

Summary
Treatment benefit in multiple myeloma (MM) patients with high‐risk cytogenetics remains suboptimal. The phase 3 ICARIA‐MM trial (NCT02990338) showed that isatuximab plus pomalidomide–dexamethasone prolongs median progression‐free survival (mPFS) in patients with relapsed/refractory MM (RRMM). This subgroup analysis of ICARIA‐MM compared the benefit of isatuximab in high‐risk [defined by the presence of del(17p), t(4;14) or t(14;16)] versus standard‐risk patients. The efficacy of isatuximab in patients with gain(1q21) abnormality was also assessed in a retrospective subgroup analysis. In ICARIA‐MM, 307 patients received isatuximab–pomalidomide–dexamethasone (n = 154) or pomalidomide–dexamethasone (n = 153). Isatuximab (10 mg/kg intravenously) was given weekly in the first 28‐day cycle, and every other week thereafter. Standard pomalidomide–dexamethasone doses were given. Isatuximab–pomalidomide–dexamethasone improved mPFS (7·5 vs 3·7 months; HR, 0·66; 95% CI, 0·33–1·28) and overall response rate (ORR, 50·0% vs 16·7%) in high‐risk patients. In patients with isolated gain(1q21), isatuximab addition improved mPFS (11·2 vs 4·6 months; HR, 0·50; 95% CI, 0·28–0·88) and ORR (53·6% vs 27·6%). More grade ≥3 adverse events occurred in high‐risk patients receiving isatuximab (95·7%) versus the control group (67·6%); however, isatuximab did not increase events leading to discontinuation or treatment‐related mortality. Isatuximab–pomalidomide–dexamethasone provides a consistent benefit over pomalidomide–dexamethasone treatment in RRMM patients regardless of cytogenetic risk.
Keywords: cytogenetics, gain (1q21), high‐risk, isatuximab, relapsed/refractory multiple myeloma
Introduction
Multiple myeloma (MM) is a haematological malignancy characterised by abnormal proliferation of plasma cells in the bone marrow, thereby leading to bone destruction and marrow failure. It is a heterogeneous disease with variable prognosis depending on host factors such as age/frailty and renal failure, disease burden and tumour biology, high‐risk cytogenetics and prior therapies.1 Despite recent advances in the treatment of MM, relapse is inevitable and the disease is considered incurable with current treatment approaches.
Risk classification based on cytogenetic profiling, and careful analysis of risk subgroups is becoming increasingly important in the evaluation of novel therapies.2 The Revised International Staging System (R‐ISS) for MM defines patients with del(17p), and/or t(4;14), and/or t(14;16) abnormalities as high‐risk.3, 4 Importantly, high‐risk patients have a median overall survival (OS) of only three years versus 10 years in low‐risk patients.1 Additionally, gain(1q21), one of the most common chromosomal abnormalities in MM, occurs in 20–50% of newly diagnosed patients with increased incidence in relapsed/refractory multiple myeloma (RRMM),5 and has been linked with inferior outcomes.6, 7, 8, 9, 10
Novel agents such as immunomodulatory (IMiD) drugs and proteasome inhibitors have led to higher response rates and lower toxicity than standard, non‐specific chemotherapy in patients with MM.11 Pomalidomide is a second‐generation IMiD, which, in combination with low‐dose dexamethasone, is approved for the treatment of RRMM in patients who previously received ≥2 lines of therapy, including lenalidomide and a proteasome inhibitor.12 Pomalidomide plus low‐dose dexamethasone compared with high‐dose dexamethasone improved progression‐free survival (PFS) in patients with RRMM who had the high‐risk abnormalities del(17p) [4·6 vs 1·1 months; hazard ratio (HR), 0·34; P < 0·001] and t(4;14) (2·8 vs 1·9 months; HR, 0·49; P = 0·028).13 While pomalidomide and other novel agents have extended the OS of MM patients, those who fail these regimens have a poor prognosis, warranting the need for new treatments.11
Among emerging therapies for MM, monoclonal antibodies targeting the CD38 receptor (expressed at high levels on malignant MM cells) represent an important class of drugs against this disease. Isatuximab, an IgG1 monoclonal antibody, binds to a different epitope on the CD38 receptor than other clinically available anti‐CD38 antibodies and mediates tumour cell death via multiple mechanisms.14, 15, 16
In the phase 3 ICARIA‐MM study involving 307 patients with RRMM, isatuximab plus pomalidomide and low‐dose dexamethasone significantly improved PFS [11·5 vs 6·5 months; HR, 0·596; 95% confidence interval (CI), 0·44–0·81; P = 0·001], overall response rate (ORR, 60% vs 35%), and the depth of response compared with pomalidomide–dexamethasone.17 This study met its primary end‐point and formed the basis for the approval of isatuximab plus pomalidomide–dexamethasone for the treatment of adult patients with RRMM who received ≥2 prior therapies, including lenalidomide and a proteasome inhibitor. Furthermore, based on the phase 3 IKEMA study, on February 26, 2021, the Committee for Medicinal Products for Human Use (CHMP) issued a positive opinion for a second indication for isatuximab, in combination with carfilzomib and dexamethasone for adult patients with MM who have received at least one prior therapy. In this prespecified subgroup analysis of ICARIA‐MM (NCT02990338), we evaluated the efficacy and safety of isatuximab plus pomalidomide–dexamethasone treatment in high‐risk versus standard‐risk cytogenetic patients with RRMM. In addition, we conducted a retrospective analysis of patient samples from ICARIA‐MM to compare the efficacy of isatuximab plus pomalidomide–dexamethasone in patients with gain(1q21) compared with those without the abnormality.
Patients and methods
Study design and patients
The ICARIA‐MM study has been previously described,11, 17 and is registered at ClinicalTrials.gov, number NCT02990338. Briefly, eligible patients had RRMM and received ≥2 prior lines of therapy and were refractory to lenalidomide and a proteasome inhibitor given alone or in combination. The protocol was approved by independent ethics committees and institutional review boards at all participating institutions. All authors had full access to the study data. Written informed consent was obtained from all patients.
Randomisation
Patients were randomly assigned in a 1:1 ratio to either isatuximab–pomalidomide–dexamethasone (154 patients), or pomalidomide–dexamethasone (153). Randomisation was stratified based on the number of prior lines of treatment (2–3 vs >3) and age (<75 vs ≥75 years).
Procedures
As described previously, patients in the isatuximab–pomalidomide–dexamethasone group received 10 mg/kg isatuximab intravenously (on days 1, 8, 15 and 22 in the first 28‐day cycle, and days 1 and 15 in subsequent cycles) plus pomalidomide (4 mg orally on days 1 to 21 in each cycle) plus dexamethasone (40 mg orally or intravenously on days 1, 8, 15 and 22 in each cycle; 20 mg for ≥75 years old). Pomalidomide and dexamethasone were administered on the same schedule in all patients.
Outcomes
The primary end‐point of the ICARIA‐MM study was PFS. Secondary end‐points included the ORR, OS and safety. Data on OS was not yet mature at the time of this analysis. The end‐points for this subgroup analysis are the same as in the primary study.
In the high‐risk cytogenetic population, PFS was a prespecified subgroup analysis. Cytogenetic risk was assessed by central laboratory fluorescence in situ hybridisation (FISH) testing after immunomagnetic isolation of CD138+ plasma cells from baseline bone marrow aspirate, interphase chromosome preparation, and hybridisation with Kreatech FISH probes [11q22.3(ATM)/17p13.1(p53), 4p16.3(FGFR3); 14q32.3(IGH); 16q23.2(MAF)]. High‐risk cytogenetic status was defined by the presence of ≥1 of del(17p), t(4;14) or t(14;16), and was considered positive if present in ≥30% plasma cells for t(4;14) and t(14;16), and in ≥50% plasma cells for del(17p). For sensitivity analyses, thresholds ranging from 5% to 60% for del(17p), and 3% to 60% for t(4;14) were used.
The gain(1q21) subgroup was evaluated in a retrospective analysis using the remaining CD138+ plasma cells collected for cytogenetic risk evaluation and was considered positive if ≥3 copies of 1q21 were present in ≥30% of analysed cells. Amplification of 1q21 was also evaluated and was considered positive if ≥4 copies of 1q21 were present in ≥30% of analysed cells.
Statistical analysis
Statistical analysis for the ICARIA‐MM study has been previously described.17 All efficacy analyses were conducted in the intent‐to‐treat population. For this subgroup analysis, the HR and the associated 95% CI for the treatment effect were estimated, and PFS was analysed using a Cox proportional hazards model with terms for the factor, treatment and their interaction. The test for the interaction was performed at the 10% alpha level. PFS was analysed using the Kaplan–Meier method by treatment group with the same censoring rules as the primary analysis, and comparisons between treatment groups were made using a one‐sided log‐rank test. The response rates were compared using the unstratified Cochran–Mantel–Haenszel test. Sensitivity analyses were conducted to assess the impact of the cytogenetic cut‐off definition used.
Results
Patients
Of the 307 patients recruited between 10 January 2017 and 2 February 2018 for the ICARIA‐MM study,17 cytogenetic risk was assessable by central laboratory in 241 patients (78·5%). Missing results in 66 (21·5%) patients were because samples were not provided or were of poor quality. Of the 241 patients with cytogenetic risk data, 60 (24·9%) were classified as high risk, according to the pre‐specified definitions. Results were compared with those from standard‐risk patients (181 out of 307).
High‐risk chromosomal abnormalities were present in fewer patients in the isatuximab treatment group [24 (15·6%)] than in the pomalidomide–dexamethasone group [36 (23·5%)], with del(17p) and t(4;14) being the most frequent abnormalities in both groups (Table I).
Table I.
Cytogenetic risk in the ITT population at baseline.
| Cytogenetic risk in the ITT population at baseline, n (%) | Isa–Pd (n = 154) | Pd (n = 153) |
|---|---|---|
| Standard‐risk | 103 (66·9) | 78 (51·0) |
| High‐risk | 24 (15·6) | 36 (23·5) |
| del(17p) | 14 (9·1) | 23 (15·0) |
| t(4;14) | 12 (7·8) | 14 (9·2) |
| t(14;16) | 1 (0·6) | 4 (2·6) |
| del(17p) and t(4;14) | 3 (1·9) | 4 (2·6) |
| del(17p) and t(14;16) | 0 | 1 (0·7) |
| Unknown/missing | 27 (17·5) | 39 (25·5) |
| 1q21, regardless of other high‐risk cytogenetic abnormalities | ||
| Gain(1q21), ≥3 copies | 76 (49·4) | 52 (34·0) |
| Amplification 1q21, ≥4 copies | 27 (17·5) | 21 (13·7) |
| Isolated 1q21, without other high‐risk cytogenetic abnormalities | ||
| Gain(1q21), ≥3 copies | 56 (36·4) | 29 (19·0) |
| Amplification 1q21, ≥4 copies | 23 (14·9) | 9 (5·9) |
High‐risk cytogenetics was prespecified as ≥1 of: del(17p) 50% cut‐off, t(4;14) 30% cut‐off, t(14;16) 30% cut‐off. The cut‐off for gain(1q21) and amplification 1q21 was 30%.
d, dexamethasone; Isa, isatuximab; ITT, intent‐to‐treat; P, pomalidomide.
Patient baseline characteristics and disease characteristics divided by cytogenetic risk are shown in Table II. More patients in the high‐risk subgroup were aged ≥75 years, had lactate dehydrogenase levels greater than the upper limit of normal, were ISS stage III, and had an Eastern Cooperative Oncology Group performance status of 2.
Table II.
Baseline demographic characteristics by risk status.
|
High‐risk CA (n = 60) |
Standard‐risk CA (n = 181) | |||
|---|---|---|---|---|
| Isa–Pd (n = 24) | Pd (n = 36) | Isa–Pd (n = 103) | Pd (n = 78) | |
| Age, y | ||||
| Mean (SD) | 68·0 (8·1) | 65·6 (10·8) | 66·5 (9·3) | 64·7 (8·5) |
| Median | 67·5 | 64·0 | 68·0 | 66·0 |
| Min; max | 52; 83 | 41; 86 | 36; 81 | 41; 85 |
| Age group (y), n (%) | ||||
| <65 | 8 (33·3) | 19 (52·8) | 36 (35·0) | 37 (47·4) |
| 65–74 | 9 (37·5) | 6 (16·7) | 47 (45·6) | 32 (41·0) |
| ≥75 | 7 (29·2) | 11 (30·6) | 20 (19·4) | 9 (11·5) |
| ECOG performance status, n (%) | ||||
| 0 | 6 (25·0) | 20 (55·6) | 38 (36·9) | 30 (38·5) |
| 1 | 14 (58·3) | 10 (27·8) | 54 (52·4) | 41 (52·6) |
| 2 | 4 (16·7) | 6 (16·7) | 11 (10·7) | 7 (9·0) |
| Geographical region, n (%) | ||||
| Western Europe | 11 (45·8) | 24 (66·7) | 34 (33·0) | 33 (42·3) |
| Eastern Europe | 4 (16·7) | 3 (8·3) | 20 (19·4) | 13 (16·7) |
| North America | 1 (4·2) | 1 (2·8) | 5 (4·9) | 2 (2·6) |
| Asia | 3 (12·5) | 2 (5·6) | 14 (13·6) | 9 (11·5) |
| Other countries* | 5 (20·8) | 6 (16·7) | 30 (29·1) | 21 (26·9) |
| MM subtype at study entry†, n (%) | ||||
| IgG | 15 (62·5) | 27 (75·0) | 71 (68·9) | 52 (66·7) |
| IgA | 7 (29·2) | 9 (25·0) | 19 (18·4) | 19 (24·4) |
| IgM | 0 | 0 | 2 (1·9) | 0 |
| IgD | 0 | 0 | 0 | 0 |
| IgE | 0 | 0 | 0 | 0 |
| Kappa light chain only | 1 (4·2) | 0 | 7 (6·8) | 4 (5·1) |
| Lambda light chain only | 1 (4·2) | 0 | 4 (3·9) | 3 (3·8) |
| Serum LDH, n (%) | ||||
| ≤ULN | 13 (54·2) | 22 (61·1) | 73 (70·9) | 55 (70·5) |
| >ULN | 11 (45·8) | 14 (38·9) | 30 (29·1) | 23 (29·5) |
| ISS stage at study entry, n (%) | ||||
| Stage I | 8 (33·3) | 9 (25·0) | 43 (41·7) | 26 (33·3) |
| Stage II | 8 (33·3) | 15 (41·7) | 36 (35·0) | 29 (37·2) |
| Stage III | 8 (33·3) | 12 (33·3) | 21 (20·4) | 22 (28·2) |
| Unknown | 0 | 0 | 3 (2·9) | 1 (1·3) |
| R‐ISS stage at study entry, n (%) | ||||
| Stage I | 0 | 0 | 31 (30·1) | 20 (25·6) |
| Stage II | 16 (66·7) | 24 (66·7) | 64 (62·1) | 51 (65·4) |
| Stage III | 8 (33·3) | 12 (33·3) | 8 (7·8) | 7 (9·0) |
| Previous lines of therapy, median (IQR) | 3 (2–6) | 3 (2–10) | 3 (2–11) | 3 (2–7) |
| Refractory status | ||||
| Relapsed and refractory‡, n (%) | 24 (100) | 36 (100) | 103 (100) | 78 (100) |
| Primary refractory | 0 | 0 | 0 | 0 |
| Relapsed | 0 | 0 | 0 | 0 |
CA, chromosomal abnormalities; d, dexamethasone; ECOG, Eastern Cooperative Oncology Group; Ig, immunoglobulin; IQR, interquartile range; Isa, isatuximab; ISS, international staging system; LDH, lactate dehydrogenase; MM, multiple myeloma; P, pomalidomide; R‐ISS, Revised International Staging System; ULN, upper limit of normal.
Other countries: Australia, New Zealand, Turkey and Russia.
As per the electronic case report form.
Excluding primary refractory.
Efficacy
Response rate based on the independent response committee assessment was determined for patients in the overall high‐risk cytogenetic population and in patients with del(17p) and t(4;14). Among high‐risk patients, the ORR was higher in patients who received isatuximab plus pomalidomide–dexamethasone than those who received pomalidomide–dexamethasone (50·0% vs 16·7%, P = 0·0031; Fig 1). This ORR benefit was maintained regardless of whether the patient had del(17p) (50·0% vs 22·0%) or t(4;14) (50·0% vs 7·0%; Fig 2). Standard‐risk patients had an ORR of 65·0% in the isatuximab group and 42·3% in the pomalidomide–dexamethasone group (P = 0·0012; Fig 1). With the addition of isatuximab, a higher percentage of patients achieved very good partial response (VGPR) in the high‐risk subgroup (29·2% vs 2·8%), and ≥VGPR rate in the standard‐risk subgroup (32·0% vs 9·0%). Among patients with both del(17p) and t(4;14), one out of three patients in the isatuximab group achieved VGPR, and one out of four patients in the control group achieved partial response. Collectively, these data show that the ORR benefit with isatuximab was maintained among patients with high‐risk cytogenetics.
Fig 1.
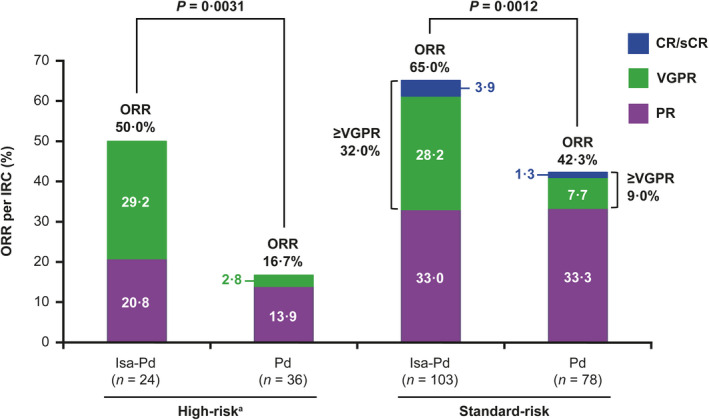
Overall response rate by cytogenetic cut‐off. P values are from the unstratified one‐sided Cochran–Mantel–Haenszel test.
a ≥ 1 of del(17p), t(4;14) or t(14;16) at study entry.
CR, complete response; d, dexamethasone; IRC, independent review committee; Isa, isatuximab; ORR, overall response rate; P, pomalidomide; PR, partial response; sCR, stringent complete response; VGPR, very good partial response.
Fig 2.
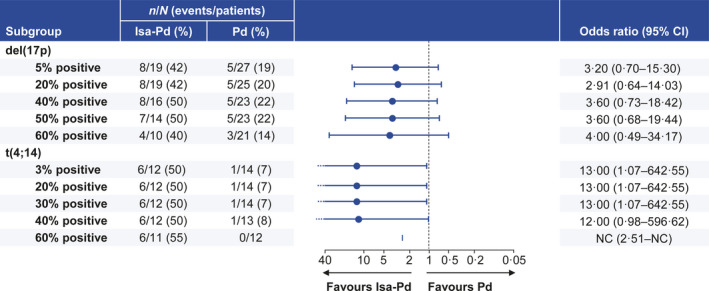
Overall response rate sensitivity analysis by cytogenetic cut‐off. CI, confidence interval; d, dexamethasone; Isa, isatuximab; NC, not calculable; P, pomalidomide.
A sensitivity analysis was conducted to determine whether the ORR benefit in the high‐risk subgroup was maintained independent of the high‐risk cytogenetic cut‐off definition used. Results demonstrated that the higher ORR observed with isatuximab compared with pomalidomide–dexamethasone backbone was consistent across all tested thresholds for both del(17p) as well as t(4;14) abnormalities. For del(17p), the percentage of overall responders with isatuximab ranged from 42% with a 5% cut‐off to 50% with a 50% cut‐off (Fig 2). Increasing the threshold to 60% decreased the ORR to 40%. The ORR in the control group ranged from 19% at 5% cut‐off to 22% at 50% cut‐off; increasing the cut‐off to 60% dropped the ORR to 14%. Similar results were obtained with t(4;14) in which the cut‐off was set at 30% in the initial analysis. An ORR of 50% in the isatuximab group was maintained across cut‐offs ranging from 3% to 40%.
PFS based on the independent response committee assessment was a prespecified subgroup analysis in the high‐risk subgroup. At the time of analysis, 14/24 (58·3%) high‐risk patients in the isatuximab group and 22/36 (61·1%) in the control group had a PFS event. Median PFS (mPFS) in high‐risk patients was longer in the isatuximab group (7·5 months) than that in the control group (3·7 months; HR, 0·66; 95% CI, 0·33–1·28; Fig 3). Among those treated with isatuximab, mPFS was lower in high‐risk patients (7·5 months) than in standard‐risk patients (11·6 months; Fig 3). However, the benefit of the addition of isatuximab was similar to that in standard‐risk patient in whom the HR was 0·62 (95% CI, 0·42–0·93; Fig 3). In high‐risk patients with t(4;14), the addition of isatuximab prolonged mPFS from 2·8 to 7·5 months (HR, 0·49; 95% CI, 0·19–1·31). In high‐risk patients with del(17p), the treatment effect of isatuximab on PFS was less pronounced (mPFS 9·1 months in the isatuximab group vs 7·4 months in the control group, HR, 0·76; 95% CI, 0·30–1·92; Fig 3). No significant interaction at the 10% level between treatment groups and cytogenetic risk status was observed. Thus, PFS benefit was observed in both high‐risk and standard‐risk patients that received isatuximab in addition to pomalidomide–dexamethasone, but was less pronounced in patients with del(17p).
Fig 3.
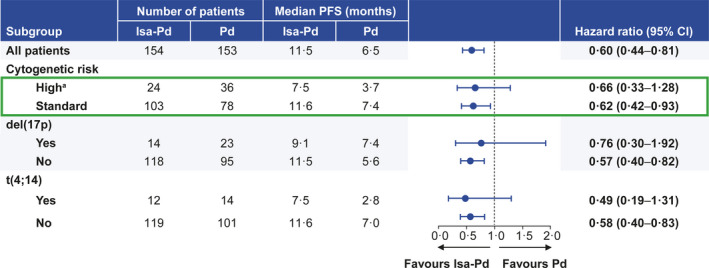
Progression‐free survival in cytogenetic subgroups. a ≥ 1 of del(17p), t(4;14) or t(14;16) at study entry. CI, confidence interval; d, dexamethasone; Isa, isatuximab; P, pomalidomide; PFS, progression‐free survival.
The sensitivity analysis demonstrated that PFS benefit in the high‐risk subgroup was maintained independent of the high‐risk cytogenetic cut‐off definition used. In high‐risk patients with del(17p), mPFS at 5% threshold was 6·5 months in the isatuximab group and 2·9 months in the control group (HR, 0·77; 95% CI, 0·35–1·70). Increasing the threshold to 50% prolonged the mPFS in both the isatuximab group (9·1 months) as well as in the control group (7·4 months; HR, 0·76; 95% CI, 0·30–1·92; Fig 4). A decrease in mPFS was observed in both groups at 60% cut‐off; however, the isatuximab group had a longer mPFS than that in the control group across all tested threshold‐cut‐offs for del(17p), with HRs between 0·75 and 0·88. Similarly, for high‐risk patients with the abnormality t(4;14), prolonged median PFS was observed with the addition of isatuximab (ranging from 7·5 to 8·1 months) compared with the control group (2·8 months). Overall, PFS benefit was maintained across threshold values ranging from 3% (HR, 0·49; 95% CI, 0·19–1·31) to 60% (HR, 0·19; 95% CI, 0·05–0·74), favouring the addition of isatuximab to pomalidomide–dexamethasone (Fig 4). Sensitivity analyses across threshold values for translocations may be less meaningful as variations between patients may be a reflection of the enrichment purity as well as potential contamination with polyclonal plasma cells.
Fig 4.
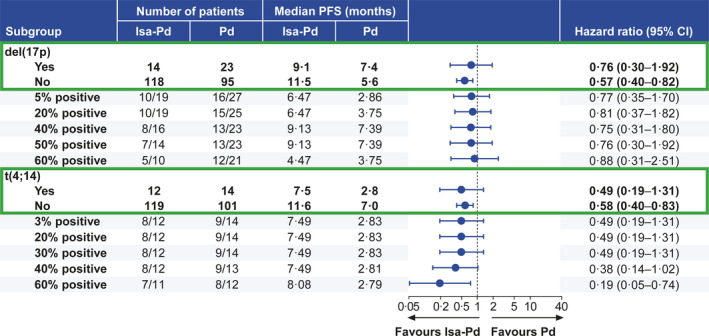
Progression‐free survival sensitivity analysis by cytogenetic cut‐off. CI, confidence interval; d, dexamethasone; Isa, isatuximab; P, pomalidomide; PFS, progression‐free survival.
Consistent with the time to progression (TTP) results reported in the primary analysis,17 the TTP benefit with isatuximab was maintained in high‐risk cytogenetic patients (Figure S1). The HRs for del(17p) were 0·62 (95% CI, 0·25–1·53) at 5% cut‐off and 0·68 (95% CI, 0·19–2·48) at 60% cut‐off (Figure S1). For t(4;14), HRs were 0·34 (95% CI, 0·11–1·04) at 3% cut‐off and 0·14 (95% CI, 0·03–0·67) at 60% cut‐off; indicating that the TTP benefit with isatuximab in high‐risk cytogenetic patients was independent of the cut‐off definition used.
Efficacy in patients with gain(1q21)
To evaluate the efficacy of isatuximab addition in patients with gain(1q21) abnormality, we conducted a retrospective analysis of plasma cells collected for cytogenetic risk evaluation. FISH testing revealed that 41·7% (128/307) of the patients had gain(1q21) abnormality irrespective of other high‐risk cytogenetic abnormalities. Of the randomised population, 26·1% (80/307) had three copies of gain(1q21) and 15·6% (48/307) had ≥4 copies (reported later as 1q21 amplification). More patients had gain(1q21) in the isatuximab group (49·4%) than in the control group (34·0%). Overall, isatuximab treatment had a positive effect on efficacy in patients both with and without gain (1q21; Table SI) as well as in patients with 1q21 amplification (Table SII).
Considering the challenge of interpreting outcomes in the context of overlapping cytogenetics with other high‐risk abnormalities, we performed a separate subgroup analysis of patients with isolated gain(1q21), defined as patients with no other high‐risk chromosomal abnormalities [del(17p), t(4;14) and t(14;16)]. Overall, 27·7% (85/307) of patients had isolated gain(1q21) with ≥3 copies, with more patients in the isatuximab group (36·4% vs 19·0%). The mPFS was higher in the isatuximab group than in the control group in patients with isolated gain(1q21) (11·2 vs 4·6 months; HR, 0·50; 95% CI, 0·28–0·88; Table III), and in those without gain(1q21) regardless of the presence of other high‐risk cytogenetic abnormalities (11·6 vs 9·8 months; HR, 0·75; 95% CI, 0·40–1·41; Table SI). Furthermore, the ORR was higher in the isatuximab group than in the control group for patients with isolated gain(1q21) (53·6% vs 27·6%; P = 0·0116), as well as those without gain(1q21) regardless of the presence of other high‐risk cytogenetic abnormalities (68·4% vs 43·5%; P = 0·0115). Compared with the control group, the isatuximab group had a higher rate of ≥VGPR both in patients with isolated gain(1q21) (25·0% vs 3·4%; P = 0·0070) and without gain(1q21) regardless of the presence of other high‐risk cytogenetic abnormalities (31·6% vs 10·9%; P = 0·0097).
Table III.
Efficacy in the 1q21 subtype without other high‐risk chromosomal abnormalities (isolated 1q21).
| ≥3 copies of gain(1q21) | Amplification with ≥4 copies of 1q21 | ||||||
|---|---|---|---|---|---|---|---|
|
Isa‐Pd (n = 56) |
Pd (n = 29) |
P value |
Isa‐Pd (n = 23) |
Pd (n = 9) |
P value | ||
| mPFS, mo[95% CI] |
11·2 [6·5–13·3] |
4·6 [2·3–7·9] |
8·9 [3·7–12·3] |
2·3 [0·1–NC] |
|||
| HR, 0·50 [0·28–0·88] | HR, 0·55 [0·21–1·43] | ||||||
| ORR, n (%)[95% CI] | 30 (53·6)[0·4–0·7] | 8 (27·6)[0·1–0·5] | 0·0116 | 12 (52·2)[0·3–0·7] | 1 (11·1)[<0·1–0·5] | 0·0182 | |
| ≥VGPR, n (%)[95% CI] | 14 (25·0)[0·1–0·4] | 1 (3·4)[<0·1–0·2] | 0·0070 | 7 (30·4)[0·1–0·5] | 0[NC] | 0·0327 | |
mPFS, median progression‐free survival; CI, confidence interval; ORR, overall response rate; VGPR, very good partial response; Isa, isatuximab; P, pomalidomide; d, dexamethasone; HR hazard ratio; NC, not calculated.
P values are from the unstratified one‐sided Cochran–Mantel–Haenszel test.
Improved mPFS in the isatuximab group versus control group was observed both in patients with isolated 1q21 amplification (≥4 copies 1q21; 8·9 vs 2·3 months; HR, 0·55; 95% CI, 0·21–1·43; Table III) as well as those with 1q21 amplification, regardless of the presence of other high‐risk cytogenetic abnormalities (8·9 vs 2·3 months; HR, 0·49; 95% CI, 0·24–0·99; Table SII). The addition of isatuximab also improved ORR [52·2% vs 11·1%, P = 0·0182, isolated 1q21 amplification with ≥4 copies (Table III); 51·9% vs 9·5%, P = 0·0011, 1q21 amplification regardless of the presence of other high‐risk cytogenetic abnormalities (Table SII)] and ≥VGPR rates [30·4% vs 0%, P = 0·0327, isolated 1q21 amplification with ≥4 copies (Table III); 33·3% vs 0%, P = 0·0018, 1q21 amplification with ≥4 copies, regardless of the presence of other high‐risk cytogenetic abnormalities (Table SII)] compared with the control group.
Safety
The overall safety with the addition of isatuximab in cytogenetic subgroups is summarised in Table IV. The median duration of treatment was higher in the isatuximab group than in the control group in both high‐risk (32 vs 18 weeks) and standard‐risk (42 vs 31 weeks) subgroups. In patients treated with isatuximab plus pomalidomide–dexamethasone, the incidence of grade ≥3 adverse events [AEs; 22 (95·7%) vs 88 (85·4%)] and serious adverse events [SAEs; 17 (73·9%) vs 60 (58·3%)] was higher in high‐risk patients than in standard‐risk patients. This differed from the control group in which the incidence of grade ≥3 AEs [23 (67·6%) vs 58 (76·3%)] and SAEs [17 (50·0%) vs 47 (61·8%)] was lower in high‐risk patients than in standard‐risk patients. All patients in the high‐risk isatuximab group had treatment‐emergent adverse events (TEAEs) of any grade compared with 94·1% in the high‐risk control group. However, isatuximab addition did not increase TEAEs leading to treatment‐discontinuation in the high‐risk [two events (8·7%) isatuximab vs 8 (23·5%) control] or standard‐risk subgroups [7 events (6·8%) isatuximab vs 6 (7·9%) control]. These results are consistent with the findings from the primary analysis, which demonstrated that isatuximab plus pomalidomide–dexamethasone was well tolerated, with no increase in treatment discontinuations due to AEs or the incidence of fatal events during study treatment compared with control group.17
Table IV.
Safety in cytogenetic subgroups.
| High‐risk | Standard‐risk | |||
|---|---|---|---|---|
| Isa‐Pd (n = 23) | Pd (n = 34) | Isa‐Pd (n = 103) | Pd (n = 76) | |
| Median duration of treatment exposure, wk (range) | 32·0 (1·3–60·1) | 18·0 (1·0–56·0) | 42·0 (4·0–76·7) | 31·3 (2·0–69·0) |
| Any TEAE | 23 (100) | 32 (94·1) | 102 (99·0) | 75 (98·7) |
| Grade ≥3 TEAE | 22 (95·7) | 23 (67·6) | 88 (85·4) | 58 (76·3) |
| Serious TEAE | 17 (73·9) | 17 (50·0) | 60 (58·3) | 47 (61·8) |
| TEAE leading to definitive discontinuation | 2 (8·7) | 8 (23·5) | 7 (6·8) | 6 (7·9) |
| Death due to adverse event | 1 (4·3) | 3 (8·8) | 1 (1·0) | 1 (1·3) |
| Treatment‐related | – | 1 (2·9) | – | 1 (1·3) |
| Grade ≥3 events in >5% of patients with Isa‐Pd in either subgroup, n (%) | ||||
| Laboratory abnormalities | ||||
| Neutropenia | 19 (82·6) | 25 (75·8)* | 88 (85·4) | 53 (69·7) |
| Thrombocytopenia | 11 (47·8) | 9 (27·3)* | 27 (26·2) | 19 (25·0) |
| TEAEs | ||||
| Febrile neutropenia | 3 (13·0) | 0 | 12 (11·7) | 2 (2·6) |
| Pneumonia | 5 (21·7) | 6 (17·6) | 16 (15·5) | 14 (18·4) |
| Influenzal pneumonia | 2 (8·7) | 0 | 0 | 2 (2·6) |
| Urinary tract infection | 2 (8·7) | 1 (2·9) | 4 (3·9) | 1 (1·3) |
| Lower respiratory tract infection | 2 (8·7) | 0 | 3 (2·9) | 4 (5·3) |
| Asthenia | 2 (8·7) | 1 (2·9) | 2 (1·9) | 3 (3·9) |
| Fatigue | 2 (8·7) | 0 | 3 (2·9) | 0 |
| Infusion reaction | 2 (8·7) | 0 | 1 (1·0) | 0 |
| Pulmonary embolism | 2 (8·7) | 0 | 1 (1·0) | 3 (3·9) |
| Vomiting | 2 (8·7) | 0 | 0 | 0 |
d, dexamethasone; Isa, isatuximab; P, pomalidomide; TEAE, treatment‐emergent adverse event.
n = 33.
Isatuximab had a manageable safety profile in high‐risk and standard‐risk patients (Table IV). In the high‐risk subgroup, analysis from laboratory values reported during study treatment showed that neutropenia [19 (82·6%) vs 25 (75·8%)] and thrombocytopenia [11 (47·8%) vs 9 (27·3%)] occurred more frequently in the isatuximab group than in the control group. In high‐risk patients, granulocyte‐colony stimulating factor (G‐CSF) was used in 16 patients (28·1%) in the isatuximab group and 21 (36·8%) in the control group. A higher incidence of neutropenia with isatuximab was also observed in standard‐risk patients [88 (85·4%) vs 53 (69·7%)]. In standard‐risk patients, G‐CSF was used in 70 patients (39·1%) in the isatuximab group and 35 (19·6%) in the control group.
The most frequent TEAEs in the high‐risk isatuximab versus control group included febrile neutropenia [3 (13·0%) vs 0] and pneumonia [5 (21·7%) vs 6 (17·6%)]. The incidence rate of both febrile neutropenia and pneumonia in the high‐risk group was generally consistent with the incidence rate in the standard‐risk group. Grade ≥3 infusion reactions occurred only in the isatuximab group in high‐risk and standard‐risk patients: two patients (8·7%) in the high‐risk and one patient (1·0%) in the standard‐risk group.
Discussion
Recent years have witnessed significant improvements in survival in MM but such benefits are not always seen in high‐risk patients, whose treatment remains a challenge. Thus, any new treatment regimen that benefits this group of patients, identified by specific cytogenetic abnormalities, has the potential to make a real difference to survival outcomes in MM.
ICARIA‐MM was the first phase 3 study to show positive outcomes when adding an anti‐CD38 antibody (isatuximab) to pomalidomide–dexamethasone therapy for RRMM in both high‐risk and standard‐risk patients.17 In this prespecified subgroup analysis of patients with high‐risk cytogenetics, the PFS and the ORR benefit with the addition of isatuximab to pomalidomide–dexamethasone was not only maintained but was also independent of the cytogenetic cut‐off definition used and also resulted in a manageable safety profile (consistent with data from the overall analysis).
Currently, there are no treatments that can overcome the poor prognosis of high‐risk cytogenetics in MM. IMiDs have varying effects on high‐risk status. While lenalidomide moderately improves TTP in high‐risk patients with t(4;14), less effect was observed in high‐risk patients with del(17p).18, 19, 20 Previous studies have demonstrated a response benefit of pomalidomide–dexamethasone in patients with del(17p),13, 21 while other trials have reported minimal effect on high‐risk patients.22 Proteasome inhibitors, such as bortezomib, carfilzomib and ixazomib, have demonstrated the ability to improve outcomes in high‐risk patients when combined with lenalidomide or pomalidomide, but still, these treatments do not overcome the poor prognosis of high‐risk cytogenetics.23, 24, 25
Targeted therapies against specific cell surface antigens expressed on MM cells represent an important class of therapy for the treatment of patients with MM. Novel targeted therapies approved for the treatment of MM include the monoclonal antibodies, elotuzumab (targets signalling lymphocyte activating molecule) and daratumumab (targets CD38), which exert their effects via direct and indirect immune‐mediated cytotoxicity.11
In triplet combination studies with daratumumab, outcomes were improved in high‐risk cytogenetic patients with RRMM, but the treatment effect versus standard of care was variable.26, 27, 28 In the updated four‐year follow‐up of the phase 3 POLLUX study, PFS benefit with daratumumab plus lenalidomide–dexamethasone was maintained in high‐risk patients (26·8 vs 8·3 months), but was lower than the PFS observed in standard‐risk patients (52·0 vs 18·6 months).28 Additionally, in the phase 3 CASTOR trial, PFS was improved in the high‐risk group receiving daratumumab plus bortezomib‐dexamethasone (11·2 vs 7·2 months). However, PFS in the high‐risk group receiving daratumumab remained much shorter in comparison to the standard‐risk group receiving daratumumab (11·2 vs 19·6 months) while the PFS for patients receiving bortezomib–dexamethasone remained nearly the same regardless of risk status [7·2 (high‐risk) vs 7·0 months (standard‐risk)].27
Results from this subgroup analysis indicate that the treatment benefit with isatuximab is similar both in high‐risk and standard‐risk cytogenetic patients; however, it does not completely overcome the negative prognosis associated with high‐risk cytogenetics [mPFS 7·5 (high‐risk) vs 11·6 months (standard‐risk)]. Notably, with the addition of isatuximab, a higher percentage of patients achieved VGPR in the high‐risk group (vs in the control group). Additionally, the PFS and ORR benefit in high‐risk cytogenetic patients treated with isatuximab was maintained irrespective of the cytogenetic cut‐off definition used. This is particularly important as differences in cytogenetic thresholds used to determine high‐risk status are inconsistently used and reported in other clinical trials. Although the negative impact of high‐risk status was not overcome, isatuximab plus pomalidomide–dexamethasone benefits both high‐risk patients and standard‐risk patients, and clinical benefit was obtained in high‐risk patients without any emergent safety issues.
The positive outcome of isatuximab addition was also observed in patients with gain(1q21) abnormality, the most common chromosomal abnormality. Importantly, the mPFS with isatuximab was 11·2 months in patients with gain(1q21), similar to that in the overall study population treated with isatuximab plus pomalidomide–dexamethasone (11·5 months). This is in contrast to the pomalidomide–dexamethasone group where patients with gain(1q21) had shorter mPFS (4·6 months) than the overall study population treated with Pd (6·5 months). This observation is of particular relevance in light of recent reports of significantly worse PFS outcomes in patients with gain(1q21) treated with daratumumab.29 The gene coding for complement‐inhibitory protein CD55 is localised to 1q32·2 and increased expression of CD55 on myeloma plasma cells has been hypothesised as a resistance mechanism to daratumumab therapy.29, 30 Interestingly, while daratumumab was selected based on its superior CDC activity,31 the plasma cell kill by isatuximab is more dependent on ADCC than on CDC.32, 33
In conclusion, the results of this subgroup analysis showed that isatuximab plus pomalidomide–dexamethasone treatment provides a consistent benefit over pomalidomide–dexamethasone treatment in patients with RRMM regardless of cytogenetic risk.
Role of the funding source
The sponsor (Sanofi, Cambridge, MA, USA) together with the investigator steering committee developed the protocol. Data collected by investigator research teams were compiled and maintained by the sponsor. Unblinded safety data were periodically reviewed by an independent Data Monitoring Committee. The sponsor’s clinical study director (FC) was responsible for study oversight. The sponsor authors participated in designing this study, data analysis, data interpretation and writing of the manuscript.
Author contributions
SJH, AP, AA, DS, MCW, AS, SD, CH, KS, TF, PV, KY and PR contributed to data acquisition and data interpretation; FC contributed to study design, data analysis and interpretation; MI contributed to statistical analysis; SM contributed to data analysis and data interpretation; M‐LR contributed to data analysis and data interpretation; HvdV contributed to study design, data analysis and interpretation. All authors contributed to critical revision and final approval of the manuscript.
Conflict of interest
SJH reports a consultancy/advisory role for AbbVie, Amgen, Celgene, GSK, Janssen, Novartis, Roche/Genentech, Sanofi and Takeda; honoraria from Amgen, Celgene, Janssen, Novartis, Roche/Genentech and Takeda; study investigator for AbbVie, Amgen, Celgene, Janssen, Novartis, Roche/Genentech and Haemalogiz; and research funding from Amgen, Celgene, GSK, Janssen, Novartis and Haemalogix. AP reports honoraria/congress support from Amgen, Celgene, Janssen, Sanofi; and reports grant support from Takeda. AA reports honoraria from Amgen, Celgene, Janssen, Sanofi and Takeda; membership on an entity’s Board of Directors or advisory committees at Amgen, Celgene, Janssen, Sanofi and Takeda. DS reports honoraria from AbbVie, Janssen, Roche; employment, stock and research funding from Beigene; research funding from Amgen, Celgene and Merch Shape Dohme, Acerta, Pharmacyclics, Sanofi and GSK. MCW has no disclosures. AS reports a consultancy role for Secura Bio, Celgene, Takeda, Janssen, Specialised Therapeutics Australia, AbbVie, Servier, Haemalogix and Sanofi; honoraria from Secure Bio, Celgene, Takeda, Janssen, Specialised Therapeutics Australie, AbbVie, Servier, Haemalogix, Sanofi and Amgen; personal fees associated with the Speaker’s Bureau for Celgene, Takeda and Janssen. SD reports personal fees associated with the Speaker’s Bureau for Amgen and Janssen. CH reports personal fees from Sanofi, Janssen and Celgene. TF reports data monitoring board participation for Sanofi; advisory board role for Celgene, Janssen, Takeda, Roche, Oncopeptides, Karyopharm, Amgen. PV reports advisory board and travel support from Celgene and Takeda, and travel support from Amgen. KY reports a consultancy/advisory role with Amgen, Janssen and Takeda; is in the Speaker’s bureau for Amgen and Takeda; received research funding from Amgen and Sanofi. FC, MI, SM, MLR, HvdV are employees of Sanofi and may hold shares and/or stock options in the company.
Supporting information
Table SI. Efficacy in the subgroup with gain(1q21) regardless of the presence of other high‐risk cytogenetic abnormalities.
Table SII. Efficacy in the subgroup with 1q amplification with four or more copies 1q21, regardless of the presence of other high‐risk cytogenetic abnormalities.
Fig S1. Time to progression sensitivity analysis by cytogenetic cut‐off.
Acknowledgements
The ICARIA‐MM study was sponsored by Sanofi. The authors thank the participating patients and their families, and the study centres and investigators, for their contributions to the study. We thank Solenn Le Guennec for her contributions to the statistical analysis. Medical writing support was provided by Stephanie Brillhart and Smitha Reddy of Elevate Medical Affairs (Fairfield, CT, USA), and funded by Sanofi (Cambridge, MA, USA).
Data availability statement
Qualified researchers can request access to patient‐level data and related study documents including the clinical study report, study protocol with any amendments, blank case report forms, statistical analysis plan and data set specifications. Patient‐level data will be anonymised, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data‐sharing criteria, eligible studies and process for requesting access are at: https://www.clinicalstudydatarequest.com.
References
- 1.Lancman G, Tremblay D, Barley K, Barlogie B, Cho HJ, Jagannath S, et al. The effect of novel therapies in high‐molecular‐risk multiple myeloma. Clin Adv Hematol Oncol. 2017;15:870–9. [PMC free article] [PubMed] [Google Scholar]
- 2.Rasche L, Kortum KM, Raab MS, Weinhold N. The impact of tumor heterogeneity on diagnostics and novel therapeutic strategies in multiple myeloma. Int J Mol Sci. 2019;20:1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23:2210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palumbo A, Avet‐Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, et al. Revised international staging system for multiple myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33:2863–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanamura I, Stewart JP, Huang Y, Zhan F, Santra M, Sawyer JR, et al. Frequent gain of chromosome band 1q21 in plasma‐cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem‐cell transplantation. Blood. 2006;108:1724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biran N, Malhotra J, Bagiella E, Cho HJ, Jagannath S, Chari A. Patients with newly diagnosed multiple myeloma and chromosome 1 amplification have poor outcomes despite the use of novel triplet regimens. Am J Hematol. 2014;89:616–20. [DOI] [PubMed] [Google Scholar]
- 7.An G, Xu Y, Shi L, Shizhen Z, Deng S, Xie Z, et al. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica. 2014;99:353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waheed S, Shaughnessy JD, van Rhee F, Alsayed Y, Nair B, Anaissie E, et al. International staging system and metaphase cytogenetic abnormalities in the era of gene expression profiling data in multiple myeloma treated with total therapy 2 and 3 protocols. Cancer. 2011;117:1001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood. 2006;108:2020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high‐risk, Double‐Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33:159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson PG, Attal M, Campana F, Le‐Guennec S, Hui A‐M, Risse M‐L, et al. Isatuximab plus pomalidomide/dexamethasone versus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma: ICARIA Phase III study design. Future Oncol. 2018;14:1035–47. [DOI] [PubMed] [Google Scholar]
- 12.Dimopoulos MA, Swern AS, Li JS, Hussein M, Weiss L, Nagarwala Y, et al. Efficacy and safety of long‐term treatment with lenalidomide and dexamethasone in patients with relapsed/refractory multiple myeloma. Blood Cancer J. 2014;4:e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimopoulos MA, Weisel KC, Song KW, Delforge M, Karlin L, Goldschmidt H, et al. Cytogenetics and long‐term survival of patients with refractory or relapsed and refractory multiple myeloma treated with pomalidomide and low‐dose dexamethasone. Haematologica. 2015;100:1327–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deckert J, Wetzel MC, Bartle LM, Skaletskaya A, Goldmacher VS, Vallee F, et al. SAR650984, a novel humanized CD38‐targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20:4574–83. [DOI] [PubMed] [Google Scholar]
- 15.Jiang H, Acharya C, An G, Zhong M, Feng X, Wang L, et al. SAR650984 directly induces multiple myeloma cell death via lysosomal‐associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia. 2016;30:399–408. [DOI] [PubMed] [Google Scholar]
- 16.Moreno L, Perez C, Zabaleta A, Manrique I, Alignani D, Ajona D, et al. The mechanism of action of the anti‐CD38 monoclonal antibody isatuximab in multiple myeloma. Clin Cancer Res. 2019;25:3176–87. [DOI] [PubMed] [Google Scholar]
- 17.Attal M, Richardson PG, Rajkumar SV, San‐Miguel J, Beksac M, Spicka I, et al. Isatuximab plus pomalidomide and low‐dose dexamethasone versus pomalidomide and low‐dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA‐MM): a randomised, multicentre, open‐label, phase 3 study. Lancet. 2019;394:2096–107. [DOI] [PubMed] [Google Scholar]
- 18.Reece D, Song KW, Fu T, Roland B, Chang H, Horsman DE, et al. Influence of cytogenetics in patients with relapsed or refractory multiple myeloma treated with lenalidomide plus dexamethasone: adverse effect of deletion 17p13. Blood. 2009;114:522–5. [DOI] [PubMed] [Google Scholar]
- 19.Chang H, Jiang A, Qi C, Trieu Y, Chen C, Reece D. Impact of genomic aberrations including chromosome 1 abnormalities on the outcome of patients with relapsed or refractory multiple myeloma treated with lenalidomide and dexamethasone. Leuk Lymphoma. 2010;51:2084–91. [DOI] [PubMed] [Google Scholar]
- 20.Avet‐Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109:3489–95. [DOI] [PubMed] [Google Scholar]
- 21.Leleu X, Karlin L, Macro M, Hulin C, Garderet L, Roussel M, et al. Pomalidomide plus low‐dose dexamethasone in multiple myeloma with deletion 17p and/or translocation (4;14): IFM 2010–02 trial results. Blood. 2015;125:1411–7. [DOI] [PubMed] [Google Scholar]
- 22.Leleu X, Attal M, Arnulf B, Moreau P, Traulle C, Marit G, et al. Pomalidomide plus low‐dose dexamethasone is active and well tolerated in bortezomib and lenalidomide‐refractory multiple myeloma: Intergroupe Francophone du Myelome 2009–02. Blood. 2013;121:1968–75. [DOI] [PubMed] [Google Scholar]
- 23.Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142–52. [DOI] [PubMed] [Google Scholar]
- 24.Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hájek R, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open‐label, multicentre study. Lancet Oncol. 2016;17:27–38. [DOI] [PubMed] [Google Scholar]
- 25.Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374:1621–34. [DOI] [PubMed] [Google Scholar]
- 26.Dimopoulos MA, San‐Miguel J, Belch A, White D, Benboubker L, Cook G, et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of POLLUX. Haematologica. 2018;103:2088–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spencer A, Lentzsch S, Weisel K, Avet‐Loiseau H, Mark TM, Spicka I, et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of CASTOR. Haematologica. 2018;103:2079–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaufman JL, Usmani SZ, San‐Miguel J, Bahlis N, White DJ, Benboubker L, et al. Four‐year follow‐up of the phase 3 pollux study of daratumumab plus lenalidomide and dexamethasone (D‐Rd) versus lenalidomide and dexamethasone (Rd) alone in relapsed or refractory multiple myeloma (RRMM). Blood. 2019;134:1866. [Google Scholar]
- 29.Mohan M, Weinhold N, Schinke C, Thanedrarajan S, Rasche L, Sawyer JR, et al. Daratumumab in high‐risk relapsed/refractory multiple myeloma patients: adverse effect of chromosome 1q21 gain/amplification and GEP70 status on outcome. Br J Haematol. 2020;189:67–71. [DOI] [PubMed] [Google Scholar]
- 30.Nijhof IS, Casneuf T, van Velzen J, van Kessel B, Axel AE, Syed K, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood. 2016;128:959–70. [DOI] [PubMed] [Google Scholar]
- 31.de WeersM,Tai Y‐T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186:1840–8. [DOI] [PubMed] [Google Scholar]
- 32.van de Donk N, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood. 2018;131:13–29. [DOI] [PubMed] [Google Scholar]
- 33.Martin TG, Corzo K, Chiron M, van de Velde H, Abbadessa G, Campana F, et al. Therapeutic opportunities with pharmacological inhibition of CD38 with isatuximab. Cells. 2019;8:1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table SI. Efficacy in the subgroup with gain(1q21) regardless of the presence of other high‐risk cytogenetic abnormalities.
Table SII. Efficacy in the subgroup with 1q amplification with four or more copies 1q21, regardless of the presence of other high‐risk cytogenetic abnormalities.
Fig S1. Time to progression sensitivity analysis by cytogenetic cut‐off.
Data Availability Statement
Qualified researchers can request access to patient‐level data and related study documents including the clinical study report, study protocol with any amendments, blank case report forms, statistical analysis plan and data set specifications. Patient‐level data will be anonymised, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data‐sharing criteria, eligible studies and process for requesting access are at: https://www.clinicalstudydatarequest.com.