

Abstract
The first liquid‐phase synthesis of high‐quality, small‐sized rare‐earth metal nanoparticles (1–3 nm)—ranging from lanthanum as one of the largest (187 pm) to scandium as the smallest (161 pm) rare‐earth metal—is shown. Size, oxidation state, and reactivity of the nanoparticles are examined (e.g., electron microscopy, electron spectroscopy, X‐ray absorption spectroscopy, selected reactions). Whereas the nanoparticles are highly reactive (e.g. in contact to air and water), they are chemically stable as THF suspensions and powders under inert conditions. The reactivity can be controlled to obtain inorganic and metal–organic compounds at room temperature.
Keywords: follow-up reactions, liquid-phase synthesis, nanoparticles, rare-earth metals
Reactive but easy to handle: For the first time, rare‐earth metals (La, Ce, Sm, Eu, Tm, Y, Sc) are obtained as high‐quality nanoparticles (1–3 nm in size) in the liquid phase. They can be highly interesting as reactive starting materials to realize new rare‐earth metal compounds and/or phase‐pure rare‐earth‐metal‐based materials.

Rare‐earth metals are highly reactive, in principle.[1] Performing reactions with rare‐earth metals in a controlled manner is nevertheless difficult. On the one hand, the surface area of the bulk metals is small and crushing them into pieces is difficult due to their hardness and sensitivity. On the other hand, the surface of rare‐earth metals is usually passivated by hydroxides, oxides, and/or carbonates, which kinetically inhibit reactions at low temperature. In chemical syntheses, these passivation layers naturally also cause impurities. However, a purification of rare‐earth metals is elaborate and requires high‐temperature distillation (>800 °C), special equipment (e.g. steel cylinders), and profound experience.[2] In contrast to alkali metals and alkali earth metals, rare‐earth metals also show only low solubility in liquid ammonia.[3] As a result, reactions with zerovalent rare‐earth metals are often limited to high‐temperature solid‐state reactions (>500 °C).[1, 4]
The availability of rare‐earth metals in the form of small‐sized nanoparticles in suspension or as powder sample could significantly change the aforementioned situation and allow an easy handling with reactions of rare‐earth metals near room temperature (≤100 °C) and/or directly in the liquid phase. A liquid‐phase synthesis of rare‐earth metal nanoparticles, however, is largely unknown. This can be ascribed to the reactivity of such nanoparticles, which needs to be expected as considerably higher than for the respective bulk rare‐earth metals. So far, few reports address physical methods to obtain rare‐earth metal particles (e.g., γ‐radiation, laser ablation, plasma treatment, electrodeposition),[5] which, however, result in heavily agglomerated particles with broad size distribution and significant oxygen contamination. Some reports claim aqueous syntheses or even biosyntheses,[6] which seems hardly traceable when considering the position of the rare‐earth metals in the voltage series with electrochemical potentials of about −2.4 V.[7] Our experience is the observation of violent reactions with explosion if the rare‐earth metal nanoparticles come into contact with water.
The dilemma of the high reactivity can be exemplarily illustrated for Gd0 nanoparticles. Here, the only reliable liquid‐phase synthesis relates to the reduction of GdCl3 with crown‐ether‐stabilized alkalides (e.g. [K(18‐crown‐6]+[K]−) as powerful reducing agents.[8] Due to the high reactivity of the Gd0 nanoparticles, the authors could only identify Gd2O3 nanoparticles via electron microscopy. Recently, they could validate the formation of Gd0 for Gd@Au core–shell nanoparticles with a dense gold shell to protect the Gd0 core from oxidation.[8a] With La0, Ce0, Sm0, Eu0, Tm0, Y0, and Sc0, we now present a reliable liquid‐phase synthesis of high‐quality, small‐sized rare‐earth metal nanoparticles ranging from lanthanum as a large (187 pm) to scandium as the smallest (161 pm) rare‐earth metal for the first time (Figure 1 a).[9]
Figure 1.
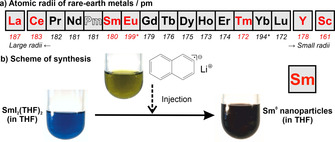
Rare‐earth metals: a) Row of metals with atomic radii (red‐marked metals prepared here, *aperiodic radii due to half‐filled f 7 and filled f 14 electron shell). b) Scheme of the exemplary liquid‐phase synthesis of Sm0 nanoparticles.
Several prerequisites need to be addressed to obtain high‐quality nanoparticles of such highly reactive metals. This includes a powerful reducing agent as well as a sample handling under strict inert conditions, starting from synthesis to all analytical characterization. Based on our experience with base metal nanoparticles (e.g., Ti0, Mo0, W0, Re0, Fe0, Zn0) and different synthesis strategies (e.g., microemulsions with liquid ammonia, sodium‐driven reduction in liquid ammonia, lithium/sodium naphthalenide‐driven reduction in tetrahydrofuran, lithium/sodium pyridinyl‐driven reduction in pyridine),[10] we have selected lithium naphthalenide ([LiNaph]) as the reducing agent of choice to obtain the even more reactive rare‐earth metals. Simple metal halides were used as low‐cost, well‐soluble starting materials (see SI). All syntheses were performed in Schlenk‐type vessels; sample handling and centrifugation were carried out in gloveboxes; suitable transfer systems were used for all analytical characterization (SI: Figure S1).
The synthesis strategy is exemplarily illustrated for Sm0 nanoparticles (Figure 1 b). Accordingly, SmI2(THF)2 was dissolved in tetrahydrofuran (THF). Moreover, lithium and naphthalene were dissolved in THF. Thereafter, the resulting [LiNaph] solution was injected with intense stirring into the SmI2(THF)2 solution. The reduction and nucleation of Sm0 nanoparticles are indicated by the immediate formation of a deep black suspension after injection of [LiNaph]. Electron paramagnetic resonance (EPR) spectra confirm the completeness of the reaction (SI: Table S1, Figure S2). Thus, the ESR signal of the paramagnetic [LiNaph] vanishes completely upon reduction of SmI2(THF)2. Subsequent to the reaction, the Sm0 nanoparticles were separated by high‐power centrifugation (25.000 rpm, 55.200×g), purified by resuspension/centrifugation in/from THF or toluene. The as‐prepared nanoparticles can be easily redispersed in THF or toluene, or they can be dried in vacuum at room temperature to obtain powder samples.
Size, shape, and size distribution of the as‐prepared Sm0 nanoparticles were evaluated by transmission electron microscopy (TEM) (Figure 2). Overview TEM images show uniform, non‐agglomerated nanoparticles with narrow size distribution (Figure 2 a,b). A statistical evaluation of 250 nanoparticles on TEM images results in a mean diameter of 1.7±0.2 nm (Figure 2 c). High‐resolution (HR)TEM images confirm a size of 1–3 nm (Figure 2 d). Moreover, HRTEM indicates the crystallinity of the as‐prepared Sm0 nanoparticles and shows highly parallel lattice fringes (Figure 2 d). The observed lattice fringe distance of 1.9 Å is well compatibel with hexagonal β‐Sm0 (d105 with 1.9 Å).[11] The presence of zerovalent Sm is further confirmed by Fourier transformation (FT) analysis of HRTEM images of single particles (Figure 2 e), which agrees with the calculated diffraction pattern of hexagonal bulk‐Sm0 (space group: P63/mmc, a=3.682, c=11.043 Å) in the [‐551] zone axis.[11] We also observed few particles with disordered structure (Figure 2 b), which could be caused by electron‐beam‐induced knock‐on damage. The formation of very small Sm0 nanoparticles might be surprising at first sight, but is well in agreement with the LaMer‐Dinegar model of particle nucleation and growth.[12] Thus, the injection of [LiNaph] initiates a fast reduction and formation of metal nanoparticles, which are highly insoluble in THF. The resulting high oversaturation promotes a fast nucleation and, therefore, very small particles.
Figure 2.
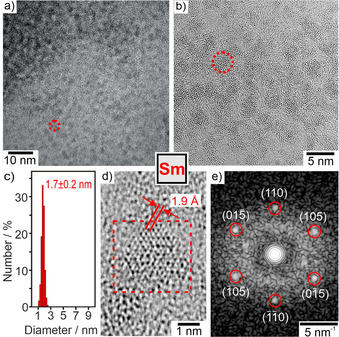
Electron microscopy of the as‐prepared Sm0 with a,b) TEM images at different levels of magnification (red dots indicate single nanoparticles), c) size distribution (based on statistical evaluation of 250 nanoparticles on TEM images), d) HRTEM image with lattice fringes, e) FT‐HRTEM image in (c) with reflection positions of calculated diffraction pattern in [‐551] zone axis indicated by red circles.
In addition to HRTEM and FFT analysis, the oxidation state of the as‐prepared Sm0 nanoparticles was validated by X‐ray absorption near edge structure (XANES) spectroscopy, electron energy loss spectroscopy (EELS), and energy‐dispersive X‐ray spectroscopy (EDXS). Sm‐XANES L3 ‐edge spectra of the as‐prepared nanoparticles were compared to bulk samarium, SmI2(THF)2, and literature data of Sm0 clusters isolated in argon matrices (Figure 3, SI: Figure S3,S4). Particularly striking is the high intensity pre‐edge peak of SmI2(THF)2 in the L3 ‐edge spectra, which can be ascribed to a 2p 6…4f 6 6s0 → 2p 5…4f 7 6s0 transition on Sm+II (Figure 3 a). This pre‐edge peak is totally missing for the nanoparticles. In comparison to bulk Sm0, however, the Sm0 nanoparticles exhibit a double‐peak structure (6708, 6716 eV). This double‐peak structure was similarly reported for Ar‐matrix‐isolated samarium clusters and ascribed to a different valence electron configuration of volume and surface Sm atoms (Figure 3 b).[13] Thus, Sm atoms in the cluster volume exhibit a 4f 5(5d 6s)3 configuration, whereas Sm atoms located at the cluster surface have a 4f 6 6s2 configuration with a shift of 8 eV between the two peaks (Figure 3 a,b).[13] The intensity of both peaks depends on the size of the cluster. For the as‐prepared Sm0 nanoparticles with an average size of 1.7±0.2 nm about 40 % of the Sm atoms are located at the surface, so that a significant contribution of surface atoms in the XANES spectra is not a surprise. Taken together, XANES spectra prove the presence of small‐sized, zerovalent Sm nanoparticles, which was also confirmed by EELS and EDXS (SI: Figure S5,S6).
Figure 3.
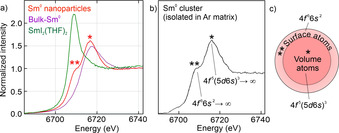
XANES spectra of the as‐prepared Sm0 nanoparticles: a) Sm‐L3 spectra with bulk metal and SmI2(THF)2 as references. b) Comparison with Ar‐matrix‐isolated Sm clusters.[13a] c) Scheme of Sm0 nanoparticle with different electron configuration of volume atoms (indicated by *) and surface atoms (indicated by **) according to literature data.[13]
Similar to Sm0, size, size distribution, and crystallinity of the as‐prepared La0, Ce0, Eu0, Tm0, Y0, and Sc0 nanoparticles were also examined by TEM (Figure 4, SI: Figure S7–S12). Overview TEM images again show uniform, non‐agglomerated nanoparticles with predominantly spherical shape (Figure 4 a–f) and mean diameters of 2–3 nm at very narrow size distribution (±0.3 nm) (Figure 5). HRTEM images prove the crystallinity of the as‐prepared nanoparticles and show highly parallel lattice fringes (Figure 4 a–f). The observed lattice fringe distances are well compatible with bulk metals (La0: d 01‐1 with 3.265 Å, Ce0: d 1‐1‐1 with 3.071 Å, Eu0: d002 with 2.695 Å, Tm0: d 11‐1 with 2.693 Å, Y0: d 1‐1‐1 with 2.785 Å, Sc0: d 1‐1‐1 with 2.51 Å).[14] Crystallinity and crystal structure of the as‐prepared rare‐earth metal nanoparticles were also confirmed by FT analysis (SI: Figure S7–S12). The crystallographic data are not only consistent with the bulk metals, they also differ clearly from the corresponding metal oxides (SI: Table S2).
Figure 4.

Electron microscopy of the as‐prepared La0, Ce0, Eu0, Tm0, Y0, Sc0 with overview TEM images (red dots indicate single nanoparticles) and HRTEM images with lattice fringes, see SI for more data: Figure S7–S12).
Figure 5.
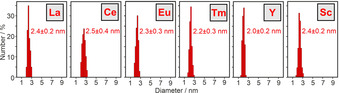
Size distribution of the as‐prepared La0, Ce0, Eu0, Tm0, Y0, and Sc0 nanoparticles (statistical evaluation of 250 nanoparticles on TEM images).
With La0 as a large and Sc0 as the smallest rare‐earth metal, the synthesis strategy is suitable to obtain all rare‐earth metals as nanoparticles in the liquid phase. Having high‐quality nanoparticles as a new form of rare‐earth metals available for the first time, we also got a first glimpse on their reactivity and their use as starting materials in chemical synthesis. Under inert conditions (argon, nitrogen, vacuum), the as‐prepared suspensions (THF, toluene) and powder samples are chemically very stable. Attention must be paid if the nanoparticles get into contact with oxidizing agents. Again, we take the Sm0 nanoparticles as example. Here, direct contact of a powder sample (≈20 mg) to air causes an immediate reaction with sting flame (Figure 6 a).
Figure 6.

Reactivity of the as‐prepared Sm0 nanoparticles: a) uncontrolled, violent reaction in air (20 mg Sm0 powder), b) controlled reactions with NH3, N2H4, S8, I2, EtOH, HCp at 25 °C in the liquid phase (Sm0 suspensions in THF).
Beside the uncontrolled reaction of the as‐prepared Sm0 powders in air, we have exemplarily reacted the Sm0 nanoparticles with NH3, N2H4, S8, I2, ethanol (EtOH), and cyclopentadiene (HCp) at room temperature (25 °C) in the liquid phase (Figure 6 b, SI: Figure S13–S26). The respective reactions are already indicated by the change of the deep black Sm0 suspension to colorless, yellowish or red solutions/suspensions and result in different inorganic and metal‐organic ocmpounds, including Sm(NH2)3(THF), Sm(N2H3)3(THF), SmS1.8, SmI3(THF)3, Sm(OEt)3, and Sm(Cp)3 (SI: Figure S14–S26). After separation from the liquid‐phase, Sm(NH2)3(THF) and SmS1.8 can be thermally converted to SmN and Sm2S3 (SI: Figure S15,S16,S19,S20). Depending on the respective reaction and compound, nanoparticles (SmS1.8, SI: Figure S18) or even single crystals (SmI3(THF)3, Sm(Cp)3, SI: Table S3, Figure S21,S22,S25) can be obtained. Moreover, Sm(N2H3)3(THF) and SmI3(THF)3 are already new compounds (SI: Figure S17,S22). The reaction of the Sm0 nanoparticles with N2H4 resulted in an unusual metal hydrazide, which is even more reactive than the intial Sm0 (see SI). Such reaction was yet only reported for alkali metals[15] and points to the reactivity of the rare‐earth metal nanoparticles. Even more interesting than each of the aforementioned follow‐up reactions, however, might be the general option to use the as‐prepared rare‐earth metal nanoparticles in a controlled manner as starting materials in chemical syntheses near room temperature (≤100 °C) and/or instantaneously in the liquid phase.
For all kinds of liquid‐phase syntheses, it is a matter of course that the as‐prepared nanoparticles contain at least the solvent as surface capping. In this regard, FT‐IR spectra show THF to be adsorbed on the surface of the as‐prepared nanoparticles (SI: Figure S13). Taking the high reactivity of the rare‐earth metal nanoparticles into account, however, it is a surprise as such that the nanoparticles are chemically stable in suspension. Moreover, a small‐sized, low‐weight molecule like THF as the surface capping is beneficial for follow‐up reactions as it can be easily exchanged by other, stronger‐binding ligands, which then can be chosen independent from the original nanoparticle synthesis. Our exemplary reactions already indicate products that do not contain THF (e.g., SmS1.8, Sm(OEt)3, Sm(Cp)3).
In sum, a reliable liquid‐phase synthesis of rare‐earth metal nanoparticles is shown for the first time with La0, Ce0, Sm0, Eu0, Tm0, Y0, and Sc0 as examples. The synthesis uses metal halides as low‐cost starting materials, THF as conventional solvent, and [LiNaph] as a powerful, easy‐to‐handle reducing agent. The as‐prepared rare‐earth metal nanoparticles exhibit very small sizes (1–3 nm) and narrow size distributions (±0.3 nm). They are monocrystalline and well‐dispersible in THF or toluene. Whereas the rare‐earth metal nanoparticles are chemically stable under inert conditions, they show heavy reactivity, for instance, in contact to air. However, the reactivity can be well‐controlled, so that the rare‐earth metal nanoparticles can be exemplarily reacted with NH3, N2H4, S8, I2, EtOH and HCp to obtain different inorganic and metal‐organic compounds at room temperature and instantaneously in the liquid phase. Having the first liquid‐phase synthesis of rare‐earth metal nanoparticles established, these nanoparticles might become most interesting as starting materials to prepare new and/or high‐purity rare‐earth metal compounds as well as rare‐earth‐metal‐based materials.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
C.F. and D.G. are grateful to the Deutsche Forschungsgemeinschaft (DFG) for funding of personnel (FE911/11‐1, GE 841/29‐1: “NanoMet”) and TEM equipment (INST 121384/33‐1 FUGG). C.F. and J.D.G. also acknowledge the DFG Collaborative Research Centre on catalysis (CRC1441: “TrackAct”). Open access funding enabled and organized by Projekt DEAL.
D. Bartenbach, O. Wenzel, R. Popescu, L.-P. Faden, A. Reiß, M. Kaiser, A. Zimina, J.-D. Grunwaldt, D. Gerthsen, C. Feldmann, Angew. Chem. Int. Ed. 2021, 60, 17373.
Dedicated to Professor Hansgeorg Schnöckel on the occasion of his 80th birthday
References
- 1.Atwood D. A., The Rare Earth Elements: Fundamentals and Applications, Wiley-VCH, Weinheim, 2012. [Google Scholar]
- 2.Brauer G., Handbuch der präparativen Anorganischen Chemie, Vol. 2, Enke, Stuttgart, 1978, pp. 1071. [Google Scholar]
- 3.
- 3a.Quitmann C. C., Mueller-Buschbaum K., Angew. Chem. Int. Ed. 2004, 43, 5994; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 6120; [Google Scholar]
- 3b.Fischer E. O., Fischer H., Angew. Chem. Int. Ed. Engl. 1964, 3, 132; [Google Scholar]; Angew. Chem. 1964, 76, 52. [Google Scholar]
- 4.
- 4a.Cloke F. G. N., Chem. Soc. Rev. 1993, 22, 17; [Google Scholar]
- 4b.Simon A., Pure Appl. Chem. 1995, 67, 311. [Google Scholar]
- 5.
- 5a.Urumese A., Jenjeti R. N., Sampath S., Jagirda B. R., J. Colloid Interface Sci. 2016, 476, 177; [DOI] [PubMed] [Google Scholar]
- 5b.Petinov V. I., Russ. J. Phys. Chem. A 2016, 90, 1413; [Google Scholar]
- 5c.Xu Q., Hu S., Wang W., Wang Y., Ju H., Zhu X., Appl. Surf. Sci. B 2018, 432, 115; [Google Scholar]
- 5d.Ertas Y. N., Jarenwattananon N. N., Bouchard L.-S., Chem. Mater. 2015, 27, 5371; [Google Scholar]
- 5e.Chall S., Saha A., Biswas S. K., Datta A., Bhattacharya S. C., J. Mater. Chem. 2012, 22, 12538; [Google Scholar]
- 5f.Tarasenko N. V., Buthsen A. V., Nevar A. A., Appl. Phys. A 2008, 93, 837; [Google Scholar]
- 5g.Aruna I., Metha B. R., Malhotra L. K., Shivaprasad S. M., Adv. Funct. Mater. 2005, 15, 131; [Google Scholar]
- 5h.Yan Z. C., Huang Y. H., Zhang Y., Okumara H., Xiao J. Q., Stoyanov S., Skumryev V., Hadjipanayis G. C., Phys. Rev. B 2003, 67, 054403. [Google Scholar]
- 6.
- 6a.Manoj Kumar K. A., Hemananthan E., Devi P. R., Kumar S. V., Hariharan R., Mater. Today 2017, 21, 887; [Google Scholar]
- 6b.Chatterjee A., Archana L., Niroshinee V., Abraham J., Res. J. Pharmaceut. Biol. Chem. Sci. 2016, 7, 1462; [Google Scholar]
- 6c.Sankar V., SalinRaj P., Athira R., Sreenivasan Soumya R., Gopalan Raghu K., RSC Adv. 2015, 5, 21074; [Google Scholar]
- 6d.Ascencio J. A., Rincon A. C., Canizal G., J. Phys. Chem. B 2005, 109, 8806. [DOI] [PubMed] [Google Scholar]
- 7.Holleman A. F., Wiberg E., Anorganische Chemie, Vol. 1, 103. ed., de Gruyter, Berlin, 2017. Annex III/IV. [Google Scholar]
- 8.
- 8a.Yan C., Wagner M. J., Nano Lett. 2013, 13, 2611; [DOI] [PubMed] [Google Scholar]
- 8b.Nelson J. A., Bennet L. H., Wagner J. J., J. Am. Chem. Soc. 2002, 124, 2979. [DOI] [PubMed] [Google Scholar]
- 9.http://www.webelements.com (scandium and lanthanum).
- 10.
- 10a.Schöttle C., Bockstaller P., Gerthsen D., Feldmann C., Chem. Commun. 2014, 50, 4547; [DOI] [PubMed] [Google Scholar]
- 10b.Gyger F., Bockstaller P., Gerthsen D., Feldmann C., Angew. Chem. Int. Ed. 2013, 52, 12443; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12671; [Google Scholar]
- 10c.Schöttle C., Bockstaller P., Popescu R., Gerthsen D., Feldmann C., Angew. Chem. Int. Ed. 2015, 54, 9866; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10004; [Google Scholar]
- 10d.Egeberg A., Block T., Janka O., Wenzel O., Gerthsen D., Pöttgen R., Feldmann C., Small 2019, 15, 1902321. [DOI] [PubMed] [Google Scholar]
- 11.Shi N. L., Fort D., J. Less-Common Met. 1985, 113, 21. [Google Scholar]
- 12.LaMer V. K., Dinegar R. H. J., J. Am. Chem. Soc. 1950, 72, 4847. [Google Scholar]
- 13.
- 13a.Lübcke M., Sonntag B., Niemann W., Rabe P., Phys. Rev. B 1986, 34, 5184; [DOI] [PubMed] [Google Scholar]
- 13b.Mason M. G., Lee S. T., Apai G., Davies R. F., Shirley D. A., Franciosi A., Weaver J. A., Phys. Rev. Lett. 1981, 47, 730. [Google Scholar]
- 14.
- 14a.Spedding F. H., Hanak J. J., Daane A. H., J. Less-Common Met. 1961, 3, 110; [Google Scholar]
- 14b.Gschneidner K. A., Elliott R. O., McDonald R. R., J. Phys. Chem. Solids 1962, 23, 555; [Google Scholar]
- 14c.Demchyna R. O., Chykhrij S. I., Kuz'ma Y. B., J. Alloys Compd. 2002, 345, 170; [Google Scholar]
- 14d.Hájek B., Brožek V., Duvigneaud P. H., J. Less-Common Met. 1973, 33, 385. [Google Scholar]
- 15.Goubeau J., Kull U., Z. Anorg. Allg. Chem. 1962, 316, 182. [Google Scholar]
- 16.Evans W. J., Gummersheimer T. S., Ziller J. W., J. Am. Chem. Soc. 1995, 117, 8999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information