

Abstract
BACKGROUND:
Identifying the mechanistic pathways potentially associated with incident heart failure (HF) may provide a basis for novel preventive strategies.
METHODS AND RESULTS:
To identify proteomic biomarkers and the potential underlying mechanistic pathways that may be associated with incident HF defined as the first hospitalization for HF, a nested-matched case-control design was used with cases (incident HF) and controls (without HF) selected from 3 cohorts (>20 000 individuals). Controls were matched on cohort, follow-up time, age, and sex. Two independent sample sets (a discovery set with 286 cases and 591 controls and a replication set with 276 cases and 280 controls) were used to discover and replicate the findings. Two hundred fifty-two circulating proteins in the plasma were studied. Adjusting for the matching variables age, sex, and follow-up time (and correcting for multiplicity of tests), 89 proteins were found to be associated with incident HF in the discovery phase, of which 38 were also associated with incident HF in the replication phase. These 38 proteins pointed to 4 main network clusters underlying incident HF: (1) inflammation and apoptosis, indicated by the expression of the TNF (tumor necrosis factor)-family members; (2) extracellular matrix remodeling, angiogenesis and growth, indicated by the expression of proteins associated with collagen metabolism, endothelial function, and vascular homeostasis; (3) blood pressure regulation, indicated by the expression of natriuretic peptides and proteins related to the renin-angiotensin-aldosterone system; and (4) metabolism, associated with cholesterol and atherosclerosis.
CONCLUSIONS:
Clusters of biomarkers associated with mechanistic pathways leading to HF were identified linking inflammation, apoptosis, vascular function, matrix remodeling, blood pressure control, and metabolism. These findings provide important insight on the pathophysiological mechanisms leading to HF.
CLINICAL TRIAL REGISTRATION:
URL: https://www.clinicaltrials.gov. Unique identifier: NCT02556450.
Keywords: apoptosis, atherosclerosis, blood pressure, heart failure, proteomics
Heart failure (HF) is a major cause of morbidity and mortality worldwide and the most frequent cause of hospitalization for patients over 65 years of age.1–3 The incidence and prevalence of HF are increasing because of the aging of the population as well as rising rates of HF risk factors such as diabetes mellitus, obesity, and hypertension.3–6 Identifying mechanistic pathways leading to HF may help improve preventive strategies.7
In the last decade, circulating biomarkers, such as NT-proBNP (N-terminal pro-B-type natriuretic peptide), have been studied for prediction of incident HF.7–10 A recently published study,10 also investigated the association of multiple proteins with incident HF for prediction purposes; these proteins (n=80) added little gain to the prognostic model, including natriuretic peptides. However, above and beyond prediction, biomarkers may reflect pathophysiological processes and thus may help in assessing the underlying pathways that contribute to the progression towards HF. Investigating the pathophysiological processes may provide potential targets for future therapies. For this purpose, knowledge-based network analysis with induced network approach may help identify the links among the identified protein bio-markers, providing the basis for the identification of the underlying pathways leading to HF.11
The HOMAGE (Heart Omics in Ageing consortium; URL: https://www.clinicaltrials.gov. Unique identifier: NCT02556450) is an EU funded program that aims to identify and validate omics biomarkers associated with incident HF to potentially develop new and personalized preventive strategies. We report proteomics results, based on assays of 252 plasma proteins related to cardiovascular disease (CVD) and inflammation, testing the associations of these proteins with incident HF, applying knowledge-based network analysis to identify mechanistic pathways underlying the progression to HF.
METHODS
Study Population
The HOMAGE consortium included 20 completed and ongoing studies conducted in 8 European countries that enrolled healthy subjects, patients with HF and patients at high risk of CVD, all of which were pooled in a common database.12 From the HOMAGE population with >20 000 patients, we identified cohorts in whom individuals had been followed-up until first hospitalization for HF. Patients from 2 suitable cohorts and one clinical trial population were identified: PREDICTOR,13 HEALTH-ABC,14 and PROSPER.12,15 Patients with a history of HF at baseline were excluded. We then used a nested matched case-control design were individuals who developed HF were considered to be at risk, that is, eligible to be selected as controls up until the time they became a case16: a total of 852 incident HF cases were identified (574 in HEALTH-ABC, 234 in PROSPER, and 44 in PREDICTOR); within the respective cohorts controls were selected, matched age, sex, and follow-up time (defined as time of incident HF from entry to the cohort). The final numbers after the matching procedures are provided below.
The data that support the findings of this study are available from the corresponding author on reasonable request.
Discovery and Replication
The HOMAGE study had 2 independent phases: discovery and replication. For the discovery phase, we selected 300 cases and 599 controls (1 case only had 1 match) randomly selected without replacement in a 1:2 proportion17–19; because of 22 missing or poor-quality samples, the final match was 286 cases to 591 controls. For replication, we selected 315 cases and 315 controls randomly selected without replacement in a 1:1 proportion; because of 74 missing or poor-quality samples, the final match was 276 cases to 280 controls.
The study was conducted in accordance with the Declaration of Helsinki and approved by each site ethics committees. All participants provided written informed consent.
Outcome
The outcome was incident HF which was defined as first hospitalization for HF as primary admission diagnosis (adjudicated by the investigators of the respective cohorts).
Sample Handling
All sample shipments and sample data acquisition within the HOMAGE consortium are according to predefined standard operating procedures and material transfer agreements to maintain uniformity. Figure I in the Data Supplement shows the sample handling and storage per cohort and the sample flow until protein measurement at the TATAA Biocenter (Gothenburg, Sweden). Aliquoting of the samples at Biobank Maastricht was performed using a multipipette in 1 run to reduce freeze/thaw cycles and batch effects. The entire sampling handling/protein measurement was performed fully blind to case-control status. The cases and controls were separately identified and selected by the study statistician. All patient information was then removed and a randomly sorted list of patient/sample IDs for each cohort was sent to Maastricht University Medical Center.
Assays and Studied Biomarkers
Baseline plasma samples were analyzed for protein biomarkers by the TATAA biocenter using the Olink Proseek Multiplex cardiovascular (CVD) II, CVD III, and inflammation panels. These panels were selected by the well-balanced inclusion of proteins with already established associations with CVD and HF (eg, BNP, ST2, and GDF [growth differentiation factor]15) and others with less well-established associations (eg, TWEAK [tumor necrosis factor ligand superfamily member 12] and PON3 [paraoxonase]). The assay uses a proximity extension assay technology,20 where 92 oligonucleotide-labeled antibody probe pairs per panel are allowed to bind to their respective targets in the sample in 96-well plate format. When binding to their correct targets, they give rise to new DNA amplicons with each ID-barcoding their respective antigens. The amplicons are subsequently quantified using a Fluidigm BioMark HD real-time polymerase chain reaction platform. The platform provides log2-NPX (normalized protein expression) data. A detailed description of the Olink technology is depicted in Addenda I in the Data Supplement. For 9 proteins measured in both the inflammation panel and CVD panels, the one from CVD panels was used for further data analyses (the results for these proteins were strongly correlated ≥0.9). In addition, 15 proteins that were below the limit of detection, were not included in the analysis. The Olink quality control samples are considered as flagged if they deviate >0.3 NPX from the median of all samples in one of 2 control assays for incubation and detection. The limit of detection is defined by the 3 negative controls run on each plate and set to 3 SDs above the measured background. Patients with missing or unusable samples (22 samples in the discovery phase and 74 samples in the replication phase) were not considered for the analyses. Where the assay results were partially missing, that is, results were missing for 1 or 2 of the 3 plates (83 patients in the discovery phase and 4 patients in the replication phase) then multiple imputation using chained equations was used.21
The abbreviations, full names, and respective Olink multiplex panels of the studied proteins are described in Table I in the Data Supplement.
The assays were performed blinded to case/control status with cases and controls randomly distributed across plates. The proteomic results were then merged with the baseline data, which included the case-control status, matching variables, and the clinical risk factors.
Statistical and Bioinformatics Considerations
For the baseline clinical characteristics, continuous variables are expressed as means and respective SD. Categorical variables are presented as frequencies and percentages. Patient baseline characteristics were compared between cases and controls using χ2 tests for categorical variables and t tests for continuous variables.
The main aim of this study was to test multiple proteins with regards to their association with incident HF and the respective underlying mechanistic pathways. Logistic regression models adjusting for the matching variables (age, sex, cohort, and follow-up time) were used to identify protein biomarkers associated with incident HF in the discovery and replication phases22 (Table II in the Data Supplement). Only those proteins which were found to be statistically significant (after correction for false discoveries) in the discovery phase (n=89) were taken forward for consideration in the replication phase. In both phases, we corrected for multiple testing using a false discovery rate of 1%.23 Additional adjustment for the prespecified clinical risk factors previously found to represent the best clinical prognostic model for incident HF in the HOMAGE population24 (smoking, diabetes mellitus, history of coronary artery disease, serum creatinine, body mass index, systolic blood pressure, use of antihypertensive medication, and heart rate) was also performed, providing similar results (Table III in the Data Supplement). Since proteins were measured using NPX values on a log2 scale, the odds ratio for each protein estimates the increase in the odds of HF associated with a doubling in the protein concentration. After the identification of the top proteins, common to the derivation and replication phases, we performed a multivariable a stepwise forward model adjusted on age, sex, cohort, phase, follow-up time, smoking, diabetes mellitus, history of coronary artery disease, serum creatinine, body mass index, systolic blood pressure, use of antihypertensive medication, and heart rate forced into the model with a P value for inclusion set at 0.05. This set of analyses was performed using STATA version 15 software (StataCorp 2017, Stata Statistical Software: Release 15; College Station, TX: StataCorp LP).
We used knowledge-based network analysis with induced network approach by consensusPathDB online server (accessed on January 29, 2019) from Max Planck Institute for Molecular Genetics to identify the links among the protein biomarkers selected in the previous step (discovery and replication with adjustment on the matching variables), based on known knowledge of interaction networks (protein interactions, genetic interactions, biochemical interactions, and gene regulatory interactions).11 The network analysis also identifies additional proteins (intermediate nodes) based on knowledge-based interactions (with the exclusion of low-confidence interactions quantified by a Z score ≤20 calculated for each intermediate node). As a validation step, network analysis was repeated using ClueGO network analysis (version 2.5.3), using implemented biological GO processes.25 Extra known connection between BNP and angiotensin was added to the network manually because of their well-described interplay on blood pressure and hydroelectrolytic regulation.26,27 The generated network was reorganized in Cytoscape (version 3.5) to merge genes with their expressed proteins and visualize the results. An additional overrepresentation analysis was performed using only the GO-biological processes and molecular function enriched by selected proteins against proteins on the OLINK panels, introducing an adjustment for the clustering of proteins on the network and consolidating the strength of true enrichment.
RESULTS
Study Population
The baseline characteristics of the studied population for both discovery (IA) and replication phases (IB) is depicted in Table 1. Cases and controls were well matched for age, sex, cohort, and follow-up time (ie, the matching variables) in both phases. Cases had higher body mass index, creatinine, were more often hypertensive (with antihypertensive medications), diabetic, and had more often coronary artery disease. All these variables were included in the HOMAGE prognostic model24 and were used for further adjustment in the models (please see below).
Table 1.
Characteristics of the Study Population for Both Discovery and Replication Phases
| Characteristics | Discovery | Replication | ||||
|---|---|---|---|---|---|---|
| Cases | Controls | P Value | Cases | Controls | P Value | |
| N=286 | N=591 | N=276 | N=280 | |||
| Age, y | 74.4 (3.5) | 74.4 (3.5) | 0.94 | 75.1 (3.5) | 75.2 (3.6) | 0.81 |
| Male sex, n (%) | 154 (53.8) | 320 (54.1) | 0.93 | 157 (56.9) | 158 (56.4) | 0.91 |
| Cohort, n (%) | 0.74 | 0.57 | ||||
| HEALTH-ABC | 215 (75.2) | 433 (73.3) | 109 (39.5) | 99 (35.4) | ||
| PREDICTOR | 15 (5.2) | 29 (4.9) | 29 (10.5) | 29 (10.4) | ||
| PROSPER | 56 (19.6) | 129 (21.8) | 138 (50.0) | 152 (54.3) | ||
| Smoking status, n (%) | 0.29 | 0.50 | ||||
| Never | 139 (48.6) | 309 (52.5) | 157 (56.9) | 169 (60.4) | ||
| Current | 30 (10.5) | 71 (12.1) | 57 (20.7) | 47 (16.8) | ||
| Past | 117 (40.9) | 209 (35.5) | 62 (22.5) | 64 (22.9) | ||
| Body mass index, kg/m2 | 28.1 (4.8) | 27.0 (4.4) | <0.001 | 27.6 (4.7) | 26.7 (4.2) | 0.02 |
| Systolic blood pressure, mm Hg | 142.4 (23.4) | 141.2 (23.0) | 0.48 | 148.2 (22.9) | 145.9 (22.7) | 0.25 |
| Diastolic blood pressure, mm Hg | 75.2 (13.7) | 74.6 (12.1) | 0.45 | 78.5 (13.7) | 78.2 (12.6) | 0.76 |
| Heart rate, bpm | 67 (11.8) | 64 (11.1) | 0.004 | 68 (11.9) | 66 (10.9) | 0.09 |
| Serum creatinine, µmol/L | 98.8 (27.7) | 93.5 (21.3) | 0.002 | 102.8 (28.2) | 95.5 (21.5) | <0.001 |
| Total cholesterol, mmol/L | 5.33 (1.00) | 5.31 (0.99) | 0.88 | 5.34 (0.97) | 5.49 (0.95) | 0.08 |
| HDL-cholesterol, mmol/L | 1.28 (0.39) | 1.34 (0.41) | 0.04 | 1.29 (0.37) | 1.36 (0.42) | 0.02 |
| LDL-cholesterol, mmol/L | 3.32 (0.89) | 3.29 (0.92) | 0.64 | 3.41 (0.91) | 3.48 (0.85) | 0.35 |
| Triglycerides, mmol/L | 1.61 (0.83) | 1.55 (0.96) | 0.33 | 1.53 (0.77) | 1.45 (0.66) | 0.19 |
| Blood glucose, mmol/L | 5.99 (2.18) | 5.60 (1.56) | 0.002 | 5.92 (1.99) | 5.49 (1.34) | 0.003 |
| Comorbidities, n (%) | ||||||
| Hypertension | 219 (76.8) | 408 (69.2) | 0.02 | 184 (66.9) | 177 (63.4) | 0.39 |
| Diabetes mellitus | 55 (19.2) | 68 (11.5) | 0.002 | 49 (17.8) | 32 (11.4) | 0.03 |
| CVD | 124 (44.4) | 168 (28.9) | <0.001 | 149 (54.0) | 105 (37.5) | <0.001 |
| CAD | 103 (36.5) | 126 (21.6) | <0.001 | 114 (41.3) | 78 (27.9) | <0.001 |
| PAD | 14 (5.0) | 22 (3.8) | 0.40 | 14 (5.1) | 4 (1.4) | 0.01 |
| Cerebrovascular disease | 25 (8.9) | 49 (8.3) | 0.76 | 34 (12.3) | 25 (8.9) | 0.19 |
| Medications, n (%) | ||||||
| Antihypertensives | 192 (67.1) | 318 (54.0) | <0.001 | 198 (71.7) | 174 (62.1) | 0.02 |
| ACE inhibitor | 64 (22.4) | 74 (12.6) | <0.001 | 63 (22.8) | 47 (16.8) | 0.07 |
| CCB | 86 (30.1) | 114 (19.4) | <0.001 | 77 (27.9) | 53 (18.9) | 0.01 |
| Diuretics | 88 (30.8) | 141 (23.9) | 0.03 | 102 (37.0) | 89 (31.8) | 0.20 |
| β-blockers | 51 (17.8) | 97 (16.5) | 0.61 | 62 (22.5) | 54 (19.3) | 0.36 |
| ARBs | 13 (4.5) | 14 (2.4) | 0.08 | 17 (6.2) | 13 (4.6) | 0.43 |
| Antiplatelet | 128 (44.8) | 228 (38.7) | 0.09 | 126 (45.7) | 102 (36.4) | 0.03 |
Numbers are mean (SD) unless otherwise specified. ACE indicates angiotensin-converting enzyme; ARBs, angiotensin receptors blockers; CAD, coronary artery diseases; CCB, calcium channel blockers; CVD, cardiovascular diseases; HDL, high-density lipoprotein; LDL, low-density lipoprotein; and PAD, peripheral arterial diseases.
Biomarkers Associated With Incident HF
Of the 252 proteins studied, adjusting for the matching variables age, sex, and follow-up time, 89 proteins were found to be associated with incident HF in the discovery phase, of which 38 were also associated with incident HF in the replication phase, Table 2. All 38 proteins were positively associated with incident HF, except for TWEAK and PON3, where patients with higher concentrations of these proteins were less likely to develop HF. Further adjusting for the clinical risk factors (smoking, diabetes mellitus, history of coronary artery disease, serum creatinine, body mass index, systolic blood pressure, use of antihypertensive medication, and heart rate) previously determined in the well-calibrated HOMAGE clinical risk model,24 provides similar associations to those presented in Table 2, suggesting that these associations were independent of the patients’ clinical risk (also supported by the weak correlation between the study proteins and the clinical risk factors), Tables III and IV in the Data Supplement.
Table 2.
Odds Ratios (OR) and 95% Confidence Intervals for the Selected Proteins With Regards to Their Association With Incident Heart Failure After Adjustment for the Matching Variables and Correction for Multiple Comparisons in Both Discovery and Replication Sets
| Protein Name | Discovery | Replication | ||||
|---|---|---|---|---|---|---|
| OR | (95% CI) | P Value | OR | (95% CI) | P Value | |
| BNP | 1.62 | (1.42–1.86) | <0.0001 | 1.73 | (1.50–2.01) | <0.0001 |
| NT-proBNP | 1.74 | (1.46–2.06) | <0.0001 | 2.00 | (1.64–2.44) | <0.0001 |
| TRAILR2 | 3.26 | (2.32–4.60) | <0.0001 | 3.22 | (2.21–4.69) | <0.0001 |
| TNFRSF13B | 1.74 | (1.34–2.24) | <0.0001 | 2.61 | (1.82–3.74) | <0.0001 |
| GAL9 | 2.96 | (1.95–4.49) | <0.0001 | 3.14 | (2.01–4.91) | <0.0001 |
| FGF23 | 1.70 | (1.40–2.07) | <0.0001 | 1.84 | (1.44–2.36) | <0.0001 |
| TNFRSF10A | 2.63 | (1.88–3.69) | <0.0001 | 2.56 | (1.74–3.77) | <0.0001 |
| REN | 1.31 | (1.11–1.55) | 0.0018 | 1.54 | (1.29–1.85) | <0.0001 |
| TNFRSF11A | 2.09 | (1.61–2.72) | <0.0001 | 1.95 | (1.47–2.59) | <0.0001 |
| GDF-15 | 2.74 | (2.08–3.61) | <0.0001 | 1.89 | (1.44–2.47) | <0.0001 |
| FABP4 | 1.82 | (1.51–2.19) | <0.0001 | 1.57 | (1.29–1.92) | <0.0001 |
| SLAMF7 | 1.55 | (1.26–1.91) | <0.0001 | 2.16 | (1.52–3.06) | <0.0001 |
| CCL16 | 1.47 | (1.21–1.79) | <0.0001 | 1.72 | (1.34–2.22) | <0.0001 |
| TWEAK | 0.51 | (0.36–0.73) | 0.0003 | 0.53 | (0.39–0.71) | <0.0001 |
| KIM1 | 1.41 | (1.19–1.68) | <0.0001 | 1.51 | (1.23–1.84) | <0.0001 |
| CD4 | 2.06 | (1.49–2.85) | <0.0001 | 2.17 | (1.49–3.16) | <0.0001 |
| VSIG2 | 1.43 | (1.14–1.81) | 0.0025 | 1.76 | (1.33–2.32) | <0.0001 |
| PON3 | 0.71 | (0.58–0.87) | 0.0011 | 0.64 | (0.51–0.80) | <0.0001 |
| PLGF | 2.17 | (1.54–3.06) | <0.0001 | 1.95 | (1.38–2.75) | 0.0002 |
| MMP-12 | 1.64 | (1.32–2.03) | <0.0001 | 1.54 | (1.23–1.92) | 0.0002 |
| ADM | 2.56 | (1.82–3.59) | <0.0001 | 1.80 | (1.32–2.45) | 0.0002 |
| RARRES2 | 3.33 | (2.02–5.51) | <0.0001 | 2.05 | (1.38–3.05) | 0.0004 |
| CEACAM8 | 1.51 | (1.22–1.86) | 0.0001 | 1.49 | (1.19–1.86) | 0.0005 |
| SLAMF1 | 1.44 | (1.17–1.76) | 0.0005 | 1.62 | (1.23–2.13) | 0.0005 |
| TNFR1 | 2.40 | (1.75–3.28) | <0.0001 | 1.63 | (1.23–2.16) | 0.0007 |
| AGRP | 1.82 | (1.38–2.40) | <0.0001 | 1.90 | (1.31–2.76) | 0.0007 |
| TNFR2 | 2.11 | (1.58–2.82) | <0.0001 | 1.55 | (1.20–2.00) | 0.0008 |
| IGFBP7 | 1.66 | (1.24–2.23) | 0.0006 | 1.60 | (1.21–2.11) | 0.0010 |
| UPAR | 2.70 | (1.94–3.75) | <0.0001 | 1.67 | (1.23–2.28) | 0.0011 |
| PAR1 | 1.86 | (1.33–2.60) | 0.0003 | 1.74 | (1.24–2.45) | 0.0014 |
| PLC | 2.26 | (1.59–3.21) | <0.0001 | 1.76 | (1.24–2.49) | 0.0015 |
| ACE2 | 1.60 | (1.27–2.02) | <0.0001 | 1.52 | (1.17–1.98) | 0.0016 |
| IL-16 | 1.54 | (1.20–1.97) | 0.0007 | 1.46 | (1.15–1.86) | 0.0019 |
| TFF3 | 1.61 | (1.32–1.95) | <0.0001 | 1.45 | (1.15–1.84) | 0.0019 |
| OPN | 2.00 | (1.52–2.63) | <0.0001 | 1.50 | (1.16–1.95) | 0.0021 |
| SPON2 | 2.86 | (1.50–5.48) | 0.0015 | 2.40 | (1.36–4.25) | 0.0026 |
| IL4RA | 1.52 | (1.15–2.02) | 0.0033 | 1.62 | (1.18–2.22) | 0.0029 |
| TNFRSF14 | 2.09 | (1.55–2.82) | <0.0001 | 1.52 | (1.15–2.01) | 0.0034 |
ACE2 indicates angiotensin-converting enzyme 2; ADM, adrenomedullin; AGRP, agouti-related protein; BNP, brain natriuretic peptide; CCL16, C-C motif chemokine 16; CD4, T-cell surface glycoprotein CD4; CEACAM8, carcinoembryonic antigen-related cell adhesion molecule 8; FABP4, fatty acid-binding protein; FGF23, fibroblast growth factor 23; GAL9, galectin 9; GDF-15, growth/differentiation factor 15; IGFBP7, insulin-like growth factor-binding protein 7; IL-16, pro-interleukin-16; IL4RA, interleukin 4 receptor subunit alpha; KIM1, kidney injury molecule 1; MMP-12, matrix metalloproteinase 12; NT-proBNP, N-terminal pro-B-type natriuretic peptide; OPN, osteopontin; PAR1, proteinase-activated receptor 1; PLC, perlecan; PLGF, placenta growth factor; PON3, paraoxonase; RARRES2, retinoic acid receptor responder protein 2; REN, renin; SLAMF1, signaling lymphocytic activation molecule; SLAMF7, SLAM family member 7; SPON2, spondin-2; TFF3, trefoil factor 3; TNFR1, tumor necrosis factor receptor 1; TNFR2, tumor necrosis factor receptor 2; TNFRSF10A, tumor necrosis factor receptor superfamily member 10A; TNFRSF11A, tumor necrosis factor receptor superfamily member 11A; TNFRSF13B, tumor necrosis factor receptor superfamily member 13B; TNFRSF14, tumor necrosis factor receptor superfamily member 14; TRAILR2, TNF-related apoptosis-inducing ligand receptor 2; TWEAK, tumor necrosis factor (ligand) superfamily, member 12; UPAR, urokinase plasminogen activator surface receptor; and VSIG2, v-set and immunoglobulin domain-containing protein 2.
The multivariable stepwise model including the matching variables and the clinical risk factors forced into the model, plus the 38 proteins independently identified in both the discovery and replication phases, retained BNP, TWEAK, NT-proBNP, REN (renin), TRAILR2 (trail receptor 2), PON3, CCL16 (C-C motif chemokine 16), and SLAMF1 (signalling lymphocytic activation molecule family member 1) as the biomarkers with stronger association with incident HF, Table V in the Data Supplement.
Induced Network Results
The 38 incident HF-associated protein biomarkers were linked with each other by known protein interactions, biochemical interactions, and gene regulatory interactions (Figure), directly or via intermediate nodes (Table VI in the Data Supplement). Our results pointed to 4 clusters with clearly defined functions: (1) inflammatory/apoptosis of mainly TNF-family members, (2) extracellular matrix remodeling, angiogenesis, and cell growth, (3) a renin-angiotensin system associated with blood pressure regulation and one minor cluster including metabolic proteins, and (4) metabolism, associated with cholesterol and atherosclerosis. The 2 major clusters inflammatory/apoptosis and blood pressure regulation were also detected as the main groups using the ClueGO network analysis (Figure II in the Data Supplement). The TNF-family members, their representative pathways and blood pressure regulation remained significantly enriched after adjustment for the preselection of proteins (Table VII in the Data Supplement).
Figure.
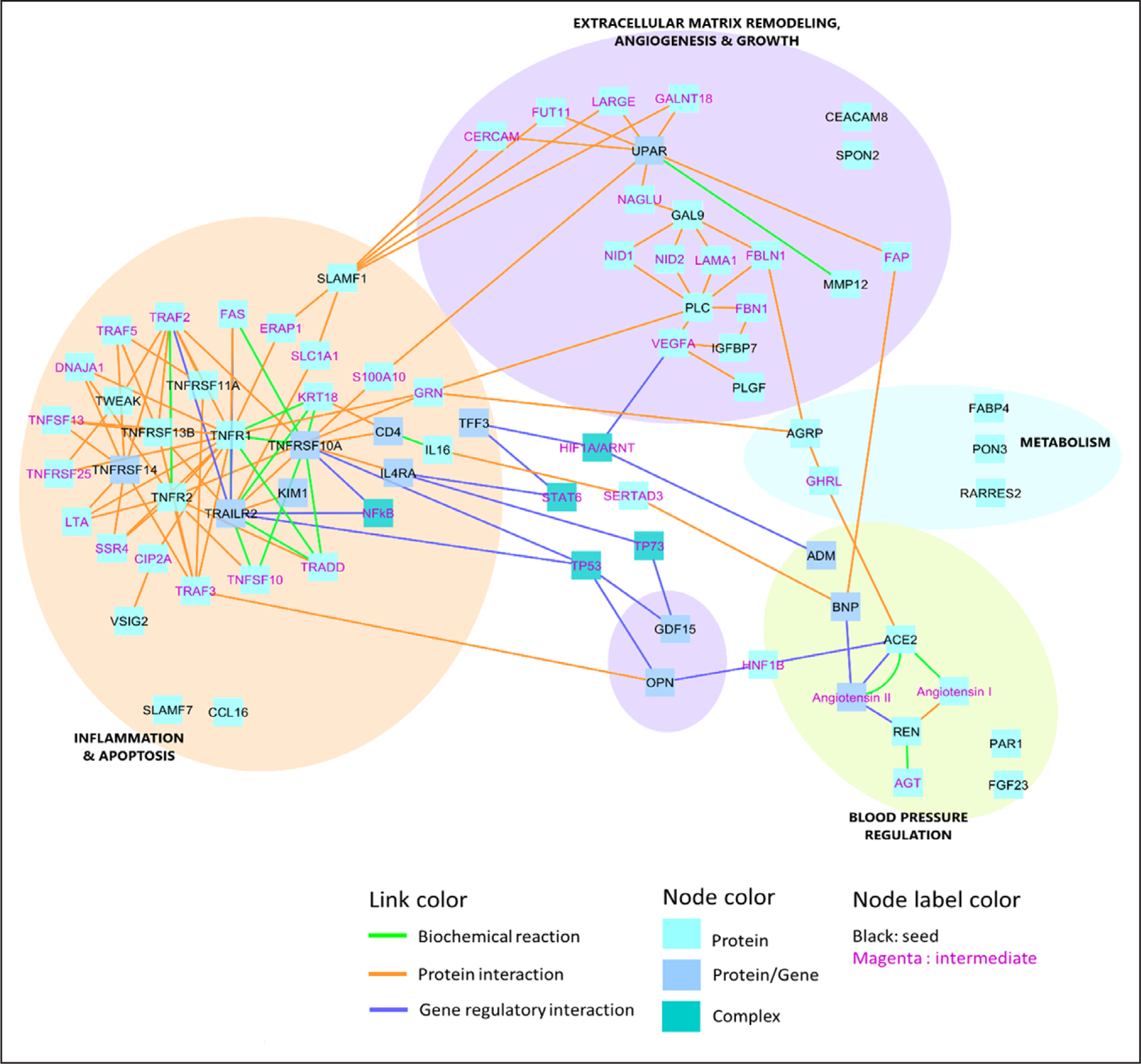
Induced network analysis: protein interactions, biochemical interactions, and gene regulatory interactions. Please see Table IV in the Data Supplement for the full names of the biomarkers and intermediates. ACE indicates angiotensin-converting enzyme; ADM, adrenomedullin; AGRP, agouti-related protein; BNP, B-type natriuretic peptide; AGT, angiotensinogen; CCL16, C-C motif chemokine 16; CEACAM8, carcinoembryonic antigen-related cell adhesion molecule 8; CERCAM, cerebral endothelial cell adhesion molecule; FAP, fibroblast activation protein alpha; FBLN1, fibulin-1; FGF23, fibroblast growth factor 23; GAL9, galectin 9; GALNT18, polypeptide N-acetylgalactosaminyltransferase 18; GDF-15, growth differentiation factor 15; GHRL, ghrelin; HNF1B, hepatocyte nuclear factor-1-β; IGFBP7, insulin-like growth factor-binding protein 7; LAMA1, laminin subunit alpha 1; LARGE, LARGE xylosyl- and glucuronyltransferase 1; LTA, lymphotoxin alpha; MMP-12, matrix metalloproteinase 12; NAGLU, N-acetyl-alpha-glucosaminidase; NF-κB, nuclear factor κB; OPN, osteopontin; PAR1, proteinase-activated receptor 1; PGF, placenta growth factor; PLC, perlecan; PLGF, placenta growth factor; RARRES2, retinoic acid receptor responder protein 2; SLAMF1, signalling lymphocytic activation molecule family member 1; SPON2, spondin-2; TNFRSF, tumor necrosis factor receptor superfamily member; TRAF, TNF receptor associated factor 1; TRAILR2, trail receptor 2; TWEAK, tumor necrosis factor ligand superfamily member 12; UPAR, urokinase plasminogen activator surface receptor; VEGFA, vascular endothelial growth factor A; and VSIG2, v-set and immunoglobulin domain-containing protein 2.
In addition, this analysis revealed multiple intracellular transcription factors: TP53 (tumor protein 53), HNF1B (hepatocyte nuclear factor-1-β), HIF1A/ARNT (hypoxia-inducible factor α/aryl hydrocarbon receptor nuclear translocator), and STAT6 (signal transducer and activator of transcription 6), which are not detected with our plasma protein panels. However, these transcription factors may supplement the biomarker profile of patients at high risk for incident HF, providing additional perspective on the interpretation of the pathophysiological processes driving HF. The role of each biomarker linked to the identified network clusters is furtherly detailed in the discussion section.
DISCUSSION
In the present study, we identified 38 plasma proteins associated with incident HF (in both the discovery and replication phases). The selected proteins allowed the identification of 4 main network clusters underpinning incident HF: (1) inflammation and apoptosis; (2) extracellular matrix remodeling, angiogenesis, and growth; (3) blood pressure regulation; and (4) metabolism. These findings are original and provide important insight on the pathophysiological mechanisms leading to HF, potentially creating the basis for the development of new HF prevention strategies, personalized to each individual patient underlying mechanism.
A recently published study10 investigated the longitudinal association between high-throughput proteomics (also using OLINK technology) and HF risk in 2 community-based prospective cohorts of elderly individuals without HF at baseline. To some extent, the proteins identified in that study overlapped with ours. Specifically, TRAILR2, GDF-15, and MMP-12 (matrix metalloproteinase 12) were identified in both studies across all discovery steps. However, the study by Stenemo et al10 studied 80 proteins, whereas ours analyzed 252. Moreover, our study was aimed to identify the biological signatures leading to HF.
Inflammation and Apoptosis Cluster
Inflammation and apoptosis, as pointed by the expression of TNF-family members, may be an important pathway leading to HF that may be identified by the expression of circulation proteins such as TRAILR2, IL-16 (interleukin 16), IL4RA (interleukin 4 receptor α), CD4 (T-cell surface glycoprotein CD4), TNFRSF (tumor necrosis factor receptor superfamily member) 10A, TNFRSF11A, TNFR1, TNFR2 (tumor necrosis factor receptor 2), TNFRS-F13B, TNFRSF14, CCL16, SLAMF1, and TWEAK. Elevated TNF signaling restrains cardiomyocyte differentiation of resident cardiac stem cells and enhances adrenergic activation, promoting adverse cardiac remodeling (also reflected by the elevated remodeling markers).28 The TRAILR2 protein (otherwise known as death receptor 5) is encoded by the TNFSF10 gene and is a receptor belonging to the TNF superfamily that preferentially induces apoptosis after binding of its ligand TRAIL.29,30 Increased levels of TRAILR2 have been associated with adverse cardiovascular events in patients with myocardial infarction, probably because of intensified apoptotic activity.31 ILs, as upstream biomarkers of inflammation converge on the central TNF signaling pathway, having major infl ence on atherosclerosis, and consequently on the risk of CVD.32 Another TNF superfamily member—TWEAK—activates the NF-κB (nuclear factor κB) and regulates several cell functions, such as proliferation, migration, differentiation, cell death, inflammation, angiogenesis, and collagen synthesis of cardiac fibroblasts.33–35 Low TWEAK has been associated with increased risk of death in patients with overt HF36 and patients with HF and reduced ejection fraction had lower TWEAK levels compared with controls.34 The TWEAK-induced proliferation of cardiomyocytes and its immunomodulatory effects may provide a basis to these findings.36 Apart from binding to its active receptor Fn14, TWEAK can also bind to a scavenger receptor CD163, which was shown to be upregulated in HF, explaining the decreased levels and activity of TWEAK in HF.34
The inflammation/apoptosis cluster grouped many proteins with a strong and independent association with HF: TWEAK, TRAILR2, CCL16, and SLAMF1.
Extracellular Matrix Remodeling, Angiogenesis, and Growth Cluster
Another major pathway identified as leading to HF was, extracellular matrix remodeling, angiogenesis, and growth supported by the expression of ADM (adreno-medullin), IGFBP7 (insulin-like growth factor-binding protein 7), PGF (placenta growth factor), PLC (perlecan), GAL9 (galectin 9), MMP-12, UPAR (urokinase plasminogen activator surface receptor), SLAMF1, CEACAM8 (carcinoembryonic antigen-related cell adhesion molecule 8), GDF-15, FGF23 (fibroblast growth factor 23), and OPN (osteopontin). ADM is a vasodilator peptide predominantly produced by the vascular endothelium and smooth muscle that increases in response to hemodynamic stress.37 IGFBP7 participates in the regulation of the availability of insulin growth factor in body fluids and tissues. IGFBP7 has been found to be associated with diastolic dysfunction and is also a strong prognosticator in HF.38 PGF is increased by pressure overload in the heart where it is expressed in both myocytes and other cells infiltrating the heart.39 PLC is constituent of the extracellular matrix that regulates angiogenesis and cell autophagy.40 PLC proangiogenic effects may be used for the treatment of ischemic diseases.41 GAL9 is produced by the extracellular matrix and may be increased in patients with ischemic stroke, its role in HF requires further study.42 MMPs degrade extracellular matrix proteins and play important roles in development and tissue repair. MMP-12 contributes to plaque growth and destabilization and increased levels of this proteins have been associated with higher atherosclerotic disease burden.43 SLAMF1 is expressed in the surface of lymphocytes and is involved in the control of infectious and neoplastic processes.44 CEACAM8 is released by granulocytes and is also involved in immune regulation.45 The role of SLAMF1 and CEACAM8 in HF requires further investigation. However, they are both related to UPAR that induces cardiac fibrosis and macrophage accumulation and is associated with worse prognosis in HF.46,47 GDF-15 regulates inflammation and apoptosis, both key mechanisms in cardiac remodeling that are potentially associated with incident HF.48,49 FGF23 is released by the osteocytes and is essential for the regulation of the metabolism of phosphate, calcium, and vitamin D. Importantly, FGF23 promotes myocardial fibrosis and has been associated with coronary heart disease and HF.50,51 OPN is a member of the extracellular matrix protein family. OPN expression increases under a variety of pathophysiological conditions affecting the heart and has been associated with an increased incidence of CVDs, including HF.52
Blood Pressure Regulation Cluster
Another major pathway identified as leading to HF was associated with blood pressure regulation, supported by the increased expression of renin, angiotensin-converting enzyme, and BNP/NT-proBNP. The renin-angiotensin-aldosterone and the natriuretic peptide systems have been thoroughly associated with CVD, including hypertension and HF.53 Natriuretic peptides (BNP and NT-proBNP) are produced by the cardiomyocytes, endothelial cells, T cells, and macrophages infiltrating the heart in response to cardiac overload.54 BNP/NT-proBNP are recommended in the current guidelines for diagnostic and prognostic assessment in HF.55,56 Natriuretic peptides have been associated with incident HF,57 and a natriuretic peptide-based strategies for preventing HF have reduced the rates of both systolic and diastolic dysfunction.58 The renin-angiotensin-aldosterone and BNP are closely related by inhibition on to each other by having counterbalanced regulatory functions on blood pressure.26 Moreover, angiotensin can upregulate cardiac BNP gene expression.27 Enhanced renin-angiotensin-aldosterone under high BNP may reflect a dysregulation on blood pressure leading to HF development. Natriuretic peptides and renin were strongly and independently associated with incident HF.
Metabolism Cluster
The other pathway identified as leading to HF was associated with metabolism, supported by the identification of PON3, FABP4 (fatty acid-binding protein 4), and RARRES2 (retinoic acid receptor responder protein 2). PON3 has been associated with HDL (high-density lipoprotein) increase and with the inhibition of LDL (low-density lipoprotein) oxidation, thus PON3 expression might be protective in the cardiovascular setting.59 In our study, PON3 was negatively associated with incident HF risk, suggesting that antioxidation may play a role in the mechanisms associated with HF development. In line with TWEAK, preclinical evidence supports a cardioprotective role for PON3 (which was also independently associated with incident HF in the multivariable model). FABP4 is predominantly expressed in macrophages and adipose tissue where it regulates fatty acids storage and lipolysis; FABP4 is also an important mediator of inflammation that has been associated with a higher risk of cardiovascular events.60,61 RARRES2 (or chemerin) is an adipose-derived signaling molecule that regulates adipogenesis and adipocyte metabolism, and it has also been associated with a higher risk of cardiovascular events.62,63
Overall, the results of our proteomic biomarker assessments in patients at risk of developing HF suggest that progression towards HF is likely to involve the interplay of several pathophysiological mechanisms, such as heart stress, blood pressure regulation, apoptosis, inflammation, and metabolism-related mechanisms. A previous report identified cytokine response, extracellular matrix organization, and inflammation as major pathways underlining HF with preserved ejection fraction.64 The next step of this important data is to determine the activity of these processes in different HF stages and eventually per individual. This will provide the basis for further development strategies in preventing HF and focusing on these specific pathways at early stages of the disease for an individual treatment approach. The intracellular transcription factors TP53, HNF1B, HIF1A/ARNT, and STAT6, which are not measured in our plasma protein panels, may complement the biomarker profile of patients at high risk for incident HF,65 suggesting a combined multi-omics approach currently being investigated within the HOMAGE consortium.
Limitations
Several limitations should be highlighted in the present study. First, this is an observational case-control study, hence causality cannot be ascertained. The bioinformatics approach also does not allow causality assessment but allows for the generation of hypothesis on the underlying pathways associated with this proteomic expression. Also, we must be aware of the biases and oversimplification in network topology. For example, most network databases are overrepresented by well-studied proteins and their interactions. This will lead to overrepresentation of these interactions in the analysis as there are more interactions known for these proteins.66 Second, incident HF was defined as first HF hospitalization, which does not exclude patients that might already have HF but without previous hospitalizations. Also, for the avoidance of competing risk, we excluded patients who died during follow-up. Therefore, it is possible that we missed patients where death was the first (and last) manifestation of HF. Third, we did not have access to the reported ejection fraction at the time of hospitalization, therefore, we cannot assess the potential value of these biomarkers in distinguishing progression to HF with reduced ejection fraction from HF with preserved ejection fraction and the HF cause. Fourth, clinical detail (signs, symptoms, ECG, and other complementary exams), troponin, and natriuretic peptides at the time of hospitalization are not available in the dataset. This information would help in further phenotyping these patients and in differentiating the cases from the controls. Fifth, the proteomics assay does not provide standard concentration units, making comparisons with clinically applied cutoffs difficult, however, the Olink standard procedures reassure a good correlation with the standard measurement methodologies. In addition, we did not use large unbiased screens but rather selected protein biomarkers based on mechanistic hypotheses. The 3 studied Olink panels (CVD II, III, and inflammation) contain circulating proteins previously found to be associated with cardiovascular and inflammatory diseases. Many other pathways are missing, for example, metabolism(omics), that could enrich our networks. Therefore, we cannot exclude the role of other mechanisms not targeted with our proteomics screen. Finally, prospective validation of these biomarkers in other populations is required to improve the external validation of these results.
Conclusions
After adjustment for the matching variables age, sex, follow-up time, and correction for multiplicity of tests, we identified 38 proteins in 2 independent sets associated with incident HF. Cluster of the selected proteins allowed the identification of 4 main networks leading to HF: (1) inflammation and apoptosis; (2) extracellular matrix remodeling, angiogenesis, and growth; (3) blood pressure regulation; and (4) metabolism. These findings provide important insight on the pathophysiological mechanisms leading to HF.
Supplementary Material
WHAT IS NEW?
We present a nested case:control study (with derivation and replication cohorts) to study 252 circulation proteins and their association with new-onset heart failure, to assess the mechanistic pathways that may lead to the development of heart failure.
WHAT ARE THE CLINICAL IMPLICATIONS?
We identified 4 main networks that may lead to heart failure. These include inflammation and apoptosis; extracellular matrix remodeling, angio-genesis and growth; blood pressure regulation; and metabolism.
These findings provide important insight on the mechanisms leading to heart failure and may help in the development of future personalized therapies.
Acknowledgments
Sources of Funding
The research leading to these results has received funding from the European Union Commission’s Seventh Framework Programme under grant agreement No. 305507 (HOMAGE [Heart Omics in Ageing consortium]). We acknowledge the support from the Netherlands Cardiovascular Research Initiative, an initiative with the support of the Dutch Heart Foundation CVON2016-Early HFPEF, and CVON 2017, ShePREDICTS.
Footnotes
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCHEARTFAILURE.118.005897.
Disclosures
Drs Ferreira, Rossignol, and Zannad are supported by the French National Research Agency Fighting Heart Failure (ANR-15-RHU-0004), by the French PIA project Lorraine Université d’Excellence GEENAGE (ANR-15-IDEX-04-LUE) programs, and the Contrat de Plan Etat Région Lorraine and FEDER IT2MP. Dr Mischak is the co-founder and co-owner of Mosaiques Diagnostics. Dr Mebazaa received honoraria for lectures from Roche and Abbott, consultation fees from Sanofi and Servier, and research grants from Adrenomed and Sphyngotec. The other authors report no conflicts.
Contributor Information
João Pedro Ferreira, Université de Lorraine, Inserm, Centre d’Investigations Cliniques-Plurithématique 14–33, and Inserm U1116, CHRU, F-CRIN INI-CRCT (Cardiovascular and Renal Clinical Trialists), Nancy, France, Department of Physiology and Cardiothoracic Surgery, Cardiovascular Research and Development Unit, Faculty of Medicine, University of Porto, Portugal.
Job Verdonschot, Department of Cardiology, Maastricht University Medical Centre, Center for Heart Failure Research, Cardiovascular Research Institute Maastricht (CARIM), University Hospital Maastricht, the Netherlands, Department of Clinical Genetics, Maastricht University Medical Center, the Netherlands.
Timothy Collier, London School of Hygiene and Tropical Medicine, United Kingdom.
Ping Wang, Department of Clinical Genetics, Maastricht University Medical Center, the Netherlands.
Anne Pizard, Université de Lorraine, Inserm, Centre d’Investigations Cliniques-Plurithématique 14–33, and Inserm U1116, CHRU, F-CRIN INI-CRCT (Cardiovascular and Renal Clinical Trialists), Nancy, France, Inserm 1024, Institut de Biologie de l’École Normale Supérieure (IBENS), PSL University of Paris, France.
Christian Bär, Institute of Molecular and Translational Therapeutic Strategies (IMTTS), Hannover Medical School, Germany.
Jens Björkman, Department of Medicine, University of Mississippi School of Medicine, Jackson, Excellence Cluster REBIRTH, Hannover Medical School, Germany.
Alessandro Boccanelli, Casa di Cura Quisisana, Rome, Italy.
Javed Butler, TATAA Biocenter AB, Gothenburg, Sweden.
Andrew Clark, Hull York Medical School, Castle Hill Hospital, Cottingham, United Kingdom.
John G. Cleland, Robertson Centre for Biostatistics and Clinical Trials, Institute of Health and Wellbeing, Glasgow, United Kingdom, National Heart and Lung Institute, Royal Brompton and Harefield Hospitals, Imperial College, University of Glasgow, London, United Kingdom.
Christian Delles, Institute of Cardiovascular and Medical Sciences, University of Glasgow, Scotland, United Kingdom.
Javier Diez, Program of Cardiovascular Diseases, Centre for Applied Medical Research, University of Navarra, Pamplona, Spain, CIBERCV, Carlos III Institute of Health, Madrid, Spain, Instituto de Investigación Sanitaria de Navarra (IdiSNA), Spain, Departments of Nephrology, and Cardiology and Cardiac Surgery, University of Navarra Clinic, Pamplona, Spain.
Nicolas Girerd, Université de Lorraine, Inserm, Centre d’Investigations Cliniques-Plurithématique 14–33, and Inserm U1116, CHRU, F-CRIN INI-CRCT (Cardiovascular and Renal Clinical Trialists), Nancy, France.
Arantxa González, Program of Cardiovascular Diseases, Centre for Applied Medical Research, University of Navarra, Pamplona, Spain, CIBERCV, Carlos III Institute of Health, Madrid, Spain, Instituto de Investigación Sanitaria de Navarra (IdiSNA), Spain.
Mark Hazebroek, Department of Cardiology, Maastricht University Medical Centre, Center for Heart Failure Research, Cardiovascular Research Institute Maastricht (CARIM), University Hospital Maastricht, the Netherlands.
Anne-Cécile Huby, Université de Lorraine, Inserm, Centre d’Investigations Cliniques-Plurithématique 14–33, and Inserm U1116, CHRU, F-CRIN INI-CRCT (Cardiovascular and Renal Clinical Trialists), Nancy, France.
Wouter Jukema, Department of Cardiology, Leiden University Medical Center, the Netherlands.
Roberto Latini, IRCCS-Istituto di Ricerche Farmacologiche Mario Negri, Milano, Italy.
Joost Leenders, ACS Biomarker BV, Amsterdam, the Netherlands.
Daniel Levy, National Heart, Lung, and Blood Institute’s and Boston University’s Framingham Heart Study, MA, Population Sciences Branch, Division of Intramural Research, National Heart, Lung, and Blood Institute, Bethesda, MD.
Alexandre Mebazaa, UMRS 942, University Paris Diderot; APHP, University Hospitals Saint Louis Lariboisière, France.
Harald Mischak, Mosaiques-diagnostics, Hannover, Germany.
Florence Pinet, Inserm U1167, Institut Pasteur de Lille, Université de Lille, FHU-REMOD-VHF, France.
Patrick Rossignol, Université de Lorraine, Inserm, Centre d’Investigations Cliniques-Plurithématique 14–33, and Inserm U1116, CHRU, F-CRIN INI-CRCT (Cardiovascular and Renal Clinical Trialists), Nancy, France.
Naveed Sattar, Institute of Cardiovascular and Medical Sciences, University of Glasgow, United Kingdom.
Peter Sever, International Centre for Circulatory Health, National Heart and Lung Institute, Imperial College London, England.
Jan A. Staessen, Studies Coordinating Centre, Research Unit Hypertension and Cardiovascular Epidemiology, KU Leuven Department of Cardiovascular Sciences, University of Leuven, Belgium, Cardiovascular Research Institute Maastricht (CARIM), Maastricht University, the Netherlands.
Thomas Thum, Institute of Molecular and Translational Therapeutic Strategies (IMTTS), Hannover Medical School, Germany, National Heart and Lung Institute, Imperial College London, United Kingdom.
Nicolas Vodovar, UMRS 942, University Paris Diderot; APHP, University Hospitals Saint Louis Lariboisière, France.
Zhen-Yu Zhang, Studies Coordinating Centre, Research Unit Hypertension and Cardiovascular Epidemiology, KU Leuven Department of Cardiovascular Sciences, University of Leuven, Belgium.
Stephane Heymans, Department of Cardiology, Maastricht University Medical Centre, Center for Heart Failure Research, Cardiovascular Research Institute Maastricht (CARIM), University Hospital Maastricht, the Netherlands, Department of Cardiovascular Research, University of Leuven, Belgium, Netherlands Heart Institute (ICIN), Utrecht, the Netherlands.
Faiez Zannad, Université de Lorraine, Inserm, Centre d’Investigations Cliniques-Plurithématique 14–33, and Inserm U1116, CHRU, F-CRIN INI-CRCT (Cardiovascular and Renal Clinical Trialists), Nancy, France.
REFERENCES
- 1.Zannad F. Rising incidence of heart failure demands action. Lancet 2018;391:518–519. doi: 10.1016/S0140-6736(17)32873-8 [DOI] [PubMed] [Google Scholar]
- 2.McMurray JJ, Petrie MC, Murdoch DR, Davie AP. Clinical epidemiology of heart failure: public and private health burden. Eur Heart J 1998;19(suppl P):P9–P16. [PubMed] [Google Scholar]
- 3.McMurray JJ, Stewart S. Epidemiology, aetiology, and prognosis of heart failure. Heart 2000;83:596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrie M, McMurray J. Changes in notions about heart failure. Lancet 2001;358:432–434. doi: 10.1016/S0140-6736(01)05664-1 [DOI] [PubMed] [Google Scholar]
- 5.Conrad N, Judge A, Tran J, Mohseni H, Hedgecott D, Crespillo AP, Allison M, Hemingway H, Cleland JG, McMurray JJV, Rahimi K. Temporal trends and patterns in heart failure incidence: a population-based study of 4 million individuals. Lancet 2018;391:572–580. doi: 10.1016/S0140-6736(17)32520-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manzano L, Babalis D, Roughton M, Shibata M, Anker SD, Ghio S, van Veldhuisen DJ, Cohen-Solal A, Coats AJ, Poole-Wilson PP, Flather MD; SENIORS Investigators. Predictors of clinical outcomes in elderly patients with heart failure. Eur J Heart Fail 2011;13:528–536. doi: 10.1093/eurjhf/hfr030 [DOI] [PubMed] [Google Scholar]
- 7.Echouffo-Tcheugui JB, Greene SJ, Papadimitriou L, Zannad F, Yancy CW, Gheorghiade M, Butler J. Population risk prediction models for incident heart failure: a systematic review. Circ Heart Fail 2015;8:438–447. doi: 10.1161/CIRCHEARTFAILURE.114.001896 [DOI] [PubMed] [Google Scholar]
- 8.Wang TJ, Wollert KC, Larson MG, Coglianese E, McCabe EL, Cheng S, Ho JE, Fradley MG, Ghorbani A, Xanthakis V, Kempf T, Benjamin EJ, Levy D, Vasan RS, Januzzi JL. Prognostic utility of novel biomarkers of cardiovascular stress: the Framingham Heart Study. Circulation 2012;126:1596–1604. doi: 10.1161/CIRCULATIONAHA.112.129437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borné Y, Persson M, Melander O, Smith JG, Engström G. Increased plasma level of soluble urokinase plasminogen activator receptor is associated with incidence of heart failure but not atrial fibrillation. Eur J Heart Fail 2014;16:377–383. doi: 10.1002/ejhf.49 [DOI] [PubMed] [Google Scholar]
- 10.Stenemo M, Nowak C, Byberg L, Sundström J, Giedraitis V, Lind L, Ingelsson E, Fall T, Ärnlöv J. Circulating proteins as predictors of incident heart failure in the elderly. Eur J Heart Fail 2018;20:55–62. doi: 10.1002/ejhf.980 [DOI] [PubMed] [Google Scholar]
- 11.Herwig R, Hardt C, Lienhard M, Kamburov A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc 2016;11:1889–1907. doi: 10.1038/nprot.2016.117 [DOI] [PubMed] [Google Scholar]
- 12.Jacobs L, Thijs L, Jin Y, Zannad F, Mebazaa A, Rouet P, Pinet F, Bauters C, Pieske B, Tomaschitz A, Mamas M, Diez J, McDonald K, Cleland JG, Brunner-La Rocca HP, Heymans S, Latini R, Masson S, Sever P, Delles C, Pocock S, Collier T, Kuznetsova T, Staessen JA. Heart ‘omics’ in AGEing (HOMAGE): design, research objectives and characteristics of the common database. J Biomed Res 2014;28:349–359. doi: 10.7555/JBR.28.20140045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mureddu GF, Agabiti N, Rizzello V, Forastiere F, Latini R, Cesaroni G, Masson S, Cacciatore G, Colivicchi F, Uguccioni M, Perucci CA, Boccanelli A; PREDICTOR Study Group. Prevalence of preclinical and clinical heart failure in the elderly. A population-based study in Central Italy. Eur J Heart Fail 2012;14:718–729. doi: 10.1093/eurjhf/hfs052 [DOI] [PubMed] [Google Scholar]
- 14.Beavers KM, Hsu FC, Houston DK, Beavers DP, Harris TB, Hue TF, Kim LJ, Koster A, Penninx BW, Simonsick EM, Strotmeyer ES, Kritchevsky SB, Nicklas BJ; Health ABC Study. The role of metabolic syndrome, adiposity, and inflammation in physical performance in the Health ABC Study. J Gerontol A Biol Sci Med Sci 2013;68:617–623. doi: 10.1093/gerona/gls213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shepherd J, Blauw GJ, Murphy MB, Cobbe SM, Bollen EL, Buckley BM, Ford I, Jukema JW, Hyland M, Gaw A, Lagaay AM, Perry IJ, Macfarlane PW, Meinders AE, Sweeney BJ, Packard CJ, Westendorp RG, Twomey C, Stott DJ. The design of a prospective study of Pravastatin in the Elderly at Risk (PROSPER). PROSPER Study Group. PROspective Study of Pravastatin in the Elderly at Risk. Am J Cardiol 1999;84:1192–1197. [DOI] [PubMed] [Google Scholar]
- 16.Essebag V, Genest J Jr, Suissa S, Pilote L. The nested case-control study in cardiology. Am Heart J 2003;146:581–590. doi: 10.1016/S0002-8703(03)00512-X [DOI] [PubMed] [Google Scholar]
- 17.Wacholder S, McLaughlin JK, Silverman DT, Mandel JS. Selection of controls in case-control studies. I. Principles. Am J Epidemiol 1992;135:1019–1028. [DOI] [PubMed] [Google Scholar]
- 18.Wacholder S, Silverman DT, McLaughlin JK, Mandel JS. Selection of controls in case-control studies. II. Types of controls. Am J Epidemiol 1992;135:1029–1041. [DOI] [PubMed] [Google Scholar]
- 19.Wacholder S, Silverman DT, McLaughlin JK, Mandel JS. Selection of controls in case-control studies. III. Design options. Am J Epidemiol 1992;135:1042–1050. [DOI] [PubMed] [Google Scholar]
- 20.Lundberg M, Eriksson A, Tran B, Assarsson E, Fredriksson S. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res 2011;39:e102. doi: 10.1093/nar/gkr424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Resche-Rigon M, White IR. Multiple imputation by chained equations for systematically and sporadically missing multilevel data. Stat Methods Med Res 2018;27:1634–1649. doi: 10.1177/0962280216666564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearce N. Analysis of matched case-control studies. BMJ 2016;352:i969. doi: 10.1136/bmj.i969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green GH, Diggle PJ. On the operational characteristics of the Benjamini and Hochberg false discovery rate procedure. Stat Appl Genet Mol Biol 2007;6. doi: 10.2202/1544-6115.1302 [DOI] [PubMed] [Google Scholar]
- 24.Jacobs L, Efremov L, Ferreira JP, Thijs L, Yang WY, Zhang ZY, Latini R, Masson S, Agabiti N, Sever P, Delles C, Sattar N, Butler J, Cleland JGF, Kuznetsova T, Staessen JA, Zannad F. Risk for incident heart failure: a subject-level meta-analysis from the heart “OMics” in Ageing (HOMAGE) study. J Am Heart Assoc 2017;6:e005231. doi: 10.1161/JAHA.116.005231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plugin to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richards AM. The renin-angiotensin-aldosterone system and the cardiac natriuretic peptides. Heart 1996;76(3 suppl 3):36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Majalahti T, Suo-Palosaari M, Sármán B, Hautala N, Pikkarainen S, Tokola H, Vuolteenaho O, Wang J, Paradis P, Nemer M, Ruskoaho H. Cardiac BNP gene activation by angiotensin II in vivo. Mol Cell Endocrinol 2007;273:59–67. doi: 10.1016/j.mce.2007.05.003 [DOI] [PubMed] [Google Scholar]
- 28.Hamid T, Xu Y, Ismahil MA, Li Q, Jones SP, Bhatnagar A, Bolli R, Prabhu SD. TNF receptor signaling inhibits cardiomyogenic differentiation of cardiac stem cells and promotes a neuroadrenergic-like fate. Am J Physiol Heart Circ Physiol 2016;311:H1189–H1201. doi: 10.1152/ajpheart.00904.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramamurthy V, Yamniuk AP, Lawrence EJ, Yong W, Schneeweis LA, Cheng L, Murdock M, Corbett MJ, Doyle ML, Sheriff S. The structure of the death receptor 4-TNF-related apoptosis-inducing ligand (DR4-TRAIL) complex. Acta Crystallogr F Struct Biol Commun 2015;71(pt 10):1273–1281. doi: 10.1107/S2053230X15016416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hymowitz SG, Christinger HW, Fuh G, Ultsch M, O’Connell M, Kelley RF, Ashkenazi A, de Vos AM. Triggering cell death: the crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol Cell 1999;4: 563–571. [DOI] [PubMed] [Google Scholar]
- 31.Skau E, Henriksen E, Wagner P, Hedberg P, Siegbahn A, Leppert J. GDF-15 and TRAIL-R2 are powerful predictors of long-term mortality in patients with acute myocardial infarction. Eur J Prev Cardiol 2017;24:1576–1583. doi: 10.1177/2047487317725017 [DOI] [PubMed] [Google Scholar]
- 32.Ridker PM, Lüscher TF. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J 2014;35:1782–1791. doi: 10.1093/eurheartj/ehu203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blanco-Colio LM. TWEAK/Fn14 Axis: a promising target for the treatment of cardiovascular diseases. Front Immunol 2014;5:3. doi: 10.3389/fimmu.2014.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ptaszynska-Kopczynska K, Marcinkiewicz-Siemion M, Lisowska A, Waszkiewicz E, Witkowski M, Jasiewicz M, Miklasz P, Jakim P, Galar B, Musial WJ, Kaminski KA. Alterations of soluble TWEAK and CD163 concentrations in patients with chronic heart failure. Cytokine 2016;80:7–12. doi: 10.1016/j.cyto.2016.02.005 [DOI] [PubMed] [Google Scholar]
- 35.Chen HN, Wang DJ, Ren MY, Wang QL, Sui SJ. TWEAK/Fn14 promotes the proliferation and collagen synthesis of rat cardiac fibroblasts via the NF-кB pathway. Mol Biol Rep 2012;39:8231–8241. doi: 10.1007/s11033-012-1671-3 [DOI] [PubMed] [Google Scholar]
- 36.Richter B, Rychli K, Hohensinner PJ, Berger R, Mörtl D, Neuhold S, Zorn G, Huber K, Maurer G, Wojta J, Pacher R, Hülsmann M, Niessner A. Differences in the predictive value of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in advanced ischemic and non-ischemic heart failure. Atherosclerosis 2010;213:545–548. doi: 10.1016/j.atherosclerosis.2010.08.061 [DOI] [PubMed] [Google Scholar]
- 37.Nishikimi T, Nakagawa Y. Adrenomedullin as a biomarker of heart failure. Heart Fail Clin 2018;14:49–55. doi: 10.1016/j.hfc.2017.08.006 [DOI] [PubMed] [Google Scholar]
- 38.Gandhi PU, Gaggin HK, Redfield MM, Chen HH, Stevens SR, Anstrom KJ, Semigran MJ, Liu P, Januzzi JL Jr. Insulin-like growth factor-binding protein-7 as a biomarker of diastolic dysfunction and functional capacity in heart failure with preserved ejection fraction: results from the RELAX trial. JACC Heart Fail 2016;4:860–869. doi: 10.1016/j.jchf.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Accornero F, van Berlo JH, Benard MJ, Lorenz JN, Carmeliet P, Molkentin JD. Placental growth factor regulates cardiac adaptation and hypertrophy through a paracrine mechanism. Circ Res 2011;109:272–280. doi: 10.1161/CIRCRESAHA.111.240820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gubbiotti MA, Neill T, Iozzo RV. A current view of perlecan in physiology and pathology: a mosaic of functions. Matrix Biol 2017;57–58:285–298. doi: 10.1016/j.matbio.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Segev A, Nili N, Strauss BH. The role of perlecan in arterial injury and angiogenesis. Cardiovasc Res 2004;63:603–610. doi: 10.1016/j.cardiores.2004.03.028 [DOI] [PubMed] [Google Scholar]
- 42.He XW, Li WL, Li C, Liu P, Shen YG, Zhu M, Jin XP. Serum levels of galectin-1, galectin-3, and galectin-9 are associated with large artery atherosclerotic stroke. Sci Rep 2017;7:40994. doi: 10.1038/srep40994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goncalves I, Bengtsson E, Colhoun HM, Shore AC, Palombo C, Natali A, Edsfeldt A, Dunér P, Fredrikson GN, Björkbacka H, Östling G, Aizawa K, Casanova F, Persson M, Gooding K, Strain D, Khan F, Looker HC, Adams F, Belch J, Pinnoli S, Venturi E, Kozakova M, Gan LM, Schnecke V, Nilsson J; SUMMIT Consortium. Elevated plasma levels of MMP-12 are associated with atherosclerotic burden and symptomatic cardiovascular disease in subjects with type 2 diabetes. Arterioscler Thromb Vasc Biol 2015;35:1723–1731. doi: 10.1161/ATVBAHA.115.305631 [DOI] [PubMed] [Google Scholar]
- 44.Wang N, Halibozek PJ, Yigit B, Zhao H, O’Keeffe MS, Sage P, Sharpe A, Terhorst C. Negative regulation of humoral immunity due to interplay between the SLAMF1, SLAMF5, and SLAMF6 receptors. Front Immunol 2015;6:158. doi: 10.3389/fimmu.2015.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singer BB, Opp L, Heinrich A, Schreiber F, Binding-Liermann R, Berrocal-Almanza LC, Heyl KA, Müller MM, Weimann A, Zweigner J, Slevogt H. Soluble CEACAM8 interacts with CEACAM1 inhibiting TLR2-triggered immune responses. PLoS One 2014;9:e94106. doi: 10.1371/journal.pone.0094106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stempien-Otero A, Plawman A, Meznarich J, Dyamenahalli T, Otsuka G, Dichek DA. Mechanisms of cardiac fibrosis induced by urokinase plasminogen activator. J Biol Chem 2006;281:15345–15351. doi: 10.1074/jbc.M512818200 [DOI] [PubMed] [Google Scholar]
- 47.Lichtenauer M, Jirak P, Wernly B, Paar V, Rohm I, Jung C, Schernthaner C, Kraus J, Motloch LJ, Yilmaz A, Hoppe UC, Christian Schulze P, Kretzschmar D, Pistulli R. A comparative analysis of novel cardiovascular biomarkers in patients with chronic heart failure. Eur J Intern Med 2017;44:31–38. doi: 10.1016/j.ejim.2017.05.027 [DOI] [PubMed] [Google Scholar]
- 48.Wollert KC, Kempf T, Wallentin L. Growth differentiation factor 15 as a biomarker in cardiovascular disease. Clin Chem 2017;63:140–151. doi: 10.1373/clinchem.2016.255174 [DOI] [PubMed] [Google Scholar]
- 49.Seropian IM, Toldo S, Van Tassell BW, Abbate A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol 2014;63:1593–1603. doi: 10.1016/j.jacc.2014.01.014 [DOI] [PubMed] [Google Scholar]
- 50.Panwar B, Judd SE, Wadley VG, Jenny NS, Howard VJ, Safford MM, Gutiérrez OM. Association of fibroblast growth factor 23 with risk of incident coronary heart disease in community-living adults. JAMA Cardiol 2018;3:318–325. doi: 10.1001/jamacardio.2018.0139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hao H, Li X, Li Q, Lin H, Chen Z, Xie J, Xuan W, Liao W, Bin J, Huang X, Kitakaze M, Liao Y. FGF23 promotes myocardial fibrosis in mice through activation of β-catenin. Oncotarget 2016;7:64649–64664. doi: 10.18632/oncotarget.11623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singh M, Dalal S, Singh K. Osteopontin: at the cross-roads of myocyte survival and myocardial function. Life Sci 2014;118:1–6. doi: 10.1016/j.lfs.2014.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sayer G, Bhat G. The renin-angiotensin-aldosterone system and heart failure. Cardiol Clin 2014;32:21–32, vii. doi: 10.1016/j.ccl.2013.09.002 [DOI] [PubMed] [Google Scholar]
- 54.Bruggink AH, de Jonge N, van Oosterhout MF, Van Wichen DF, de Koning E, Lahpor JR, Kemperman H, Gmelig-Meyling FH, de Weger RA. Brain natriuretic peptide is produced both by cardiomyocytes and cells infiltrating the heart in patients with severe heart failure supported by a left ventricular assist device. J Heart Lung Transplant 2006;25:174–180. doi: 10.1016/j.healun.2005.09.007 [DOI] [PubMed] [Google Scholar]
- 55.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 56.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos G, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA Guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017;136:e137–e161. doi: 10.1161/CIR.0000000000000509 [DOI] [PubMed] [Google Scholar]
- 57.de Boer RA, Nayor M, deFilippi CR, Enserro D, Bhambhani V, Kizer JR, Blaha MJ, Brouwers FP, Cushman M, Lima JAC, Bahrami H, van der Harst P, Wang TJ, Gansevoort RT, Fox CS, Gaggin HK, Kop WJ, Liu K, Vasan RS, Psaty BM, Lee DS, Hillege HL, Bartz TM, Benjamin EJ, Chan C, Allison M, Gardin JM, Januzzi JL Jr, Shah SJ, Levy D, Herrington DM, Larson MG, van Gilst WH, Gottdiener JS, Bertoni AG, Ho JE. Association of cardiovascular biomarkers with incident heart failure with preserved and reduced ejection fraction. JAMA Cardiol 2018;3:215–224. doi: 10.1001/jamacardio.2017.4987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ledwidge M, Gallagher J, Conlon C, Tallon E, O’Connell E, Dawkins I, Watson C, O’Hanlon R, Bermingham M, Patle A, Badabhagni MR, Murtagh G, Voon V, Tilson L, Barry M, McDonald L, Maurer B, McDonald K. Natriuretic peptide-based screening and collaborative care for heart failure: the STOP-HF randomized trial. JAMA 2013;310:66–74. doi: 10.1001/jama.2013.7588 [DOI] [PubMed] [Google Scholar]
- 59.Shen Y, Ding FH, Sun JT, Pu LJ, Zhang RY, Zhang Q, Chen QJ, Shen WF, Lu L. Association of elevated apoA-I glycation and reduced HDL-associated paraoxonase1, 3 activity, and their interaction with angiographic severity of coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc Diabetol 2015;14:52. doi: 10.1186/s12933-015-0221-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Floresta G, Pistarà V, Amata E, Dichiara M, Marrazzo A, Prezzavento O, Rescifina A. Adipocyte fatty acid binding protein 4 (FABP4) inhibitors. A comprehensive systematic review. Eur J Med Chem 2017;138:854–873. doi: 10.1016/j.ejmech.2017.07.022 [DOI] [PubMed] [Google Scholar]
- 61.Höbaus C, Herz CT, Pesau G, Wrba T, Koppensteiner R, Schernthaner GH. FABP4 and cardiovascular events in peripheral arterial disease. Angiology 2018;69:424–430. doi: 10.1177/0003319717728226 [DOI] [PubMed] [Google Scholar]
- 62.Leiherer A, Muendlein A, Kinz E, Vonbank A, Rein P, Fraunberger P, Malin C, Saely CH, Drexel H. High plasma chemerin is associated with renal dysfunction and predictive for cardiovascular events - Insights from phenotype and genotype characterization. Vascul Pharmacol 2016;77:60–68. doi: 10.1016/j.vph.2015.08.010 [DOI] [PubMed] [Google Scholar]
- 63.Goralski KB, McCarthy TC, Hanniman EA, Zabel BA, Butcher EC, Parlee SD, Muruganandan S, Sinal CJ. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J Biol Chem 2007;282:28175–28188. doi: 10.1074/jbc.M700793200 [DOI] [PubMed] [Google Scholar]
- 64.Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, Metra M, Anker SD, Cleland JG, Dickstein K, Filippatos G, van der Harst P, Lang CC, Ng LL, Zannad F, Zwinderman AH, Hillege HL, van der Meer P, Voors AA. Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol 2018;72:1081–1090. doi: 10.1016/j.jacc.2018.06.050 [DOI] [PubMed] [Google Scholar]
- 65.Mak TW, Hauck L, Grothe D, Billia F. p53 regulates the cardiac transcriptome. Proc Natl Acad Sci U S A 2017;114:2331–2336. doi: 10.1073/pnas.1621436114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sama IE, Huynen MA. Measuring the physical cohesiveness of proteins using physical interaction enrichment. Bioinformatics 2010;26:2737–2743. doi: 10.1093/bioinformatics/btq474 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.