λ = 460 nm, using potassium ferrioxalate as the actinometer; see section D.5 in Supporting Information for details). This experiment further supports the ability of donor A to catalyze the photochemical generation of alkyl radicals while triggering the overall Minisci process in the absence of external oxidants.
Finally, we envisioned that our EDA catalytic platform could be compatible with the enantioselective Minisci protocol recently reported by Phipps and co-workers,29 who used a chiral phosphoric acid [(R)-TRIP, Scheme 3] to direct the stereoselective addition of prochiral radicals, generated using an external iridium-based photocatalyst, to heteroarenes. The EDA donor catalyst A was successfully applied in this asymmetric radical process, affording the chiral products 11q and 11r in high yield and stereocontrol.
Scheme 3. Application in Enantioselective Radical Catalysis.

Abbreviations: Ac, acetyl; NPhth, phthalimide.
Further Synthetic Applications of the EDA Catalytic System
The general applicability of a chemical strategy is a suitable parameter for evaluating its usefulness. To further explore the potential of our EDA catalytic radical generation approach, we investigated the activation of other radical precursors that can form an EDA complex. For example, we found that catalyst A can effectively promote the formation of a trifluoromethyl radical upon EDA complex activation of the Togni reagent 12 (Scheme 4).30 An effective radical trap by silyl enol ether 7a afforded the α-trifluoromethyl ketone product 13.
Scheme 4. Trifluoromethylation of Ketones via EDA Complex Catalysis.

We also used the xanthate catalyst B to generate an amidyl radical upon EDA complex activation of the dinitrophenoxy amide 14 (Scheme 5).31 Radical cyclization afforded the lactam product 15 in high yield.
Scheme 5. Amidyl Radical Formation and Cyclization.

Conclusion
In summary, we have reported a modular class of organic catalysts that, acting as donors, can readily form photoactive electron donor–acceptor (EDA) complexes with a wide variety of radical precursors. Excitation with weak visible light grants access to stabilized and nonstabilized alkyl radicals under mild experimental conditions. The generated radicals were then leveraged to design synthetically useful transformations. The modular nature of the commercially available organocatalysts served to develop mechanistically distinct photoinduced redox-neutral and net-reductive radical transformations. For all the developed processes, we established, by means of quantum yield determination, that a closed catalytic cycle is operational, highlighting the ability of the EDA catalysts to turn over and iteratively drive every catalytic cycle. We also highlighted how the catalysts’ stability and the method’s high functional group tolerance could be advantageous for the direct radical functionalization of abundant functional groups, including aliphatic carboxylic acids and amines, and for applications in the late-stage elaboration of biorelevant compounds and enantioselective radical catalysis. All these features showcase the versatility of this EDA complex catalytic platform, which may be useful for developing further radical processes.
Acknowledgments
Financial support was provided by Agencia Estatal de Investigación (PID2019-106278GB-I00), the AGAUR (Grant 2017 SGR 981), and the European Research Council (ERC-2015-CoG 681840-CATA-LUX). E.P.B. thanks MINECO (CTQ2016-75520-P) for a predoctoral fellowship. W.Z. thanks the China Scholarship Council (CSC201908310093) for a predoctoral fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c05607.
Details of experimental procedures and full characterization data and copies of NMR and HPLC spectra for synthesized compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Foster R. Electron Donor-Acceptor Complexes. J. Phys. Chem. 1980, 84, 2135–2141. 10.1021/j100454a006. [DOI] [Google Scholar]; b Rosokha S. V.; Kochi J. K. Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex. Acc. Chem. Res. 2008, 41, 641–653. 10.1021/ar700256a. [DOI] [PubMed] [Google Scholar]
- Crisenza G. E. M.; Mazzarella D.; Melchiorre P. Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shaw M. H.; Twilton J.; MacMillan D. W. C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wei Y.; Zhou Q.-Q.; Tan F.; Lu L.-Q.; Xiao W.-J. Visible-Light-Driven Organic Photochemical Reactions in the Absence of External Photocatalysts. Synthesis 2019, 51, 3021–3054. 10.1055/s-0037-1611812. [DOI] [Google Scholar]
- Selected synthetic applications of stoichiometric EDA complexes:; a Sankararaman S.; Haney W. A.; Kochi J. K. Annihilation of Aromatic Cation Radicals by Ion-Pair and Radical-Pair Collapse. Unusual Solvent and Salt Effects in the Competition for Aromatic Substitution. J. Am. Chem. Soc. 1987, 109, 7824–7838. 10.1021/ja00259a035. [DOI] [Google Scholar]; b Russell G. A.; Wang K. Homolytic Alkylation of Enamines by Electrophilic Radicals. J. Org. Chem. 1991, 56, 3475–3479. 10.1021/jo00011a007. [DOI] [Google Scholar]; c Tobisu M.; Furukawa T.; Chatani N. Visible Light-mediated Direct Arylation of Arenes and Heteroarenes Using Diaryliodonium Salts in the Presence and Absence of a Photocatalyst. Chem. Lett. 2013, 42, 1203–1205. 10.1246/cl.130547. [DOI] [Google Scholar]; d Kandukuri S. R.; Bahamonde A.; Chatterjee I.; Jurberg I. D.; Escudero-Adán E. C.; Melchiorre P. X-Ray Characterization of an Electron Donor-Acceptor Complex Drives the Photochemical Alkylation of Indoles. Angew. Chem., Int. Ed. 2015, 54, 1485–1489. 10.1002/anie.201409529. [DOI] [PubMed] [Google Scholar]; e Liu B.; Lim C.-H.; Miyake G. M. Visible-Light-Promoted C–S Cross-Coupling via Intermolecular Charge Transfer. J. Am. Chem. Soc. 2017, 139, 13616–13619. 10.1021/jacs.7b07390. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Börgel J.; Tanwar L.; Berger F.; Ritter T. Late-Stage Aromatic C–H Oxygenation. J. Am. Chem. Soc. 2018, 140, 16026–16031. 10.1021/jacs.8b09208. [DOI] [PubMed] [Google Scholar]; g Xie S.; Li D.; Huang H.; Zhang F.; Chen Y. Intermolecular Radical Addition to Ketoacids Enabled by Boron Activation. J. Am. Chem. Soc. 2019, 141, 16237–16242. 10.1021/jacs.9b09099. [DOI] [PubMed] [Google Scholar]; h Lübbesmeyer M.; Mackay E. G.; Raycroft M. A. R.; Elfert J.; Pratt D. A.; Studer A. Base-Promoted C–C Bond Activation Enables Radical Allylation with Homoallylic Alcohols. J. Am. Chem. Soc. 2020, 142, 2609–2616. 10.1021/jacs.9b12343. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Kammer L. M.; Badir S. O.; Hu R.-M.; Molander G. A. Photoactive electron donor–acceptor complex platform for Ni-mediated C(sp3)–C(sp2) bond formation. Chem. Sci. 2021, 12, 5450–5457. 10.1039/D1SC00943E. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Liu Y.-Y.; Yu X.-Y.; Chen J.-R.; Qiao M.-M.; Qi X.; Shi D.-Q.; Xiao W.-J. Visible-Light-Driven Aza-ortho-quinone Methide Generation for the Synthesis of Indoles in a Multicomponent Reaction. Angew. Chem., Int. Ed. 2017, 56, 9527–9531. 10.1002/anie.201704690. [DOI] [PubMed] [Google Scholar]
- For photochemical enzymatic processes that use cofactors as catalytic donors in EDA complexes, see the following:; a Emmanuel M. A.; Greenberg N. R.; Oblinsky D. G.; Hyster T. K. Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 2016, 540, 414–417. 10.1038/nature20569. [DOI] [PubMed] [Google Scholar]; b Clayman P. D.; Hyster T. K. Photoenzymatic Generation of Unstabilized Alkyl Radicals: An Asymmetric Reductive Cyclization. J. Am. Chem. Soc. 2020, 142, 15673–15677. 10.1021/jacs.0c07918. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Page C. G.; Cooper S. J.; DeHovitz J. S.; Oblinsky D. G.; Biegasiewicz K. F.; Antropow A. H.; Armbrust K. W.; Ellis J. M.; Hamann L. G.; Horn E. J.; Oberg K. M.; Scholes G. D.; Hyster T. K. Quaternary Charge-Transfer Complex Enables Photoenzymatic Intermolecular Hydroalkylation of Olefins. J. Am. Chem. Soc. 2021, 143, 97–102. 10.1021/jacs.0c11462. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a distinct EDA complex catalytic approach, see the following:; d Quint V.; Morlet-Savary F.; Lohier J.-F.; Lalevée J.; Gaumont A.-C.; Lakhdar S. Metal-Free, Visible Light-Photocatalyzed Synthesis of Benzo[b]phosphole Oxides: Synthetic and Mechanistic Investigations. J. Am. Chem. Soc. 2016, 138, 7436–7441. 10.1021/jacs.6b04069. [DOI] [PubMed] [Google Scholar]
- a Arceo E.; Jurberg I. D.; Álvarez-Fernández A.; Melchiorre P. Photochemical activity of a key donor-acceptor complex can drive stereoselective catalytic α-alkylation of aldehydes. Nat. Chem. 2013, 5, 750–756. 10.1038/nchem.1727. [DOI] [PubMed] [Google Scholar]; b Bahamonde A.; Melchiorre P. Mechanism of the Stereoselective α-Alkylation of Aldehydes Driven by the Photochemical Activity of Enamines. J. Am. Chem. Soc. 2016, 138, 8019–8030. 10.1021/jacs.6b04871. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Arceo E.; Bahamonde A.; Bergonzini G.; Melchiorre P. Enantioselective direct α-alkylation of cyclic ketones by means of photo-organocatalysis. Chem. Sci. 2014, 5, 2438–2442. 10.1039/c4sc00315b. [DOI] [Google Scholar]; d Cao Z.-Y.; Ghosh T.; Melchiorre P. Enantioselective radical conjugate additions driven by a photoactive intramolecular iminium-ion-based EDA complex. Nat. Commun. 2018, 9, 3274. 10.1038/s41467-018-05375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an example that does not proceed via a radical chain mechanism, see the following:; e Morack T.; Mück-Lichtenfeld C.; Gilmour R. Bioinspired Radical Stetter Reaction: Radical Umpolung Enabled by Ion-Pair Photocatalysis. Angew. Chem., Int. Ed. 2019, 58, 1208–1212. 10.1002/anie.201809601. [DOI] [PubMed] [Google Scholar]
- Woźniak Ł.; Murphy J. J.; Melchiorre P. Photo-organocatalytic Enantioselective Perfluoroalkylation of β-Ketoesters. J. Am. Chem. Soc. 2015, 137, 5678–5681. 10.1021/jacs.5b03243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque I.; Bach T. 3-Acetoxyquinuclidine as Catalyst in Electron Donor-Acceptor Complex-Mediated Reactions Triggered by Visible Light. ACS Catal. 2019, 9, 9103–9109. 10.1021/acscatal.9b01039. [DOI] [Google Scholar]
- McClain E. J.; Monos T. M.; Mori M.; Beatty J. W.; Stephenson C. R. J. Design and Implementation of a Catalytic Electron Donor–Acceptor Complex Platform for Radical Trifluoromethylation and Alkylation. ACS Catal. 2020, 10, 12636–12641. 10.1021/acscatal.0c03837. [DOI] [Google Scholar]
- a Fu M.-C.; Shang R.; Zhao B.; Wang B.; Fu Y. Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide. Science 2019, 363, 1429–1434. 10.1126/science.aav3200. [DOI] [PubMed] [Google Scholar]; b Wang Y.-T.; Fu M.-C.; Zhao B.; Shang R.; Fu Y. Photocatalytic decarboxylative alkenylation of α-amino and α-hydroxy acid-derived redox active esters by NaI/PPh3 catalysis. Chem. Commun. 2020, 56, 2495–2498. 10.1039/C9CC09654J. [DOI] [PubMed] [Google Scholar]; c Fu M.-C.; Wang J. X.; Shang R. Triphenylphosphine-Catalyzed Alkylative Iododecarboxylation with Lithium Iodide under Visible Light. Org. Lett. 2020, 22, 8572–8577. 10.1021/acs.orglett.0c03173. [DOI] [PubMed] [Google Scholar]
- Yan M.; Lo J. C.; Edwards J. T.; Baran P. S. Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer-Chaput B.; Horwitz M. A.; de Pedro Beato E.; Melchiorre P. Photochemical generation of radicals from alkyl electrophiles using a nucleophilic organic catalyst. Nat. Chem. 2019, 11, 129–135. 10.1038/s41557-018-0173-x. [DOI] [PubMed] [Google Scholar]
- de Pedro Beato E.; Mazzarella D.; Balletti M.; Melchiorre P. Photochemical generation of acyl and carbamoyl radicals using a nucleophilic organic catalyst: applications and mechanism thereof. Chem. Sci. 2020, 11, 6312–6324. 10.1039/D0SC02313B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Spinnato D.; Schweitzer-Chaput B.; Goti G.; Ošeka M.; Melchiorre P. A Photochemical Organocatalytic Strategy for the α-Alkylation of Ketones by using Radicals. Angew. Chem., Int. Ed. 2020, 59, 9485–9490. 10.1002/anie.201915814. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cuadros S.; Horwitz M. A.; Schweitzer-Chaput B.; Melchiorre P. A visible-light mediated three-component radical process using dithiocarbamate anion catalysis. Chem. Sci. 2019, 10, 5484–5488. 10.1039/C9SC00833K. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mazzarella D.; Magagnano G.; Schweitzer-Chaput B.; Melchiorre P. Photochemical Organocatalytic Borylation of Alkyl Chlorides, Bromides, and Sulfonates. ACS Catal. 2019, 9, 5876–5880. 10.1021/acscatal.9b01482. [DOI] [Google Scholar]
- a Frenette M.; Aliaga C.; Font-Sanchis E.; Scaiano J. C. Bond Dissociation Energies for Radical Dimers Derived from Highly Stabilized Carbon-Centered Radicals. Org. Lett. 2004, 6, 2579–2582. 10.1021/ol049111j. [DOI] [PubMed] [Google Scholar]; b Leifert D.; Studer A. The Persistent Radical Effect in Organic Synthesis. Angew. Chem., Int. Ed. 2020, 59, 74–108. 10.1002/anie.201903726. [DOI] [PubMed] [Google Scholar]
- a Fawcett A.; Pradeilles J.; Wang Y.; Mutsuga T.; Myers E. L.; Aggarwal V. K. Photoinduced decarboxylative borylation of carboxylic acids. Science 2017, 357, 283–286. 10.1126/science.aan3679. [DOI] [PubMed] [Google Scholar]; b Murarka S. N-Acyloxy)phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal. 2018, 360, 1735–1753. 10.1002/adsc.201701615. [DOI] [Google Scholar]
- a Giese B. Formation of CC Bonds by Addition of Free Radicals to Alkenes. Angew. Chem., Int. Ed. Engl. 1983, 22, 753–764. 10.1002/anie.198307531. [DOI] [Google Scholar]; b Kanegusuku A. L. G.; Roizen J. L. Recent Advances in Photoredox-Mediated Radical Conjugate Addition Reactions: An Expanding Toolkit for the Giese Reaction. Angew. Chem., Int. Ed. 2021, 10.1002/anie.202016666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liu B.; Lim C.-H.; Miyake G. M. Visible-Light-Promoted C–S Cross-Coupling via Intermolecular Charge Transfer. J. Am. Chem. Soc. 2017, 139, 13616–13619. 10.1021/jacs.7b07390. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yang M.; Cao T.; Xu T.; Liao S. Visible-Light-Induced Deaminative Thioesterification of Amino Acid Derived Katritzky Salts via Electron Donor–Acceptor Complex Formation. Org. Lett. 2019, 21, 8673–8678. 10.1021/acs.orglett.9b03284. [DOI] [PubMed] [Google Scholar]; c Li G.; Yan Q.; Gan Z.; Li Q.; Dou X.; Yang D. Photocatalyst-Free Visible-Light-Promoted C(sp2)–S Coupling: A Strategy for the Preparation of S-Aryl Dithiocarbamates. Org. Lett. 2019, 21, 7938–7942. 10.1021/acs.orglett.9b02921. [DOI] [PubMed] [Google Scholar]; d Sundaravelu N.; Nandy A.; Sekar G. Visible Light Mediated Photocatalyst Free C–S Cross Coupling: Domino Synthesis of Thiochromane Derivatives via Photoinduced Electron Transfer. Org. Lett. 2021, 23, 3115–3119. 10.1021/acs.orglett.1c00806. [DOI] [PubMed] [Google Scholar]
- a Wu J.; He L.; Noble A.; Aggarwal V. K. Photoinduced Deaminative Borylation of Alkylamines. J. Am. Chem. Soc. 2018, 140, 10700–10704. 10.1021/jacs.8b07103. [DOI] [PubMed] [Google Scholar]; b Sandfort F.; Strieth-Kalthoff F.; Klauck F. J. R.; James M. J.; Glorius F. Deaminative Borylation of Aliphatic Amines Enabled by Visible Light Excitation of an Electron Donor–Acceptor Complex. Chem. - Eur. J. 2018, 24, 17210–17214. 10.1002/chem.201804246. [DOI] [PubMed] [Google Scholar]; c Wu J.; Grant P. S.; Li X.; Noble A.; Aggarwal V. K. Catalyst-Free Deaminative Functionalizations of Primary Amines by Photoinduced Single-Electron Transfer. Angew. Chem., Int. Ed. 2019, 58, 5697–5701. 10.1002/anie.201814452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In general, the potassium ethyl xanthate catalyst B is a suitable EDA donor catalyst for phthalimide estersubstrates 1. In contrast, the indole-based dithiocarbamate anion catalyst A consistently offers better results in the activation of pyridinium salts 4. This is mainly due to the relative instability of catalyst B toward the acidic environment generated by the formation of the protonated pyridine, which arises upon SET reduction of the pyridinium substrate 4 (see section D1.4 in Supporting Information for details on catalyst stability). The greater stability of catalyst A toward slightly acidic conditions also explains its superior performance in the Minisci reaction discussed in Figure 13
- a Barton D. H. R.; Crich D.; Motherwell W. B. New and Improved Methods for the Radical Decarboxylation of Acids. J. Chem. Soc., Chem. Commun. 1983, 939–941. 10.1039/c39830000939. [DOI] [Google Scholar]; b Barton D. H. R.; Crich D.; Motherwell W. B. The invention of new radical chain reactions. Part VIII. Radical chemistry of thiohydroxamic esters; A new method for the generation of carbon radicals from carboxylic acids. Tetrahedron 1985, 41, 3901–3924. 10.1016/S0040-4020(01)97173-X. [DOI] [Google Scholar]; For a recent application, see the following:; c Qin T.; Malins L. R.; Edwards J. T.; Merchant R. R.; Novak A. J. E.; Zhong J. Z.; Mills R. B.; Yan M.; Yuan C.; Eastgate M. D.; Baran P. S. Nickel-Catalyzed Barton Decarboxylation and Giese Reactions: A Practical Take on Classic Transforms. Angew. Chem., Int. Ed. 2017, 56, 260–265. 10.1002/anie.201609662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ito H.; Takeguchi S.; Kawagishi T.; Iguchi K. Total Synthesis of (±)-Clavubicyclone. Org. Lett. 2006, 8, 4883–4885. 10.1021/ol061947u. [DOI] [PubMed] [Google Scholar]; b Xu Z.; Hu W.; Liu Q.; Zhang L.; Jia Y. Total Synthesis of Clavicipitic Acid and Aurantioclavine: Stereochemistry of Clavicipitic Acid Revisited. J. Org. Chem. 2010, 75, 7626–7635. 10.1021/jo101506c. [DOI] [PubMed] [Google Scholar]
- Kong W.; Yu C.; An H.; Song Q. Photoredox-Catalyzed Decarboxylative Alkylation of Silyl Enol Ethers To Synthesize Functionalized Aryl Alkyl Ketones. Org. Lett. 2018, 20, 349–352. 10.1021/acs.orglett.7b03587. [DOI] [PubMed] [Google Scholar]
- a Minisci F.; Bernardi R.; Bertini F.; Galli R.; Perchinummo M. Nucleophilic character of alkyl radicals—VI: A new convenient selective alkylation of heteroaromatic bases. Tetrahedron 1971, 27, 3575–3580. 10.1016/S0040-4020(01)97768-3. [DOI] [Google Scholar]; b Minisci F.; Fontana F.; Vismara E. Substitutions by nucleophilic free radicals: A new general reaction of heteroaromatic bases. J. Heterocycl. Chem. 1990, 27, 79. 10.1002/jhet.5570270107. [DOI] [Google Scholar]; c Duncton M. A. J. Minisci reactions: Versatile CH-functionalizations for medicinal chemists. MedChemComm 2011, 2, 1135–1161. 10.1039/c1md00134e. [DOI] [Google Scholar]; d Wang W. G.; Wang S. F. Recent Advances in Minisci-type Reactions and Applications in Organic Synthesis. Curr. Org. Chem. 2021, 25, 894–934. 10.2174/1385272824999201230211157. [DOI] [Google Scholar]
- For a review, see:; a Proctor R. S. J.; Phipps R. J. Recent Advances in Minisci-Type Reactions. Angew. Chem., Int. Ed. 2019, 58, 13666–13699. and references therein 10.1002/anie.201900977. [DOI] [PubMed] [Google Scholar]; For selected examples, see:; b Seiple I. B.; Su S.; Rodriguez R. A.; Gianatassio R.; Fujiwara Y.; Sobel L. A.; Baran P. S. Direct C–H Arylation of Electron-Deficient Heterocycles with Arylboronic Acids. J. Am. Chem. Soc. 2010, 132, 13194–13196. 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]; c O’Hara F.; Blackmond D. G.; Baran P. S. Radical-Based Regioselective C–H Functionalization of Electron-Deficient Heteroarenes: Scope, Tunability, and Predictability. J. Am. Chem. Soc. 2013, 135, 12122–12134. 10.1021/ja406223k. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gutiérrez-Bonet A.; Remeur C.; Matsui J. K.; Molander G. A. Late-Stage C–H Alkylation of Heterocycles and 1,4-Quinones via Oxidative Homolysis of 1,4-Dihydropyridines. J. Am. Chem. Soc. 2017, 139, 12251–12258. 10.1021/jacs.7b05899. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Matsui J. K.; Primer D. N.; Molander G. A. Metal-free C-H alkylation of heteroarenes with alkyltrifluoroborates: a general protocol for 1°, 2° and 3° alkylation. Chem. Sci. 2017, 8, 3512–3522. 10.1039/C7SC00283A. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Jin J.; MacMillan D. W. C. Direct α-Arylation of Ethers through the Combination of Photoredox-Mediated C-H Functionalization and the Minisci Reaction. Angew. Chem., Int. Ed. 2015, 54, 1565–1569. 10.1002/anie.201410432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieszczad B.; Perego L. A.; Melchiorre P. Photochemical C–H Hydroxyalkylation of Quinolines and Isoquinolines. Angew. Chem., Int. Ed. 2019, 58, 16878–16883. 10.1002/anie.201910641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeres E.; Debacher N. A.; Sierra M. M. d. S.; Franco J. D.; Schutz A. Mechanisms of Acid Decomposition of Dithiocarbamates. 1. Alkyl Dithiocarbamates. J. Org. Chem. 1998, 63, 1598–1603. 10.1021/jo971869b. [DOI] [Google Scholar]
- Aynetdinova D.; Callens M. C.; Hicks H. B.; Poh C. Y. X.; Shennan B. D. A.; Boyd A. M.; Lim Z. H.; Leitch J. A.; Dixon D. J. Installing the “magic methyl” – C–H methylation in synthesis. Chem. Soc. Rev. 2021, 50, 5517–5563. 10.1039/D0CS00973C. [DOI] [PubMed] [Google Scholar]
- Proctor R. S. J.; Davis H. J.; Phipps R. J. Catalytic enantioselective Minisci-type addition to heteroarenes. Science 2018, 360, 419–422. 10.1126/science.aar6376. [DOI] [PubMed] [Google Scholar]
- a Cheng Y.; Yu S. Hydrotrifluoromethylation of Unactivated Alkenes and Alkynes Enabled by an Electron-Donor–Acceptor Complex of Togni’s Reagent with a Tertiary Amine. Org. Lett. 2016, 18, 2962–2965. 10.1021/acs.orglett.6b01301. [DOI] [PubMed] [Google Scholar]; b Tu H.-Y.; Zhu S.; Qing F.-L.; Chu L. A four-component radical cascade trifluoromethylation reaction of alkenes enabled by an electron-donor–acceptor complex. Chem. Commun. 2018, 54, 12710–12713. 10.1039/C8CC07344A. [DOI] [PubMed] [Google Scholar]
- Davies J.; Svejstrup T. D.; Fernandez Reina D.; Sheikh N. S.; Leonori D. Visible-Light-Mediated Synthesis of Amidyl Radicals: Transition-Metal-Free Hydroamination and N-Arylation Reactions. J. Am. Chem. Soc. 2016, 138, 8092–8095. 10.1021/jacs.6b04920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.