

Abstract
Biodegradable nanostructures displaying aggregation‐induced emission (AIE) are desirable from a biomedical point of view, due to the advantageous features of loading capacity, emission brightness, and fluorescence stability. Herein, biodegradable polymers comprising poly (ethylene glycol)‐block‐poly(caprolactone‐gradient‐trimethylene carbonate) (PEG‐P(CLgTMC)), with tetraphenylethylene pyridinium‐TMC (PAIE) side chains have been developed, which self‐assembled into well‐defined polymersomes. The resultant AIEgenic polymersomes are intrinsically fluorescent delivery vehicles. The presence of the pyridinium moiety endows the polymersomes with mitochondrial targeting ability, which improves the efficiency of co‐encapsulated photosensitizers and improves therapeutic index against cancer cells both in vitro and in vivo. This contribution showcases the ability to engineer AIEgenic polymersomes with structure inherent fluorescence and targeting capacity for enhanced photodynamic therapy.
Keywords: aggregation-induced emission, biodegradable polymersomes, mitochondria targeting, photo-dynamic therapy, polycarbonates
Polymersomes with inherent aggregation‐induced emission are presented that have mitochondrial targeting capacity due to the presence of pyridinium groups. The AIEgenic polymersomes, upon encapsulation of photosensitizers, show efficient energy transfer to enable photodynamic therapy, which improves the therapeutic index against cancer cells both in vitro and in vivo.
Introduction
Polymersomes are recognized as attractive candidates for delivery vehicles in the field of nanomedicine.[1] With a shell structure consisting of a bilayer membrane self‐assembled from amphiphilic block copolymers, polymersomes can encapsulate not only hydrophilic cargoes in their inner aqueous lumen but also accommodate a considerable quantity of hydrophobic molecules in the membrane pocket.[2] Besides that, the chemically versatile nature of the block copolymer constituents allows much freedom concerning tuning the physicochemical properties of the polymersomes, including the incorporation of cell‐targeting capacity and stimulus‐responsiveness.[3] Fluorescent‐labeled polymersomes are interesting systems for in vivo tracking of nanoparticle transport, monitoring cargo distribution, and cell/tissue imaging.[4] Traditionally, fluorescence is introduced by physically loading conventional fluorescent materials, or by chemical conjugation approaches.[5] While frequently used, these conventional approaches are generally recognized to suffer from low loading/conjugation efficiency, leakage of the dye, aggregation‐induced quenching (ACQ), and/or photo‐bleaching behavior.[5a, 6] A strategy that circumvents the abovementioned drawbacks is to incorporate the luminogens with the feature of aggregation‐induced emission (AIEgen) into the polymer building blocks, which upon self‐assembly/aggregation become fluorescent and afford a nanosized, bright fluorescent system.[6b, 7] This is a consequence of the restricted intramolecular motion and non‐radiative energy dissipation of the AIEgenic molecules when they are in an assembled state.[8] These moieties have been widely explored in nanoscience and have also been integrated into block copolymers; very recently, they have been employed to construct AIEgenic polymersomes.[9] Until now, however, the application potential of these polymersomes in the biomedical field has been underexplored. Polymersomes that accommodate AIEgenic moieties are potentially very useful as alternative fluorescent materials for both cellular and in vivo imaging as a consequence of their high contrast, enhanced photo‐stability, and intrinsic chemical versatility.
Their intrinsic fluorescence can furthermore also be employed more actively in therapies that rely on photochemical processes, such as photodynamic therapy (PDT).[10] Upon irradiation with light of the appropriate wavelength, highly active radicals are formed via the action of photosensitizers.[11] This results in irreversible damage to cells in the close vicinity of the PDT agent.[12] Combining AIEgenic features with photosensitizer capacity in a polymersome system would allow to locally create Förster resonance energy transfer (FRET) that would lead to highly selective singlet oxygen production.[13] To improve the selectivity of the PDT process, the polymersomes should be targeted to the correct microenvironment, which is normally achieved via the conjugation of biological target ligands, such as antibodies, to the polymersome surface.[14] This is often however not trivial. Engineering polymersomes with structure‐inherent targeting capacity would provide an opportunity to realize a more straightforward manner of delivering the agents to the desired site of action.[15] Intracellular targeting of certain essential organelles, such as the mitochondria, could furthermore even improve the therapeutic effect by initiating and executing various apoptotic and necrotic processes.[16]
A versatile class of block copolymers for the engineering of AIE‐polymersomes (AIE‐Ps) with PDT and structure‐inherent targeting capacity are based on poly(ethylene glycol)‐poly(trimethylene carbonate) (PEG‐PTMC).[17] Synthetic aliphatic polycarbonates have already been extensively used in biomedicine, because of their inherent low toxicity and biodegradability.[18] These block copolymers are very useful for the construction of a range of polymer assemblies, including morphologies such as worm‐like micelles and vesicles.[19] Exploration of AIEgenic PEG‐PTMC based polymersomes with PDT and intracellular targeting capacity has however not been demonstrated, which would provide a potential platform for bioimaging and theranostics.
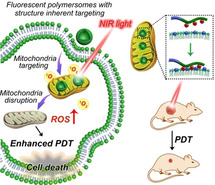
Herein, we report a strategy for the controlled formation of biodegradable fluorescent polymersomes with aggregation‐induced emission (AIE) features prepared from amphiphilic block copolymers composed of poly (ethylene glycol)‐block‐poly(caprolactone‐gradient‐trimethylene carbonate) (PEG‐P(CLgTMC)), with a terminal block of tetraphenylethylene pyridinium modified PTMC as a functional unit (PAIE) (Figure 1). By controlling the self‐assembly conditions (varying the NaCl concentration), well‐defined AIE‐polymersomes were prepared, which displayed typical aggregation‐induced emission behavior. Owing to the positively charged pyridinium moieties on the polymersome surface, the polymersomes preferentially accumulated intracellularly around the mitochondria.[20] The incorporation of a hydrophobic BODIPY photosensitizer resulted in an intrinsic Förster resonance energy transfer (FRET) between the AIE moieties and BODIPY. Under NIR light irradiation, abundant reactive oxygen species (ROS) were therefore generated, leading to fast cancer cell necrosis in vitro; this process could also effectively be translated in vivo to inhibit tumor growth.
Figure 1.

Programming the formation of biodegradable fluorescent polymersomes via a salt‐induced self‐assembly process toward mitochondria‐targeted photo‐dynamic therapy. a) Terpolymer chemical structure and self‐assembly into fluorescent polymersomes. The presence of the quaternary pyridinium block enables preferential accumulation in mitochondria; b) photosensitizer‐loaded polymersomes with structure‐inherent targeting behavior are delivered to mitochondria; upon NIR light irradiation ROS is produced which causes mitochondrial damage, leading to an enhanced PDT that kills cancer cells in vitro and which inhibits tumor growth in vivo.
Results and Discussion
The designed block copolymers were synthesized via a modular polymerization approach, as presented in the literature for similar macromolecules (Supporting Information, Scheme S1). Poly(ethylene glycol)‐block‐poly(caprolactone‐gradient‐trimethylene carbonate) (PEG‐PCLgTMC) copolymers were utilized as the structural basis for this system due to their intrinsic biodegradability/biocompatibility and well‐established capacity in generating well‐defined polymer assemblies via the direct hydration process.[19b] Using PEG22‐PCL30gTMC30 (ca. 7.5 kDa, termed as TerP22) as a macro‐initiator, a bromide‐functional TMC derivative was polymerized onto the end of the PTMC chain.[21] To functionalize the block copolymer with the AIE and pyridinium moieties, a pyridine‐modified tetraphenylethylene derivative was firstly synthesized, following well‐established literature procedures.[22] Via a nucleophilic substitution reaction, the bromides were replaced by quaternary pyridinium groups to provide the block copolymers with both AIE capacity and pyridinium targeting moieties to effectively localize the nanoparticles intracellularly to the mitochondria.
A series of terpolymers (termed TerP22‐PAIE) was prepared in which the length of the ternary block was systematically varied yielding TerP22‐PAIEn (where n=5, 8, and 16 repeats) to investigate the effect of the hydrophilic‐hydrophobic balance and charge density on the self‐assembly and size/morphology of the resulting particles (Scheme S1, Table S2). As we demonstrated previously, copolymers of PCL and PTMC can form polymersome‐like nano‐structures; however, we also observed that when upon modification the hydrophilic volume was increased, the chain packing changed, leading to the generation of micellar architectures.[19a] Retaining the polymersome structure with the current terpolymer series was therefore challenging and required an additional strategy. Because electrostatic repulsions between the highly charged PTMC‐PAIE blocks tend to induce the formation of high surface curvature nanoparticles (i.e., micelles), the concentration of salt present during formation to screen electrostatic repulsions was expected to be a key factor in controlling the assembly of these TerP22‐PAIE copolymers.[19a]
The self‐assembly of TerP22‐PAIE block copolymers was induced via a direct hydration process, whereby a 10 wt % polymer solution was prepared firstly in oligo (ethylene glycol) (OEG) and then directly hydrated with a buffer solution by stirring at room temperature for 5 min.[19b, 23] To determine the effect of ionic strength on the self‐assembly process, solutions of increasing [NaCl] from 0–100 mM were used for particle formation. Dynamic light scattering (DLS) data provided evidence that the assembly process of the three copolymers was significantly different. Terpolymers bearing the longer PAIE terminal blocks (TerP22‐PAIE16 and TerP22‐PAIE8) appeared to form uniform micelles under low and high salt solutions (i.e. 0 and 100 mM NaCl solution) with a hydrodynamic radius of ca. 30–50 nm (Figure S24 & S25), which was further verified by cryo‐TEM imaging. Such consistent micellar formation (rather than vesicular) can be attributed to the long positively charged domains, which result in strong repulsive forces leading to a relatively large hydrophilic volume.
To reduce the strength of electrostatic repulsions between PAIE blocks, we decreased the chain length of the pyridinium block. The self‐assembly of TerP22‐PAIE5 clearly showed a response to ionic strength during the hydration step. When hydrated in Milli‐Q water, TerP22‐PAIE5 formed uniform spherical micelles (SMs) with hydrodynamic radius (R h) of ca. 20 nm (Figure 2 a,d, and Figure S26). Quantitative insight was obtained using asymmetric flow field‐flow fractionation (AF4) coupled with multi‐angle light scattering (MALS) and DLS. Measurements of the shape factor (ρ) provided insight into nanoparticle topology by comparison of the radius of gyration (R g) and hydrodynamic radius R h (ρ=R g/R h). Indeed, assemblies of TerP22‐PAIE5 upon hydration into Milli‐Q water eluted after 10–12 minutes with ρ values ranging from 0.7–0.75, indicative of micelles that possess a dense core and hydrated corona (Figure 2 b). Cryo‐TEM microscopy further confirmed their micellar nanostructure (Figure 2 e). When the polymer was hydrated with 50 mM or higher NaCl concentration (Figure 2 a,d, and Figure S26), DLS measurements identified a change in nanoparticle morphology with a hydrodynamic size increase. While polymersome formation was still partial when hydrated with 50 mM NaCl solution, as identified clearly by cryo‐TEM, a higher salt solution (≥100 mM NaCl) during the self‐assembly led mainly to polymersomes (Figure 2 f and Figure S26). AF4 measurements further proved that the resulting polymersomes eluted later than micelles (after 20–22 mins) and possessed characteristic shape factor (ρ) values of ca. 1.0, indicative of vesicular structures (Figure 2 c). To further determine the role of salt concentration on the resultant morphology, NaCl was added to a micellar solution of TerP22‐PAIE5 in Milli‐Q water to a final concentration of 200 mM NaCl, followed by 24 h incubation. Cryo‐TEM imaging analysis showed that the micellar nature was not affected at all, which indicated kinetically stable self‐assembly behavior via the direct hydration approach (Figure S27). Self‐assembly of control polymers without PCLgTMC block was found to give micellar nanoparticles in MQ water, while the presence of salt (100 mM NaCl) resulted in micron‐sized particles and visible aggregates, possibly because of the strong interactions between the pyridinium moieties and the electrolyte that disrupted the self‐assembly process (Figure S28). These results indicate the important role of the hydrophobic flexible PCLgTMC block and the presence of salt to enable effective chain packing to generate well‐defined polymersome nanostructures.
Figure 2.

Characterization of the polymersomes from TerP22‐PAIE5 with self‐assembly‐induced emission. a) DLS curve of TerP22‐PAIE5 hydrated in a different salt solution. AF4 fractogram (scattering profile in black) of b) micelles hydrated in Milli‐Q and c) polymersomes hydrated in 100 mM NaCl, comparing the radius of gyration (R g, blue) to the hydrodynamic radius (R h, red). d) Hydrodynamic size determined by DLS measurements of TerP22‐PAIE5 terpolymers hydrated in different concentrations of NaCl. Cryo‐TEM images of e) micelles (in MQ) and f) polymersomes (in 100 mM NaCl) (scale bars=200 nm) (inset: zoom‐in images of the formed micelles and polymersomes, scale bars: 20 nm). g) Fluorescence emission curve of TerP22‐PAIE5 polymersomes in the self‐assembled state (1 % DMSO) and a dissolved state (99 % DMSO), λ ex=405 nm. h) Fluorescence intensity of TerP22‐PAIE5 polymersomes in a mixture of water and DMSO, λ ex=405 nm, inset: TerP22‐PAIE5 polymersome solution in 1 % and 99 % DMSO under the UV‐lamp (25 μg mL−1). i) A typical confocal image of the TerP22‐PAIE5 polymersome solution (25 μg mL−1) (scale bar: 10 μm).
Having confirmed that TerP22‐PAIE5 was a suitable polymer for the controlled formation of polymersomes, its photophysical behavior in the molecularly dissolved and assembled states was next evaluated. The TerP22‐PAIE5 polymer solution in DMSO was not fluorescent at all, while polymers self‐assembled into polymersomes displayed a significantly stronger fluorescence, as clearly observed under the UV‐lamp and via confocal laser scanning microscopy (CLSM) imaging (Figure 2 g, h, and i). When the amount of water in DMSO was 20 % (volume) or more, the fluorescence emission intensity (ca. 650 nm) increased, which coincided with the assembly of the polymers in polymersomes (Figure 2 h and Figure S29), displaying a typical aggregation‐induced emission. Although both PEG and pyridinium blocks induce water solubility, the long hydrophobic chain (i.e. PCLgTMC) effectively drives their self‐assembly in water. This was further confirmed by DLS measurements (Figure S29), To ensure applicability in biomedical research, the stability of AIE‐polymersomes (AIE‐Ps) in PBS was assessed, which was indeed confirmed using DLS (Figure S30a and S30b). Zeta‐potential measurements of AIE‐polymersomes highlighted the positive charge characteristics with values of 16.5±3.1 mV (Figure S30c).
The pyridinium moieties present in the polymersome structure would allow selective interaction with mitochondria.[20] Various mitochondrial drug delivery systems have been functionalized with the pyridinium group, a polar lipophilic cation that enables molecules/nanoparticles to penetrate and accumulate selectively in the phospholipid bilayer of the mitochondria.[20, 24] As central organelles in the cell's energy supply, damaging mitochondria can directly activate the intrinsic mitochondrial pathway of apoptosis, making mitochondria an appealing target site for cancer therapy.
To investigate the targeting capacity of the AIE‐polymersomes toward mitochondria, a time‐dependent co‐localization experiment was performed in a range of cell lines. CLSM imaging revealed that upon incubation with A549 cells for 1 h, the red fluorescence signal from AIE‐polymersomes and the green signal from Mito‐tracker green (MTG) co‐localized well and showed yellow fluorescent spots within mitochondria, demonstrating the specific mitochondria targeting ability of AIE‐polymersomes (Figure 3 a, and Figure S31). Pearson correlation coefficient analysis confirmed the co‐localization of the polymersomes within the mitochondrial compartments over time (Pearson correlation coefficient, r 0.25h=0.41, r 1h=0.73) (Figure S33). There was furthermore little overlap between the AIE‐polymersomes and Lyso‐tracker (Pearson correlation co‐efficiency, r 0.25h=0.13, r 1h=0.17) (Figure S32), and cell membrane mask (Pearson correlation coefficient, r 1h=0.15) (Figure S34).
Figure 3.

Mitochondrial targeting capacity of AIE‐polymersomes (AIE‐Ps) comprising TerP22‐PAIE5. a) Confocal images of cancer cell lines (HepG2, HeLa, and A549 cells) and healthy fibroblast cells (3T3 cells) treated with AIE‐polymersomes (50 μg mL−1) for 1 h. (scale bar: 50 μm, blue: Hoechst (nucleus); green: mitochondria tracker green (MTG); red: AIE‐polymersomes). b) Relative fluorescence intensity analysis of green channel (MTG) and red channel (AIE‐polymersomes) via continuous confocal scanning, the analysis is achieved via image J. c) Confocal images of A549 cells incubated for 1 h with AIE‐polymersomes (50 μg mL−1) obtained after different numbers of scans, green channel: MTG. Red channel: AIE‐polymersomes. Scale bar: 20 μm.
The same behaviors were also observed in HeLa and HepG2 cancer cell lines (Figure 3 a). The main distinction was the higher fluorescence accumulation in cancer cells (>3.5 fold) compared to healthy fibroblast 3T3 cells (Figure S35). The enhanced co‐localization with cancer cell mitochondria was likely because the mitochondria membrane potential (MMP) in cancer cell lines is more negative compared with that in healthy fibroblast cells. To prove this, a mitochondria permeable dye and membrane potential probe JC‐1 assay (5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetraethylbenzimidazolylcarbocyanine iodide) was used. This dye is sensitive to the MMP and displays green emission at low MMP (in normal healthy cells), but red emission at high MMP (in cancer cells).[25] Indeed, cancer cell lines stained with JC‐1 displayed a higher red/green emission ratio (1.5 in HepG2 cells, 2.5 in HeLa cells, 3.2 in A549 cells) than NIH 3T3 cells (0.6) (Figure S36). An interesting additional feature of the AIE polymersomes is their high photo‐stability. Compared to the commercial MTG the red fluorescence of AIE‐polymersomes was more resistant to photo‐bleaching (Figure 3 b and c). The MTG signal was attenuated >80 % after 60 scans with the CLSM, while the red channel retained approximately 70 % of its original value, which is of importance for imaging and tracking in the nano‐biomedical field.
As mentioned above photodynamic therapy (PDT) employs photosensitizers to produce reactive oxygen species when exposed to a localized (NIR) light source, allowing spatiotemporal control of the treatment. In order to make these intrinsically fluorescent AIE polymersomes suitable for PDT, an appropriate photosensitizer had to be installed that was capable of providing FRET with the AIE fluorescent moieties. For this purpose, a BODIPY photosensitizer (termed BPP) was synthesized according to reported procedures and loaded into AIE‐polymersomes via the direct hydration process (Scheme S2, and Figure S37).[26] Loading efficiency of 93±3 % was reached, measured by UV/Vis absorption with λ abs=668 nm (Figure S37). It was subsequently investigated if the photosensitizer could be targeted to the mitochondria using the pyridinium moieties similarly displayed on the polymersomes as the unloaded particles. CLSM imaging revealed that the co‐localization was gradually enhanced as the incubation time increased. Upon incubation for 2 to 8 h, the fluorescence signal from the AIE‐polymersomes (green as false color) and the red signal from the BPP photosensitizer overlapped and showed yellow fluorescent spots (Figure 4 a, and Figure S39). The Pearson correlation analysis also denoted the efficient co‐localization of AIE‐polymersomes and BPP with prolonged incubation times (Pearson correlation coefficient, r 2h=0.57, r 8h=0.76) (Figure 4 c). One thing worth noticing is that the initial fluorescence of AIE‐polymersomes (upon 2 h incubation) was partially quenched by the BPP photosensitizer due to the Förster resonance energy transfer (FRET) taking place in the BPP loaded polymersomes (Figure 5 a, Figure S38). While the AIE fluorescence increased significantly over time (Figure 4 a,b, and Figure S39), and was 11.4 fold higher after 8 h compared to the initial fluorescence emission at 2 h incubation, the BPP photosensitizer fluorescence only showed a 2.6 fold increase after 8 h (Figure 4 b). Such enhanced AIE emission could be ascribed to the increased uptake and accumulation of AIEgenic polymersomes in mitochondria: a consequence of prolonged incubation time. Furthermore, the red signals of the BPP photosensitizer loaded polymersomes coincided well with the green fluorescence of MTG (Pearson's correlation coefficient, r 8h=0.66), owing to the mitochondria targeting ability of the polymersomes (Figure S40). The photosensitizer‐loaded polymersomes furthermore showed a ca. 42 fold higher photosensitizer signal compared to the freely administered photosensitizer, demonstrating that the delivery via the polymersomes is highly efficient and robust (Figure S41).
Figure 4.
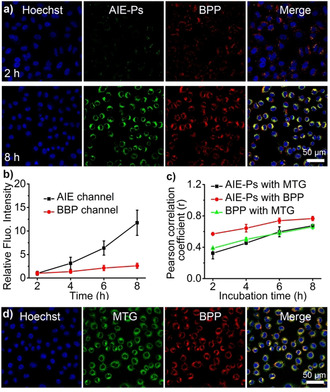
In vitro performance of BPP photosensitizer loaded AIE‐Ps (termed AIE‐BPP). a) Confocal images of A549 cells treated with BPP photosensitizer loaded AIE‐Ps (50 μg mL−1 AIE‐Ps) for 2 h and 8 h. (scale bar: 50 μm, blue: Hoechst; green: AIE‐Ps; red: BPP). b) Relative fluorescence intensity analysis of green channel (AIE‐Ps) and red channel (BPP) in (a) as a function of incubation time; the analysis was performed with image J. c) Pearson's correlation coefficient among AIE‐Ps, MTG, and BPP at different incubation times. d) Confocal images of A549 cells incubated for 8 h with AIE‐BPP (50 μg mL−1 AIE‐Ps). (Blue: Hoechst; green channel: MTG. Red channel: BPP. Scale bar: 50 μm.
Figure 5.

In vitro performance of BPP photosensitizer loaded AIE‐Ps (AIE‐BPP). a) Fluorescence intensity of SOSG assay at 530 nm treated with different groups (PBS, AIE‐Ps, free BPP, and AIE‐BPP) (50 μg mL−1 AIE‐Ps) with or without NIR light irradiation. b) Confocal images of A549 cells treated with AIE‐BPP with or without NIR light irradiation (blue: Hoechst; green: DCFH, scare bar=50 μm). c) Microplate reader analysis of fluorescence intensity in a DCFH‐DA assay of A549 cells treated with different groups (PBS, AIE‐Ps, free BPP, and BPP loaded AIE‐polymersomes) with or without NIR light irradiation. d) Relative A549 cell viability treated with BPP loaded AIE‐polymersomes with or without light irradiation. e) Bright‐field images of A549 cells treated with AIE‐BPP with or without light irradiation, scale bar=100 μm. f) Microplate reader analysis of PI intensity in A549 cells treated with different groups (PBS, AIE‐Ps, free BPP, and AIE‐BPP) with or without NIR light irradiation. g) Confocal images of A549 cells treated with PBS, AIE‐Ps, free BPP, and AIE‐BPP with light irradiation after calcein and PI stain, (green: calcein, red: PI, scale bar: 100 μm). Significance was assessed using one‐way ANOVA, followed by Tukey's multiple comparisons tests (***p<0.001, ****p<0.0001).
Having established that the photosensitizer‐loaded AIE‐polymersomes were effectively internalized and co‐localized with mitochondria, ROS production upon light irradiation was investigated next and evaluated with the indicators Singlet Oxygen Sensor Green (SOSG) and 2, 7‐dichlorodihydrofluorescein diacetate (DCFH‐DA) assays. Under 405 nm light irradiation of BPP photosensitizer loaded AIE‐Ps (absorption peak of AIE‐Ps), a significantly enhanced fluorescence at 530 nm was observed, which indicated abundant 1O2 was generated compared with AIE‐polymersomes and free BPP photosensitizer (Figure S42); this further resulted in enhanced toxicity to cancer cells upon light irradiation (Figure S42). To further extend photosensitizer options with enhanced absorption in the NIR window, two‐photon irradiation (810 nm) using a confocal setup was applied to irradiate the photosensitizer loaded AIE‐polymersomes to activate ROS production. Indeed 1O2 was effectively produced as indicated by the increase of green fluorescence of DCFH‐DA, compared with PBS blank and controls (AIE‐polymersomes, photosensitizer loaded neutral polymersomes), which can be ascribed to the FRET behaviour between AIE‐Ps and photosensitizer to activate the generation of reactive radicals (Figure S43 and S44). To further prove and demonstrate photo‐therapeutic potential of the photosensitizer loaded AIEgenic polymersome with structure inherent targeting capacity, a standard NIR laser (660 nm irradiation) was then used both in vitro and in vivo. Firstly NIR irradiation (660 nm and 100 mW cm−2) of a BPP photosensitizer‐loaded AIE‐polymersome solution containing SOSG for 5 min showed a significantly enhanced fluorescence at 530 nm indicative of abundant ROS production, compared to the control groups including AIE‐polymersomes, free BPP photosensitizer, and PBS with or without NIR light irradiation (Figure 5 a, and Figure S45). Intracellular ROS production was then confirmed in A549 cancer cells via the DCFH‐DA probe. Cancer cells incubated for 6 h with BPP photosensitizer‐loaded AIE‐polymersomes and irradiated with NIR light irradiation for 5 min displayed a significantly stronger green fluorescence, and more efficient ROS production compared to the other groups (Figure 5 b,c and Figure S46). Then the in vitro toxicity of this PDT system was investigated.
The AIE‐polymersomes displayed more toxicity to cancer cell lines, indicated by the lower IC50 in A549 cells (IC50=403 μg mL−1), HepG2 cells (IC50=180 μg mL−1), and HeLa cells (IC50=14 μg mL−1), compared to NIH 3T3 cells (IC50>500 μg mL−1), which could be ascribed to the fact that the polymersomes were more effectively taken up by cancer cells (Figure S47). The photo‐therapeutic efficiency of the BPP photosensitizer‐loaded AIE‐polymersomes was then investigated. With a NIR 660 nm laser (100 mW, 5 min), bubble formation and membrane destabilization were observed (Figure 5 e), which could be ascribed to the 1O2 generation that alters the cellular osmotic pressure, also contributing to killing cancer cells. NIR irradiation lowered the IC50 from 171 μg mL−1 (no irradiation) down to 0.71 μg mL−1 (0.031 μg mL−1 based on the amount of BPP photosensitizer) (Figure 5 d and e). After laser irradiation, a reduction of ca. 95 % in A549 cell viability was seen with BPP photosensitizer‐loaded AIE‐polymersomes at 0.2 μg mL−1 BPP photosensitizer (Figure 5 d), while free BPP photosensitizer under the same conditions only led to a 5 % viability reduction (IC50>50 μg mL−1) (Figure S48). The enhanced cancer cell killing efficiency of photosensitizer‐loaded AIE‐polymersomes with NIR light irradiation could be ascribed to the mitochondria‐targeted effective delivery. PDT‐induced cytotoxicity was further investigated by measuring apoptosis of A549 cells with calcein‐AM as a live stain, alongside propidium iodide (PI) staining dead cells. Under control conditions (cells only, AIE‐polymersomes, and free BPP photosensitizer) with light irradiation, or BPP photosensitizer‐loaded AIE‐polymersomes without light irradiation, no significant cellular apoptosis was observed; however, in the presence of BPP photosensitizer‐loaded AIE‐polymersomes and NIR light, significant cell death was found (Figure 5 f and g). Again, the mitochondria‐targeted photosensitizer‐loaded AIE‐polymersomes displayed enhanced PDT and were most effective (Figure S49). These results suggest that mitochondria‐targeted delivery and targeted photodynamic therapy could greatly improve cancer cell killing efficiency. Neutral polymersomes (from PEG‐PCLgPTMC polymersomes) loaded with the photosensitizer did not cause significant cell death, highlighting the importance of mitochondrial targeting of the AIE‐polymersomes to achieve an effective photo‐therapeutic performance (Figure S50).
To test the PDT efficacy of the BPP photosensitizer‐loaded AIE‐polymersomes in an in vivo model, subcutaneous A549 tumor‐bearing nude mice were used. As shown in Figure S51, the in vivo imaging of photosensitizer fluorescence demonstrated that the photosensitizer loaded in AIE‐polymersomes after intratumoral injection was effectively retained in the tumor site and remained strong up to 96 h, which could be ascribed to the efficient tumor cell uptake and sub‐organelle targeting. The ex vivo fluorescence images of the tumor and major organs (heart, liver, spleen, lung, and kidney) further confirmed that most fluorescence of AIE‐polymersomes and photosensitizer effectively remained in the tumor region up to 96 h, highlighting the tumor retention ability of the targeting delivery system (Figure S52 and S53). For in vivo therapy, A549 tumor‐bearing nude mice were randomly divided into five groups: control PBS, AIE‐polymersomes with light, free photosensitizer with light, photosensitizer‐loaded AIE‐polymersomes without light and photosensitizer‐loaded AIE‐polymersomes with light. Then the different groups of mice were treated with the corresponding agents by intratumoral injection. After 6 h, the groups selected for light irradiation were then irradiated under a standard NIR laser irradiation (660 nm, 0.5 W cm−2) for 5 min. After 24 h, these groups were subjected to another 5 min irradiation. The relative tumor volume was monitored every other day during the following 18 d. It is noteworthy to mention that using light irradiation (660 nm, 0.5 W cm−2 for 5 min) solely results in generation of heat that is insufficient for tumour growth inhibition.[27] Indeed, the mice treated with PBS, AIE‐polymersomes with light, and photosensitizer‐loaded AIE‐polymersomes without light displayed a similar tumor growth rate, which indicated that AIE‐polymersomes themselves in combination with light irradiation is not able to inhibit tumor growth (Figure 6 a). The mice treated with photosensitizer‐loaded AIE‐polymersomes and NIR light demonstrated a more efficient inhibition of tumor growth and reduced tumor volume compared with free photosensitizer with light irradiation (Figure 6 a). The enhanced inhibition efficacy toward tumor growth compared to the free photosensitizer group should be primarily assigned to the improved tumor accumulation and organelle targeting capability of the photosensitizer‐loaded AIE‐polymersomes. After treatment for 18 d, the dissected tumor volume, the weight of the mice, and TdT‐mediated dUTP nick end‐labeling (TUNEL) staining provided further evidence of the efficient in vivo tumor growth inhibition by photosensitizer‐loaded AIE‐polymersomes with NIR light irradiation (Figure 6 b and d). The major organs were collected and analyzed by hematoxylin/eosin (H&E) staining after treatment for 18 d. For the group of photosensitizer‐loaded AIE polymersome with light irradiation, the H&E stained images of major organs showed no obvious inflammation lesions or impairment compared with the control groups (Figure S54), suggesting good biocompatibility and low side effects of photosensitizer‐loaded AIE polymersomes. Moreover, no obvious variations were observed for the mice's body weights for all treatments (Figure 6 c), which indicated that light irradiation, bare AIE‐polymersomes, and the photosensitizer itself displayed an inherent low in vivo toxicity and good biocompatibility. Collectively, these results verified that photosensitizer‐loaded AIE‐polymersomes are effectively taken up by tumor cells and their effective intracellular delivery enables ROS production efficiently, thereby achieving optimal antitumor efficacy in vivo.
Figure 6.

Therapeutic performance of photosensitizer loaded AIE‐polymersomes (AIE‐BPP) in subcutaneous A549 lung tumor‐bearing nude mice. a) The mice treated with BPP photosensitizer‐loaded AIE‐polymersomes (AIE‐BPP) (8 mg kg−1 AIE‐Ps, 0.9 mg kg−1 BPP) displayed a more pronounced antitumor effect compared with the PBS group and other negative controls. b) Tumor weight separated from the mice after the treatment, in which BPP loaded AIE‐polymersomes (AIE‐BPP) group, displayed a minimum tumor weight compared with the other groups. c) After treatment with PBS, the negative controls, and the AIE‐BPP with light irradiation, the mice all still retained comparable body weight. d) Typical TUNEL staining of tumor sections excised from the tumor‐bearing mice following 18 days of treatment with different formulations, apoptotic cells were stained by TUNEL (green) and the nucleus was stained with DAPI (blue), scale bar: 100 μm. Significance was assessed using one‐way ANOVA, followed by Tukey's multiple comparisons test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Conclusion
We applied a simple direct hydration approach to afford a biodegradable, intrinsically fluorescent polymersome system by aggregation‐induced emission. The intrinsic fluorescence allows facile tracking and co‐localization of the polymersomes and their (fluorescent) cargo upon cell uptake. The pyridinium moieties which are incorporated in the polymersome mediate fast cellular integration and active targeted intracellular delivery of the particles toward mitochondria. When loaded with a conventional sensitizer (e.g. BODIPY photosensitizer) their (mitochondria)‐targeted delivery provides these polymersomes with a strongly boosted therapeutic index against cancer cells both in vitro and in vivo, which makes this an exciting system with great potential for biomedical research.
Associated content: The Supporting Information is available free of charge. Included are details of materials, methods, synthesis, and other supporting data utilized in this work.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors would like to acknowledge the ERC Advanced Grant Artisym 694120, the Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035), the NWO‐NSFC Advanced Materials (project 792.001.015), and the European Union's Horizon 2020 research and innovation program Marie Sklodowska‐Curie Innovative Training Networks Nanomed (No. 676137) for funding. We thank the Ser Cymru II program for support of DSW; this project received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement No. 663830. This work was also supported by research funding from the National Natural Science Foundation of China (NSFC 51561135010). We also thank Alex Mason for help with Cryo‐TEM measurements.
S. Cao, Y. Xia, J. Shao, B. Guo, Y. Dong, I. A. B. Pijpers, Z. Zhong, F. Meng, L. K. E. A. Abdelmohsen, D. S. Williams, J. C. M. van Hest, Angew. Chem. Int. Ed. 2021, 60, 17629.
Contributor Information
Prof. Dr. Fenghua Meng, Email: fhmeng@suda.edu.cn.
Dr. Loai K. E. A. Abdelmohsen, Email: l.k.e.a.abdelmohsen@tue.nl.
Dr. David S. Williams, Email: d.s.williams@bristol.ac.uk.
Prof. Dr. Jan C. M. van Hest, Email: J.C.M.v.Hest@tue.nl.
References
- 1.
- 1a.Palivan C. G., Goers R., Najer A., Zhang X. Y., Car A., Meier W., Chem. Soc. Rev. 2016, 45, 377–411; [DOI] [PubMed] [Google Scholar]
- 1b.Zhu Y. Q., Yang B., Chen S., Du J. Z., Prog. Polym. Sci. 2017, 64, 1–22; [Google Scholar]
- 1c.Nishimura T., Sasaki Y., Akiyoshi K., Adv. Mater. 2017, 29, 1702406; [DOI] [PubMed] [Google Scholar]
- 1d.Gu W. X., An J. N., Meng H., Yu N., Zhong Y. A., Meng F. H., Xu Y., Cornelissen J., Zhong Z. Y., Adv. Mater. 2019, 31, 1904742. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a.Discher D. E., Eisenberg A., Science 2002, 297, 967–973; [DOI] [PubMed] [Google Scholar]
- 2b.Mai Y. Y., Eisenberg A., Chem. Soc. Rev. 2012, 41, 5969–5985; [DOI] [PubMed] [Google Scholar]
- 2c.Wang F. Y. K., Xiao J. G., Chen S., Sun H., Yang B., Jiang J. H., Zhou X., Du J. Z., Adv. Mater. 2018, 30, 1705674; [DOI] [PubMed] [Google Scholar]
- 2d.Discher B. M., Won Y. Y., Ege D. S., Lee J. C. M., Bates F. S., Discher D. E., Hammer D. A., Science 1999, 284, 1143–1146. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a.Williams D. S., Pijpers I. A. B., Ridolfo R., van Hest J. C. M., J. Controlled Release 2017, 259, 29–39; [DOI] [PubMed] [Google Scholar]
- 3b.Pijpers I. A. B., Abdelmohsen L. K. E. A., Xia Y., Cao S., Williams D. S., Meng F., van Hest J. C. M., Zhong Z., Adv. Ther. 2018, 1, 1800068; [Google Scholar]
- 3c.Altay Y., Cao S., Che H., Abdelmohsen L. K. E. A., van Hest J. C. M., Biomacromolecules 2019, 20, 4053–4064; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d.Zhao Y., Sakai F., Su L., Liu Y. J., Wei K. C., Chen G. S., Jiang M., Adv. Mater. 2013, 25, 5215–5256; [DOI] [PubMed] [Google Scholar]
- 3e.Jiang Y., Zhang J., Meng F. H., Zhong Z. Y., ACS Nano 2018, 12, 11070–11079. [DOI] [PubMed] [Google Scholar]
- 4.Ghoroghchian P. P., Frail P. R., Susumu K., Blessington D., Brannan A. K., Bates F. S., Chance B., Hammer D. A., Therien M. J., Proc. Natl. Acad. Sci. USA 2005, 102, 2922–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a.Chen M. J., Yin M. Z., Prog. Polym. Sci. 2014, 39, 365–395; [Google Scholar]
- 5b.Wong C. K., Laos A. J., Soeriyadi A. H., Wiedenmann J., Curmi P. M. G., Gooding J. J., Marquis C. P., Stenzel M. H., Thordarson P., Angew. Chem. Int. Ed. 2015, 54, 5317–5322; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5407–5412. [Google Scholar]
- 6.
- 6a.Hu F., Xu S. D., Liu B., Adv. Mater. 2018, 30, 1801350; [DOI] [PubMed] [Google Scholar]
- 6b.Hong Y. N., Lam J. W. Y., Tang B. Z., Chem. Soc. Rev. 2011, 40, 5361–5388. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a.Cai X. L., Liu B., Angew. Chem. Int. Ed. 2020, 59, 9868–9886; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 9952–9970; [Google Scholar]
- 7b.Mei J., Hong Y. N., Lam J. W. Y., Qin A. J., Tang Y. H., Tang B. Z., Adv. Mater. 2014, 26, 5429–5479; [DOI] [PubMed] [Google Scholar]
- 7c.Wang S. W., Wu W. B., Manghnani P., Xu S. D., Wang Y. B., Goh C. C., Ng L. G., Liu B., ACS Nano 2019, 13, 3095–3105. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a.Wang H., Zhao E. G., Lam J. W. Y., Tang B. Z., Mater. Today 2015, 18, 365–377; [Google Scholar]
- 8b.Mei J., Leung N. L. C., Kwok R. T. K., Lam J. W. Y., Tang B. Z., Chem. Rev. 2015, 115, 11718–11940. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a.Zhang N., Chen H., Fan Y., Zhou L., Trepout S., Guo J., Li M. H., ACS Nano 2018, 12, 4025–4035; [DOI] [PubMed] [Google Scholar]
- 9b.Hu R., Leung N. L. C., Tang B. Z., Chem. Soc. Rev. 2014, 43, 4494–4562; [DOI] [PubMed] [Google Scholar]
- 9c.Zhang D. P., Fan Y. J., Chen H., Trepout S., Li M. H., Angew. Chem. Int. Ed. 2019, 58, 10260–10265; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10366–10371; [Google Scholar]
- 9d.Cao S., Shao J., Wu H., Song S., De Martino M. T., Pijpers I. A. B., Friedrich H., Abdelmohsen L. K. E. A., Williams D. S., van Hest J. C. M., Nat. Commun. 2021, 12, 2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a.Lucky S. S., Soo K. C., Zhang Y., Chem. Rev. 2015, 115, 1990–2042; [DOI] [PubMed] [Google Scholar]
- 10b.Elsabahy M., Heo G. S., Lim S. M., Sun G. R., Wooley K. L., Chem. Rev. 2015, 115, 10967–11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Z. J., Song J. B., Nie L. M., Chen X. Y., Chem. Soc. Rev. 2016, 45, 6597–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang B. W., Chen Y., Shi J. L., Chem. Rev. 2019, 119, 4881–4985. [DOI] [PubMed] [Google Scholar]
- 13.Huang L., Li Z. J., Zhao Y., Yang J. Y., Yang Y. C., Pendharkar A. I., Zhang Y. W., Kelmar S., Chen L. Y., Wu W. T., Zhao J. Z., Han G., Adv. Mater. 2017, 29, 1604789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J. S., Feijen J., J. Controlled Release 2012, 161, 473–483. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a.Li M. L., Long S. R., Kang Y., Guo L. Y., Wang J. Y., Fan J. L., Du J. J., Peng X. J., J. Am. Chem. Soc. 2018, 140, 15820–15826; [DOI] [PubMed] [Google Scholar]
- 15b.Hyun H., Park M. H., Owens E. A., Wada H., Henary M., Handgraaf H. J. M., Vahrmeijer A. L., Frangioni J. V., Choi H. S., Nat. Med. 2015, 21, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W. H., Luo G. F., Zhang X. Z., Adv. Mater. 2019, 31, 1802725. [Google Scholar]
- 17.
- 17a.Tempelaar S., Mespouille L., Coulembier O., Dubois P., Dove A. P., Chem. Soc. Rev. 2013, 42, 1312–1336; [DOI] [PubMed] [Google Scholar]
- 17b.Chen W., Meng F. H., Cheng R., Deng C., Feijen J., Zhong Z. Y., J. Controlled Release 2014, 190, 398–414. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a.Mespouille L., Coulembier O., Kawalec M., Dove A. P., Dubois P., Prog. Polym. Sci. 2014, 39, 1144–1164; [Google Scholar]
- 18b.Deng C., Jiang Y. J., Cheng R., Meng F. H., Zhong Z. Y., Nano Today 2012, 7, 467–480; [Google Scholar]
- 18c.Nederberg F., Zhang Y., Tan J. P. K., Xu K. J., Wang H. Y., Yang C., Gao S. J., Guo X. D., Fukushima K., Li L. J., Hedrick J. L., Yang Y. Y., Nat. Chem. 2011, 3, 409–414; [DOI] [PubMed] [Google Scholar]
- 18d.Wang M. L., Zhai Y. L., Ye H., Lv Q. Z., Sun B. J., Luo C., Jiang Q. K., Zhang H. T., Xu Y. J., Jing Y. K., Huang L., Sun J., He Z. G., ACS Nano 2019, 13, 7010–7023. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a.Cao S., Shao J., Xia Y., Che H., Zhong Z., Meng F., van Hest J. C. M., Abdelmohsen L. K. E. A., Williams D. S., Small 2019, 15, 1901849; [DOI] [PubMed] [Google Scholar]
- 19b.van Oppen L., Abdelmohsen L. K. E. A., van Emst-de Vries S. E., Welzen P. L. W., Wilson D. A., Smeitink J. A. M., Koopman W. J. H., Brock R., Willems P., Williams D. S., van Hest J. C. M., ACS Cent. Sci. 2018, 4, 917–928; [Google Scholar]
- 19c.Su L., Li R. C., Khan S., Clanton R., Zhang F. W., Lin Y. N., Song Y., Wang H., Fan J. W., Hernandez S., Butters A. S., Akabani G., MacLoughlin R., Smolen J., Wooley K. L., J. Am. Chem. Soc. 2018, 140, 1438–1446. [DOI] [PubMed] [Google Scholar]
- 20.Hu F., Cai X. L., Manghnani P. N., Kenry, Wu W. B., Liu B., Chem. Sci. 2018, 9, 2756–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S. Q., Yang C., Huang Y., Ding X., Li Y., Fan W. M., Hedrick J. L., Yang Y. Y., Adv. Mater. 2012, 24, 6484–6489. [DOI] [PubMed] [Google Scholar]
- 22.Wang S. W., Hu F., Pan Y. T., Ng L. G., Liu B., Adv. Funct. Mater. 2019, 29, 1902717. [Google Scholar]
- 23.O'Neil C. P., Suzuki T., Demurtas D., Finka A., Hubbell J. A., Langmuir 2009, 25, 9025–9029. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a.Yamada Y., Satrialdi, Hibino M., Sasaki D., Abe J., Harashima H., Adv. Drug Delivery Rev. 2020, 154, 187–209; [DOI] [PubMed] [Google Scholar]
- 24b.Huang Y. Y., Zhang G. X., Zhao R., Zhang D. Q., ChemMedChem 2020, 15, 2220–2227. [DOI] [PubMed] [Google Scholar]
- 25.Zhang T. F., Li Y. Y., Zheng Z., Ye R. Q., Zhang Y. R., Kwok R. T. K., Lam J. W. Y., Tang B. Z., J. Am. Chem. Soc. 2019, 141, 5612–5616. [DOI] [PubMed] [Google Scholar]
- 26.Jung H. S., Han J., Shi H., Koo S., Singh H., Kim H. J., Sessler J. L., Lee J. Y., Kim J. H., Kim J. S., J. Am. Chem. Soc. 2017, 139, 7595–7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a.Guo Z. Q., Zou Y. L., He H., Rao J. M., Ji S. S., Cui X. N., Ke H. T., Deng Y. B., Yang H., Chen C. Y., Zhao Y. L., Chen H. B., Adv. Mater. 2016, 28, 10155–10164; [DOI] [PubMed] [Google Scholar]
- 27b.Ye S. Y., Rao J. M., Qiu S. H., Zhao J. L., He H., Yan Z. L., Yang T., Deng Y. B., Ke H. T., Yang H., Zhao Y. L., Guo Z. Q., Chen H. B., Adv. Mater. 2018, 30, 1801216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information