

Abstract
Background and purpose
Spinocerebellar ataxia 21 (SCA21) is a rare autosomal dominant neurodegenerative disorder caused by TMEM240 gene mutations. To date, SCA21 has been reported only in a limited number of families worldwide. Here, we describe clinical and molecular findings in five additional SCA21 patients from four unrelated families, diagnosed through a multicentre next generation sequencing‐based molecular screening project on a large cohort of patients with degenerative and congenital ataxias.
Methods
A cohort of 393 patients with ataxia of unknown aetiology was selected. Following the identification of heterozygous pathogenic TMEM240 variants using a target resequencing panel, we carried out an in‐depth phenotyping of the novel SCA21 patients.
Results
Five patients from four unrelated families, three of Italian and one of Libyan origin, were identified. These patients were carriers of previously reported TMEM240 mutations. Clinically, our SCA21 cohort includes both adult onset, slowly progressive cerebellar ataxias associated with cognitive impairment resembling cerebellar cognitive affective syndrome and early onset forms associated with cognitive delay, neuropsychiatric features, or evidence of hypomyelination on brain magnetic resonance imaging. None of our patients exhibited signs of extrapyramidal involvement. The so‐called “recurrent” c.509C>T (p.Pro170Leu) mutation was detected in two of four families, corroborating its role as a hot spot.
Conclusions
Our results confirm that SCA21 is present also in Italy, suggesting that it might not be as rare as previously thought. The phenotype of these novel SCA21 patients indicates that slowly progressive cerebellar ataxia, and cognitive and psychiatric symptoms are the most typical clinical features associated with mutations in the TMEM240 gene.
Keywords: CCAS, NGS, SCA21, spinocerebellar ataxia, TMEM240
Spinocerebellar ataxia 21 (SCA21), caused by heterozygous TMEM240 mutations, so far has been rarely reported. By screening a cohort of 393, mostly Italian, ataxic patients with ataxia of unknown aetiology using a next generation sequencing‐based targeted resequencing approach, we identified five novel SCA21 patients from four unrelated families. In all of them, we detected previously described TMEM240 mutations, with the c.509C>T (p.Pro170Leu) mutation confirmed as a mutational hot spot. Thus, SCA21 might not be as rare as previously thought; slowly progressive cerebellar ataxia, and cognitive and psychiatric symptoms appear to be the most typically associated clinical features.
INTRODUCTION
Autosomal dominant spinocerebellar ataxias (SCAs) are rare neurodegenerative disorders, clinically manifesting with the core features of slowly progressive gait instability, limb incoordination, and cerebellar dysarthria. Up to 48 distinct causative SCA genes have been identified so far [1, 2] repeat expansions are a major aetiology of SCA, accounting for about 60% of cases, and conventional mutations are reported in 3%–6%, so a significant number of cases still remain undiagnosed [3].
Recently, the large‐scale application of next generation sequencing (NGS) diagnostic approaches has improved both the molecular and phenotypic characterisation of SCAs [2]. In this regard, mutations in the transmembrane 240 (TMEM240) gene have recently been associated with SCA type 21 (SCA21) [4].
So far, only 13 unrelated SCA21 families have been reported [4, 5, 6, 7], diagnosed mostly through NGS‐based targeted multigene panel (TRP) screening. A recurrent mutation (c.509C>T; p. Pro170Leu) was detected in seven of 13 families, with no evidence of a common ancestor origin. The emerging phenotype in SCA21 includes a slowly progressive cerebellar ataxia, often associated with cognitive impairment and parkinsonism, and in some cases associated with hyperkinetic movement disorders [7].
Here, we report the results of a collaborative study, based on NGS‐TRP molecular screening of a large SCA cohort that has allowed us to identify four additional SCA21 families and gather further information on its clinical manifestations.
METHODS
The study was carried out in line with the principles of the Declaration of Helsinki, and ethical approval was obtained at all three participating centres (Fondazione Policlinico Universitario A. Gemelli IRCCS, Università la Sapienza, and IRCCS Fondazione Stella Maris, Italy). We enrolled 393 subjects (326 sporadic, 67 familial) who were examined at movement disorders specialist centres and diagnosed with degenerative SCA, after ruling out other acquired aetiologies. Except for one proband coming from North Africa, two Brazilian siblings with Italian step‐parents, and one patient from Peru, all the other patients were of Italian origin. These patients tested negative for pathological repeat expansions at Friedreich's ataxia (FRDA), SCA 1, 2, 3, 6, 7, 17, and Fragile X ‐tremor ataxia syndrome (FXTAS) loci. All patients gave their informed consent to participate to the study.
The annual disease progression rate was estimated as (Scale for the Assessment and Rating of Ataxia [SARA] score at the last examination – SARA score at the first examination)/years of follow‐up. In adult patients, the neuropsychological examination included Mini‐Mental State Examination and a test battery exploring in detail all cognitive domains [8].
DNA samples were analysed at the neurogenetic laboratory of the IRCCS Fondazione Stella Maris (Pisa) through a TRP methodology (SureSelect, Agilent) that includes 273 genes related to hereditary ataxias using an Illumina NextSeq500 platform, previously described [9]. The Ingenuity Variant Analysis suite (QIAGEN) was used for variant annotation according to an in‐house validated pipeline; the impact of mutations on TMEM240 was determined in silico using 11 prediction tools [10]. Gene variants were confirmed by Sanger sequencing, followed by segregation studies whenever possible and by an in‐depth phenotyping of the five identified carriers of pathogenic variants in TMEM240.
Apart from patients with SCA21 mutations herein described, in this cohort we also identified cases presenting with SCA14 (manuscript in preparation), SCA15, SCA5, SCA19, SCA28, and SCA48 [11] mutations.
RESULTS
Demographic, clinical, and neuroimaging characteristics of the SCA21 patients diagnosed in this study are summarised in Table 1. Figure 1 illustrates their pedigrees.
TABLE 1.
Demographics and main diagnostic features of SCA21 patients
| Family | A | B | C | D | |
|---|---|---|---|---|---|
| Patient | I‐1 | II‐2 | II‐3 | II‐4 | II‐5 |
| Sex | M | M | M | M | M |
| Origin | Libya | Libya | Italy | Italy | Italy |
| Age of onset, years | 62 | 40 | 30 | 3 | 0 |
| Age at diagnosis, years | 79 | 46 | 52 | 12 | 7 |
| Symptom of onset | GA | GA | GA | Clumsiness | DD |
| Gait ataxia | +++ | + | ++ | ++ | ++ |
| Dysarthria | ++ | + | – | – | – |
| Hypokinetic signs | – | – | – | – | – |
| Hyperkinetic signs | – | – | – | – | +(DY) |
| Ocular findings | Ny | Ny | HS | HS | OP |
| Tremor | – | + (postural) | + (postural) | + (intention) | + (intention and postural) |
| Psychomotor delay | – | – | – | + | ++ |
| Cognitive decline | ++ | + | + | – | – |
| SARA at first visit | 15 | 6.5 | 10 | 5 | n.d. |
| SARA at last visit | 18 | 6.5 | 23 | 6 | n.d. |
| Timespan of FU | 4 years | – | 7 years | 7 years | 6 years |
| Progression rate | 0.6/year | – | 0.8/year | 0.16/year | – |
| Neuroimaging | CA | n.d. | CA | CA | DM |
| Behaviour disorder | – | – | – | + | ++ |
| Others | – | REM‐BD | Strabismus | Hearing loss | – |
| Mutation |
c.509C>T p.Pro170Leu |
c.509C>T p.Pro170Leu |
c.509C>T p.Pro170Leu |
c.239C>T p.Thr80Met |
c.196G>A p.Gly66Arg |
Abbreviations: –, not present; +, mild; ++, moderate; +++, severe; CA, cerebellar atrophy; DY, dystonia; FU, follow‐up; GA, gait ataxia; HS, hypermetric saccades; ID, developmental delay; M, male; n.d., not determined; Ny, nystagmus; OP, Opsoclonus; REM‐BD, REM sleep‐behavior disorder; SARA, Scale for the Assessment and Rating of Ataxia.
FIGURE 1.
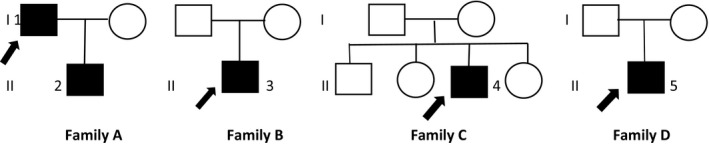
Family trees of Families A, B, C, and D are displayed. Numbers indicate patients as listed in the text. Black arrows indicate probands
The index case of Family A (Patient 1) is a Libyan 80‐year‐old male who developed progressive gait ataxia and speech problems during his 60s, later complicated by mild signs of cognitive impairment. His first neurological examination, at the age of 74 years, showed slurred speech, moderate trunk and limb ataxia (SARA score = 15), and a gaze‐evoked nystagmus. Clinical revaluation 4 years later documented a mild progression of ataxic symptoms (SARA score = 18, annual progression rate = 0.6 points/year).
His 45‐year‐old son (Family A, Patient 2) referred for mild gait imbalance and rapid eye movement sleep behaviour disorder, both started around 40 years of age. His examination showed very mild cerebellar signs (SARA score = 6.5) together with slight upper limb postural tremor and mild executive and verbal cognitive impairment.
Patient 3 (Family B), was reported to have strabismus, mild hand tremor, and clumsiness causing frequent falls when he was a child, with a subsequent progressive improvement of symptoms during adolescence. In his 30s, he noticed progressive gait difficulties. Clinical examination at the age of 45 years showed cerebellar ataxia and postural upper limb tremor. After a 7‐year follow‐up, a progression in SARA score from 10 to 23 (1.8 points/year) was documented.
Patient 4 (Family C) is a 13‐year‐old boy who presented at the age of 3 years with clumsiness. A first neurological examination at 6 years showed mild gait and limb ataxia (SARA score = 5) and mild intellectual disability (global intelligence quotient score = 65); at the age of 13 years, his SARA score was 6 (0.16 points/year).
The earliest onset of symptoms occurred in Patient 5 (Family D), a 7‐year‐old child born of unrelated healthy parents, with a global developmental delay (trunk control at 12 months, walking at 24 months, and first words by 18 months). By 1 year of age, he showed truncal ataxia, dysmetria, and intention tremor. During the following 4 years, his motor functioning progressively improved and his clinical picture was dominated by anxiety, hyperactivity, attention problems, irritability, and impulse control disorder. His last examination, at the age of 7 years (Video S1), showed a prominent ocular movement disorder characterised by opsoclonus, associated with slight appendicular ataxia and distal dystonic posturing of the hands associated with jerky movements when arms were held extended.
Overall, in all our probands brain magnetic resonance imaging (MRI) showed cerebellar atrophy, more pronounced in the vermis (Figure 2); moreover, a brain MRI performed at 2 years of age in Patient 5 displayed signs of delayed myelination (not shown).
FIGURE 2.
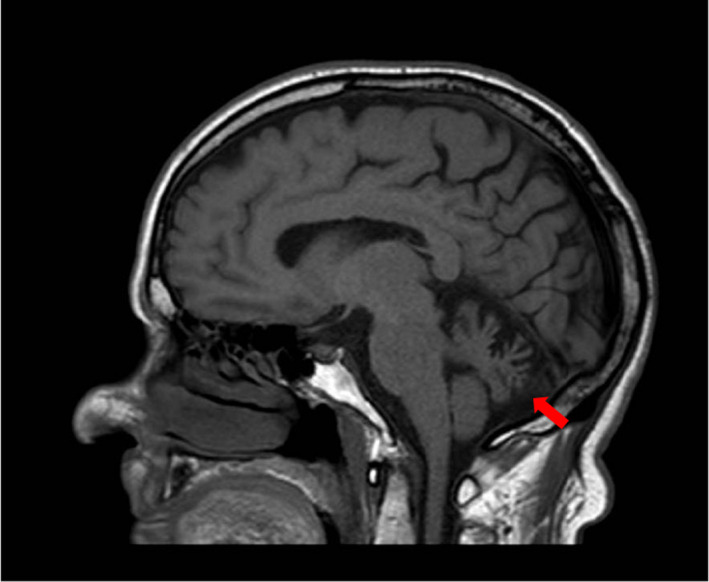
T1‐weighted, sagittal brain magnetic resonance imaging sequence of Patient 1, showing cerebellar atrophy (red arrow) as the main common neuroimaging finding in our spinocerebellar ataxia 21 cohort [Colour figure can be viewed at wileyonlinelibrary.com]
Three previously described pathogenic missense mutations in TMEM240 were detected in the four unrelated SCA families of this study: c.509C>T (p. Pro170Leu), c.239C>T (p. Thr80Met), and c.196G>A (p. Gly66Arg). Family segregation studies indicated that c.196G>A occurred as a de novo event in Patient 5, whereas they could not be performed for Patient 4 to confirm the sporadic occurrence of the c.239C>T variant.
DISCUSSION
We report five novel SCA21 cases belonging to three Italian and one Libyan family. Overall, 17 families from Europe, Asia, Africa, and South America have been described, indicating that SCA21 occurs worldwide.
Regarding the clinical presentation of SCA21, various reports expanded its phenotypic spectrum from the initial report [4] including various movement disorders, neuropsychiatric symptoms, and childhood onset forms [5, 6, 7] A recent review of SCA21 case series [7] has pointed out that the disease course of childhood cases is characterised by a temporary improvement of the neurological symptoms during the first 5–10 years of life, and then becomes a slowly progressive ataxia during adulthood. Accordingly, in our Cases 3 and 5, a clear improvement was evident from a motor standpoint during childhood.
Our data confirm the wide heterogeneity of SCA21 and its variable age of onset; two of five patients showed a very early onset (<3 years), whereas symptoms occurred during adulthood in the other three cases. All patients manifested with a prevalent cerebellar motor phenotype and related ocular abnormalities (illustrated in Table 1), either associated with cognitive delay or cerebellar cognitive affected syndrome (CCAS), depending on the disease onset (infantile or adult, respectively). The occurrence of CCAS in SCA21 was first reported by Braga‐Neto et al. [12]; similar dysexecutive cognitive symptoms were detected by neuropsychological assessment in our Patients 1 and 2. Moreover, as previously described in early onset SCA21 patients [6, 7] both Patients 4 and 5 showed a developmental cognitive defect and severe impairment in emotions and affective control.
In line with current literature [4, 7] we observed a relatively benign course of cerebellar symptoms in SCA21, with a slow progression of ataxia in late onset and a relative stability in early onset cases. Conversely, only Patient 5 in our SCA21 cohort manifested mild extrapyramidal symptoms, mainly consisting of slight dystonic posturing of the hands (Video S1).
All brain MRI available of our adult or adolescent patients showed cerebellar atrophy, more pronounced in the vermis; of note, Patient 5, on whom brain MRI was performed at 2 years, displayed signs of delayed myelination, which represents a novel described feature in SCA21.
All detected pathogenic variants reported in this study have already been described previously; our results confirm that c.509C>T (p.Pro170Leu) is a mutational hot spot in TMEM240. Interestingly, regarding the missense variant c.196G>A, the two siblings who were carriers of the same mutation originally described [7] manifested with important neuropsychiatric disorders, similarly to our Patient 5, thus suggesting a possible genotype–phenotype correlation for this particular mutation.
TMEM240 encodes a transmembrane protein of unknown function; recently, it has been shown [13] that its product is expressed in the whole mouse and human brain and is localised in all the afferents fibres projecting to Purkinje cells and in glutamatergic synaptic terminals, suggesting a probable primary role of TMEM240 protein in the whole cerebellar network and thus explaining the motor, cognitive, and social impairment of SCA21 patients.
In conclusion, we described four additional SCA21 families, documenting the presence of this mutation also in Italy and in North Africa. Our study also confirms that patients with early onset cerebellar ataxias associated with mental delay and behavioural problems and with adult onset slowly progressive degenerative cerebellar ataxias associated with CCAS, with or without extrapyramidal features, should be screened for SCA21 mutations.
CONFLICT OF INTEREST
S.R. received a fellowship partially granted by the nonprofit organization Associazione Italiana Vivere la Paraparesi Spastica. F.M.S. received funds from the E‐Rare‐3 Joint Transnational Call grant PREPARE (Ministry of Health Project 3398) and from the Italian Ministry of Health, Ricerca Finalizzata RF‐2016–02361610.
AUTHOR CONTRIBUTIONS
Vittorio Riso: data curation (equal), formal analysis (equal), writing–original draft (equal). Daniele Galatolo: formal analysis (equal), methodology (equal). Melissa Barghigiani: formal analysis (equal), validation (equal). Serena Galosi: data curation (equal), writing–review & editing (equal). Alessandra Tessa: methodology (equal), supervision (equal). Ivana Ricca: data curation (equal), formal analysis (equal). Salvatore Rossi: data curation (equal), writing–review & editing (equal). Caterina Caputi: investigation (equal), visualisation (equal). Ettore Cioffi: data curation (equal), investigation (equal). Vincenzo Leuzzi: conceptualisation (equal), writing–review & editing (equal). Carlo Casali: conceptualisation (equal), supervision (equal). Filippo M. Santorelli: conceptualisation (equal), supervision (equal), writing–review & editing (equal). Gabriella Silvestri: conceptualisation (equal), writing–review & editing, supervision (equal).
Supporting information
Video S1. Segment 1–3: Saccadic intrusions such as opsoclonus are particularly evident in segment 1 superimposed during smooth pursuit and in segment 2, occurring spontaneously while the child is talking with the operator. Outside of saccadic intrusions he has normal saccades initiation and velocity and full range ocular pursuit with only mild fixation instability. Segment 4: Slight distal dystonic posturing of the hands associated with jerky movements when arms are held extended. Segment 5, 6: Mild appendicular ataxia manifesting as dysdiadochokinesia during rapid alternating hand movement test and upper limb dysmetria at the finger to nose test. Segment 7, 8: The child is able to stand with feet together in parallel with only minimal body sway and to stand on one foot; he has no difficulties in walking, turning, running, and jumping.
ACKNOWLEDGMENTS
S.R. received a fellowship partially granted by the nonprofit organization Associazione Italiana Vivere la Paraparesi Spastica. The molecular studies in Pisa were partially supported by the Italian Ministry of Health, Ricerca Finalizzata RF‐2016‐02361610 (to F.M.S.) and the E‐Rare‐3 Joint Transnational Call grant PREPARE (Ministry of Health Project 3398 to F.M.S.). We acknowledge Dr. Anna Maino for revision of the English manuscript.
See letter by C. H. F. Camargo et al. on page e63.
Contributor Information
Filippo M. Santorelli, Email: filippo3364@gmail.com.
Gabriella Silvestri, Email: gabriella.silvestri@unicatt.it.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available upon request from the corresponding authors.
REFERENCES
- 1.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885‐894. 10.1016/S1474-4422(10)70183-6 [DOI] [PubMed] [Google Scholar]
- 2.Klockgether MC, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019;5:24. 10.1038/s41572-019-0074-3 [DOI] [PubMed] [Google Scholar]
- 3.Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42:174‐183. 10.1159/000358801 [DOI] [PubMed] [Google Scholar]
- 4.Delplanque J, Devos D, Huin V, et al. TMEM240 mutations cause spinocerebellar ataxia 21 with mental retardation and severe cognitive impairment. Brain. 2014;137:2657‐2663. 10.1093/brain/awu202 [DOI] [PubMed] [Google Scholar]
- 5.Zeng S, Zeng J, He M, et al. Spinocerebellar ataxia type 21 exists in the Chinese Han population. Sci Rep. 2016;6:19897. 10.1038/srep19897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yahikozawa H, Miyatake S, Sakai T, et al. A Japanese family of spinocerebellar ataxia type 21: clinical and neuropathological studies. Cerebellum. 2018;17:525‐530. 10.1007/s12311-018-0941-6 [DOI] [PubMed] [Google Scholar]
- 7.Traschütz A, van Gaalen J, Oosterloo M, et al. The movement disorder spectrum of SCA21 (ATX‐TMEM240): 3 novel families and systematic review of the literature. Parkinsonism Relat Disord. 2019;62:215‐220. 10.1016/j.parkreldis.2018.11.027 [DOI] [PubMed] [Google Scholar]
- 8.Carlesimo GA, Caltagirone C, Gainotti G. Group for the standardization of the mental deterioration battery. The mental deterioration battery. Eur Neurol. 1996;36:378‐384. 10.1159/000117297 [DOI] [PubMed] [Google Scholar]
- 9.Ricca I, Morani F, Bacci GM, et al. Clinical and molecular studies in two new cases of ARSACS. Neurogenetics. 2019;20:45. 10.1007/s10048-019-00564-7 [DOI] [PubMed] [Google Scholar]
- 10.D'Amore A, Tessa A, Casali C, et al. Next generation molecular diagnosis of Hereditary Spastic Paraplegias: an Italian cross‐sectional study. Front Neurol. 2018;9:981. 10.3389/fneur.2018.00981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lieto M, Riso V, Galatolo D, et al. The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. Eur J Neurol. 2020;27(3):498‐505. 10.1111/ene.14094 [DOI] [PubMed] [Google Scholar]
- 12.Braga‐Neto P, Pedroso J, Barsottini O, Schmahmann JD. Cognition in SCA21 reflects developmental and adult onset cerebellar cognitive affective syndrome. Brain. 2015;138(Pt 7):e364. 10.1093/brain/awu382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Homa M, Loyens A, Eddarkaoui S. The TMEM240 protein, mutated in SCA21, is expressed in Purkinje cells and synaptic terminals. Cerebellum. 2020;19(3):358‐369. 10.1007/s12311-020-01112-y [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Segment 1–3: Saccadic intrusions such as opsoclonus are particularly evident in segment 1 superimposed during smooth pursuit and in segment 2, occurring spontaneously while the child is talking with the operator. Outside of saccadic intrusions he has normal saccades initiation and velocity and full range ocular pursuit with only mild fixation instability. Segment 4: Slight distal dystonic posturing of the hands associated with jerky movements when arms are held extended. Segment 5, 6: Mild appendicular ataxia manifesting as dysdiadochokinesia during rapid alternating hand movement test and upper limb dysmetria at the finger to nose test. Segment 7, 8: The child is able to stand with feet together in parallel with only minimal body sway and to stand on one foot; he has no difficulties in walking, turning, running, and jumping.
Data Availability Statement
The data that support the findings of this study are available upon request from the corresponding authors.