

Abstract

The pathogen Mycobacterium tuberculosis (Mtb), causing tuberculosis disease, features an extraordinary thick cell envelope, rich in Mtb-specific lipids, glycolipids, and glycans. These cell wall components are often directly involved in host–pathogen interaction and recognition, intracellular survival, and virulence. For decades, these mycobacterial natural products have been of great interest for immunology and synthetic chemistry alike, due to their complex molecular structure and the biological functions arising from it. The synthesis of many of these constituents has been achieved and aided the elucidation of their function by utilizing the synthetic material to study Mtb immunology. This review summarizes the synthetic efforts of a quarter century of total synthesis and highlights how the synthesis layed the foundation for immunological studies as well as drove the field of organic synthesis and catalysis to efficiently access these complex natural products.
1. Introduction
The pathogen Mycobacterium tuberculosis (Mtb), causing the tuberculosis (Tb) disease, has been a scourge for mankind since ancient times. As a matter of fact, the oldest confirmed “case” of the disease can be dated back to a Neolithic settlement in the Eastern Mediterranean, 9000 years ago.1 But also other civilizations, such as ancient Egypt, suffered deaths caused by Tb.2 Detection of mycobacterial DNA in a number of Egyptian mummies is evidence for the early “success” of Mtb as a global pathogen.3−5 Throughout the millennia, the cause of this disease was a mystery to humankind until the pioneering, and Nobel Prize winning, work of Robert Koch, who in 1882 found that Mtb is the source of this pest that caused countless fatalities.6 To this day, Mtb continues to be a major global health threat.7 Contrary to any other pathogen, Mtb managed to infect an estimated 1.7 billion people worldwide, which is over 20% of the world population. Every year, approximately 10 million people fall ill from this infectious disease which is therefore classified as a pandemic. Moreover, Tb is the world’s most deadly bacterial disease with a dead toll exceeding 1.5 million annually. Despite the fact that a large part of infected individuals might clear latent infection over time,8 Tb is listed in the World Health Organization top-10 causes of death.
Tb is primarily a lung pathogen (80–85% of the cases) and therefore an airborne disease which is transmittable by coughing; however, extrapulmonary Tb is also frequent.9 These forms of Tb are non-infectious, and most notably manifest themselves in the pleural cavities just outside the lung (tuberculous pleurisy), central nervous system (tuberculous meningitis), genitourinary system (in urogenital tuberculosis), lymphatic system (mycobacterial cervical lymphadenitis), and bones and joints (in Pott’s disease). Extrapulmonary Tb is observed mostly in children and immunocompromised humans, in particular HIV patients.10 Other Tb risk factors are malnutrition, diabetes, and substance abuse (i.e., smoking and alcohol). An HIV/Tb coinfection is particularly worth highlighting since the HIV/Tb synergy dramatically impacts the progression of the Tb disease.7 Tb is an opportunistic infection meaning that the likelihood of HIV patients progressing into active Tb is 18 times(!) higher than those who are not HIV infected, even for those using antiretroviral treatment. In 2019, 8.2% of recorded Tb patients were also diagnosed with HIV, and in the same year 15% of the tuberculosis deaths were ascribed to HIV-infected persons. Therefore, HIV prevention alone can significantly impact attenuation of the Tb pandemic.
Taking into account the aforementioned risk factors, it is not surprising that Tb is mostly prevalent in developing countries, where 95% of the Tb deaths are situated, and thus disproportionately affects the poor. Due to the enormous efforts of raising Tb awareness, prevention, developments in point-of-care diagnostics, and the subsequent rapid treatment, the number of Tb deaths decreased significantly until the year 2000, although the incidence rate has been stable since.11 Despite these encouraging developments in controlling the Tb disease, there is a dramatic rise in multidrug resistant tuberculosis (MDR Tb), meaning the bacilli are resistant to at least both of the first-line antibiotics isoniazid and rifampicin, two of the most powerful anti-Tb drugs.12 From the clinical perspective the patient is practically incurable with a standard first-line treatment meaning the clinician has to resort to the second-line antibiotics (mostly weak in activity and toxic) including the aminoglycosides (e.g. capreomycin, amikacin, and kanamycin), fluoroquinolones (e.g. levofloxacin, moxifloxacin, and ciprofloxacin), and the most recently approved antibiotics bedaquiline (2012), delamanid (2014), and pretomanid (2019). An even worse development is the emergence of extensively drug resistant tuberculosis (XDR Tb) tuberculosis strains which, besides being resistant to isoniazid and rifampicin, are also resistant to fluoroquinolones and at least one of the three second-line injectable drugs (i.e., capreomycin, amikacin, and kanamycin).12 Needless to say, both MDR Tb and XDR Tb complicate treatment and also reduce positive treatment outcomes.13 Surprisingly, in contrast to where tuberculosis causes the most disturbance, these drug-resistant forms of Tb are mostly prevalent in eastern European countries (i.e., Ukraine, Belarus, west Russia, Azerbaijan, Republic of Moldova) and gradually make their way into western Europe.14,15
The “success” of Mtb as the world’s leading pathogen can largely be attributed to its parasitic nature. Mtb is able to persist in its human host up to decades by evading host immune responses and surviving within macrophages (= immune cells). By creating this intracellular niche, the bacterium is largely protected from other immune responses while having access to host nutrients such as lipids and iron.16−22 Moreover, residence in the macrophage also provides partial cloaking from antibiotics.
Another physical protector of Mtb is its thick lipophilic cell wall that provides a fortress against the hostile, bactericidal environment within macrophages as well as against antibiotics, consequently complicating tuberculosis treatment.23 The cell wall consists of a complex array of (glyco)lipids, polysaccharides, and peptidoglycans, which exhibits low permeability of drugs into the mycobacterial cell.24−27 As a result, a typical Tb drug regimen against drug susceptible Tb takes up to one year, whereas treatment of drug resistant forms of Tb can take several years. In both cases a cocktail of antibiotics is administered and patient compliance with the lengthy therapy is of utmost importance for successful treatment.28
Figure 1 depicts a molecular representation of the cell envelope of Mtb, showing an exquisite, and highly complex, architecture buildup of many different layers.
Figure 1.
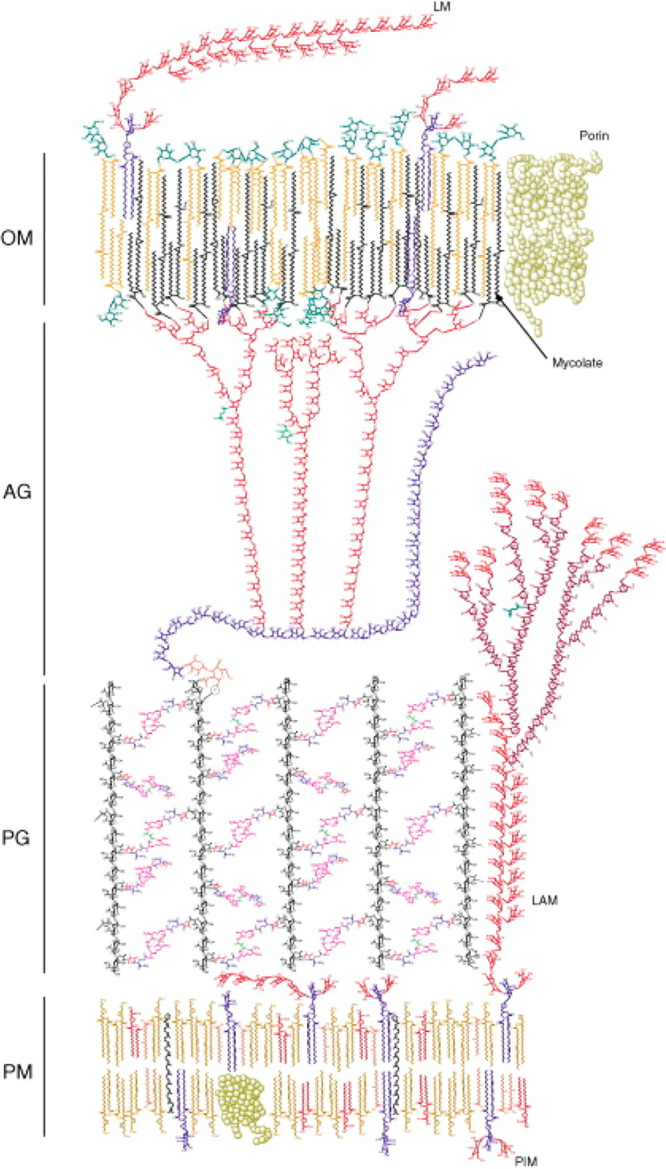
Schematic representation of the cell wall of Mtb (Reproduced with permission from ref (25). Copyright Elsevier 2009).
On the inside of the cell envelope lays the so-called mycobacterial inner membrane or plasma membrane (PM). This lipid bilayer consists primarily of glycerol-based phospholipids, mainly phosphatidylethanolamines (cephalins). Here one also finds phosphatidylinositol mannosides (PIMs),29,30 the first of the many biologically active glycoconjugates in the Mtb cell wall.31 Connected to the plasma membrane is the periplasm, which consists of lipomannan (LM) and lipoarabinomannan (LAM), with the latter being a complex oligosaccharide which is a known virulence factor of tuberculosis.32 Noncovalently bound to the periplasm is the arabinogalactan peptidoglycan (AGP) complex of which the peptidoglycan (PG) provides shape and osmotic stability.33,34 PG is cross-linked with peptide bonds, which gives rigidity to the cell wall. Covalently connected to the PG sugar moieties is the arabinogalactan which is a polymeric saccharide that spans a large part of the overall cell wall and eventually branches out to connect with mycolic acids. The mycolic acids are the major constituent of the mycobacterial outer membrane (MOM), forming the thick lipophilic membrane that is characteristic for mycobacteria. The mycolic acid-based glycolipids form an interwoven network of the long aliphatic (C70–C90) mycolic acid chains that contribute to the fluidity of the cell wall.35 The outer membrane also hosts another wide range of lipophilic molecules such as sulfoglycolipids (SGL), diacyltrehaloses (DAT), polyacyltrehaloses (PAT), phthiocerol dimycocerosates (PDIM), and others. The cell-wall is topped by an outermost compartment, a loosely bound structure called the capsule (not shown in Figure 1) which primarily consists of polysaccharides and peptides.36
The cell wall morphology of Mtb is well understood, and a considerable number of constituents that make up the cell wall are known.24,37 However, it is surprising how little is known about the antigenic properties and other biological functions of the majority of these lipids. Since the mycobacterial cell envelope is at the interface with human host cells,38 its constituents play a key role in Mtb pathogenicity but also in host immune responses.39−45 The identification of new lipids and their exact, molecular, function is therefore instrumental to understand Mtb survival and virulence mechanisms but also to understand basic human immune responses. Moreover, studies on Mtb cell wall components can significantly aid the discovery of new antibiotic targets,43 vaccine adjuvants,46 and biomarkers47−50 for tuberculosis diagnosis.
Throughout evolution, humans acquired two sets of defense strategies against unwanted invaders of the body.51,52 The most primitive one is the innate immune system which is a nonspecific defense mechanism that acts within hours of invasion by the unwanted guest (e.g., pathogens such as bacteria, fungi, viruses).53 Parts that make up the innate immune system are anatomical barriers such as skin and chemicals in the blood, gastrointestinal tract (gut bacteria), and eyes (tears) but also phagocytic cells such as macrophages, neutrophils, and dendritic cells that can devour and digest these foreign invaders.
The second defense mechanism is that of adaptive (i.e., acquired) immunity, which is more complex than the innate immune system.54 The adaptive immune system is a cellular defense mechanism that first has to recognize the foreign invader and process it prior to defending against it. Once recognized, “an army of immune cells” is created that specifically attack the invader. The adaptive immune system is thus an antigen-specific immune response and also includes “memory” that makes future response against the specific invader more efficient. Antibodies are an important part of adaptive immunity.
The adaptive immune response is regulated by a collection of cell surface glycoproteins which is called the major histocompatibility complex (MHC). These proteins are able to bind small peptides (antigens) from pathogens and present these on the cell surface for recognition by T-cells which either kill the foreign invader (killer T-cells) or orchestrate an immune response (helper T-cells).
One other class of cell surface proteins that is able to present antigens to T-cells are CD (cluster of differentiation) proteins. Instead of small peptides, the CD1 protein subclass is able to recognize and present lipids.55,56 This part of the acquired immunity significantly broadens the diversity of molecules recognized by the immune system creating a tighter defense network against foreign invaders. Recognition of lipids by CD1 proteins is particularly interesting in the context of Mtb, since the mycobacterial outer membrane is largely comprised of a wide array of lipids and thus creates a plethora of opportunities for CD1 mediated immunity against Mtb. Throughout the past 30 years a variety of Mtb lipids, of which many are presented in this review, have been shown to be recognized by CD1 proteins.57
Besides recognition by CD1 proteins, mycobacterial cell wall components such as, and most notably, trehalose dimycolate are also recognized by the immune receptor Mincle (macrophage-inducible C-type lectin).58,59 This transmembrane protein is expressed on different immune cells such as macrophages, dendritic cells, and neutrophils. Mincle activation, by binding of extracellular lipids, leads to signaling through FcR-γ and finally activation of the transcription factor NF-κB. Ultimately this signaling pathway induces expression of chemokines, cytokines, and growth factors resulting in an overall pro-inflammatory response. Consequently, as key part of the innate immune response, inflammatory cells move in to the site of activation. Mincle is a potent receptor and unique in its ability to recognize a wide array of low molecular weight (glyco)lipids derived from microorganisms (bacterial and fungal) as well as those from self-damage.60,61 Since its discovery, Mincle and its agonists have received great attention, and Mincle is suggested to be a promising target for the development of subunit vaccines and vaccine adjuvants.62 In particular for Mtb, such a development would mark a scientific breakthrough, as to this date the only used vaccine is the BCG vaccine which shows varying levels of efficacy.63
The discovery of biologically active small molecules (such as lipids) from Mtb and the investigation of their biological, and more specific immunogenic, role is significantly hampered by tedious isolation procedures and, when successful, small isolated quantities. Also, one should not forget that Mtb is a slow growing bacterium, dividing only once every ∼24 h, and also requires culturing in biosafety 3 level laboratories due to its pathogenicity. Synthetic organic chemistry has the great potential to circumvent these problems by total synthesis of sufficient and (stereochemically) pure material. Moreover, an often-underestimated aspect of synthetic efforts is that the chemically synthesized product is void of biological contaminants which might be part of the natural isolate below the limit of detection of (bio)analytical tools. Such impurities, how minute these might be, can be a source of data misinterpretation by causing hard to prove false positive (enhanced bioactivity) or false negative (toxicity) results. Therefore, the pure synthetic material can be viewed as a 'gold standard' in biological evaluations.
Total synthesis of complex natural products can also be used to unequivocally confirm or revise the proposed chemical structure, by comparison of spectroscopic and chromatographic data of natural isolate and synthetic material. It is noteworthy that isolation of these mycobacterial membrane cell wall components only provides small amounts of purified natural product. Thus, researchers often have to rely on collisional HPLC-MS or GC-MS data since isolated quantities are not sufficient for extensive NMR structural analysis. Often this is sufficient to propose the structure, and sometimes also the stereochemistry, but it does not provide ultimate proof of the molecular structure of the isolated material.
Furthermore, development of synthetic routes to prepare natural products opens up possibilities to access various synthetic analogues and non-natural modifications. These can be used as chemical probes or to gain insight into structure–activity relationships which will serve to ultimately expand the fundamental molecular understanding of the biological and immunological processes in question.
Besides aiding biological and immunological investigations, and as we will see, many Mtb cell wall components exhibit an exquisite molecular architecture which forms a playground for synthetic chemists to develop and showcase novel methodology. In this review, we discuss a quarter century of stereoselective total syntheses of natural products from Mtb (from 1995 up to June 2021) focusing on cell wall components and pathogen-shed lipids. The emphasis lies on the construction of complete natural products of Mtb and fragments thereof, not simplified analogues, highlighting the stereoinducing steps. We will also discuss the stereoselective synthesis of complex optically pure constituents, of lipidic nature, of the natural products. Additionally, where applicable, a brief description on how the synthetic material was used in biological or immunological studies to address fundamental questions on virulence mechanisms and Mtb immunology is provided. The membrane components discussed contain lipids, such as mycolic acids, phthiocerol-based lipids, 1,3-multimethyl-branched lipids and their trehalose glycolipids, lipopeptides, terpene nucleosides, as well as oligo- and polysaccharides.
2. Mycolic Acids
An important class of mycobacterial cell envelope lipids comprises the mycolic acids, which make up for a large part of the cell membrane of Mtb. Mycolic acids were isolated by Anderson64 nearly 100 years ago, but it was in the 1960s that the overall structure of these fatty acids was elucidated.65−67 Mycolic acids are long-chain α-alkyl-β-hydroxy fatty acids, which show considerable structural diversity with respect to their functional groups and carbon chain length, depending on the mycobacterial species and strain.68 Mycolic acids occur as free acids as well as esterified to the arabinogalactan layer of the cell envelope. Furthermore, mycolate esters of trehalose, trehalose mono- and dimycolate (TMM and TDM, respectively) and glycerol monomycolate (GroMM) are produced by Mtb as well.35 A special case of mycolate esters is glucose monomycolate (GMM). Mycobacteria do not produce GMM by themselves; however, when M. smegmatis, M. Phlei, and M. Avium were cultured in the presence of glucose, their total lipid extracts exhibited a GMM-specific T cell response of LDN5.69 Moreover, crude sonicates of M. Leprae (leprosy causing baceterium), isolated from infected armadillo liver, also showed stimulation of LDN5 indicating a mammalian source of glucose suffices in GMM biosynthesis. From these experiments it was concluded that exogenous glucose is needed to produce GMM. Thus, GMM is a 'biochimeric molecule'70 in which mycobacteria-produced mycolate is coupled with host-produced glucose.69
Depending on the functional groups present in their main chain, mycolic acids are divided into three categories (Figure 2).71 α-Mycolic acids are the most abundant group of mycolic acids and form around 60% of the total isolatable mycolic acids from Mtb depending on the bacterial strain.68 They contain two strictly cis-configured cyclopropyl groups, of unknown absolute configuration, and their alkyl chains range from 74 to 80 carbons. The other two classes of mycobacterial mycolic acids comprise methoxy- and keto-mycolic acids. These contain α-methyl-methoxy and α-methyl-keto moieties distal, and cyclopropyl groups proximal, to the carboxylic acid moiety. Contrary to the α-mycolic acids the cyclopropyl groups found in keto- and methoxy-mycolic acids can be both cis- or trans-configured. The trans-cyclopropyl mycolic acids additionally contain an α-methyl group.66
Figure 2.

Classification of mycolic acids based on their functional groups.
The (R,R)-configuration of the α-alkyl-β-hydroxy carboxylic acid unit has been shown to be crucial for T-cell recognition of mycolic acids when presented by CD1b.69 Furthermore, the stereochemistry of the distal α-methyl-methoxy unit is S,S, as proven by synthesis of both diastereomeric forms followed by comparison of their optical rotation with the natural product.67,72,73 The stereochemistry is relevant as it influences the binding of the mycolic acid to CD1b.73 To date, the absolute stereochemistry of the cis-cyclopropyl group has not been indisputably elucidated, although evidence from CD1b binding studies points toward the (R,S)-configuration.73
Mycolic acids have been found to exhibit various biological functions.74 Mycolic acids form a tight hydrophobic barrier (called mycomembrane), which serves as physical protection and fortification against the harsh environment within macrophages as well as antibiotics due to its low permeability.75,76 Mycolic acids are essential for Mtb viability, a feature that is exploited in treatment of Tb. Enzymes involved in the biosynthesis of mycolic acids are targets of small molecule inhibitors77 such as the well-established first-line drug isoniazid78−80 and the more recently developed drugs delamanid81 and pretomanid.82 Furthermore, mycolic acids are linked to Mtb virulence83 and play a role in host–pathogen interactions. Free mycolic acids induce host innate immune responses such as alveolar macrophage activation and differentiation to foamy macrophages (=lipid-laden macrophages).84,85 Mycolic acids also act as antigens in serological assays,86 and mycolic acids and their carbohydrate esters are recognized by T-cells.69,87−89 Mycolates and synthetic analogues thereof are therefore of interest in the field of vaccine (adjuvant) development and have been identified as potent activators of the immune receptor Mincle.58,90
To aid such immunological studies, most notably, the Prandi laboratory developed efficient semisynthesis of glycerol and glucose monomycolates by connecting the glycerol and glucose precursors with mycolic acid isolates.90−92 Although the synthetic products, which are a mixture of mycolate esters, were of great importance in the immunological studies, in this review we focus on total syntheses of well-defined chemical structures.
2.1. α-Mycolic Acid
The first asymmetric synthesis of an α-mycolic acid as its methyl ester was reported by the group of Baird.93,94 The synthesis was initialized by the conversion of meso-methyl ester 1 into meso-diester 2 (Scheme 1). This molecule was subjected to enzymatic desymmetrization using porcine pancreas lipase type II providing alcohol 3 in 94% and >99 ee.95 Further elaboration set the stage for a hydrogenation of the rather sensitive allyl cyclopropane 4, which was carried out with diimide, to avoid ring opening, delivering 5. A set of straightforward transformations to extend the carbon chain gave rise to building block 6.
Scheme 1. Synthesis of Methyl α-Mycolate Building Blocks 6 and 10.

A second building block 10 was synthesized from enantiopure epoxide 7 by regioselective ring-opening to furnish chiral alcohol 8. Silylation, hydrogenolysis followed by RuCl3·H2O oxidation of the alcohol, and subsequent esterification/deprotection gave diol 9, which was converted into 10 using a five steps sequence involving protection, α-alkylation, acetylation, deprotection, and oxidation.
To complete the synthesis of the protected α-mycolic acid, another cyclopropane fragment 11 had to be constructed which could then be joined with 6 and 10 (Scheme 2). A Mitsunobu/oxidation sequence initiated on enantiopure alcohol 3 gave sulfone 11. Next, a Julia–Kocienski olefination of 11 with 6 and two subsequent reductions gave alcohol 12 in 33% over the three steps. 12 was then reacted to give the appropriate sulfone for another Julia–Kocienski olefination, but then with building block 10. A diimide reduction provided the acetate protected α-mycolic acid methyl ester, the end-stage of the synthesis.
Scheme 2. Completion of the Synthesis of Methyl α-Mycolate.

The synthetic material was compared to isolated α-mycolic acid (a mixture of homologues with 13 as the predominant compound) after esterification by means of 1H and 13C NMR. The spectra of natural and synthetic materials were found to be virtually identical, but it was also recognized that in this case NMR analysis does not provide ultimate proof of structure, in particular of the absolute and relative stereochemistry of the cyclopropane rings. The specific rotation of synthetic and natural protected α-mycolic acid was found to be very similar as well, but it has to be noted that the rotation is likely determined by the β-acetoxy ester part. Therefore, no conclusion on the absolute stereochemistry of the cyclopropane moieties could be drawn. MALDI- and ESI-MS spectra of synthetic methyl α-mycolate corresponded to those of the natural isolate. Taken together, this total synthesis confirmed the overall, gross, molecular structure of naturally occurring α-mycolic acid.
2.2. cis- and trans-Configured Keto- and Methoxy-mycolic Acids
In 2007, the Baird laboratory reported the first total synthesis of two enantiomers 55 and 57 (with regard to the methoxy methyl motif) of methoxy-mycolic acid as well as two enantiomers 42 and 44 (with regard to the cis-cyclopropane motif) of methyl methoxy-mycolate.72 The synthesis strategy is based on the preparation of various aldehyde- and sulfone-equipped fragments and their subsequent connection through a Julia–Kocienski olefination/reduction sequence in analogy to their previous synthesis of α-mycolic acid.94
The synthesis commenced with the preparation of two enantiomeric methoxy methyl building blocks 24 and 25 (Scheme 3). Synthesis of 25 was initiated with the conversion of L-gulono-1,4-lactone to α,β-unsaturated ester 14 through an one-pot acetal protection, oxidative cleavage, and esterification sequence. Diastereoselective conjugate addition of methyllithium followed by LiAlH4 reduction to the alcohol and subsequent oxidation gave aldehyde 15 in 53% over three steps. The enantiomeric aldehyde 17 was accessed in two steps from ethyl ester 16 (prepared from D-mannitol). Julia–Kocienski olefination of 17 with sulfone 18 followed by double bond reduction provided acetonide 19 in 87% over two steps, which after four more synthetic steps led to THP ether 21. Deprotection of the THP moiety followed by PCC oxidation furnished aldehyde 22, which was subjected to another Julia–Kocienski olefination/reduction sequence with sulfone 23, which completed the synthesis of the methoxy methyl intermediate 24. The enantiomeric 25 was prepared from aldehyde 15 in analogy to 24.
Scheme 3. Baird’s Methoxy-Mycolic Acid Building Block Synthesis Part 1.

Next, the two methoxy methyl building blocks 24 and 25 were used to prepare three stereoisomeric cis-cyclopropyl building blocks 33, 35, and 40 (Scheme 4).
Scheme 4. Baird’s Methoxy-Mycolic Acid Building Block Synthesis Part 2.

Cyclopropanecarboxaldehyde 26 was subjected to a Julia–Kocienski olefination with sulfone 27 giving alkene 28 in 78% yield. Further synthetic steps provided cyclopropanecarboxaldehyde 29, which underwent Julia–Kocienski olefination with sulfone 30 (generated from 24) followed by alkene reduction to give bromide 31. Subsequent treatment of 31 with thiol 32 under basic conditions furnished thioether 33. The diastereomeric thiol 35 was prepared in analogy using aldehyde 29 and sulfone 34 (prepared from 25) in 63% yield over three steps. The synthesis of the (S,S)-cyclopropyl intermediate 40 was initiated by the synthesis of sulfone 37 in two steps from alcohol 36. Julia–Kocienski olefination of 37 with 6-bromohexanal and subsequent double bond reduction with trisyl hydrazide provided bromide 38 in 59% over two steps. A series of synthetic transformations then delivered bromide 39, which was subjected to nucleophilic substitution with thiol 32 to yield thioether intermediate 40.
Next, sulfones 41 and 43 (prepared from 33 and 40) were subjected to another sequence of Julia–Kocienski olefination with aldehyde 10 and diimide double bond reduction to give the diastereomeric protected methoxy-mycolic acid methyl esters 42 and 44 in 56% and 54% yield, respectively (Scheme 5). In order to also access the free mycolic acids, aldehyde building block 52 carrying a silyl protected β-hydroxy ester moiety was prepared (Scheme 5). Starting from iodide 45, a series of steps provided aldehyde 46, which was subjected to a Wittig reaction followed by Sharpless asymmetric dihydroxylation to give diol 47 in 82% over two steps. The α-hydroxyl of 47 was removed by preparing a cyclic sulfate followed by its regioselective reduction to give alcohol 48 in 63% yield over two steps. Stereoselective alkylation of allyl iodide with the enolate generated from 48 and subsequent silyl protection furnished alkene 49 in 50% yield over two steps. Further synthetic steps then provided pivaloyl ester 51, which was hydrolyzed and oxidized to the desired aldehyde building block 52.
Scheme 5. Baird’s Protected Methyl Methoxy-Mycolate Assembly and Building Block Synthesis Part 3.

In the final assembly (Scheme 6), Julia–Kocienski olefination of sulfone 53 or 41 with aldehyde 52 followed by diimide reduction provided 54 and 56 in 68% and 62% yield over two steps, respectively. Lastly, 54 and 56 were both subjected to desilylation and ester hydrolysis to give the two diastereomeric methoxy-mycolic acids 55 and 57.
Scheme 6. Baird’s Methoxy-Mycolic Acid Assembly.

Four mycolic acid methyl esters 42, 44, 54, and 56 were used to assess the proposed stereochemistry of the methoxy methyl motif by comparison of the optical rotation with the methyl ester of the natural sample ([α]D22 −0.1). Acetyl protected mycolic acid methyl esters 42 and 44 showed [α]D +7.2 and +7.7, respectively, indicating that in these cases the methoxy methyl motif is opposite to that in the natural mycolic acid. Methyl esters 56 and 54 exhibited [α]D22 values of +6.0 and −1.0, respectively. This led to the conclusion that 54 has the same stereochemistry in the methoxy methyl motif as natural mycolic acid, which is in agreement with the stereochemistry inferred by Asselineau in 1970.67
Following the total synthesis of α- and methoxy-mycolic acid, the Baird laboratory communicated the synthesis of a cis-cyclopropyl containing keto mycolic acid96 as well as keto- and methoxy-mycolic acids bearing trans-cyclopropyl units.97 In the total syntheses of these mycolic acids (Figure 3), the same synthetic strategy was followed as in their previous synthesis of methoxy-mycolic acid,72 involving various Julia–Kocienski olefination/reduction sequences for the coupling of the fragments.
Figure 3.

Mycolic acid representatives synthesized by the Baird laboratory.
The preparation of various stereoisomers of α-, keto-, and methoxy-mycolic acids72,94,96,97 containing cis- or trans-cyclopropyl motifs aided the understanding of the structure–activity relationship of antibody binding to mycolic acids.86 Antibody binding using ELISA (enzyme-linked immunosorbent assay) showed that the methyl esters of the tested synthetic mycolic acids possess no antigenicity, whereas the free mycolic acids show different levels of antibody binding depending on the type of functional groups. It was found that synthetic methoxy-mycolic acid binds the strongest followed by hydroxy-, keto-, and α-mycolic acids. Furthermore, the stereochemistry of the methoxy methyl fragment was shown to significantly influence the binding ability of the synthetic mycolic acids. The tested natural and synthetic mycolic acids were found to bind to antibodies in sera from Tb positive and negative patients. Yet surprisingly, no mycolic acid was capable of reliably discriminating between Tb positive or negative sera. It remains unknown as to why the tested synthetic mycolic acids showed cross-reactivity, yet the lipophilic (or amphiphilic) nature of the tested lipids could be a source for these results.
Furthermore, the synthetic mycolic acids prepared in the Baird laboratory have been assessed for their role in airway immune responses in mouse pulmonary inflammation models.98 Methoxy-mycolic acids were found to be inflammatory and to activate alveolar macrophages. Keto-mycolic acid exerted opposite effects, being anti-inflammatory and suppressing inflammatory response. In contrast, α-mycolic acids exhibited no inflammatory effects, indicating that they are not involved in promoting or suppressing the innate immune responses of the host. On the other hand, the oxygenated mycolic acid species (keto and methoxy) seem to possess different regulatory roles in the inflammatory responses of the host. These results hint toward the fact that Mtb is able to balance host immune responses by altering the expression levels of different oxygenated mycolic acid structures, as suggested previously.99
The Baird laboratory also synthesized a range of different glycerol monomycolates (GroMM) from Mtb.100 with both (R)- and (S)-configuration on the glycerol unit (Figure 4). Starting from commercially available (R)- or (S)-solketal, a total of nine different GroMMs 61–69 were prepared. These were then evaluated for their ability to induce cytokine production in bone marrow-derived dendritic cells, yet none of the synthetic GroMMs displayed any effect. Besides that, in a follow-up report the GroMMs were shown to selectively induce CD1b-restricted germline-encoded mycolyl lipid-reactive (GEM) T-cell responses, similar to the response caused by free mycolic acids.101
Figure 4.

Synthetic (R)- and (S)-GroMM prepared by the Baird group.
2.3. Methoxy-mycolic Acid
The most recent total synthesis of four diastereoisomers 92a–d of methoxy-mycolic acid was communicated by our laboratory.73 In order to access the target mycolic acids, a new synthetic strategy was developed involving the synthesis of three main fragments (A–C). For the assembly of the fragments to the desired mycolic acids, a Suzuki–Fu cross-coupling was incorporated in the synthesis to increase yield and to reduce the step count of the total synthesis. Two enantiomers of fragment B and C and the natural enantiomer of fragment A were prepared to access the four diastereomeric mycolic acids 92a–d.
The synthesis of fragment A was initiated by the DIBAL-H reduction of lactone 70 followed by Wittig reaction providing α,β-unsaturated thioester 72 (Scheme 7). Subjecting 72 to a Fukuyama reduction produced saturated aldehyde 73 in 82% yield, notably with in situ protection of the free hydroxyl as its TES silyl ether. Next, chiral auxiliary 74 was esterified with the acyl bromide derived from 10-bromodecanoic acid delivering ester 75 in 94% yield. With 73 and 75 in hand, an Abiko–Masamune aldol reaction was performed, constructing the desired α-alkyl β-hydroxy-ester 76 in 55% yield with a dr of 97:3. Silyl protection of 76 followed by a Pd-catalyzed Suzuki–Fu cross-coupling with 1-hexadecene provided 77. Selective silyl deprotection of the primary TBS ether followed by Dess–Martin oxidation concluded the synthesis of fragment A in 91% yield over two steps.
Scheme 7. Minnaard’s Methoxy-Mycolic Acid Fragment A and B Synthesis.

The synthesis of fragment B started with an alkyne-zipper reaction of 78 and subsequent silyl protection of the primary hydroxyl giving terminal alkyne 79 (Scheme 7). Next, 79 was deprotonated and treated with paraformaldehyde, and the product was subjected to a P-2 nickel reduction providing the desired cis allylic alcohol 80 in 51% yield over two steps. Charette asymmetric cyclopropanation of 80 yielded the desired cyclopropane 82 in 95% ee, which was protected as pivaloyl ester, desilylated, and brominated giving fragment B in 87% yield over four steps. The other enantiomer of fragment B was synthesized in analogy.
The synthesis of fragment C was initiated with a diastereoselective conjugate addition of methyl lithium to 83 producing ester 84 (Scheme 8). Reduction of the ester moiety with LiAlH4 to the corresponding alcohol followed by Appel bromination provided bromide 85. Next, Suzuki–Fu cross-coupling between bromide 85 and 1-hexadecene furnished 86 in good yield. Acetonide deprotection of 86 and subsequent tosylation of the primary alcohol under basic conditions gave epoxide 87. The epoxide moiety of 87 was then opened by reaction with Grignard reagent 88 in the presence of copper(I) followed by methylation of the alcohol providing fragment C in 81% yield over two steps. The enantiomer of fragment C was synthesized in the same manner. Notably all fragments were synthesized on (multi)gram scale, highlighting the efficiency and scalability of the synthetic route.
Scheme 8. Minnaard’s Methoxy-mycolic Acid Fragment C Synthesis and Fragment Unification.

With all required fragments in hand, the final assembly was initiated by Suzuki–Fu cross-coupling of fragment C with fragment B (Scheme 8). After pivaloyl deprotection, the desired coupling product 89 was obtained in 78% yield over two steps. Next, the hydroxyl group of 89 was converted into the corresponding sulfone 90 by Misunobu reaction with 32 and oxidation. Sulfone 90 was then subjected to a Julia–Kocienski olefination with fragment A furnishing alkene 91 in 90% yield. The desired methoxy-mycolic acid 92a was accessed from 91 by a three-step sequence. This strategy enabled the preparation of four methoxy-mycolic acid diastereomers 92a–d (Scheme 9). In addition to the free mycolic acids, four diastereomers of the corresponding 6′-glucose- and methyl esters 93a–d and 94a–d were prepared for biological evaluation and comparison of optical rotations (Scheme 9).
Scheme 9. Minnaard’s Methoxy-mycolic Acid Derivatization.

The four synthetic mycolic acid diastereomers enabled confirmation of the stereochemical assignment of the methoxy methyl moiety. This was achieved by comparison of the specific molar rotation, and the stereochemistry of the methoxy methyl unit was confirmed to be (S,S), as inferred by Asselineau and Baird (vide supra).67,94 The synthetic glucose monomycolates 93a–d were tested for their T-cell antigenicity. It was found that all four diastereomers were able to activate LDN5 T-cell lines in the presence of CD1b-expressing antigen presenting cells in a dose dependent manner. Furthermore, the free mycolic acids showed the ability to activate T-cells. Differences in T-cell response dependent on the stereochemistry of the methoxy methyl unit were observed, with the (S,S)-isomer 93a and 93d being the most potent over the natural and remaining synthetic mycolic acids. CD1b-tetramer staining experiments performed with synthetic glucose monomycolates 93a–d showed minor differences dependent on the stereochemistry of the methoxy methyl moiety. Mycolates 93b and 93c with the natural (S,S)-configuration showed the strongest interaction between T-cell receptor and CD1b-mycolate, but no difference in interaction was observed regarding the cyclopropyl stereochemistry. However, in the case of free mycolic acids, significant differences based on the methoxy methyl stereochemistry were observed. Furthermore, mycolic acids 92a and 92d with the (R,S)-configuration on the cyclopropane ring show stronger interaction than the corresponding (S,R)-configured mycolic acids 92b and 92c. Assuming that the mycolic acids with the natural cyclopropane stereochemistry exert the highest T-cell receptor affinity, these results allowed the authors to propose that the naturally occurring stereochemistry of the cis-cyclopropane is (R,S).
3. β-D-Mannosyl Phosphomycoketide
Mannosyl phosphomycoketide (MPM) is a glycolipid antigen from Mtb that was found to be presented by CD1c. In 2000 Moody and co-workers reported the isolation of this molecule from Mtb and M. avium.102 MPM was postulated to be presented by CD1c by binding the alkyl chain within a hydrophobic groove. The moiety presented and recognized by the T-cell receptor is the hydrophilic mannosyl phosphate functionality. Interestingly, it was shown that the T-cell response was dependent on the chain length of the aliphatic moiety in MPM, with an optimum around C35. Also, the hydrophilic headgroup was shown to be crucial for the T-cell response as glucose, instead of mannose, was not recognized.
The structure of MPM was elucidated by mass spectrometry since only minute quantities of MPM were isolated. As a consequence, the assignment of the stereochemical elements present in the MPM chain and anomeric center remained unsolved. To elucidate the stereochemistry of MPM, a total synthesis was required followed by biological assays. In 2002, Crich and Dudkin set out to unravel the stereochemistry of the anomeric center of MPM by performing a stereorandom total synthesis of the mycoketide chain but stereoselectively synthesizing the glycosidic bond with both α- and β-stereochemisty.103 The anomeric stereocenter was found to be a β-glycosidic bond by comparison of the MS fragmentation pattern with that in the original report. Despite these efforts, however, the stereochemistry of the methyl branches in the alkyl chain remained unknown.
In 2006, Minnaard and Feringa communicated the first asymmetric total synthesis of MPM (Schemes 10 and 11).104,105 At the start of the synthesis it was hypothesized that the biosynthesis of the mycoketide proceeds through an iterative sequence in which the methyl groups are introduced all-syn by polyketide synthase pks12.106 Arbitrarily, all-(S)-MPM 115 was chosen as the target.
Scheme 10. β-Mannosyl Phosphomycoketide Building Block Synthesis.

Scheme 11. Completion of β-Mannosyl Phosphomycoketide.

The total synthesis started with dienone 95 which was prepared by means of a double IBX oxidation of cyclooctanone (Scheme 10).107 Dienone 95 was used for the introduction of the first two stereogenic centers via sequential conjugate addition reactions of dimethylzinc. The first conjugate addition, in which a relatively high catalyst loading of 5 mol% of Cu(OTf)2/10 mol% of phosphoramidite L1 was used to avoid Michael addition of the resulting zinc enolate, proceeded in an excellent ee of >99%, yielding compound 96 in 85% yield. The second asymmetric conjugate addition was performed using half the catalyst loading and using ent-L1. The enolate produced after conjugate addition was trapped in situ as its TMS enol ether 97 and subsequently converted into alcohol 98 using an ozonolysis-esterification protocol. The four-step sequence from 96 to 98 was performed with an overall yield of 45% and an excellent de of 98% and ee exceeding 99%. Alcohol 98 was subjected to silyl protection and ester reduction to provide building block 99. Alcohol 99 was converted to the corresponding sulfone 90 by Mitsunobu reaction with tetrazole 32 followed by oxidation with m-CPBA in excellent yield. Furthermore, alcohol 99 was tosylated and then used in a Grignard alkylation in the presence of copper(I) to give silyl ether 102 in 83% over two steps. After desilylation of 91 and Ley–Griffith oxidation, the corresponding aldehyde was coupled to sulfone 101 by means of Julia–Kocienski olefination. Alkene 103 was obtained in 53% over three steps.
To complete the mycoketide chain, a fifth methyl-branched stereocenter had to be introduced (Scheme 11). This was realized by constructing sulfone building block 109 from 1,4-butanediol derived aldehyde 104. This molecule was converted into pure (E)-α,β-unsaturated thioester 105 by performing a Wittig olefination (E/Z = 9:1) and subsequent DMAP-catalyzed isomerization. The fifth, and final, stereocenter of the MPM chain was introduced by means of an asymmetric copper-catalyzed conjugate addition with methylmagnesium bromide. The combination of 5 mol % of CuBr·SMe2 with Josiphos ligand L2 provided 106 in 92% yield with 93% ee. The thioester of 106 was fully reduced by LiAlH4 and further functionalized to the sulfone 109. A Julia–Kocienski olefination of the aldehyde generated from 103 and 109 was performed which produced, after orthogonal benzyl group hydrogenolysis and alkene hydrogenation, mycoketide 110 in a respectable 54% over the four steps.
The total synthesis of MPM was completed by connecting the mycoketide chain to the mannose headgroup (Scheme 11). This was realized by reacting 111 with diphenyl chlorophosphate to give predominantly the β-anomer of mannosyl phosphate 112 in 79% yield. The phenyl substituents were removed using Adams’ catalyst which, after quenching with pyridine, afforded pyridinium mannosyl phosphate 113. This fragment was reacted with mycoketide 110 to install the glycosidic bond with β-selectivity giving 114. Removal of the acetate groups of the mannose concluded the first asymmetric total synthesis of all-(S)-β-MPM 115.
Having achieved synthesis of milligram quantities of all-(S)-MPM 115, the relative and absolute stereochemistry could be scrutinized in biological assays. Together with MPM, a set of analogues (not shown) were synthesized to perform a structure–activity relationship in order to find out what determines antigen binding by the T-cells. The synthetic compounds were tested and compared to natural isolate in an IL-2 release assay by subjecting CD1c-presenting cells to the MPMs.108 It was found that all-(S)-MPM 115 exhibited a similar level of IL-2 release upon T-cell activation compared to the natural isolate. Notably, the stereorandom mixture of MPMs was significantly less potent. Also, when subjecting simplified MPM analogs with only a stereogenic C4-methyl (Scheme 11) to the T-cell activation assay, the molecule with (S)-stereochemistry showed much stronger activity than the (R)-isomer. These results thus showed that T-cell activation is highly sensitive to the stereochemistry of the MPM chain. In addition, synthetic α-MPM, phosphomycoketide (i.e., MPM without mannose), and mycoketide 110 were investigated, all showing no IL-2 release. All in all, as a result of the synthetic efforts and following biological evaluation, the overall structure of MPM was determined to have all-(S) stereochemistry for the methyl groups. The synthetic material was used to investigate the antigenicity of β-mannosyl phosphomycoketide. It was demonstrated that 115 is recognized by CD1c and that the lipid-branching pattern and (S)-stereochemistry were crucial for recognition.109
The second total synthesis of all-(S)-β-mannosyl phosphomycoketide 115 was reported by the Piccirilli group (Scheme 12).110 Whereas in our synthesis asymmetric catalysis stood central, the Piccirilli laboratory mined from the chiral pool. Commercial (S)-Roche ester was used to construct sulfone 118 (11 steps) and sulfone 122 (13 steps from 119). The sulfone building blocks came together in the assembly, by means of Julia–Kocienski olefinations, furnishing eventually MPM 115 with very high stereopurity (>96%). One other difference to our approach was the multiple reductions of the formed double bonds. Three diimide mediated reductions were employed as an alternative to our final Pd-catalyzed hydrogenation/debenzylation reaction. This methodology excludes potential epimerization of the nearby stereocenters, whereas the Pd-catalyst can cause this phenomenon by an isomerization/hydrogenation mechanism.
Scheme 12. Piccirilli’s Synthesis of β-Mannosyl Phosphomycoketide.

The MPM, as well as phosphomycoketide, synthesized was shared with immunologists who used it to establish the molecular basis of presentation by CD1c and its recognition by αβ T cells.111 A cocrystal structure of phosphomycoketide in CD1c was obtained and provided molecular insight in antigen binding and presenting to αβ T cells.
Very recently the Minnaard lab executed a more streamlined and efficient total synthesis of the mycoketide lipid (Scheme 13).112 When carefully examining the mycoketide structure, one can envision the potential of geranylgeraniol 125 as a retrosynthetic precursor. Although geranylgeraniol is commercially available, its price (85 EUR/100 mg) limited its use for the development of a new total synthesis. This problem was overcome by isolation of geranylgeraniol from commercially available annatto seeds (Bixa orellana). Ten kilogram (∼200 euro) of seeds was extracted with heptane followed by fractional distillation and column chromatography, affording 35 g of pure geranylgeraniol 125. Initial investigations into a one-step syn-selective asymmetric reduction of the double bonds of 125 using Pfaltz catalyst C2 showed that the reduction of the alkene proximal to the hydroxyl functionality had an inferior diastereoselectivity. To obtain maximum selectivity, albeit at the cost of the number of steps, the first stereocenter was introduced using a Noyori asymmetric hydrogenation with 0.3 mol % of catalyst C1, providing alcohol 126 in an excellent 94% yield with 98% ee. The alcohol was protected whereafter the terminal alkene was epoxidized, affording 127 in 47% over the three steps. The epoxide was then hydrolyzed to the vicinal diol as well as cleaved with periodic acid, with the resulting aldehyde subjected to a Horner–Wadworth–Emmons olefination to afford ester 128. After removal of the protecting group and oxidation to the aldehyde, an additional stereocenter was introduced by means of a Julia–Kocienski olefination with stereochemical pure sulfone 129, leaving the ester untouched. The ester was reduced, and compound 130 was subjected to an asymmetric hydrogenation with 2 mol% of Pfaltz catalyst C2. The stereocenters were introduced with excellent diastereoselectivities, producing all-syn alcohol 131 in 98% yield. Mycoketide 110 was then crafted out of this building block by a four-step sequence. This mycoketide total synthesis was completed with a longest linear sequence of 15 steps in 16% overall yield, which is a doubling of the overall yield compared to the previous 17 step synthesis which proceeded with 8% overall yield.
Scheme 13. Minnaard’s Second Synthesis of Mycoketide.

4. 1,3-Methyl-Branched Lipids
The mycobacterial cell envelope accommodates a variety of complex lipids and in particular a large number of 1,3-methyl-branched lipids and glycolipids in the outer membrane.44,113 These methyl-branched lipids form a tight, hydrophobic barrier by interaction with the covalently attached mycolic acid layer ultimately resulting in very low permeability of toxic molecules such as antibiotics.75,114,115 Apart from forming a thick physical barrier, these lipids are involved in receptor-mediated uptake by macrophages as well as modulation of the host immune response.17,40,116 Consequently, the 1,3-methyl-branched mycobacterial (glyco)lipids are of great interest to gain further insight into the immunology and pathophysiology of Mtb. Besides that, there has been growing interest by the organic chemistry community in constructing these chiral 1,3-methyl units with a high degree of stereocontrol and synthetic efficiency. The following section describes the synthesis of various 1,3-methyl-branched mycobacterial cell wall components and their use in biological studies. Furthermore, the accomplishments in synthetic method development are highlighted.
4.1. Catalytic Asymmetric Deoxypropionate Synthesis
After the synthesis of MPM, bearing a chiral 1,5-methyl array, our group gained interest in other Mtb lipids with repeating chiral 1,3-methyl units.113 Of particular interest are the complex glycolipids diacylated sulfoglycolipid (Ac2SGL) and its “big brother” sulfolipid-1 (SL-1) (vide infra). These natural products contain lipid chains with up to eight repeating methyl groups, which is challenging to construct in an enantio- and diastereoselective fashion. Also, somewhat smaller molecules with shorter 1,3-methyl arrays were of synthetic and biological interest, which among others included PDIM-A, mycoside B, and diacyl trehaloses.
Since the deoxypropionate functionality is a repeating 1,3-methyl unit, at the time, an iterative synthesis seemed especially appealing. One iterative asymmetric synthesis of deoxypropinates that was used in several syntheses of Mtb lipids is based on the copper-catalyzed asymmetric conjugate addition (Cu-cat. ACA) of methylmagnesium bromide to α,β-unsaturated thioesters, by Feringa and Minnaard in 2005.117 The synthetic precursor for the iterative sequence is α,β-unsaturated thioester 135 (Scheme 14). A Cu-cat. ACA with methyl Grignard, using Josiphos L2 as the chiral ligand, proceeded in 94% yield and an excellent 98% ee. Thioester 136 was reduced to aldehyde 137 by DIBAL-H (or initially Fukuyama reduction), which was subjected to a Horner–Wadsworth–Emmons (HWE) olefination to provide α,β-unsaturated thioester 139 in high yields over the two steps. This sequence can be repeated until the desired number of methyl groups is installed. An attractive feature of this strategy is that the synthetic steps are easy to execute and high yielding, and the sequence proved to be highly stereoselective as each consecutive methyl introduction led to increased diastereoselectivity. An obvious downside is the lack of convergence.
Scheme 14. General Iterative Deoxypropionate Synthesis Strategy Used in the Construction of Mtb Lipids118.

4.2. Phthiocerol Dimycocerosate A
The first Mtb lipid synthesized using the aforementioned iterative asymmetric conjugate addition protocol was phthiocerol dimycocerosate A (PDIM A).119 In 1999 two independent studies found that PDIM A is only present in virulent mycobacteria and that mutant strains deficient in PDIM A showed attenuated virulence.120,121 This finding thus suggested an important role for PDIM A as a virulence factor although its exact biological role remained elusive. In a study by Jackson and co-workers in 2004, it was found that PDIM production protects Mtb in its initial in vivo growth phase by protecting it from nitric oxide dependent killing mechanisms by macrophages and modulating the early immune response to infection.122 10 years later, in connection to this finding, the Ramakrishnan laboratory reported that PDIM acts as a protective cloak for pathogen-associated molecular patterns (PAMPs).123 These PAMPs can signal a Toll-like receptor dependent recruitment of microbicidal macrophages that produces reactive nitrogen species. This helps Mtb to evade these microbicidal macrophages but infect the permissive ones. PDIM has thus been linked to an immune-evasion mechanism.
In 2001 the Guilhot laboratory showed that transposon mutants unable to synthesize or translocate PDIM A exhibit higher cell wall permeability and thus play a role in cell wall envelope architecture.124 In further work from Guilhot and co-workers, it was demonstrated that PDIM is transferred from the Mtb cell wall into macrophage membranes to confer a lipid organization to modulate phagocytosis and infect macrophages.125 Interestingly this biological function was attributed to a specific molecular conformation in which PDIM adopts a conical shape that inserts itself in the two opposing lipid bilayers of macrophage membranes.126 This work, thus, shows that molecular shape confers biological activity and potentially explains previous work from the Briken lab, who revealed that the levels of PDIM A are in direct correlation with the ability of Mtb to escape the host phagosome and induce host cell necrosis.127 Ramakrishnan and co-workers showed that this is likely caused by enhancing phagosomal permeabilization and that PDIM causes membrane damage,128 which might be attributed to the conformation of PDIM.
Preceding the discovery of this intriguing biology, the structure and absolute configuration of the phthiocerol and mycocerosic acid chains were meticulously determined by the Polgar laboratory, by means of chemical degradation, in a series of publications ranging from 1954 to 1973.129−133 Confirmation of the structure was provided by Guilhot and co-workers using MALDI and 1H NMR analysis.124 Despite the fact that these analyses were performed with care and did not contain any obvious flaws, misinterpretation of NMR data of complex lipids is easily made. The best way to confirm molecular structure is by means of chemical synthesis of a well-defined structure, which our group embarked upon by performing an asymmetric total synthesis of PDIM A (Schemes 15 and 16).
Scheme 15. Asymmetric Synthesis of Mycocerosic Acid by Cu-cat. ACA.

Scheme 16. Asymmetric Total Synthesis of PDIM A.

PDIM A consists of two parts: phthiocerol 153 which contains four stereogenic centers and mycocerosic acid 144 being the deoxypropionate moiety with four all-syn stereogenic methyl groups. Mycocerosic acid 144 was synthesized by the copper-catalyzed iterative conjugate addition sequence (vide supra).134 The synthesis started from α,β-unsaturated thioester 135, and the four stereogenic methyl groups were installed providing thioester 143 in 15% yield over 11 steps and in excellent de >98% (Scheme 15). The thioester moiety of 143 was then reduced to the alcohol, followed by tosylation, Grignard alkylation, deprotection, and oxidation gave mycocerosic acid 144 in 55% yield over five steps.
Synthesis of the stereogenic methyl branch with a vicinal methoxy stereocenter of the phtiocerol moiety started with an asymmetric conjugate addition of Me2Zn to cycloheptenone 145 in the presence of Cu(OTf)2 and phosphoramidite L3 (Scheme 16). The enolate intermediate was trapped in situ with ethyl iodide to provide 146 with 95% ee and >20:1 de. The functionalized cycloheptanone 146 was ring-opened through a Baeyer–Villiger oxidation/hydrolysis sequence, which after three subsequent transformations provided aldehyde 147 in 43% over the five steps. The first stereogenic secondary alcohol was introduced by means of an asymmetric 1,2-addition of 2-methyl-3-butyn-2-ol to aldehyde 147, employing (+)-N,N-dimethylephedrine 148 as the chiral ligand, to produce propargylic alcohol 149 with an excellent selectivity of 95% de. A four-step sequence converted 149 into alkyne 150, which was hydrosilylated to yield vinyl silane 151 in 86%. The vinyl silane was oxidized to ketone 152 by means of a Tamao–Fleming oxidation which was diastereoselectively reduced with tetramethylammonium triacetoxyborohydride to yield phthiocerol 153.
Ultimately, the hydroxy groups of the anti-1,3-diol 153 were esterified with mycocerosic acid 144 to produce the target molecule, PDIM A 154. The NMR data and MALDI spectrum of the synthetic material were compared with the natural isolate and matched perfectly, thereby providing absolute confirmation of its proposed chemical structure. The synthetic material also aided in the development of an analytical procedure to detect PDIM A in Mtb samples.135
In 2016 the Hosokawa laboratory reported the second stereoselective total synthesis of PDIM A.136 The synthesis of mycocerosic acid 144 started with a vinylogous Mukaiyama alkylation of TBS enolate 155 and bisbenzyloxymethane (Scheme 17).136 Stereoinduction was efficiently achieved producing acyloxazolidinone 156 in 71% yield with an excellent dr > 20:1. Reductive auxiliary cleavage followed by iodination provided alkylating agent 157. This reagent was used in another vinylogous alkylation with vinylogous TBS enol ether 155, providing 158 in 59% over three steps, again with a diastereomeric ratio exceeding 20:1. The benzyl protecting group in 158 was removed by a Birch reduction, which also reduced the α,β-unsaturation to form the corresponding enolate. A diastereoselective protonation was performed with methylbenzimidazole as the proton source. More conventional proton sources (e.g., NH4Cl and BHT) were shown to provide product 159 with poor diastereoselectivities (dr ∼ 2:1), but methylbenzimidazole proved to be the reagent of choice, yielding 159 in 81% with a dr > 20:1. The last methyl-branched stereocenter was installed by means of a diastereoselective hydrogenation with 15 mol% Schrock–Osbourne catalyst, again providing an excellent diastereoselectivity (dr > 20:1), to furnish all-syn compound 160. The synthesis of mycocerosic acid 144 was completed in four additional steps.
Scheme 17. Hosokawa Total Synthesis of Mycocerosic Acid.

For the synthesis of PDIM A 154, phthiocerol 153 had to be constructed (Scheme 18).137 This time a vinylogous Mukaiyama aldol reaction was performed by reaction of propionaldehyde with vinylogous TBS enol ether 155. The desired vinylogous aldol product 161 was isolated in near quantitative yield with a dr > 20:1. After methylation of the free alcohol, the chiral auxiliary was removed by means of ozonolysis, followed by a reductive workup. The intermediary alcohol was then converted into a good leaving group and substituted with an acetylene group providing 162 in 69% yield over five steps. The alkyne functionality of 162 was coupled with C2-symmetric bis-epoxide 164 (prepared from diol 163, TPsO = triisopropylbenzenesulfonate) to provide compound 165. The epoxide was opened with an alkyl nucleophile, and the triple bond was completely reduced to produce the desired phthiocerol 153. As for the final step of the total synthesis, phthiocerol was esterified with mycocerosic acid 144 to give PDIM A 154 in its second total synthesis to date.
Scheme 18. Stereoselective Total Synthesis of PDIM A by the Hosokawa Laboratory.

4.3. Phenolic Glycolipid
A molecule bearing structural similarities to PDIM A is phenolic glycolipid, PGL-tb1. Whereas PDIM A is purely lipidic, PGL-tb1 is essentially PDIM A linked to a phenol bearing a trisaccharide on the para-position. The first report about a PGL-tb1 came from the hands of Hunter and Brennan in 1981 in which they reported the isolation of PGL-tb1 from an M. leprae infected armadillo liver.138 The structure was elucidated in following reports,139,140 but more importantly, PGL-tb1 was shown to be an antigen binding antibodies in both rabbit and human blood sera.139
Although phenol-phthiocerol monosaccharides were known to be present in M. bovis since 1963,141 and later found to be present in M. marinum(142) and M. kansasii,143 no phthiocerol-based phenolic glycolipids were reported for Mtb. In 1989 this changed as the Daffé laboratory managed to show the existence of PGL-tb1 in the outer layer of the cell envelope in the Canetti strain of Mtb. A beautiful combination of chemical degradation with mass spectrometry and NMR analysis showed that PGL-tb1 from Mtb deviated in the trisaccharide part of the molecule from that of M. leprae,144,145 making the molecule somewhat species-specific and therefore a potential Mtb antigen.
Despite having isolated PGL-tb1 for the first time from Mtb, Daffé and co-workers did not investigate the biological role of PGL-tb1. Yet, in 2004 it was shown that PGL-tb1 plays a major role in infection by conferring a “hyper lethal” phenotype, in murine disease models, in Mtb isolates belonging to the W-Beijing family.146 This phenotype was shown to be the result of inhibiting the release of key inflammatory effector molecules (i.e., pro-inflammatory cytokines, tumor-necrosis factor-a, and interleukins 6 and 12) by cells of the host’s innate immune response. Knockout of the pks1-15 gene, which is responsible for PGL-tb1 biosynthesis, resulted in loss of this hypervirulent phenotype.
The hypervirulent phenotype was further investigated by Sinsimer et al., and they showed the biological activity of PGL-tb1 is not caused by the molecule itself.147Mtb H37Rv (lab adapted strain), which does not produce PGL-tb1, was transformed to produce PGL-tb1 and subjected to in vitro immunoassays. Also, here, suppressed induction of several pro- and anti-inflammatory cytokines in human monocytes was observed; however, the production of PGL-tb1 did lead to increased virulence in mice and rabbits compared to the wild-type H37Rv strain. It was therefore concluded that PGL-tb1 probably acts in synergy with other strain dependent factors. Still, PGL-tb1 is regarded as a virulence factor of Mtb.148
A recent study in zebrafish addressed the role of PGL-tb1 in Mtb macrophage escapement. It was uncovered that the STING cytosolic sensing pathway in macrophages is activated by PGL-tb1, leading to chemokine production and recruitment of monocytes toward the site of infection. The resulting fusion of infected macrophages with monocytes enables transfer and further spreading of the bacterium. Thus, interruption of this PGL-tb1 dependent process could prove beneficial for clearing of Mtb infection.149
The fact that PGL-tb1 can act as a target antigen for tuberculosis diagnosis was shown by the development of an ELISA based on PGL-tb1.150 Since PGL-tb1 is produced in specific strains, in relatively small quantities, its isolation is tedious. Chemically synthesized PGL-tb1 can replace isolation procedures and assist the development of diagnostic tests and other biological assays.
In 2012 our group reported the first, and up to date only, asymmetric synthesis of PGL-tb1 (Schemes 19–21).151 The synthesis started with the preparation of aldehyde 147 which was also used in the synthesis of PDIM A (Scheme 16).119 The aldehyde was converted into keto-ester 167 which was stereoselectively hydrogenated using Noyori’s catalyst with an excellent diastereoselectivity of >99% in 76% yield providing 168 (Scheme 19). After installation of this third stereocenter, the ester functionality in 147 was transformed into Weinreb amide 169. Compound 169 was alkylated with the alkyllithium reagent generated from compound 171. The β-hydroxy ketone functionality formed by alkylation was then reduced to the 1,3-anti diol using the diastereoselective Saksena–Evans reduction using NH4BH(OAc)3. Deprotection of the alkyne provided key phthiocerol building block 172.
Scheme 19. Asymmetric Synthesis of the Phthiocerol Backbone in PGL-tb1.

Scheme 21. Completion of the Total Synthesis of PGL-tb1.

The trisaccharide building block 179 was constructed by conversion of L-rhamnose into glycoside acceptor 173 (Scheme 20). Glycosylation of 173 with glycosyl donor 174, using N-thiophenyl-ε-caprolactam and Tf2O in a tri-tert-butyl pyrimidine (TTBP) buffered solution, provided the α-linked dirhamnoside 175 with full stereocontrol in 71% yield. After a three-step deprotection–protection sequence, the fucose functionality (177) was introduced to yield trisaccharide 179 after two subsequent steps (deacetylation and methylation).
Scheme 20. Construction of the Trisaccharide Building Block for the Total Synthesis of PGL-tb1.

Unification of the phthiocerol backbone 172 with trisaccharide 179 was achieved using a carefully optimized Sonagashira coupling, providing the desired cross-coupled product 180 in 51% yield, recovering 35% of precious phthiocerol building block 172 (Scheme 21). The mycocerosic acid esters were introduced followed by a benzylether hydrogenolysis to deprotect the trisaccharide and reduce the alkyne functionality. This concluded the first asymmetric total synthesis of PGL-tb1 181.
4.4. Phthioceranic and Hydroxyphthioceranic Acid
After the successful synthesis of mycocerosic acid and PGL-tb1, our group became interested in the synthesis of the even more complex lipids phthioceranic and hydroxyphthioceranic acid, which carry a total of seven to eight methyl branches as 1,3-syn arrays and an additional hydroxyl stereocenter. These two mycobacterial lipids are found as trehalose esters in the cell wall glycolipids Ac2SGL and SL-1. By utilizing the in house developed Cu-cat. ACA strategy, phthioceranic acid Phthioceranic acid183 was first synthesized in 2007 by ter Horst et al.118 The seven methyl units were installed in a linear fashion giving 182 in 8% yield over 18 steps (Scheme 22). With thioester 182 in hand, a five-step sequence gave phthioceranic acid in 61% yield over these steps.
Scheme 22. Asymmetric Total Synthesis of Phthioceranic Acid.

The hydroxyphthioceranic acid lipid was synthesized using two different approaches as part of the total synthesis of Ac2SGL (vide infra).152 In the first-generation synthesis (Scheme 23) the chiral hydroxyl moiety was constructed first using a copper/taniaphos L4-catalyzed allylic alkylation of ester 184 with C15H31MgBr producing desired olefin 185 in 76% yield with an excellent ee of >98%. Cyclization, by means of a ring-closing metathesis reaction, provided lactone 186 which was diastereoselectively methylated using a Gillman reaction to produce 187 in 94% yield. The α,β-unsaturated thioester 188 was synthesized in five steps from 187, which set the stage for the iterative introduction of the remaining seven methyl groups. The stereogenic TBDPS protected alcohol in 188 proved not to be of influence on the asymmetric conjugate addition outcome as the methyl group was installed succesfully providing 189 in 91% yield with de of 98%. Six iterations were performed to eventually provide deoxypropionate 190 in an 14% overall yield over the 18 steps from α,β-unsaturated thioester 188. The thioester moiety in 190 was reacted with methyl lithium cuprate to yield the corresponding methyl ketone 191, which in five steps was converted into hydroxyphthioceranic methyl ester 192. Since the relative stereochemistry of the hydroxy functionality, compared to the chiral methyl ramification, was unknown, an NMR comparison with natural hydroxyphthioceranic acid was performed which showed the hydroxyl function had opposite stereochemistry. This inconveniency was readily corrected by inversion of stereochemistry using a Mitsunobu reaction. The obtained p-nitrobenzoyl ester 193 was transesterified using MeOH and catalytic NaCN, affording hydroxyphthioceranic acid methyl ester 194 with the naturally occurring stereochemistry in place.
Scheme 23. First Generation Asymmetric Synthesis of Hydroxyphthioceranic Acid Methyl Ester.

Critically reviewing our synthetic efforts, it was realized that the first-generation synthesis lacks the flexibility to also access phthioceranic acid from this route, which is required for the total synthesis of SL-1, the “big brother” of Ac2SGL (vide infra). This is caused by first introducing the stereogenic hydroxyl functionality, which preferably is introduced in a late stage so that one route can provide both lipids. In addition, as the stereochemistry of this hydroxy group had been elucidated, the steps required for stereoinversion should be avoided. We achieved these goals (Scheme 24)152 by synthesis of compound 195, the synthetic precursor to phthioceranic acid (Scheme 22), which was converted into allylic bromide 196 in four steps. Subjecting 196 to an asymmetric allylic alkylation with 7 mol% of CuBr·SMe2 and 8 mol% of (R,RFe)-taniaphos L4 in the presence of MeMgBr, octamethyl alkene 197 was obtained in 88% yield with an excellent diastereomeric excess of >95%. The alkene functionality served as an excellent scaffold for the introduction of the stereogenic hydroxyl group. The most obvious reaction to do so, meanwhile also creating a functional group on the terminal carbon for introduction of the alkyl chain, is a Sharpless asymmetric dihydroxylation. Despite producing the desired product 198, this reaction unfortunately provided a moderate diastereomeric excess of 70%.153 This problem was eventually solved by performing an asymmetric diborylation reaction, which after oxidation provided the desired diol 198. So, alkene 197 was reacted with bis(pinacolato)diboron (B2pin2) catalyzed by Pt2(dba)3 and taddol-based ligand L5. The diborylated product was oxidized to provide the desired diol 198 in 98% yield with a diastereomeric excess >95%. Having all stereocenters set, the aliphatic side chain and carboxylic acid functionality were installed to yield protected hydroxyphthioceranic acid 199 in 38% over six steps.
Scheme 24. Second Generation Asymmetric Synthesis of Protected Hydroxyphthioceranic Acid.

Since their first reported total syntheses118,152 phthioceranic and hydroxyphthioceranic acids have attracted a lot of attention. Over the past years, due to their exquisite and synthetically challenging 1,3-methyl ramification, five more stereoselective syntheses were reported by four different groups.154−158 This intriguing target motif has sparked the creativity of the chemical community to develop highly efficient and elegant solutions to the synthesis of the 1,3-methyl array.
In 2013, Schneider communicated a total synthesis in which the methyl branches were largely installed via iridium-catalyzed substrate controlled asymmetric hydrogenation (Scheme 25–27).154 In contrast to ours, the Schneider synthesis is based on a more convergent approach in which three building blocks are unified. The synthesis started with the preparation of enantiopure (R)-200 by an enzymatic kinetic resolution of propargylic alcohol rac-200 employing commercially available Novozym 435 (Scheme 25). Both enantiomers were obtained in good yields with very high stereoselectivity (96% ee for (S)-200 and 91% ee for (R)-200). Both of these products could be converted into benzoylated alcohol 201 which after multiple crystallizations from hexane, and subsequent deprotection, afforded (R)-200 (>99% ee). Regioselective and substrate-controlled hydrozirconation employing a [Cp2ZrHCl]/ZnCl2 complex, followed by iodination, gave vinyl iodide 202 in 70% yield with no observable terminal iodide.
Scheme 25. Schneider’s Hydroxyphthioceranic Acid Synthesis Part 1.

Scheme 27. Completion of the Total Synthesis of Hydroxyphthioceranic Acid by the Schneider Laboratory.

The other two building blocks to be crafted before assembly were alkyl iodide 211 and vinyl iodide 213, which were reckoned to be available from common precursor 210 (Scheme 26).154,159,160 Starting with an Evans’ aldol reaction using 203 and methacrolein 204, aldol product 205 was obtained with an excellent syn:anti ratio of >98:2. Benzoylation of 205 was followed by an oxy-Cope rearrangement yielding 207. With the first chiral methyl branch set, the second methyl branch was installed by means of an asymmetric hydrogenation using 2 mol% of iridium catalyst C3 delivering 208 in 98% yield and an excellent dr of 97:3. Further elaboration, by means of a stereoselective enolate alkylation, gave rise to trideoxypropionate 209 in 93% yield with a syn:anti ratio of >98:2. Auxiliary removal, silylation, and benzyl removal gave common intermediate 210 which could be easily converted into the building blocks 211 and 213.
Scheme 26. Schneider’s Hydroxyphthioceranic Acid Synthesis Part 2.

With the three desired building blocks in hand, the assembly of hydroxyphthioceranic acid commenced with a Suzuki cross-coupling of vinyl iodide 202 and trideoxypropionate 211, yielding allylic alcohol 214 in 79% (Scheme 27). Diastereoselective hydrogenation of this substrate however failed due to accompanied alkene isomerization to the corresponding ketone. Therefore, 214 was benzoylated and desilylated in 94% yield over four steps before the diastereoselective hydrogenation with modified Crabtree’s catalyst C4. Obtained in 98% yield and a 98:2 syn:anti ratio, 216 was easily converted to 217 in five steps. A Suzuki cross-coupling with building block 213 was performed to give rise to alkene 218 after desilylation. To complete the construction of the deoxypropionate unit, another asymmetric iridium-catalyzed hydrogenation was performed affording diol 219 in 98% yield with an excellent syn:anti selectivity of >99:1. The synthesis of hydroxyphthioceranic acid 220 was finalized by a TEMPO oxidation of 219 followed by a Pinnick oxidation. Using this strategy, phthioceranic acid was also constructed (not shown).
The Aggarwal laboratory reported their hydroxyphthioceranic acid total synthesis in 2014, which was based on their in-house developed stereoselective lithiation–borylation–protodeboronation strategy.155 At first, two main building blocks were crafted from commercially available acetate 221 (Scheme 28). Standard protecting group manipulations furnished the 1,3-dimethyl carbamates 222 and 223, of which the latter was borylated using standard conditions giving 224. In order to obtain homologated product 226 with the correct stereochemistry, ethyl carbamate was deprotonated using s-BuLi/(+)-sparteine 225 followed by the addition of boronic ester 224. Trimethylated boronic ester 226 was accessed in an excellent yield of 92% with a diastereomeric ratio of 99:1.
Scheme 28. Synthesis of Hydroxyphthioceranic Acid Building Blocks.

With sufficient quantities of the two key building blocks 222 and 226 synthesized, both were united by treatment of carbamate 222 with s-BuLi/TMEDA followed by addition of the boronic ester 226 (Scheme 29). Pentamethylated product 227 was obtained smoothly in 91% yield with a diastereomeric ratio of 1:1 of the secondary boronic ester functionality. Protodeborylation of 227 was achieved by first treatment with n-BuLi (boronate formation) and subsequently Mn(OAc)3·2H2O (oxidant) and 4-tert-butylcatechol (TBC, H-donor) to provide 228 in a good yield of 68%. The two-step sequence described was also demonstrated to be feasible using a one-pot procedure providing the desired product 228 in effectively the same yield. Straightforward protecting group manipulations gave carbamate 229 which was subjected to the described lithiation/borylation/protodeboronation strategy to give 231 in 51% yield over the two steps. Again, this sequence could be executed in one-pot as well, giving the product 231 in virtually the same yield. With the deoxypropionate functionality set, the final stereogenic center could be installed. After a two-step procedure, carbamate 232 was obtained, which by stereoselective deprotonation with s-BuLi/(+)-sparteine was borylated and subsequently oxidized to furnish alcohol 219 in 85% yield and an excellent diastereomeric ratio >95:5. Deprotection of the MOM group followed by chemoselective primary alcohol oxidation then gave hydroxyphthioceranic acid 220 in just 14 steps, an impressively small number and a considerable improvement compared to the Minnaard and Schneider routes (vide supra).
Scheme 29. Aggarwal’s Hydroxyphthioceranic Acid Synthesis.

More recently the Aggarwal group reported the use of this methodology in an iterative fashion by an, even more efficient, “assembly-line synthesis” of hydroxyphthioceranic acid (Scheme 30).156 This second-generation strategy improved on their previous synthesis in terms of step count (from 14 to 8, based on purification steps). To achieve this, the deoxypropionate motif was introduced by reaction of 4-methoxy aryl boronic ester 241 with lithium reagent 235, followed by coupling with (chloromethyl)lithium 237. After every two methyl installations, the corresponding boronic ester product was purified, which was realized in an average of 75% yield for two methyl group incorporations. After a total of seven iterations (31% overall yield), all desired stereogenic methyl groups were installed. Boronic ester 242 was then subjected to a reaction with lithium reagent 240, which after hydrolysis of the intermediate boronate furnished alcohol 243, in 85% yield and a diastereomeric ratio >95:5. Silyl protection, oxidative arene cleavage, and silyl deprotection then concluded the synthesis of hydroxyphthioceranic acid 220.
Scheme 30. Aggarwal’s “Assembly-Line” Synthesis of Hydroxyphthioceranic Acid.

In 2015 Negishi and co-workers reported the iterative synthesis of phthioceranic acid based on the Zr-catalyzed asymmetric carboalumination (ZACA) of alkenes to install the chiral methyl-branched groups.157 The synthesis also proved to be convergent, requiring the synthesis of three optically pure building blocks (Scheme 31). Two of these building blocks were crafted by means of the ZACA reaction. To construct building block 249, octadec-1-ene 244 was subjected to the asymmetric carboalumination reaction to produce the corresponding aluminate 246. This intermediate was transmetalated to the organozinc reagent, a substrate used in the Negishi cross-coupling with vinyl bromide. The terminal alkene 247 obtained was once more subjected to the ZACA reaction and subsequently oxidized with molecular oxygen to provide alcohol 248 in 44% over the three steps with excellent stereoselectivities (dr > 50:1, 97% ee). Bromination completed the synthesis of building block 249.
Scheme 31. Synthesis of Phthioceranic Acid Building Blocks.

Building block 255 was crafted out of styrene which also efficiently reacted under the ZACA conditions to provide the desired stereogenic center. After vinylation of the intermediary aluminate, two subsequent ZACA reactions were performed to install the remaining two stereogenic centers, affording, after bromination, bromide 255 with a diastereomeric ratio of >50:1 with an enantiomeric excess >99%.
The third building block 258 was created out of enantiomerically enriched, commercially available, diol 257. A Mitsunobu reaction and subsequent tosylation produced building block 258 in 80% yield with a dr of >50:1.
The building blocks were unified in only five steps to furnish phthioceranic acid (Scheme 32). Building block 249 was converted into the corresponding Grignard reagent and coupled with tosylate 258 in a SN2 reaction by using cuprate chemistry. The diastereoselectivity proved to be excellent (dr > 40:1) providing the desired compound in 58% yield. The ester functionality in 260 was hydrolyzed, and the obtained alcohol was tosylated, in 90% over the two steps, to form 261. Again, a Grignard reagent was formed, this time from building block 255, and coupled under similar conditions as previously mentioned to provide, after oxidative cleavage of the arene group, phthioceranic acid 183 with a diastereomeric ratio of >50:1.
Scheme 32. Completion of the Synthesis of Phthioceranic Acid.

The most recent dissemination of an iterative synthesis of phthioceranic acid was published by the groups of Zhu and Zhou in 2018 (Scheme 33).158 Their total synthesis started with the nucleophilic displacement of the bromide in ethyl 2-(bromomethyl)acrylate 264 with a, from pentadecyl Grignard prepared, Gillman reagent, followed by hydrolysis of the ethyl ester. With the free carboxylic acid 266 in hand, the first stereogenic methyl group was installed by means of an asymmetric hydrogenation using only 0.1 mol% of chiral spiro P,N-ligand-based iridium catalyst C6, which proceeded in 94% ee and gave 267 near quantitative yield. The carboxylic acid was homologated by means of a carboxymethylation with Meldrum’s acid, followed by alkenylation with Eschenmoser’s salt, and subsequent in situ hydrolysis to yield acrylic acid 269. This two-step sequence proceeded in a yield of 81%, setting the stage for the first iteration in which the second stereogenic methyl branch was introduced, delivering 270 in 99% yield and a diastereomeric ratio of 96:4. Five more iterations provided phthioceranic acid 183 in an impressive overall yield of 34%. The most attractive feature of this iterative sequence is, besides the low catalyst loading, very good yields, and excellent stereoselectivities, that no purification by column chromatography was required during the iterative steps. This makes this sequence even more practical and scalable compared to the previously presented strategies.
Scheme 33. Zhu and Zhou’s 19-Step Phthioceranic Acid Synthesis.

5. Trehalose Glycolipids
5.1. Ac2SGL
In 2004, Gilleron and co-workers isolated a previously unknown, sulfated, Mtb-specific lipid called diacylated sulfoglycolipid (Ac2SGL).161 The molecule resides in the outer membrane where it can induce immune responses. This indeed proved to be the case as Ac2SGL was shown to induce T-cell responses, in clinical isolates, by stimulating the CD1b protein. More specifically, Ac2SGL-activated T-cells release the cytokine interferon-γ, but more interestingly the activated T-cells were able to recognize Mtb infected antigen-presenting cells and were able to kill intracellularly residing Mtb in vitro. These studies showed that Ac2SGL is a potent antigen and thus is a promising candidate for the development of vaccines against tuberculosis.
During their studies, Gilleron et al. showed that the immunogenicity of Ac2SGL is dependent on the sulfate, with desulfated Ac2SGL treated T-cells not able to produce interferon-γ.161 Further structural insight into the role of the lipid appendages on the antigenicity of Ac2SGL was provided by Prandi and co-workers, who performed a broad structure–activity relationship study in which the trehalose-sulfate functionality was modified with varying acyl side chains.162,163 It was shown that T-cell receptor recognition and T-lymphocyte activation was dependent on (1) the number of stereogenic methyl substituents, (2) their stereochemistry, and (3) the respective location of the acyl chains on the trehalose core. Some simplified Ac2SGL analogues were able to activate T-cells at low concentrations (<100 nM, ∼0.1 μg mL–1); however, nature proved to have the upper hand as antigenicity was significantly attenuated for the synthetic analogues compared to the naturally occurring Ac2SGL.
Although synthetic analogues could be used for Mtb vaccine development, Ac2SGL is desired for such studies due to its improved bioactivity. The low abundancy of Ac2SGL (∼1 mg/L bacterial culture), however, makes chemical synthesis a viable tool for producing sufficient quantities for vaccine development studies. Our group set out to achieve the first complete total synthesis of naturally occurring Ac2SGL,152 which started with the synthesis of the deoxypropionates phthioceranic acid118 and hydroxyphthioceranic acid (see Schemes 22–24).
The synthesis of Ac2SGL was achieved by coupling these to the trehalose core (Scheme 34). After protection and acylation of trehalose, protected hydroxyphthioceranic acid 199 (Scheme 24) was introduced by means of a Yamaguchi esterification, which proceeded in 76% yield to generate 272. The bis-silylether 272 was deprotected under buffered conditions and thereafter regioselectively sulfated at the C2′-position using 2,2,2-trichloroethyl sulfuryl imidazolium salt 274 providing protected sulfate 275. Hydrogenolysis of the 2,2,2-trichloroethyl protecting group and the benzylidene acetals of 275 yielded the target molecule, Ac2SGL 276. All analytic data were in agreement with those of the natural isolate, and cytokine release assays showed that the synthetic material was equally potent as natural Ac2SGL, thereby concluding the first asymmetric total synthesis of Ac2SGL and allowing the confirmation of its proposed chemical structure.
Scheme 34. Completion of the Total Synthesis of Naturally Occurring Ac2SGL.

In 2018, another total synthesis of Ac2SGL was reported (Scheme 35).164 Here, the synthesis of the hydroxyphthioceranic acid was shortened by merging the asymmetric conjugate addition strategy used in Minnaard’s total synthesis of Ac2SGL with the stereoselective lithiation–borylation–protodeboronation strategy developed by Aggarwal (vide supra). Starting from thioester 135, the first three chiral methyl units were installed by three iterations of Cu-cat. ACA, DIBAL-H reduction, and Horner–Wadsworth–Emmons olefination, providing ketone 277.165 A sequence of steps gave boronic ester 280, which set the stage for the installation of the last chiral methyl branch of 281 by stereoselective lithiation–borylation. A second building block 284 was synthesized by first introducing four chiral methyl units by means of Cu-cat. asymmetric alkylation producing ketone 282.165 A series of functional group manipulations and protection/deprotection steps gave carbamate 284. Building blocks 281 and 284 were then coupled by Aggarwal’s lithiation–borylation–protodeborylation approach providing, after protodeborylation of compound 285, MOM ether 231. Deprotection of the MOM ether followed by carbamoylation provided 286. The hydroxyl stereocenter was introduced by subjecting carbamate 286 to another lithiation–borylation reaction with linear alkyl boronic ester. The synthesis of hydroxyphthioceranic acid 220 was completed by deprotection and oxidation of silyl ether 287. The efficiency of the total synthesis was further improved by omitting hydroxyl protection in the hydroxyphthioceranic acid side chain prior to esterification, and thus, Ac2SGL 276 was accessed in fewer steps and higher overall yield analogous to the previous total synthesis reported by Geerdink and Minnaard.152 Synthetic and natural Ac2SGL as well as various saturated and unsaturated analogues lacking the hydroxyl in the lipid chain were used to generate Ac2SGL-loaded CD1b tetramers providing a tool for the ex vivo study of T-cell function and reactivity in the future. This was anticipated to be useful for the design and evaluation of synthetic Ac2SGL analogues as Mtb vaccine subunits.
Scheme 35. Hybrid Total Synthesis of Ac2SGL.

5.2. Sulfolipid-1
After the first successful total synthesis of Ac2SGL, our laboratory set out to synthesize SL-1, arguably Mtb’s most complex glycolipid to date.166 SL-1 was first isolated by the Goren laboratory, who in 1970 managed to elucidate its molecular architecture by meticulous degradation studies.167−169 In contrast to Ac2SGL containing two acyl chains, sulfolipid-1 contains four acyl chains on the sulfated trehalose core, two times hydroxyphthioceranic acid, one phthioceranic acid, and one palmitic acid. Being a prominent cell wall constituent, sulfolipid-1 was postulated to be a virulence factor that mediates host–pathogen interaction. Early studies, before its structure elucidation, did show that levels of sulfolipid-1 correlated positively with virulence.170 In the late 80s and early 90s it was demonstrated that sulfolipid-1 inhibits several key host responses to mycobacterial infection such as phagosome acidification and phagosome–lysosome fusion in cultured macrophages, activation of human neutrophil and macrophages, and cytokine production in human monocytes and neutrophils.170−174
In contrast to these findings, the Bertozzi group, however, showed that knockout mutants of sulfotransferase stf0, therefore unable to produce sulfolipid-1, exhibit augmented survival in human macrophages.175 Moreover, in vitro studies showed that the knockout mutants also proved to be more resistant against killing by the antimicrobial peptide LL-37, showing increased survival for Δstf0 in human macrophages. These contradictory findings show there is still a lot unknown about the exact role of sulfolipid-1 in Mtb pathogenesis and virulence.
Recently, however, a landmark publication reported a biological role for sulfolipid-1 that has profound influence on Mtb as the world’s leading pathogen. The Shiloh lab discovered that SL-1 activates nociceptive neurons, present in the lung, and induces cough. Not only is cough a primary tuberculosis symptom, but more importantly, it is the primary mechanism of transmission of infection.176 So, 50 years after its initial isolation, a major role of virulence of Mtb has been attributed to this impressive glycolipid.
The total synthesis of SL-1 borrowed largely from the expertise gained during the total synthesis of Ac2SGL.152 To prepare SL-1, protected and acylated trehalose 271 was reacted with phthioceranic acid 183 in a Yamaguchi esterification to provide 288 in 76% yield (Scheme 36). The benzylidene acetals were regioselectivity ring-opened to liberate the free 6-OH and 6′-OH groups. To achieve this, several reductive ring-opening conditions were scrutinized, among them the Lewis acids CoCl2, Cu(OTf)2, and TMSOTf in combination with BH3·THF. The latter reaction conditions proved to be the best providing 289 in a still moderate but satisfactory yield of 59%.
Scheme 36. Total Synthesis of SL-1.

The liberated alcohol functionalities were both acylated with benzyl protected hydroxyphthioceranic acid 199 using EDC as the coupling agent rather than a Yamaguchi esterification which only provided monoacylated product. Compound 290 was desilylated and then attempted to be sulfated as in the Ac2SGL synthesis (Scheme 34). Unfortunately, the sulfation did not only take place at the desired C2′ position, as in the Ac2SGL synthesis, but also at the C3′ position. This result was hypothesized to be caused by the reduced rigidity in compound 290 compared to that of compound 273 (Scheme 34) used in the Ac2SGL synthesis. Whereas compound 273 had an intact benzylidene acetal, compound 290 is free of this conformational strain which might lead to exposing the C3′ position to sulfation. The desired C2′ sulfated product could be isolated in pure form which was then subjected to the final hydrogenolysis conditions described for the Ac2SGL synthesis. Using atmospheric H2 pressure, no conversion was observed. Increasing the hydrogen pressure to 17 bar did not lead to any product formation either. Further increase to 34 bar ultimately cleaved the benzyl protecting groups; however, the relatively labile sulfate group was partly cleaved as well, as confirmed by isolation of “desulfated SL-1”. The problematic hydrogenolysis was attributed to the long-tailed lipids that create a “coat of armor” around the trehalose core, impeding the reagents from accessing the benzyl reaction site. All in all, SL-1 291 was synthesized, in a total of 46 steps, for the first time after its isolation 40 years ago, confirming the chemical structure of this impressive molecule.
5.3. Acyl Trehaloses
The family of acyl trehaloses forms another class of mycobacterial cell wall glycolipids and is composed of di-, tri-, and poly acyl (or penta-acyl) trehaloses (DAT, TAT, and PAT, respectively) bearing linear fatty acid residues and the methyl-branched acyl residues mycosanoic, mycolipenic, and mycolipanolic acids.177−180 Mycobacterial acyl trehaloses have been the subject of various studies since their first isolation in the 1980s and 90s, but to this date, their role in Mtb pathogenesis remains largely unknown. Mtb mutants deficient of DAT, TAT, and PAT show no changes in survival and replication, yet possess an altered cell surface composition and enhanced phagocytosis efficiency in mouse models.181 Thus, it was hypothesized that the function of these acyl trehaloses lies in the regulation of the cell envelope architecture by anchoring the capsule to the outer membrane leading to prevention of recognition and phagocytosis by host macrophages.181−184
Among the acyl trehaloses, DAT has received by far the most attention, and various studies indicated that DAT exerts various immunomodulatory effects.185−187 DAT is able to inhibit monocyte production of pro-inflammatory cytokines and thus negatively regulates the immune response of the host.188 Furthermore, it was demonstrated that DAT inhibits murine T-cell proliferation in vitro.189
In the past, DAT (and to some extent TAT) has received attention for its antigenic properties and was regarded as a promising candidate for the development of antibody-based tests for Tb serodiagnosis.180,190−193 Due to difficulties in developing a reliable ELISA, little progress has been made in utilizing acyl trehaloses as diagnostic markers.194 In recent years, DAT and TAT have been identified among other mycobacterial glycolipids as agonists of the immune receptor Mincle90 suggesting that they have potential for application in the development of Mtb vaccine adjuvants. Thus far, natural DAT has been used in immunological assays, often as a mixture of structurally different DATs. Therefore, the biological effect of the individual DAT components, with well-defined chemical structure, was not ascertained.
In order to gain more insight into the effect of the individual DAT components on host immune responses, our group embarked on the total synthesis of three representatives of the DAT family (DAT1–3).195 By this, synthetic standards for immunological testing can be provided and the observed biological effects in functional assays can be without doubt attributed to the tested compounds rather than to minute amounts of contaminants or isomers/related compounds resulting from isolation.
The three target DATs differ in the nature of the mycobacterial acyl residues. DAT1 is esterified with mycosanoic acid on the 3-OH of trehalose, whereas DAT2 and DAT3 carry mycolipanolic and mycolipenic acid esters on that position, respectively. For the synthesis of these methyl-branched lipids, a synthetic route was optimized, which was published by us previously.196
The synthesis of the mycobacterial lipids (Scheme 37) commenced with the previously discussed asymmetric Cu-cat ACA of methylmagnesium bromide to α,β-unsaturated thioester 135 providing 136 in 81% yield and 98% ee. DIBAL-H reduction of 136 followed by Horner–Wadsworth–Emmons olefination and a second asymmetric conjugate addition furnished 292 containing two methyl branches in 86% yield over three steps and dr > 20:1. The thioester moiety of 292 was then converted to the corresponding alcohol by two rounds of DIBAL-H reduction. After tosylation, the corresponding tosylate 293 was obtained in 88% yield over three steps. The C16 alkyl tail of 294 was installed by copper-mediated Grignard alkylation of 293 in 96% yield. Desilylation of 294 followed by Dess–Martin periodinane oxidation then provided aldehyde 295 in 97% yield over two steps. From 295 the three target mycobacterial acids could be obtained in a few steps. Pinnick oxidation of 295 furnished mycosanoic acid 296 in 92% yield. The two remaining stereocenters of mycolipanolic acid 299 were installed by diastereoselective Evans’ aldol condensation of 295 with chiral auxiliary 297 and provided 298 in 82% yield and excellent diastereoselectivity exceeding 20:1. Cleavage of the chiral auxiliary of 298 then provided mycolipanolic acid 299 in quantitative yield. Lastly, mycolipenic acid 300 was accessed from aldehyde 295 by Wittig olefination and ester hydrolysis in 80% yield over two steps. Notably, by careful optimization of the reactions, the overall yield of the mycobacterial lipids was significantly improved, rendering the synthesis highly efficient. For mycosanoic acid an overall yield of 53% over 10 steps was achieved. Mycolipanolic and mycolipenic acid were obtained in a total yield of 47% (previously 2%) and 46% (previously 5%)196 over 11 steps.
Scheme 37. Asymmetric Synthesis of Mycosanoic, Mycolipanolic, and Mycolipenic Acids.

With the three enantiopure lipids in hand, the synthesis of DAT1–3 was finalized by Yamaguchi esterification of protected trehalose 271 (Scheme 38). The diacylated trehaloses 301a–c were obtained in good yields ranging from 66% to 75%. In the case of mycolipanolic acid 299, the procedure was optimized by lowering the equivalents of base and changing solvent to THF in order to suppress elimination of the unprotected β-hydroxyl moiety. After desilylation, under buffered conditions, and removal of the benzylidene protecting groups, the total synthesis of DAT1 (303), DAT2 (304), and DAT3 (305) was completed.
Scheme 38. Completion of the Total Synthesis of DAT1, DAT2, and DAT3.

In order to assess if the synthesized DATs indeed corresponded to the structures of the mycobacterial natural products, lipid extracts of Mtb strain H37Rv and three clinical isolates were analyzed by HPLC-MS. Comparison with the synthetic DATs showed coelution and identical mass spectra for DAT1 and DAT3, but for DAT2 the retention times differed albeit with identical observed collisional mass-spectra. This confirms that synthetic DAT1 and DAT3 are identical to the natural glycolipids, whereas synthetic DAT2 is likely an isomer of natural DAT2, of which the exact structure remains to be elucidated. These results highlight the importance of chemical synthesis of natural products to confirm and/or disprove proposed chemical structures of natural isolates.
In addition, the synthetic DATs were tested for activation of Mincle and compared to the known potent Mincle agonist trehalose dimycolate (TDM). Whereas DAT1 and DAT2 showed only weak binding to Mincle, DAT3 was found to be a strong Mincle agonist (in the potency range of TDM), indicating that the nature of the chiral lipid strongly influences Mincle binding and activation.
To further gain insight into their immunomodulatory properties, the synthetic DATs, with their well-defined structures, were investigated for their potential to be recognized by human T cells.197 Tetramers of the lipid binding antigen presenting molecule CD1b were constructed and tested for their ability to present DAT molecules to human T cells. Interestingly, whereas DAT3 was a potent agonist for Mincle, DAT3 was not an antigen recognized by T cells. In contrast, DAT1 and DAT2, which were both weak binding Mincle ligands, were presented to T cells by CD1b and are thus lipid antigens. This intriguing result showed that the structure of the lipid in the C3 position is a determinant for the immunogenic properties of the DATs. More specifically, the α-stereocenter in the mycosanoic lipid of DAT1 and mycolipanoic lipid of DAT2 seems to be important for recognition by T cells, a structural feature missing from the myolipenic lipid in DAT3. A broader conclusion from this work was that small structural differences determine recognition of mycobacterial lipids by different parts of the immune system and that DAT can be both an agonist for the Mincle receptor and an antigen for T cells. It should be mentioned that such conclusions can only be reached with very well-defined chemical structures from synthesis rather than heterogeneous natural isolates, which is the case for the DATs, and thus highlight the importance of total synthesis in determining the immunomodulatory properties of Mtb lipids.
6. Glycerolipids
6.1. β-Gentiobiosyl Diacylglycerides
The plethora of mycobacterial membrane lipids and glycolipids also includes the family of β-gentiobiosyl diacylglycerides. This series of glycerolipids was first described by Hunter et al. in 1986 and contains a common gentiobiosylglycerol motif.198 The glycerol moiety carries long-chain iso- and anteiso-fatty acid ester homologues. The exact molecular structure of this class of glycoglycerolipids has not been elucidated or confirmed yet, but upon first isolation it was demonstrated that the mycobacterial β-gentiobiosyl diacylglycerides are serologically active. Yet, β-gentiobiosyl diacylglycerides are not limited to Mtb and are found to be of fungal origin (containing anteiso-fatty acyl groups) as well. The fungal β-gentiobiosyl diacylglycerides were found to be capable of activating the immune receptor Mincle, though only with moderate potency.199 For decades the mycobacterial diglycosyl glycerolipids have not received much attention of researchers. It was only in 2015, when the Williams group embarked on the synthesis of a whole array of candidate structures and analogues to assess their Mincle signaling potential and to gain insight on structural requirements for Mincle activation.200
The synthesis of the target structures was initiated by the perbenzoylation of readily available amygdalin 306 to give heptabenzoate 307 (Scheme 39). Treatment of 307 with ZnCl2 and dichloromethyl methyl ether gave rise to the corresponding gentiobiosyl chloride which was treated with potassium thiocresolate to cleanly furnish thioglycoside 308 in 80% yield over two steps. Next, NIS/TfOH-promoted glycosylation of 308 with p-bromobenzyl (PBB) protected glycerol 309 provided gentiobiosyl glyceride 310 in excellent yield. The benzoyl groups of 310 were removed, and the corresponding heptol was benzylated. Selective deprotection of the PBB groups was brought about by palladium-catalyzed amination followed by treatment with TFA providing diol 311 in 80% yield over four steps. Various ester groups were installed by either direct acylation with the corresponding acyl chlorides or COMU/DMAP/DIPEA-mediated esterification. Lastly, palladium-catalyzed hydrogenolysis of the benzyl ethers provided a series of β-gentiobiosyl diacylglycerides. Compounds 312a–d are candidate structures of mycobacterial β-gentiobiosyl diacylglycerides. In addition, various β-gentiobiosyl diacylglyceride analogues 312e–g, 313, 314, and 315a–b were synthesized, in order to assess their Mincle binding and activation potential.
Scheme 39. Synthesis of Mycobacterial β-Gentiobiosyl Diglyceride Candidates.

Mincle activation of the synthesized compounds was assessed by functional assays, and binding was confirmed by ELISA. The study revealed a striking dependence of signaling on the length of the acyl chain. The β-gentiobiosyl diacylglyceride candidates 312a–d provided only weak stimulation of murine but not of human Mincle. Among the linear chain β-gentiobiosyl diacylglycerides 312d–g only the C12 analogue 312e was able to induce weak activation of human and moderate activation of murine Mincle. The glucose analogue 315a was significantly more potent than the gentiobiosyl derivatives, whereas the truncated analogues 313 and 314 failed to stimulate Mincle. Based on this the authors speculated that Mtb elaborates glucosyl diacylglycerides (the biosynthetic precursors of β-gentiobiosyl diacylglycerides)201 to β-gentiobiosyl diacylglycerides to mitigate the host immune response mediated by Mincle.
In 2019, a second synthesis of a mycobacterial β-gentiobiosyl diacylglyceride candidate was reported.202 In this approach, the gentiobiosyl unit was constructed by glycosylation of glycosyl donors and acceptors bearing stereodirecting cyclic silyl protecting groups. The glycerol moiety was introduced by glycosylation with commercially available (S)-solketal, which was esterified after isopropylidene hydrolysis with palmitic and pentadecanoic acid to provide the desired β-gentiobiosyl diacylglyceride candidate structure for the use in future biological assays.
6.2. Tuberculostearic Acid Containing Phosphoglycerolipids
Phosphatidylethanolamines (cephalins) constitute yet another family of glycerophospholipids which contribute to the structure and function of cell membranes.203 An at the time unknown yet abundant glycerophospholipid was isolated from the reference strain H37Rv of Mtb by the Moody laboratory.204 In an initial study the molecular composition of the isolated material was investigated using collisional mass spectrometry, which resulted in the identification of cephalin, palmitoyl, and tuberculostearoyl fragments.204 The exact connectivity of the fragments could not be deduced by mass-spectrometric analysis, and the quantities of the isolate were too low to allow for detailed NMR analysis. Consequently, full characterization of the natural products was not possible at this point. To elucidate the regiochemistry of the palmitoyl and tuberculostearoyl fragments on the glycerol core, both isomers were targeted for total synthesis (Scheme 40, only one product shown).
Scheme 40. Total Synthesis of Mycobacterial TBSA-Based Phosphatidylethanolamine.

The synthesis commenced with an alkene cross-metathesis of terminal alkene 316 with thioacrylate 317 to give α,β-unsaturated thioester 318 (Scheme 40). Copper-catalyzed asymmetric conjugate addition of 318 as novel substrate provided methyl-branched thioester 319 in 85% yield with an enantiomeric excess of 90%. Fukuyama reduction of the thioester moiety and subsequent Wittig olefination then gave alkene 320 in 92% over the two steps. Next, the double bond of 320 was reduced by the in-house developed flavin-catalyzed diimide reduction205 followed by ester hydrolysis providing tuberculostearic acid (TBSA) 322. Coupling of the TBSA fragment 322 with (R)-benzyl glycidyl ether 323, in the presence of Et4NBr as the catalyst, led to opening of the epoxide producing glycerol monoester 324. Esterification with palmitic acid followed by debenzylation furnished alcohol 325, notably with no significant acyl migration. The phosphatidylethanolamine moiety was installed through reaction of 325 with phosphoramidite reagent 326 followed by oxidation of the intermediate product which gave, after deprotection of the benzyl and carboxybenzyl groups, the target phosphatidylethanolamine 327.
Comparing the mass spectral data of the synthetic TBSA-based phosphatidylethanolamine327 and its regioisomer (1-O-palmitoyl-2-O-TBSA-sn-phospholipid, structure not shown) with the natural isolate led to the conclusion that the structure of the natural isolate is that of 327. The total synthesis of this natural product was not only crucial for structure elucidation but also provided material which can be utilized as LC-MS reference for the application as molecular marker.
In 2013, an improved synthesis of phospholipid 327 was reported as part of our studies on stereoselective synthesis of glycerolipids (Scheme 41).206 The previous synthesis of tuberculostearic acid was shortened by two steps. Here, aldehyde 328 was subjected to a Wittig olefination with ylide 71 using “on water” conditions. The corresponding unsaturated thioester 329 was obtained in 92% yield with an E:Z ratio >95:5. Introduction of the chiral methyl branch was accomplished in 90% yield and 90% ee delivering 330. Thioester reduction of 330 followed by olefination with the free carboxylic acid and double bond reduction gave TBSA 321.
Scheme 41. Improved Total Synthesis of the TBSA-Based Phosphatidylethanolamine.

The glycidyl part was synthesized starting from silyl protected glycidol 331 using Co-salen catalyst C7, Hüniǵs base, and TBSA 322. In the same pot the palmitate moiety was introduced by means of a classic Steglich esterification, producing diester 332 in 90% over the two steps. The delicate desilylation of diacyl glycerol 332 was accomplished by using BF3·CH3CN, and the free alcohol was obtained without migration of the palmitoyl ester. The phosphate group was installed followed by deprotection as previously described giving phosphatidylethanolamine 327.
Next to phosphatidylethanolamines, theres is also a class of molecules present in animals, plants, fungi, and bacteria in which the ethanolamine moiety is replaced by a glycerol. This class is called phosphatidylglycerols,207 and in mycobacteria these molecules are thought to be phosphatidyl carriers for the biosynthesis of, for instance, cardiolipin208 and phosphatidylinositol.209 The Moody and Brenner laboratories found that phosphatidylglycerols can be presented by the antigen presenting proteins CD1b210 and CD1d.211,212 Due to their role as a biosynthetic intermediate, their relative abundance is low which hampers isolation and further studies.
In 2017, the Williams group reported the total synthesis of the CD1d presented TBSA-containing phosphatidylglycerol (Scheme 42).213 The tuberculostearic acid 322 was synthesized in just two steps from (S)-citronellyl bromide 333, by first executing a copper-catalyzed Grignard alkylation followed by a one-pot alkene cross-metathesis with 6-heptenoic acid and subsequent double bond hydrogenation. This sequence proceeded with an overall yield of 72% and is the shortest route of tuberculostearic acid 322 to date. The phosphatidyl moiety was constructed by using the protocol we developed for the later described phosphatidylethanolamine synthesis. Thus, epoxide 323 was opened using the Salen catalyst C7 and TBSA 322, followed by a Steglich esterification with palmitic acid. The benzyl group was removed by hydrogenolysis to provide diacylated glycerol 325 in 76% over the described steps. In order to install the phosphoglycerol moiety, 325 was converted into the phosphoramidite 335 mediated by diisopropylammonium tetrazolide (DIPAT). The unstable phosphoramidite 335 was then reacted with dibenzylglycerol 336, to form after hydrogenolysis the desired TBSA-based phosphatidylglycerol 337 in 53% over the last two steps.
Scheme 42. Total Synthesis of the TBSA-Based Phosphatidylglycerol.

7. Terpene Nucleosides
In 2004 the Russel laboratory disseminated a landmark publication reporting that transposon knockouts of the Rv3377c or Rv3378c gene led to reduced survival of Mtb in mice compared to wild-type Mtb.214 More specifically, it was found that the phagosomal compartment, which forms after a macrophage internalizes a foreign invader, does not acidify (i.e., mature) to levels in which fusion with lysosomes, organelles containing acid and hydrolases, takes place. This means that pathogen digestion is inhibited, allowing the Mtb to escape the phagosomal compartment,16 enabling survival of Mtb within the harmless, yet nutritious, cytosol of immune cells. Although intracellular survival of Mtb cannot be pinpointed to one specific event, blockade of phagosomal maturation is paramount to the persistence of Mtb in its human host. Despite the fact that the ability of Mtb to stop phagosome maturation has been recognized for decades,215,216 fundamental understanding of the molecular mechanism remained unknown.
In 2005 the Hoshino group expressed the Mtb Rv3377c-gene in E. coli, establishing that geranylgeranyl pyrophosphate 338 was cyclized by Rv3377c to produce a halimane skeleton.217−219 The cyclized product was named tuberculosinyl pyrophosphate 339 and postulated to be hydrolyzed by Rv3378c to produce tuberculosinol 340 (Scheme 43).218 In 2009, Peters reported that the enzyme Rv3378c does not act merely as a hydrolase but as a phosphatase that cyclizes tuberculosinylpyrophosphate 339 to form edaxadiene 341.220 Importantly, these investigations, although contradictory, tied the Rv3378c gene to a molecular structure which was accessible by total synthesis. Since the structure produced by the important Rv3377c-Rv3378c locus was uncertain at this stage, there was a driving force for synthetic chemists to solve this structural puzzle.
Scheme 43. Original Proposed Biosynthetic Pathways of the Rv3377c-Rv3378c Locus of Mtb.

In 2010 the research groups of Snider221 and Sorensen222 reported the total synthesis of tuberculosinol 340. Furthermore, both groups communicated the structural revision of the in 2009 proposed edaxadiene 341. Both groups questioned the proposed structure upon careful inspection of the NMR spectra reported for edaxadiene 341. It was concluded that the NMR spectra resembled that of isotuberculosinol 350, an isomer of tuberculosinol, corresponding to SN2′ hydrolysis of tuberculosinyl pyrophosphate 339.
The structure of (iso)tuberculosinol exhibits a cyclohexene ring which inevitably led to retrosynthetic disconnections, by both groups, involving a Diels–Alder cycloaddition. The target Diels–Alder adduct however is unusual in the sense that it arises from exo-attack of the diene. This product is generally disfavored as a consequence of the inherent endo-preference (the Alder endo rule).223 The endo-preference could be overcome by the introduction of steric hindrance in the dienophile.224 The Snider laboratory performed the Diels–Alder cycloaddition of diene 342 with oxazolidinone tiglate 343 furnishing the cyclization product 344 with an exo:endo ratio of 10:1 in 54% yield (Scheme 44).221 Reduction of the oxazolidinone moiety and subsequent Dess–Martin oxidation gave the corresponding aldehyde 346. The aldehyde was subjected to an aldol condensation with acetone, followed by a conjugate reduction/oxidation sequence to furnish ketone 348 in 54% yield over the three steps. A Grignard reaction on ketone 348, with vinyl Grignard, provided isotuberculosinol 350. By performing a Horner–Wadsworth–Emmons olefination on ketone 348, followed by reduction of the freshly installed ester functionality, tuberculosinol 340 was obtained.
Scheme 44. Snider’s Total Synthesis of Tuberculosinol and Isotuberculosinol.

A similar, yet slightly different, approach to tuberculosinol was reported by Sorensen, who started with a Diels–Alder reaction between diene 342 and ethyl tiglate providing the desired exo product in a 2:1 ratio over the endo product (Scheme 45).222 Enantiomeric separation by preparative supercritical fluid chromatography provided optically pure Diels–Alder adduct 344. Reduction and oxidation gave aldehyde 346 which served as a common intermediate to construct both tuberculosinol 340 and isotuberculosinol 350. A Wittig olefination provided alkene 351 which was subjected to a hydroboration with 9-BBN, to then be cross-coupled with vinyl iodide 352 in a Suzuki reaction, providing tuberculosinol 340. CuCl2-mediated isomerization converted tuberculosinol 340 directly into isotuberculosinol 350, albeit in only 20% yield. An alternative route was performed, along the same lines as in the Snider synthesis, to give isotuberculosinol 350 in only three steps from aldehyde 346.
Scheme 45. Sorensen’s Total Synthesis of Tuberculosinol and Isotuberculosinol.

The Sorensen lab also synthesized the edaxadiene substructure 359 proposed by Peters and co-workers (Scheme 46). This synthesis started with a Diels–Alder reaction between tiglic aldehyde and oxazolidinone-based modified Rawal’s diene 353, forming Diels–Alder adduct 354 in 80% yield. A Wittig homologation set the stage for a Suzuki coupling that was initiated by hydroboration of the terminal alkene functionality in 355 and subsequent cross-coupling with (Z)-2-bromo-2-butene. The silyl ether was cleaved with TBAF which initiated an E1cB elimination of the oxazolidinone, furnishing enone 356. To create the all-carbon quaternary stereocenter, an intramolecular oxidative ketone allylation was performed using MnIII(OAc)3. The reaction proceeded via the intermediate conformer 356a in which the allylic 1,3-strain was minimized, leading to a single stereoisomer of [3.3.1] bicyclic compound 357 in 57% yield. Full reduction of the enone functionality, via a two-step protocol, provided alcohol 358 which was deoxygenated using Martin’s sulfurane to provide edaxadiene subunit 359 after semipreparative gas chromatography.
Scheme 46. Sorensen’s Total Synthesis of Edaxadiene.

NMR analysis of both isotuberculosinol 350 and edaxadiene subunit 359 undoubtably showed that the natural isolate from Peters and co-workers corresponded with that of isotuberculosinol 350 and not edaxadiene 341 as proposed. Therefore, it was reasoned that Rv3378c acts as tuberculosinyl pyrophosphate hydrolase (Scheme 43, pathway A) rather than as cyclase (Scheme 43, pathway B).
In 2014 the Moody laboratory reported the isolation of an unknown compound from Mtb.225 The molecule was found during extensive lipidomics226 studies investigating the chemotaxonomy of the virulent Mtb versus the nonvirulent M. bovis BCG. It was found that this new molecule was abundant in Mtb, whereas it was completely absent in M. bovis BCG. This initial result prompted investigation into the nature of the molecule, its biosynthesis, and presence in other medically relevant bacteria.
Isolation and subsequent structure elucidation by NMR and collisional mass analysis resulted in the formulation of its structure being 1-tuberculosinyladenosine (1-TbAd), which is the tuberculosinyl lipid connected to the 1-position of adenosine, a structural feature rarely seen in nature. Since 1-TbAd contains the tuberculosinyl core, the cyclization product of geranylgeranyl pyrophosphate 339 by Rv3377c, it was hypothesized that Rv3378c is not a hydrolase as postulated earlier, but rather acts as an adenosine transferase (Scheme 47).
Scheme 47. Revised Biosynthetic Product of the Rv3378c Gene in Mtb.

This biosynthesis proposal was put to the test by providing recombinant Rv3378c enzyme with synthetic racemic tuberculosinyl pyrophosphate 339 and adenosine. This experiment yielded 1-TbAd 360 (Scheme 47, pathway C) while the hydrolyzed products, (iso)tuberculosinols 340 and 350, were not detected (Scheme 47, pathway A). Further evidence was provided by transformation of Mycobacterium smegmatis (Msmeg) with the Rv3377c-Rv3378c locus. Whereas Msmeg does not produce 1-TbAd, installation of the Rv3377c-Rv3378c genes did lead to 1-Tbad production. It was therefore concluded that the function of the Rv3778c enzyme is an adenosine transferase.227
1-TbAd was thus found to be abundantly produced (∼1 w/w% of the total extractable lipids), and in a subsequent communication it was reported that 1-TbAd is specifically produced by Mtb and no other (myco)bacteria.227 Moreover, it was also discovered that 1-TbAd can rearrange into N6-TbAd via a nucleophile-promoted Dimroth rearrangement (vide infra).228,229 Both 1-TbAd and N6-TbAd were shown to be suitable biomarkers of Mtb infection in mice lung homogenates and prompt further investigation for its use in tuberculosis diagnostics.
The abundancy, specificity, and association to Rv3377c and Rv3378c genes, involved in phagosome maturation arrest,214 hinted that 1-TbAd might be a molecule that attributes to the intracellular survival (i.e., parasitic nature) of Mtb. To investigate its role, sufficient amounts of material had to be generated to aid the biological assays. Due to tedious isolation, which only provided microgram quantities of semipure 1-TbAd, chemical synthesis was the way to continue. The aforementioned reasons, and the potential of 1-TbAd and N6-TbAd as diagnostic molecules for tuberculosis infection, provided a strong rationale to embark on a stereoselective synthesis of 1-TbAd.
In 2016 we reported the first asymmetric total synthesis of 1-TbAd, providing 2.4 g of enantiopure compound.230 This multigram scale synthesis was initiated by an asymmetric Diels–Alder reaction (Scheme 48). Despite numerous attempts to construct the unsaturated decalin core employing an asymmetric copper-catalyzed exo-selective Diels–Alder reaction,231 the successful approach proved to be the use of a chiral auxiliary. So, the reaction between diene 342 and chiral oxazolidinone tigloyl-based dienophile 361 was performed with excellent stereoinduction (>98% ee, 9:1 exo:endo). In silico studies showed that the Diels–Alder reaction proceeded stepwise from the Si-face (TS) according to the Curtin–Hammett principle via reaction from the thermodynamically less stable s-cis conformer. The chiral auxiliary of 362 was removed by the aid of ethanethiolate which smoothly produced thioester 363 in 96% yield. Reduction followed by oxidation provided aldehyde 346, which was functionalized using an aldol reaction with acetone to provide enone 347. Conjugate reduction by the aid of Wilkinson’s catalyst with triethylsilane yielded ketone 348. A Horner–Wadsworth–Emmons olefination was performed to install the alkene functionality, to produce, after reduction of the ester intermediate, tuberculosinol 340 in 82% over the two steps. Conversion of tuberculosinol 340 into allylic chloride 364 set the stage for the final coupling with adenosine. Alkylation under Finkelstein conditions in DMF smoothly provided the desired natural product, 1-TbAd 360, in 76% yield. 1-TbAd 360 was also quantitatively converted into N6-TbAd 365 by a Dimroth rearrangement induced by aqueous dimethylamine.
Scheme 48. Asymmetric Total Synthesis of 1-TbAd and N6–TbAd.

Recently we reported biological investigations with synthetic 1-TbAd 360 to elucidate its biological function.232 We discovered that 1-TbAd is cell wall associated and acts as a mycobacterial antacid which neutralizes acidic organelles. We showed 1-TbAd uses mechanisms independent of macrophage function since 1-TbAd producing M. kansasii can grow (= survive) in acidic culture media (pH 5.1), whereas wild-type M. kansasii (which does not produce 1-TbAd) cannot grow.
In the context of the macrophage, as alluded to earlier, phagosomal maturation is driven by acidification to induce fusion with the lysosome. This fusion leads to digestion of the bacterium by subjecting it to acid hydrolases. Due to the pKa of 1-TbAd of ∼8.5 (= acidic NH2), it is in equilibrium with its neutral conjugate base, which can traverse biological membranes (in contrast to positively charged 1-TbAd!), in a process called lysosomotropism.233 The conjugate base of 1-TbAd can passively enter the acidified phagosomal space and block the acidification process. The protonation of the conjugate base reintroduces the positive charge in 1-TbAd which traps the molecule inside the phagosome, preventing it from leaving this organelle. Whereas Russell and co-workers showed that a Rv3378c knockout Mtb strain failed to sufficiently neutralize phagosomes,214 we demonstrated this phenomenon is caused by 1-TbAd and that it functions as a lysosomotropic antacid. The consequence of this mechanism is that phagosomal maturation, and thus phago-lysosome fusion, is blocked which stops pathogen digestion by the macrophage and leads to enhanced intracellular survival of Mtb.
The trapping of 1-TbAd in lysosomes was observed using electron microscopy. Treatment of macrophages with chemically synthesized 1-TbAd led to an enormous osmotic swelling, increasing the average lysosome radius by a factor 3–4. Furthermore, comparing electron microscopy images of Mtb and Mtb Rv3378c knockout (= no 1-TbAd production) infected macrophages showed this lysosomal swelling as well, together with an osmotic swelling of the phagosomal space. Other, nonacidic, organelles such as the Golgi, and mitochondria, remained unaltered by 1-TbAd, showing a specificity for acidic compartments.
A more recent study into 1-TbAd’s biological function was reported by the Behr laboratory in 2020.234 1-TbAd producing M. kansasii was used in infection of mice and alveolar macrophages (= lung macrophages), and it was found that in comparison to wild-type M. kansasii the bacterial burden in both the mice (in vivo) and alveolar macrophages (ex vivo) was greater during the first 24 h of infection. These results might imply that 1-TbAd is a “ready-to-use” antacid which allows the bacterium to quickly respond to hostile acidic environments encountered in macrophages. This might give the bacterium time to activate other survival mechanisms to ensure the long-term intracellular survival.
8. Mycobactins
Upon infection of its host, Mtb has to create a microenvironment in which it can survive. By infection of macrophages, Mtb creates such an environment which protects it in part from antibiotics and immune responses. However, in order to ensure years long survival, a continuous supply of “food” is a requisite. One food source which Mtb, and other pathogenic bacteria, cannot live without is iron (in the form of Fe3+).235,236 In order to acquire iron while residing in immune cells, Mtb has to compete with its host to gather a sufficient supply.237 In both the host and Mtb, iron plays a crucial role, either as cofactor or as prosthetic group, in the functioning of essential enzymes that regulate, for instance, metabolism and basic cellular functions such as energy production by oxidative phosphorylation and the production of ATP. To fulfill its iron requirements, Mtb has to “steal” iron from its host, which it does using so-called mycobactins,238 intracellularly produced siderophores by mycobacteria. Siderophores239−242 are low-molecular-weight iron chelators that sequester and transport iron for survival. Two well-known siderophores of Mtb are mycobactin T and carboxymycobactin T, and they have the role of chelating iron and shuttling it into the cytoplasm of the tubercular cell.243
The mycobactins were first isolated from M. Phlei by Francis, Snow, and co-workers back in 1953,244 after being recognized as essential mycobacterial growth factors four decades earlier.245 Meticulous degradation studies by Snow led to the identification of the mycobactin fragments246,247 and eventually full structure elucidation including the stereochemistry.248 The first mycobactin from Mtb was isolated in 1965 and was named mycobactin T.249 In 1995 another class of mycobactins was discovered by the Horwitz laboratory, who coined these molecules carboxymycobactins.250 It was found that carboxymycobactin is a secreted siderophore that recruits iron from the extracellular space and donates the iron to cell wall associated mycobactins, which in turn transport it to its destiny in the cytoplasm.251
It was prior to the isolation of the mycobactins that Francis recognized the therapeutic potential of the mycobactins.238 His ingenious hypothesis was that mycobactins (read mycobactin analogues) can actually be used to inhibit intracellular growth of Mtb. Since Mtb is scavenging iron out of its microenvironment, one can imagine that prebinding this iron with an administered siderophore disables Mtb from assimilating this much needed iron. As a consequence, the depletion of iron in Mtb leads to iron starvation and thus growth inhibition. Since minor changes in siderophore structure can significantly alter the iron binding affinities252 or transport abilities,253 being able to make structurally very similar natural siderophores becomes even more important.
Another strong incentive for the synthesis of siderophores, and thus mycobactins, is its use as a “Trojan Horse” for carrying drugs into bacteria.254−258 Since mycobactins transfer iron through the mycobacterial cell wall, their access to the mycobacterial cytosol can be exploited by synthesis of mycobactin–drug conjugates. One particular example worth highlighting is the synthesis of a mycobactin T analogue conjugated with artemisinin reported by Miller and co-workers.259 Artemisinin is a potent antimalarial drug which by itself does not exhibit antituberculosis activity; however, conjugated to a mycobactin T analogue, the construct showed selective antituberculosis activity even against MDR and XDRT Mtb. Although for drug conjugation, analogues of the mycobactins are needed, which are not discussed in this review, the naturally occurring architectures serve as an indispensable template for such drug designs.
Mycobactin T is thus known for over 55 years but, surprisingly, no total synthesis of precisely this molecule has been undertaken. In 1997 the Miller laboratory, however, performed the total synthesis of mycobactin S.260 Although this molecule is formally from M. smegmatis, mycobactin S differs from mycobactin T in only one stereogenic center and thus resembles mycobactin T. Moreover, to further justify its inclusion in this review, mycobactin S was chosen as the target to test Francis’ hypothesis that alternative forms of my mycobactin might possess inhibitory activity against Mtb.
The Miller synthesis of mycobactin S (Scheme 49) started with construction of mycobactic acid, the oxazoline containing fragment. Methyl salicate 366 was amidated with L-serine benzyl ester after a benzylation ester hydrolysis sequence. The oxazoline was installed by treatment of 367 with Burgess reagent 368, which rather than as a dehydrating agent functioned as a leaving group. Hydrogenolysis then provided mycobactic acid 369 in 50% yield over the five steps.
Scheme 49. Total Synthesis of Mycobactin S by Hu and Miller.

The other two necessary fragments, cobactin T 372 and the SEM protected hydroxamic acid 373, were both constructed starting from amino acid derivative 370. The lactam was constructed in a six-step sequence via the intermediacy of a hydroxylamine (product from nitrone hydrolysis). The amine functionality in 371 was hydrogenolytically liberated after which it was reacted with β-hydroxybutanoic acid, to produce cobactin T 372 after removal of the silyl protecting group. Via a similar pathway SEM-protected hydroxamic acid 373 was produced. After removal of the Cbz-group, the fusion with mycobactic acid 369 was executed. Hydrolysis of the methyl ester set the stage for the Mitsunobu reaction between 374 and cobactin T 372. The silyl groups were removed which provided mycobactin S 375 in a longest linear sequence of 10 steps.
The obtained mycobactin S was subjected to biological tests and showed that at a concentration of 12.5 μg/mL the growth of Mtb H37Rv was inhibited by 99% and that the minimum inhibitory concentration of mycobactin was 3.13 μg/mL. This result confirmed the 50-year-old hypothesis from Francis that alternative forms of my mycobactin might possess inhibitory activity against Mtb. It is remarkable that inversion of a single stereogenic center converts a growth factor for Mtb, mycobactin T, into a growth inhibitor.
In 2003, Poreddy et al. embarked on the solid-phase synthesis of methyl carboxymycobactin T 386 and several analogues (Scheme 50).261 For their synthesis, hydroxyl amine O-functionalized with Wang resin was chosen as the solid-phase. The amine moiety was nosylated in order to be monofunctionalized with protected L-6-hydroxynorleucine 381 by means of a Mitsunobu reaction. The lactam was then introduced in three steps to, after removal of the Bpoc group, react with (S)-β-hydroxybutanoic acid to produce 383. 384 was then synthesized by first performing a Mitsunobu reaction between the carboxylic acid moiety in L-6-hydroxynorleucine 382 and the hydroxyl unit of 383, followed by removal of the 2-(p-biphenylyl)-2-propyloxycarbonyl (Bpoc) protecting group. This set the stage for introducing the mycobactic acid 369 onto 384 using standard amidation chemistry. The desired methyl carboxymycobactin T 386 was then obtained in 14% over four steps.
Scheme 50. Solid Phase Synthesis of Methyl Carboxymycobactin T by Poreddy et al.

The obtained carboxymycobactin catalogue was investigated for its iron binding properties, as well as their inhibitory activity of M. avium. While at different concentrations, growth was inhibited in the first week to about 10–15% of the control, the following weeks the inhibition diminished and was negligible compared to the control by week 3 and 4. These results were attributed to slow degradation of the carboxymycobactin by the bacteria.
In 2004 the Moody laboratory discovered a new member of the mycobactin family as part of a screening for candidate antigens of CD1a.262 Subjecting chromatographic fractions of Mtb cell wall extracts to a CD1a-restricted T-cell line showed that these were activated by a molecule with a nominal mass of 838 Da. To gain more information about the molecular structure that causes T-cell activation, collisional mass spectrometry and NMR analysis were performed revealing the structure to be congruent with the lipopeptide dideoxymycobactin (DDM). DDM is a depsipeptide with four stereogenic centers which are part of an unusual α-methyl serine that is cyclized to an oxazoline, a 3-hydroxy butyric acid unit, and two lysines of which one is cyclized as a caprolactam. The stereochemistry of the lysine amino acids was known, but the former two stereogenic residues were of unknown stereochemistry. Since NMR and mass spectrometry analyses were unable to address the question of the stereochemistry of DDM, together with insufficient quantities being isolated for studies of its role in iron acquisition and testing T-cell responses in human populations, there was sufficient incentive to embark on a total synthesis.
The first total synthesis of DDM-838 393 was performed in the Moody laboratory and consisted of a solid-phase synthesis (Scheme 51).263 The synthesis was rather straightforward in the sense that standard solid-phase deprotection–coupling sequences were employed. The synthesis was built-up by first introducing the hydroxy butyramide functionality providing 389, followed by a HATU-mediated esterification. The cis-eicosenonic amide moiety was constructed and then coupled with oxazolinic acid 391 to provide 392. The 4-methyltrityl (Mtt) group in 392 was cleaved with simultaneous release from the solid support, after which the caprolactam was formed by intramolecular amide coupling. DDM-838 393 was purified by preparative HPLC to provide submilligram quantities which were sufficient for further studies.
Scheme 51. Solid-Phase Synthesis of Dideoxymycobactins.

Using the solid-phase synthesis, all four diastereoisomers (varying the stereocenters designated as * in Scheme 51) were synthesized to elucidate the relative stereochemistry of DDM-838. The synthetic compounds were tested in a T-cell response assay using intact dendritic cells by subjecting CD1a-restricted T cells to the diastereomers. Interleukin-2 release was measured, and it was found that only DDM-838 with the (S,R)-stereochemistry on the α-methyl serine and 3-hydroxy butyric acid (as drawn in Scheme 51), respectively, exhibited the exact same bioactivity as the natural isolate. The diastereomeric compounds, with alterations at either stereogenic center, exhibited a 1000-fold or greater loss of T-cell activation, showcasing the importance of enantiopure compounds in immunoassays.
A second total synthesis of DDM was reported in 2017 by the Williams group.264 Their synthesis was designed to optimize the synthesis by Young et al.263 in terms of overall yield and potentially, undetected, epimerization during esterification or peptide coupling. The Williams synthesis was initiated by construction of the two building blocks 398 and 400 (Scheme 52, first approach). Amine 394 was condensed with 395 using COMU as the peptide coupling agent to produce amide 396. The α-methyl serine moiety was cyclized by the action of XtalFluor-M to introduce the oxazoline group in 397. The ester of 397 was hydrolyzed, and the formed carboxylic acid was reacted with a protected lysine to form the peptide bond. The ester from the protected lysine was then hydrolyzed to give 398, which was esterified with amide 400, formed by reaction of caprolactam 399 with hydroxybutyric acid 388. The esterification proceeded in a moderate 51% yield, and in addition, the stereocenter in 401 was fully epimerized under the coupling conditions providing a 1:1 mixture of S (desired) and R (undesired) isomers of 401. A three-step sequence comprising benzyl hydrogenolysis, Boc deprotection, and EDC mediated coupling of 402 gave rise to DDM-838 393 in 15% yield, together with its stereoisomer in 29% yield. An unexpected finding was that during the EDC-mediated coupling with pure (Z)-402, alkene isomerization took place as well, thereby also providing two alkene isomers (not shown) of DDM-838 393 and its stereoisomer.
Scheme 52. Williams’ Synthesis of Dideoxymycobactins.

To address the undesired epimerization in the esterification of 398 and 400, and to circumvent the alkene isomerization in the last step of the DDM total synthesis, the Williams group designed a second synthetic strategy (Scheme 52, second approach). In this linear synthesis, the esterification reaction was performed with less complex reagents by employing a Mitsunobu reaction between 403 and Fmoc-l-lysine(Z)-OH. Despite a significant amount of elimination in this reaction, no epimerization was observed. Deprotection of the Fmoc moiety provided 404 in 26% over the two steps. The synthesis of DDM-838 393 was finalized in a three-step sequence of which the last step was a DIC-mediated amide coupling with acid 402. In this amidation reaction, no alkene isomerization took place. The overall yield for approach two was not higher than that of the first approach. However, despite the sacrifice of synthetic convergence in the second strategy, no unwanted isomerization reactions took place that hampered isolation procedures.
The synthetic compounds from this total synthesis were also used in biological assays to gain more insight into the structure–activity of DDM-838. Although not favored during synthesis, the stereoisomers with inverted stereochemistry at the lysine and the alkene geometrical isomer provided a unique opportunity to investigate the bioactivity of structurally related DDM-838 structures. In this immunoassay, CD1a restricted T-cells derived from unrelated human donors were subjected to the DDM-838 molecules. It was found that the molecules exhibited similar reactivity for each donor. Whereas in the study by Young et al. (vide supra)263 only DDM-838 showed bioactivity and other diastereomers not, the DDM-838 isomer with E-alkene geometry exhibited a significant but attenuated activity. Also, the DDM-838 isomer with opposite stereochemistry at the central lysine unit showed some activity. Bioactivity was not seen using DDM-838 having unnatural stereochemistry at the lysine and the acyl lipid. This structure–activity relationship, together with the results obtained by Young et al.,263 highlighted the eminent sensitivity of the CD1a restricted lipopeptide-reactive T-cells for naturally occurring DDM-838 stereochemistry.
9. Cell Wall Oligo- and Polysaccharides
Another major component of the mycobacterial cell wall is the mycolyl arabinogalactan peptidoglycan complex (mAGP), which is interspersed with the lipoglycans phosphatidylinositol mannosides (PIM), lipomannan (LM), lipoarabinomannan (LAM), and mannose-capped LAM (ManLAM) and anchored by non-covalent interactions with their phosphatidyl-myo-inositol (PI) moieties (Figure 5).265−267 LM, LAM, and ManLAM all contain a conserved PIM motif.
Figure 5.
Chemical architecture of mycobacterial cell wall oligo- and polysaccharides.268 Reproduced with permission from ref (268). Copyright 2021 Springer Nature.
One can imagine that dissection and elucidation of this complex array of lipidated oligo- and polysaccharides has been a monumental task. Considering that even with modern analytical techniques deciphering these molecular architectures is challenging269 makes the pioneering work by Anderson, who identified the structural features of the PIMs, even more impressive.270 By saponification of phosphatide fractions from several mycobacteria, including Mtb, Anderson was able to identify all the structural elements belonging to the PIMs, namely fatty acids, an organophosphoric acid, phosphorylated glycan, and mannositose which hydrolyzed to mannose and inositol. It was Ballou and co-workers who in a series of publications meticulously pieced together the fragments to arrive at the chemical structures we now know as the PIMs.271−274 There are multiple different PIMs with a varying number of mannose units (PIM1–6) and acyl residues (up to four). In PIM1, the inositol core of phosphatidylinositol is mannosylated at C-2 and PIM2 contains an additional mannose at C-6. Further mannosylations give rise to PIM3 to PIM6. PIMs are known to carry different fatty acyl chains, with palmitic and tuberculostearic acids being the most abundant acyl residues. Thirty years after the impressive structure elucidation by Ballou, the position of the fatty acid moiety in the acetylated PIMs (Ac1PIMs) was determined to be at the C6 position of the mannose linked to the C2-position of the innositol.275 Work by Gilleron et al. showed that another fatty acid, that in Ac2PIMs, is present on the C3-position of the inositol core.276
The exact role of PIMs in Mtb pathogenesis remains unclear as studies describe various different biological effects of PIMs triggering divergent host responses, at times, also depending on the exact chemical identity of these lipoglycans.277 PIMs have been shown to induce early endosomal fusion within macrophages as such supplying nutrients to the pathogen.278 Furthermore, PIMs are involved in infection establishment by participation in receptor-mediated internalization in macrophages279 and inhibit macrophage activation and host inflammatory responses.280 Higher order acylated PIMs preferentially interact with mannose receptors on macrophages inhibiting phagosome–lysosome fusion,281 and lower order PIMs from Mtb preferentially interact with the dendritic cell specific intercellular adhesion molecule-grabbing nonintegrin (DC-SIGN) protein.282 PIMs are also involved in T-cell activation by presentation through CD1e-CD1b-coexpressing antigen presenting cells.283 In addition, PIMs have been shown to directly bind to VLA-5 on CD4+ T-cells.284 It is thought that PIM2 (and potentially PIM3 or PIM4) is the biosynthetic precursor for LM, LAM, and ManLAM. Further extension of position 6 of the PIM core by introduction of an α-1,6 linked mannose backbone carrying varying amounts of single α-mannose residues at the 2-position gives rise to LM. To this date, it is not established if LM is an end-product or merely a biosynthetic intermediate for the formation of LAM. Formation of LAM is achieved by further attachment of arabinans (repeating units of α-1,5 arabinose). The arabinan skeleton of LAM can be capped with one or two α-mannoses at position 5 producing ManLAM. LAM itself is a highly heterogeneous entity with varying degree and pattern of branching in the mannan and arabinan subunits.266 The elucidation of the exact structure of the cell wall oligo- and polysaccharides has still not been concluded as evident from a very recent report by Jackson and co-workers, who showed a structural refinement of the LAM structure.268 The structure elucidation of LAM has considerable history, and before this structural revision took place, other groups were dedicated to elucidating the structure. This started with the initial structure analysis of PIMs in 1965.274 A few years later, the attachment of arabinan to the α-1,6-mannan backbone was unraveled285 followed by the discovery that arabinomannan is linked to PIM.286,287 The position of the linkage was then unveiled to be at the 6-O of the mannosyl residue, which itself is attached at its 2-position to the 6-O of inositol.288 In 1999, the exact location of the acyl substituents in PIM and LAM of M. bovis was reported.289 Furthermore, structural research also shed light on the architecture of the six-linked mannan backbone, carrying terminal α-Manp residues at the 2-position.288,290,291 In addition, various structural studies on arabinan292,293 and its terminal caps290,294−296 have been communicated over the years. In a recent report, investigations of the LAM biosynthesis revealed that the first five to seven Manp units attached to inositol are present as a linear saccharide chain.297 Ultimately, a report from the Jackson laboratory then discovered the presence of extended secondary mannan side chains linked to the internal mannan chain, as well as that the attachment of the arabinan unit is at the nonreducing end of mannan and not at the internal regions as thought previously.268
The biological effects exerted by LM, LAM, and ManLAM have been reviewed extensively.32,267,298,299 In brief, ManLAM has been shown to engage the mannose receptor of macrophages thus inducing phagocytosis, whereas uncapped LAM (also referred to as AraLAM) did not produce this effect.300 This selectivity in biological effect was also observed in the activation of DC-SIGN on dendritic cells by ManLAM but not AraLAM, leading to the hypothesis that LAM capping evolved as the mechanism to attenuate host inflammatory responses.301,302 ManLAM also inhibits phagosome–lysosome fusion by engaging mannose receptors.303,304 Furthermore, LAM has been found to be an antigen and is presented in a mannose receptor mediated process to T-cells by loading on CD1b molecules.305 LAM is also an urine biomarker for active TB and utilized in point-of-care clinical tests.306
9.1. Phosphatidylinositol Mannosides
Several synthetic studies and total syntheses of PIMs have been reported up to 1995 but fall outside the scope of this review.307−309
In 1996 Watanabe and co-workers reported a synthesis of a PIM2 (Scheme 53).310 Their synthetic approach relied heavily on in-house developed phosphite chemistry for both the glycosidations and the phosphorylation. Starting with such a phosphite glycosidation, rac-405 was reacted with glycoside donor 406 yielding α-anomers 407a and 407b as an inseparable mixture (45%, 1:1) together with the easily removable β-anomer (15%). Acetal deprotection gave, after separation of the diastereomers, 408 in 45% yield. Triol 408 was subjected to a phosphorylation with glyceryl dimethyl phosphite 412, derived from sn-glycerol 1,2-carbonate 409 and dimethyl N,N-diisopropylphosphoramidite 410. Treatment of 408 with phosphite 412 in the presence of pyridinium tribromide yielded phosphorylated glycoside 411 in 83% (dr = 1:1, due to the asymmetric P-atom) with excellent regioselectivity. The diastereomeric mixture of 411 was then stereo- and regioselectively glycosidated in 74% using 406 to furnish trisaccharide 413. Carbonate cleavage by treatment with EtMgBr and regioselective diacylation were followed by a three step deprotection sequence furnished PIM2414 in 43% over the five steps.
Scheme 53. Watanabe’s PIM2 Total Synthesis.

In 2003, Schmidt and co-workers communicated the total synthesis of PIM1 (Scheme 54).311 Their endeavor started with the acetal protection of myo-inositol 415 with cyclohexanone. This uncontrolled reaction provided, besides the desired product 416, also isomeric side-products. These were easily removed by crystallization and flash column chromatography affording clean 416. In order to get the isomerically pure D-myo-inositol scaffold, a resolution was performed after selective installation of a menthol-derived chiral auxiliary. With the resolution performed, the allyl protecting group was installed affording 417. Selective removal of the 4,5-O-cyclohexylidene acetal by treatment with a pyridinium p-toluenesulfonate (PPTS) p-toluenesulfonic acid (PTSA) mixture furnished diol 418 in 68% yield. A sequence of protecting group manipulations then set the stage for the glycosidation of myo-inositol 419 with trichloroacetimidate 420. Using 5 mol% of Sn(OTf)2 as the catalyst, smooth glycosidation occurred providing pseudodisaccharide 421 with complete α-selectivity in 95% yield. With this key-step performed, a straightforward sequence of protecting group removal and phosphorylation provided the desired PIM1423.
Scheme 54. Total Synthesis of PIM1 by the Schmidt Laboratory.

In 2006, the Larsen laboratory communicated their synthesis of two PIM1 regioisomers 427 and 432 carrying a mannosyl group at the 2- and 6-positions of the inositol core, respectively, as well as PIM2414 (Scheme 55).312 Contrary to previous syntheses, their prepared PIMs are equipped with stearyl esters at the phosphatidyl unit, as these have been identified to be present in biologically active PIM extracts. The synthesis of the 2-O-mannosylated PIM1427 was initiated by the TMSOTf-promoted glycosylation of inositol acceptor 424 with mannosyl phoshite 406 providing an inseparable mixture of α- and β-configured pseudodisaccharides, which were separated after PMB removal, giving 425 in 41% yield. The phosphate group was then installed by coupling of 425 to phosphonate 426 in the presence of pivaloyl chloride, followed by iodine-mediated oxidation. The desired PIM1427 was obtained as the triethylammonium salt in 76% yield over two steps. For the synthesis of 6-O-mannosyl PIM1432, diol 429 was selectively glycosylated with mannosyl imidate 428 to provide 430 in 50% yield. After a sequence of protecting group manipulations, pseudodisaccharide 431 was coupled to phosphonate 426 as done previously, giving 6-O-mannosyl-PIM1432 in 60% yield over two steps. PIM2 was also accessed starting from diol 429. After double glycosylation with excess mannosyl imidate 428, 433 was obtained in 57% yield. Again, a series of protecting group manipulations was performed arriving at 434, which allowed access of PIM2 by phosphonate-coupling and global deprotecting in 27% yield over two steps. The authors used the synthetic PIMs to assess their ability to suppress eosinophil (= type of white blood cell) recruitment in an asthma mouse model. After intranasal administration of the synthetic PIMs, a significant decrease of the number of eosinophils were observed, with PIM1432 generating the strongest immunosuppression. Furthermore, this effect was not only limited to eosinophils, but a decrease in the levels of both lymphocyte and macrophage was observed, demonstrating an immunosuppressive effect of mycobacterial PIMs that is not restricted to a single immune cell type. Following up on this synthesis, the crystal structure of murine CD1d bound to synthetic PIM2414 was characterized. By gaining structural information on the binding mode of PIMs to CD1d, the authors anticipate that based on this, new agonists for NKT cells presented by CD1d could be designed in a rational fashion.313
Scheme 55. Total Synthesis of Two Regioisomers of PIM1 and Synthesis of PIM2.

Shortly after their synthesis of PIM1427 and 432, and PIM2414, the same group reported the synthesis of PIM4443 and an improved synthesis of PIM2436 in order to investigate their immune adjuvant properties (Scheme 56).314 The construction of the known pseudotrisaccharide 434 was performed in analogy to Scheme 55, yet the yield of the double glycosylation reaction could be improved to 70% by changing the solvent from dichloromethane to diethyl ether. The phosphate moiety was then installed by coupling of phosporamidite 435 to 434 and in situ oxidation. After deprotection, the desired PIM2436 was obtained in 66% yield. In the synthesis of PIM4443, disaccharide donor 439 was accessed in 58% over three steps by TMSOTf-promoted glycosylation of alcohol 437 with trichloroacetimidate 438, followed by hydrolysis and imidate formation. Next, pseudotrisaccharide 433 was prepared by selective glycosylation of diol 429, first with donor 438 followed by donor 440. Subsequent desilylation provided then 433 in 27% yield over three steps. Building blocks 433 was then glycosylated with 439 were then coupled by glycosylation to give 441 in 80% yield, and subsequent routine protecting group manipulations delivered alcohol 442. Pseudopentasaccharide 442 was then coupled, as done before, to phosphoramidite 435 and delivered after deprotection PIM4443 in 46% yield over two steps. Synthetic PIM2436 and PIM4443 were then tested as potential adjuvants in an ovalbumin-specific mouse model. Injection of 436 and 443 with a model antigen resulted in a significant immune response as measured by INF-γ production compared to the negative control, with PIM2436 causing a stronger response than PIM4443. Notably, both synthetic PIMs did not cause significant local lesions or edema compared to Complete Freud’s Adjuvant (CFA), which was used as positive control, indicating that PIMs possess adjuvant properties without the toxic effects of CFA.
Scheme 56. Improved Synthesis of PIM2 and Total Synthesis of PIM4.

Building on their synthesis of various PIMs, in 2011 Harper and Painter (collaborating with Larsen) reported the synthesis of PIM6460.315 The synthesis was initiated with the preparation of tetramannosyl building block 454 (Scheme 57). Starting from benzylidene protected mannose thioglycoside 444, the 2- and 3-OH groups were protected in 73% yield, followed by reductive opening of the benzylidene group providing alcohol 446 in 90% yield. The primary hydroxyl of 446 was then silylated giving thioglycoside 447. Next, donor 450 was prepared by protection of the 2-OH of 448 as chemically orthogonal 2-(azidomethyl)benzoyl (AZMB) ester and subsequent thioglycoside hydrolysis and trichloroacetimidate formation. With these monosaccharide units in hand, the desired tetrasaccharide building block 454 was assembled. For this, TMSOTf-promoted glycosidation of acceptor 446 with 2-OAZMB donor 450 provided disaccharide 451 in the excellent yield of 94%. The 2-OH was then liberated by removal of the AZMB ester with tributyl phosphine delivering acceptor 452 in 83% yield. Again, glycosylation of 452 with donor 450, subsequent AZMB removal, and last glycosylation with donor 453 then provided the desired tetrasaccharide 454 in 36% yield over three steps.
Scheme 57. Synthesis of PIM6 Building Blocks.

Next in the synthesis of PIM6460, the inositol core was prepared (Scheme 58). First, regioselective glycosylation of diol 429 with thioglycoside donor 447 provided 455 in 57%. Pseudodisaccharide 455 was then subjected to glycosylation with 456 followed by desilylation with acetyl chloride giving pseudotrisaccharide 457 in 75% yield over two steps. Coupling of 457 with tetrasaccharide 454 by glycosylation then delivered 458 in 76% yield which contained the complete carbohydrate skeleton of PIM6460. A sequence of protecting group manipulations then set the stage for the attachment of the phospholipid unit, which was brought about by coupling of 459 with phosphoramidite 435 and in situ oxidation. After global deprotection, the desired PIM6460 was obtained as triethylammonium salt in 67% yield over two steps. The 1H NMR spectra of natural and synthetic PIM6 were compared and revealed that synthetic and natural material are in good agreement. With synthetic PIM6, the authors saught to answer the question whether PIM6 enhances or suppresses T-cell immune responses. In a human mixed lymphocyte reaction assay, synthetic PIM6 caused dendritic cell dependent suppression of CD8+ T cell expansion in a dose dependent manner. These results demonstrate the immunosuppressive effects of PIM6in vitro, an effect well-known for Mtb residing in human host cells.
Scheme 58. PIM6 Completion.

After their efforts to access PIM2, PIM4, and PIM6, the synthetic PIMs were tested as ligands for DC-SIGN, the major receptor for Mtb on dendritic cells. The study revealed that DC-SIGN has a higher affinity for PIM6 compared to its lesser mannosylated counterparts PIM2 and PIM4. Yet, in experiments with M. bovis mutants lacking PIMs, DC-SIGN interactions were still intact, indicating that DC-SIGN interactions are far more complex and that mycobacteria express other DC-SIGN ligands beyond PIMs, potentially even mannosylated proteins.282
The group of Seeberger reported the combined synthesis of both natural acetylated PIM2 and acetylated PIM6.316 Whereas previous syntheses focused merely on efficient introduction of the glycosidic bonds, in this study, the phospholipid moiety was installed mimicking the natural scenario. The nature of the phospholipid composition in antigenic glycosides can play a crucial role in immune response induction.317
The lipid component of the phospholipid moiety is tuberculostearic acid 322. Seeberger’s synthesis of this lipid started with a THP protection of (S)-Roche ester followed by reduction of the ester functionality and subsequent tosylation, giving 461 (Scheme 59). A cuprate coupling/THP deprotection sequence, and again tosylation/cuprate coupling, gave after a TEMPO oxidation TBSA 322 in 57% over the four steps. A Steglich esterification of benzyl protected glycerol 462 provided 463 in 75% isolated yield, notably without acyl migration. A second acylation and debenzylation (with minor acyl 2 to 3 migration) giving 464 set the stage for the phosphorylation event yielding H-phosphonate 324.
Scheme 59. Tuberculostearic Acid Bearing Phospholipid Synthesis by Seeberger and co-workers.

In order to maximize the synthetic convergence of the PIM2 and PIM6 syntheses, three mannose building blocks 428, 467, and 470, from two mannose orthoesters 465 and 466, were crafted (Scheme 60). Orthoester 465 was smoothly converted into trichloroacetimidate 428 using a hydrolysis/acetimidate formation sequence. Allylation and subsequent desilylation of 466 gave 467, a component in the synthesis of PIM2.
Scheme 60. Seeberger’s Synthesis of AcPIM2 and AcPIM6 Building Blocks.

Glycoside donor 428 was reacted with glycoside acceptor 467 promoted by TMSOTf, followed by the cleavage of the acetate to furnish 468 in 80% over two steps. A second iteration of the glycosidation event using glycoside donor 428 furnished 469. This was followed by glycosylation, deallylation, and trichloroacetimidate formation to give tetramannoside 471, a key-component in the construction of PIM6.
With all the building blocks in hand, the assembly of the components was performed by first constructing PIM2 starting from enantiopure myo-inositol derivative 472 (Scheme 61).318 A three-step sequence performed on 472 gave 473, which was glycosidated with trichloroacetimidate 470 and subsequently desilylated to furnish diglycoside 474. The free hydroxyl group of 474 was esterified with levulinic acid, and the naphthyl group was removed by treatment with DDQ yielding 475 in 75% over the two steps. Mannosylation of pseudodisaccharide 475 with 470 stereoselectively furnished 476. A protection/deprotection sequence, including palmitoylation, was executed with consecutive phosphorylation employing H-phosphonate 464 to obtain fully protected PIM2 . Deprotection, however, proved less than trivial since employing standard debenzylation conditions with Pd(OH)2/C in CHCl3/MeOH/H2O gave unexpected partial cleavage of the lipids from the carbohydrate backbone. Careful screening of the reaction conditions led to the debenzylation employing Pd/C in EtOAc/THF/PrOH/H2O, yielding AcPIM2478 in 82%.
Scheme 61. Seeberger’s AcPIM2 Synthesis.

The syntheses outlined above provided the authors with tetrasaccharide 471 (Scheme 60) and trisaccharide 479 (obtained by delevulinoylation of 476, Scheme 61) needed for the construction of AcPIM6 (Scheme 62). Thus, the union of saccharides 471 and 479 by treatment with catalytic TMSOTf gave 480 in 91% yield. A protection/deprotection strategy was employed comprising a palmitoylation to furnish heptasaccharide 481. The final deallylation, however, as in the PIM2 synthesis (not discussed) using PdCl2 as the catalyst, proved to be problematic. This deallylation reaction provided 481 in only 40% yield with an equal amount of Wacker oxidation byproduct (30% in the AcPIM2 synthesis). The solution was found in the use of a cationic CpRuII complex with quinaldic acid in MeOH319 which cleanly afforded heptasaccharide 481 in 72% yield, albeit that a stoichiometric amount of catalyst was required. Phosphorylation and full debenzylation of 481 as performed in the PIM2 synthesis smoothly provide acetylated PIM6482 in 62% over the two steps.
Scheme 62. Completion of the Total Synthesis of AcPIM6.

The second group to report on the synthesis of a tuberculostearic-based PIM2 (and not shown PIM1) was the Larsen laboratory.320 Their synthesis of the tuberculostearic acid 322 was straightforwardly achieved in eight steps from commercially available (S)-citronellol (Scheme 63). The phospholipid 484 was accessed in four steps starting from (R)-benzylglycidol obtained by a catalytic kinetic resolution as described in Scheme 40.
Scheme 63. Larsen’s Tuberculostearic Acid and Phosphoramidite Synthesis.

The construction of the trisaccharide moiety was cleverly achieved by a double glycosidation of unsymmetrical myo-inositol building block 485 (Scheme 64) which was obtained via a Ferrier rearrangement of a glucose derivative.318 The double glycosidation smoothly provided α,α-pseudotrisaccharide 486 with minor formation of the α,β-isomer (10%) when performed in ether as the solvent. Routine protecting group manipulations gave alcohol 434 in 43% over three steps. Treatment of 405 with phosphoramidite 484 in the presence of 1H-imidazole followed by in situ oxidation gave, after hydrogenolysis, the desired PIM2487.
Scheme 64. Larsen’s Total Synthesis of PIM2.

It should be noted that the synthesized PIM2 has the TBSA lipid on the sn-2 position of the glycerol moiety, whereas in the previous described synthesis by Seeberger the TBSA lipid was on the sn-1 position. The reason was that both acylation patterns were reported to be present in the PIMs. In 2001 Gilleron et al. reported that PIM2 comes in a 68:32 mixture of acylation patterns with the major species having the TBSA moiety on the sn-2 position (synthesized by Larsen and co-workers) and the minor species bearing two palmitoyl groups.321 In 2003, however, it was reported by the same group that AcPIM6 has the TBSA lipid on the sn-1 position322 (the PIM2isomer synthesized by Seeberger and co-workers), thereby creating some structural ambiguity. Unfortunately for the Larsen lab this ambiguity was solved during the course of their synthetic efforts, as Gilleron and co-workers revised their previous structural characterization of the PIMs, concluding the TBSA lipid is located at the sn-1 position.276 Larsen and co-workers thus, unintentionally, synthesized an unnatural isomer.
Nevertheless, not putting their synthesized materials to waste, the natural PIM1, unnatural PIM2 (with TBSA on the sn-2 positions), and natural PIM2 (containing two palmityl fatty acids) were subjected to a biological evolution. Due to their immunomodulatory properties, it was studied whether these PIMs modulate the release of the pro-inflammatory Th1 cytokine interleukin-12 (IL-12) by treatment of dendritic cells (DCs) from mice. Interestingly, in this in vitro experiment, the PIM2 possessing two palmitoyl residues showed more IL-12 release than PIM1 and the tuberculostearyl containing PIM2487. This result shows that stimulation of the dendritic cell is very sensitive to minor structural changes in the fatty acid residue on the sn-2 position.
Merely two years after their first synthesis of PIM2 and PIM6, the Seeberger group communicated the synthesis of the complete catalogue of PIM analogues (PIM1 to PIM6).323 Importantly, in this synthesis the phosphate group was equipped with a thiol linker which allows immobilization to, for example, carrier proteins, beads, quantum dots, microarrays, or surface plasmon resonance (SPR) surfaces. In this way, the Seeberger group performed biochemical analysis of the complete PIM catalogue, something missing in previous synthetic studies.
In their first-generation PIM synthesis, the Seeberger laboratory used mannosyl trichloroacetimidates as glycosyl donors. This synthesis relied on the use of phosphate mannosyl donors 488 and 490 which were obtained from mannose (Scheme 65). Mannosides 490 and 467 were constructed via tricyclic orthoester 489, efficiently obtained in 70% overall yield, in six steps from mannose, notably with only one purification step. Phosphate 488 was obtained in six steps via orthoester intermediate 465.
Scheme 65. Seeberger’s Building Block Synthesis for the Entire PIM Catalogue.

The myo-inositol building block 473 was prepared from methyl glucopyranoside 491. The pyranoside functionality 492 was converted into the myo-inositol moiety via a Ferrier carbocyclization giving 472. Various protecting/deprotection reactions eventually furnished compound 473. Glycosidation of myo-inositol 473 with phosphate 490 provided the desired α-glycosidic bond in high yield. Protecting group manipulations gave pseudodisaccharide 475 which was glycosidated, again, using mannosyl phosphate 488. This glycosidation was found to be nontrivial since reaction under the standard conditions (TMSOTf as promotor at −40 °C) led to decomposition of 488 to form the corresponding anomeric alcohol. Changing the promotor TMSOTf to the milder activator TBSOTf had a significant effect, providing the glycoside in 27% yield. Changing the temperature to 0 °C led to pseudotrisaccharide 493 in 85% yield after cleavage of the Lev protecting group. 493 served as the key component for the construction of PIM2 to PIM6.
The building blocks 467 and 488 were used to construct the three oligomannosylating agents 495, 497, and 471 for the synthesis of PIM4 to PIM6 (Scheme 66). Glycosidation of 467 with 488 gave disaccharide 494. Deacylation of 494 followed by another glycosidation with 488 then gave trisaccharide 496. Repetition of this sequence on 496 also provided tetrasaccharide 498. All the oligosaccharides were deallylated and converted to the anomeric trichloroacetimidates to efficiently provide 495, 497, and 471.
Scheme 66. Synthesis of the Oligomannosylating Agents for the Synthesis of the PIM Catalogue.

The final stages of the PIM2 to PIM6 synthesis consisted of the unification of myo-inositol 499 with glycosyl donors 488, 495, 497, and 471 (Scheme 67). To construct PIM1, phosphate 488 was reacted with myo-inositol 400 (synthesized from 472) to furnish 500 in 69% yield after deacetylation and benzylation. Pseudotrisaccharide 493 was glycosidated with donors 488, 495, 497, and 471 followed by debenzoylation and benzylation to give the protected PIM2 to PIM6 backbones 501–505. The protected PIMs 500–505 were deallylated using an [Ir(COD)(PCH3Ph2)2]PF6-catalyzed isomerization to the corresponding enol ether which was subsequently hydrolyzed by treatment with p-TsOH. Installation of the phosphate moiety, with the thiohexyl linker, was followed by complete debenzylation through Birch reduction to furnish the complete PIM catalogue. The PIMs were formed as a mixture of free thiols and disulfide dimers. Before immobilization of the PIMs on a maleimide activated glass slide, they were treated with tris(carboxyethyl) phosphine hydrochloride to reduce the dimeric product into the monomeric species.
Scheme 67. Seeberger’s Completion of the Synthesis of the Entire Catalogue of PIM Analogues.

Interestingly, after the synthetic efforts, the catalogue of PIM analogues was biologically evaluated for its activity in binding to DC-SIGN. This protein functions as a receptor on dendritic cells and is able to recognize evolutionary conserved pathogenic structures on the surface of either viruses or bacteria. Binding of the pathogen to the DC-SIGN protein sets into motion a mechanism in which the antigens are internalized, processed, and presented, together with costimulatory molecules, on the surface of the dendritic cells. After binding of the DC-SIGN protein with the immobilized PIM molecules and incubation with fluorescein-conjugated anti-DC-SIGN antibody, the fluorescence intensity was monitored. It was found that the greatest binding was observed for PIM5 and PIM6, underlining the importance of the α-1,2-mannose motif in the PIMs and ManLAM structures. Furthermore, the adjuvant activity of the PIMs was also assessed, and it was shown that the PIMs act as immune stimulators during in vivo immunization experiments when coupled to the model antigen keyhole limpet hemocyanin (KHL). PIMs are thus potential candidates as adjuvants for vaccine development.
Three other recent synthetic endeavors into the synthesis of PIM2 were reported by Hung and Lear in 2009 and 2010. Hung’s synthesis was based on the deracemization of benzylated meso-myo-inositol 1,3,5-orthoformate 507 by reaction with D-mannose-derived donor 420 (Scheme 68).324 With four diastereomeric products possible, a series of experiments had to be conducted to find optimal conditions for the reaction. It was found that BF3·OEt2 was the best promotor when carrying out the reaction at an initial temperature of −78 °C and gradually warming to −20 °C. The reaction exclusively produced the α-anomers 508 and 509 in 64% and 12% yield, respectively, which were separable by flash chromatography. Debenzylation set the stage for a mannosylation of 510 with donor 420 to furnish pseudotrisaccharide 511 regioselectively as a single diastereomer in 87% yield. Cleavage of the orthoacetate in 511 produced a tetraol which had to be regioselectively benzylated at C3–5 to give alcohol 434. Williamson ether synthesis with NaH and BnBr gave a complicated mixture of isomers, an outcome which was circumvented by first a complete silylation giving 512 and subsequent regioselective desilyation/benzylation by the aid of benzaldehyde, TMSOTf, and Et3SiH. With alcohol 434 in hand, a phosphorylation with H-phosphonate 513 and removal of the benzyl protecting groups smoothly afforded PIM2414.
Scheme 68. Hung’s Desymmetrization Strategy for the Synthesis of PIM2.

Just one year after their synthesis of PIM2, the Hung laboratory communicated the synthesis of triacylated PIM2 (Scheme 69).325 Here, their previously employed desymmetrization approach was evolved to a stereoselective one-pot double glycosylation of triol 514 with D-mannosyl trichloroacetimidate 420. A mixture of three separable products was formed, and the desired pseudotrisaccharide 511 was isolated in 64% yield. With 511 in hand, the orthoester was hydrolyzed followed by per-O-trimethylsilylation providing 512 in excellent yield. Regioselective benzyl protection of two out of four available hydroxyl groups was achieved by utilizing their in-house developed two-step reductive benzylation giving diol 517 in 72% yield. Regioselective Steglich esterification of 517 with stearic acid gave rise to 518 in 86% yield. The remaining free hydroxyl was coupled to H-phosphonate 513 mediated by pivaloyl chloride followed by ion exchange to provide, after benzyl hydrogenolysis, the sodium salt of the triacylated PIM2519 in just seven steps.
Scheme 69. Hung’s Desymmetrization Strategy for the Synthesis of Triacylated PIM2.

In 2016, the Hung laboratory reported a follow-up study constructing Ac2PIM6, which was evaluated, together with PIM1, PIM2, and PIM6 isolated from Mtb (H37Rv strain), for its immunomodulatory activity.326 As in their PIM2 synthesis, the key building block was meso-myo-inositol 1,3,5-orthoformate 520 (Scheme 70). Glycosidation, as previously mentioned, provided pseudotrisaccharide 522 which, after a four-step sequence, led to diol 523. The primary alcohol functionality in 523 was selectively glycosylated by reaction with tetra-saccharide 524 in 52% yield. The naphthyl protecting group of 525 was removed, and acylation was performed with stearic acid to install the linear fatty acid chain. Phosphonate 526 was then coupled, to give after a global deprotection triacylated PIM6527.
Scheme 70. Hung’s Synthesis of Acylated PIM6.

Assessment of the in vivo immunomodulatory activity was performed by coadministration of the PIMs (synthetic PIM6527 and isolated PIM1, PIM2, and PIM6) with ovalbumin or tetanus toxoid. The synthetic PIM was shown to exhibit comparable adjuvant activity, as evident from the induced production of interleukin-4 and interferon-γ, to that of the PIMs from the natural source. The acylated PIMs are thus considered to be candidate adjuvants for vaccine design.
Lear and co-workers reported a very efficient synthesis of PIM2 based on a resolution strategy using D-mannoside 529 as a “permanent resolving agent” (Scheme 71).327 Mannosylation of racemic 528 with 529 under standard α-selective conditions, and subsequent deallylation, gave rise to a separable mixture of the diastereomers 530 and 531 formed in a 1:1 mixture. This direct resolution of C2-coupled myo-inositides 530 and 531 was speculated to be viable due to D-mannosyl donor 529 bearing the bulky TBDPS protecting group at C6. Further mannosylation of 530 gave pseudotrisaccharide 532 in 96% yield which was smoothly converted into PIM2487 using established chemistry from previous synthetic endeavors of PIM2.
Scheme 71. Lear’s PIM2 Synthesis by Resolution.

Another group who set out to synthesize several PIMs was the Tanaka laboratory. Their first PIM synthesis was that of AcPIM2 using 2-(azidomethyl)benzoyl (AZMB) mannosyl donors for formation of the glycosidic bonds (Scheme 72).328 This particular protecting group for the C2-position was chosen because glycosylation with benzoyl esters is known to proceed with complete α-selectivity due to anchimeric assistance. The problem with conventional benzoyl esters, however, is that selective removal without affecting the fatty acid-based acyl motifs is nearly impossible. In order to avoid this problem, generally, protecting group shuffling is performed after the glycosylation. To circumvent such an undesired and nonideal sequence, the AZMB group was used. This group can be removed under hydrogenolytic conditions, which converts the azide into the corresponding amine which in turn will cyclize to form the lactam and liberate the sugar. Two AZMB C2-protected mannosyl donors 534 and 535 were used to mannosylate inositol 533. The reaction was performed in a one-pot two-step procedure to afford the mannosylated inositol 536 in 88% yield with complete α-selectivity for both anomeric centers. The chloroacetyl group was removed using thiourea, and the now free C6-OH was acylated with stearic acid. After oxidative removal of the naphthyl unit, the phosphate group was installed, and final hydrogenolysis provided acylated PIM2538.
Scheme 72. Acylated PIM2 Synthesis by the Tanaka Laboratory.

Importantly, the obtained synthetic material was used to confirm the binding of AcPIM2 to a C-type lectin (like Mincle) named DCAR, which stands for dendritic cell immunoactivating receptor. Upon binding of a ligand, DCAR can trigger monocyte-derived inflammatory cell accumulation at the site of infection and promote a protective Th1 response. It was the Yamasaki laboratory which discovered that DCAR binds mycobacterial cell wall constituents, specifically AcPIM2 and Ac2PIM2, and initiates the orchestration of host protective immunity.329,330 The PIMs used in the DCAR binding assay were isolated from M. bovis BCG, and it was recognized by the Yamasaki group that minute, unobservable, impurities might be responsible for the obtained result. To exclude such a false positive result in which potential impurities bind to DCAR, the synthetic AcPIM2 from the Tanaka laboratory was used, which confirmed that AcPIM2 is an active ligand for DCAR.
A year later, in 2015, the Tanaka group communicated yet another synthesis of several PIMs (Scheme 73). This time AcPIM3546 and AcPIM4547 were constructed via the intermediacy of a protected form of AcPIM2544 synthesized using a modified procedure.331 The synthesis commenced with o-nitrobenzyl protected inositol 539 which was bis-mannosylated in a one-pot procedure to provide 542 in 80% yield. After introduction of the stearyl group, in 72% over two steps, the acetal in 543 was site-selectively opened to liberate the C6-OH. This hydroxyl function was silylated, followed by photodeprotection of the o-nitrobenzyl protecting group, which then set the stage for the phosphatidylation. The phosphorylation of the inositol in this stage of the synthesis is the highlight of the synthesis. In other syntheses of the PIMs, the phosphatidyl glycerol moiety was introduced in the final stage of the synthesis, right before the global deprotection. Tanaka and co-workers introduced the phosphatidyl glycerol to then desilylate and subject this protected form of AcPIM2544 to mannosylation with mannosyl donor 540 and 545 to produce, after hydrogenolysis, acylated PIM3546 and acylated PIM4547.
Scheme 73. Total Synthesis of AcPIM3 and AcPIM4 by Tanaka and Co-workers.

In 2014 the Larsen group communicated the first synthesis of the acylated myo-inositol PIM1 via an enantioconvergent route from myo-inositols (−)-549 and (+)-550 (Scheme 74).332 The enantiopure myo-inositols were obtained by a desymmetrization employing the enzyme Candida antarctica lipase B (Novozym 435) as previously reported by Simas and co-workers.333,334 A wide variety of racemic myo-inositols were synthesized and subjected to the enzymatic desymmetrization reaction. In many cases the reaction was performed with near full conversion (∼50%) leading to enantioselectivities, for both enantiomers, > 98% ee.
Scheme 74. Larsen’s Enantioconvergent Synthesis of Triacylated PIM1.

For the synthesis of Ac3PIM1, the authors chose to work further with myo-inositols (−)-549 and (+)-550. It was realized that both enantiomers could be converted into the target inositol, thereby making optimal use of the racemic myo-inositol starting material. Thus, after acetate cleavage from (−)-549, both enantiomers were converted via standard protection and deprotection sequences into myo-inositols 551 and ent-551. Glycosidation with donors 552 or 553 then gave the PIM1 core structures 554 and 555 which, via slightly different protecting group manipulations, afforded common pseudodisaccharide intermediate 556. Phosphatidylation using H-phosphonate 557, followed by oxidation, ion-exchange, and deprotection concluded the enantioconvergent total synthesis of acylated PIM1558.
The latest PIM synthesis was reported in 2020 by Fujimoto and co-workers (Scheme 75).335 They set out to be the first to synthesize AcPIM1568 and employed a regioselective phosphorylation of protected myo-inositol 559 with BINOL-derived selenophosphoryl chlorides. Several selenophosphoryl chlorides with alternative BINOL structures were screened, but it was BINOL-derived selenophosphoryl chloride 560 that provided the best regioselectivity (88:12) while giving a satisfactory 74% yield as well. The obtained phosphorylated inositol regioisomers (as well as diastereomers) were then protected with an alloc group to enable separation. The alloc group was then swapped for an allyl group to form 563. The BINOL moiety was removed by means of a transesterification with allyl alcohol, and three further steps provided inositol 564. This compound was mannosylated by reaction with mannosyl donor 565 bearing the N-phenyl-2,2,2-trifluoroacetoimidate activating group to yield the α-anomer of mannosylated inositol 567 in 48% yield. One allyl group was removed from the phosphate diester which was subsequently esterified under Mitsunobu conditions with isopropylideneglycerol to furnish 568 in 51% over the two steps. The synthesis was finalized with a five-step sequence initiated by TBS and acetonide removal to be able to install the palmityl esters. The remaining phosphate allyl group was removed whereafter a global deprotecting gave rise to acylated PIM1558.
Scheme 75. First Synthesis of AcPIM1 Reported by Fujimoto and Co-workers.

In collaboration with the Yamasaki laboratory, the synthesized AcPIM1568 was tested for its ability to bind to DCAR. Interestingly, AcPIM1 proved to be a stronger agonist of DCAR compared to the previously known ligand AcPIM2. This result implies that the AcPIM1 moiety featured in AcPIM2 was causing recognition by DCAR. Furthermore, it was shown that AcPIM1 bound weakly to TLR2 (Toll-like receptor) and CD1d. The study of the immunomodulatory function of AcPIM1 helps to understand host–pathogen interaction on a molecular level, as well as forming a basis for the design of novel vaccine adjuvants recognized by DCAR.
9.2. Higher Order Oligosaccharides
The Fraser-Reid laboratory was the first to report on the synthesis of the complex lipomannan (LM) domain 582, part of the lipoarabinomannan capsule.336 Their overall strategy did not rely on skillful deployment of a wide variety of protecting groups but instead was based on Paulsen’s concept of donor/acceptor “match”.337 In its basic form this concept tells that a poor yield obtained from a given donor/acceptor pair could be enhanced by switching donors. The issue of regioselectivity was later addressed by Fraser-Reid to further elaborate Paulsen’s concept of “match”.
In their synthesis of the LM domain 582 (Scheme 76), known diol acceptor 569 was treated, under Lewis-acidic conditions, with n-pentenylorthoester donor 570. Pseudodisaccharide 571 was formed with complete stereo- and regioselectivity in an almost quantitative yield of 97%. 571 was then converted with high selectivity (α/β = 8:1, 94% yield) into pseudotrisaccharide 573 by reaction with armed338 glycosyl donor 572.
Scheme 76. Fraser-Reid’s Total Synthesis of the Lipomannan Component.

A straightforward protection/deprotection sequence followed, providing glycoside acceptor 575. Treatment of 575 with n-pentenyl orthoester 576 in the presence of NIS and Sc(OTf)3, followed by debenzoylation, set the stage for further elaboration using an iterative strategy. The change of Sc(OTf)3 to Yb(OTf)3 proved to be a necessity, since the generated diol (and later triol and tetraol) had to be regioselectively glycosidated at the primary hydroxyl group. After the iterative glycosidation/debenzoylation sequence, pentol 577 was glycosidated with trichloroacetimidate 578 to give pseudododecasaccharide 579 in 86% yield.
The final stages of the LM domain synthesis were relatively straightforward (Scheme 77). A three-step protection/deprotection sequence on 579 gave after acylation and deallylation alcohol 580. Introduction of the phosphonate moiety using standard chemistry and subsequent debenzylation then provided LM component 582.
Scheme 77. Completion of the Total Synthesis of the Lipomannan Component by Fraser-Reid.

The synthesis of a LAM component was reported by the Seeberger laboratory in 2006.339 Whereas Frasier-Reid and co-workers reported the synthesis of the lipomannan (LM) backbone, Seeberger focused on the arabinomannan (AM) part. Key differences between the two LAM components are that the LM domain consists of a myo-inositol anchor with α-D-mannoside linkages, whereas the AM domain not only bears multiple α-D-mannoside linkages but also α-D-arabinofuranosyl linkages. Seeberger’s strategy therefore relied on synthesis of the mannoside and arabinose fragments and subsequent coupling of the two oligosaccharides, maximizing synthetic convergence.
The endeavor started with the synthesis of multiple mannoside building blocks (583, 470, 584, and 578) using known chemistry. With these building blocks in hand, the mannohexasaccharide 586 was quickly and efficiently assembled by multiple deprotections and subsequent glycosidations using catalytic amounts of TMSOTf (Scheme 78). The glycosidation sequence was initialized with mannoside donor A followed by four iterations using B, C, B, and once again C, providing mannanhexasaccharide 585 in an impressive yield of 28% over 10 steps (!). A six-step protection/deprotection sequence (four steps to form the imidate) then furnished key intermediate 586, for coupling with the arabinose component. For the purpose of utilizing the molecules in bioassays, the thio-functionality was installed for further coupling to a microarray or a protein carrier.
Scheme 78. Seeberger’s Synthesis of Mannan Hexasaccharide and Hexa-Arabinose Building Blocks.

The arabinose motif was constructed in a similar approach as the mannan hexasaccharide 586. First several arabinose building blocks (587, 588, 589, and 590) were synthesized which were then coupled using an iterative approach (Scheme 78). Thus, four deprotection/glycosidation sequences (order: A, B, A, C) were performed, after which pentenyl deprotection and subsequent acetimidate formation produced hexaarabinose 591, in 34% over 10 steps (!).
With the two hexasaccharide building blocks 586 and 591 in hand, unification could be achieved through a [6 + 6] glycosidation (Scheme 79). The glycosylation smoothly provided exclusively the α-anomer in 70% yield. Debenzoylation was followed by debenzylation under Birch conditions, affording AM domain 592 as a disulfide linked dimer.
Scheme 79. Seeberger’s Completion of the Arabinomannan Dodecasaccharide.

Shortly after the disclosure of their LM domain synthesis, the Fraser-Reid laboratory reported the ambitious synthesis of the complete oligosaccharide core of the mycobacterial LAM domain.340 The synthesis of this 28-mer was regarded as a milestone in glycochemistry since at the time it was the largest hetero-oligosaccharide synthesized in a laboratory.
The synthesis started with the elaboration of the branched arabinan domain 603 which possesses furanoside units, linked α-1,5-linearly, with occasional C3 branches (Scheme 80). As in their synthesis of the complex LM domain 582 (Scheme 77), the strategy relied on the successful use of n-pentenyl orthoester chemistry. First n-pentenyl furanoside 593 was reacted under standard conditions with glycoside donor 594 followed by a deprotection of the silyl protecting group. Iteration of this sequence led to trisaccharide 595, which was glycosidated under the same conditions with glycoside donor 596. Tetrasaccharide 597 was obtained after deprotection of the acetyl chloride group using thiourea. The first branch of the arabinan motif was introduced by subjecting 597 to, once again, glycosylation with donor 594, to give after desilylation hexasaccharide 598.
Scheme 80. Fraser-Reid’s Synthesis of the Branched Arabinan Unit Part 1.

Elaboration of the branches in hexasaccharide 598 came about by the construction of heteropentasaccharide 602 from a glycosidation/deprotection sequence employing donors 594 and 601. Reaction of hexasaccharide 598 with pentasaccharide 602 smoothly resulted in double glycosidation to furnish, after trichloroacetimidate installation, 16-mer donor 603.
With the 16-mer arabinan motif 603 constructed, the remaining 12-mer mannan domain 606 was synthesized (Scheme 81). Heptasaccharide 604 (synthesis analogous to that of 577; see Scheme 76) was first regioselectively glycosidated at C2 using orthoester 605 followed by glycosidation of the remaining free hydroxyl moieties with trichloroacetimidate donor 578. Removal of the trityl protecting group gave rise to dodecasaccharide acceptor 606.
Scheme 81. Fraser-Reid’s Synthesis of the Dodecasaccharide Acceptor.

With the synthesis of arabinan motif 603 and dodecasaccharide 606 realized, unification of both advanced building blocks was carried out in the presence of NIS and Yb(OTf)3 to give the desired 28-mer in 35% yield (Scheme 82). To validate that the right product was obtained, a debenzoylation/benzylation sequence was performed giving the desired protected lipoarabinomannan domain 607, of which the mass-spectroscopic data clearly indicated the right decrease of mass (−224 u).
Scheme 82. Completion of the Synthesis of the Protected 28-mer Lipoarabinomannan Component.

Another very important contribution to the synthesis of mycobacterial glycolipids, and to the field of glycochemistry in general, was communicated by Lowary and co-workers.341 They reported the synthesis of an impressive 22-mer oligosaccharide from the arabinogalactan domain bearing the synthetically challenging 1,2-cis linked β-arabinofuranoside motif.
The synthesis of this molecule was tackled using a highly convergent approach, in which three main building blocks were united in a late stage of the synthesis. The first building block to be constructed was pentasaccharide 614 starting from thioglycoside 608 (Scheme 83), prepared from d-arabinose in six steps as previously reported.342 Thioglycoside 608 also served as a precursor for multiple small building blocks such as azide 609, which was crafted in only two straightforward steps. 608 was converted into trichloroacetimidate 611 which was reacted with thioglycoside 610 under standard conditions to furnish disaccharide 612. Glycosidation of 612 with 609 smoothly provided trisaccharide 613 in 84% yield. Desilylation of the trisaccharide was followed by a glycosidation with disaccharide 612 followed by again a desilylation, furnishing pentasaccharide 614 in 78% over three steps.
Scheme 83. Lowary’s Synthesis of the Pentasaccharide Building Block.

After construction of the first building block, the Lowary group focused on the construction of heptasaccharide 618 (Scheme 84). First, thioglycoside 615 was converted into diol 616 using a five-step protection/deprotection sequence in 60% yield. Diol 616 was then doubly glycosylated with thioglycoside 608, giving trisaccharide 617 in 74% yield. Desilylation of 617 set the stage for another double glycosylation reaction but this time with disaccharide 612 (see Scheme 83). Subsequent PMP deprotection then provided building block 618.
Scheme 84. Lowary’s Synthesis of Hepta- and Pentasaccharide Building Blocks.

The third building block to be synthesized was pentasaccharide 623, bearing the β-arabinofuranose (β-Araf) ramification (Scheme 84). This synthesis started by glycosylation of thioglycoside 619 with acceptor 616. With trisaccharide 620 in hand the stage was set for the introduction of the β-Araf motif. It is the stereoselective introduction of this moiety which complicates the synthesis of the arabinogalactan domain. Several methods had been developed shortly before the synthesis of the arabinogalactan domain, and it was the strategy developed by the Boons laboratory which was ultimately utilized.343 It was shown that conformational constraints introduced by the silyl protecting group lock the intermediate oxacarbenium ion in such a way (E3 conformer) that the β-face is more accessible. Having such protecting groups in trisaccharide 620, after levulinoyl deprotection, the formation of the β-Araf functionality proceeded smoothly. The desired pentasaccharide 622 was obtained in 67% yield, together with a small amount (<8%) of inseparable isomers. A set of straightforward protecting group manipulations ultimately led to the completion of pentasaccharide 623.
With the three building blocks 614, 618, and 623 in hand, the unification sequence was initiated by coupling of the first two building blocks, 618 and 614 (Scheme 85). Thus, after conversion of 618 into the corresponding trichloroacetimidate, glycosidation with 614 was performed promoted by TMSOTf, furnishing after desilylation, dodecasaccharide 624. This molecule was coupled under the same conditions as before with the acetimidate of 623 to give, after debenzoylation and subsequent azide reduction, the desired arabinogalactan domain 625. In addition to this synthesis, Lowary and co-workers also synthesized the proposed biosynthetic octadecasaccharide (Araf22–β-Araf motif) precursor (synthesis not shown).
Scheme 85. Completion of the Total Synthesis of the Arabinogalactan 22-mer.

After Lowary’s first report of the synthesis of the mycobacterial Araf22 motif,341 the Ito laboratory communicated their synthesis of this impressive oligosaccharide.344 Contrary to the Lowary synthesis, their strategy relied on fragment couplings in linear subunits rather than at the sites of branching. Their synthesis relied heavily on the use of thioglycoside donors built up by a convergent synthesis to access a series of tri- and pentasaccharide building blocks. These were coupled to arrive at two key fragments, Araf7 donor 640 and Araf8 acceptor 637 to ultimately construct the Araf22 target structure. As part of their synthesis, various methods to stereoselectively access the β-Araf linkages were investigated.
The total synthesis of the Araf22 motif was initiated by the synthesis of trisaccharide 629 and pentasaccharides 632 and 635 (Scheme 86). The NIS/AgOTf-promoted glycosylation of 626 and 627 followed by a series of protecting group manipulations gave disaccharide 628 in excellent yield of 91% over four steps. Disaccharide 628 was then subjected to the same series of glycosylation and protecting group changes giving trisaccharide 629. Next, glycosylation of disaccharide 628 with donor 630 gave trisaccharide 631 in very good yield. From 631, pentasaccharide building block 632 was synthesized by a reaction sequence of desilylation, double glycosylation with 626, and further protecting group manipulations, in 73% yield over four steps. To access pentasaccharide 635, trisaccharide 631 was first subjected to debenzoylation, benzyl protection, and desilylation to give trisaccharide 634. Double glycosylation promoted by methyl triflate and subsequent deacetylation cleanly provided pentasaccharide 635 in 93% yield over two steps.
Scheme 86. Ito’s Synthesis of Tri- and Pentasaccharide Building Blocks.

Next, with the necessary building blocks in hand, the key fragments donor 640 and acceptor 637 were crafted (Scheme 87). Pentasaccharide 632 was subjected to a sequence of protecting group modifications giving 636. NIS/AgOTf-promoted glycosylation of 636 with trisaccharide acceptor 629 followed by deacetylation, benzylation, and desilylation produced the desired Araf8 acceptor 637 in 61% yield over four steps. For the synthesis of Araf7 donor 640, the β-Araf linkages were constructed by NIS/AgOTf-promoted double glycosylation of diol 635 with donor 638, delivering heptasaccharide 639 in good stereoselectivity of 8.5:1 (β,β-639:other isomers). Desilylation then delivered the desired β,β-isomer 639 after purification in 87% yield over two steps. From 639, a series of protecting group manipulations finally delivered Araf7 donor 640.
Scheme 87. Synthesis of Araf7 Donor and Araf8 Acceptor and Completion of the Docosaccharide Arabinan Motif.

With the two key fragments 637 and 640 in hand, the target Araf22 unit was constructed by glycosylation promoted by NIS/AgOTf in excellent yield of 96%. Global deprotection of the benzyl, acetyl, and silyl ether moieties then completed the synthesis of the docosaccharide arabinan motif 641.
In 2017, the Hotha laboratory reported the synthesis of a mycobacterial heneicosafuranosyl arabinogalactan (HAG), consisting of 21 furanosyl monomers.345 Contrary to the oligosaccharides synthesized by the Lowary341 and Ito344 groups, their target contains Araf as well as Galf units. For the synthesis, a convergent strategy was developed, involving the synthesis of six monosaccharide building blocks (Scheme 88, in boxes), which were used to access four key oligosaccharide fragments (Scheme 89, in boxes). In order to construct these and unify them to arrive at the target HAG, their in-house developed gold-catalyzed glycosylation346 using alkynyl 1,2-orthoester donors was utilized.
Scheme 88. Hotha’s Furanoside Monomer Synthesis.

Scheme 89. Hotha’s Synthesis of Four Key Oligosaccharide Fragments.

The synthesis of HAG commenced with the synthesis of six furanosyl monomers (Scheme 88). Starting from 1,2-orthoester 642, a series of protecting group manipulations provided monomers 643 and 645 in 61% and 74% yield over three steps, respectively. Orthoester 643 was then converted to n-pentenyl furanoside monomer 644 by gold-catalyzed glycosylation followed by removal of the TBDPS ether in 66% yield over two steps. Furanosyl monomer 645 was subjected to gold-catalyzed glycosylation providing n-pentenyl furanoside 646. Intermediate 646 was then subjected to a series of protecting group manipulations and ultimately was converted into 1,2-orthoester monomer 647 by preparation of the anomeric bromide and treatment with propargyl alcohol. The two Galf monomers were readily accessed from n-pentenyl furanoside 648. 1,2-Orthoester monomer 649 was synthesized, again, by preparation of the anomeric bromide followed by treatment with propargyl alcohol in 81% yield over two steps. A series of synthetic steps then provided Galf monomer 650 in 56% yield over five steps.
With the furanosyl monomers in hand, the four key oligosaccharide fragments 652, 656, 658, and 660 were synthesized (Scheme 89). Au/Ag-catalyzed glycosylation of Galf donor 649 with acceptor 650 provided disaccharide 651 in 70% yield. A second glycosylation of 651 with disaccharide acceptor 643 followed by TBDPS deprotection then delivered advanced building block 652 in 71% yield over two steps. Synthesis of building block 656 was initiated by Au/Ag-catalyzed glycosylation of 1,2-orthoester 643 with acceptor 644 giving n-pentenyl disaccharide 653 in 70% yield. From 653, two intermediates were accessed. Desilylation of 653 gave primary alcohol 654, and 1,2-orthoester formation from 653 via the anomeric bromide delivered disaccharide 655. Coupling of disaccharides 654 and 655 by Au–Ag-catalyzed glycosylation and 1,2-orthoester synthesis concluded the synthesis of building block 656 in 74% over two steps. Next, gold-catalyzed glycosylation of 1,2-orthoester 645 with acceptor 644 furnished disaccharide 657. Another glycosylation of 657 with 1,2-orthoester 647 followed by installation of the 1,2-orthoester moiety delivered building block 658. Disaccharide 657 was glycosylated with 643 under gold catalysis followed by desilylation to give diol 659. The synthesis of hexasaccharide building block 660 was concluded by double gold-catalyzed glycosylation with 643 and introduction of the 1,2-orthoester unit in 43% yield over two steps.
In the end game of the synthesis (Scheme 90), building blocks 652 and 656 were unified by Au/Ag-catalyzed glycosylation followed by liberation of the primary alcohol, giving 661 in 60% yield over two steps. Another Au–Ag-catalyzed glycosylation of 661 with 660 and subsequent desilylation gave diol 662. After coupling of 662 with 658 by Au/Ag-catalyzed glycosylation, and global deprotection, the stereoselective total synthesis of HAG 667 was completed in 0.09% overall yield. It is expected that this modular synthesis allows access of various synthetic analogues for further biological studies.
Scheme 90. Fragment Assembly and Completion of the Heneicosafuranosyl Arabinogalactan.

Shortly after the Hotha laboratory reported their synthesis of HAG,345 the group of Ye communicated the impressive first total synthesis of the mycobacterial arabinogalactan 92-mer(!).347 Contrary to the previous syntheses by Lowary341 and Ito,344 this total synthesis features the complete construction of arabinogalactan rather than truncated oligosaccharide fragments. In order to achieve the preparation of the arabinogalactan 92-mer, a highly convergent synthesis route was designed. Their strategy was built on various preactivation-based one-pot glycosylation reactions to access various oligosaccharide building blocks, ultimately leading to two key fragments: a linear Galf30 acceptor 673 and a branched Araf31 donor 686, to be unified giving the target polysaccharide.
The synthesis of Galf30 acceptor 673 was initiated by the synthesis of linear hexasaccharide 667 (Scheme 91). Through a one-pot triple glycosylation of donor 664 with 665 (2×) and 666 in 79% yield followed by desilylation, 667 was obtained, carrying two orthogonal levulinoyl protecting groups for later coupling to the Araf31 donor 686. A series of additional hexasaccharide building blocks was synthesized by another one-pot glycosylation of 664 with monosaccharides 666 (3×) and 668 (2×) giving 669 in 63% yield. Desilylation of 669 then gave 670, which was benzoylated providing 671, whereas glycosylation of 669 with 1-octanol followed by desilylation gave building block 672. The desired Galf30 acceptor 673 was then constructed by a remarkable one-pot [6 + 6 + 6 + 6 + 6] glycosylation in 64% yield and subsequent liberation of the levulinoyl esters.
Scheme 91. Ye’s Synthesis of Galf30 Acceptor.

Next, for the synthesis of Araf31 donor 686, three oligosaccharide building blocks 677, 681, and 685 were prepared (Scheme 92). Starting from 674, the linear hexasaccharide 676 was accessed in 73% yield by five consecutive glycosylations with monosaccharide 675 in one pot. The TBS ether of 676 was then removed providing intermediate 677. The next two branched intermediates 680 and 682 were crafted by triple one-pot glycosylation of 674 or 679 with 678 and 675 (2×) in 78% and 76% yield, respectively. Desilylation of 680 provided pentasaccharide 681, whereas 683 was synthesized by delevulinoylation of 682. Glycosylation of diol 683 with donor 684 delivered the 1,2-cis linked heptasaccharide in excellent stereoselectivity. Next, removal of the silyl ether and benzoyl protection gave branched oligosaccharide building block 685. With all necessary building blocks in hand, the desired Araf31 donor 686 was assembled by one-pot triple preactivation glycosylation of 685 with 681 and 677 (2×) giving arabinofuranoside 31-mer 686 in 70% yield.
Scheme 92. Ye’s Synthesis of the Araf31 Donor.

After complete synthesis of Araf31 donor 686 and Galf30 acceptor 673, the two polysaccharide building blocks were coupled to arrive at the arabinogalactan 92-mer (Scheme 93). For this double glycosylation, a number of different reaction conditions were studied, eventually identifying the benzenesulfinyl morpholine/Tf2O promotor system as most successful. These conditions cleanly provided the coupled product in an impressive yield of 84%. Lastly, global deprotection involving debenzoylation and debenzylation concluded the first total synthesis of the mycobacterial arabinogalactan 92-mer 687 in 75% yield over two steps.
Scheme 93. Completion of the Total Synthesis of the Arabinogalactan 92-mer.

10. Conclusion and Outlook
In this review we have compiled a quarter century of total syntheses of Mtb cell wall components ranging from lipids, glycolipids, terpenes, and lipopeptides to oligo- and even polysaccharides. It is evident from the variety and complexity of these molecular architectures, highlighted in this review, that Mtb can be considered a natural product “powerhouse”, showing these molecular entities to be of great interest and value to both synthetic chemists and immunologists. From an organic synthesis standpoint, these total syntheses have contributed significantly to the progress of the field of asymmetric synthesis and the development of synthesis methodology. A key example of this is the synthesis of (hydroxy)phthioceranic acid, which commenced with a robust linear synthesis and transitioned into remarkably convergent and efficient synthesis routes. This evolution of chemical methodology is also nicely illustrated by the synthetic efforts toward mycobacterial arabinogalactan, set in motion by the first reports of fragments to ultimately enable total synthesis of the whole polysaccharide. Each of these reports is an example of pioneering work in organic synthesis.
Apart from pushing the boundaries of organic chemistry, total synthesis of mycobacterial natural products (as with any other natural product) allowed ultimate confirmation or elucidation of structure and stereochemistry. This has also very practical consequences. Research on the biosynthesis and immunology of Mtb components, either with molecular biology tools or using these components from cultured bacteria, can to a large extent take place successfully without knowledge of their precise molecular structure. Once such compounds are chemically synthesized, however, the structure including stereochemistry must be known. Indeed, any deviation from the structure of the natural compound leads to differences in biological activity. This was shown clearly in the synthesis of MPM and Ac2SGL, where all attempts to change structure, stereochemistry, and even chain length of the lipid components led invariably to a diminished activity.
The syntheses of DAT and Ac2SGL highlight the necessity for chemical synthesis for structure elucidation. The synthesis of three DAT structures ultimately revealed through direct comparison with natural material from cell wall extracts that synthetic DAT2 does not correspond to the proposed structure of the natural material which needs to be revised. In the first synthesis of hydroxyphthioceranic acid as part of the synthesis of Ac2SGL, the stereochemistry of its hydroxy group was unknown. Only by chemical synthesis and NMR comparison with natural material could the question of the stereochemistry be answered. Furthermore, the synthetic material obtained by total synthesis of elaborate mycobacterial natural products serves as an invaluable reference material for biological studies to answer fundamental questions on Mtb virulence and host immunology. A prime example is the asymmetric total synthesis of TbAd, which enabled detailed and extensive studies on the survival mechanisms of Mtb within macrophages. This total synthesis ultimately contributed to the elucidation of the role of TbAd as another virulence factor of Mtb.
The World Health Organization set the ambitious goal to eradicate Tb by 2035. Although certainly significant steps have been taken, judging the current yearly mortality and infection statistics, this target is not in sight yet. After all, despite the positive tone in the scientific literature, this review included, there is no effective vaccine against Tb, drug therapy is cumbersome and long, and there is an urgent need for low-cost diagnostics in developing countries. To reach the goals set by the WHO, the scientific community, e.g. academia, industry, and funding agencies, should make Tb eradication a top priority. Pertaining to this review, the authors believe that chemical synthesis endeavors greatly benefit from joint scientific projects in which compounds, knowledge, and inventions can be converted into actual point-of-care diagnosis, treatment and, ultimately, a vaccine. Such collaborations do require building bridges between seemingly distant fields of research, i.e. organic chemistry, bacteriology, immunology, and medicine. This is easier said than done, but with the common goal of ending Tb in mind, and the willingness to collaborate, a foundation can be laid from which exciting and eventually breakthrough discoveries can emerge.
After a large number of reported total syntheses, the field now approaches a turning point. Inherently it is not clear how many mycobacterial (cell wall) components are awaiting discovery. In order to drive the field further and to expand the knowledge on how Mtb utilizes cell wall constituents in virulence and host-immunology, we need continuing efforts to discover new Mtb-specific molecules. Most known compounds have been accessed by synthesis. This by no means, however, makes them readily available. It is very important that many of the mycobacterial antigens become available to immunology as readily as antibodies and other “reagents”. This means that cutting-edge synthesis tools and creativity are required to develop the next generation syntheses of these antigens.
All in all, discoveries from interdisciplinary collaborations in this field can lead to the identification of new drug targets, vaccine adjuvants, or even new diagnostic platforms which would benefit millions of people.
Acknowledgments
The Dutch Research Council (NWO) is acknowledged for funding. J.B. specifcally acknowledges NWO for funding through the NWO-VENI program (VI.Veni.192.122). The authors thank N. Marinus, N. R. M. Reintjens, N. Duindam, and C. H. M. van der Loo for proofreading the manuscript and B. L. H. Andringa for providing synthesis schemes of the mycoketide total synthesis.
Biographies
Mira Holzheimer was born in Schweinfurt, Germany, in 1991. She completed her Bachelor in Pharmaceutal Sciences at the Ludwig-Maximilians University in Munich in 2013. After that, she moved to Leiden, The Netherlands, to pursue her Master studies in Bio-Pharmaceutical Sciences and obtained her Master’s degree in 2016 with honors (cum laude). In the same year, she started her Ph.D. research in the group of Prof. Dr. Ir. Adriaan J. Minnaard which focuses on the asymmetric total synthesis of the complex archaeal membrane lipid crenarchaeol and on mycobacterial trehalose glycolipids (2016–2020). After receiving her Ph.D. degree in 2021 and a short postdoctoral stay in the Minnaard group, she is a now a postdoctoral researcher with Prof. Alois Fürstner at the Max-Planck Institut für Kohlenforschung in Mülheim an der Ruhr, Germany, continuing in the field of total synthesis.
Jeffrey Buter studied molecular chemistry at the University of Groningen, The Netherlands. At the same university, he obtained his Ph.D. in the group of Prof. Adriaan J. Minnaard, working on the stereoselective total synthesis of terpenes containing quaternary stereocenters (2016). He then joined the laboratory of Prof. D. Branch Moody at Brigham and Women’s Hospital/Harvard Medical School (United States of America) as a postdoctoral fellow (2016–2018). Here he investigated a survival mechanism of Mycobacterium tuberculosis (Mtb) and codiscovered a novel immunogenic lipid from Salmonella Typhi. In 2018 he joined the laboratory of Prof. Ben L. Feringa at the University of Groningen, where he is leading the catalysis group. Jeffrey secured his own research funding to unravel survival mechanisms of Mtb and for the development of a new therapeutic strategy against tuberculosis disease (NWO-VENI and NWO-NWA idea generator grant). Together with Prof. Adriaan J. Minnaard, Prof. D. Branch Moody, and I. van Rhijn, he is the corecipient of the NWO Team Science Award (2020) for the collaborative effort involving discovery of novel tuberculosis lipids.
Adriaan J. Minnaard studied Molecular Sciences at Wageningen Agricultural University, The Netherlands. At the same university, he obtained his Ph.D. degree with Prof. Æ. De Groot in the field of natural product synthesis. He subsequently joined Royal DSM as a research scientist. In 1999 he joined the department of Prof. B. L. Feringa at the University of Groningen as an assistant professor. After a year as guest researcher at the Max Planck Institut für Molekulare Physiologie, Dortmund, Germany, in the department of Prof. H. Waldmann, he was appointed in 2009 professor in Bio-organic Chemistry at the University of Groningen. In 2016 he was appointed director of the Stratingh Institute for Chemistry. The main focus areas in his research are the chemistry of mycobacterial glycolipids, the synthesis of archaeal lipids, novel methods in homogeneous catalysis, and the selective modification of unprotected carbohydrates.
The authors declare no competing financial interest.
References
- Hershkovitz I.; Donoghue H. D.; Minnikin D. E.; Besra G. S.; Lee O. Y.-C.; Gernaey A. M.; Galili E.; Eshed V.; Greenblatt C. L.; Lemma E.; Bar-Gal G. K.; Spigelman M. Detection and Molecular Characterization of 9000-Year-Old Mycobacterium tuberculosis from a Neolithic Settlement in the Eastern Mediterranean. PLoS One 2008, 3, e3426 10.1371/journal.pone.0003426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberis I.; Bragazzi N. L.; Galluzzo L.; Martini M. The History of Tuberculosis: From the First Historical Records to the Isolation of Koch’s Bacillus. J. Prev. Med. Hyg. 2017, 58, E9–E12. [PMC free article] [PubMed] [Google Scholar]
- Cave A. J. E. The Evidence for the Incidence of Tuberculosis in Ancient Egypt. Br. J. Tuberc. 1939, 33, 142–152. 10.1016/S0366-0850(39)80016-3. [DOI] [Google Scholar]
- Nerlich A. G.; Haas C. J.; Zink A.; Szeimies U.; Hagedorn H. G. Molecular Evidence for Tuberculosis in an Ancient Egyptian Mummy. Lancet 1997, 350, 1404. 10.1016/S0140-6736(05)65185-9. [DOI] [PubMed] [Google Scholar]
- Zink A. R.; Sola C.; Reischl U.; Grabner W.; Rastogi N.; Wolf H.; Nerlich A. G. Characterization of Mycobacterium tuberculosis Complex DNAs from Egyptian Mummies by Spoligotyping. J. Clin. Microbiol. 2003, 41, 359–367. 10.1128/JCM.41.1.359-367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann S. H. E. Robert Koch, the Nobel Prize, and the Ongoing Threat of Tuberculosis. N. Engl. J. Med. 2005, 353, 2423–2426. 10.1056/NEJMp058131. [DOI] [PubMed] [Google Scholar]
- Global tuberculosis report; World Health Organization: Geneva, 2020.
- Behr M. A.; Edelstein P. H.; Ramakrishnan L. Is Mycobacterium tuberculosis Infection Life Long?. BMJ. 2019, 367, l5770. 10.1136/bmj.l5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantres-Fonseca O. J.; Rodriguez-Cintrón W.; Del Olmo-Arroyo F.; Baez-Corujo S. In Extra Pulmonary Tuberculosis: An Overview; Chauhan N. S., Ed.; 2018. [Google Scholar]
- Bruchfeld J.; Correia-Neves M.; Källenius G. Tuberculosis and HIV Coinfection. Cold Spring Harbor Perspect. Med. 2015, 5, a017871 10.1101/cshperspect.a017871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd K.; Glaziou P.; Zumla A.; Raviglione M. The Global Tuberculosis Epidemic and Progress in Care, Prevention, and Research: An Overview in Year 3 of the End TB Era. Lancet Respir. Med. 2018, 6, 299–314. 10.1016/S2213-2600(18)30057-2. [DOI] [PubMed] [Google Scholar]
- Seung K. J.; Keshavjee S.; Rich M. L. Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harbor Perspect. Med. 2015, 5, a017863 10.1101/cshperspect.a017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontali E.; Raviglione M. C.; Migliori G. B.; Committee w. g. m. o. t. G. T. N. C. T. Regimens to Treat Multidrug-Resistant Tuberculosis: Past, Present and Future Perspectives. Eur. Respir. Rev. 2019, 28, 190035. 10.1183/16000617.0035-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliori G. B.; Dara M.; de Colombani P.; Kluge H.; Raviglione M. C. Multidrug-Resistant Tuberculosis in Eastern Europe: Still on the Increase?. Eur. Respir. J. 2012, 39, 1290–1291. 10.1183/09031936.00214411. [DOI] [PubMed] [Google Scholar]
- Acosta C. D.; Dadu A.; Ramsay A.; Dara M. Drug-Resistant Tuberculosis in Eastern Europe: Challenges and Ways Forward. Public Health Action 2014, 4, S3–S12. 10.5588/pha.14.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel N.; Hava D.; Houben D.; Fluitsma D.; van Zon M.; Pierson J.; Brenner M.; Peters P. J. M. tuberculosis and M. leprae Translocate from the Phagolysosome to the Cytosol in Myeloid Cells. Cell 2007, 129, 1287–1298. 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- Pieters J. Mycobacterium tuberculosis and the Macrophage: Maintaining a Balance. Cell Host Microbe 2008, 3, 399–407. 10.1016/j.chom.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Vandal O. H.; Nathan C. F.; Ehrt S. Acid Resistance in Mycobacterium tuberculosis. J. Bacteriol. 2009, 191, 4714–4721. 10.1128/JB.00305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meena L. S.; Rajni Survival Mechanisms of Pathogenic Mycobacterium tuberculosis H37Rv. FEBS J. 2010, 277, 2416–2427. 10.1111/j.1742-4658.2010.07666.x. [DOI] [PubMed] [Google Scholar]
- Jayachandran R.; BoseDasgupta S.; Pieters J.. Surviving the Macrophage: Tools and Tricks Employed by Mycobacterium tuberculosis. In Curr. Top. Microbiol. Immunol.; Pieters J., McKinney J. D., Eds.; Springer-Verlag: Berlin Heidelberg, 2013; Vol. 374, pp 189–209. [DOI] [PubMed] [Google Scholar]
- Cambier C. J.; Falkow S.; Ramakrishnan L. Host Evasion and Exploitation Schemes of Mycobacterium tuberculosis. Cell 2014, 159, 1497–1509. 10.1016/j.cell.2014.11.024. [DOI] [PubMed] [Google Scholar]
- Zhai W.; Wu F.; Zhang Y.; Fu Y.; Liu Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. 10.3390/ijms20020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batt S. M.; Minnikin D. E.; Besra G. S. The Thick Waxy Coat of Mycobacteria, a Protective Layer Against Antibiotics and the Host’s Immune System. Biochem. J. 2020, 477, 1983–2006. 10.1042/BCJ20200194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffé M.; Reyrat J.-M.. The Mycobacterial Cell Envelope; ASM Press: Washington, D.C., 2008. [Google Scholar]
- Kaur D.; Guerin M. E.; Škovierová H.; Brennan P. J.; Jackson M.. Biogenesis of the Cell Wall and Other Glycoconjugates of Mycobacterium tuberculosis. In Advances in Applied Microbiology; Elsevier: 2009; pp 23–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankute M.; Cox J. A. G.; Harrison J.; Besra G. S. Assembly of the Mycobacterial Cell Wall. Annu. Rev. Microbiol. 2015, 69, 405–423. 10.1146/annurev-micro-091014-104121. [DOI] [PubMed] [Google Scholar]
- Ghazaei C. Mycobacterium tuberculosis and Lipids: Insights into Molecular Mechanisms from Persistence to Virulence. J. Res. Med. Sci. 2018, 23, 63. 10.4103/jrms.JRMS_904_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgiu G.; Centis R.; D’ambrosio L.; Migliori G. B. Tuberculosis Treatment and Drug Regimens. Cold Spring Harbor Perspect. Med. 2015, 5, a017822 10.1101/cshperspect.a017822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B.; Williams S. J. Chemical Approaches for the Study of the Mycobacterial Glycolipids Phosphatidylinositol Mannosides, Lipomannan and Lipoarabinomannan. Nat. Prod. Rep. 2010, 27, 919–947. 10.1039/c000604a. [DOI] [PubMed] [Google Scholar]
- Shashidhar M. S.; Patil N. T. Recent Developments in the Synthesis of Biologically Relevant Inositol Derivatives. In Carbohydrates in Drug Discovery and Development 2020, 283–329. 10.1016/B978-0-12-816675-8.00007-5. [DOI] [Google Scholar]
- Daffé M.; Lemassu A.. Glycobiology of the Mycobacterial Surface. In Glycomicrobiology; Doyle R. J., Ed.; Kluwer Academic/Plenum Publishers: New York, 2002. [Google Scholar]
- Chatterjee D.; Khoo K.-H. Mycobacterial Lipoarabinomannan: An Extraordinary Lipoheteroglycan with Profound Physiological Effects. Glycobiology 1998, 8, 113–120. 10.1093/glycob/8.2.113. [DOI] [PubMed] [Google Scholar]
- Alderwick L. J.; Harrison J.; Lloyd G. S.; Birch H. L. The Mycobacterial Cell Wall - Peptidoglycan and Arabinogalactan. Cold Spring Harbor Perspect. Med. 2015, 5, a021113 10.1101/cshperspect.a021113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A.; Munshi T.; Healy J.; Martin L. T.; Vollmer W.; Keep N. H.; Bhakta S. Cell Wall Peptidoglycan in Mycobacterium tuberculosis: An Achilles’ Heel for the TB-causing Pathogen. FEMS Microbiol. Rev. 2019, 43, 548–575. 10.1093/femsre/fuz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrakchi H.; Laneélle M. A.; Daffé M. Mycolic Acids: Structures, Biosynthesis, and Beyond. Chem. Biol. 2014, 21, 67–85. 10.1016/j.chembiol.2013.11.011. [DOI] [PubMed] [Google Scholar]
- Kalscheuer R.; Palacios A.; Anso I.; Cifuente J.; Anguita J.; Jacobs W. R. Jr.; Guerin M. E.; Prados-Rosales R. The Mycobacterium tuberculosis Capsule: A Cell Structure with Key Implications in Pathogenesis. Biochem. J. 2019, 476, 1995–2016. 10.1042/BCJ20190324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnikin D. E.; Brennan P. J.. Lipids of Clinically Significant Mycobacteria. In Health Consequences of Microbial Interactions with Hydrocarbons, Oils, and Lipids; Goldfine H., Ed.; Springer International Publishing: Cham, 2020; pp 33–108. [Google Scholar]
- Stanley S. A.; Cox J. S.. Host-Pathogen Interactions During Mycobacterium tuberculosis infections. In Curr. Top. Microbiol. Immunol.; Pieters J., McKinney J. D., Eds.; Springer-Verlag: Berlin Heidelberg, 2013; Vol. 374, pp 211–241. [DOI] [PubMed] [Google Scholar]
- Ryll R.; Kumazawa Y.; Yano I. Immunological Properties of Trehalose Dimycolate (Cord Factor) and Other Mycolic Acid-Containing Glycolipids-A Review. Microbiol. Immunol. 2001, 45, 801–811. 10.1111/j.1348-0421.2001.tb01319.x. [DOI] [PubMed] [Google Scholar]
- Guenin-Mace L.; Simeone R.; Demangel C. Lipids of Pathogenic Mycobacteria: Contributions to Virulence and Host Immune Suppression. Transboundary Emerging Dis. 2009, 56, 255–268. 10.1111/j.1865-1682.2009.01072.x. [DOI] [PubMed] [Google Scholar]
- Astarie-Dequeker C.; Nigou J.; Passemar C.; Guilhot C. The Role of Mycobacterial Lipids in Host Pathogenesis. Drug Discovery Today: Dis. Mech. 2010, 7, e33–e41. 10.1016/j.ddmec.2010.09.003. [DOI] [Google Scholar]
- Jackson M. The Mycobacterial Cell Envelope—Lipids. Cold Spring Harbor Perspect. Med. 2014, 4, a021105 10.1101/cshperspect.a021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queiroz A.; Riley L. W. Bacterial Immunostat: Mycobacterium tuberculosis Lipids and Their Role in the Host Immune Response. Rev. Soc. Bras. Med. Trop. 2017, 50, 9–18. 10.1590/0037-8682-0230-2016. [DOI] [PubMed] [Google Scholar]
- Singh P.; Rameshwaram N. R.; Ghosh S.; Mukhopadhyay S. Cell Envelope Lipids in the Pathophysiology of Mycobacterium tuberculosis. Future Microbiol. 2018, 13, 689–710. 10.2217/fmb-2017-0135. [DOI] [PubMed] [Google Scholar]
- Kinsella R. L.; Zhu D. X.; Harrison G. A.; Mayer Bridwell A. E.; Prusa J.; Chavez S. M.; Stallings C. L. Perspectives and Advances in the Understanding of Tuberculosis. Annu. Rev. Pathol.: Mech. Dis. 2021, 16, 377–408. 10.1146/annurev-pathol-042120-032916. [DOI] [PubMed] [Google Scholar]
- Moreno-Mendieta S. A.; Rocha-Zavaleta L.; Rodriguez-Sanoja R. Adjuvants in Tuberculosis Vaccine Development. FEMS Immunol. Med. Microbiol. 2010, 58, 75–84. 10.1111/j.1574-695X.2009.00629.x. [DOI] [PubMed] [Google Scholar]
- Wallis R. S.; Kim P.; Cole S.; Hanna D.; Andrade B. B.; Maeurer M.; Schito M.; Zumla A. Tuberculosis Biomarkers Discovery: Developments, Needs, and Challenges. Lancet Infect. Dis. 2013, 13, 362–372. 10.1016/S1473-3099(13)70034-3. [DOI] [PubMed] [Google Scholar]
- Tucci P.; Gonzalez-Sapienza G.; Marin M. Pathogen-derived Biomarkers for Active Tuberculosis Diagnosis. Front. Microbiol. 2014, 5, 549. 10.3389/fmicb.2014.00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goletti D.; Petruccioli E.; Joosten S. A.; Ottenhoff T. H. Tuberculosis Biomarkers: From Diagnosis to Protection. Infect. Dis. Rep. 2016, 8, 6568. 10.4081/idr.2016.6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goletti D.; Lee M. R.; Wang J. Y.; Walter N.; Ottenhoff T. H. M. Update on Tuberculosis Biomarkers: From Correlates of Risk, to Correlates of Active Disease and of Cure from Disease. Respirology 2018, 23, 455–466. 10.1111/resp.13272. [DOI] [PubMed] [Google Scholar]
- Murphy K.; Weaver C.. Janeway’s Immunobiology. 9th ed.; Garland Science: New York, 2016. [Google Scholar]
- Sompayrac L.How the ImmuneSystem Works, 5th ed.; John Wiley & Sons, Ltd.: Chichester, 2016. [Google Scholar]
- Monie T. P.Section 1 - A Snapshot of the Innate Immune System. In The Innate Immune System; Monie T. P., Ed.; Academic Press, 2017; pp 1–40. [Google Scholar]
- Monie T. P.Section 5 - Connecting the Innate and Adaptive Immune Responses. In The Innate Immune System; Monie T. P., Ed.; Academic Press, 2017; pp 171–187. [Google Scholar]
- Barral D. C.; Brenner M. B. CD1 Antigen Presentation: How it Works. Nat. Rev. Immunol. 2007, 7, 929–941. 10.1038/nri2191. [DOI] [PubMed] [Google Scholar]
- Siddiqui S.; Visvabharathy L.; Wang C.-R. Role of Group 1 CD1-restricted T Cells in Infectious Disease. Front. Immunol. 2015, 6, 337. 10.3389/fimmu.2015.00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rhijn I.; Moody D. B. CD1 and Mycobacterial Lipids Activate Human T Cells. Immunol. Rev. 2015, 264, 138–153. 10.1111/imr.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa E.; Ishikawa T.; Morita Y. S.; Toyonaga K.; Yamada H.; Takeuchi O.; Kinoshita T.; Akira S.; Yoshikai Y.; Yamasaki S. Direct Recognition of the Mycobacterial Glycolipid, Trehalose Dimycolate, by C-type Lectin Mincle. J. Exp. Med. 2009, 206, 2879–2888. 10.1084/jem.20091750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga I.; Moody D. B. Mincle is a Long Sought Receptor for Mycobacterial Cord Factor. J. Exp. Med. 2009, 206, 2865–2868. 10.1084/jem.20092533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. J. Sensing Lipids with Mincle: Structure and Function. Front. Immunol. 2017, 8, 1662. 10.3389/fimmu.2017.01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Nagata M.; Yamasaki S. Mincle: 20 Years of a Versatile Sensor of Insults. Int. Immunol. 2018, 30, 233–239. 10.1093/intimm/dxy028. [DOI] [PubMed] [Google Scholar]
- Patin E. C.; Orr S. J.; Schaible U. E. Macrophage Inducible C-Type Lectin As a Multifunctional Player in Immunity. Front. Immunol. 2017, 8, 861. 10.3389/fimmu.2017.00861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockrell H. M.; Smith S. G. What Have We Learnt about BCG Vaccination in the Last 20 Years?. Front. Immunol. 2017, 8, 1134. 10.3389/fimmu.2017.01134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R. J. The Separation of Lipiod Fractions from Tubercle Bacilli. J. Biol. Chem. 1927, 74, 525–535. 10.1016/S0021-9258(20)74044-5. [DOI] [Google Scholar]
- Minnikin D. E.; Polgar N. Studies on the Mycolid Acids from Human Tubercle Bacilli. Tetrahedron Lett. 1966, 23, 2643–2647. 10.1016/S0040-4039(01)84131-9. [DOI] [PubMed] [Google Scholar]
- Minnikin D. E.; Polgar N. The Methocymycolic and Ketomycolic Acids from Human Tubercle Bacilli. Chem. Commun. 1967, 1172–1174. 10.1039/c19670001172. [DOI] [Google Scholar]
- Asselineau J.; Tocanne G.; Tocanne J. F. Stéréochimie des Acides Mycoliques. Bull. Soc. Chim. Fr. 1970, 4, 1455–1459. [PubMed] [Google Scholar]
- Watanabe M.; Aoyagi Y.; Ridell M.; Minnikin D. E. Separation and Characterization of Individual Mycolic Acids in Representative Mycobacteria. Microbiology 2001, 147, 1825–1837. 10.1099/00221287-147-7-1825. [DOI] [PubMed] [Google Scholar]
- Moody D. B.; Guy M. R.; Grant E.; Cheng T.-Y.; Brenner M. B.; Besra G. S.; Porcelli S. A. CD1b-mediated T Cell Recognition of a Glycolipid Antigen Generated from Mycobacterial Lipid and Host Carbohydrate during Infection. J. Exp. Med. 2000, 192, 965–976. 10.1084/jem.192.7.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenslager G. T.; Simpson L. L. Chimeric Molecules Constructed with Endogenous Substances. Adv. Mol. Cell Biol. 1994, 9, 233–262. 10.1016/S1569-2558(08)60387-2. [DOI] [Google Scholar]
- Watanabe M.; Aoyagi Y.; Mitome H.; Fujita T.; Naoki H.; Ridell M.; Minnikin D. E. Location of Functional Groups in Mycobacterial Meromycolate Chains; The Recognition of new Structural Principles in Mycolic Acids. Microbiology 2002, 148, 1881–1902. 10.1099/00221287-148-6-1881. [DOI] [PubMed] [Google Scholar]
- Al Dulayymi J. R.; Baird M. S.; Roberts E.; Deysel M.; Verschoor J. The First Syntheses of Single Enantiomers of the Major Methoxymycolic Acid of Mycobacterium tuberculosis. Tetrahedron 2007, 63, 2571–2592. 10.1016/j.tet.2007.01.007. [DOI] [Google Scholar]
- Tahiri N.; Fodran P.; Jayaraman D.; Buter J.; Witte M. D.; Ocampo T. A.; Moody D. B.; Van Rhijn I.; Minnaard A. J. Total Synthesis of a Mycolic Acid from Mycobacterium tuberculosis. Angew. Chem., Int. Ed. 2020, 59, 7555–7560. 10.1002/anie.202000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry III C. E.; Lee R. E.; Khisimusi M.; Sampson A. E.; Schroeder B. G.; Slayden R. A.; Yuan Y. Mycolic Acids: Structure, Biosynthesis and Physiological Functions. Prog. Lipid Res. 1998, 37, 143–179. 10.1016/S0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Prevention of Drug Access to Bacterial Targets: Permeability Barriers and Active Efflux. Science 1994, 264, 382–388. 10.1126/science.8153625. [DOI] [PubMed] [Google Scholar]
- Kremer L. S.; Besra G. S. Current Status and Future Development of Antitubercular Chemotherapy. Expert Opin. Invest. Drugs 2002, 11, 1033–1049. 10.1517/13543784.11.8.1033. [DOI] [PubMed] [Google Scholar]
- Nataraj V.; Varela C.; Javid A.; Singh A.; Besra G. S.; Bhatt A. Mycolic Acids: Deciphering and Targeting the Achilles’ Heel of the Tubercle Bacillus. Mol. Microbiol. 2015, 98, 7–16. 10.1111/mmi.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder F. G.; Collins P. B. Inhibition by Isoniazid of Synthesis of Mycolic Acids in Mycobacterium tuberculosis. J. Gen. Microbiol. 1970, 63, 41–48. 10.1099/00221287-63-1-41. [DOI] [PubMed] [Google Scholar]
- Takayama K.; Wang L.; David H. L. Effect of Isoniazid on the In Vivo Mycolic Acid Synthesis, Cell Growth, and Viability of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1972, 2, 29–35. 10.1128/AAC.2.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilcheze C.; Jacobs W. R. Jr. The Isoniazid Paradigm of Killing, Resistance, and Persistence in Mycobacterium tuberculosis. J. Mol. Biol. 2019, 431, 3450–3461. 10.1016/j.jmb.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M.; Hashizume H.; Tomishige T.; Kawasaki M.; Tsubouchi H.; Sasaki H.; Shimokawa Y.; Komatsu M. OPC-67683, a Nitro-Dihydro-Imidazooxazole Derivative with Promising Action against Tuberculosis In Vitro and In Mice. PLoS Med. 2006, 3, e466 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover C. K.; Warrener P.; VanDevanter D. R.; Sherman D. R.; Arain T. M.; Langhorne M. H.; Anderson S. W.; Towell J. A.; Yuan Y.; McMurray D. N.; Kreiswirth B. N.; Barry C. E.; Baker W. R. A Small-Molecule Nitroimidazopyran Drug Candidate for the Treatment of Tuberculosis. Nature 2000, 405, 962–966. 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- Dubnau E.; Chan J.; Raynaud C.; Mohan V. P.; Lanéelle M.-A.; Keming Y.; Quémard A.; Smith I.; Daffé M. Oxygenated Mycolic Acids are Necessary for Virulence of Mycobacterium tuberculosis in Mice. Mol. Microbiol. 2000, 36, 630–637. 10.1046/j.1365-2958.2000.01882.x. [DOI] [PubMed] [Google Scholar]
- Korf J.; Stoltz A.; Verschoor J.; De Baetselier P.; Grooten J. The Mycobacterium tuberculosis Cell Wall Component Mycolic Acid Elicits aathogen-Associated Host Innate Immune Responses. Eur. J. Immunol. 2005, 35, 890–900. 10.1002/eji.200425332. [DOI] [PubMed] [Google Scholar]
- Russell D. G.; Cardona P.-J.; Kim M.-J.; Allain S.; Altare F. Foamy Macrophages and the Progression of the Human Tuberculosis Granuloma. Nat. Immunol. 2009, 10, 943–948. 10.1038/ni.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beukes M.; Lemmer Y.; Deysel M.; Al Dulayymi J. R.; Baird M. S.; Koza G.; Iglesias M. M.; Rowles R. R.; Theunissen C.; Grooten J.; Toschi G.; Roberts V. V.; Pilcher L.; Van Wyngaardt S.; Mathebula N.; Balogun M.; Stoltz A. C.; Verschoor J. A. Structure-Function Relationships of the Antigenicity of Mycolic Acids in Tuberculosis Patients. Chem. Phys. Lipids 2010, 163, 800–808. 10.1016/j.chemphyslip.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody D. B.; Reinhold B. B.; Guy M. R.; Backman E. M.; Frederique D. E.; Furlong S. T.; Ye S.; Reinhold V. N.; Sieling P. A.; Modlin R. L.; Besra G. S.; Porcelli S. A. Structural Requirements for Glycolipid Antigen Recognition by CD1b-Restricted T Cells. Science 1997, 278, 283–286. 10.1126/science.278.5336.283. [DOI] [PubMed] [Google Scholar]
- Grant E. P.; Beckman E. M.; Behar S. M.; Degano M.; Frederique D.; Besra G. S.; Wilson I. A.; Porcelli S. A.; Furlong S. T.; Brenner M. B. Fine Specificity of TCR Complementarity-Determining Region Residues and Lipid Antigen Hydrophilic Moieties in the Recognition of a CD1-Lipid Complex. J. Immunol. 2002, 168, 3933–3940. 10.4049/jimmunol.168.8.3933. [DOI] [PubMed] [Google Scholar]
- Van Rhijn I.; Iwany S. K.; Fodran P.; Cheng T.-Y.; Gapin L.; Minnaard A. J.; Moody D. B. CD1b-Mycolic Acid Tetramers Demonstrate T-Cell Fine Specificity for Mycobacterial Lipid Tails. Eur. J. Immunol. 2017, 47, 1525–1534. 10.1002/eji.201747062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decout A.; Silva-Gomes S.; Drocourt D.; Barbe S.; Andre I.; Cueto F. J.; Lioux T.; Sancho D.; Perouzel E.; Vercellone A.; Prandi J.; Gilleron M.; Tiraby G.; Nigou J. Rational Design of Adjuvants Targeting the C-type Lectin Mincle. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 2675–2680. 10.1073/pnas.1612421114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layre E.; Collmann A.; Bastian M.; Mariotti S.; Czaplicki J.; Prandi J.; Mori L.; Stenger S.; De Libero G.; Puzo G.; Gilleron M. Mycolic Acids Constitute a Scaffold for Mycobacterial Lipid Antigens Stimulating CD1-Restricted T Cells. Chem. Biol. 2009, 16, 82–92. 10.1016/j.chembiol.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Prandi J. A Convenient Synthesis of Glucose Monomycolate. Carbohydr. Res. 2012, 347, 151–154. 10.1016/j.carres.2011.11.007. [DOI] [PubMed] [Google Scholar]
- Al Dulayymi J. R.; Baird M. S.; Roberts E. The Synthesis of a Single Enantiomer of a Major Alpha-Mycolic Acid of Mycobacterium tuberculosis. Chem. Commun. 2003, 228–229. 10.1039/b209876h. [DOI] [PubMed] [Google Scholar]
- Al Dulayymi J. R.; Baird M. S.; Roberts E. The Synthesis of a Single enantiomer of a Major α-Mycolic Acid of M. tuberculosis. Tetrahedron 2005, 61, 11939–11951. 10.1016/j.tet.2005.09.056. [DOI] [Google Scholar]
- Grandjean D.; Pale P.; Chuche J. Enzymatic Hydrolysis of Cyclopropanes. Total Synthesis of Optically Pure Dictyoterpenes A and C’. Tetrahedron 1991, 47, 1215–1230. 10.1016/S0040-4020(01)86378-X. [DOI] [Google Scholar]
- Koza G.; Theunissen C.; Al Dulayymi J. R.; Baird M. S. The Synthesis of Single Enantiomers of Mycobacterial Ketomycolic Acids Containing cis-Cyclopropanes. Tetrahedron 2009, 65, 10214–10229. 10.1016/j.tet.2009.09.099. [DOI] [Google Scholar]
- Koza G.; Muzael M.; Schubert-Rowles R. R.; Theunissen C.; Al Dulayymi J. R.; Baird M. S. The Synthesis of Methoxy and Keto Mycolic Acids Containing Methyl-trans-cyclopropanes. Tetrahedron 2013, 69, 6285–6296. 10.1016/j.tet.2013.04.134. [DOI] [Google Scholar]
- Vander Beken S.; Al Dulayymi J. R.; Naessens T.; Koza G.; Maza-Iglesias M.; Rowles R.; Theunissen C.; De Medts J.; Lanckacker E.; Baird M. S.; Grooten J. Molecular Structure of the Mycobacterium tuberculosis Virulence Factor, Mycolic Acid, Determines the Elicited Inflammatory Pattern. Eur. J. Immunol. 2011, 41, 450–460. 10.1002/eji.201040719. [DOI] [PubMed] [Google Scholar]
- Riley L. W. Of Mice, Men and Elephants: Mycobacterium tuberculosis Cell Envelope Lipids and Pathogenesis. J. Clin. Invest. 2006, 116, 1475–1478. 10.1172/JCI28734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali O. T.; Sahb M. M.; Al Dulayymi J. R.; Baird M. S. Glycerol Mycolates from Synthetic Mycolic Acids. Carbohydr. Res. 2017, 448, 67–73. 10.1016/j.carres.2017.04.023. [DOI] [PubMed] [Google Scholar]
- Chancellor A.; Tocheva A. S.; Cave-Ayland C.; Tezera L.; White A.; Al Dulayymi J. R.; Bridgeman J. S.; Tews I.; Wilson S.; Lissin N. M.; Tebruegge M.; Marshall B.; Sharpe S.; Elliott T.; Skylaris C. K.; Essex J. W.; Baird M. S.; Gadola S.; Elkington P.; Mansour S. CD1b-restricted GEM T Cell Responses are Modulated by Mycobacterium tuberculosis Mycolic Acid Meromycolate Chains. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E10956–E10964. 10.1073/pnas.1708252114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody D. B.; Ulrichs T.; Mühlecker W.; Young D. C.; Gurcha S. S.; Grant E.; Rosat J.-P.; Brenner M. B.; Costello C. E.; Besra G. S.; Porcelli S. A. CD1c-mediated T-cell Recognition of Isoprenoid Glycolipids in Mycobacterium tuberculosis Infection. Nature 2000, 404, 884–888. 10.1038/35009119. [DOI] [PubMed] [Google Scholar]
- Crich D.; Dudkin V. Confirmation of the Connectivity of 4,8,12,16,20-Pentamethylpentacosylphoshoryl β-D-Mannopyranoside, an Unusual β-Mannosyl Phosphoisoprenoid from Mycobacterium avium, through Synthesis. J. Am. Chem. Soc. 2002, 124, 2263–2266. 10.1021/ja0123958. [DOI] [PubMed] [Google Scholar]
- van Summeren R. P.; Reijmer S. J.; Feringa B. L.; Minnaard A. J. Catalytic Asymmetric Synthesis of Enantiopure Isoprenoid Building Blocks: Application in the Synthesis of Apple Leafminer Pheromones. Chem. Commun. 2005, 1387–1389. 10.1039/b419268k. [DOI] [PubMed] [Google Scholar]
- van Summeren R. P.; Moody D. B.; Feringa B. L.; Minnaard A. J. Total Synthesis of Enantiopure β-D-Mannosyl Phosphomycoketides from Mycobacterium tuberculosis. J. Am. Chem. Soc. 2006, 128, 4546–4547. 10.1021/ja060499i. [DOI] [PubMed] [Google Scholar]
- Matsunaga I.; Bhatt A.; Young D. C.; Cheng T. Y.; Eyles S. J.; Besra G. S.; Briken V.; Porcelli S. A.; Costello C. E.; Jacobs W. R. Jr.; Moody D. B. Mycobacterium tuberculosis pks12 Produces a Novel Polyketide Presented by CD1c to T Cells. J. Exp. Med. 2004, 200, 1559–1569. 10.1084/jem.20041429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C.; Montagnon T.; Baran P. S.; Zhong Y.-L. Iodine(V) Reagents in Organic Synthesis. Part 4. o-Iodoxybenzoic Acid as a Chemospecific Tool for Single Electron Transfer-Based Oxidation Processes. J. Am. Chem. Soc. 2002, 124, 2245–2258. 10.1021/ja012127+. [DOI] [PubMed] [Google Scholar]
- de Jong A.; Arce E. C.; Cheng T.-Y.; van Summeren R. P.; Feringa B. L.; Dudkin V.; Crich D.; Matsunaga I.; Minnaard A. J.; Moody D. B. CD1c Presentation of Synthetic Glycolipid Antigens with Foreign Alkyl Branching Motifs. Chem. Biol. 2007, 14, 1232–1242. 10.1016/j.chembiol.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly D.; Kasmar A. G.; Cheng T.-Y.; de Jong A.; Huang S.; Roy S.; Bhatt A.; van Summeren R. P.; Altman J. D.; Jacobs W. R. Jr.; Adams E. J.; Minnaard A. J.; Porcelli S. A.; Moody D. B. CD1c Tetramers Detect ex vivo T Cell Responses to Processed Phosphomycoketide Antigens. J. Exp. Med. 2013, 210, 729–741. 10.1084/jem.20120624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.-S.; Scharf L.; Adams E. J.; Piccirilli J. A. Highly Stereocontrolled Total Synthesis of β-D-Mannosyl Phosphomycoketide: A Natural Product from Mycobacterium tuberculosis. J. Org. Chem. 2013, 78, 5970–5986. 10.1021/jo4006602. [DOI] [PubMed] [Google Scholar]
- Roy S.; Ly D.; Li N.-S.; Altman J. D.; Piccirilli J. A.; Moody D. B.; Adams E. J. Molecular Basis of Mycobacterial Lipid Antigen Presentation by CD1c and its Recognition by αβ T Cells. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E4648–E4657. 10.1073/pnas.1408549111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnaard A. J.; Andringa R.; de Kok N.; Driessen A.. A Unified Approach for the Total Synthesis of cyclo-Archaeol, iso-Caldarchaeol, Caldarchaeol, and Mycoketide. Angew. Chem., Int. Ed. 2021, 10.1002/anie.202104759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnikin D. E.; Kremer L.; Dover L. G.; Besra G. S. The Methyl-Branched Fortifications of Mycobacterium tuberculosis. Chem. Biol. 2002, 9, 545–553. 10.1016/S1074-5521(02)00142-4. [DOI] [PubMed] [Google Scholar]
- Jarlier V.; Nikaido H. Mycobacterial Cell Wall: Structure and Role in Natural Resistance to Antibiotics. FEMS Microbiol. Lett. 1994, 123, 11–18. 10.1111/j.1574-6968.1994.tb07194.x. [DOI] [PubMed] [Google Scholar]
- Dmitriev B. A.; Ehlers S.; Rietschel E. T.; Brennan P. J. Molecular Mechanics of the Mycobacterial Cell Wall: From Horizontal Layers to Vertical Scaffolds. Int. J. Med. Microbiol. 2000, 290, 251–258. 10.1016/S1438-4221(00)80122-8. [DOI] [PubMed] [Google Scholar]
- Jackson M.; Stadthagen G.; Gicquel B. Long-chain Multiple Methyl-branched Fatty Acid-containing Lipids of Mycobacterium tuberculosis: Biosynthesis, Transport, Regulation and Biological Activities. Tuberculosis 2007, 87, 78–86. 10.1016/j.tube.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Des Mazery R.; Pullez M.; López F.; Harutyunyan S. R.; Minnaard A. J.; Feringa B. L. An Iterative Catalytic Route to Enantiopure Deoxypropionate Subunits: Asymmetric Conjugate Addition of Grignard Reagents to α,β-Unsaturated Thioesters. J. Am. Chem. Soc. 2005, 127, 9966–9967. 10.1021/ja053020f. [DOI] [PubMed] [Google Scholar]
- Ter Horst B.; Feringa B. L.; Minnaard A. J. Catalytic Asymmetric Synthesis of Phthioceranic Acid, a Heptamethyl-Branched Acid from Mycobacterium tuberculosis. Org. Lett. 2007, 9, 3013–3015. 10.1021/ol071078o. [DOI] [PubMed] [Google Scholar]
- Casas-Arce E.; Ter Horst B.; Feringa B. L.; Minnaard A. J. Asymmetric Total Synthesis of PDIM A: A Virulence Factor of Mycobacterium tuberculosis. Chem. - Eur. J. 2008, 14, 4157–4159. 10.1002/chem.200800243. [DOI] [PubMed] [Google Scholar]
- Camacho L. R.; Ensergueix D.; Perez E.; Gicquel B.; Guilhot C. Identification of a Virulence Gene Cluster of Mycobacterium tuberculosis by Signature-Tagged Transposon Mutagenesis. Mol. Microbiol. 1999, 34, 257–267. 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- Cox J. S.; Chen B.; McNeil M.; Jacobs W. R. Jr. Complex Lipid Determines Tissue-specific Replication of Mycobacterium tuberculosis in Mice. Nature 1999, 402, 79–83. 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- Rousseau C.; Winter N.; Pivert E.; Bordat Y.; Neyrolles O.; Avé P.; Huerre M.; Gicquel B.; Jackson M. Production of Phthiocerol Dimycocerosates Protects Mycobacterium tuberculosis from the Cidal Activity of Reactive Nitrogen Intermediates Produced by Macrophages and Modulates the Early Immune Response to Infection. Cell. Microbiol. 2004, 6, 277–287. 10.1046/j.1462-5822.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- Cambier C. J.; Takaki K. K.; Larson R. P.; Hernandez R. E.; Tobin D. M.; Urdahl K. B.; Cosma C. L.; Ramakrishnan L. Mycobacteria Manipulate Macrophage Recruitment Through Coordinated Use of Membrane Lipids. Nature 2014, 505, 218–222. 10.1038/nature12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho L. R.; Constant P.; Raynaud C.; Laneelle M. A.; Triccas J. A.; Gicquel B.; Daffe M.; Guilhot C. Analysis of the Phthiocerol Dimycocerosate Locus of Mycobacterium tuberculosis. J. Biol. Chem. 2001, 276, 19845–19854. 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- Astarie-Dequeker C.; Le Guyader L.; Malaga W.; Seaphanh F.-K.; Chalut C.; Lopez A.; Guilhot C. Phthiocerol Dimycocerosates of M. tuberculosis Participate in Macrophage Invasion by Inducing Changes in the Organization of Plasma Membrane Lipids. PLoS Pathog. 2009, 5, e1000289 10.1371/journal.ppat.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augenstreich J.; Haanappel E.; Ferré G.; Czaplicki G.; Jolibois F.; Destainville N.; Guilhot C.; Milon A.; Astarie-Dequeker C.; Chavent M. The Conical Shape of DIM Lipids Promotes Mycobacterium tuberculosis Infection of Macrophages. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 25649–25658. 10.1073/pnas.1910368116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley J.; Hughitt V. K.; Velikovsky C. A.; Mariuzza R. A.; El-Sayed N. M.; Briken V. The Cell Wall Lipid PDIM Contributes to Phagosomal Escape and Host Cell Exit of Mycobacterium tuberculosis. mBio 2017, 8, e00148-17 10.1128/mBio.00148-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman M. M.; Pagán A. J.; Shanahan J. K.; Ramakrishnan L. Mycobacterium marinum phthiocerol dimycocerosates enhance macrophage phagosomal permeabilization and membrane damage. PLoS One 2020, 15 (7), e0233252 10.1371/journal.pone.0233252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar N. Constituents of the Lipids of Tubercle Bacili. Part IV. Mycoceranic Acid. J. Chem. Soc. 1954, 1011–1012. 10.1039/jr9540001011. [DOI] [Google Scholar]
- Marks G. S.; Polgar N. Mycoceranic Acid. Part II. J. Chem. Soc. 1955, 3851–3857. 10.1039/jr9550003851. [DOI] [Google Scholar]
- Polgar N.; Smith W. Mycoceranic Acid. Part III. J. Chem. Soc. 1963, 3081–3085. 10.1039/jr9630003081. [DOI] [Google Scholar]
- Maskens K.; Polgar N. Stereochemisty of the Phtiocerols. J. Chem. Soc. D 1970, 340. 10.1039/c29700000340. [DOI] [Google Scholar]
- Maskens K.; Polgar N. Absolute Configuration of the -CH(OMe)-CHMe- System in the Phtiocerols. J. Chem. Soc., Perkin Trans. 1 1973, 1909–1912. 10.1039/P19730001909. [DOI] [Google Scholar]
- Ter Horst B.; Feringa B. L.; Minnaard A. J. Catalytic Asymmetric Synthesis of Mycocerosic Acid. Chem. Commun. 2007, 489–491. 10.1039/B612593J. [DOI] [PubMed] [Google Scholar]
- Flentie K. N.; Stallings C. L.; Turk J.; Minnaard A. J.; Hsu F.-F. Characterization of Phthiocerol and Phthiodiolone Dimycocerosate Esters of M. tuberculosis by Multiple-stage Linear Ion-trap MS. J. Lipid Res. 2016, 57, 142–155. 10.1194/jlr.D063735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T.; Nakagome H.; Sano S.; Sadayuki T.; Hosokawa S. Total Synthesis of PDIM A. Chem. Lett. 2016, 45, 550–551. 10.1246/cl.160152. [DOI] [Google Scholar]
- Nakamura T.; Kubota K.; Ieki T.; Hosokawa S. Stereoselective Alkylation of the Vinylketene Silyl N,O-Acetal and Its Application to the Synthesis of Mycocerosic Acid. Org. Lett. 2016, 18, 132–135. 10.1021/acs.orglett.5b03422. [DOI] [PubMed] [Google Scholar]
- Hunter S. W.; Brennan P. J. A Novel Phenolic Glycolipid from Mycobacterium leprae Possibly Involved in Immunogenicity and Pathogenicity. J. Bacteriol. 1981, 147, 728–735. 10.1128/jb.147.3.728-735.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S. W.; Fujiwara T.; Brennan P. J. Structure and Antigenicity of the Major Specific Glycolipid Antigen of Mycobacterium leprae. J. Biol. Chem. 1982, 257, 15072–15078. 10.1016/S0021-9258(18)33395-7. [DOI] [PubMed] [Google Scholar]
- Tarelli E.; Draper P.; Payne S. N. Structure of the Oligosaccharide Component of a Serologically Active Phenolic Glycolipid of Mycobacterium leprae. Carbohydr. Res. 1984, 131, 346–352. 10.1016/0008-6215(84)85258-1. [DOI] [Google Scholar]
- Demarteau-Ginsburg H.; Lederer E. Sur la Structure Chimique du Mycoside B. Biochim. Biophys. Acta 1963, 70, 442–451. 10.1016/0006-3002(63)90774-1. [DOI] [PubMed] [Google Scholar]
- Villé C.; Gastambide-Odier M. Le 3-O-méthyl-l-rhamnose, sucre isolé du mycoside G de Mycobacterium marinum. Carbohydr. Res. 1970, 12, 97–107. 10.1016/S0008-6215(00)80229-3. [DOI] [Google Scholar]
- Gastambide-Odier M.; Sarda P. Contribution à l’étude de la structure et de la biosynthèse de glycolipides spécifiques isolés de mycobactéries: les mycosides A et B. Lung 1970, 142, 241–255. 10.1007/BF02095222. [DOI] [PubMed] [Google Scholar]
- Daffé M.; Lacave C.; Lanéelle M.-A.; Lanéelle G. Structure of the Major Triglycosyl Phenol-phthiocerol of Mycobacterium tuberculosis (Strain Canetti). Eur. J. Biochem. 1987, 167, 155–160. 10.1111/j.1432-1033.1987.tb13317.x. [DOI] [PubMed] [Google Scholar]
- Daffé M.; Servin P. Scalar, Dipolar-correlated and J-resolved 2D-NMR Spectroscopy of the Specific Phenolic Mycoside of Mycobacterium tuberculosis. Eur. J. Biochem. 1989, 185, 157–162. 10.1111/j.1432-1033.1989.tb15097.x. [DOI] [PubMed] [Google Scholar]
- Reed M. B.; Domenech P.; Manca C.; Su H.; Barczak A. K.; Kreiswirth B. N.; Kaplan G.; Barry III C. E. A Glycolipid of Hypervirulent Tuberculosis Strains that Inhibits the Innate Immune Response. Nature 2004, 431, 84–87. 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- Sinsimer D.; Huet G.; Manca C.; Tsenova L.; Koo M. S.; Kurepina N.; Kana B.; Mathema B.; Marras S. A.; Kreiswirth B. N.; Guilhot C.; Kaplan G. The Phenolic Glycolipid of Mycobacterium tuberculosis Differentially Modulates the Early Host Cytokine Response but Does Not in Itself Confer Hypervirulence. Infect. Immun. 2008, 76, 3027–3036. 10.1128/IAI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes D. D.; Lundahl M. L. E.; Lavelle E. C.; Scanlan E. M. The Emergence of Phenolic Glycans as Virulence Factors in Mycobacterium tuberculosis. ACS Chem. Biol. 2017, 12, 1969–1979. 10.1021/acschembio.7b00394. [DOI] [PubMed] [Google Scholar]
- Cambier C. J.; O’Leary S. M.; O’Sullivan M. P.; Keane J.; Ramakrishnan L. Phenolic Glycolipid Facilitates Mycobacterial Escape from Microbicidal Tissue-Resident Macrophages. Immunity 2017, 47, 552–565. 10.1016/j.immuni.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonney N.; Chavanet P.; Perronne C.; Leportier M.; Revol F.; Herrmann J. L.; Lagrange P. H. B-cell Immune Responses in HIV Positive and HIV Negative Patients with Tuberculosis Evaluated with an ELISA Using a Glycolipid Antigen. Tuberculosis 2007, 87, 109–122. 10.1016/j.tube.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Barroso S.; Castelli R.; Baggelaar M. P.; Geerdink D.; Ter Horst B.; Casas-Arce E.; Overkleeft H. S.; van der Marel G. A.; Codee J. D.; Minnaard A. J. Total Synthesis of the Triglycosyl Phenolic Glycolipid PGL-tb1 from Mycobacterium tuberculosis. Angew. Chem., Int. Ed. 2012, 51 (47), 11774–11777. 10.1002/anie.201206221. [DOI] [PubMed] [Google Scholar]
- Geerdink D.; Horst B. t.; Lepore M.; Mori L.; Puzo G.; Hirsch A. K. H.; Gilleron M.; de Libero G.; Minnaard A. J. Total Synthesis, Stereochemical Elucidation and Biological Evaluation of Ac2SGL; a 1,3-Methyl Branched Sulfoglycolipid from Mycobacterium tuberculosis. Chem. Sci. 2013, 4, 709–716. 10.1039/C2SC21620E. [DOI] [Google Scholar]
- Geerdink D.Total Synthesis of Enantiopure Lipids: On Mycobacterial Glycolipids and a Putative Sex Pheromone of Trichogramma turkestanica. PhD Thesis, University of Groningen, Groningen, 2013. [Google Scholar]
- Pischl M. C.; Weise C. F.; Muller M. A.; Pfaltz A.; Schneider C. A Convergent and Stereoselective Synthesis of the Glycolipid Components Phthioceranic Acid and Hydroxyphthioceranic Acid. Angew. Chem., Int. Ed. 2013, 52, 8968–8972. 10.1002/anie.201303776. [DOI] [PubMed] [Google Scholar]
- Rasappan R.; Aggarwal V. K. Synthesis of Hydroxyphthioceranic Acid Using a Traceless Lithiation-borylation-protodeboronation Strategy. Nat. Chem. 2014, 6, 810–814. 10.1038/nchem.2010. [DOI] [PubMed] [Google Scholar]
- Balieu S.; Hallett G. E.; Burns M.; Bootwicha T.; Studley J.; Aggarwal V. K. Toward Ideality: The Synthesis of (+)-Kalkitoxin and (+)-Hydroxyphthioceranic Acid by Assembly-Line Synthesis. J. Am. Chem. Soc. 2015, 137, 4398–4403. 10.1021/ja512875g. [DOI] [PubMed] [Google Scholar]
- Xu S.; Oda A.; Bobinski T.; Li H.; Matsueda Y.; Negishi E. Highly Efficient, Convergent, and Enantioselective Synthesis of Phthioceranic Acid. Angew. Chem., Int. Ed. 2015, 54, 9319–9322. 10.1002/anie.201503818. [DOI] [PubMed] [Google Scholar]
- Che W.; Wen D. C.; Zhu S. F.; Zhou Q. L. Iterative Synthesis of Polydeoxypropionates Based on Iridium-Catalyzed Asymmetric Hydrogenation of α-Substituted Acrylic Acids. Org. Lett. 2018, 20, 3305–3309. 10.1021/acs.orglett.8b01193. [DOI] [PubMed] [Google Scholar]
- Weise C. F.; Pischl M.; Pfaltz A.; Schneider C. A Non-Iterative, Flexible, and Highly Stereoselective Synthesis of Polydeoxypropionates-Synthesis of (+)-Vittatalactone. Chem. Commun. 2011, 47, 3248–3250. 10.1039/c0cc05215a. [DOI] [PubMed] [Google Scholar]
- Weise C. F.; Pischl M. C.; Pfaltz A.; Schneider C. A General, Asymmetric, and Noniterative Synthesis of Trideoxypropionates. Straightforward Syntheses of the Pheromones (+)-Vittatalactone and (+)-Norvittatalactone. J. Org. Chem. 2012, 77, 1477–1488. 10.1021/jo202330b. [DOI] [PubMed] [Google Scholar]
- Gilleron M.; Stenger S.; Mazorra Z.; Wittke F.; Mariotti S.; Böhmer G.; Prandi J.; Mori L.; Puzo G.; De Libero G. Diacylated Sulfoglycolipids Are Novel Mycobacterial Antigens Stimulating CD1-restricted T Cells during Infection with Mycobacterium tuberculosis. J. Exp. Med. 2004, 199, 649–659. 10.1084/jem.20031097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiard J.; Collmann A.; Gilleron M.; Mori L.; De Libero G.; Prandi J.; Puzo G. Synthesis of Diacylated Trehalose Sulfates: Candidates for a Tuberculosis Vaccine. Angew. Chem., Int. Ed. 2008, 47, 9734–9738. 10.1002/anie.200803835. [DOI] [PubMed] [Google Scholar]
- Guiard J.; Collmann A.; Garcia-Alles L. F.; Mourey L.; Brando T.; Mori L.; Gilleron M.; Prandi J.; De Libero G.; Puzo G. Fatty Acyl Structures of Mycobacterium tuberculosis Sulfoglycolipid Govern T Cell Response. J. Immunol. 2009, 182, 7030–7037. 10.4049/jimmunol.0804044. [DOI] [PubMed] [Google Scholar]
- James C. A.; Yu K. K. Q.; Gilleron M.; Prandi J.; Yedulla V. R.; Moleda Z. Z.; Diamanti E.; Khan M.; Aggarwal V. K.; Reijneveld J. F.; Reinink P.; Lenz S.; Emerson R. O.; Scriba T. J.; Souter M. N. T.; Godfrey D. I.; Pellicci D. G.; Moody D. B.; Minnaard A. J.; Seshadri C.; Van Rhijn I. CD1b Tetramers Identify T Cells that Recognize Natural and Synthetic Diacylated Sulfoglycolipids from Mycobacterium tuberculosis. Cell Chem. Biol. 2018, 25, 392–402. 10.1016/j.chembiol.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madduri A. V. R.; Minnaard A. J. Formal Synthesis of the Anti-Angiogenic Polyketide (−)-Borrelidin under Asymmetric Catalytic Control. Chem. - Eur. J. 2010, 16, 11726–11731. 10.1002/chem.201001284. [DOI] [PubMed] [Google Scholar]
- Geerdink D.; Minnaard A. J. Total Synthesis of Sulfolipid-1. Chem. Commun. 2014, 50, 2286–2288. 10.1039/C3CC48087A. [DOI] [PubMed] [Google Scholar]
- Goren M. B. Sulfolipid I of Mycobacterium Tuberculosis, Strain H37Rv II. Structural Studies. Biochim. Biophys. Acta, Lipids Lipid Metab. 1970, 210, 127–138. 10.1016/0005-2760(70)90068-8. [DOI] [PubMed] [Google Scholar]
- Goren M. B. Sulfolipid I of Mycobacterium Tuberculosis, Strain H37Rv I. Purification and Propoerties. Biochim. Biophys. Acta, Lipids Lipid Metab. 1970, 210, 116–126. 10.1016/0005-2760(70)90067-6. [DOI] [PubMed] [Google Scholar]
- Goren M. B.; Brokl O.; Das B. C.; Lederer E. Sulfolipid I of Mycobacterium tuberculosis, Strain H37Rv. Nature of the Acyl Substituents. Biochemistry 1971, 10, 72–81. 10.1021/bi00777a012. [DOI] [PubMed] [Google Scholar]
- Gangadharam P. R. J.; Cohn M. L.; Middlerrook G. Infectivity, Pathogenicity and Sulpholipid Fraction of Some Indian and British Strains of Tubercle Bacilli. Tubercle 1963, 44, 452–455. 10.1016/S0041-3879(63)80087-2. [DOI] [PubMed] [Google Scholar]
- Pabst M. J.; Gross J. M.; Brozna J. P.; Goren M. B. Inhibition of Macrophage Priming by Sulfatide from Mycobacterium tuberculosis. J. Immunol. 1988, 140, 634–640. [PubMed] [Google Scholar]
- Zhang L.; Goren M. B.; Holzer T. J.; Andersen B. R. Effect of Mycobacterium tuberculosis-Derived Sulfolipid I on Human Phagocytic Cells. Infect. Immun. 1988, 56, 2876–2883. 10.1128/iai.56.11.2876-2883.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozna J. P.; Horan M.; Rademacher J. M.; Pabst K. M.; Pabst M. J. Monocyte Responses to Sulfatide from Mycobacterium tuberculosis: Inhibition of Priming for Enhanced Release of Superoxide, Associated with Increased Secretion of Interleukin-1 and Tumor Necrosis Factor Alpha, and Altered Protein Phosphorylation. Infect. Immun. 1991, 59, 2542–2548. 10.1128/iai.59.8.2542-2548.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; English D.; Andersen B. R. Activation of Human Neutrophils by Mycobacterium tuberculosis-derived Sulfolipid-1. J. Immunol. 1991, 146, 2730–2736. [PubMed] [Google Scholar]
- Gilmore S. A.; Schelle M. W.; Holsclaw C. M.; Leigh C. D.; Jain M.; Cox J. S.; Leary J. A.; Bertozzi C. R. Sulfolipid-1 Biosynthesis Restricts Mycobacterium tuberculosis Growth in Human Macrophages. ACS Chem. Biol. 2012, 7, 863–870. 10.1021/cb200311s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl C. R.; Pasko B. L.; Khan H. S.; Kindt L. M.; Stamm C. E.; Franco L. H.; Hsia C. C.; Zhou M.; Davis C. R.; Qin T.; Gautron L.; Burton M. D.; Mejia G. L.; Naik D. K.; Dussor G.; Price T. J.; Shiloh M. U. Mycobacterium tuberculosis Sulfolipid-1 Activates Nociceptive Neurons and Induces Cough. Cell 2020, 181, 293–305. 10.1016/j.cell.2020.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnikin D. E.; Dobson G.; Sesardic D.; Ridell M. Mycolipenates and Mycolipanolates of Trehalose from Mycobacterium tuberculosis. Microbiology 1985, 131, 1369–1374. 10.1099/00221287-131-6-1369. [DOI] [PubMed] [Google Scholar]
- Daffé M.; Lacave C.; Lanéelle M.-A.; Gillois M.; Lanéelle G. Polyphthienoyl Trehalose, Glycolipids for Virulent Strains of the Tubercle Bacillus. Eur. J. Biochem. 1988, 172, 579–584. 10.1111/j.1432-1033.1988.tb13928.x. [DOI] [PubMed] [Google Scholar]
- Daffé M.; Papa F.; Laszlo A.; David H. L. Glycolipids of Recent Clinical Isolates of Mycobacterium tuberculosis: Chemical Characterization and Immunoreactivity. Microbiology 1989, 135, 2759–2766. 10.1099/00221287-135-10-2759. [DOI] [PubMed] [Google Scholar]
- Besra G. S.; Bolton R. C.; McNeil M. R.; Ridell M.; Simpson K. E.; Glushka J.; van Halbeek H.; Brennan P. J.; Minnikin D. E. Structural Elucidation of a Novel Family of Acyltrehaloses from Mycobacterium tuberculosis. Biochemistry 1992, 31, 9832–9837. 10.1021/bi00155a040. [DOI] [PubMed] [Google Scholar]
- Rousseau C.; Neyrolles O.; Bordat Y.; Giroux S.; Sirakova T. D.; Prevost M.-C.; Kolattukudy P. E.; Gicquel B.; Jackson M. Deficiency in Mycolipenate- and Mycosanoate-Derived Acyltrehaloses Enhances Early Interactions of Mycobacterium tuberculosis with Host Cells. Cell. Microbiol. 2003, 5, 405–415. 10.1046/j.1462-5822.2003.00289.x. [DOI] [PubMed] [Google Scholar]
- Dubey V. S.; Sirakova P. E.; Kolattukude P. E. Disruption of msl3 Abolishes the Synthesis of Mycolipanoic and Mycolipenic Acids Required for Polyacyltrehalose Synthesis in Mycobacterium tuberculosis H37Rv and Causes Cell Aggregation. Mol. Microbiol. 2002, 45, 1451–1459. 10.1046/j.1365-2958.2002.03119.x. [DOI] [PubMed] [Google Scholar]
- Gonzalo Asensio J.; Maia C.; Ferrer N. L.; Barilone N.; Laval F.; Soto C. Y.; Winter N.; Daffe M.; Gicquel B.; Martin C.; Jackson M. The Virulence-associated Two-component PhoP-PhoR System Controls the Biosynthesis of Polyketide-derived Lipids in Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 1313–1316. 10.1074/jbc.C500388200. [DOI] [PubMed] [Google Scholar]
- Walters S. B.; Dubnau E.; Kolesnikova I.; Laval F.; Daffe M.; Smith I. The Mycobacterium tuberculosis PhoPR Two-Component System Regulates Genes Essential for Virulence and Complex Lipid Biosynthesis. Mol. Microbiol. 2006, 60, 312–330. 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- Gonzalo-Asensio J.; Mostowy S.; Harders-Westerveen J.; Huygen K.; Hernandez-Pando R.; Thole J.; Behr M.; Gicquel B.; Martin C. PhoP: A Missing Piece in the Intricate Puzzle of Mycobacterium tuberculosis Virulence. PLoS One 2008, 3, e3496 10.1371/journal.pone.0003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzios S. K.; Schelle M. W.; Holsclaw C. M.; Behrens C. R.; Botyanszki Z.; Lin F. L.; Carlson B. L.; Kumar P.; Leary J. A.; Bertozzi C. R. PapA3 is an Acyltransferase Required for Polyacyltrehalose Biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 2009, 284, 12745–12751. 10.1074/jbc.M809088200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J. E.; Ramirez A. S.; Salas L. P.; Helguera-Repetto C.; Gonzalez-Y-Merchand J.; Soto C. Y.; Hernandez-Pando R. Transcription of Genes Involved in Sulfolipid and Polyacyltrehalose Biosynthesis of Mycobacterium tuberculosis in Experimental Latent Tuberculosis Infection. PLoS One 2013, 8, e58378 10.1371/journal.pone.0058378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.-S.; Dubey V. S.; Kolattukudy P. E.; Song C.-H.; Shin A.-R.; Jung S.-B.; Yang C.-S.; Kim S.-Y.; Jo E.-K.; Park J.-K.; Kim H.-J. Diacyltrehalose of Mycobacterium tuberculosis Inhibits Lipopolysaccharide- and Mycobacteria-Induced Proinflammatory Cytokine Production in Human Monocytic Cells. FEMS Microbiol. Lett. 2007, 267, 121–128. 10.1111/j.1574-6968.2006.00553.x. [DOI] [PubMed] [Google Scholar]
- Saavedra R.; Segura E.; Leyva R.; Esparza L. A.; Lopez-Marin L. M. Mycobacterial Di-O-Acyl-Trehalose Inhibits Mitogen- and Antigen-Induced Proliferation of Murine T Cells In Vitro. Clin. Diagn. Lab. Immunol. 2001, 8, 1081–1088. 10.1128/CDLI.8.6.1-91-1088.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Casabona N.; Gonzales Fuente T.; Papa F.; Rosselló Urgell J.; Vidal Plá R.; Codina Gau G.; Ruiz Camps I. Time Course of Anti-SL-IV Immunoglobin G Antibodies in Patients with Tuberculosis-Associated AIDS. J. Clin. Microbiol. 1992, 30, 1089–1093. 10.1128/jcm.30.5.1089-1093.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa F.; Luquin M.; David H. L. DOT-ELISA for Detection of Phenolic Glycolipid PGL-Tb1 and Diacyl-Trehalose Antigens of Mycobacterium tuberculosis. Res. Microbiol. 1992, 143, 327–331. 10.1016/0923-2508(92)90024-I. [DOI] [PubMed] [Google Scholar]
- Tórtola M. T.; Lanéelle M. A.; Martín-Casabona N. Comparison of Two 2,3-Diacyl Trehalose Antigens from Mycobacterium tuberculosis and Mycobacterium fortuitum for Serology in Tuberculosis Patients. Clin. Diagn. Lab. Immunol. 1996, 3, 563–566. 10.1128/cdli.3.5.563-566.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz M.; Lanéelle M.-A.; Luquin M.; Torrelles J.; Julián E.; Ausina V.; Daffé M. Occurrence of an Antigenic Triacyl Trehalose in Clinical Isolated and Reference Strains of Mycobacterium tuberculosis. FEMS Microbiol. Lett. 1997, 157, 251–259. 10.1111/j.1574-6968.1997.tb12781.x. [DOI] [PubMed] [Google Scholar]
- Julián E.; Cama M.; Martínez P.; Luquin M. An ELISA for Five Glycolipids from the Cell Wall of Mycobacterium tuberculosis: Tween 20 Interference in the Assay. J. Immunol. Methods 2001, 251, 21–30. 10.1016/S0022-1759(01)00313-1. [DOI] [PubMed] [Google Scholar]
- Holzheimer M.; Reijneveld J. F.; Ramnarine A. K.; Misiakos G.; Young D. C.; Ishikawa E.; Cheng T. Y.; Yamasaki S.; Moody D. B.; Van Rhijn I.; Minnaard A. J. Asymmetric Total Synthesis of Mycobacterial Diacyl Trehaloses Demonstrates a Role for Lipid Structure in Immunogenicity. ACS Chem. Biol. 2020, 15, 1835–1841. 10.1021/acschembio.0c00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Horst B.; van Wermeskerken J.; Feringa B. L.; Minnaard A. J. Catalytic Asymmetric Synthesis of Mycolipenic and Mycolipanolic Acid. Eur. J. Org. Chem. 2010, 2010, 38–41. 10.1002/ejoc.200901120. [DOI] [Google Scholar]
- Reijneveld J. F.; Holzheimer M.; Young D. C.; Lopez K.; Suliman S.; Jimenez J.; Calderon R.; Lecca L.; Murray M. B.; Ishikawa E.; Yamasaki S.; Minnaard A. J.; Moody D. B.; Van Rhijn I. Synthetic Mycobacterial Diacyl Trehaloses Reveal Differential Recognition by Human T Cell Receptors and the C-type Lectin Mincle. Sci. Rep. 2021, 11, 2010. 10.1038/s41598-021-81474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S. W.; McNeil M. R.; Brennan P. J. Diglycosyl Diacylglycerol of Mycobacterium tuberculosis. J. Bacteriol. 1986, 168, 917–922. 10.1128/jb.168.2.917-922.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T.; Itoh F.; Yoshida S.; Saijo S.; Matsuzawa T.; Gonoi T.; Saito T.; Okawa Y.; Shibata N.; Miyamoto T.; Yamasaki S. Identification of Distinct Ligands for the C-type Lectin Receptors Mincle and Dectin-2 in the Pathogenic Fungus Malassezia. Cell Host Microbe 2013, 13, 477–488. 10.1016/j.chom.2013.03.008. [DOI] [PubMed] [Google Scholar]
- Richardson M. B.; Torigoe S.; Yamasaki S.; Williams S. J. Mycobacterium tuberculosis β-Gentiobiosyl Diacylglycerides Signal through the Pattern Recognition Receptor Mincle: Total Synthesis and Structure Activity Relationships. Chem. Commun. 2015, 51, 15027–15030. 10.1039/C5CC04773K. [DOI] [PubMed] [Google Scholar]
- Andrés E.; Martínez N.; Planas A. Expression and Characterization of a Mycoplasma genitalium Glycosyltransferase in Membrane Glycolipid Biosynthesis. J. Biol. Chem. 2011, 286, 35367–35379. 10.1074/jbc.M110.214148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takato K.; Kurita M.; Yagami N.; Tanaka H.-N.; Ando H.; Imamura A.; Ishida H. Chemical Synthesis of Diglucosyl Diacylglycerols Utilizing Glycosyl Donors with Stereodirecting Cyclic Silyl Protective Groups. Carbohydr. Res. 2019, 483, 107748. 10.1016/j.carres.2019.107748. [DOI] [PubMed] [Google Scholar]
- Vance J. E. Phosphatidylserine and Phosphatidylethanolamine in Mammalian Cells: Two Metabolically Related Aminophospholipids. J. Lipid Res. 2008, 49, 1377–1387. 10.1194/jlr.R700020-JLR200. [DOI] [PubMed] [Google Scholar]
- Ter Horst B.; Seshadri C.; Sweet L.; Young D. C.; Feringa B. L.; Moody D. B.; Minnaard A. J. Asymmetric Synthesis and Structure Elucidation of a Glycerophospholipid from Mycobacterium tuberculosis. J. Lipid Res. 2010, 51, 1017–1022. 10.1194/jlr.M001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit C.; Fraaije M. W.; Minnaard A. J. Reduction of Carbon-Carbon Double Bonds Using Organocatalytically Generated Diimide. J. Org. Chem. 2008, 73, 9482–9485. 10.1021/jo801588d. [DOI] [PubMed] [Google Scholar]
- Fodran P.; Minnaard A. J. Catalytic Synthesis of Enantiopure Mixed Diacylglycerols - Synthesis of a Major M. tuberculosis Phospholipid and Platelet Activating Factor. Org. Biomol. Chem. 2013, 11, 6919–6928. 10.1039/c3ob41483c. [DOI] [PubMed] [Google Scholar]
- Furse S. Is Phosphatidylglycerol Essential for Terrestrial Life?. J. Chem. Biol. 2017, 10, 1–9. 10.1007/s12154-016-0159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma P. V. G. K.; Srikanth L.; Venkatesh K.; Murthy P. S.; Sarma P. U. Isolation, Purification and Characterization of Cardiolipin Synthase from Mycobacterium phlei. Bioinformation 2013, 9, 690–695. 10.6026/97320630009690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M.; Crick D. C.; Brennan P. J. Phosphatidylinositol is an Essential Phospholipid of Mycobacteria. J. Biol. Chem. 2000, 275, 30092–30099. 10.1074/jbc.M004658200. [DOI] [PubMed] [Google Scholar]
- Van Rhijn I.; van Berlo T.; Hilmenyuk T.; Cheng T. Y.; Wolf B. J.; Tatituri R. V.; Uldrich A. P.; Napolitani G.; Cerundolo V.; Altman J. D.; Willemsen P.; Huang S.; Rossjohn J.; Besra G. S.; Brenner M. B.; Godfrey D. I.; Moody D. B. Human Autoreactive T Cells Recognize CD1b and Phospholipids. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 380–385. 10.1073/pnas.1520947112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatituri R. V.; Watts G. F.; Bhowruth V.; Barton N.; Rothchild A.; Hsu F. F.; Almeida C. F.; Cox L. R.; Eggeling L.; Cardell S.; Rossjohn J.; Godfrey D. I.; Behar S. M.; Besra G. S.; Brenner M. B.; Brigl M. Recognition of Microbial and Mammalian Phospholipid Antigens by NKT Cells with Diverse TCRs. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1827–1832. 10.1073/pnas.1220601110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf B. J.; Tatituri R. V.; Almeida C. F.; Le Nours J.; Bhowruth V.; Johnson D.; Uldrich A. P.; Hsu F. F.; Brigl M.; Besra G. S.; Rossjohn J.; Godfrey D. I.; Brenner M. B. Identification of a Potent Microbial Lipid Antigen for Diverse NKT Cells. J. Immunol. 2015, 195, 2540–2551. 10.4049/jimmunol.1501019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burugupalli S.; Richardson M. B.; Williams S. J. Total Synthesis and Mass Spectrometric Analysis of a Mycobacterium tuberculosis Phosphatidylglycerol Featuring a Two-Step Synthesis of (R)-Tuberculostearic Acid. Org. Biomol. Chem. 2017, 15, 7422–7429. 10.1039/C7OB01786C. [DOI] [PubMed] [Google Scholar]
- Pethe K.; Swenson D. L.; Alonso S.; Anderson J.; Wang C.; Russell D. G. Isolation of Mycobacterium tuberculosis Mutants Defective in the Arrest of Phagosome Maturation. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 13642–13647. 10.1073/pnas.0401657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill-Kszycki S.; Schlesinger P. H.; Chakraborty P.; Haddix P. L.; Collins H. L.; Fok A. K.; Allen R. D.; Gluck S. L.; Heuser J.; Russel D. G. Lack of Acidification in Mycobacterium Phagosomes Produced by Exclusion of the Vesicular Proton-ATPase. Science 1994, 263, 678–681. 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- Oh Y.-K.; Straubinger R. M. Intracellular Fate of Mycobacterium avium: Use of Dual-Label Spectrofluorometry To Investigate the Influence of Bacterial Viability and Opsonization on Phagosomal pH and Phagosome-Lysosome Interaction. Infect. Immun. 1996, 64, 319–325. 10.1128/iai.64.1.319-325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano C.; Okamura T.; Sato T.; Dairi T.; Hoshino T. Mycobacterium tuberculosis H37Rv3377c Encodes the Diterpene Cyclase for Producing the Halimane Skeleton. Chem. Commun. 2005, 1016–1018. 10.1039/b415346d. [DOI] [PubMed] [Google Scholar]
- Nakano C.; Hoshino T. Characterization of the Rv3377c Gene Product, a Type-B Diterpene Cyclase, from the Mycobacterium tuberculosis H37 Genome. ChemBioChem 2009, 10, 2060–2071. 10.1002/cbic.200900248. [DOI] [PubMed] [Google Scholar]
- Roncero A. M.; Tobal I. E.; Moro R. F.; Díez D.; Marcos I. S. Halimane Diterpenoids: Sources, Structures, Nomenclature and Biological Activities. Nat. Prod. Rep. 2018, 35, 955–991. 10.1039/C8NP00016F. [DOI] [PubMed] [Google Scholar]
- Mann F. M.; Xu M.; Chen X.; Fulton D. B.; Russel D. G.; Peters R. J. Edaxadiene: A New Bioactive Diterpene from Mycobacterium tuberculosis. J. Am. Chem. Soc. 2009, 131, 17526–17527. 10.1021/ja9019287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugel N.; Mann F. M.; Hillwig M. L.; Peters R. J.; Snider B. B. Synthesis of (±)-Nosyberkol (Isotuberculosinol, Revised Structure of Edaxadiene) and (±)-Tuberculosinol. Org. Lett. 2010, 12, 2626–2629. 10.1021/ol100832h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler J. E.; Carson C. A.; Sorensen E. J. Synthesis Enables a Structural Revision of the Mycobacterium tuberculosis-produced Diterpene, Edaxadiene. Chem. Sci. 2010, 1, 202–205. 10.1039/c0sc00284d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder K.; Stein G. Untersuchungen über den Verlauf der Diensynthese. Angew. Chem. 1937, 50, 510–519. 10.1002/ange.19370502804. [DOI] [Google Scholar]
- Yoon T.; Danishefsky S. J.; de Gala S. A Concise Total Synthesis of (±)-Mamanuthaquinone by Using an exo-Diels-Alder Reaction. Angew. Chem., Int. Ed. Engl. 1994, 33, 853–855. 10.1002/anie.199408531. [DOI] [Google Scholar]
- Layre E.; Lee H. J.; Young D. C.; Martinot A. J.; Buter J.; Minnaard A. J.; Annand J. W.; Fortune S. M.; Snider B. B.; Matsunaga I.; Rubin E. J.; Alber T.; Moody D. B. Molecular Profiling of Mycobacterium tuberculosis Identifies Tuberculosinyl Nucleoside Products of the Virulence-associated Enzyme Rv3378c. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 2978–2983. 10.1073/pnas.1315883111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layre E.; Sweet L.; Hong S.; Madigan C. A.; Desjardins D.; Young D. C.; Cheng T. Y.; Annand J. W.; Kim K.; Shamputa I. C.; McConnell M. J.; Debono C. A.; Behar S. M.; Minnaard A. J.; Murray M.; Barry C. E. 3rd; Matsunaga I.; Moody D. B. A Comparative Lipidomics Platform for Chemotaxonomic Analysis of Mycobacterium tuberculosis. Chem. Biol. 2011, 18, 1537–1549. 10.1016/j.chembiol.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young D. C.; Layre E.; Pan S. J.; Tapley A.; Adamson J.; Seshadri C.; Wu Z.; Buter J.; Minnaard A. J.; Coscolla M.; Gagneux S.; Copin R.; Ernst J. D.; Bishai W. R.; Snider B. B.; Moody D. B. In Vivo Biosynthesis of Terpene Nucleosides Provides Unique Chemical Markers of Mycobacterium tuberculosis Infection. Chem. Biol. 2015, 22, 516–526. 10.1016/j.chembiol.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ashry E. S. H.; Nadeem S.; Shah M. R.; El Kilany Y.. Chapter 5 - Recent Advances in the Dimroth Rearrangement: A Valuable Tool for the Synthesis of Heterocycles. In Advances in Heterocyclic Chemsitry; Katritzky A. R., Ed.; Elsevier Inc.: 2010; Vol. 101, pp 161–228. [Google Scholar]
- Krajczyk A.; Boryski J. Dimroth Rearrangement-Old but not Outdated. Curr. Org. Chem. 2017, 21, 2515–2529. 10.2174/1385272821666170427125720. [DOI] [Google Scholar]
- Buter J.; Heijnen D.; Wan I. C.; Bickelhaupt F. M.; Young D. C.; Otten E.; Moody D. B.; Minnaard A. J. Stereoselective Synthesis of 1-Tuberculosinyl Adenosine; a Virulence Factor of Mycobacterium tuberculosis. J. Org. Chem. 2016, 81, 6686–6696. 10.1021/acs.joc.6b01332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buter J.On the Total Synthesis of Terpenes Containing Quaternary Stereocenters: Stereoselective Synthesis of the Taiwaniaquinoids, Mastigophorene A, and Tuberculosinyl Adenosine. PhD Thesis, University of Groningen, Groningen, 2016. [Google Scholar]
- Buter J.; Cheng T.-Y.; Ghanem M.; Grootemaat A. E.; Raman S.; Feng X.; Plantijn A. R.; Ennis T.; Wang J.; Cotton R. N.; Layre E.; Ramnarine A. K.; Mayfield J. A.; Young D. C.; Jezek Martinot A.; Siddiqi N.; Wakabayashi S.; Botella H.; Calderon R.; Murray M.; Ehrt S.; Snider B. B.; Reed M. B.; Oldfield E.; Tan S.; Rubin E. J.; Behr M. A.; van der Wel N. N.; Minnaard A. J.; Moody D. B. Mycobacterium tuberculosis Releases an Antacid that Remodels Phagosomes. Nat. Chem. Biol. 2019, 15, 889–899. 10.1038/s41589-019-0336-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Duve C.; de Barsy T.; Poole B.; Trouet A.; Tulkens P.; Van Hoof F. Lysosomotropic Agents. Biochem. Pharmacol. 1974, 23, 2495–2531. 10.1016/0006-2952(74)90174-9. [DOI] [PubMed] [Google Scholar]
- Ghanem M.; Dubé J.-Y.; Wang J.; McIntosh F.; Houle D.; Domenech P.; Reed M. B.; Raman S.; Buter J.; Minnaard A. J.; Moody D. B.; Behr M. A. Heterologous Production of 1-Tuberculosinyladenosine in Mycobacterium kansasii Models Pathoevolution towards the Transcellular Lifestyle of Mycobacterium tuberculosis. mBio 2020, 11, e02654-20 10.1128/mBio.02645-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratledge C. Iron, Mycobacteria and Tuberculosis. Tuberculosis 2004, 84, 110–130. 10.1016/j.tube.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Schaible U. E.; Kaufmann S. H. Iron and Microbial Infection. Nat. Rev. Microbiol. 2004, 2, 946–953. 10.1038/nrmicro1046. [DOI] [PubMed] [Google Scholar]
- Fang Z.; Sampson S. L.; Warren R. M.; Gey van Pittius N. C.; Newton-Foot M. Iron Acquisition Strategies in Mycobacteria. Tuberculosis 2015, 95, 123–130. 10.1016/j.tube.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Snow G. A. Mycobactins: Iron-Chelating Growth Factors from Mycobacteria. Bacteriol. Rev. 1970, 34, 99–125. 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilands J. B. Siderophores: Structure and Fuction of Microbial Iron Transport Compounds. J. Biol. Chem. 1995, 270, 26723–26726. 10.1074/jbc.270.45.26723. [DOI] [PubMed] [Google Scholar]
- Saha R.; Saha N.; Donofrio R. S.; Bestervelt L. L. Microbial Siderophores: A Mini Review. J. Basic Microbiol. 2013, 53, 303–317. 10.1002/jobm.201100552. [DOI] [PubMed] [Google Scholar]
- Wilson B. R.; Bogdan A. R.; Miyazawa M.; Hashimoto K.; Tsuji Y. Siderophores in Iron Metabolism: From Mechanism to Therapy Potential. Trends Mol. Med. 2016, 22, 1077–1090. 10.1016/j.molmed.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A.; Singh P.; Srivastava A. Synthesis, Nature and Utility of Universal Iron Chelator - Siderophore: A Review. Microbiol. Res. 2018, 212–213, 103–111. 10.1016/j.micres.2017.10.012. [DOI] [PubMed] [Google Scholar]
- Patel K.; Butala S.; Khan T.; Suvarna V.; Sherje A.; Dravyakar B. Mycobacterial Siderophore: A Review on Chemistry and Biology of Siderophore and its Potential as a Target for Tuberculosis. Eur. J. Med. Chem. 2018, 157, 783–790. 10.1016/j.ejmech.2018.08.030. [DOI] [PubMed] [Google Scholar]
- Francis J.; Macturk H. M.; Madinavetta J.; Snow G. A. Mycobactin, a Growth Factor for Mycobacterium johnei. Biochem. J. 1953, 55, 596–607. 10.1042/bj0550596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twort F. W.; Ingram G. L. Y.. A monograph on Johne’s disease (enteritis chronica pseudotuberculosa bovis); Baillière, Tindall and Cox: London, 1913. [Google Scholar]
- Snow G. A.; Mycobactin A Growth Factor for Mycobacterium johnei. Part II. Degradation, and Identification of Fragments. J. Chem. Soc. 1954, 2588–2596. 10.1039/jr9540002588. [DOI] [Google Scholar]
- Snow G. A.; Mycobactin A Growth Factor for Mycobacterium johnei. Part III. Degradation and Tentative Structure. J. Chem. Soc. 1954, 4080–4093. 10.1039/jr9540004080. [DOI] [Google Scholar]
- Snow G. A. The Structure of Mycobactin P, a Growth Factor for Mycobacterium johnei, and the Significance of its Iron Complex. Biochem. J. 1965, 94, 160–165. 10.1042/bj0940160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow G. A. Isolation and Structure of Mycobactin T, a Growth Factor from Mycobacterium tuberculosis. Biochem. J. 1965, 97, 166–175. 10.1042/bj0970166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobin J.; Moore C. H.; Reeve Jr J. R.; Wong D. K.; Gibson B. W.; Horwitz M. A. Iron Acquisition by Mycobacterium tuberculosis: Isolation and Characterization of a Family of Iron-Binding Exochelins. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 5189–5193. 10.1073/pnas.92.11.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobin J.; Horwitz M. A. Exochelins of Mycobacterium tuberculosis Remove Iron from Human Iron-binding Proteins and Donate Iron to Mycobactins in the M. tuberculosis Cell Wall. J. Exp. Med. 1996, 183, 1527–1532. 10.1084/jem.183.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter J. G. D.; Moore J. W. Synthesis of an Analogue of Mycobactin. J. Chem. Soc. C 1969, 12, 1610–1611. 10.1039/j39690001610. [DOI] [PubMed] [Google Scholar]
- Maurer P. J.; Miller M. J. Total Synthesis of a Mycobactin: Mycobactin S2. J. Am. Chem. Soc. 1983, 105, 240–245. 10.1021/ja00340a017. [DOI] [Google Scholar]
- Roosenberg J. M.; Lin Y.-M.; Lu Y.; Miller M. J. Studies and Syntheses of Siderophores, Microbial Iron Chelators, and Analogs as Potential Drug Delivery Agents. Curr. Med. Chem. 2000, 7, 159–197. 10.2174/0929867003375353. [DOI] [PubMed] [Google Scholar]
- He J.-L.; Xie J.-O. Advances in Mycobacterium Siderophore-Based drug Discovery. Acta Pharm. Sin. B 2011, 1, 8–13. 10.1016/j.apsb.2011.04.008. [DOI] [Google Scholar]
- Ji C.; Juárez-Hernández R. E.; Miller M. J. Exploiting Bacterial Iron Acquisition: Siderophore Conjugates. Future Med. Chem. 2012, 4, 297–313. 10.4155/fmc.11.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Górska A.; Sloderbach A.; Marszall M. P. Siderophore-Drug Complexes: Potential Medicinal Applications of the ‘Trojan Horse’ Strategy. Trends Pharmacol. Sci. 2014, 35, 442–449. 10.1016/j.tips.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Negash K. H.; Norris J. K. S.; Hodgkinson J. T. Siderophore-Antibiotic Conjugate Design: New Drugs for Bad Bugs?. Molecules 2019, 24, 3314. 10.3390/molecules24183314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. J.; Walz A. J.; Zhu H.; Wu C.; Moraski G.; Möllmann U.; Tristani E. M.; Crumbliss A. L.; Ferdig M. T.; Checkley L.; Edwards R. L.; Boshoff H. I. Design, Synthesis, and Study of a Mycobactin-Artemisinin Conjugate That Has Selective and Potent Activity against Tuberculosis and Malaria. J. Am. Chem. Soc. 2011, 133, 2076–2079. 10.1021/ja109665t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Miller M. J. Total Synthesis of a Mycobactin S, a Siderophore and Growth Promoter of Mycobacterium Smegmatis, and Determination of its Growth Inhibitory Activity against Mycobacterium tuberculosis. J. Am. Chem. Soc. 1997, 119, 3462–3468. 10.1021/ja963968x. [DOI] [Google Scholar]
- Poreddy A. R.; Schall O. F.; Marshall G. R.; Ratledge C.; Slomczynska U. Solid-Phase Synthesis of Methyl Carboxymycobactin T 7 and Analogues as Potential Antimycobacterial Agents. Bioorg. Med. Chem. Lett. 2003, 13, 2553–2556. 10.1016/S0960-894X(03)00473-6. [DOI] [PubMed] [Google Scholar]
- Moody D. B.; Young D. C.; Cheng T.-Y.; Rosat J.-P.; Roura-Mir C.; O’Connor P. B.; Zajonc D. M.; Walz A.; Miller M. J.; Levery S. B.; Wilson I. A.; Costello C. E.; Brenner M. B. T Cell Activation by Lipopeptide Antigens. Science 2004, 303, 527–531. 10.1126/science.1089353. [DOI] [PubMed] [Google Scholar]
- Young D. C.; Kasmar A.; Moraski G.; Cheng T.-Y.; Walz A. J.; Hu J.; Xu Y.; Endres G. W.; Uzieblo A.; Zajonc D.; Costello C. E.; Miller M. J.; Moody D. B. Synthesis of Dideoxymycobactin Antigens Presented by CD1a Reveals T Cell Fine Specificity for Natural Lipopeptide Structures. J. Biol. Chem. 2009, 284, 25087–25096. 10.1074/jbc.M109.000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J. M. H.; Liu L.; Pellicci D. G.; Reddiex S. J. J.; Cotton R. N.; Cheng T.-Y.; Young D. C.; Van Rhijn I.; Moody D. B.; Rossjohn J.; Fairlie D. P.; Godfrey D. I.; Williams S. J. Total Synthesis of Mycobacterium tuberculosis Dideoxymycobactin-838 and Stereoisomers: Diverse CD1a-Restricted T Cells Display a Common Hierarchy of Lipopeptide Recognition. Chem. - Eur. J. 2017, 23, 1694–1701. 10.1002/chem.201605287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankute M.; Grover S.; Rana A. K.; Besra G. S. Arabinogalactan and Lipoarabinomannan Biosynthesis: Structure, Biogenesis and Their Potential as Drug Targets. Future Microbiol. 2012, 7, 129–147. 10.2217/fmb.11.123. [DOI] [PubMed] [Google Scholar]
- Angala S. K.; Belardinelli J. M.; Huc-Claustre E.; Wheat W. H.; Jackson M. The Cell Envelope Glycoconjugates of Mycobacterium tuberculosis. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 361–399. 10.3109/10409238.2014.925420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K.-L.; Li X.; Zhang X.-L.; Pan Q. Mycobacterial Mannose-Capped Lipoarabinomannan: A Modulator Bridging Innate and Adaptive Immunity. Emerging Microbes Infect. 2019, 8, 1168–1177. 10.1080/22221751.2019.1649097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angala S. K.; Li W.; Boot C. M.; Jackson M.; McNeil M. R. Secondary Extended Mannan Side Chains and Attachment of the Arabinan in Mycobacterial Lipoarabinomannan. Commun. Chem. 2020, 3, 3. 10.1038/s42004-020-00356-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigou J.; Gilleron M.; Brando T.; Puzo G. Structural Analysis of Mycobacterial Lipoglycans. Appl. Biochem. Biotechnol. 2004, 118, 253–267. 10.1385/ABAB:118:1-3:253. [DOI] [PubMed] [Google Scholar]
- Anderson R. J. The Chemistry of the Lipids of the Tubercle Bacillus and Certain Other Microorganisms. Prog. Chem. Org. Nat. Prod. 1939, 3, 145–202. 10.1007/978-3-7091-7186-8_4. [DOI] [Google Scholar]
- Ballou C. E.; Vilkas E.; Lederer E. Structural Studies on the Myo-inositol Phospholipids of Mycobacterium tuberculosis (var. bovis, strain BCG). J. Biol. Chem. 1963, 238, 69–76. 10.1016/S0021-9258(19)83963-7. [DOI] [PubMed] [Google Scholar]
- Ballou C. E.; Lee Y. C. The Structure of a Myoinositol Mannoside from Mycobacterium tuberculosis Glycolipid. Biochemistry 1964, 3, 682–685. 10.1021/bi00893a014. [DOI] [PubMed] [Google Scholar]
- Lee Y. C.; Ballou C. E. Structural Studies on the Myo-inositol Mannosides from the Glycolipids of Mycobacterium tuberculosis and Mycobacterium phlei. J. Biol. Chem. 1964, 239, 1316–1327. 10.1016/S0021-9258(18)91317-7. [DOI] [PubMed] [Google Scholar]
- Lee Y. C.; Ballou C. E. Complete Structures of the Glycophospholipids of Mycobacteria. Biochemistry 1965, 4, 1395–1404. 10.1021/bi00883a026. [DOI] [PubMed] [Google Scholar]
- Khoo K.-H.; Dell A.; Morris H. R.; Brennan P. J.; Chatterjee D. Structural Definition of Acylated Phosphatidylinositol Mannosides from Mycobacterium tuberculosis: Definition of a Common Anchor for Lipomannan and Lipoarabinomannan. Glycobiology 1995, 5, 117–127. 10.1093/glycob/5.1.117. [DOI] [PubMed] [Google Scholar]
- Gilleron M.; Lindner B.; Puzo G. MS/MS Approach for Characterization of the Fatty Acid Distribution on Mycobacterial Phosphatidyl-myo-inositol Mannosides. Anal. Chem. 2006, 78, 8543–8548. 10.1021/ac061574a. [DOI] [PubMed] [Google Scholar]
- Garcia-Vilanova A.; Chan J.; Torrelles J. B. Underestimated Manipulative Roles of Mycobacterium tuberculosis Cell Envelope Glycolipids During Infection. Front. Immunol. 2019, 10, 2909. 10.3389/fimmu.2019.02909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergne I.; Fratti R. A.; Hill P. J.; Chua J.; Belisle J.; Deretic V. Mycobacterium tuberculosis Phagosome Maturation Arrest: Mycobacterial Phosphatidylinositol Analog Phosphatidylinositol Mannoside Stimulates Early Endosomal Fusion. Mol. Biol. Cell 2004, 15, 751–760. 10.1091/mbc.e03-05-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve C.; Gilleron M.; Maridonneau-Parini I.; Daffé M.; Astarie-Dequeker C.; Etienne G. Mycobacteria Use Their Surface-Exposed Glycolipids to Infect Human Macrophages Through a Receptor-Dependent Process. J. Lipid Res. 2005, 46, 475–483. 10.1194/jlr.M400308-JLR200. [DOI] [PubMed] [Google Scholar]
- Court N.; Rose S.; Bourigault M.-L.; Front S.; Martin O. R.; Dowling J. K.; Kenny E. F.; O’Neill L.; Erard F.; Quesniaux V. F. J. Mycobacterial PIMs Inhibit Host Inflammatory Responses through CD14-Dependent and CD14-Independent Mechanisms. PLoS One 2011, 6, e24631 10.1371/journal.pone.0024631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrelles J. B.; Azad A. K.; Schlesinger L. S. Fine Discrimination in the Recognition of Individual Species of Phosphatidyl-myo-Inositol Mannosides from Mycobacterium tuberculosis by C-Type Lectin Pattern Recognition Receptors. J. Immunol. 2006, 177, 1805–1816. 10.4049/jimmunol.177.3.1805. [DOI] [PubMed] [Google Scholar]
- Driessen N. N.; Ummels R.; Maaskant J. J.; Gurcha S. S.; Besra G. S.; Ainge G. D.; Larsen D. S.; Painter G. F.; Vandenbroucke-Grauls C. M. J. E.; Geurtsen J.; Appelmelk B. J. Role of Phosphatidylinositol Mannosides in the Interaction between Mycobacteria and DC-SIGN. Infect. Immun. 2009, 77, 4538–4547. 10.1128/IAI.01256-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Salle H.; Mariotti S.; Angenieux C.; Gilleron M.; Garcia-Alles L.-F.; Malm D.; Berg T.; Paoletti S.; Maître B.; Mourey L.; Salamero J.; Cazenave J. P.; Hanau D.; Mori L.; Puzo G.; De Libero G. Assistance of Microbial Glycolipid Antigen Processing by CD1e. Science 2005, 310, 1321–1324. 10.1126/science.1115301. [DOI] [PubMed] [Google Scholar]
- Rojas R. E.; Thomas J. J.; Gehring A. J.; Hill P. J.; Belisle J. T.; Harding C. V.; Boom W. H. Phosphatidylinositol Mannoside from Mycobacterium tuberculosis Binds α5β1 Integrin (VLA-5) on CD4 + T Cells and Induces Adhesion to Fibronectin. J. Immunol. 2006, 177, 2959–2968. 10.4049/jimmunol.177.5.2959. [DOI] [PubMed] [Google Scholar]
- Misaki A.; Azuma I.; Yamamura Y. Structural and Immunochemical Studies on D-Arabino-D-Mannans and D-Mannans of Mycobacterium tuberculosis and Other Mycobacterium Species. J. Biochem. 1977, 82, 1759–1770. 10.1093/oxfordjournals.jbchem.a131874. [DOI] [PubMed] [Google Scholar]
- Hunter S. W.; Gaylord H.; Brennan P. J. Structure and Antigenicity of the Phosphorylated Lipopolysaccharide Antigens from the Leprosy and Tubercle Bacilli. J. Biol. Chem. 1986, 261, 12345–12351. 10.1016/S0021-9258(18)67246-1. [DOI] [PubMed] [Google Scholar]
- Hunter S. W.; Brennan P. J. Evidence for the Presence of a Phosphatidylinositol Anchor on the Lipoarabinomannan and Lipomannan of Mycobacterium tuberculosis. J. Biol. Chem. 1990, 265, 9272–9279. 10.1016/S0021-9258(19)38843-X. [DOI] [PubMed] [Google Scholar]
- Chatterjee D.; Hunter S. W.; McNeil M.; Brennan P. J.; Lipoarabinomannan Multiglycosylated Form of the Mycobacterial Mannosylphosphatidylinositols. J. Biol. Chem. 1992, 267, 6228–6233. [PubMed] [Google Scholar]
- Gilleron M.; Nigou J.; Cahuzac B.; Puzo G. Structural Study of the LipoMannans from Mycobacterium bovis BCG: Characterisation of Multiacylated Forms of the Phosphatidyl-myo- inositol Anchor. J. Mol. Biol. 1999, 285, 2147–2160. 10.1006/jmbi.1998.2438. [DOI] [PubMed] [Google Scholar]
- Chatterjee D.; Khoo K.-H.; McNeil M.; Dell A.; Morris H. R.; Brennan P. J. Structural Definition of the Non-Reducing Termini of Mannose-Capped LAM from Mycobacterium tuberculosis Through Selective Enzymatic Degradation and Fast Atom Bombardment-Mass Spectrometry. Glycobiology 1993, 3, 497–506. 10.1093/glycob/3.5.497. [DOI] [PubMed] [Google Scholar]
- Nigou J.; Vercellone A.; Puzo G. New Structural Insights into the Molecular Deciphering of Mycobacterial Lipoglycan Binding to C-type Lectins: Lipoarabinomannan Glycoform Characterization and Quantification by Capillary Electrophoresis at the Subnanomole Level. J. Mol. Biol. 2000, 299, 1353–1362. 10.1006/jmbi.2000.3821. [DOI] [PubMed] [Google Scholar]
- Chatterjee D.; Bozic C. M.; McNeil M.; Brennan P. J. Structural Features of the Arabinan Component of the Lipoarabinomannan of Mycobacterium tuberculosis. J. Biol. Chem. 1991, 266, 9652–9660. 10.1016/S0021-9258(18)92870-X. [DOI] [PubMed] [Google Scholar]
- Shi L.; Berg S.; Lee A.; Spencer J. S.; Zhang J.; Vissa V.; McNeil M. R.; Khoo K. H.; Chatterjee D. The Carboxy Terminus of EmbC from Mycobacterium smegmatis Mediates Chain Length Extension of the Arabinan in Lipoarabinomannan. J. Biol. Chem. 2006, 281, 19512–19526. 10.1074/jbc.M513846200. [DOI] [PubMed] [Google Scholar]
- Monsarrat B.; Brando T.; Condouret P.; Nigou J.; Puzo G. Characterization of Mannooligosaccharide Caps in Mycobacterial Lipoarabinomannan by Capillary Electrophoresis/Electrospray Mass Spectrometry. Glycobiology 1999, 9, 335–342. 10.1093/glycob/9.4.335. [DOI] [PubMed] [Google Scholar]
- Treumann A.; Xidong F.; McDonnell L.; Derrick P. J.; Ashcroft A. E.; Chatterjee D.; Homans S. W. 5-Methylthiopentose: A New Substituent on Lipoarabinomannan in Mycobacterium tuberculosis. J. Mol. Biol. 2002, 316, 89–100. 10.1006/jmbi.2001.5317. [DOI] [PubMed] [Google Scholar]
- Turnbull W. B.; Shimizu K. H.; Chatterjee D.; Homans S. W.; Treumann A. Identification of the 5-Methylthiopentosyl Substituent in Mycobacterium tuberculosis Lipoarabinomannan. Angew. Chem., Int. Ed. 2004, 43, 3918–3922. 10.1002/anie.200454119. [DOI] [PubMed] [Google Scholar]
- Kaur D.; Angala S. K.; Wu S. W.; Khoo K. H.; Chatterjee D.; Brennan P. J.; Jackson M.; McNeil M. R. A Single Arabinan Chain Is Attached to the Phosphatidylinositol Mannosyl Core of the Major Immunomodulatory Mycobacterial Cell EnvelopecGlycoconjugate, Lipoarabinomannan. J. Biol. Chem. 2014, 289, 30249–30256. 10.1074/jbc.M114.599415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmeier G. R.; Fenton M. J. Roles of Lipoarabinomannan in the Pathogenesis of Tuberculosis. Microbes Infect. 1999, 1, 709–717. 10.1016/S1286-4579(99)80072-0. [DOI] [PubMed] [Google Scholar]
- Nigou J.; Gilleron M.; Rojas M.; García L. F.; Thurnher M.; Puzo G. Mycobacterial Lipoarabinomannans: Modulators of Dendritic Cell Function and the Apoptotic Response. Microbes Infect. 2002, 4, 945–953. 10.1016/S1286-4579(02)01621-0. [DOI] [PubMed] [Google Scholar]
- Schlesinger L. S.; Hull S. R.; Kaufmann T. M. Binding of the Terminal Mannosyl Units of Lipoarabinomannan From a Virulent Strain of Mycobacterium tuberculosis to Human Macrophages. J. Immunol. 1994, 152, 4070–4079. [PubMed] [Google Scholar]
- Geijtenbeek T. B.; Van Vliet S. J.; Koppel E. A.; Sanchez-Hernandez M.; Vandenbroucke-Grauls C. M. J. E.; Appelmelk B.; Van Kooyk Y. Mycobacteria Target DC-SIGN to Suppress Dendritic Cell Function. J. Exp. Med. 2003, 197, 7–17. 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N.; Nigou J.; Herrmann J.-L.; Jackson M.; Amara A.; Lagrange P. H.; Puzo G.; Gicquel B.; Neyrolles O. The Cell Surface Receptor DC-SIGN Discriminates between Mycobacterium Species through Selective Recognition of the Mannose Caps on Lipoarabinomannan. J. Biol. Chem. 2003, 278, 5513–5516. 10.1074/jbc.C200586200. [DOI] [PubMed] [Google Scholar]
- Vergne I.; Chua J.; Deretic V. Tuberculosis Toxin Blocking Phagosome Maturation Inhibits a Novel Ca2+/Calmodulin-PI3K hVPS34 Cascade. J. Exp. Med. 2003, 198, 653–659. 10.1084/jem.20030527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang P. B.; Azad A. K.; Torrelles J. B.; Kaufman T. M.; Beharka A.; Tibesar E.; DesJardin L. E.; Schlesinger L. S. The Human Macrophage Mannose Receptor Directs Mycobacterium tuberculosis Lipoarabinomannan-Mediated Phagosome Biogenesis. J. Exp. Med. 2005, 202, 987–999. 10.1084/jem.20051239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigozy T. I.; Sieling P. A.; Clemens D.; Stewart P. L.; Behar S. M.; Porcelli S. A.; Brenner M. B.; Modlin R. L.; Kronenberg M. The Mannose Receptor Delivers Lipoglycan Antigens to Endosomes for Presentation to T Cells by CD1b Molecules. Immunity 1997, 6, 187–197. 10.1016/S1074-7613(00)80425-2. [DOI] [PubMed] [Google Scholar]
- Bulterys M. A.; Wagner B.; Redard-Jacot M.; Suresh A.; Pollock N. R.; Moreau E.; Denkinger C. M.; Drain P. K.; Broger T. Point-Of-Care Urine LAM Tests for Tuberculosis Diagnosis: A Status Update. J. Clin. Med. 2020, 9, 111. 10.3390/jcm9010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elie C. J. J.; Dreef C. E.; Verduyn R.; Van der Marel G. A.; Van Boom J. H. Synthesis of 1-O-(1,2-Di-O-Palmitoyl-sn-glycero-3-phosphoryl)-2-O-α-D-mannopyranosyl-D-myo-inositol: A Fragment of Mycobacterial Phospholipids. Tetrahedron 1989, 45, 3477–3486. 10.1016/S0040-4020(01)81026-7. [DOI] [Google Scholar]
- Elie C. J. J.; Verduyn R.; Dreef C. E.; Van der Marel G. A.; Van Boom J. H. Iodonium Ion-Mediated Mannosylations of Myo-Inositol: Synthesis of a Mycobacteria Phospholipid Fragment. J. Carbohydr. Chem. 1992, 11, 715–739. 10.1080/07328309208020088. [DOI] [Google Scholar]
- Zamyatina A. Y.; Shvets V. I. The Synthesis of 1-O-(2-N-Stearoyl-D-erythro-sphinganine-1-phosphoryl)-2-O-(α-D-mannopyranosyl)-D-myo-inositol: A Fragment of the Naturally Occurring Inositol-Containing Glycophosphosphingolipids. Chem. Phys. Lipids 1995, 76, 225–240. 10.1016/0009-3084(95)02446-P. [DOI] [PubMed] [Google Scholar]
- Watanabe Y.; Yamamoto T.; Ozaki S. Regiospecific Synthesis of 2,6-Di-O- (α-d-mannopyranosyl)phosphatidyl-d-myo-inositol. J. Org. Chem. 1996, 61, 14–15. 10.1021/jo951511k. [DOI] [Google Scholar]
- Stadelmaier A.; Schmidt R. R. Synthesis of Phosphatidylinositol Mannosides (PIMs). Carbohydr. Res. 2003, 338, 2557–2569. 10.1016/j.carres.2003.06.002. [DOI] [PubMed] [Google Scholar]
- Ainge G. D.; Hudson J.; Larsen D. S.; Painter G. F.; Gill G. S.; Harper J. L. Phosphatidylinositol Mannosides: Synthesis and Suppression of Allergic Airway Disease. Bioorg. Med. Chem. 2006, 14, 5632–5642. 10.1016/j.bmc.2006.04.037. [DOI] [PubMed] [Google Scholar]
- Zajonc D. M.; Ainge G. D.; Painter G. F.; Severn W. B.; Wilson I. A. Structural Characterization of Mycobacterial Phosphatidylinositol Mannoside Binding to Mouse CD1d. J. Immunol. 2006, 177, 4577–4583. 10.4049/jimmunol.177.7.4577. [DOI] [PubMed] [Google Scholar]
- Ainge G. D.; Parlane N. A.; Denis M.; Hayman C. M.; Larsen D. S.; Painter G. F. Phosphatidylinositol Mannosides: Synthesis and Adjuvant Properties of Phosphatidylinositol Di- and Tetramannosides. Bioorg. Med. Chem. 2006, 14, 7615–7624. 10.1016/j.bmc.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Ainge G. D.; Compton B. J.; Hayman C. M.; Martin W. J.; Toms S. M.; Larsen D. S.; Harper J. L.; Painter G. F. Chemical Synthesis and Immunosuppressive Activity of Dipalmitoyl Phosphatidylinositol Hexamannoside. J. Org. Chem. 2011, 76, 4941–4951. 10.1021/jo200588u. [DOI] [PubMed] [Google Scholar]
- Liu X.; Stocker B. L.; Seeberger P. H. Total Synthesis of Phosphatidylinositol Mannosides of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2006, 128, 3638–3648. 10.1021/ja0565368. [DOI] [PubMed] [Google Scholar]
- Wu D.; Xing G.-W.; Poles M. A.; Horowitz A.; Kinjo Y.; Sullivan B.; Bodmer-Narkevitch V.; Plettenburg O.; Kronenberg M.; Tsuji M.; Ho D. D.; Wong C.-H. Bacterial Glycolipids and Analogs as Antigens for CD1d-Restricted NKT Cells. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 1351–1356. 10.1073/pnas.0408696102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z. J.; Olsson L.; Fraser-Reid B. Ready Routes to Key myo-Inositol Component of GPIs Employing Microbial Arene Oxidation or Ferrier Reaction. J. Chem. Soc., Perkin Trans. 1 1998, 631–632. 10.1039/a800282g. [DOI] [Google Scholar]
- Tanaka S.; Saburi H.; Ishibashi Y.; Kitamura M. CpRuIIPF6/Quinaldic Acid-Catalyzed Chemoselective Allyl Ether Cleavage. A Simple and Practical Method for Hydroxyl Deprotection. Org. Lett. 2004, 6, 1873–1875. 10.1021/ol0493397. [DOI] [PubMed] [Google Scholar]
- Dyer B. S.; Jones J. D.; Ainge G. D.; Denis M.; Larsen D. S.; Painter G. F. Synthesis and Structure of Phosphatidylinositol Dimannoside. J. Org. Chem. 2007, 72, 3282–3288. 10.1021/jo0625599. [DOI] [PubMed] [Google Scholar]
- Gilleron M.; Ronet C.; Mempel M.; Monsarrat B.; Gachelin G.; Puzo G. Acylation State of the Phosphatidylinositol Mannosides from Mycobacterium bovis Bacillus Calmette Guérin and Ability to Induce Granuloma and Recruit Natural Killer T Cells. J. Biol. Chem. 2001, 276, 34896–34904. 10.1074/jbc.M103908200. [DOI] [PubMed] [Google Scholar]
- Gilleron M.; Quesniaux V. F.; Puzo G. Acylation State of the Phosphatidylinositol Hexamannosides from Mycobacterium bovis Bacillus Calmette Guérin and Mycobacterium tuberculosis H37Rv and Its Implication in Toll-like Receptor Response. J. Biol. Chem. 2003, 278, 29880–29889. 10.1074/jbc.M303446200. [DOI] [PubMed] [Google Scholar]
- Boonyarattanakalin S.; Liu X.; Michieletti M.; Lepenies B.; Seeberger P. H. Chemical Synthesis of All Phosphatidylinositol Mannoside (PIM) Glycans from Mycobacterium tuberculosis. J. Am. Chem. Soc. 2008, 130, 16791–16799. 10.1021/ja806283e. [DOI] [PubMed] [Google Scholar]
- Patil P. S.; Hung S.-C. Total Synthesis of Phosphatidylinositol Dimannoside: A Cell-Envelope Component of Mycobacterium tuberculosis. Chem. - Eur. J. 2009, 15, 1091–1094. 10.1002/chem.200802189. [DOI] [PubMed] [Google Scholar]
- Patil P. S.; Hung S.-C. Synthesis of Mycobacterial Triacylated Phosphatidylinositol Dimannoside Containing an Acyl Lipid Chain at 3-O of Inositol. Org. Lett. 2010, 12, 2618–2621. 10.1021/ol1008137. [DOI] [PubMed] [Google Scholar]
- Patil P. S.; Cheng T.-J. R.; Zulueta M. M. L.; Yang S.-T.; Lico L. S.; Hung S.-C. Total Synthesis of Tetraacylated Phosphatidylinositol Hexamannoside and Evaluation of its Immunomodulatory Activity. Nat. Commun. 2015, 6, 7239. 10.1038/ncomms8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A.; Wenk M. R.; Lear M. J. Total Synthesis of a Fully Lipidated Form of Phosphatidyl-myo-Inositol Dimannoside (PIM-2) of Mycobacterium tuberculosis. Tetrahedron Lett. 2009, 50, 5664–5666. 10.1016/j.tetlet.2009.07.109. [DOI] [Google Scholar]
- Ohira S.; Yamaguchi Y.; T T.; H T. Synthesis of a Phosphatidylinositol Dimannoside Using 2-(Azidomethyl)benzoate Mannosyl Donors. Heterocycles 2014, 89, 763–774. 10.3987/COM-14-12944. [DOI] [Google Scholar]
- Toyonaga K.; Torigoe S.; Motomura Y.; Kamichi T.; Hayashi J. M.; Morita Y. S.; Noguchi N.; Chuma Y.; Kiyohara H.; Matsuo K.; Tanaka H.; Nakagawa Y.; Sakuma T.; Ohmuraya M.; Yamamoto T.; Umemura M.; Matsuzaki G.; Yoshikai Y.; Yano I.; Miyamoto T.; Yamasaki S. C-Type Lectin Receptor DCAR Recognizes Mycobacterial Phosphatidyl-Inositol Mannosides to Promote a Th1 Response during Infection. Immunity 2016, 45, 1245–1257. 10.1016/j.immuni.2016.10.012. [DOI] [PubMed] [Google Scholar]
- Omahdi Z.; Horikawa Y.; Nagae M.; Toyonaga K.; Imamura A.; Takato K.; Teramoto T.; Ishida H.; Kakuta Y.; Yamasaki S. Structural Insight Into the Recognition of Pathogen-Derived Phosphoglycolipids by C-type Lectin Receptor DCAR. J. Biol. Chem. 2020, 295, 5807–5817. 10.1074/jbc.RA120.012491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira S.; Yamaguchi Y.; Takahashi T.; Tanaka H. The Chemoselective O-glycosylation of Alcohols in the Presence of a Phosphate Diester and its Application to the Synthesis of Oligomannosylated Phosphatidyl Inositols. Tetrahedron 2015, 71, 6602–6611. 10.1016/j.tet.2015.06.041. [DOI] [Google Scholar]
- Lee A. M. M.; Painter G. F.; Compton B. J.; Larsen D. S. Resolution of Orthogonally Protected myo-Inositols with Novozym 435 Providing an Enantioconvergent Pathway to Ac2PIM1. J. Org. Chem. 2014, 79, 10916–10931. 10.1021/jo5019188. [DOI] [PubMed] [Google Scholar]
- Cunha A. G.; da Silva A. A. T.; da Silva A. J. R.; Tinoco L. W.; Almeida R. V.; de Alencastro R. B.; Simas A. B. C.; Freire D. M. G. Efficient Kinetic Resolution of (±)-1,2-O-isopropylidene-3,6-di-O-benzyl-myo-inositol with the Lipase B of Candida antarctica. Tetrahedron: Asymmetry 2010, 21, 2899–2903. 10.1016/j.tetasy.2010.12.006. [DOI] [Google Scholar]
- Simas A. B. C.; da Silva A. A. T.; Cunha A. G.; Assumpção R. S.; Hoelz L. V. B.; Neves B. C.; Galvão T. C.; Almeida R. V.; Albuquerque M. G.; Freire D. M. G.; de Alencastro R. B. Kinetic Resolution of (±)-1,2-O-isopropylidene-3,6-di-O-benzyl-myo-inositol by Lipases: An Experimental and Theoretical Study on the Reaction of a Key Precursor of Chiral Inositols. J. Mol. Catal. B: Enzym. 2011, 70, 32–40. 10.1016/j.molcatb.2011.02.001. [DOI] [Google Scholar]
- Arai Y.; Torigoe S.; Matsumaru T.; Yamasaki S.; Fujimoto Y. The Key Entity of a DCAR Agonist, Phosphatidylinositol Mannoside Ac1PIM1: Its Synthesis and Immunomodulatory Function. Org. Biomol. Chem. 2020, 18, 3659–3663. 10.1039/C9OB02724F. [DOI] [PubMed] [Google Scholar]
- Jayaprakash K. N.; Lu J.; Fraser-Reid B. Synthesis of a Lipomannan Component of the Cell-Wall Complex of Mycobacterium tuberculosis Is Based on Paulsen’s Concept of Donor/Acceptor “Match. Angew. Chem., Int. Ed. 2005, 44, 5894–5898. 10.1002/anie.200500505. [DOI] [PubMed] [Google Scholar]
- Paulsen H.Selectivity: A goal for synthetic efficiency; Verlag Chemie: Basel, 1985. [Google Scholar]
- Fraser-Reid B.; Wu Z.; Udodong U. E.; Ottosson H. Armed/Disarmed Effects in Glycosyl Donors: Rationalization and Sidetracking. J. Org. Chem. 1990, 55, 6068–6070. 10.1021/jo00312a004. [DOI] [Google Scholar]
- Hölemann A.; Stocker B. L.; Seeberger P. H. Synthesis of a Core Arabinomannan Oligosaccharide of Mycobacterium tuberculosis. J. Org. Chem. 2006, 71, 8071–8088. 10.1021/jo061233x. [DOI] [PubMed] [Google Scholar]
- Fraser-Reid B.; Lu J.; Jayaprakash K. N.; López J. C. Synthesis of a 28-mer Oligosaccharide Core of Mycobacterial Lipoarabinomannan (LAM) Requires only Two n-Pentenyl Orthoester Progenitors. Tetrahedron: Asymmetry 2006, 17, 2449–2463. 10.1016/j.tetasy.2006.09.008. [DOI] [Google Scholar]
- Joe M.; Bai Y.; Nacario R. C.; Lowary T. L. Synthesis of the Docosanasaccharide Arabinan Domain of Mycobacterial Arabinogalactan and a Proposed Octadecasaccharide Biosynthetic Precursor. J. Am. Chem. Soc. 2007, 129, 9885–9901. 10.1021/ja072892+. [DOI] [PubMed] [Google Scholar]
- Callam C. S.; Gadikota R. R.; Lowary T. L. Sensitivity of 1JC1-H1Magnitudes to Anomeric Stereochemistry in 2,3-Anhydro-O-furanosides. J. Org. Chem. 2001, 66, 4549–4558. 10.1021/jo001747a. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Kawatkar S.; Rao Y.; Boons G.-J. Practical Approach for the Stereoselective Introduction of β-Arabinofuranosides. J. Am. Chem. Soc. 2006, 128, 11948–11957. 10.1021/ja0629817. [DOI] [PubMed] [Google Scholar]
- Ishiwata A.; Ito Y. Synthesis of Docosasaccharide Arabinan Motif of Mycobacterial Cell Wall. J. Am. Chem. Soc. 2011, 133 (7), 2275–22791. 10.1021/ja109932t. [DOI] [PubMed] [Google Scholar]
- Thadke S. A.; Mishra B.; Islam M.; Pasari S.; Manmode S.; Rao B. V.; Neralkar M.; Shinde G. P.; Walke G.; Hotha S. [Au]/[Ag]-Catalysed Expedient Synthesis of Branched Heneicosafuranosyl Arabinogalactan Motif of Mycobacterium tuberculosis Cell Wall. Nat. Commun. 2017, 8, 14019. 10.1038/ncomms14019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thadke S. A.; Mishra B.; Hotha S. Gold(III)-Catalyzed Glycosidations for 1,2-trans and 1,2-cis Furanosides. J. Org. Chem. 2014, 79, 7358–7371. 10.1021/jo501052y. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Xiong D. C.; Chen S. C.; Wang Y. S.; Ye X. S. Total Synthesis of Mycobacterial Arabinogalactan Containing 92 Monosaccharide Units. Nat. Commun. 2017, 8, 14851. 10.1038/ncomms14851. [DOI] [PMC free article] [PubMed] [Google Scholar]