

Abstract
Biallelic loss‐of‐function variants in the thrombospondin‐type laminin G domain and epilepsy‐associated repeats (TSPEAR) gene have recently been associated with ectodermal dysplasia and hearing loss. The first reports describing a TSPEAR disease association identified this gene is a cause of nonsyndromic hearing loss, but subsequent reports involving additional affected families have questioned this evidence and suggested a stronger association with ectodermal dysplasia. To clarify genotype–phenotype associations for TSPEAR variants, we characterized 13 individuals with biallelic TSPEAR variants. Individuals underwent either exome sequencing or panel‐based genetic testing. Nearly all of these newly reported individuals (11/13) have phenotypes that include tooth agenesis or ectodermal dysplasia, while three newly reported individuals have hearing loss. Of the individuals displaying hearing loss, all have additional variants in other hearing‐loss‐associated genes, specifically TMPRSS3, GJB2, and GJB6, that present competing candidates for their hearing loss phenotype. When presented alongside previous reports, the overall evidence supports the association of TSPEAR variants with ectodermal dysplasia and tooth agenesis features but creates significant doubt as to whether TSPEAR variants are a monogenic cause of hearing loss. Further functional evidence is needed to evaluate this phenotypic association.
Keywords: autosomal recessive deafness, ectodermal dysplasia, hearing loss, tooth agenesis, TSPEAR
1. INTRODUCTION
Variants in the thrombospondin‐type laminin G domain and epilepsy‐associated repeats (TSPEAR) gene have been associated with two different autosomal recessive phenotypes; individuals with biallelic TSPEAR variants have presented either with sensorineural hearing loss (SNHL) or with a variety of phenotypes that affect ectodermal‐derived structures such as skin, hair, nails, and teeth and which span a range of severity from isolated tooth agenesis to ectodermal dysplasia (ED; Delmaghani et al., 2012; Du et al., 2018; Peled et al., 2016; Sloan‐Heggen et al., 2016; Song et al., 2020). This divergence of phenotypes is interesting. At one extreme, individuals have been reported to have profound hearing loss and otherwise purportedly normal features, and at another extreme have demonstrated striking ED and completely normal hearing (Delmaghani et al., 2012; Peled et al., 2016). There is currently little information available to explain such a wide divergence in TSPEAR‐related clinical features.
The TSPEAR hearing loss association originates from a 2012 study which identified three consanguineous Iranian siblings affected by profound SNHL (Delmaghani et al., 2012). Whole exome sequencing (WES) for these siblings identified a homozygous c.1726_1728delGTCinsTT (p.Val576Leufs*38) TSPEAR variant segregating with hearing loss. No other hearing loss variants were identified and the authors concluded that TSPEAR is a cause of nonsyndromic hearing loss. One subsequent report used panel‐based sequencing to identify additional TSPEAR individuals with hearing loss: two siblings with compound heterozygous variants, a synonymous c.1566G>A (p.Pro522Pro) variant with a potential impact on splicing and a frameshift c.1676_1677delAT (p.Tyr559Cysfs*134) variant, which together were the sole candidate hearing loss variants (Sloan‐Heggen et al., 2016). Both siblings displayed bilateral SNHL and enlargement of the vestibular aqueduct (EVA). Neither of these studies reported any additional features that were evocative of ED.
In contrast to these two hearing loss reports, recent studies have described TSPEAR subjects with either isolated, nonsyndromic tooth agenesis or ED with tooth agenesis as a component of the subject phenotype (Du et al., 2018; Peled et al., 2016; Song et al., 2020). A 2016 report identified three separate families affected by dysmorphic facial features, scalp hypotrichosis, and hypodontia (Peled et al., 2016). These findings have been partially corroborated by subsequent studies which have identified additional individuals displaying normal hearing and nonsyndromic oligodontia (Du et al., 2018; Song et al., 2020). Of note, these reports have also identified several hearing‐normal individuals with the same c.1726_1728delGTCinsTT (p.Val576Leufs*38) variant initially associated with hearing loss. Based on the current literature, the majority of individuals with biallelic TSPEAR variants have displayed either isolated, nonsyndromic tooth agenesis or ED with tooth agenesis (Du et al., 2018; Peled et al., 2016; Song et al., 2020). The evidence around the TSPEAR hearing loss is less clear, raising the question of whether hearing loss displays variable penetrance in individuals with TSPEAR variants, or if hearing loss has been misattributed to TSPEAR variants in prior studies.
There is conflicting biological evidence to suggest a role for TSPEAR in hearing function. Prior studies have proposed that TSPEAR may regulate inner ear hair cell fate decisions during development by interacting with the Notch signaling pathway (Peled et al., 2016). Additionally, TSPEAR exons 2–5 contain a laminin G domain, commonly found in extracellular matrix proteins, and immunohistochemistry (IHC) of healthy mouse cochlear sections suggests that TSPEAR may localize to the base of mammalian inner ear hair cells (Delmaghani et al., 2012; Mills et al., 2018; Mitchell et al., 2018). However, RNA‐sequencing data from the gEAR portal (gene Expression Analysis Resource, umgear.org) indicates extremely low or absent expression of TSPEAR transcripts within murine cochlea and no studies have directly examined the effects of TSPEAR loss‐of‐function variants on hair cell morphology (Elkon et al., 2015; Scheffer et al., 2015; Waldhaus et al., 2015). While TSPEAR knockout mice are slated for development through the International Mouse Phenotyping Consortium, no TSPEAR knockout animal models have been produced thus far. Furthermore, when considering the evidence supporting a role for TSPEAR in hearing loss, one must consider that hearing loss is extremely heterogeneous. It is possible unidentified variants in genes other than TSPEAR may be causally related to an individual's hearing loss phenotype; for example, the nearby TMPRSS3 locus has been implicated in autosomal recessive, nonsyndromic hearing loss and variants at this locus can be co‐inherited alongside TSPEAR variants (Masmoudi et al., 2001). Lastly, hearing loss often has a significant environmental or prenatal component, with diseases such as cytomegalovirus and rubella representing the leading environmental causes of acquired childhood sensorineural hearing loss worldwide (Goderis et al., 2014; Korver et al., 2011; Korver et al., 2017).
In summary, the existing literature presents limited evidence to argue that TSPEAR variants are a direct cause of hearing loss, and previously identified individuals present with conflicting phenotypes of hearing loss, isolated tooth agenesis, or ED with tooth agenesis as a component of the subject phenotype. While the connection between TSPEAR variants and tooth agenesis or ectodermal dysplasia appears to be well‐supported by previous cohort studies, we aim to clarify the association between TSPEAR and hearing loss phenotypes by evaluating specific variant/phenotype associations in additional individuals. In this text, we describe 13 newly reported individuals with biallelic, suspected pathogenic TSPEAR variants, three of whom have hearing loss phenotypes, and compare these data with previous reports.
2. METHODS
2.1. Editorial policies and ethical considerations
All individuals provided informed consent to be included in this study through protocols approved by their corresponding Institutional Review Boards.
2.2. Cohort inclusion criteria and subject recruitment
Individuals were included in this study if they presented with biallelic, confirmed trans, TSPEAR variants. Individuals in this cohort were identified by the Mayo Clinic Department of Clinical Genetics, the Molecular Otolaryngology and Renal Research Laboratories (MORL) at the University of Iowa Carver College of Medicine, and through collaborations facilitated by GeneMatcher (Sobreira et al., 2015). For retrospectively identified individuals, clinical descriptions were abstracted from data collected by the MORL.
2.3. Phenotype definitions
For all clinical descriptions, hypodontia is defined as the absence of one to five teeth, while oligodontia is defined as the absence of six or more teeth (de La Dure‐Molla et al., 2019). “Isolated tooth agenesis” is used to describe a subset of cohort subjects with nonsyndromic tooth agenesis who do not meet the diagnostic criteria for ectodermal dysplasia. Individuals were classified as having ectodermal dysplasia if they presented with phenotypes affecting two or more ectodermal structures (e.g., teeth, hair, nails, or skin) (Wright et al., 2019). To ensure standard reporting between subjects, phenotypes have been described using Human Phenotype Ontology (HPO) terms when possible (Köhler et al., 2018).
2.4. Subject phenotyping
Cohort individuals 1, 2, and 5 presented with hearing loss and were referred for genetic hearing loss testing; for these subjects, de‐identified phenotypic information has been provided by the sequencing lab rather than the ordering clinicians. This information indicates that cohort individuals 1 and 2 presented with hearing loss and did not receive clinical or radiographic evaluation for dental phenotypes as a part of their visits with healthcare providers, while cohort subject 5 received clinical evaluation for dental phenotypes that did not include dental radiography. The remaining subjects reported in this study were phenotyped through visits with study clinicians, during which time the subjects were evaluated for ectodermal dysplasia including dental phenotypes. During this phenotyping, clinicians noted when subjects satisfied the diagnostic criteria for ectodermal dysplasia and differentiated between syndromic and isolated tooth agenesis. As a part of their clinical visits, cohort individuals 7, 8, 10, 15, 16, 17, and 18 received dental X‐rays, while cohort individuals 6, 9, and 14 did not receive dental X‐rays. Individuals 3, 4, 11, 12, 13, 19, 20, and 21 have been described in prior publications, but are reported in this article in order to fully detail the spectrum of phenotypes for individuals with biallelic TSPEAR variants. Individuals 3 and 4 did not receive dental radiography as a part of their clinical evaluation (Delmaghani et al., 2012; Sloan‐Heggen et al., 2016), while the publications describing individuals 11, 12, 13, 19, 20, and 21 reported dental radiography as a part of the subject phenotyping process (Du et al., 2018; Peled et al., 2016; Song et al., 2020). For newly identified individuals presenting with tooth agenesis, providers confirmed that all missing teeth were developmentally absent rather than extracted.
2.5. Subject sequencing
Individuals were evaluated using a variety of sequencing methods, including WES (N = 6), hearing loss gene panels (N = 3), ectodermal dysplasia or tooth agenesis gene panels (N = 3), and SNP array followed by Sanger sequencing (N = 1). Sequencing methods are summarized for each newly reported case in Table 2, while sequencing chromatograms and information on variant sequencing depth are presented further in Table S2, Supporting Information. Hearing loss testing used the targeted hearing loss gene panel (OtoSCOPE v4‐v6) performed at the MORL. OtoSCOPE is a comprehensive gene panel designed for nonsyndromic hearing loss and includes TSPEAR, but does not include the full spectrum of potential ectodermal dysplasia genes (Shearer et al., 2010; Sloan‐Heggen et al., 2016). OtoSCOPE versions 4–6 include 66, 89, and 109 genes, respectively. All nucleotide changes are reported according to the GRh37 reference genome and the NM_144991.2 TSPEAR transcript, which is the longest transcript and is indicated by the GTEx Portal database to be the dominant transcript in skin. Variants in GJB2, TMPRSS3, and ATP1A3 are reported using the NM_004004.5, NM_024022.2, and NM_152296.4 RefSeq transcripts, respectively. Per the clinical report, all variants were confirmed with Sanger sequencing, and all available chromatograms are included in Supporting Information.
2.6. Variant classification
All variants, including those previously reported in the literature, were classified based on ACMG/AMP 2015 criteria (Table 2) (Richards et al., 2015). Alamut® Visual 2.11 (Interactive Biosoftware, Rouen, France) was used to aid in variant interpretation. The allele frequency for each variant in general population was obtained from GnomAD v2.1.1 (Karczewski et al., 2019). The AlignGVD, SIFT, MutationTaster, and PolyPhen‐2 tools were used to generate in silico predictions of variant pathogenicity (Adzhubei et al., 2010; Ng & Henikoff, 2001; Schwarz et al., 2014; Tavtigian et al., 2006). CADD scores are reported for all missense variants using the GRCh37/hg19 genome assembly (Rentzsch et al., 2018). The SpliceAI, SpliceSiteFinder, MaxEntScan, and GeneSplicer tools were used to evaluate variant impact on splicing (Jaganathan et al., 2019; Pertea et al., 2001; Shapiro & Senapathy, 1987; Yeo & Burge, 2004). Frameshift and nonsense variants were evaluated for the likelihood of undergoing nonsense mediated decay using the NMDEscPredictor tool (Coban‐Akdemir et al., 2018). Individuals 17 and 18 were investigated using the Diagnosing Dental Defects Database (D4) and the Phenodent phenotyping tool (www.phenodent.org) (CNIL No. 908416; MES, Commission Bioéthique Collection Biologique “Manifestations bucco‐dentaires des maladies rares” DC‐2012‐1677 and DC‐2012‐1002; https://clinicaltrials.gov: NCT01746121 and NCT02397824) and genotyped with GenoDENT (Rey et al., 2019).
2.7. Variant burden calculation
To identify TSPEAR gene regions that are less tolerant to variation than others, a variant concentration (V) calculation is presented which normalizes the variant number (N) within each exon or protein domain to the length of the gene‐region in base‐pairs (L),
The score was calculated for all exons as well as for the laminin G and EAR protein domains and is intended to provide a simple metric to determine the variant concentration in these gene regions. As an additional measure of genotype/phenotype correlation, a Mann–Whitney U test used to examine the association between variant CADD score and phenotype (hearing loss vs. hearing normal, α = 0.05).
2.8. Data availability
All variants described in this study are openly available for access within the ClinVar variant database (https://www.ncbi.nlm.nih.gov/clinvar/).
3. RESULTS
3.1. Cohort features
Newly reported individuals (N = 13) are aggregated alongside previously reported individuals (N = 8) in Table 1 to provide a comprehensive overview of reported TSPEAR phenotypes. When grouped by phenotype, individuals with TSPEAR variants fall into three broad categories: individuals with reported hearing loss (individuals 1–5), individuals with ectodermal dysplasia (with or without tooth agenesis, individuals 5–14), and individuals with isolated, nonsyndromic tooth agenesis (individuals 18, 20, and 21). Individual 5 presents with both hearing loss and ectodermal dysplasia without tooth agenesis, while individuals 15, 16, and 17 have nonsyndromic tooth agenesis accompanied by additional dental features such as conical teeth or taurodontism which do not satisfy diagnostic criteria for ectodermal dysplasia. All variants were scored according to ACMG 2015 criteria (Table 2) (Richards et al., 2015). For the three newly reported individuals with reported hearing loss, sequencing identified additional variants in the hearing‐loss‐associated genes GJB2, GJB6, and TMPRSS3, none of which were classified as benign according to ACMG 2015 criteria (Table 2, individuals 1, 2, 6). Full descriptions and family histories for each individual are reported in Appendix A.
TABLE 1.
Expanded cohort of TSPEAR individuals
| Variant | Phenotype | Reference | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Gene | Nucleotide | Amino acid | Zygosity | Hearing loss | ED | Dentition | Skin, nails, and hair | Miscellaneous | ||||||
| Hypodontia | Oligodontia | Conical teeth | Nail dysplasia | Sparse hair | Hypohidrosis | Hypotrichosis | |||||||||
| 1 | TSPEAR | c.533C>T | p.Pro178Leu | Hom | Yes | None | Newly reported | ||||||||
| TMPRSS3 | c.1204G>A | p.Gly402Arg | Hom | ||||||||||||
| 2 | TSPEAR | c.1163T>C | p.Val388Ala | Hom | Yes | None | Newly reported | ||||||||
| TMPRSS3 | c.1211C>T | p.Pro404Leu | Hom | ||||||||||||
| 3 | TSPEAR | c.1726_1728delGTCinsTTa | p.Val576Leufs*38 | Hom | Yes | None | Delmaghani et al. (2012) | ||||||||
| 4 | TSPEAR | c.1566G>A | p.Pro522Pro | Het | Yes | Enlarged vestibular aqueduct, proteinuria | Sloan‐Heggen et al. (2016) | ||||||||
| TSPEAR | c.1676_1677delAT | p.Tyr559Cysfs*134 | Het | ||||||||||||
| 5 | TSPEAR | c.38delT | p.Leu13Argfs*38 | Het | Yes | Yes | Yes | Yes | Yes | None | Newly reported | ||||
| TSPEAR | c.589C>T | p.Arg197* | Het | ||||||||||||
| GJB2 | c.35delG | p.Gly12Valfs*2 | Het | ||||||||||||
| GJB6 | del(GJB6‐D13S1854) | NA | Het | ||||||||||||
| 6 | TSPEAR | c.1915G>A | p.Asp639Asn | Het | Yes | Yes | Yes | Yes | Aplasia/hypoplasia of the corpus callosum, brachydactyly syndrome, cortical visual impairment, global developmental delay, nystagmus, seizures | Newly reported | |||||
| TSPEAR | c.589C>T | p.Arg197* | Het | ||||||||||||
| 7 | TSPEAR | c.1915G>A | p.Asp639Asn | Het | Yes | Yes | Yes | Yes | Abnormality of movement, intellectual disability, profound global developmental delay, self‐injurious behavior, strabismus | Newly reported | |||||
| TSPEAR | c.1754G>T | p.Ser585Ile | Het | ||||||||||||
| 8 | TSPEAR | c.589C>T | p.Arg197* | Het | Yes | Yes | Yes | Attention deficit hyperactivity disorder, abnormal heart morphology, basilar artery fenestration, recurrent otitis media, resting tremor, strabismus, tall stature | Newly reported | ||||||
| TSPEAR | c.83‐13708_1566+248del | NA | Het | ||||||||||||
| 9 | TSPEAR | c.1574G>A | p.Gly525Asp | Het | Yes | Yes | Yes | Yes | Yes | Yes | Microdontia | Newly reported | |||
| TSPEAR | c.543‐1G>A | NA | Het | ||||||||||||
| 10 | TSPEAR | c.942C>G | p.Tyr314* | Het | Yes | Yes | Yes | Yes | Yes | Yes | Absent meibomian glands, conductive hearing impairment, eczema, recurrent otitis media, Turner syndrome | Newly reported | |||
| TSPEAR | c.1469T>A | p.Leu490Gln | Het | ||||||||||||
| 11 | TSPEAR | c.1726_1728delGTCinsTT | p.Val576Leufs*38 | Hom | Yes | Yes | Yes | Yes | Abnormal facial shape | Peled et al. (2016) | |||||
| 12 | TSPEAR | c.1726_1728delGTCinsTT | p.Val576Leufs*38 | Het | Yes | Yes | Yes | Yes | Abnormal facial shape | Peled et al. (2016) | |||||
| TSPEAR | c.454_457delCTGG | p.Leu152Trpfs*29 | Het | ||||||||||||
| 13 | TSPEAR | c.1852T>A | p.Tyr618Asn | Het | Yes | Yes | Yes | Yes | Abnormal facial shape | Peled et al. (2016) | |||||
| TSPEAR | c.1915G>A | p.Asp639Asn | Het | ||||||||||||
| 14 | TSPEAR | c.240T>G | p.Cys80Trp | Het | Yes | Yes | Sparse hair in childhood | Newly reported | |||||||
| TSPEAR | c.633+2C>A | NA | Het | ||||||||||||
| 15 | TSPEAR | c.1505delA | p.Lys502Argfs*67 | Hom | Yes | Yes | Attention disorder, large anteverted ears, prognathism, speech difficulties | Newly reported | |||||||
| 16 | TSPEAR | c.1726_1728delinsTT | p.Val576Leufs*38 | Het | Yes | Yes | Depressed nasal bridge, developmental delay, hypertelorism, paroxysmal dyskinesia, primary molar taurodontism, sagittal craniosynostosis, small and cupped ears | Newly reported | |||||||
| TSPEAR | c.589C>T | p.Arg197* | Het | ||||||||||||
| ATP1A3 | c.2767G>A | p.Asp923Asn | Het | ||||||||||||
| 17 | TSPEAR | c.1915G>A | p.Asp639Asn | Het | Yes | Dry skin, eczema, taurodontism | Newly reported | ||||||||
| TSPEAR | c.1331G>A | p.Arg444Gln | Het | ||||||||||||
| 18 | TSPEAR | c.1663C>T | p.Gln555* | Het | Yes | Scoliosis | Newly reported | ||||||||
| TSPEAR | c.1899dup | p.Thr634Hisfs*60 | Het | ||||||||||||
| 19 | TSPEAR | c.1726_1728delGTCinsTT | p.Val576Leufs*38 | Hom | Yes | Abnormality of the antitragus, high‐arched palate, low‐set ears, microcephaly, narrow forehead | Du et al. (2018) | ||||||||
| 20 | TSPEAR | c. 1877T>C | p.Phe626Ser | Hom | Yes | None | Du et al. (2018) | ||||||||
| 21 | TSPEAR | c.1528C>T | p.Arg510* | Het | Yes | None | Song et al. (2020) | ||||||||
| TSPEAR | c.1330C>T | p.Arg444Trp | Het | ||||||||||||
Note: 13 newly reported TSPEAR individuals are listed alongside eight previously reported cases. The majority of these individuals segregate into three broad phenotypic categories: those who display hearing loss (#1–5), those who display ectodermal dysplasia (ED) with and without tooth agenesis (#5–21), and those who display isolated, nonsyndromic tooth agenesis (#18, 20, 21). Individual 5 presents with both hearing loss and ectodermal dysplasia without tooth agenesis, while individuals 15, 16, and 17 have nonsyndromic tooth agenesis accompanied by additional dental features such as conical teeth or taurodontism which do not satisfy diagnostic criteria for ectodermal dysplasia. Full case descriptions can be found in appendices 1 and 2.
Abbreviations: del, deletion; delins, deletion/insertion; dup, duplication; ED, ectodermal dsyplasia.
Variants for individual 3 were originally reported as homozygous c.1726G>T and c.1728delC variants in cis, which can alternately be reported as c.1726_1728delGTCinsTT (p.Val576Leufs*38).
TABLE 2.
TSPEAR variant ACMG classification
| Variant | Variant interpretation | Sequencing method | References | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Gene | Nucleotide change | Amino acid | Zygosity | CADD score | GnomAD AF | ClinVar clinical significance | ACMG classification | Evidence | ||
| 1 | TSPEAR | c.533C>T | p.Pro178Leu | Hom | 17.86 | 0.00003668 | NA | VUS | PM2, PP3, BP5 | Hearing loss panel | Newly reported |
| TMPRSS3 | c.1204G>A | p.Gly402Arg | Hom | 31 | 0 | NA | VUS | PM2, PP3, BP5 | |||
| 2 | TSPEAR | c.1163T>C | p.Val388Ala | Hom | 33 | 0 | NA | VUS | PM2, PP3, BP5 | Hearing loss panel | Newly reported |
| TMPRSS3 | c.1211C>T | p.Pro404Leu | Hom | 26.1 | 0.00001193 | Pathogenic | Likely pathogenic | PP1, PM2, PM3, PP3, PM1, BP5 | |||
| 3 | TSPEAR | c.1726_1728delGTCinsTTa | p.Val576Leufs*38 | Hom | 33 | 0 | Likely pathogenic | Pathogenic | PVS1, PM2, PP3 | WES | Delmaghani et al. (2012) |
| 4 | TSPEAR | c.1566G>A | p.Pro522Pro | Het | 33 | 0.00007454 | NA | VUS | PM2, PM3, PP3 | Hearing loss panel | Sloan‐Heggen et al. (2016) |
| TSPEAR | c.1676_1677delAT | p.Tyr559Cysfs*134 | Het | 31 | 0 | Pathogenic/Likely pathogenic | Pathogenic | PVS1, PM2, PP3 | |||
| 5 | TSPEAR | c.38delT | p.Leu13Argfs*38 | Het | 24.6 | 0.00008334 | NA | Pathogenic | PVS1, PM2, PM3, PP3, BP5 | Hearing loss panel | Newly reported |
| TSPEAR | c.589C>T | p.Arg197* | Het | 36 | 0.000305 | Conflicting | Pathogenic | PVS1, PM2, PM3, PP3, BP5 | |||
| GJB2 | c.35delG | p.Gly12Valfs*2 | Het | 25.3 | 0.006188 | Pathogenic | Pathogenic | PVS1, PS3, PS4, PM2, PP1‐S, PP3, BP5 | |||
| GJB6 | del(GJB6‐D13S1854) | NA | Het | NA | NA | Pathogenic | Pathogenic | PVS1, PS3, PS4, PP1‐S, PM2, BP5 | |||
| 6 | TSPEAR | c.1915G>A | p.Asp639Asn | Het | 28.2 | 0.002251 | Conflicting | VUS | PM2, PM3, PP3, BS2 | WES | Newly reported |
| TSPEAR | c.589C>T | p.Arg197* | Het | 36 | 0.000305 | Conflicting | Pathogenic | PVS1, PM2, PP3 | |||
| 7 | TSPEAR | c.1915G>A | p.Asp639Asn | Het | 28.2 | 0.002251 | Conflicting | VUS | PM2, PP3, BS2 | WES | Newly reported |
| TSPEAR | c.1754G>T | p.Ser585Ile | Het | 25.6 | 0.00006734 | Uncertain significance | VUS | PM2, PP3 | |||
| 8 | TSPEAR | c.589C>T | p.Arg197* | Het | 36 | 0.000305 | Conflicting | Pathogenic | PVS1, PM2, PM3, PP3 | WES | Newly reported |
| TSPEAR | c.83‐13708_1566+248del | NA | Het | NA | NA | NA | Pathogenic | PVS1, PM2, PM3, PP3 | |||
| 9 | TSPEAR | c.1574G>A | p.Gly525Asp | Het | 24.2 | 0.00003199 | NA | VUS | PM2, PM3, PP3 | WES | Newly reported |
| TSPEAR | c.543‐1G>A | NA | Het | 23.9 | 0.000007119 | NA | Pathogenic | PVS1, PM2, PP3 | |||
| 10 | TSPEAR | c.942C>G | p.Tyr314* | Het | 36 | 0 | Likely pathogenic | Pathogenic | PVS1, PM2, PP3 | WES | Newly reported |
| TSPEAR | c.1469T>A | p.Leu490Gln | Het | 29.7 | 0.00005569 | Uncertain significance | VUS | PM2, PM3, PP3 | |||
| 11 | TSPEAR | c.1726_1728delGTCinsTT | p.Val576Leufs*38 | Hom | 33 | 0 | Likely pathogenic | Pathogenic | PVS1, PM2, PP3 | WES | Peled et al. (2016) |
| 12 | TSPEAR | c.1726_1728delGTCinsTT | p.Val576Leufs*38 | Het | 33 | 0 | Likely pathogenic | Pathogenic | PVS1, PM2, PP3 | WES | Peled et al. (2016) |
| TSPEAR | c.454_457delCTGG | p.Leu152Trpfs*29 | Het | 23.3 | 0.000004063 | Pathogenic | Pathogenic | PVS1, PM2, PP3 | |||
| 13 | TSPEAR | c.1852T>A | p.Tyr618Asn | Het | 32 | 0.0000199 | Pathogenic | VUS | PM2, PP3 | WES | Peled et al. (2016) |
| TSPEAR | c.1915G>A | p.Asp639Asn | Het | 28.2 | 0.002251 | Conflicting | VUS | PM2, PP3, BS2 | |||
| 14 | TSPEAR | c.240T>G | p.Cys80Trp | Het | 23.3 | 0 | NA | VUS | PM2, PM3, PP3 | Ectodermal dysplasia panel; Sanger sequencing | Newly reported |
| TSPEAR | c.633+2C>A | NA | Het | 22.7 | 0.000004001 | NA | Pathogenic | PVS1, PM2, PP3 | |||
| 15 | TSPEAR | c.1505delA | p.Lys502Argfs*67 | Hom | 18.1 | 0.00001592 | NA | Pathogenic | PVS1, PM2, PP3 | SNP array; Sanger sequencing | Newly reported |
| 16 | TSPEAR | c.1726_1728delinsTT | p.Val576Leufs*38 | Het | 33 | 0 | Likely pathogenic | Pathogenic | PVS1, PM2, PM3, PP3 | WES | Newly reported |
| TSPEAR | c.589C>T | p.Arg197* | Het | 36 | 0.000305 | Conflicting | Pathogenic | PVS1, PM2, PP3, PM3 | |||
| ATP1A3 | c.2767G>A | p.Asp923Asn | Het | 23.8 | 0 | Pathogenic | Likely pathogenic | PS1, PM1, PM2, PP3 | |||
| 17 | TSPEAR | c.1915G>A | p.Asp639Asn | Het | 28.2 | 0.002251 | Conflicting | VUS | PM2, PP3, BS2 | GenoDENT: dental anomalies panel; Sanger sequencing | Newly reported |
| TSPEAR | c.1331G>A | p.Arg444Gln | Het | 28.5 | 0.00008862 | NA | VUS | PM2, PP3 | |||
| 18 | TSPEAR | c.1663C>T | p.Gln555* | Het | 39 | 0 | NA | Pathogenic | PVS1, PM2, PM3, PP3 | GenoDENT: dental anomalies panel; Sanger sequencing | Newly reported |
| TSPEAR | c.1899dup | p.Thr634Hisfs*60 | Het | 34 | 0 | NA | Pathogenic | PVS1, PM2, PM3, PP3 | |||
| 19 | TSPEAR | c.1726_1728delGTCinsTT | p.Val576Leufs*38 | Hom | 33 | 0 | Likely pathogenic | Pathogenic | PVS1, PM2, PP3 | WES | Du et al. (2018) |
| 20 | TSPEAR | c. 1877T>C | p.Phe626Ser | Hom | 28.5 | 0.0001401 | NA | VUS | PM2, PP3 | WES | Du et al. (2018) |
| 21 | TSPEAR | c.1528C>T | p.Arg510* | Het | 37 | 0.0003256 | Uncertain significance | Pathogenic | PVS1, PM2, PP3 | WES | Song et al. (2020) |
| TSPEAR | c.1330C>T | p.Arg444Trp | Het | 25.8 | 0.00001595 | Uncertain significance | VUS | PM2, PM3, PP3 | |||
Note: All variants in this study were classified according to American College of Medical Genetics (ACMG) 2015 criteria. Classifications are shown here, along with the evidence used to support each classification such as combined annotation dependent depletion (CADD) score and genome aggregation database (GnomAD) allele frequency. While numerous suspected pathogenic TSPEAR variants have been reported in the literature, these studies did not report ACMG scoring criteria. To provide a means of comparing newly reported TSPEAR variants to published TSPEAR variants, these published variants have been scored here using the available evidence. ClinVar accessed January 12, 2021, GnomAD v2.1.1 accessed January 13, 2021.
Abbreviations: AF, allele frequency; CADD, combined annotation dependent depletion; del, deletion; delins, deletion/insertion; dup, duplication; GnomAD, genome aggregation database; NA, not available; VUS, variant of uncertain significance.
Variants for individual 3 were originally reported as homozygous c.1726G>T and c.1728delC variants in cis, which can alternately be reported as c.1726_1728delGTCinsTT (p.Val576Leufs*38).
3.2. Hearing loss
Individuals with hearing loss presented with variable phenotypes that included syndromic, congenital‐onset SNHL and nonsyndromic adolescent‐onset hearing loss. For newly reported individuals, we observed a lower prevalence of hearing loss compared to phenotypes such as isolated tooth agenesis or ectodermal dysplasia with and without tooth agenesis (3/13 individuals versus 11/13 individuals). In each of the three newly reported individuals with hearing loss, variants in other genes associated with hearing loss were also identified. Detailed descriptions of these newly reported hearing loss individuals are presented below and summarized in Table 1.
Individual 1 is an Asian‐ancestry male who presented with SNHL and no features evocative of ED or tooth agenesis. The individual carries a newly reported c.533C>T (p.Pro178Leu) missense TSPEAR variant in a homozygous state (Table 1). The individual's hearing loss diagnosis is complicated by a c.1204G>A (p.Gly402Arg) homozygous missense variant in the nearby TMPRSS3 gene. While loss‐of‐function variants in TMPRSS3 have been related to autosomal recessive, childhood‐onset, neurosensory deafness (Masmoudi et al., 2001), this variant is novel. The TSPEAR and TMPRSS3 variants are currently classified as variants of uncertain significance (VUS; Table 2).
Individual 2 is an Iranian male and one of two affected children born to consanguineous parents presenting with deafness and a homozygous c.1163C>T (p.Val388Ala) TSPEAR missense variant (Table 1). This variant has not been previously reported and is currently considered to be a VUS. This individual also carries a homozygous c.1211C>T TMPRSS3 (p.Pro404Leu) variant previously reported to be pathogenic in ClinVar, though no assertion criteria were provided for this entry (Accession number RCV000005233.4). Using the current available evidence, this TMPRSS3 variant was classified as likely pathogenic following review in this study (Table 2).
Individuals 3–4 have been previously reported, are presented in Table 1 to provide a complete overview of TSPEAR phenotypes, and are summarized in greater detail in Appendix B. Individual 3 was reported by Delmaghani et al. to have profound sensorineural hearing loss and an otherwise normal phenotype (Delmaghani et al., 2012). WES identified two sets of homozygous TSPEAR variants, c.1726G>T and c.1728delC (which can alternatively be presented as a single c.1726_1728delGTCinsTT, p.Val576Leufs*38 indel). Individual 4 has been described by Sloan‐Heggen et al. to have a phenotype that includes childhood onset SNHL, EVA, and proteinuria (Sloan‐Heggen et al., 2016). This individual underwent gene panel‐based hearing loss evaluation (OtoScope v5, 89 genes) which identified a c.1566G>A (p.Pro522Pro) TSPEAR variant which is predicted to impact splicing and a c.1676_1677delAT (p.Tyr559Cysfs*134) frame‐shift TSPEAR variant.
Individual 5 is a Caucasian male presenting with features that include sparse hair, sparse eyebrows, dysplastic nails, cone‐shaped teeth, and severe congenital SNHL. This individual's phenotype satisfies the diagnostic criteria for ectodermal dysplasia without tooth agenesis. Sequencing identified a c.589C>T (p.Arg197*) TSPEAR variant which results in a premature stop codon, inherited alongside a likely pathogenic c.38delT (p.Leu13Argfs*38) frame‐shift variant (Table 1). In addition to these TSPEAR variants, predicted to result in biallelic loss of function, the individual carries a GJB2 variant (c.35delG, p.Gly12Valfs*2) and a 309 kb GJB6‐D13S1854 deletion. GJB2 is an established cause of autosomal dominant hearing loss with or without skin findings, while the GJB6‐D13S1854 deletion spans multiple gene loci and is a common cause of nonsyndromic hearing loss (del Castillo et al., 2005; Rodriguez‐Paris et al., 2011; Tayoun et al., 2016). Together, the individual's GJB2 and TSPEAR variants, alongside the GJB6‐D13S1854 deletion, present competing genetic candidates to explain their phenotype.
3.3. Additional cohort phenotypes
Individuals with normal hearing displayed varied phenotypes that most commonly included tooth agenesis: five individuals displayed isolated tooth agenesis (nonsyndromic hypodontia or oligodontia), while seven presented with additional evidence of nail dysplasia, sweating difficulties, or sparse hair that satisfied diagnostic criteria for ectodermal dysplasia (Table 1). More detailed dental findings were reported for three individuals: individual 9 was noted to have microdontia, while individuals 16 and 17 were reported to have taurodontism. Orthopantomographs for TSPEAR subjects 15, 17, and 18 are detailed further in Figure 1 to illustrate a subset of TSPEAR dental phenotypes including oligodontia, conical teeth, and taurodontism.
FIGURE 1.
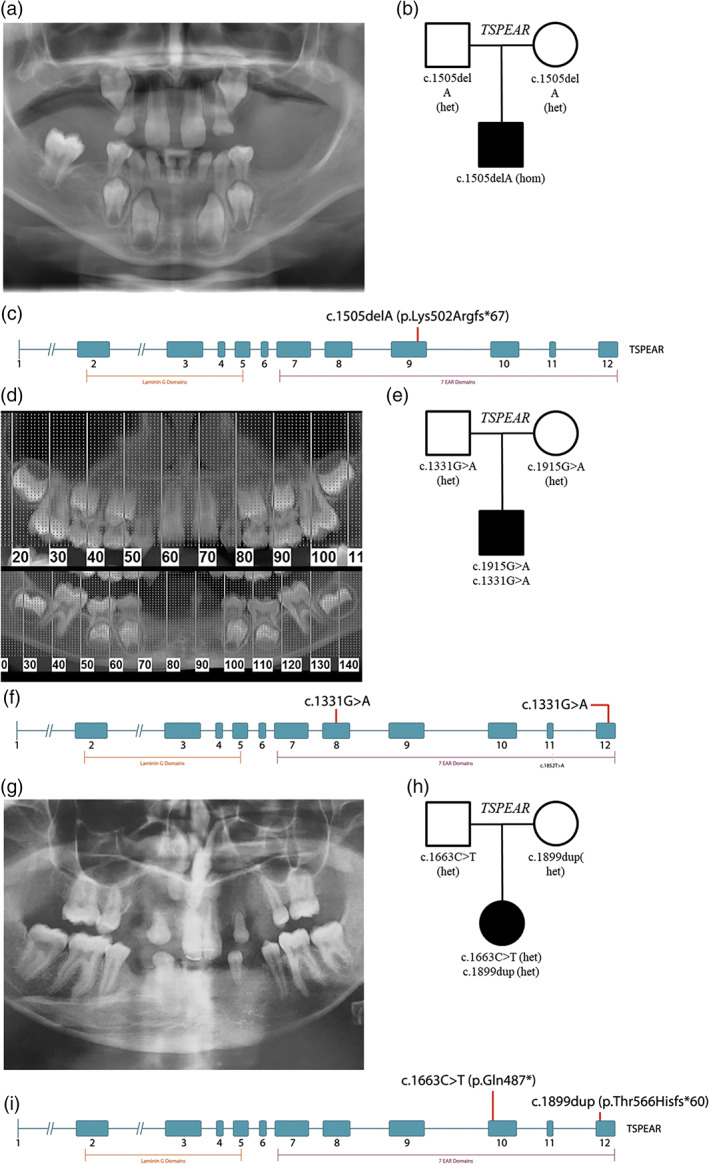
Representative oligodontia phenotypes for TSPEAR cohort subjects. Eighteen of the cohort subjects detailed in this article presented with tooth agenesis, which varied in severity from hypodontia to oligodontia, while a subset of study individuals presented with additional features such as taurodontism and conical teeth. (a) Orthopantomographs for cohort subject 15 show the absence of 19 permanent teeth and cone‐shaped canines that are characteristic of a subset of TSPEAR subjects detailed in this report. (b) The subject presented at 11 years of age and was born to unaffected, nonconsanguineous parents of Peruvian ancestry. Sequencing indicates that the subject is homozygous for c.1505delA (p.Lys502Argfs*67) variants. (c) The identified deletion is located in exon 9 within the epilepsy‐associated repeat (EAR) protein motif. (d) Orthopantomographs for cohort subject 17 show oligodontia with agenesis of 12, 13, 22, 23, 31, 32, 33, 41, 42, 43, as well as taurodontism of 16 and 26. (e) The subject was born to unaffected parents of European ancestry and presented for sequencing at age 9 with oligodontia, eczema, dry skin, and taurodontism. Sequencing identified two missense variants, c.1915G>A (p.Asp639Asn) and c.1331G>A (p.Arg444Gln). (f) These variants fall within exons 8 and 12, both of which form EAR protein motifs. (g) Orthopantomographs for cohort subject 18 show oligontia with agenesis of teeth 12, 13, 14, 17, 18, 22, 23, 24, 27, 28, 31, 32, 33, 34, 38, 41, 42, 43, 44, and 48. (h) The subject was born to unaffected parents of European ancestry and presented for sequencing at age 18 with tooth agenesis and scoliosis. Sequencing indicated the subject is heterozygous for two c.1633C>T and c.1899dup variants. (i) These variants are located within exons 10 and 12 and occur within EAR protein motifs. del, deletion; dup, duplication [Color figure can be viewed at wileyonlinelibrary.com]
Eight of twelve individuals presented with additional findings that are believed to be unrelated to their TSPEAR diagnosis. Various neurological findings were identified in four individuals: individual 6 displayed hypoplasia of the corpus callosum, cortical visual impairment, global developmental delay and seizures while individual 7 demonstrated movement abnormalities, profound global developmental delay, and self‐injurious behavior. Individual 8 was reported to have attention deficit hyperactivity disorder and resting tremor, in addition to abnormal heart morphology, basilar artery fenestration, recurrent otitis media, strabismus and tall stature. Individual 15 was described to have attention disorder, speech difficulties, large anteverted ears, and prognathism.
Individual 16 presented with facial features that included a depressed nasal bridge, hypertelorism, and small cupped ears in addition to sagittal craniosynostosis, paroxysmal dyskinesia and developmental delays that are believed to be related to a pathogenic ATP1A3 variant identified via WES. While subtle facial dysmorphisms (such as down slanting palpebral fissures or low columella insertion) have been reported in TSPEAR subjects (Peled et al., 2016), they do not match these newly reported subject descriptions and no causative link has been established thus far between TSPEAR variants and these phenotypes. Two remaining individuals reported with non‐neurological phenotypes: individual 10 was diagnosed with Turner syndrome in addition to their TSPEAR diagnosis, and presented with additional features including absent Meibomian glands, conductive hearing loss associated with otitis media, and eczema. Lastly, individual 18 was diagnosed with scoliosis. Full clinical descriptions for these individuals are available in Appendix A.
3.4. TSPEAR structure and variant distribution
All TSPEAR variants were mapped using the NM_144991.2 transcript and evaluated to determine if they concentrate within particular exons or predicted protein motifs (Figure 2). Variant burden scores, which normalize the variant concentration in each exon or predicted protein domain by the length of the region, indicate that the majority of reported variants concentrate in exons 4, 10, and 12, and within the EAR protein domains (Table S1). While variants are concentrated in particular regions of the gene, there was no observable correlation between variant location and phenotype, or between variant CADD score and presence of hearing loss (Mann–Whitney U test, α = 0.05, p = 0.91).
FIGURE 2.
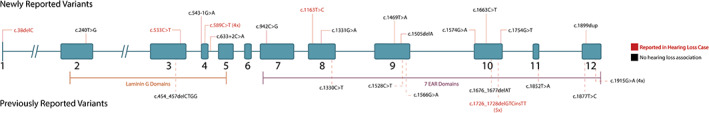
Variant location and gene structure. All variants associated with tooth agenesis or ectodermal dysplasia (with and without tooth agenesis, black) and variants associated with hearing loss (red) were mapped to their location within the NM_144991.2 TSPEAR transcript to provide an indication of exons or protein motifs that may be intolerant to variation. Newly reported variants are shown above the diagram, while previously reported variants are shown below the diagram. Large gene deletions are not shown. The c.1726_1728delGTCinsTT variant (p.Val576Leufs*38) in exon 10 has been separately reported in association with both hearing loss and ectodermal dysplasia. TSPEAR contains a laminin G structural domain and seven epilepsy‐associated repeat (EAR) domains which may mediate protein–protein interactions; both domains are indicated below the gene diagram. EAR, epilepsy‐associated repeat; del, deletion; dup, duplication [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
This cohort significantly expands the number of reported individuals with TSPEAR alterations and confirms that the most prevalent phenotypes associated with biallelic TSPEAR variants include tooth agenesis (18/21 individuals) and ectodermal dysplasia with tooth agenesis (7/21 individuals). Only 3/13 newly reported individuals presented with hearing loss, and all possessed variants in additional genes that may explain the hearing loss phenotype. Two of the probands affected by hearing loss (individuals 1 and 2) do not seem to show any sign of ED despite the large body of evidence linking TSPEAR variants to tooth agenesis and ED. It is possible that TSPEAR phenotypes such as hearing loss and tooth agenesis show variable penetrance in subjects with different genetic backgrounds. For example, FGF3 polymorphisms have been associated with variable levels of tooth agenesis in prior cohorts (Vieira et al., 2013), while multilocus inheritance has been well documented in Bardet‐Biedl syndrome, where up to 23 loci have been identified that initiate or modify disease (McConnachie et al., 2020; McKusick‐Nathans Institute of Genetic Medicine, 2020). While polygenic inheritance is one potential explanation for differences in TSPEAR phenotype penetrance, it is also possible that more subtle findings such as nail dysplasia or tooth agenesis were overlooked during clinical evaluation. The fact that hearing loss was a phenotype in less than 25% of the newly reported cases, and in each instance there was another reported plausible genetic cause for the hearing loss, weakens the evidence associating TSPEAR variants with hearing loss. It is reasonable to question the assertion that TSPEAR variants are a monogenic cause of hearing loss, or to at least recognize the reduced penetrance of this phenotype.
In light of these findings, it is worth revisiting the first studies to connect TSPEAR variants to hearing loss. Initial descriptions of the TSPEAR hearing loss association emerged from a 2012 study by Delmaghani et al. that used WES to identify biallelic TSPEAR variants which segregated with a SNHL phenotype (Delmaghani et al., 2012). Subsequent reports in 2016 and 2018 identified individuals with the same genotype with normal hearing, which casts doubt on the original hearing loss report (Du et al., 2018; Peled et al., 2016). While no other hearing loss variants were identified in the Delmaghani study, hearing loss is an extremely heterogeneous condition with numerous genetic contributors—over 100 different genes have been associated with nonsyndromic hearing loss and it is possible that the true genetic cause was not recognized due to test limitations (Abou Tayoun et al., 2016). For example, WES has limitations in ability to detect copy number variants such as single or multiple exon deletions, which are known to be an important contributor to genetic hearing loss (Shearer et al., 2014). Additionally, negative findings on panel or WES‐based approaches may simply reflect that the causal variant is not contained within the capture region, as can occur with deep intronic or noncoding variants; this is a situation potentially remedied through the use of next generation sequencing methods such as whole genome sequencing or RNA sequencing (Bodle et al., 2021; Di Scipio et al., 2020; Larrue et al., 2020). Furthermore, in the nearly 9 years since this first TSPEAR hearing loss publication, numerous additional hearing loss associations have emerged in the literature; variants in these hearing loss genes may have been overlooked at the time of publication (DiStefano et al., 2020). Lastly, hearing loss often has a significant environmental component, with pathogens such as cytomegalovirus accounting for a significant proportion of acquired hearing loss, particularly in underserved health systems (Goderis et al., 2014; Korver et al., 2017; Sloan‐Heggen et al., 2016). These same limitations may apply to the second study to identify individuals with TSPEAR variants and nonsyndromic hearing loss: a 2016 report describing siblings with childhood onset, nonsyndromic, bilateral hearing loss, EVA, and compound heterozygous TSPEAR variants (Sloan‐Heggen et al., 2016). These siblings were sequenced using a hearing loss gene panel—no other hearing loss variants were identified. These two reports represent the only cases of TSPEAR‐related hearing loss that have been described in the literature. While these cases did not identify any alternate genetic explanations for subject hearing loss, they represent the minority of publications to date and there is an emerging consensus in the literature that the primary phenotypes associated with TSPEAR variants are tooth agenesis and ED rather than hearing loss.
While we have identified new individuals in this study who present with biallelic TSPEAR variants, these individuals carry variants in other hearing loss genes such as TMPRSS3 that may present more compelling explanations for their hearing loss phenotype. Our ability to more comprehensively explore the cause of hearing loss for these individuals is limited by an absence of complete clinical histories; the hearing loss descriptions collected in this article have been abstracted from limited clinical reports, and more complete information on the degree of hearing loss is therefore unavailable. Similar limitations apply to normal hearing cohort subjects, as dental radiographs and complete clinical histories were only available for a subset of these individuals. This makes it difficult to comprehensively evaluate how variable tooth agenesis phenotypes are in TSPEAR subjects, and complicates the identification of more subtle TSPEAR‐associated phenotypes such as nail dysplasia. Despite this, this article strengthens evidence around the ED and tooth agenesis TSPEAR phenotypes, adds additional variants to the literature, and delivers insight into gene function. Missense variants appear to concentrate in exons 4, 10, and 12, and within the EAR domain of TSPEAR, indicating that these regions may have greater significance for normal gene function. The sub‐RVIS score, a measure of tolerance to variation within protein domains and exons, lists exons 4, 9, and 10 as the least tolerant in this gene and describes exon 8 and 12 as the most tolerant, in partial agreement with results from this study (Gussow et al., 2016). Additionally, exons 7–12 correspond to the EAR protein motif, which is believed to mediate protein/protein interactions (The UniProt Consortium, 2019). If TSPEAR interacts with NOTCH1, as has been previously suggested by Peled et al., the EAR domains may be the primary protein regions responsible for accomplishing this (Peled et al., 2016).
Lastly, this cohort presents several additional occurrences of the c.1726_1728delGTCinsTT (p.Val576Leufs*38) variant which has been consistently reported in the literature, almost entirely within individuals of Middle‐Eastern ancestry (Delmaghani et al., 2012; Du et al., 2018; Peled et al., 2016). This variant is absent from the GnomAD database, and while this change can also be reported as two separate c.1726G>C (p.Val576Leu) and c.1728delC (p.Lys577Serfs*37) variants in cis with each other, neither variant has been observed as of the GnomAD v3 release (Karczewski et al., 2019). This c.1726_1728delGTCinsTT (p.Val576Leufs*38) variant may be highly prevalent in Middle‐Eastern populations given its consistent observance in individuals of Middle‐Eastern ancestry; the GnomAD database does not currently contain a separate population category for Middle‐Eastern individuals and these individuals are instead classified as “other” (N = 3070 exomes and 1077 genomes). The Greater Middle East (GME) Variome Project (Scott et al., 2016), which reports whole exome data for 2497 individuals across the GME, also does not report any individuals with this same indel variant, although these sample sizes are low in comparison to the GnomAD database. Collectively, this delineates a continued need for the increased representation of diverse populations within publicly‐available sequencing databases.
In conclusion, this cohort study significantly expands the body of reported individuals with TSPEAR variants, and brings into question whether loss‐of‐function of TSPEAR is a monogenic cause of hearing loss. Future work, featuring either larger TSPEAR cohorts or experimental approaches to analyze gene function will be needed to conclusively examine the putative TSPEAR hearing loss association.
CONFLICT OF INTEREST
IMW and KMc are employees of GeneDx, Inc.
AUTHOR CONTRIBUTIONS
BB collected and analyzed patient variant data. EWK conceived and directed the study. BB, AF, FPV, and EWK performed the variant interpretation and wrote the article. CJN, RJS, JAH, AA, and ABZ providing editing suggestions for the article. NS, TR, KM, and IMW provided genetic data and assisted with variant interpretation. KM and IMW facilitated contacts between study authors. CJN, TR, BL, JS, KS, EA, DC, AW, AC, SL, SG, CR, CL, DBV, BCL, MLD, AA, JAH, BD, FCR, VS, BG, ABZ, and RJS provided clinical and phenotypic relevant information included in the article. All authors reviewed and approved the final submitted text.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
We would like to acknowledge the assistance of patients and their families, in addition to Drs. Nathalie Seivert and Denise Halter who assisted with accruing and evaluating TSPEAR individuals in this cohort. We would also like to thank members of the NIH Undiagnosed Diseases Network who assisted with evaluation patients within this cohort: Maria T. Acosta, Margaret Adam, David R. Adams, Pankaj B. Agrawal, Mercedes E. Alejandro, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A. Ashley, Mahshid S. Azamian, Carlos A. Bacino, Guney Bademci, Eva Baker, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak‐Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Jimmy Bennet, Beverly Berg‐Rood, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Carsten Bonnenmann, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C. Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Olveen Carrasquillo, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao‐Tuan Chao, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Cynthia M. Cooper, Heidi Cope, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Precilla D'Souza, Hongzheng Dai, Surendra Dasari, Mariska Davids, Jyoti G. Dayal, Matthew Deardorff, Esteban C. Dell'Angelica, Shweta U. Dhar, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Emilie D. Douine, David D. Draper, Laura Duncan, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Cecilia Esteves, Tyra Estwick, Marni Falk, Liliana Fernandez, Carlos Ferreira, Elizabeth L. Fieg, Laurie C. Findley, Paul G. Fisher, Brent L. Fogel, Irman Forghani, Laure Fresard, William A. Gahl, Ian Glass, Rena A. Godfrey, Katie Golden‐Grant, Alica M. Goldman, David B. Goldstein, Alana Grajewski, Catherine A. Groden, Andrea L. Gropman, Irma Gutierrez, Sihoun Hahn, Rizwan Hamid, Neil A. Hanchard, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike‐Pyne, Alden Huang, Yong Huang, Rosario Isasi, Fariha Jamal, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Jean M. Johnston, Lefkothea Karaviti, Emily G. Kelley, Jennifer Kennedy, Dana Kiley, Isaac S. Kohane, Jennefer N. Kohler, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Susan Korrick, Mary Koziura, Joel B. Krier, Seema R. Lalani, Byron Lam, Christina Lam, Brendan C. Lanpher, Ian R. Lanza, C. Christopher Lau, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Roy Levitt, Richard A. Lewis, Sharyn A. Lincoln, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Marta M. Majcherska, Bryan Mak, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Kenneth Maravilla, Thomas C. Markello, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez‐Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie‐Rosell, Colleen E. McCormack, Alexa T. McCray, Elisabeth McGee, Heather Mefford, J. Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo M. Moretti, Marie Morimoto, John J. Mulvihill, David R. Murdock, Mariko Nakano‐Okuno, Avi Nath, Stan F. Nelson, John H. Newman, Sarah K. Nicholas, Deborah Nickerson, Shirley Nieves‐Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen Pak, J. Carl Pallais, Christina GS. Palmer, Jeanette C. Papp, Neil H. Parker, John A. Phillips III, Jennifer E. Posey, Lorraine Potocki, Barbara N. Pusey, Aaron Quinlan, Wendy Raskind, Archana N. Raja, Deepak A. Rao, Genecee Renteria, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Natalie Rosenwasser, Maura Ruzhnikov, Ralph Sacco, Jacinda B. Sampson, Susan L. Samson, Mario Saporta, C. Ron Scott, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A. Scott, Prashant Sharma, Vandana Shashi, Jimann Shin, Rebecca Signer, Catherine H. Sillari, Edwin K. Silverman, Janet S. Sinsheimer, Kathy Sisco, Edward C. Smith, Kevin S. Smith, Emily Solem, Lilianna Solnica‐Krezel, Rebecca C. Spillmann, Joan M. Stoler, Nicholas Stong, Jennifer A. Sullivan, Kathleen Sullivan, Angela Sun, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Cecelia P. Tamburro, Queenie K.‐G. Tan, Mustafa Tekin, Fred Telischi, Willa Thorson, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Brianna M. Tucker, Tiina K. Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Stephanie Wallace, Nicole M. Walley, Chris A. Walsh, Melissa Walker, Jennifer Wambach, Jijun Wan, Lee‐kai Wang, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Mark Wener, Tara Wenger, Katherine Wesseling Perry, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Jeremy D. Woods, Shinya Yamamoto, John Yang, Guoyun Yu, Diane B. Zastrow, Chunli Zhao, and Stephan Zuchner. This study was funded in part by NIDCDs R01s DC002842 and DC012049 to RJS. Individual 7 was ascertained in the Duke Genome Sequencing Clinic as a participant in a research study (Duke Protocol 00032301). Funding for the Duke Genome Sequencing Clinic was provided by Duke University Health System. Research reported in this article was also supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number U01HG007672 (Duke University). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This material is also based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. NSF1744557. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. For ABZ, BG, and TR this work was supported by the project ARS (Agence Régionale de Santé Grand EST) Appel à projets 2018 «Innovations en santé»: e_diagnostic des maladies rares orales et dentaires «e‐GenoDENT» and the project No. 1.7 “RARENET: a trinational network for education, research and management of complex and rare disorders in the Upper Rhine” co‐financed by the European Regional Development Fund (ERDF) of the European Union in the framework of the INTERREG V Upper Rhine program. ABZ is a USIAS 2015 Fellow of the Institute of Advanced Studies (Institut d'Etudes Avancées) de l'Université de Strasbourg, France.
NEWLY REPORTED PHENOTYPIC DESCRIPTIONS
1.
All newly reported individuals with hearing loss are detailed in the main text as individuals 1, 2, and 5. The clinical histories for the remaining individuals are detailed below. The main text and Table 1 also reference individuals who have been previously published in other studies; these cases are summarized in Appendix B.
Individual 6 is a European ancestry male who displays phenotypic features such as nail dysplasia, conical teeth, and hypodontia that are believed to be connected to the observed TSPEAR variants and that satisfy diagnostic criteria for ectodermal dysplasia with tooth agenesis. In addition, the individual presented with nystagmus and brachydactyly, as well as significant neurological features including global developmental delay, hypoplasia of the corpus callosum, and cortical visual impairment. WES detected a maternally inherited, c.1915G>A (p.Asp639Asn) missense variant and a paternally inherited c.589C>T (p.Arg197*) nonsense variant. This individual has a brother with cone‐shaped teeth and tooth agenesis, while a maternal aunt was noted to have a supernumerary tooth and a paternal grandfather was reported to have tooth agenesis.
Individual 7 is a European ancestry male presenting with a phenotype that includes oligodontia, conical teeth, and poor sweating. This phenotype satisfies the diagnostic criteria for ectodermal dysplasia with tooth agenesis. This individual also displayed strabismus in addition to significant neurological features such as intellectual disability, movement disorder, dissociative disorder, profound developmental delay, and self‐injurious behavior. This individual harbors a c.1915G>A (p.Asp639Asn) variant in addition to a c.1754G>A (p.Ser585Ile) missense variant which is located at the 3′ end of exon 10 and is predicted to disrupt splicing. Exon 10, if skipped, will result in an out‐of‐frame product. This individual's family history was otherwise uninformative.
Individual 8 is a male who presents with oligodontia, nail bed pitting, and thin nails. This phenotype satisfies the diagnostic criteria for ectodermal dysplasia with tooth agenesis. Additionally, this individual's phenotype consists of complex congenital heart disease, left strabismus, attention deficit hyperactivity disorder, tall stature, fenestration of the basilar artery, bilateral resting tremor, and recurrent otitis media infections. More specifically, this individual's heart disease encompasses coarctation of the aorta, a bicuspid aortic valve, an atrial septal defect, and patent ductus arteriosus. This individual has an identified c.589C>T (p.Arg197*) nonsense variant which results in a premature stop. In addition, this individual has a c.83‐13708_1566+248 deletion that encompasses exons 2–9 of TSPEAR. This individual did not have similarly affected relatives.
Individual 9 is a European ancestry male who was referred for cone‐shaped teeth, and who also displayed dysplastic nails, hypopigmented hair, sparse hair, sparse eyebrows, hypodontia, and misshapen teeth. This individual was also reported to “turn red” when hot, which suggests possible hypohidrosis. This phenotype satisfies the diagnostic criteria for ectodermal dysplasia with tooth agenesis. This individual's family history was otherwise uninformative. WES detected a c.1574G>A (p.Gly525Asp) paternally inherited missense alteration and a maternally inherited c.543‐1G>A splicing variant which affects the −1 nucleotide of intron 3. This is predicted to result in the loss of a splice acceptor site, and the formation of an out of frame product due to skipping of exon 4.
Individual 10 is a three year old female of European ancestry who presented with hypotrichosis, conical teeth, eczema with mottling of skin, absence of the Meibomian glands, abnormal nail growth, hypohidrosis, and conductive hearing loss associated with otitis media. This phenotype satisfies the diagnostic criteria for ectodermal dysplasia without tooth agenesis. WES detected two novel compound heterozygous variants in TSPEAR, a c.942C>G (p.Tyr314*) nonsense change and c.1469T>A (p.Leu490Gln) variant that is predicted to activate a cryptic splice acceptor in the center of exon 9. Variant segregation was confirmed by parental testing. Her diagnostic evaluation was complicated by incidental detection of variant Turner syndrome with a karyotype of 46,X,i(X)(q10)(46)/45,X(4). Her infant sister was subsequently found to have the same TSPEAR genotype and similar clinical features without Turner syndrome.
Individual 14 is a European ancestry male initially evaluated at 18 months and presenting with ectodermal dysplasia and conical teeth. The individual's hearing was normal and, upon initial evaluation, he also displayed sparse hair which resolved later in childhood. This phenotype satisfies the diagnostic criteria for ectodermal dysplasia without tooth agenesis. Ectodermal dysplasia panel‐based sequencing identified compound heterozygous TSPEAR variants: a paternal c.240T>G (p.Cys80Trp) variant and a maternal c.633+2C>A variant with a possible effect on exon 4 splicing. The family history for this individual was otherwise uninformative.
Individual 15 is a European ancestry male presenting at age 11 years with oligodontia, conical‐shaped canine teeth, attention disorder, anteverted ears, prognathism, and speech difficulties. Audiometric testing confirmed that the individual has normal hearing. The individual was sequenced using a SNP array which identified multiple regions of homozygosity, one of which overlaps the TSPEAR locus. Targeted Sanger sequencing of the chromosome 21 region containing TSPEAR revealed the presence of likely pathogenic, homozygous c.1505delA (p.Lys502Argfs*67) variants. While the parents are both of Peruvian ancestry, there was no reported history of consanguinity and the individual's family history was otherwise uninformative.
Individual 16 is an African American ancestry male who presented at age 7 years with oligodontia, conical teeth, primary molar taurodontism, sagittal craniosynostosis, and dysmorphisms that included hypertelorism, small and cupped ears, and a depressed nasal bridge. This individual also presented with a neurological phenotype that includes paroxysmal dyskinesia and developmental delays, which are believed to be caused by a pathogenic ATP1A3 variant (c.2767G>A, p.D923N). This particular variant has been identified in several probands with variable features that included rapid‐onset dystonia parkinsonism, alternating hemiplegia, and developmental delays (Anselm, Sweadner, Gollamudi, Ozelius, & Darras, 2009; Roubergue et al., 2013; Zanotti‐Fregonara et al., 2008). Whole exome sequencing identified additional findings in TSPEAR: a c.1726_1728delGTCinsTT (p.Val576Leufs*38) and a c.589C>T (p.Arg197*) in trans which present a compelling explanation for the subject's dental phenotypes. Audiology evaluation indicated normal hearing. The subject's parents were unaffected, but his brother (who did not receive genetic testing) was reported to have missing permanent teeth as well.
Individual 17 is a 9‐year‐old European ancestry male presenting with ectodermal dysplasia, oligodontia, and conical teeth. He also displayed taurodontism and had no reported history of hearing loss. Sequencing identified compound heterozygous TSPEAR variants: c.1915G>A (p.Asp639Asn) and c.1331G>A (p.Arg444Gln) missense changes, the latter of which occurs near the end of the exon 8 and is predicted to create a weak splice acceptor site. The individual's family history was otherwise uninformative.
Individual 18 is a European ancestry female who presented at age 18 with oligodontia and severe scoliosis. This individual carries a c.1663C>T (p.Gln555*) nonsense variant. Additionally, this individual harbors a c.1899dup (p.Thr634Hisfs*60) variant which is predicted to create an out‐of‐frame product. This individual had no relevant family history and their hearing was not impacted.
SUMMARY AND SCORING OF PREVIOUSLY REPORTED TSPEAR CASES
Previously reported TSPEAR cases are described below. For the original manuscripts detailing these individuals, please refer to Delmaghani et al. (2012), Peled et al. (2016), and Du et al. (2018). Subject numbering refers to their order in Table 1 of the main text.
Individual 3 is an Iranian‐ancestry male who was reported by Delmaghani et al. to have bilateral, profound SNHL, as confirmed by audiology (Delmaghani et al., 2012). No other clinical features or variants of interest were reported. This individual was initially reported to have biallelic c.1726G>T and c.1728delC TSPEAR variants, which can alternatively be presented as biallelic c.1726_1728delGTCinsTT (p.Val576Leufs*38) complex variants. This individual was born to consanguineous parents and had two affected brothers presenting with the same TSPEAR variants.
Individual 4 has been briefly described as part of a large hearing loss study, but is being reported here in greater detail (Sloan‐Heggen et al., 2016). This individual is a Caucasian male with a phenotype that includes childhood onset SNHL, enlargement of the vestibular aqueduct, and proteinuria. The individual carries two TSPEAR variants: a c.1676_1677delAT (p.Tyr559Cysfs*134) deletion, and an additional c.1566G>A (p.Pro522Pro) missense variant affecting the last nucleotide of exon 9. Splicing prediction tools delivered conflicting reports for this variant—Alamut Visual tools (SpliceSiteFinder, MaxEntScan, and GeneSplice) suggested a reduction in splice donor strength, while the SpliceAI predicted a lesser impact on splice donor efficiency (DS_DL=0.4613) (Jaganathan et al., 2019). Disrupted splicing at exon 9 and subsequent skipping of the exon is predicted to result in an out‐of‐frame product. The individual has a sister who is similarly affected with bilateral SNHL and EVA.
Individuals 11–13 have been previously reported by Peled et al. (2016). These individuals presented with hypodontia, hypotrichosis, and subtle facial dysmorphisms that included a long oval face, square chin, down slanting palpebral features, low insertion of columella, and thick lips. These phenotypic features satisfy the diagnostic criteria for ectodermal dysplasia with tooth agenesis. Individual 11 is of Middle‐Eastern ancestry and carries two c.1726_1728delGTCinsTT (p.Val576Leufs*38) TSPEAR frameshift variants, while individual 12 is of Middle Eastern ancestry and is compound heterozygous for the same c.1726_1728delGTCinsTT (p.Val576Leufs*38) variant and a c.454_457delCTG (p.Leu152Trpfs*29) deletion. Individual 13 is of Jewish ancestry and is compound heterozygous for c.1852T>A (p.Tyr618Asn) and c.1915G>A (p.Asp639Asn) missense variants and was less severely affected. Individual 11 was born to consanguineous parents and had an affected cousin and uncle, while the family history for individuals 12 and 13 was uninformative.
Individuals 19–20 were previously reported in a study by Du et al. (2018). Individual 19 was noted to have oligodontia, microcephaly, and facial dysmorphisms including a narrow forehead, increased hair growth on the forehead, a high arched palate, low set ears, and minimal striation at the border of the ear lobules and antitragus and was homozygous for c.1726_1278delGTCinsTT (c.1726_1728delGTCinsTT*) TSPEAR variants (Du et al., 2018). Individual 20 is homozygous for c.1877T>C (p.Phe626Ser) variants and was observed to have nonsyndromic tooth agenesis—no other clinical features were identified. Both individuals were born to consanguineous parents. Family history was otherwise uninformative for individual 19, while individual 20 had a similarly affected brother and father.
Individual 21 is a male of Korean ancestry who was reported by Song et al. to have nonsyndromic oligodontia (Song et al., 2020). This individual inherited a paternal c.1528C>T (p.Arg510*) nonsense variant and a maternal c.1330C>T (p.Arg444Trp) variant. The family history was otherwise uninformative.
Bowles, B., Ferrer, A., Nishimura, C. J., Pinto e Vairo, F., Rey, T., Leheup, B.…Klee, E. W. (2021). TSPEAR variants are primarily associated with ectodermal dysplasia and tooth agenesis but not hearing loss: A novel cohort study. American Journal of Medical Genetics Part A, 185A:2417–2433. 10.1002/ajmg.a.62347
Undiagnosed Diseases Network: A list of authors appears in the acknowledgments section of this article.
Funding information National Institute on Deafness and Other Communication Disorders, Grant/Award Numbers: DC002842, DC012049; National Science Foundation, Grant/Award Number: NSF1744557; NIH Office of the Director, Grant/Award Number: U01HG007672
DATA AVAILABILITY STATEMENT
All variants described in this study are openly available for access within the ClinVar variant database (https://www.ncbi.nlm.nih.gov/clinvar/).
REFERENCES
- Abou Tayoun, A. N., Al Turki, S. H., Oza, A. M., Bowser, M. J., Hernandez, A. L., Funke, B. H., Rehm, H. L., & Amr, S. S. (2016). Improving hearing loss gene testing: A systematic review of gene evidence toward more efficient next‐generation sequencing–based diagnostic testing and interpretation. Genetics in Medicine, 18(6), 545–553. 10.1038/gim.2015.141 [DOI] [PubMed] [Google Scholar]
- Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., Kondrashov, A. S., & Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselm, I. A., Sweadner, K. J., Gollamudi, S., Ozelius, L. J., & Darras, B. T. (2009). Rapid‐onset dystonia‐parkinsonism in a child with a novel ATP1A3 gene mutation. Neurology, 73(5), 400–401. 10.1212/wnl.0b013e3181b04acd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodle, E. E., Zhu, W., Velez‐Bartolomei, F., Tesi‐Rocha, A., Liu, P., & Bernstein, J. A. (2021). Combined genome sequencing and RNA analysis reveals and characterizes a deep intronic variant in IGHMBP2 in a patient with spinal muscular atrophy with respiratory distress type 1. Pediatric Neurology, 114, 16–20. 10.1016/j.pediatrneurol.2020.09.011 [DOI] [PubMed] [Google Scholar]
- Coban‐Akdemir, Z., White, J. J., Song, X., Jhangiani, S. N., Fatih, J. M., Gambin, T., Bayram, Y., Chinn, I. K., Karaca, E., Punetha, J., Poli, C., Baylor‐Hopkins Center for Mendelian Genomics , Boerwinkle, E., Shaw, C. A., Orange, J. S., Gibbs, R. A., Lappalainen, T., Lupski, J. R., & Carvalho, C. M. B. (2018). Identifying genes whose mutant transcripts cause dominant disease traits by potential gain‐of‐function alleles. American Journal of Human Genetics, 103(2), 171–187. 10.1016/j.ajhg.2018.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Dure‐Molla, M., Fournier, B. P., Manzanares, M. C., Acevedo, A. C., Hennekam, R. C., Friedlander, L., Boy‐Lefèvre, M.‐L., Kerner, S., Toupenay, S., Garrec, P., Vi‐Fane, B., Felizardo, R., Berteretche, M.‐V., Jordan, L., Ferré, F., Clauss, F., Jung, S., de Chalendar, M., Troester, S., … Bloch‐Zupan, A. (2019). Elements of morphology: Standard terminology for the teeth and classifying genetic dental disorders. American Journal of Medical Genetics Part A, 179(10), 1913–1981. 10.1002/ajmg.a.61316 [DOI] [PubMed] [Google Scholar]
- del Castillo, F. J., Rodríguez‐Ballesteros, M., Alvarez, A., Hutchin, T., Leonardi, E., de Oliveira, C. A., Azaiez, H., Brownstein, Z., Avenarius, M. R., Marlin, S., Pandya, A., Shahin, H., Siemering, K. R., Weil, D., Wuyts, W., Aguirre, L. A., Martín, Y., Moreno‐Pelayo, M. A., Villamar, M., … del Castillo, I. (2005). A novel deletion involving the connexin‐30 gene, del(GJB6‐d13s1854), found in trans with mutations in the GJB2 gene (connexin‐26) in subjects with DFNB1 non‐syndromic hearing impairment. Journal of Medical Genetics, 42(7), 588–594. 10.1136/jmg.2004.028324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmaghani, S., Aghaie, A., Michalski, N., Bonnet, C., Weil, D., & Petit, C. (2012). Defect in the gene encoding the EAR/EPTP domain‐containing protein TSPEAR causes DFNB98 profound deafness. Human Molecular Genetics, 21(17), 3835–3844. 10.1093/hmg/dds212 [DOI] [PubMed] [Google Scholar]
- Di Scipio, M., Tavares, E., Deshmukh, S., Audo, I., Green‐Sanderson, K., Zubak, Y., Zine‐Eddine, F., Pearson, A., Vig, A., Tang, C. Y., Mollica, A., Karas, J., Tumber, A., Yu, C. W., Billingsley, G., Wilson, M. D., Zeitz, C., Héon, E., & Vincent, A. (2020). Phenotype driven analysis of whole genome sequencing identifies deep intronic variants that cause retinal dystrophies by aberrant exonization. Investigative Ophthalmology & Visual Science, 61(10), 36–36. 10.1167/iovs.61.10.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiStefano, M. T., Hughes, M. Y., Patel, M. J., Wilcox, E. H., & Oza, A. M. (2020). Expert interpretation of genes and variants in hereditary hearing loss. Medizinische Genetik, 32(2), 109–115. 10.1515/medgen-2020-2018 [DOI] [Google Scholar]
- Du, R., Dinckan, N., Song, X., Coban‐Akdemir, Z., Jhangiani, S. N., Guven, Y., Aktoren, O., Kayserili, H., Petty, L. E., Muzny, D. M., Below, J. E., Boerwinkle, E., Wu, N., Gibbs, R. A., Posey, J. E., Lupski, J. R., Letra, A., & Uyguner, Z. O. (2018). Identification of likely pathogenic and known variants in TSPEAR, LAMB3, BCOR, and WNT10A in four Turkish families with tooth agenesis. Human Genetics, 137(9), 689–703. 10.1007/s00439-018-1907-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon, R., Milon, B., Morrison, L., Shah, M., Vijayakumar, S., Racherla, M., Leitch, C. C., Silipino, L., Hadi, S., Weiss‐Gayet, M., Barras, E., Schmid, C. D., Ait‐Lounis, A., Barnes, A., Song, Y., Eisenman, D. J., Eliyahu, E., Frolenkov, G. I., Strome, S. E., … Hertzano, R. (2015). RFX transcription factors are essential for hearing in mice. Nature Communications, 6, 8549. 10.1038/ncomms9549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goderis, J., De Leenheer, E., Smets, K., Van Hoecke, H., Keymeulen, A., & Dhooge, I. (2014). Hearing loss and congenital CMV infection: A systematic review. Pediatrics, 134(5), 972–982. 10.1542/peds.2014-1173 [DOI] [PubMed] [Google Scholar]
- Gussow, A. B., Petrovski, S., Wang, Q., Allen, A. S., & Goldstein, D. B. (2016). The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biology, 17(1), 9. 10.1186/s13059-016-0869-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., Kosmicki, J. A., Arbelaez, J., Cui, W., Schwartz, G. B., Chow, E. D., Kanterakis, E., Gao, H., Kia, A., Batzoglou, S., Sanders, S. J., & Farh, K. K.‐H. (2019). Predicting splicing from primary sequence with deep learning. Cell, 176(3), 535–548.e524. 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer‐Berk M, England EM, Seaby EG, Kosmicki JA, … MacArthur DG (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. bioRxiv:531210
- Köhler, S., Carmody, L., Vasilevsky, N., Jacobsen, J. O. B., Danis, D., Gourdine, J.‐P., … Robinson, P. N. (2018). Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Research, 47(D1), D1018–D1027. 10.1093/nar/gky1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korver, A. M., Admiraal, R. J., Kant, S. G., Dekker, F. W., Wever, C. C., Kunst, H. P., Frijns, J. H. M., Oudesluys‐Murphy, A. M., & Oudesluys‐Murphy, A. M. (2011). Causes of permanent childhood hearing impairment. Laryngoscope, 121(2), 409–416. 10.1002/lary.21377 [DOI] [PubMed] [Google Scholar]
- Korver, A. M. H., Smith, R. J. H., Van Camp, G., Schleiss, M. R., Bitner‐Glindzicz, M. A. K., Lustig, L. R., Usami, S.‐I., & Boudewyns, A. N. (2017). Congenital hearing loss. Nature Reviews Disease Primers, 3(1), 16094. 10.1038/nrdp.2016.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrue, R., Chamley, P., Bardyn, T., Lionet, A., Gnemmi, V., Cauffiez, C., Glowacki, F., Pottier, N., & Broly, F. (2020). Diagnostic utility of whole‐genome sequencing for nephronophthisis. NPJ Genomic Medicine, 5, 38–38. 10.1038/s41525-020-00147-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masmoudi, S., Antonarakis, S. E., Schwede, T., Ghorbel, A. M., Gratri, M. h., Pappasavas, M.‐P., Drira, M., Elgaied‐Boulila, A., Wattenhofer, M., Rossier, C., Scott, H. S., Ayadi, H., & Guipponi, M. (2001). Novel missense mutations of TMPRSS3 in two consanguineous Tunisian families with non‐syndromic autosomal recessive deafness. Human Mutation, 18(2), 101–108. 10.1002/humu.1159 [DOI] [PubMed] [Google Scholar]
- McConnachie, D. J., Stow, J. L., & Mallett, A. J. (2020). Ciliopathies and the kidney: A review. American Journal of Kidney Diseases, 77(3), 410–419. 10.1053/j.ajkd.2020.08.012 [DOI] [PubMed] [Google Scholar]
- McKusick‐Nathans Institute of Genetic Medicine . (2020). Online Mendelian inheritance in man, OMIM®. Retrieved from https://omim.org/
- Mills, C. L., Garg, R., Lee, J. S., Tian, L., Suciu, A., Cooperman, G. D., Beuning, P. G., & Ondrechen, M. J. (2018). Functional classification of protein structures by local structure matching in graph representation. Protein Science, 27(6), 1125–1135. 10.1002/pro.3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., Brown, S. D., Chang, H.‐Y., El‐Gebali, S., Fraser, M. I., Gough, J., Haft, D. R., Huang, H., Letunic, I., Lopez, R., Luciani, A., Madeira, F., Marchler‐Bauer, A., Mi, H., … Finn, R. D. (2018). InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Research, 47(D1), D351–D360. 10.1093/nar/gky1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, P. C., & Henikoff, S. (2001). Predicting deleterious amino acid substitutions. Genome Research, 11(5), 863–874. 10.1101/gr.176601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled, A., Sarig, O., Samuelov, L., Bertolini, M., Ziv, L., Weissglas‐Volkov, D., Eskin‐Schwartz, M., Adase, C. A., Malchin, N., Bochner, R., Fainberg, G., Goldberg, I., Sugawara, K., Baniel, A., Tsuruta, D., Luxenburg, C., Adir, N., Duverger, O., Morasso, M., … Sprecher, E. (2016). Mutations in TSPEAR, encoding a regulator of notch signaling, affect tooth and hair follicle morphogenesis. PLoS Genetics, 12(10), e1006369. 10.1371/journal.pgen.1006369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, M., Lin, X., & Salzberg, S. L. (2001). GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Research, 29(5), 1185–1190. 10.1093/nar/29.5.1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch, P., Kircher, M., Witten, D., Cooper, G. M., & Shendure, J. (2018). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47(D1), D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey, T., Tarabeux, J., Gerard, B., Delbarre, M., Le Béchec, A., Stoetzel, C., Prasad, M., Laugel‐Haushalter, V., Kawczynski, M., Muller, J., Chelly, J., Dollfus, H., Manière, M.‐C., & Bloch‐Zupan, A. (2019). Protocol GenoDENT: Implementation of a new NGS panel for molecular diagnosis of genetic disorders with orodental involvement. Methods in Molecular Biology, 1922, 407–452. 10.1007/978-1-4939-9012-2_36 [DOI] [PubMed] [Google Scholar]
- Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier‐Foster, J., Grody, W. W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K., Rehm, H. L., & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Paris, J., Tamayo, M. L., Gelvez, N., & Schrijver, I. (2011). Allele‐specific impairment of GJB2 expression by GJB6 deletion del(GJB6‐D13S1854). PLoS One, 6(6), e21665. 10.1371/journal.pone.0021665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubergue, A., Roze, E., Vuillaumier‐Barrot, S., Fontenille, M.‐J., Méneret, A., Vidailhet, M., Fontaine, B., Doummar, D., Philibert, B., Riant, F., & Nicole, S. (2013). The multiple faces of the ATP1A3‐related dystonic movement disorder. Movement Disorders, 28(10), 1457–1459. 10.1002/mds.25396. [DOI] [PubMed] [Google Scholar]
- Scheffer, D. I., Shen, J., Corey, D. P., & Chen, Z. Y. (2015). Gene expression by mouse inner ear hair cells during development. Journal of Neuroscience, 35(16), 6366–6380. 10.1523/jneurosci.5126-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M., Cooper, D. N., Schuelke, M., & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Scott, E. M., Halees, A., Itan, Y., Spencer, E. G., He, Y., Azab, M. A., Gabriel, S. B., Belkadi, A., Boisson, B., Abel, L., Clark, A. G., Greater Middle East Variome Consortium , Alkuraya, F. S., Casanova, J.‐L., & Gleeson, J. G. (2016). Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nature Genetics, 48(9), 1071–1076. 10.1038/ng.3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, M. B., & Senapathy, P. (1987). RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Research, 15(17), 7155–7174. 10.1093/nar/15.17.7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer, A. E., DeLuca, A. P., Hildebrand, M. S., Taylor, K. R., Gurrola, J., Scherer, S., Scheetz, T. E., & Smith, R. J. H. (2010). Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proceedings of the National Academy of Sciences of the United States of America, 107(49), 21104. 10.1073/pnas.1012989107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer, A. E., Kolbe, D. L., Azaiez, H., Sloan, C. M., Frees, K. L., Weaver, A. E., Clark, E. T., Nishimura, C. J., Black‐Ziegelbein, E. A., & Smith, R. J. H. (2014). Copy number variants are a common cause of non‐syndromic hearing loss. Genome Medicine, 6(5), 37–37. 10.1186/gm554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan‐Heggen, C. M., Bierer, A. O., Shearer, A. E., Kolbe, D. L., Nishimura, C. J., Frees, K. L., Ephraim, S. S., Shibata, S. B., Booth, K. T., Campbell, C. A., Ranum, P. T., Weaver, A. E., Black‐Ziegelbein, E. A., Wang, D., Azaiez, H., & Smith, R. J. H. (2016). Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Human Genetics, 135, 441–450. 10.1007/s00439-016-1648-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira, N., Schiettecatte, F., Valle, D., & Hamosh, A. (2015). GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36(10), 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J.‐S., Bae, M., & Kim, J.‐W. (2020). Novel TSPEAR mutations in non‐syndromic oligodontia. Oral Diseases, 26(4), 847–849. 10.1111/odi.13316 [DOI] [PubMed] [Google Scholar]
- Tavtigian, S. V., Deffenbaugh, A. M., Yin, L., Judkins, T., Scholl, T., Samollow, P. B., de Silva, D., & Thomas, A. (2006). Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. Journal of Medical Genetics, 43(4), 295–305. 10.1136/jmg.2005.033878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayoun, A. N. A., Mason‐Suares, H., Frisella, A. L., Bowser, M., Duffy, E., Mahanta, L., Funke, B., Rehm, H. L., & Amr, S. S. (2016). Targeted droplet‐digital PCR as a tool for novel deletion discovery at the DFNB1 locus. Human Mutation, 37(1), 119–126. 10.1002/humu.22912 [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium . (2019). UniProt: A worldwide hub of protein knowledge. Nucleic Acids Research, 47(D1), D506–D515. 10.1093/nar/gky1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira, A. R., D'Souza, R. N., Mues, G., Deeley, K., Hsin, H. Y., Küchler, E. C., Meira, R., Patir, A., Tannure, P. N., Lips, A., Costa, M. C., Granjeiro, J. M., Seymen, F., & Modesto, A. (2013). Candidate gene studies in hypodontia suggest role for FGF3. European Archives of Paediatric Dentistry, 14(6), 405–410. 10.1007/s40368-013-0010-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldhaus, J., Durruthy‐Durruthy, R., & Heller, S. (2015). Quantitative high‐resolution cellular map of the organ of Corti. Cell Reports, 11(9), 1385–1399. 10.1016/j.celrep.2015.04.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, J. T., Fete, M., Schneider, H., Zinser, M., Koster, M. I., Clarke, A. J., Hadj‐Rabia, S., Tadini, G., Pagnan, N., Visinoni, A. F., Bergendal, B., Abbott, B., Fete, T., Stanford, C., Butcher, C., D'Souza, R. N., Sybert, V. P., & Morasso, M. I. (2019). Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. American Journal of Medical Genetics. Part A, 179(3), 442–447. 10.1002/ajmg.a.61045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo, G., & Burge, C. B. (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. Journal of Computational Biology, 11(2–3), 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Zanotti‐Fregonara, P., Vidailhet, M., Kas, A., Ozelius, L. J., Clot, F., Hindié, E., Ravasi, L., Devaux, J.‐Y., & Roze, E. (2008). [123I]‐FP‐CIT and [99mTc]‐HMPAO single photon emission computed tomography in a new sporadic case of rapid‐onset dystonia–parkinsonism. Journal of the Neurological Sciences, 273(1‐2), 148–151. 10.1016/j.jns.2008.06.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
All variants described in this study are openly available for access within the ClinVar variant database (https://www.ncbi.nlm.nih.gov/clinvar/).
All variants described in this study are openly available for access within the ClinVar variant database (https://www.ncbi.nlm.nih.gov/clinvar/).