

Abstract
Objective
We have previously reported that stimulation of mouse bone marrow–derived macrophages with tumor necrosis factor (TNF) and interleukin‐6 (IL‐6) induces differentiation of osteoclast‐like cells. We undertook this study to clarify the characterization and function of human TNF and IL‐6–induced osteoclasts using peripheral blood collected from patients with rheumatoid arthritis (RA) and healthy donors.
Methods
Peripheral blood monocytes were cultured with a combination of TNF and IL‐6, TNF alone, IL‐6 alone, or with RANKL, and their bone resorption ability was evaluated. Expression levels of NFATc1, proinflammatory cytokines, and matrix metalloproteinase 3 were analyzed. The effects of NFAT inhibitor and JAK inhibitor were examined. Furthermore, the relationship between the number of TNF and IL‐6–induced osteoclasts or RANKL‐induced osteoclasts differentiated from peripheral blood mononuclear cells (PBMCs) in patients with RA and the modified total Sharp score (mTSS) or whole‐body bone mineral density (BMD) was examined.
Results
Peripheral blood monocytes stimulated with a TNF and IL‐6–induced osteoclasts were shown to demonstrate the ability to absorb bone matrix. Cell differentiation was not inhibited by the addition of osteoprotegerin. Stimulation with a combination of TNF and IL‐6 promoted NFATc1 expression, whereas the NFAT and JAK inhibitors prevented TNF and IL‐6–induced osteoclast formation. Expression levels of IL1β, TNF, IL12p40, and MMP3 were significantly increased in TNF and IL‐6–induced osteoclasts, but not in RANKL‐induced osteoclasts. The number of TNF and IL‐6–induced osteoclasts differentiated from PBMCs in patients with RA positively correlated with the mTSS, whereas RANKL‐induced osteoclast numbers negatively correlated with the whole‐body BMD of the same patients.
Conclusion
Our results demonstrate that TNF and IL‐6–induced osteoclasts may contribute to the pathology of inflammatory arthritis associated with joint destruction, such as RA.
INTRODUCTION
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by polyarthritis and joint destruction. Although recent advances in treatments have enabled us to reduce disease activity among RA patients, it is still difficult to “cure” the disease and halt joint destruction. Thus, preventing bone and joint destruction in RA patients who have a poor prognosis has long been a goal for rheumatologists in the treatment of RA, and new therapeutic strategies and targets need to be urgently developed.
Osteoclasts have long been considered the only cells capable of absorbing bone matrix in vivo. They are differentiated from monocytes/macrophage‐lineage precursor cells that are derived from bone marrow hematopoietic cells. It is believed that osteoclast differentiation is firmly dependent on the receptor activator of NF‐κB ligand/RANK signaling (1). The hyperactivation of osteoclasts is implicated in osteoporosis and the pathogenesis of inflammatory arthritis with bone destruction and functional disability, such as RA (2). In fact, drugs targeting osteoclasts, such as bisphosphonates and anti‐RANKL antibodies, are used in treating osteoporosis worldwide. However, there is scarce evidence that bisphosphonates prevent bone destruction in RA. Furthermore, anti‐RANKL antibodies are approved only in Japan for the prevention of joint structural damage in patients with RA. Taken together, the role of RANKL/RANK signaling–dependent osteoclast differentiation in RA may be overstated.
A recently emerging hypothesis is that “alternative pathways of osteoclastogenesis” may be functioning during chronic inflammatory conditions such as RA (3). This hypothesis has been supported by many experimental findings showing that infiltrating inflammatory cells and the cytokine milieu provide multiple routes to bone destruction (4, 5).
In RA, tumor necrosis factor (TNF) and interleukin‐6 (IL‐6) are the two major proinflammatory cytokines that trigger bone destruction (6). TNF and IL‐6 were shown to directly induce the activation of osteoclasts by binding to their respective surface receptors and indirectly inducing the expression of RANKL on fibroblast‐like synoviocytes (7, 8). Consequently, a blockade of TNF or IL‐6 impedes or arrests the progression of bone destruction in RA (9), which is observed even when antiinflammatory responses are not induced (10, 11).
In previous studies, we have demonstrated that a combination of TNF and IL‐6 induces mouse osteoclast‐like cells with bone resorption activity both in vitro and in vivo (12). More recently, O’Brien and colleagues verified our findings by reporting that the combination of TNF and IL‐6 induced the differentiation of osteoclasts (13). They further demonstrated that synovial tartrate‐resistant acid phosphatase (TRAP)–positive multinucleated cells contributed to bone erosion in the arthritic joints of Rank‐deficient mice with K/BxN serum‐transfer arthritis. Thus, RANK‐independent osteoclastogenesis occurs in inflamed joints. However, the precise characteristics and function of human TNF and IL‐6–induced osteoclasts in RA are unknown.
In this study, we investigated the characteristics and function of human TNF and IL‐6–induced osteoclasts using peripheral blood collected from patients with RA and from healthy donors. In addition, we also analyzed the differences in the novel molecular expression patterns and functions of TNF and IL‐6–induced osteoclasts as compared to those of RANKL‐induced osteoclasts.
PATIENTS AND METHODS
Samples from patients with RA and healthy donors
All patients with RA fulfilled the 2010 American College of Rheumatology (ACR)/European Alliance of Associations for Rheumatology criteria for RA (14) or the 1987 ACR classification criteria for RA (15). Demographic and clinical characteristics of the RA patients are provided in Supplementary Table 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41666/abstract. Ten healthy donors were also enrolled in the study as a control group. All patients and healthy donors provided written informed consent prior to sample collection. The study was reviewed and approved by the Saitama Medical University Hospital Institutional Review Board (no. 15‐129) and Ethics Committee (no. 836).
In vitro assays for TNF and IL‐6–induced osteoclasts and osteoclast differentiation and function
Peripheral blood mononuclear cells (PBMCs) were isolated from the peripheral blood of patients with RA and healthy donors using Ficoll‐Paque Plus gradient centrifugation (16). Peripheral blood monocytes were isolated from whole PBMCs using the Human Monocyte Isolation Kit (StemCell Technologies) according to the manufacturer’s protocol. PBMCs or peripheral blood monocytes were cultured in α‐minimum essential medium (Gibco) supplemented with 10% fetal bovine serum, 50 units/ml of penicillin/streptomycin (Gibco), and 50 ng/ml of macrophage colony‐stimulating factor (M‐CSF) (R&D Systems). PBMCs were cultured at a cell density of 7.5 × 105 cells per well in 48‐well plates (17). In addition, peripheral blood monocytes were cultured at a cell density of 7.5 × 104 cells per well in 96‐well plates, 2.0 × 105 cells per well in 24‐well plates, and 5 × 106 cells in 6‐cm dishes for 3 days.
Following the initial 3‐day culture period, PBMCs or peripheral blood monocytes were used as monocyte‐derived macrophages and cultured in medium supplemented with 10 ng/ml or 50 ng/ml of RANKL, TNF, IL‐6, a combination of TNF/IL‐6, or IL‐1β (all from PeproTech). The medium was replenished with fresh medium every 2 days until various assays were performed. TRAP was assayed with a TRAP Staining Kit (Cosmo Bio) according to the manufacturer’s instructions. Osteoprotegerin (OPG; R&D Systems), NFAT inhibitor tacrolimus (FK‐506; Sigma‐Aldrich), the pan JAK inhibitor tofacitinib (CP690550; SelleckChem), and a rabbit anti–IL‐1β polyclonal antibody (ab9722; Abcam) were added at the same time as RANKL or proinflammatory cytokines. For the pit formation assay, monocyte‐derived macrophages cultured on dentine slices (Wako) were cultured for 14 days in the presence of the cytokines. After removing the cells, resorption pits were examined using a S‐4800 electron microscope (Hitachi). In addition, resorption pits were visualized by toluidine blue staining, and the number of resorption pits per dentin slice was counted under a BZ‐X700 microscope (Keyence).
Real‐time quantitative polymerase chain reaction (qPCR)
Total RNA was isolated with the RNeasy Micro Kit (Qiagen), and complementary DNA (cDNA) was generated by reverse transcription using random hexamers and MultiScribe reverse transcriptase (ThermoFisher). Expression levels of messenger RNAs (mRNAs) were determined by TaqMan real‐time PCR on an ABI Prism 7000 Sequence Detection System (Thermo Fisher). Primers for the detection of NFATc1, IL1β, TNF, IL12p40, IL10, MMP3, and CTSK were purchased from ThermoFisher. The expression level of the housekeeping gene GAPDH was used as an endogenous control. To calculate fold changes in expression, the comparative threshold cycle (Ct) method was used as previously described (16).
Enzyme‐linked immunosorbent assays (ELISAs) for detection of IL‐1β, TNF, and IL‐6
Levels of IL‐1β protein in the cell supernatants were detected with a DuoSet ELISA development kit (R&D Systems) according to the manufacturer’s instructions. Levels of TNF and IL‐6 proteins in the cell supernatants were detected with a human TNF and IL‐6 standard ABTS ELISA development kit (PeproTech) according to the manufacturer’s instructions.
Western blot analysis and immunofluorescence staining
For Western blot analysis, whole cell lysates from a 60‐mm polystyrene tissue culture plate were dissolved in sample buffer (62.5 mM Tris HCl buffer, 10% glycerol, 2% sodium dodecyl sulfate [SDS], 1/20 β‐mercaptoethanol, and 0.0025% bromophenol blue [BPB]) and boiled at a temperature of 95°C for 10 minutes. The samples were centrifuged for 5 minutes at 3,000 revolutions per minute, and proteins in the supernatant were separated by electrophoresis using 7.5% polyacrylamide gel. The separated proteins were transferred to a PVDF membrane using a semi‐dry blot transfer system (Bio‐Rad Laboratories). PVDF membranes were blocked with 5% nonfat dry milk in Tris buffered saline–Tween (TBST) (Tris HCl buffer, pH 7.9, 0.9% NaCl, 0.01% Tween 20) for 3 hours, and then incubated overnight at a temperature of 4°C with a primary mouse anti‐NFATc1 monoclonal antibody (7A6) (Santa Cruz Biotechnology) in a 1:500 dilution of TBST. The membranes were washed with TBS solution 3 times and then incubated with a 1:2,000 dilution of an anti‐mouse IgG peroxidase‐conjugated secondary antibody (Binding Site) for 1 hour (1). Chemiluminescence assay (GE Healthcare) was used for the detection of the target protein. Expression levels of the protein were analyzed using densitometry (Atto AE‐6920‐MF). The housekeeping protein β‐actin (Sigma‐Aldrich) with a dilution of 1:2,000 was used as a loading control.
For immunofluorescence staining, the cultured TNF and IL‐6–induced osteoclasts, as well as RANKL‐induced osteoclasts as controls, were washed twice in phosphate buffered saline (PBS) and then fixed in 4% paraformaldehyde at room temperature for 20 minutes. The fixed cells were then washed in PBS followed by 2% bovine serum albumin (BSA) in PBS with 0.1% Triton X‐100 for 10 minutes. The cells were then incubated 1 hour at a temperature of 37°C with a primary rabbit anti–IL‐1β polyclonal antibody (ab9722; Abcam) in a 1:100 dilution of PBS with 2% BSA; rabbit IgG served as the negative control. After washing the cells in PBS 3 times, the cells were incubated for 45 minutes at room temperature with a goat anti‐rabbit IgG–fluorescein isothiocyanate secondary antibody (sc‐2012; Santa Cruz Biotechnology) in a 1:100 dilution of PBS with 2% BSA. Nuclear staining was performed with DAPI. After washing the cells in PBS twice, the cells were observed using a BZ‐X700 fluorescence microscope.
Clinical assessments
Clinical information was obtained by reviewing electronic medical records. In patients with RA, the modified total Sharp score (mTSS) was evaluated by 2 rheumatologists (KY and NK) who have >20 years of experience and expertise in musculoskeletal radiology and who were blinded with regard to the clinical information and laboratory data of the study participants (18). In patients with RA, the whole‐body bone mineral density (BMD) was measured by dual‐energy x‐ray absorptiometry (Discovery DXA System; Hologic).
Statistical analysis
Values are presented as the mean ± SEM. Comparisons between 2 groups were performed using the Mann‐Whitney U test or Wilcoxon’s signed rank test for paired data. Correlations between the number of TNF and IL‐6–induced osteoclasts or RANKL‐induced osteoclasts and the mTSS or whole‐body BMD was calculated using Spearman’s rank correlation test. P values less than 0.05 were considered significant.
RESULTS
Differentiation of TRAP‐positive multinucleated TNF and IL‐6–induced osteoclasts produced by the combination of TNF and IL‐6 in vitro
TRAP expression and multinucleated cells are defining features of osteoclasts (19). IL‐6 has been demonstrated to trigger direct osteoclast formation and induce bone resorption and has been considered to play an indirect role in inducing RANKL on osteoblasts and stromal cells (20, 21). In the present study, we differentiated RANKL‐induced osteoclasts in vitro by culturing peripheral blood monocytes with M‐CSF and then adding RANKL and M‐CSF (Figures 1A–C). Our findings showed that IL‐6 induced a low number of TRAP‐positive cells and few multinucleated cells (Figures 1B and C). When another major proinflammatory cytokine, TNF, was added to this culture system, only a few multinucleated cells were observed, although TRAP‐positive cells were abundant.
Figure 1.
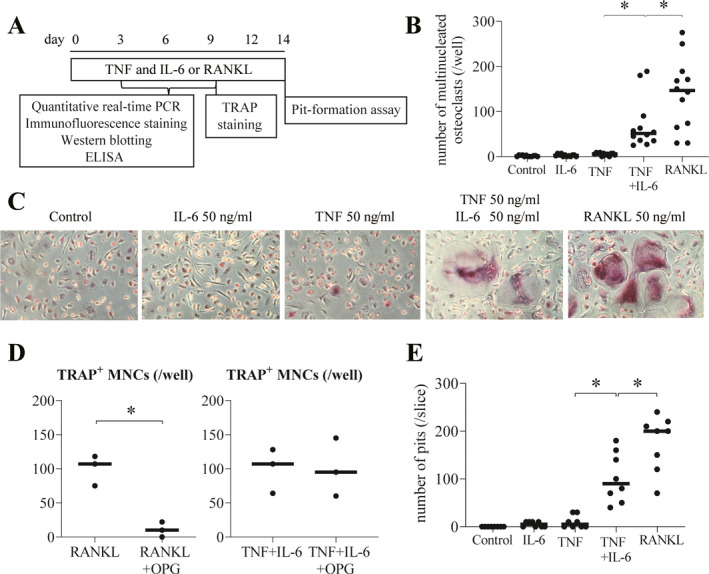
Tumor necrosis factor (TNF) and interleukin‐6 (IL‐6)–induced osteoclasts display bone resorption activity in a RANKL‐independent manner. A, Schematic representation of the culture system used in the in vitro experiments. B, Quantification of the number of tartrate‐resistant acid phosphatase–positive (TRAP+) multinucleated (i.e., those with ≥3 nuclei) osteoclasts per well (n = 12). C, Photomicrographs of TRAP‐positive multinucleated cells (MNCs). Original magnification × 100. D, Effect of osteoprotegerin (OPG) (1 mg/ml) on RANKL‐induced osteoclastogenesis and TRAP‐positive MNC differentiation induced by a combination of TNF and IL‐6 (n = 3). E, Quantification of the number of resorption pits per dentin slice (n = 4). In B, D, and E, symbols represent individual samples; values are the mean ± SEM. * = P < 0.05. PCR = polymerase chain reaction; ELISA = enzyme‐linked immunosorbent assay.
It has been reported that TNF induces cell fusion in human monocyte‐derived macrophages and causes them to differentiate into a few multinucleated cells (22). In the present study, we observed the effects of TNF stimulation on the morphology of the cells differed from those in cultures with IL‐6 stimulation (Figure 1C). Interestingly, however, the combination of TNF and IL‐6 induced the formation of multiple TRAP‐positive multinucleated cells (Figures 1B and C). These cells morphologically differ from those induced by RANKL as follows: 1) the number of osteoclasts induced by TNF and IL‐6 was less than that of osteoclasts induced by RANKL, and 2) the intensity of staining for TRAP among osteoclasts induced by TNF and IL‐6 was slightly weaker than that of osteoclasts induced by RANKL.
OPG is known as a decoy receptor for RANKL and an osteoclastogenesis inhibitory factor (23). We added OPG to the culture system to determine whether RANKL induced by TNF and IL‐6 in monocyte‐derived macrophages was involved in the differentiation of TRAP‐positive multinucleated cells. OPG inhibited osteoclastogenesis induced by RANKL, whereas OPG did not inhibit the differentiation of TRAP‐positive multinucleated cells induced by the combination of TNF and IL‐6 (Figure 1D). Moreover, in cell cultures stimulated with the combination of TNF and IL‐6, we confirmed the functional role of TRAP‐positive multinucleated cells. Stimulation of monocyte‐derived macrophages with TNF or IL‐6 alone did not generate resorption pits on the dentin slices. In contrast, stimulation with TNF plus IL‐6 strongly generated resorption pits in a manner similar to those generated by stimulation with RANKL (Figure 1E and Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41666/abstract). These findings suggest that TNF and IL‐6 induced the differentiation of TRAP‐positive multinucleated cells (named here as TNF and IL‐6–induced osteoclasts) as the bone‐resorbing cells in a RANKL‐independent manner.
Necessary role of NFATc1 and JAK activity in the differentiation of TNF and IL‐6–induced osteoclasts
We next examined the mechanisms underlying the differentiation of TNF and IL‐6–induced osteoclasts. The master regulator transcription factors involved in osteoclast differentiation are thought to be c‐Fos and NFATc1 (24). We have previously reported that the expression level and activity of c‐Fos and NFATc1 are critical for the differentiation of mouse osteoclast‐like cells induced by TNF and IL‐6 (12). During the course of TNF and IL‐6–induced osteoclast differentiation, NFATc1 protein and mRNA levels were clearly up‐regulated after stimulation with TNF and IL‐6 (Figures 2A and B). As expected, the NFAT inhibitor tacrolimus (FK‐506) inhibited the differentiation of TNF and IL‐6–induced osteoclasts (Figure 2C). Thus, we demonstrated that NFATc1 activity is necessary for the differentiation of TNF and IL–6‐induced osteoclasts.
Figure 2.
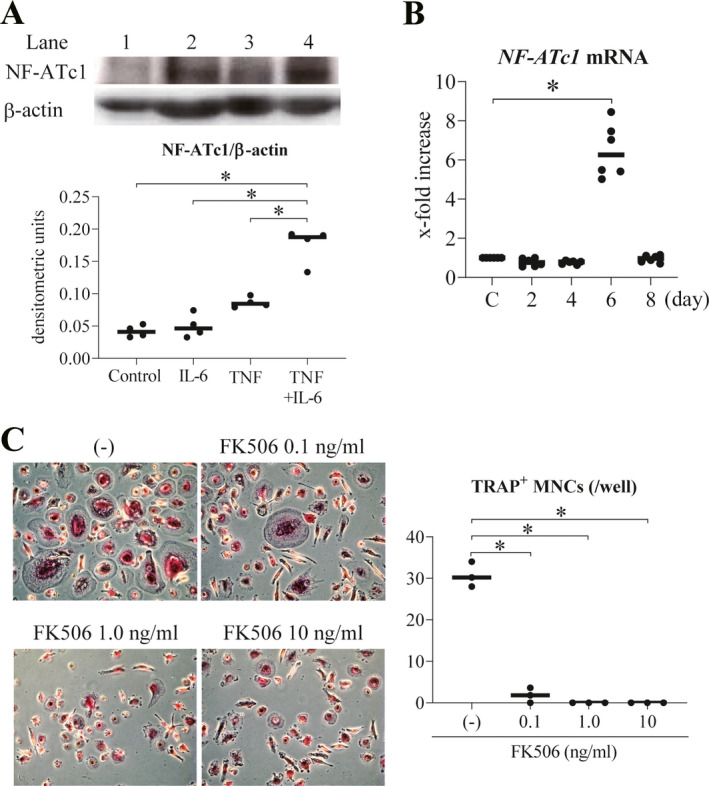
NFATc1 activity necessary for the differentiation of TNF and IL‐6–induced osteoclasts. A, Top, Western blot analysis of NFATc1 after 8 days of stimulation of peripheral blood monocyte‐derived macrophages with IL‐6, TNF, and TNF plus IL‐6 (lanes 2–4, respectively). Control cells were left untreated (lane 1). Bottom, Western blot detection of NFATc1 protein expression relative to β‐actin in macrophages (n = 4). B, Time course of the expression levels of NFATc1 mRNA in peripheral blood monocyte‐derived macrophages stimulated with TNF plus IL‐6 (n = 6). Changes in mRNA are assessed as the fold increase relative to that in unstimulated controls. C, Effects of various doses of the NFAT inhibitor tacrolimus (FK506) on the differentiation of TRAP‐positive multinucleated TNF and IL–6‐induced osteoclasts, assessed by immunofluorescence (original magnification × 50) (left), with results quantified as the mean number of TRAP‐positive MNCs per well (n = 3) (right). Symbols represent individual samples; values are the mean ± SEM. * = P < 0.05. See Figure 1 for other definitions.
Intracellular signals involving IL‐6 are largely transmitted via the JAK/STAT pathway (25). We have previously reported the dependence of mouse osteoclast‐like cells, but not RANKL‐induced osteoclasts, on JAK (12). Thus, we examined whether addition of the pan‐JAK inhibitor tofacitinib inhibited the differentiation of TNF and IL‐6–induced osteoclasts derived from PBMCs of healthy donors. Tofacitinib has recently been shown to be effective against RA (26). Here, we showed that in vitro addition of tofacitinib inhibited the differentiation of TNF and IL‐6–induced osteoclasts in a dose‐dependent manner (Figure 3A and Supplementary Figure 2A, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41666/abstract). In this case, TRAP positivity was not affected. Like our previous report on mouse osteoclast‐like cells (12), the same concentrations of tofacitinib did not inhibit osteoclastogenesis induced by RANKL (Figure 3A and Supplementary Figure 2B). Next, we confirmed these findings using cells from multiple independent donors with RA. The results show that tofacitinib inhibited the differentiation of TNF and IL‐6–induced osteoclasts derived from PBMCs in RA patients when compared to findings in PBMCs of healthy donors (Figure 3B).
Figure 3.
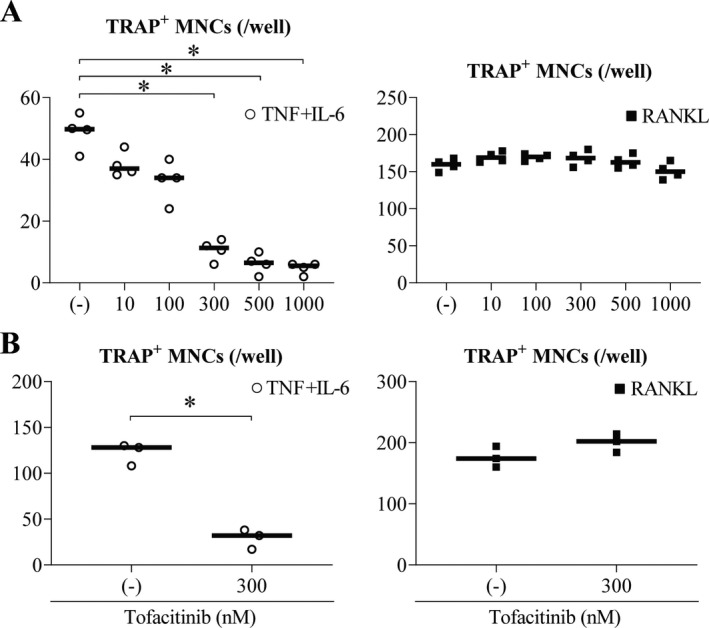
Inhibition of TNF and IL‐6–induced osteoclast differentiation by the JAK inhibitor tofacitinib. A, Various concentrations of tofacitinib were tested for their inhibitory effects on the differentiation of TRAP‐positive TNF and IL‐6–induced multinucleated osteoclasts (left) compared to TRAP‐positive RANKL‐induced osteoclasts (right) from peripheral blood monocytes of healthy donors (n = 4 each). B, A dose of 300 nM of tofacitinib was tested for its inhibitory effects on the differentiation of TRAP‐positive TNF and IL‐6–induced MNCs from peripheral blood monocytes of patients with rheumatoid arthritis (left) compared to TRAP‐positive RANKL‐induced MNCs from peripheral blood monocytes of healthy donors (right) (n = 3 each). Symbols represent individual samples; values are the mean ± SEM. * = P < 0.05. See Figure 1 for other definitions.
Up‐regulation of the differentiation of TNF and IL‐6–induced osteoclasts by IL‐1β
To clarify the characterization of TNF and IL‐6–induced osteoclasts, we examined cytokine expression by TNF and IL‐6–induced osteoclasts compared to RANKL‐induced osteoclasts. We stimulated monocyte‐derived macrophages with the combination of TNF and IL‐6 or RANKL and measured the mRNA expression level of the proinflammatory cytokines IL1β, TNF, and IL12p40 and anti‐inflammatory cytokine IL10. Expression levels of IL1β, TNF, and IL12p40 were significantly up‐regulated after 8 days of stimulation with TNF and IL‐6, whereas stimulation with RANKL failed to increase their expression (Figure 4A and Supplementary Figure 3A, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41666/abstract). Interestingly, the expression level of IL10 mRNA was significantly down‐regulated after 8 days of stimulation with TNF and IL‐6; however, IL10 expression was unaffected by RANKL (Supplementary Figure 3A). Furthermore, expression levels of IL‐1β protein were significantly up‐regulated and maintained at 9 days of stimulation with TNF and IL‐6 compared to the unstimulated control or stimulation with RANKL (Figure 4B and Supplementary Figure 4A [http://onlinelibrary.wiley.com/doi/10.1002/art.41666/abstract]).
Figure 4.
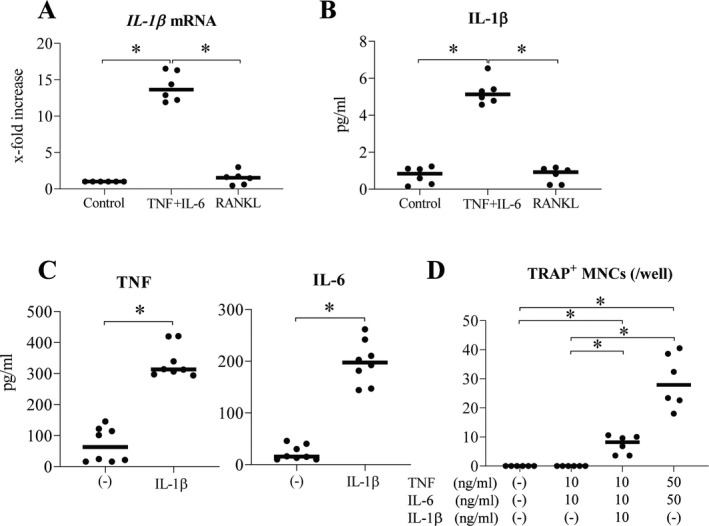
Promotion of the differentiation of TNF and IL‐6–induced osteoclasts by IL‐1β. A, Expression levels of IL‐1β mRNA in peripheral blood monocyte‐derived macrophages after 8 days of stimulation with TNF plus IL‐6 (n = 6). B, Expression levels of IL‐1β protein in the supernatant from peripheral blood monocyte‐derived macrophages after 9 days of stimulation with TNF plus IL‐6 (n = 6). C, Expression levels of TNF and IL‐6 proteins in the supernatant from peripheral blood monocyte‐derived macrophages after 3 days of stimulation with IL‐1β (n = 8). D, Quantification of TRAP‐positive MNCs in the absence versus presence of TNF, IL‐6, and IL‐1β (n = 6) at concentrations of 10 ng/ml (for all 3) and 50 ng/ml (for TNF and IL‐6). Symbols represent individual samples; values are the mean ± SEM. * = P < 0.05. See Figure 1 for other definitions.
Previously, it has been reported that stimulation with the combination of TNF and IL‐1β induced the differentiation of osteoclasts in a RANKL‐independent manner (5). In our system, the addition of an anti–IL‐1β antibody inhibited the differentiation of TNF and IL‐6–induced osteoclasts, but not that of RANKL‐induced osteoclasts (Supplementary Figures 4B and C). To determine the mechanism by which IL‐1β up‐regulated TNF and IL‐6–induced osteoclast differentiation, we stimulated monocyte‐derived macrophages with IL‐1β, and then measured expression levels of TNF and IL‐6 proteins. As expected, expression levels of these proteins were clearly up‐regulated in cells stimulated with IL‐1β (Figure 4C). Whereas the lower concentration of the combination of TNF and IL‐6 (each 10 ng/ml) alone did not induce TRAP‐positive multinucleated cells, the lower concentration of the combination of TNF, IL‐6, and IL‐1β (each 10 ng/ml) did induce this formation (Figure 4D and Supplementary Figure 4D). In addition, IL‐1β increased TNF and IL‐6–induced osteoclast differentiation even in the presence of high doses of TNF and IL‐6 (data not shown). These results suggest that IL‐1β synergistically promotes the differentiation of TNF and IL‐6–induced osteoclasts induced by the combination of TNF and IL‐6.
Functional differences between TNF and IL‐6–induced osteoclasts and RANKL‐induced osteoclasts differentiated from PBMCs in patients with RA
To understand the functional differences in TNF and IL‐6–induced osteoclasts and RANKL‐induced osteoclasts in patients with RA, we cultured PBMCs with a combination of TNF and IL‐6 or RANKL and assessed the number of TRAP‐positive multinucleated TNF and IL‐6–induced osteoclasts and RANKL‐induced osteoclasts. The number of TNF and IL‐6–induced osteoclasts and RANKL‐induced derived from PBMCs in patients with RA was significantly increased compared to that derived from PBMCs in healthy donors (Figure 5A). It should be noted that even higher doses of TNF and IL‐6 (each 100 ng/ml) did not increase the differentiation of osteoclasts derived from PBMCs in healthy donors (data not shown).
Figure 5.
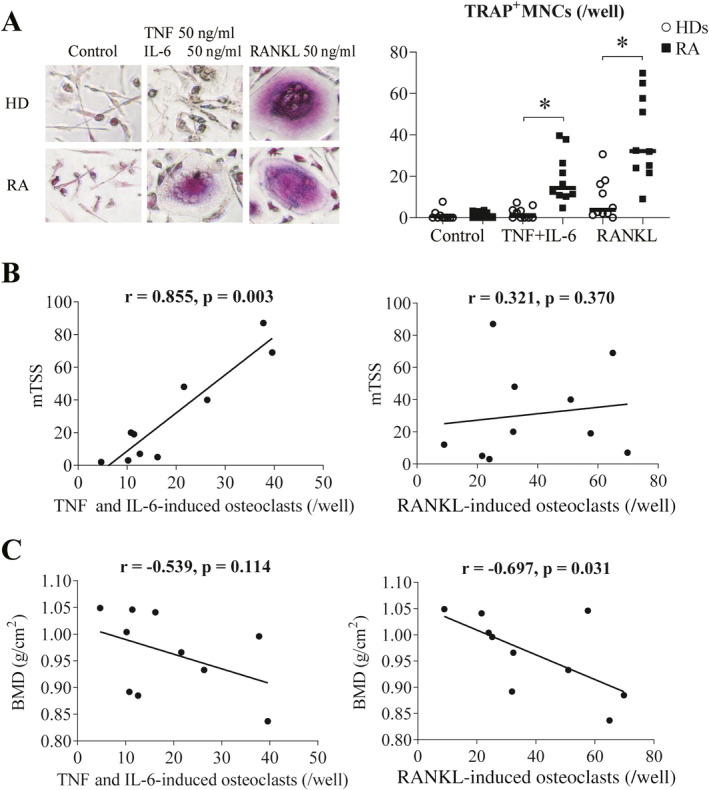
Functional differences between TNF and IL‐6–induced osteoclasts and RANKL‐induced osteoclasts differentiated from peripheral blood mononuclear cells (PBMCs) in patients with rheumatoid arthritis (RA). A, Photomicrographs (original magnification × 100) and quantification of TRAP‐positive MNCs differentiated from PBMCs from patients with RA and healthy donors (HDs) (n = 10 each). B, Correlation between the number of TNF and IL‐6–induced osteoclasts or RANKL‐induced osteoclasts and modified total Sharp score (mTSS) in patients with RA (n = 10). C, Correlation between the number of TNF and IL‐6–induced osteoclasts or RANKL‐induced osteoclasts and whole‐body bone mineral density (BMD) in patients with RA (n = 10). Symbols represent individual samples; values are the mean ± SEM. * = P < 0.05. See Figure 1 for other definitions.
To determine whether there was a correlation between the number of TNF and IL‐6–induced osteoclasts or osteoclasts induced from RA PBMCs, clinical indicators of the study participants were analyzed. There was a significant and positive correlation between the number of TNF and IL‐6–induced osteoclasts and serum levels of C‐reactive protein (CRP) in the same patients (P = 0.027, r = 0.709), whereas there was no correlation between the number of RANKL‐induced osteoclasts and serum levels of CRP (P = 0.218, r = 0.430) (data not shown). Interestingly, the number of TNF and IL‐6–induced osteoclasts also had a significant and positive correlation with mTSS values (P = 0.003, r = 0.855), whereas there was no correlation observed with mTSS values and number of RANKL‐induced osteoclasts (P = 0.370, r = 0.321) (Figure 5B). In addition, the number of TNF and IL‐6–induced osteoclasts did not correlate with whole‐body BMD values (P = 0.114, r = −0.539), whereas the number of RANKL‐induced osteoclasts had a significant negative correlation with BMD (P = 0.031, r = −0.697) (Figure 5C).
To clarify the potential role of TNF and IL‐6–induced osteoclasts in the progression of joint destruction and RANKL‐induced osteoclasts in the development of systemic osteoporosis, we stimulated monocyte‐derived macrophages with the combination of TNF and IL‐6 or RANKL and measured the mRNA expression of MMP3 and CTSK. As expected, the expression levels of MMP3 mRNA were significantly higher in TNF and IL‐6–induced osteoclasts induced by the combination of TNF and IL‐6 than in osteoclasts induced by RANKL and unstimulated controls (Supplementary Figure 3B [http://onlinelibrary.wiley.com/doi/10.1002/art.41666/abstract]). Conversely, the expression levels of CTSK mRNA were significantly higher in RANKL‐induced osteoclasts than in unstimulated controls (Supplementary Figure 3B). Consistent with these results, the TNF and IL‐6–induced osteoclasts differentiated from PBMCs in patients with RA likely contribute to the progression of joint destruction by producing proinflammatory cytokines and matrix metalloproteinase 3 (MMP‐3), whereas RANKL‐induced osteoclast activity leads to the development of systemic osteoporosis by producing cathepsin K.
DISCUSSION
In the present study, we showed that the combination of TNF and IL‐6, representative proinflammatory cytokines, induced the differentiation of TRAP‐positive multinucleated TNF and IL‐6–induced osteoclasts from human peripheral blood monocyte‐derived macrophages, and that these TNF and IL‐6–induced osteoclasts had the ability to absorb bone matrix. The differentiation of TNF and IL‐6–induced osteoclasts depends on the activation of NFATc1 and JAK, leading to a prominent increase in the production of proinflammatory cytokines and MMP‐3. Indeed, TNF and IL‐6 can induce the expression of IL‐1β in monocyte‐derived macrophages, which promotes the differentiation of TNF and IL‐6–induced osteoclasts. Moreover, PBMCs from patients with RA showed higher TNF and IL‐6–induced osteoclast differentiation potentials compared to those from healthy donors. In addition, of importance was that the differentiation potential of TNF and IL‐6–induced osteoclasts derived from PBMCs in patients with RA was positively correlated with mTSS values, and that the osteoclast differentiation potential was negatively correlated with whole‐body BMD.
We identified human proinflammatory cytokine–induced osteoclasts, which are similar to RANKL‐induced osteoclasts in that both are TRAP‐positive multinucleated cells with bone resorption activity. However, TNF and IL‐6–induced osteoclasts differ from RANKL‐induced osteoclasts as follows: 1) TNF and IL‐6–induced osteoclasts differentiate in a RANKL‐independent manner and depend on activation of JAK, 2) these cells induce proinflammatory cytokines IL‐1β, TNF, and IL‐12p40 as well as MMP‐3, and 3) the differentiation potential of TNF and IL‐6–induced osteoclasts derived from PBMCs in patients with RA was positively correlated with mTSS values, but not with whole‐body BMD. Thus, these findings suggest that TNF and IL‐6–induced osteoclasts could play an important role in joint destruction in patients with RA (Table 1).
Table 1.
Characteristics of human TNF and IL‐6–induced osteoclasts and RANKL‐induced osteoclasts*
|
TNF and IL‐6– induced osteoclasts |
RANKL‐induced osteoclasts |
|
|---|---|---|
| TRAP‐positive MNCs | ++ | +++ |
| Bone resorption activity | ++ | +++ |
| Inhibitory effect of OPG on differentiation | – | ++ |
| Inhibitory effect of JAK inhibitor on differentiation | ++ | – |
| Expression of inflammatory cytokines and MMP‐3 | ++ | – |
Results are shown as either ++ or +++ based on the intensity of staining for each characteristic, while – indicates the absence of the characteristic. TNF = tumor necrosis factor; IL‐6 = interleukin‐6; TRAP+ MNCs = tartrate‐resistant acid phosphatase–positive multinucleated cells; OPG = osteoprotegerin; MMP‐3 = matrix metalloproteinase 3.
Recently, Hasegawa et al described murine arthritis‐associated osteoclastogenic macrophages as the osteoclast precursor–containing population in the inflamed synovium, which were distinctive from RANKL‐induced osteoclast precursors (27). These arthritis‐associated osteoclastogenic macrophages differentiated into osteoclasts in a RANKL‐dependent manner, and their differentiation was promoted by TNF, but not by IL‐6. In addition, these cells were inhibited by OPG. However, the differentiation of TNF and IL‐6–induced osteoclasts was induced in a RANKL‐independent manner, and both TNF and IL‐6 were required for differentiation. This suggests that arthritis‐associated osteoclastogenic macrophages and TNF and IL‐6–induced osteoclasts are different cell lineages. Furthermore, the efficacy and safety of anti–IL‐6 receptor antibodies in the treatment of patients with RA have been well‐established. In addition, anti–IL‐6 receptor antibodies impede or arrest the progression of bone destruction in RA (9), thus indicating the importance of IL‐6–dependent osteoclastogenesis. Therefore, these findings suggest that TNF and IL‐6–induced osteoclasts are also involved in the pathologic mechanisms of inflammatory arthritis associated with joint destruction in RA.
TNF and IL‐6–induced osteoclasts may play a physiologic role in the post‐fracture healing process. In general, immediately after bone fractures, blood vessels rupture and bleed, and immune cells accumulating at the fracture site release proinflammatory cytokines that come into contact with hematopoietic precursor cells (28). During this process, the combination of TNF and IL‐6 could stimulate osteoclast precursors and induce the differentiation of TNF and IL‐6–induced osteoclasts, thereby promoting the resorption of damaged bone tissue. In the near future, further studies could be pursued to verify this hypothesis.
We investigated the molecular mechanisms driving cell differentiation. Expression levels and activities of NFATc1 are critical for the differentiation of TNF and IL‐6–induced osteoclasts, which were both elevated in response to stimulation with combined TNF and IL‐6. Previously, in mouse osteoclast‐like cells, we demonstrated that TNF activated both the canonical and noncanonical NF‐κB pathways, and TNF and IL‐6 synergistically affected the activities and expression of c‐Fos, a master regulator of osteoclastogenesis. Knockdown of c‐Fos inhibited the expression of NFATc1 and differentiation of osteoclast‐like cells. In addition, NFATc1, JAK, and ERK inhibitors blocked osteoclast‐like cell differentiation, whereas conditional knockout of STAT3 did not block osteoclast‐like cell differentiation. Taken together, the JAK‐MEK/ERK signaling pathway likely regulates the differentiation of osteoclast‐like cells (12). Based on the findings from that previous report and the results of the current study, it is likely that TNF together with IL‐6 can substitute RANKL/RANK signaling through the activation of NF‐κB/c‐Fos/NFATc1 and calcium signaling for human TNF and IL‐6–induced osteoclast differentiation (29).
We demonstrate that PBMCs from patients with RA had significantly higher osteoclastogenic potential than those from healthy donors, confirming the results of a previous study (17). It has been reported that the frequency of osteoclast precursors was significantly higher in patients with RA than in healthy donors. In addition, an enhanced frequency of peripheral osteoclast precursors from patients with RA associated with increased osteoclastogenic potential of PBMCs has been shown in other groups (30, 31). We also showed that the differentiation potential of TNF and IL‐6–induced osteoclasts derived from PBMCs in patients with RA was positively correlated with serum levels of CRP. In general, CRP levels reflect synovial inflammation, which is demonstrated by significantly increased serum levels of inflammation‐associated cytokines, including IL‐6, IL‐1β, and TNF (32). In addition, the serum levels of CRP were shown to mirror those of IL‐6 and to correlate with radiographic progression (33).
Although we did not measure serum levels of proinflammatory cytokines, it has been reported that expression levels of TNF and IL‐6 in the serum and synovial fluid of patients with RA are significantly higher than those in the serum and synovial fluid of healthy donors (1). Based on these reports, we speculate that the higher levels of TNF and IL‐6 in serum and synovial fluid originating from RA synovial inflammation induced the differentiation of TNF and IL‐6–induced osteoclasts, and that these activated cells may be implicated in joint destruction by producing proinflammatory cytokines and MMP‐3. It is suggested that TNF or IL‐6 inhibitor may suppress bone destruction by inhibiting the differentiation of bone‐resorptive TNF and IL‐6–induced osteoclasts induced by the combination of TNF and IL‐6.
Our study had several limitations. First, to identify the cell‐specific molecules of human TNF and IL‐6–induced osteoclasts and RANKL‐induced osteoclasts, we examined and compared the differences in gene and protein expression in cultured whole cells using transcriptome analysis. However, to identify more cell‐specific molecules of human TNF and IL‐6–induced osteoclasts and RANKL‐induced osteoclasts, comprehensive gene expression analyses, such as a single‐cell approach, may be required. Second, additional studies are needed to determine whether other osteoclast subsets contribute to bone destruction in RA patients.
In conclusion, the results of the present study demonstrate that TNF and IL‐6–induced osteoclasts can differentiate via RANKL‐independent pathways, and that there are functional differences between TNF and IL‐6–induced osteoclasts and RANKL‐induced osteoclasts. In particular, targeting TNF and IL‐6–induced osteoclasts as well as RANKL‐induced osteoclasts is anticipated to create new therapeutic strategies such as JAK inhibitors.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Yokota had full access to all the study data and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Yokota, Miyazaki, Tanaka, Mimura.
Acquisition of data
Yokota, Miyazaki, Kozu.
Analysis and/or interpretation of data
Yokota, Sato, Miyazaki, Aizaki, Tanaka, Sekikawa, Kadono, Oda, Mimura.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
Table S1
Acknowledgments
We are grateful to N. Murai, N. Honma, T. Kamei, and K. Yamaguchi (Saitama Medical University) for technical assistance, and we also thank H. Kajiyama and Y. Araki (Saitama Medical University) for helpful discussions.
Supported in part by grants‐in‐aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, grants from the Tokyo Biochemical Research Foundation, grant 19‐B‐1‐22 from Saitama Medical University Internal Research, and grant‐in‐aid 01‐E‐1‐04 for Young Researchers from Saitama Medical University Hospital.
No potential conflicts of interest relevant to this article were reported.
References
- 1.Sato K, Suematsu A, Nakashima T, Takemoto‐Kimura S, Aoki K, Morishita Y, et al. Regulation of osteoclast differentiation and function by the CaMK‐CREB pathway [letter]. Nat Med 2006;12:1410–6. [DOI] [PubMed] [Google Scholar]
- 2.Sato K, Takayanagi H. Osteoclasts, rheumatoid arthritis, and osteoimmunology [review]. Curr Opin Rheumatol 2006;18:419–26. [DOI] [PubMed] [Google Scholar]
- 3.Adamopoulos IE, Mellins ED. Alternative pathways of osteoclastogenesis in inflammatory arthritis [review]. Nat Rev Rheumatol 2015;11:189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao B, Grimes SN, Li S, Hu X, Ivashkiv LB. TNF‐induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP‐J. J Exp Med 2012;209:319–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim N, Kadono Y, Takami M, Lee J, Lee SH, Okada F, et al. Osteoclast differentiation independent of the TRANCE–RANK–TRAF6 axis. J Exp Med 2005;202:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schett G, Elewaut D, McInnes IB, Dayer JM, Neurath MF. How cytokine networks fuel inflammation: toward a cytokine‐based disease taxonomy. Nat Med 2013;19:822–4. [DOI] [PubMed] [Google Scholar]
- 7.Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF‐α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest 2000;106:1481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Axmann R, Böhm C, Krönke G, Zwerina J, Smolen J, Schett G. Inhibition of interleukin‐6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum 2009;60:2747–56. [DOI] [PubMed] [Google Scholar]
- 9.Finzel S, Kraus S, Figueiredo CP, Regensburger A, Kocijan R, Rech J, et al. Comparison of the effects of tocilizumab monotherapy and adalimumab in combination with methotrexate on bone erosion repair in rheumatoid arthritis. Ann Rheum Dis 2019;78:1186–91. [DOI] [PubMed] [Google Scholar]
- 10.Smolen JS, Han C, Bala M, Maini RN, Kalden JR, van der Heijde D, et al. Evidence of radiographic benefit of treatment with infliximab plus methotrexate in rheumatoid arthritis patients who had no clinical improvement: a detailed subanalysis of data from the Anti–Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy study. Arthritis Rheum 2005;52:1020–30. [DOI] [PubMed] [Google Scholar]
- 11.Smolen JS, Avila JC, Aletaha D. Tocilizumab inhibits progression of joint damage in rheumatoid arthritis irrespective of its anti‐inflammatory effects: disassociation of the link between inflammation and destruction. Ann Rheum Dis 2012;71:687–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yokota K, Sato K, Miyazaki T, Kitaura H, Kayama H, Miyoshi F, et al. Combination of tumor necrosis factor α and interleukin‐6 induces mouse osteoclast‐like cells with bone resorption activity both in vitro and in vivo. Arthritis Rheumatol 2014;66:121–9. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien W, Fissel BM, Maeda Y, Yan J, Ge X, Gravallese EM, et al. RANK‐independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol 2016;68:2889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 15.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 16.Yokota K, Miyazaki T, Hemmatazad H, Gay RE, Kolling C, Fearon U, et al. The pattern‐recognition receptor nucleotide‐binding oligomerization domain–containing protein 1 promotes production of inflammatory mediators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 2012;64:1329–37. [DOI] [PubMed] [Google Scholar]
- 17.Bozec A, Zaiss MM, Kagwiria R, Voll R, Rauh M, Chen Z, et al. T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci Transl Med 2014;6:235ra60. [DOI] [PubMed] [Google Scholar]
- 18.Van der Heijde D, Simon L, Smolen J, Strand V, Sharp J, Boers M, et al. How to report radiographic data in randomized clinical trials in rheumatoid arthritis: guidelines from a roundtable discussion. Arthritis Rheum 2002;47:215–8. [DOI] [PubMed] [Google Scholar]
- 19.Minkin C. Bone acid phosphatase: tartrate‐resistant acid phosphatase as a marker of osteoclast function. Calcif Tissue Int 1982;34:285–90. [DOI] [PubMed] [Google Scholar]
- 20.Tamura T, Udagawa N, Takahashi N, Miyaura C, Tanaka S, Yamada Y, et al. Soluble interleukin‐6 receptor triggers osteoclast formation by interleukin 6. Proc Natl Acad Sci U S A 1993;90:11924–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishimi Y, Miyaura C, Jin CH, Akatsu T, Abe E, Nakamura Y, et al. IL‐6 is produced by osteoblasts and induces bone resorption. J Immunol 1990;145:3297–303. [PubMed] [Google Scholar]
- 22.Yarilina A, Xu K, Chen J, Ivashkiv LB. TNF activates calcium‐nuclear factor of activated T cells (NFAT)c1 signaling pathways in human macrophages. Proc Natl Acad Sci U S A 2011;108:1573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998;93:165–76. [DOI] [PubMed] [Google Scholar]
- 24.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 2002;3:889–901. [DOI] [PubMed] [Google Scholar]
- 25.Garbers C, Heink S, Korn T, Rose‐John S. Interleukin‐6: designing specific therapeutics for a complex cytokine [review]. Nat Rev Drug Discov 2018;17:395–412. [DOI] [PubMed] [Google Scholar]
- 26.Van der Heijde D, Strand V, Tanaka Y, Keystone E, Kremer J, Zerbini CA, et al. Tofacitinib in combination with methotrexate in patients with rheumatoid arthritis: clinical efficacy, radiographic, and safety outcomes from a twenty‐four–month, phase III study. Arthritis Rheumatol 2019;71:878–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasegawa T, Kikuta J, Sudo T, Matsuura Y, Matsui T, Simmons S, et al. Identification of a novel arthritis‐associated osteoclast precursor macrophage regulated by FoxM1. Nat Immunol 2019;20:1631–43. [DOI] [PubMed] [Google Scholar]
- 28.Adamopoulos IE. Inflammation in bone physiology and pathology [review]. Curr Opin Rheumatol 2018;30:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung YK, Kang YM, Han S. Osteoclasts in the inflammatory arthritis: implications for pathologic osteolysis [review]. Immune Netw 2019;19:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herman S, Müller RB, Krönke G, Zwerina J, Redlich K, Hueber AJ, et al. Induction of osteoclast‐associated receptor, a key osteoclast costimulation molecule, in rheumatoid arthritis. Arthritis Rheum 2008;58:3041–50. [DOI] [PubMed] [Google Scholar]
- 31.Li P, Schwarz EM, O’Keefe RJ, Ma L, Looney RJ, Ritchlin CT, et al. Systemic tumor necrosis factor α mediates an increase in peripheral CD11bhigh osteoclast precursors in tumor necrosis factor α–transgenic mice. Arthritis Rheum 2004;50:265–76. [DOI] [PubMed] [Google Scholar]
- 32.Gabay C, Kushner I. Acute‐phase proteins and other systemic responses to inflammation. N Eng J Med 1999;340:448–54. [DOI] [PubMed] [Google Scholar]
- 33.Lindqvist E, Eberhardt K, Bendtzen K, Heinegård D, Saxne T. Prognostic laboratory markers of joint damage in rheumatoid arthritis. Ann Rheum Dis 2005;64:196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
Table S1