

Abstract
Unprecedented 3D hexa‐adducts of [60]fullerene peripherally decorated with twelve tryptophan (Trp) or tyrosine (Tyr) residues have been synthesized. Studies on the antiviral activity of these novel compounds against HIV and EV71 reveal that they are much more potent against HIV and equally active against EV71 than the previously described dendrimer prototypes AL‐385 and AL‐463, which possess the same number of Trp/Tyr residues on the periphery but attached to a smaller and more flexible pentaerythritol core. These results demonstrate the relevance of the globular 3D presentation of the peripheral groups (Trp/Tyr) as well as the length of the spacer connecting them to the central core to interact with the viral envelopes, particularly in the case of HIV, and support the hypothesis that [60]fullerene can be an alternative and attractive biocompatible carbon‐based scaffold for this type of highly symmetrical dendrimers. In addition, the functionalized fullerenes here described, which display twelve peripheral negatively charged indole moieties on their globular surface, define a new and versatile class of compounds with a promising potential in biomedical applications.
Keywords: antiviral agents, EV71, fullerenes, hexa-adduct, HIV
Hexa‐adducts of [60]fullerene with twelve tryptophan (Trp)/tyrosine (Tyr) residues in their periphery have been synthesized. They are much potent against HIV and equally active against EV71 than the prototype AL‐385, with the same number of Trp moieties but a smaller and flexible pentaerythritol scaffold. This finding showed the relevance of the globular presentation of the peripheral groups for inhibiting viral replication, particularly HIV.
Introduction
Since the discovery by Kroto, Smalley and Curl of fullerenes in 1985,[1] biological properties of these singular 3D carbon cages have attracted much attention as biocompatible, highly hydrophobic and rigid scaffolds with unique electronic properties.[2]
Most of the former studies regarding biological applications of [60]fullerene involved functionalization with ionic or polar groups to enhance solubility and to provide peripheral reactive positions to which a variety of different ligands can be attached.[3] A singular and successful type of chemically modified fullerenes are those containing biomolecules such as peptides or saccharides that have been designed in the search for biomedical applications.[4] Such functionalization of [60]fullerene usually leads to amphiphilic derivatives because of the hydrophobic fullerene surface, and this character facilitates the penetration of fullerene derivatives through the cell membrane, and also promotes self‐aggregation.[5]
This hydrophobicity can be completely masked by the formation of hexa‐adducts of [60]fullerene.[6] These hexa‐adducts can be efficiently obtained by using the Bingel‐Hirsch cyclopropanation reaction and allow the globular presentation of ligands also providing a rigid and compact spherical core. The biological properties of hexa‐adducts of [60]fullerene have been intensively investigated in the last years. As an example, hexa‐adducts bearing cationic groups in the periphery exhibit a remarkable efficiency in the complexation and transfection of DNA, thus acting as promising non‐viral vectors for transfection purposes.[7] When they are modified with single DNA strands, hexa‐adducts of C60 can function as single entities with the ability to regulate gene expression without the need of a transfection agent.[8] A particularly remarkable role has been played by hexa‐adducts appended with carbohydrates forming the so called “sugar balls”.[4b, 9] These glycofullerenes have shown interesting properties in different fields, as antibacterial derivatives,[10] enzyme inhibitors,[11] antiviral agents,[12] cancer targeting tools,[13] or protective agents against oxidative stress and inflammation caused by particulate matter.[14] In our group, we have synthesized a variety of fullerenes appended with different numbers of carbohydrate moieties (12–36) in the periphery as efficient inhibitors of the lectin DC‐SIGN in an Ebola virus (EBOV) infection assay model.[12a, 15] Furthermore, 3D giant tridecafullerenes formed by 13 [60]fullerene units have shown highly efficient inhibition of EBOV infection, with IC50 values in the subnanomolar range.[12b] Recently, tridecafullerenes bearing up to 360 disaccharides efficiently inhibited infection by emergent viruses, namely Zika and Dengue, at picomolar concentrations.[16]
On the other hand, molecules containing amino acids covalently attached to the C60 fullerene core have been known since 1993,[17] and different fullerene amino acids have been used as synthons to create larger structures in which the carbon sphere was inserted into a peptide backbone.[4c, 18] These amino acid‐ or peptide‐decorated fullerene derivatives also display antiviral, antimicrobial, or antioxidant activities, or can be used as drug delivery systems.[19] Some of them have also shown anti‐HIV activity, generally by causing impairment of virus maturation,[20] or by inhibiting the HIV reverse transcriptase involved in virus replication.[21] Only polycarboxylic [70]fullerene derivatives have been reported to act by interacting with envelope gp120 protein thus inhibiting viral entry.[22]
We have previously reported a new class of dendrimers with tryptophan (Trp) residues on the surface that show dual antiviral activity against HIV and enterovirus A71 (EV‐A71), a completely unrelated non‐polio neurotropic enterovirus with pandemic potential.[23] The prototype compound of this family, AL‐385 (Figure 1), contains a flexible pentaerythritol central scaffold appended with trivalent aliphatic branched arms and twelve tryptophan residues on the periphery. Those Trp moieties are linked to the central scaffold through their amino groups, thus the carboxylic acids are free and exposed to the surface/solvent. Replacing the Trp residue of the prototype by other aromatic amino acid (tyrosine) afforded AL‐463 that also showed significant anti‐HIV/EV71 activity (Figure 1).[24]
Figure 1.
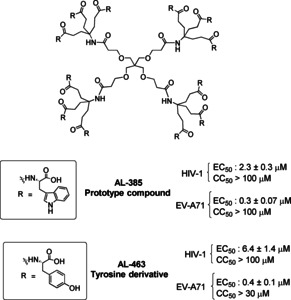
Structure of the dendrimer prototype AL‐385 and its tyrosine analogue AL‐463.
Experimental work carried out to determine the molecular mechanism by which AL‐385 inhibits HIV and EV‐A71 replication in vitro demonstrated that this dendrimer interacts with the envelopes of HIV and EV71 thereby blocking the binding of those viruses to the host cell. In the case of HIV, AL‐385 interacts with the gp120 glycoprotein of the viral envelope[23a] whereas in the case of enterovirus A71 (EV‐A71) AL‐385 targets residues of the structural viral protein VP1, particularly the region that forms the 5‐fold vertex of the viral capsid.[25]
The early steps (fusion/entry) in the replicative cycle of a virus are very attractive targets for antiviral drug development.[26] In the fight against HIV/AIDS, such type of inhibitors (viral entry inhibitors) may be advantageous over existing therapeutic approaches that target key viral enzymes (reverse transcriptase, protease or integrase) because they (i) prevent virus uptake by the uninfected target cells, thus preventing the spread of the infection, and (ii) show effectiveness against viruses resistant to existing anti‐HIV inhibitors.[26, 27] To date, only four entry inhibitors (enfuvirtide [Fuzeon®], maraviroc, ibalizumab and fostemsavir) have been approved by the FDA for the treatment of HIV infection.[28] The situation is even worse for EV71 for which there are no approved antiviral drugs. Therefore, we considered that the Trp dendrimers discovered in our group are attractive candidates for further development.
SAR studies led to the conclusion that a multivalent presentation of the Trp residues on these dendrimers is crucial for the observed activity against HIV and EV71 replication.[23, 24, 29] Therefore, in view of the above mentioned properties of the functionalized 3D fullerenes, we considered of interest the use of [60]fullerene as a multivalent scaffold to which twelve Trp residues could be attached (as is the case of AL‐385). Moreover, in order to determine the influence of the nature of the peripheral ligands on the activity, tyrosine (Tyr) residues were also attached to the [60]fullerene core. Our final aim was to study the effect on the antiviral activity of the replacement of the flexible pentaerythritol scaffold, as in dendrimer AL‐385, by a rigid [60]fullerene core in which the peripheral Trp/Tyr ligands adopt a unique globular 3D presentation, thus mimicking the virus symmetrically spherical structure.
For this purpose, in this work two series of [60]fullerene derivatives (Figure 2), all of them bearing twelve peripheral Trp/Tyr residues, were synthesized. We had previously synthesized a monodisperse hexa‐adduct of [60]fullerene appended with 12 amino acid residues by using a thiol‐ene click chemistry strategy.[30] Here, we have employed two different synthetic pathways: in series I, the peripheral amino acid residues were connected to the fullerene through amide bond formation and a five methylene alkyl spacer; in series II, a Copper‐Catalyzed Azide‐Alkyne Cycloaddition (CuAAC) strategy and a larger spacer, which includes an ethylene glycol moiety linked to a triazole ring, were used. Importantly, as in the Trp prototype AL‐385, the peripheral amino acid residues (Trp or Tyr) were linked to the central scaffold through each of their amino groups so as to leave their free carboxylic acid groups fully exposed in the periphery.
Figure 2.
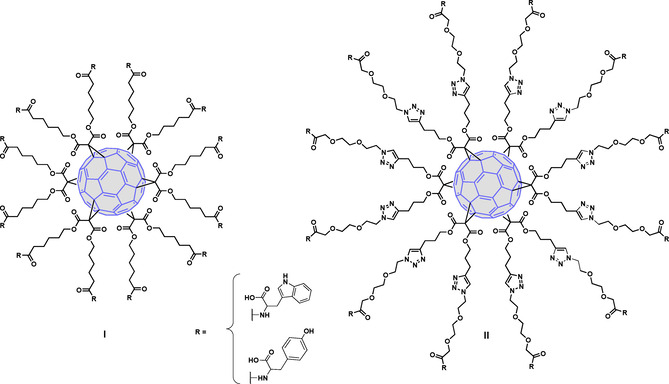
General structures of fullerene derivatives of series I and II.
Results and Discussion
Synthesis and characterization
First, fullerene derivatives of series I, in which the peripheral Trp residues were connected through five methylene units to the [60]fullerene core were prepared. The synthesis of such molecules includes the addition of the tert‐butyl protected bismalonate 2[31] to [60]fullerene under Bingel cyclopropanation reaction conditions (Scheme 1) to afford the tert‐butyl protected intermediate 3. In turn, the bismalonate intermediate 2 was obtained in 67 % yield by reaction of alcohol 1 with malonyl dichloride.[32] Subsequent deprotection of 3 in acid media (trifluoroacetic acid) afforded the carboxy fullerene 4, containing twelve carboxylic acids in the periphery in almost quantitative yield (Scheme 1). This dodecacarboxylic acid intermediate was used in the next step without further purification. Coupling of 4 with the corresponding (OBn or OMe) protected amino acid (H‐Trp‐OBn⋅HCl, H‐Trp‐OMe⋅HCl or H‐Tyr(Bn)‐OMe⋅HCl) in the presence of HATU, as the coupling agent, and a base (DIPEA) gave intermediates 5 (30 %), 6 (22 %) and 7 (14 %), respectively. Hydrogenation of 7 gave the tyrosine intermediate 8 in quantitative yield (Scheme 1).
Scheme 1.
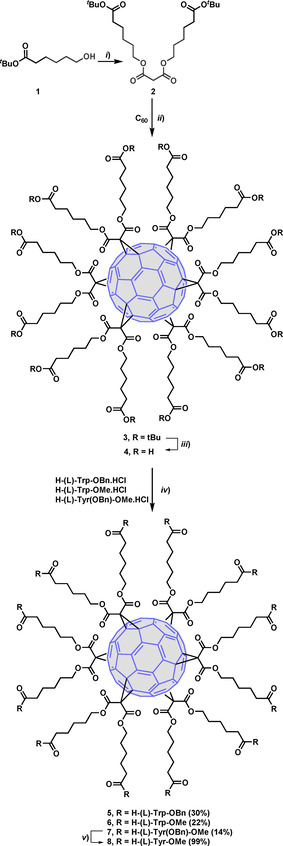
Synthesis of intermediates 5–8. Reagents and conditions: i) pyr, malonyl dichloride, 20 h, rt (67 %); ii) C60, CBr4, toluene, DBU, rt, 3 days (70 %); iii) TFA/CH2Cl2 (1 : 5), rt, overnight (quant.); iv) HATU, protected amino acid, DMF, DIPEA, 30 °C, 2 days; v) H2, Pd−C, THF/MeOH (1 : 1) (quant.).
Debenzylation of 5 by catalytic hydrogenation (H2, 10 % Pd−C) afforded a complex untreatable mixture of partially deprotected compounds, which made it necessary the synthesis of the OMe protected derivative 6. Methyl ester deprotection of 6 under basic conditions (LiOH/H2O), followed by acidification at pH=2 and purification of the crude by size‐exclusion chromatography, afforded the desired final compound 9 in good yield (81 %) (Scheme 2). This last synthetic procedure was also used for the synthesis of the Tyr final compound 10, which was obtained from 8 in quantitative yield (Scheme 2).
Scheme 2.
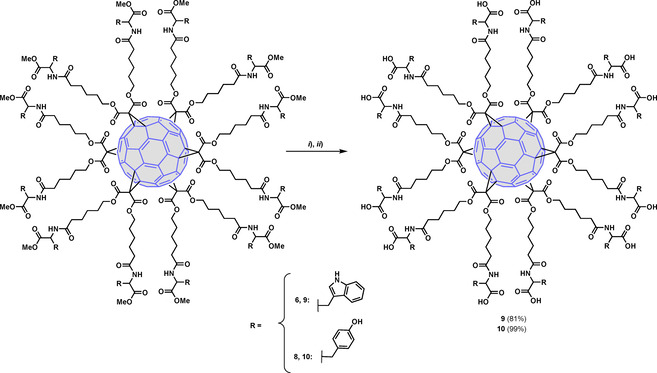
Synthesis of fullerenes 9 and 10. Reagents and conditions: i) LiOH, THF/H2O (1 : 2), rt, 5 h; ii) HCl, pH=2.
Next, compounds of series II (Figure 2), with a larger spacer that includes an ethylene glycol moiety linked to a triazole ring, were synthesized using a click chemistry reaction as the crucial step. For this purpose, suitable Trp linker molecules 13 or 14, bearing a terminal azido group, were synthesized (Scheme 3). These azide precursors were obtained from a common starting azide (12) that was prepared in four steps from the commercially available glycol 11 following a literature procedure.[33] Coupling of 12 with H‐Trp‐OBn⋅HCl or H‐Trp‐OMe⋅HCl, in the presence of HATU and DIPEA, gave compounds 13 (43 %) and 14 (23 %), respectively.
Scheme 3.
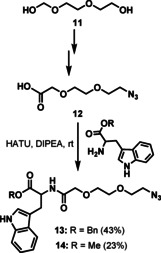
Synthesis of azide precursors 13 and 14.
The subsequent step relies on compound 15, a compact C60 hexa‐adduct scaffold that bears twelve terminal alkyne units (Scheme 4). This crucial fullerene intermediate was prepared following a previously reported protocol consisting of the addition of dipent‐4‐ynyl malonate to C60 in toluene,[9] employing an excess of CBr4.[34] 1,3‐Dipolar cycloaddition of 15 with the corresponding azide precursor 13 or 14, using CuBr⋅SMe2 and Cu (0) in DMSO, afforded crude compounds 16 and 17, respectively (Scheme 4).[12a] For further biological studies all traces of copper metal were removed with the commercially available resin (QuadraSil Mercaptopropyl) (QuadraSil® MP) obtaining pure compounds 16 and 17 in 84 and 80 % yields, respectively. Debenzylation of 16 by catalytic hydrogenation (H2, 10 % Pd−C) afforded a complex mixture of partially deprotected compounds. However, saponification of 17 using the previously mentioned basic conditions (LiOH/H2O) gave the deprotected hexa‐adduct 18 in 82 % yield (Scheme 4).
Scheme 4.
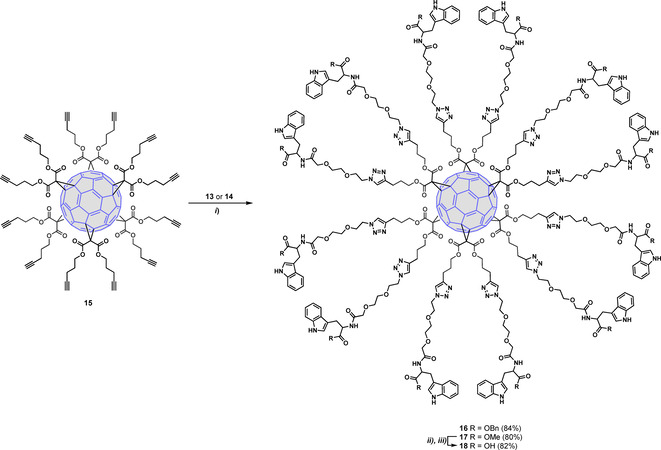
Synthesis of intermediates 16 and 17 and deprotected Trp fullerene 18. Reagents and conditions: i) CuBr⋅SMe2, Cu(0), DMSO, rt, 24 h; ii) LiOH, THF/H2O (1 : 2), rt, 5 h; iii) HCl, pH=2.
The structure of the final compounds 9, 10 and 18 was confirmed by 1H and 13C NMR spectroscopy. As shown in Figure 3, for 9 and 18 (chosen as model compounds) the presence of only two signals for the sp2 carbons of C60 at ∼145 and 141 ppm in the 13C NMR spectra confirmed the presence of the hexa‐substituted [60]fullerene core. Moreover, in the 13C NMR spectrum of 18, two signals at around 146 and 122 ppm, (l’ and m’ in Figure 3) unambiguously confirmed the formation of 1,2,3‐triazole rings, and consequently the effectiveness of the CuAAC reaction. DEPT, COSY, and HSQC experiments were also carried out to confirm the proposed structures.
Figure 3.
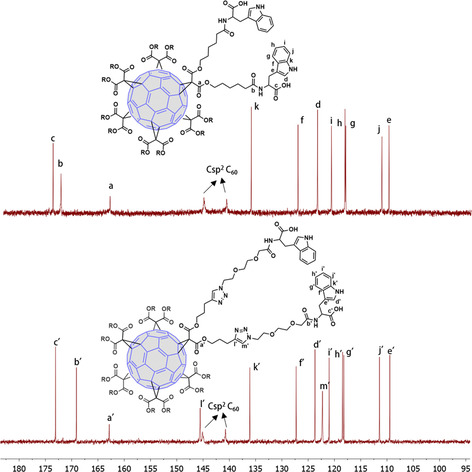
Partial view of the 13C NMR spectra of symmetric hexa‐adducts 9 (top) and 18 (down) (CDCl3, 125.8 MHz).
Antiviral evaluation
Antiviral activity against HIV: First, the new fullerene derivatives 5, 9, 10, 16 and 18 were evaluated for their inhibitory effects against HIV‐1 and HIV‐2 replication in CD4 cell cultures. Table 1 summarizes the results of this evaluation. The antiviral data of the prototype AL‐385 and its corresponding Tyr derivative AL‐463 are also included as reference compounds.[24] Tenofovir (PMPA)[35] and Pradimicin A (PRM‐A)[36] acted as positive controls. PMPA is an HIV reverse transcriptase inhibitor and PRM‐A is a gp120 carbohydrate‐binding HIV entry inhibitor. These compounds showed activities that were in the same range as previously reported.[35, 36]
Table 1.
Anti‐HIV activity of the test compounds.
|
Compound |
EC50 [μM][a] HIV‐1/NL4.3 |
EC50 [μM][a] HIV‐2/ROD |
CC50 [μM][b] |
SI[c] |
|---|---|---|---|---|
|
5 |
>50 |
>50 |
50 |
– |
|
9 |
0.24±0.04 |
0.86±0.49 |
52.13±2 |
217 |
|
10 |
0.23±0.12 |
0.76±0.064 |
45.79±6 |
200 |
|
16 |
>50 |
>50 |
50.00±5 |
– |
|
18 |
0.083±0.002 |
1.14±0.25 |
50.44±4 |
602 |
|
AL‐385 |
2.3±0.3 |
6.60±7.7 |
>100 |
44 |
|
AL‐463 |
6.4±1.4 |
20±15 |
>100 |
15.6 |
|
Tenofovir (PMPA) |
4.74 |
1.00 |
>100 |
133.3 |
|
Pradimicin A (PRM‐A) |
3.3±1.2 |
5.9±3.7 |
>100 |
30.3 |
Data are the mean ± S.D. of at least 2 to 3 independent experiments. [a] 50 % effective concentration, or concentration required to inhibit HIV‐induced cytopathicity by 50 %. [b] 50 % cytostatic concentration, or compound concentration required to inhibit CD4 T cell proliferation by 50 %. [c] SI: selectivity index (CC50/EC50).
It can be seen that the OBn‐protected derivatives 5 and 16, independently of the nature of the spacer, were inactive whereas 9, 10 and 18, endowed with free carboxylic acids on the periphery, showed anti‐HIV‐1 activity in the low micromolar range. This result confirms the importance of the free COOHs for activity, as previously observed for the AL‐385 and AL‐463 prototypes. Thus, the Tyr‐substituted fullerene 10 (EC50: 0.23 μM) resulted as active as the Trp fullerene 9 (EC50: 0.24 μM). On the other hand, fullerene 9 (EC50: 0.24 μM), with polymethylene linkers, turned out to be ∼3‐fold less potent than 18 (EC50: 0.083 μM), which has longer ethylene glycol/triazole linkers. The higher solubility and/or the longer length of the linker in 18 may account for this activity improvement. It should be pointed out that all the deprotected amino acyl fullerenes 9, 10 and 18 (EC50: 0.24, 0.23 and 0.083 μM) were 10–100‐fold more potent against HIV‐1 than the prototypes AL‐385 (EC50: 2.3 M) and AL‐463 (EC50: 6.4 μM). This result suggests that the 3D spherical shape of the C60 fullerene and consequently the globular 3D orientation of the peripheral groups, can be beneficial for the antiviral activity against HIV replication. These experimental findings highlight the efficient multivalent effect provided by the globular spatial distribution of the fullerene core as well as by the inherent flexibility and increased length of the chains linking the Tyr/Trp residues to the dendrimer's core.[37] These singular features must give rise to significantly improved interactions with the viral envelope glycosylated proteins. Interestingly, 9, 10 and 18 also showed (sub)micromolar activity against HIV‐2 at subtoxic concentrations.
Although fullerene derivatives 9, 10 and 18 were slightly more toxic than the Trp and Tyr dendrimers AL‐385 and AL‐463, their therapeutic indexes were considerably higher due to their higher activity.
Finally, according to the data shown in Table 1, it can be concluded that fullerene 18 (EC50: 0.083 M; SI >600) is the most potent compound of this series against HIV replication. 18 was 28‐ and 77‐fold, respectively, more potent than the Trp‐containing AL‐385 (EC50: 2.3 μM) and the Tyr‐derived AL‐463 (EC50: 6.4 M) prototypes, and also considerably more potent than the control compounds PMPA and PRM‐A.
As the most promising compound, 18 was also evaluated in a Surface Plasmon Resonance (SPR) assay to investigate whether the gp120 glycoprotein of the HIV‐1 envelope is the viral target, as is the case for the prototype AL‐385.[23a] Thus, recombinant HIV‐1 IIIB gp120 (produced in Chinese hamster ovary cells) was bound to a sensor chip as a monomer and the interaction of 18 with this viral envelope glycoprotein was determined (Figure 4 and Table 2). Pradimicin A (PRM‐A) was used as a positive control. The results confirm that 18, binds to gp120, as does PRM‐A, in a concentration‐dependent manner.
Figure 4.
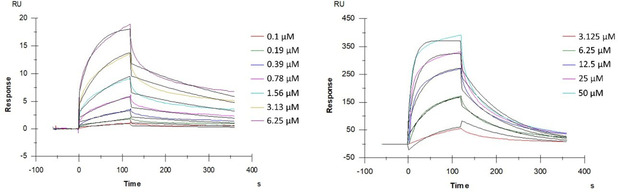
Multiple cycle kinetics of 18 (left) and PRM‐A (right). This figure shows one of four replicate experiments. The colored curves represent the real‐time binding responses, the black curves indicate the calculated 1 : 1 Langmuir model fitting. The concentration of 18 ranged between 0.1 and 6.25 μM and those of PRM‐A between 3.125 to 50 μM. The analytes were diluted using two‐fold dilution steps. For 18 the ability of binding to gp120 was confirmed.
Table 2.
Apparent KD for 18 binding to gp120.[a]
|
Compound |
ka (103)(1/M⋅s) |
kd (10−3)(1/s) |
KD (μM) |
|---|---|---|---|
|
18 |
3.45±0.29 |
2.73±0.23 |
0.80±0.13 |
|
PRM‐A |
1.93±0.42 |
12.64±2.92 |
6.52±0.11 |
[a] Average and standard deviation of kinetic parameters obtained from four replicate SPR experiments measuring the binding response between gp120 and 18/PRM‐A.
Next, detailed SPR‐directed affinity was determined using concentrations of 18 ranging from 0.1 to 6.25 μM. Our results showed that this compound binds faster and dissociates slower from gp120 than the positive control, PRM‐A. The apparent KD value for 18 (KD : 0.80 μM) binding to gp120 (Table 2) was 8‐fold smaller than that found for PRM‐A (KD : 6.52 μM), which means that the interaction of 18 with gp120 is 8‐fold more pronounced than with PRM‐A.
Antiviral activity against EV71: The new fullerene derivatives 5, 9, 10, 16 and 18 were also tested for antiviral activity (EC50) against the BrCr laboratory‐adapted strain of EV71 in a rhabdomyosarcoma (RD) cell‐based assay (Table 3).[38] Cell viability (CC50) was also assessed in a similar setup with compound‐treated, uninfected cells. Trp and Tyr dendrimer prototypes AL‐385 and AL‐463,[24] together with pirodavir, a potent EV71 capsid binder, were included in the assay as reference compounds.[39]
Table 3.
Antiviral activity of the test compounds against the BrCr lab strain of EV71 in RD cells.
|
Compound |
EC50 [μM] |
CC50 [μM] |
SI |
|---|---|---|---|
|
5 |
>17.5 |
50 |
– |
|
9 |
3.88±1.46 |
64.08±1.62 |
16 |
|
10 |
0.33±0.04 |
49±0.02 |
149 |
|
16 |
>17.5 |
>20 |
– |
|
18 |
7.11±3.55 |
>87 |
12 |
|
AL‐385 |
0.30±0.10 |
225 |
750 |
|
AL‐463 |
0.40±0.10 |
>30 |
78 |
|
Pirodavir |
0.30±0.10 |
>100 |
>333 |
All values are in micromolar (μM) concentrations and are a summary of multiple dose‐response curves (>2) in multiple independent (>1) experiments; CC50: concentration of compound at which a 50 % reduction in cell viability is observed; EC50: concentration of compound at which the virus‐induced cytopathic effect is reduced by 50 %; SI=selectivity index (CC50/EC50).
As in the case of HIV replication, the benzyl protected compounds 5 and 16 proved inactive. This result confirms the importance of the negative charges (carboxylate groups) in the molecules as a prerequisite for antiviral activity (Table 3).
In general, with a range of activities between 0.33 μM and 7.11 μM (Table 3), the fullerenes 9, 10 and 18 were less potent against EV71 replication than against HIV‐1 replication (0.083–0.24 μM, Table 1) and in contrast to what was observed against HIV‐1, the Tyr fullerene 10 (EC50: 0.33 μM) was one order of magnitude more potent than its Trp analogue 9 (EC50: 3.88 μM). Moreover, fullerene 18 (EC50: 7.11 μM), with a polyethylene glycol/triazole linker, turned out to be less potent against EV71 than fullerene 9 (EC50: 3.88 μM), with a shorter polymethylene linker. Of the three derivatives, the Tyr fullerene 10 (EC50: 0.33 μM) was found to be the most potent compound of this series against EV71 replication. In fact, this compound is as active as the Trp and Tyr dendrimer prototypes AL‐385 and AL‐463 (EC50: 0.30 μM and EC50: 0.40 μM, respectively) and the reference compound, pirodavir (EC50: 0.30 μM).
The best compound of this series, 10 (best activity/toxicity profile), was evaluated against a panel of representative enteroviruses in virus‐cell‐based assays (Table 4). Compound 10 showed antiviral activity against enterovirus D68 (EV‐D68), but this was lower than against enterovirus EV71. This indicates that, as was the case with the prototype AL‐385, 10 is quite a specific inhibitor of EV71 replication.
Table 4.
Evaluation of the broad‐spectrum antiviral effect of 10 against a representative panel of enteroviruses.
|
Species |
Virus |
Host cell |
Fullerene 10 EC50 [μM][ª] |
AL‐385 EC50 [μM] |
|---|---|---|---|---|
|
Enterovirus A |
Enterovirus A71 BrCr strain |
RD |
0.33±0.04[b] |
0.29±0.07[b] |
|
Enterovirus B |
Coxsackievirus B3 Nancy strain |
Vero A[c] |
>50 |
>28 |
|
Enterovirus D |
Enterovirus D68 CU70 strain |
HeLa Rh |
13±4[b] |
6.0±2.1[b] |
|
Rhinovirus A |
Rhinovirus 2 |
HeLa Rh |
>50 |
>28 |
|
Rhinovirus B |
Rhinovirus 14 |
HeLa Rh |
>50 |
>28 |
[a] All values were obtained from multiple (>2) independent (>1) experiments. [b] Following microscopic quality control, at least at one concentration of compound, no virus‐induced cell death was observed, and the compound did not cause any morphological changes in the host cell monolayers. [c] Vero cells, African green monkey kidney cells.
Next, the antiviral potency of 10 was also evaluated in virus‐cell‐based assays against a panel of clinical isolates representative of the different (sub)genogroups of EV71 (B2, B5, C2 and C4). The prototype AL‐385 was included as the reference compound. Except for line B2, the activity of 10 on the clinical isolates was improved with respect to that found for the BrCr laboratory strain (subgeno‐group A). This improvement was smaller (3.8, 3.5 and 5‐fold respectively) than those observed for the prototype AL‐385 (Table 5).
Table 5.
Evaluation of the broad‐spectrum antiviral effect of 10 against a representative panel of clinical EV71 isolates in RD cells.
|
EV71 genogroup |
Virus strain |
EC50 [μM][a] |
|
|---|---|---|---|
|
10 |
AL‐385 |
||
|
A |
BrCr |
334±41 |
285±70 |
|
B2 |
11316 |
517±120 |
0.4±0.0 |
|
B5 |
TW/70902/08 |
87±10 |
0.2±0.1 |
|
C2 |
H08300 461#812 |
95±7 |
1.1±0.3 |
|
C4 |
TW/1956/05 |
68±4 |
0.2±0.2 |
[a] All values are in nanomolar (nM) concentrations and were obtained in multiple (>2) independent (>1) experiments. Following microscopic quality control, at least at one concentration of compound, no virus‐induced cell death was observed, and the compound did not cause any morphological changes in the host cell monolayers.
Finally, 10 was also evaluated for its inhibitory activity against the single mutant EV‐A71 strains VP1_S184T and VP1_P246S and the double mutant VP1_S184T_P246S that are resistant to the prototype AL‐385 (Table 6).[25] As shown in Table 6, 10 retains full antiviral activity against these mutant virus strains. The lack of cross‐resistance between AL‐385 and 10 suggests that either they bind to different sites or the binding energy is still high enough in the presence of these amino acid replacements.
Table 6.
Susceptibility of reverse‐engineered EV‐A71 strains to the AL‐385 prototype and 10.
|
Virus |
EC50 [μM][a] |
|
|---|---|---|
|
AL‐385 |
10 |
|
|
EV‐A71 BrCr |
0.28±0.01 |
0.33±0.07 |
|
VP1 S184T |
1.99±0.10 (7.1) |
0.4±0.03 (1.2) |
|
VP1 P246S |
4.71±0.18 (16.8) |
0.36±0.02 (1.1) |
|
VP1_S184T_P246S |
8.93±0.53 (31.9) |
0.35±0.05 (1) |
[a] Averages and standard deviations were calculated from data obtained from three independent antiviral assays. Values in italics within the brackets indicate fold resistance.
Molecular modeling
A structural rationale for EV71 entry inhibition by AL‐385 has been put forward.[25] Briefly, a single molecule binds to each 5‐fold vertex of the viral capsid and establishes interactions with positively charged residues that are crucial for viral attachment to the P‐selectin glycoprotein ligand‐1 (PSGL‐1) receptor and cellular proteoglycans containing heparan sulfate. We envisage a similar binding mode for dendrimers 9, 10 and 18, with the differences in anti‐EV71 potencies (Table 3) being ascribed to the combined effect of increasing the linker length and replacing Tyr with Trp at the ends.
Here we have focused on HIV‐1 because of the greatly improved potency, particularly of 18, with respect to prototype AL‐385. Comparison of structural models of both compounds containing fullerene and pentaerythritol cores, respectively (Figure 5), reveals the much larger size of the former and highlights the spherical distribution of the twelve radiating long and highly flexible branches holding the terminal decorated tryptophans. Each of these tryptophan residues provides not only a flat surface for C−H⋅⋅⋅π interactions[40] with the pyranose rings, as seen at the carbohydrate‐binding sites of numerous lectins‐including that from the cyanobacteria Oscillatoria agardhii (PDB entry 3OBL)[41]‐and the antigen‐binding fragments (Fab) of broadly neutralizing antibodies (bNAbs) such as PG16 (PDB entry 6ULC),[42] but also the potential for multiple hydrogen bonds between its free carboxylic group and the sugar hydroxyls. To test this hypothesis, we modeled and simulated the dynamic behavior of (i) a membrane‐embedded and partially glycosylated HIV‐1 Env trimer (Figure 5) to gain insight into the glycan shield distribution and the motion of individual oligosaccharides attached at different sequence positions, and (ii) 18, both free in solution and in the presence of two molecules of the high‐mannose glycan Man9GlcNAc2‐OMe to study the range of interactions that are possible with this type of dendrimers. Our results show the wide protein surface coverage and high mobility of the gp120 glycans that are likely to be encountered by an approaching dendrimer (Supporting Information Movie 1) and the feasibility of multiple patterns of simultaneous hydrogen bonding and CH⋅⋅⋅π interactions between the terminal tryptophans and the sugars (Supporting Movie 2). Hence, we postulate that the more potent anti‐HIV‐1 activity in this series ‐ over that of the original prototype AL‐385 ‐ is related to improved interactions with the heavily glycosylated gp120 protein that makes up the Env trimer,[43] although additional or alternative interactions with gp41 glycans cannot be ruled out at present. Further details will have to await the results of ongoing experimental efforts directed at identifying the differential sensitivity to these dendrimers of HIV‐1 isolates with divergent glycosylation profiles.
Figure 5.
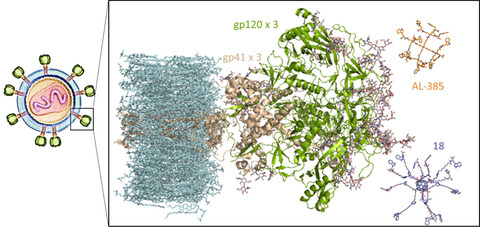
Side‐by‐side comparison (same scale) of AL‐385 (C atoms in orange) and 18 (C atoms in purple) in relation to the HIV‐1 Env glycoprotein (gp120+gp41 trimer, green and cream cartoons, respectively). Membrane lipids, including cholesterol, are colored in light blue. The oligosaccharide residues (GlcNAc2 and Man1‐9GlcNAc2) attached to several asparagine side chains in both gp120 and gp41 are displayed as sticks with C atoms colored in grey. These sugars represent only a fraction of the N‐linked mannose‐rich glycans that not only shield the HIV‐1 envelope from immune surveillance but also provide specific recognition by lectins, bNAbs and, possibly, Trp‐decorated dendrimers such as AL‐385 and 18.
Conclusion
A novel class of symmetrical hexa‐adducts of C60 fullerene decorated with twelve Trp/Tyr residues on the periphery have been synthesized that show dual antiviral activity against HIV and EV71. These compounds turned out to be more potent against HIV‐1 than the dendrimer prototype AL‐385, which possesses the same number of Trp residues on the periphery but attached to a pentaerythritol central scaffold. We suggest that the fullerene core present in the novel compounds reported here helps distribute the peripheral aromatic rings (indole/phenol) and negative charges in a spherical fashion that can be optimal for binding to the globular HIV‐1 envelope. Regarding their anti‐EV71 activity, 10 was found to be as potent as the dendrimer prototype AL‐385.
Our results support that [60]fullerene is a very attractive scaffold that ensures a unique globular 3D distribution of the peripheral Trp/Tyr ligands that appears to improve the interaction with the viral envelopes, in particular that of HIV‐1.
The fullerenes here described, which have twelve negative charges and indole/phenol moieties in their periphery, thus appear as a new and versatile class of compounds with a promising potential as antivirals that may prevent the viral infection and, therefore, become also useful as microbicides.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
This work has been supported by the Spanish MINECO/FEDER (Projects CTQ2017‐84327‐P and CTQ2017‐83531‐R), the Spanish MICINN (Projects PID2019‐104070RB‐C21 and PID2019‐104070RB‐C22), the Spanish Agencia Estatal Consejo Superior de Investigaciones Científicas (CSIC, Projects CSIC‐PIE‐201980E100 and CSIC‐PIE‐201980E028), “The Centers of Excellence” of the KU Leuven (EF‐05/15 and PF‐10/18), EU FP7 (FP7/2007–2013) Project EUVIRNA (Grant 408 Agreement 264286), EU FP7 SILVER (Contract HEALTH‐F3‐2010‐260644), a grant from the Belgian Interuniversity Attraction Poles (IAP) Phase VII‐P7/45 (BELVIR) and the EU FP7 Industry‐Academia Partnerships and Pathways Project AIROPICO. The Spanish MEC/MINECO is also acknowledged for a grant to O. M.‐M. We also thank Charlotte Vanderheydt, Caroline Collard, Kim Donckers, Sandra Claes and Evelyne Van Kerckhove for help with the processing of the antiviral data.
M. Ruiz-Santaquiteria, B. M. Illescas, R. Abdelnabi, A. Boonen, A. Mills, O. Martí-Marí, S. Noppen, J. Neyts, D. Schols, F. Gago, A. San-Félix, M.-J. Camarasa, N. Martín, Chem. Eur. J. 2021, 27, 10700.
Contributor Information
Prof. Beatriz M. Illescas, Email: beti@ucm.es.
Ana San‐Félix, Email: anarosa@iqm.csic.es.
María‐José Camarasa, Email: mj.camarasa@iqm.csic.es.
Prof. Nazario Martín, Email: nazmar@ucm.es.
References
- 1.Kroto H. W., Heath J. R., O'Brien S. C., Curl R. F., Smalley R. E., Nature 1985, 318, 162–163. [Google Scholar]
- 2.
- 2a.Bosi S., Da Ros T., Spalluto G., Prato M., Eur. J. Med. Chem. 2003, 38, 913–923; [DOI] [PubMed] [Google Scholar]
- 2b.Bianco A., Da Ros T., in Fullerenes: Principles and Applications (2), The Royal Society of Chemistry, Cambridge, 2012, pp. 507–545; [Google Scholar]
- 2c.Castro E., Hernández García A., Zavala G., Echegoyen L., J. Mater. Chem. B 2017, 5, 6523–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a.Jensen A. W., Wilson S. R., Schuster D. I., Bioorg. Med. Chem. 1996, 4, 767–779; [DOI] [PubMed] [Google Scholar]
- 3b.Bakry R., Vallant R. M., Najam-ul-Haq M., Rainer M., Szabo Z., Huck C. W., Bonn G. K., Int. J. Nanomed. 2007, 2, 639–649; [PMC free article] [PubMed] [Google Scholar]
- 3c.Djordjevic A., Srdjenovic B., Seke M., Petrovic D., Injac R., Mrdjanovic J., J. Nanomater. 2015, 2015, 567073. [Google Scholar]
- 4.
- 4a.Yang X., Ebrahimi A., Li J., Cui Q., Int. J. Nanomed. 2014, 9, 77–92; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b.Nierengarten I., Nierengarten J. F., Chem. Asian J. 2014, 9, 1436–1444; [DOI] [PubMed] [Google Scholar]
- 4c.Jennepalli S., Pyne S. G., Keller P. A., RSC Adv. 2014, 4, 46383–46398. [Google Scholar]
- 5.
- 5a.Partha R., Mitchell L. R., Lyon J. L., Joshi P. P., Conyers J. L., ACS Nano 2008, 2, 1950–1958; [DOI] [PubMed] [Google Scholar]
- 5b.Muñoz A., Illescas B. M., Sánchez-Navarro M., Rojo J., Martin N., J. Am. Chem. Soc. 2011, 133, 16758–16761; [DOI] [PubMed] [Google Scholar]
- 5c.Pérez-Ojeda M. E., Wabra I., Böttcher C., Hirsch A., Chem. Eur. J. 2018, 24, 14088–14100; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d.Muñoz A., Illescas B. M., Luczkowiak J., Lasala F., Ribeiro-Viana R., Rojo J., Delgado R., Martín N., J. Mater. Chem. B 2017, 5, 6566–6571. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a.Hirsch A., Vostrowsky O., Eur. J. Org. Chem. 2001, 829–848; [Google Scholar]
- 6b.Lamparth I., Maichle–Mössmer C., Hirsch A., Angew. Chem. Int. Ed. 1995, 34, 1607–1609; [Google Scholar]; Angew. Chem. 1995, 107, 1755–1757; [Google Scholar]
- 6c.Yan W., Seifermann S. M., Pierrat P., Bräse S., Org. Biomol. Chem. 2015, 13, 25–54. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a.Sigwalt D., Holler M., Iehl J., Nierengarten J. F., Nothisen M., Morin E., Remy J. S., Chem. Commun. 2011, 47, 4640–4642; [DOI] [PubMed] [Google Scholar]
- 7b.Illescas B. M., Pérez-Sánchez A., Mallo A., Martín-Domenech Á., Rodríguez-Crespo I., Martín N., J. Mater. Chem. B 2020, 8, 4505–4515. [DOI] [PubMed] [Google Scholar]
- 8.Li H., Zhang B., Lu X., Tan X., Jia F., Xiao Y., Cheng Z., Li Y., Silva D. O., Schrekker H. S., Zhang K., Mirkin C. A., Proc. Natl. Acad. Sci. USA 2018, 115, 4340–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nierengarten J. F., Iehl J., Oerthel V., Holler M., Illescas B. M., Munoz A., Martin N., Rojo J., Sanchez-Navarro M., Cecioni S., Vidal S., Buffet K., Durka M., Vincent S. P., Chem. Commun. 2010, 46, 3860–3862. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a.Buffet K., Gillon E., Holler M., Nierengarten J. F., Imberty A., Vincent S. P., Org. Biomol. Chem. 2015, 13, 6482–6492; [DOI] [PubMed] [Google Scholar]
- 10b.Durka M., Buffet K., Iehl J., Holler M., Nierengarten J.-F., Taganna J., Bouckaert J., Vincent S. P., Chem. Commun. 2011, 47, 1321–1323. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a.Rísquez-Cuadro R., García Fernández J. M., Nierengarten J. F., Ortiz Mellet C., Chem. Eur. J. 2013, 19, 16791–16803; [DOI] [PubMed] [Google Scholar]
- 11b.Trinh T. M. N., Holler M., Schneider J. P., García-Moreno M. I., García Fernández J. M., Bodlenner A., Compain P., Ortiz Mellet C., Nierengarten J. F., J. Mater. Chem. B 2017, 5, 6546–6556; [DOI] [PubMed] [Google Scholar]
- 11c.Tikad A., Fu H., Sevrain C. M., Laurent S., Nierengarten J. F., Vincent S. P., Chem. Eur. J. 2016, 22, 13147–13155; [DOI] [PubMed] [Google Scholar]
- 11d.Nierengarten J. F., Schneider J. P., Trinh T. M. N., Joosten A., Holler M., Lepage M. L., Bodlenner A., García-Moreno M. I., Ortiz Mellet C., Compain P., Chem. Eur. J. 2018, 24, 2483–2492. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a.Luczkowiak J., Munoz A., Sanchez-Navarro M., Ribeiro-Viana R., Ginieis A., Illescas B. M., Martin N., Delgado R., Rojo J., Biomacromolecules 2013, 14, 431–437; [DOI] [PubMed] [Google Scholar]
- 12b.Muñoz A., Sigwalt D., Illescas B. M., Luczkowiak J., Rodriguez-Perez L., Nierengarten I., Holler M., Remy J. S., Buffet K., Vincent S. P., Rojo J., Delgado R., Nierengarten J. F., Martin N., Nat. Chem. 2016, 8, 50–57. [DOI] [PubMed] [Google Scholar]
- 13.Serda M., Ware M. J., Newton J. M., Sachdeva S., Krzykawska-Serda M., Nguyen L., Law J., Anderson A. O., Curley S. A., Wilson L. J., Corr S. J., Nanomedicine 2018, 13, 2981–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee C. W., Su Y. H., Chiang Y. C., Lee I. T., Li S. Y., Lee H. C., Hsu L. F., Yan Y. L., Li H. Y., Chen M. C., Peng K. T., Lai C. H., Biomol. Eng. 2020, 10, 514. [Google Scholar]
- 15.
- 15a.Sanchez-Navarro M., Muñoz A., Illescas B. M., Rojo J., Martin N., Chem. Eur. J. 2011, 17, 766–769; [DOI] [PubMed] [Google Scholar]
- 15b.Illescas B. M., Rojo J., Delgado R., Martin N., J. Am. Chem. Soc. 2017, 139, 6018–6025; [DOI] [PubMed] [Google Scholar]
- 15c.Rodríguez-Pérez L., Ramos-Soriano J., Pérez-Sánchez A., Illescas B. M., Muñoz A., Luczkowiak J., Lasala F., Rojo J., Delgado R., Martin N., J. Am. Chem. Soc. 2018, 140, 9891–9898. [DOI] [PubMed] [Google Scholar]
- 16.Ramos-Soriano J., Reina J. J., Illescas B. M., de la Cruz N., Rodriguez-Perez L., Lasala F., Rojo J., Delgado R., Martin N., J. Am. Chem. Soc. 2019, 141, 15403–15412. [DOI] [PubMed] [Google Scholar]
- 17.An Y. Z., Anderson J. L., Rubin Y., J. Org. Chem. 1993, 58, 4799–4801. [Google Scholar]
- 18.Barron A. R., J. Enzyme Inhib. Med. Chem. 2016, 31, 164–176. [DOI] [PubMed] [Google Scholar]
- 19.Pochkaeva E. I., Podolsky N. E., Zakusilo D. N., Petrov A. V., Charykov N. A., Vlasov T. D., Penkova A. V., Vasina L. V., Murin I. V., Sharoyko V. V., Semenov K. N., Prog. Solid State Chem. 2020, 57, 100255. [Google Scholar]
- 20.
- 20a.Castro E., Cerón M. R., Garcia A. H., Kim Q., Etcheverry-Berríos A., Morel M. J., Díaz-Torres R., Qian W., Martinez Z., Mendez L., Perez F., Santoyo C. A., Gimeno-Muñoz R., Esper R., Gutierrez D. A., Varela-Ramirez A., Aguilera R. J., Llano M., Soler M., Aliaga-Alcalde N., Echegoyen L., RSC Adv. 2018, 8, 41692–41698; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b.Martinez Z. S., Castro E., Seong C.-S., Cerón M. R., Echegoyen L., Llano M., Antimicrob. Agents Chemother. 2016, 60, 5731–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a.Nakamura S., Mashino T., J. Nippon Med. Sch. 2012, 79, 248–254; [DOI] [PubMed] [Google Scholar]
- 21b.Yasuno T., Ohe T., Takahashi K., Nakamura S., Mashino T., Bioorg. Med. Chem. Lett. 2015, 25, 3226–3229. [DOI] [PubMed] [Google Scholar]
- 22.Kornev A. B., Peregudov A. S., Martynenko V. M., Balzarini J., Hoorelbeke B., Troshin P. A., Chem. Commun. 2011, 47, 8298–8300. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a.Rivero-Buceta E., Doyagüez E. G., Colomer I., Quesada E., Mathys L., Noppen S., Liekens S., Camarasa M. J., Pérez-Pérez M. J., Balzarini J., San-Félix A., Eur. J. Med. Chem. 2015, 106, 34–43; [DOI] [PubMed] [Google Scholar]
- 23b.Rivero-Buceta E., Sun L., Martínez-Gualda B., Doyagüez E. G., Donckers K., Quesada E., Camarasa M. J., Delang L., San-Félix A., Neyts J., Leyssen P., Antimicrob. Agents Chemother. 2016, 60, 5064–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martínez-Gualda B., Sun L., Rivero-Buceta E., Flores A., Quesada E., Balzarini J., Noppen S., Liekens S., Schols D., Neyts J., Leyssen P., Mirabelli C., Camarasa M. J., San-Félix A., Antiviral Res. 2017, 139, 32–40. [DOI] [PubMed] [Google Scholar]
- 25.Sun L., Lee H., Thibaut H. J., Lanko K., Rivero-Buceta E., Bator C., Martínez-Gualda B., Dallmeier K., Delang L., Leyssen P., Gago F., San-Félix A., Hafenstein S., Mirabelli C., Neyts J., PLoS Pathog. 2019, 15, e1007760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a.Esté J. A., Antiviral Drug Strategies, Methods and Principles in Medicinal Chemistry (Ed.: E. De Clercq), Wiley-VCH, Weinheim, 2011, Vol. 50, pp. 29–50; [Google Scholar]
- 26b.Kang Y., Guo J., Chen Z., Protein Cell 2013, 4, 86–102; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26c.Klasse P. J., Cell. Microbiol. 2012, 14, 1183–1192; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26d.Lu L., Yu F., Cai L., Debnath A. K., Jiang S., Curr. Top. Med. Chem. 2016, 16, 1074–1090; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26e.Micewicz E. D., Ruchala P., Curr. Pharm. Des. 2013, 19, 1784–1799; [DOI] [PubMed] [Google Scholar]
- 26f.Wilen C. B., Tilton J. C., Doms R. W., Adv. Exp. Med. Biol. 2012, 726, 223–242. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a.Haqqani A. A., Tilton J. C., Antiviral Res. 2013, 98, 158–170; [DOI] [PubMed] [Google Scholar]
- 27b.Singh I. P., Chauthe S. K., Expert Opin. Ther. Pat. 2011, 21, 399–416. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a.Zhang X., Ding X., Zhu Y., Chong H., Cui S., He J., Wang X., He Y., AIDS 2019, 33, 1–11; [DOI] [PubMed] [Google Scholar]
- 28b.Fletcher C. V., Lancet 2003, 361, 1577–1578; [DOI] [PubMed] [Google Scholar]
- 28c.Woollard S. M., Kanmogne G. D., Drug Des. Dev. Ther. 2015, 9, 5447–5468; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28d.Markham A., Drugs 2018, 78, 505–511; [DOI] [PubMed] [Google Scholar]
- 28e.Markham A., Drugs 2020, 80, 1485–1490. [DOI] [PubMed] [Google Scholar]
- 29.Martínez-Gualda B., Sun L., Martí-Marí O., Noppen S., Abdelnabi R., Bator C. M., Quesada E., Delang L., Mirabelli C., Lee H., Schols D., Neyts J., Hafenstein S., Camarasa M.-J., Gago F., San-Félix A., J. Med. Chem. 2020, 63, 349–368. [DOI] [PubMed] [Google Scholar]
- 30.Ramos-Soriano J., Reina J. J., Illescas B. M., Rojo J., Martin N., J. Org. Chem. 2018, 83, 1727–1736. [DOI] [PubMed] [Google Scholar]
- 31.Witte P., Hörmann F., Hirsch A., Chem. Eur. J. 2009, 15, 7423–7433. [DOI] [PubMed] [Google Scholar]
- 32.Witte P., Beuerle F., Hartnagel U., Lebovitz R., Savouchkina A., Sali S., Guldi D., Chronakis N., Hirsch A., Org. Biomol. Chem. 2007, 5, 3599–3613. [DOI] [PubMed] [Google Scholar]
- 33.Kato H., Böttcher C., Hirsch A., Eur. J. Org. Chem. 2007, 2659–2666. [Google Scholar]
- 34.Li H., Haque S. A., Kitaygorodskiy A., Meziani M. J., Torres-Castillo M., Sun Y.-P., Org. Lett. 2006, 8, 5641–5643. [DOI] [PubMed] [Google Scholar]
- 35.Balzarini J., Holy A., Jindrich J., Naesens L., Snoeck R., Schols D., De Clercq E., Antimicrob. Agents Chemother. 1993, 37, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balzarini J., Van Laethem K., Daelemans D., Hatse S., Bugatti A., Rusnati M., Igarashi Y., Oki T., Schols D., J. Virol. 2007, 81, 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engstrom O., Munoz A., Illescas B. M., Martin N., Ribeiro-Viana R., Rojo J., Widmalm G., Org. Biomol. Chem. 2015, 13, 8750–8755. [DOI] [PubMed] [Google Scholar]
- 38.Yamayoshi S., Yamashita Y., Li J., Hanagata N., Minowa T., Takemura T., Koike S., Nat. Med. 2009, 15, 798–801. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a.Tijsma A., Franco D., Tucker S., Hilgenfeld R., Froeyen M., Leyssen P., Neyts J., Antimicrob. Agents Chemother. 2014, 58, 6990–6992; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39b.Andries K., Dewindt B., Snoeks J., Willebrords R., van Eemeren K., Stokbroekx R., Janssen P. A., Antimicrob. Agents Chemother. 1992, 36, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumari M., Balaji P. V., Sunoj R. B., Phys. Chem. Chem. Phys. 2011, 13, 6517–6530. [DOI] [PubMed] [Google Scholar]
- 41.Koharudin L. M., Furey W., Gronenborn A. M., J. Biol. Chem. 2011, 286, 1588–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pan J., Peng H., Chen B., Harrison S. C., J. Mol. Biol. 2020, 432, 1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Behrens A.-J., Vasiljevic S., Pritchard L. K., Harvey D. J., Andev R. S., Krumm S. A., Struwe W. B., Cupo A., Kumar A., Zitzmann N., Seabright G. E., Kramer H. B., Spencer D. I. R., Royle L., Lee J. H., Klasse P. J., Burton D. R., Wilson I. A., Ward A. B., Sanders R. W., Moore J. P., Doores K. J., Crispin M., Cell Rep. 2016, 14, 2695–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information