

Abstract
Catalyst‐controlled functionalization of unmodified carbonyl compounds is a relevant operation in organic synthesis, especially when high levels of site‐ and stereoselectivity can be attained. This objective is now within reach for some subsets of enolizable substrates using various types of activation mechanisms. Recent contributions to this area include enantioselective transformations that proceed via transiently generated noncovalent di(tri)enolate‐catalyst coordination species. While relatively easier to form than simple enolate congeners, di(tri)enolates are ambifunctional in nature and so control of the reaction regioselectivity becomes an issue. This Minireview discusses in some detail this and other problems, and how noncovalent activation approaches based on metallic and metal free catalysts have been developed to advance the field.
Keywords: asymmetric catalysis, Brønsted base catalysis, C−H functionalization, dienolates, regioselectivity
Extend and divert. Extended enolates may be generated by deprotonation of β,γ‐ or α,β‐unsaturated carbonyl compounds smoothly, to react with a variety of electrophiles in situ. Easier to form than the parent simple enolates, di‐ and trienolates present, however, two nucleophilic carbons (typically Cα and Cγ) potentially leading to regioisomeric products. In recent years, methods based on both metallic and organic chiral catalysts have been developed able to address some of these site‐ and stereoselectivity issues, enabling direct asymmetric C−H functionalization of carbonyl compounds.
1. Introduction
Enolates stand as one of the most versatile reactive intermediates in organic synthesis.[1] Evenly accessible from carbonyl compounds in the ketone and the carboxylic acid oxidation states, they are rarely isolated because of their high reactivity. Instead, C−H deprotonation upon treatment with stoichiometric or catalytic base is commonly applied in situ. Enolates formed upon irreversible deprotonation by stoichiometric strong base have been investigated extensively, being instrumental to set new paradigms and concepts in asymmetric C−C and C−X bond‐forming methodology development based on chiral substrate‐, auxiliary‐ and ligand‐controlled strategies. The discovery of chiral Lewis acid‐catalyzed reactions of preformed silyl enol ethers and silyl (thio)acetal ketenes as enolate equivalents by Mukaiyama[2] represented a milestone towards generalization of catalyst‐controlled asymmetric variants.[3] More recently, direct asymmetric and catalytic approaches were shown viable using as catalysts either bifunctional metal complexes[4] or small molecule organocatalysts,[5] considerably expanding the chemistry of transiently generated enolate, or equivalent enamine, intermediates.
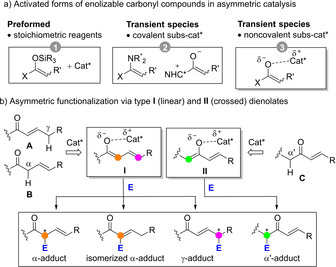
In this context, enolates with conjugated C=C double bond(s), that is di(poly)enolates or π‐extended enolates, deserve separate attention. While these species would retain the nucleophilic character ascribed to enolates in general, they present some unique additional features. Namely, they (i) may be generated under comparatively smoother reaction conditions, (ii) present two or more nucleophilic carbon centers (multivalence reactivity), (iii) may act in tandem reaction processes characterized by sequential donor/acceptor reactivity (ambivalent reactivity), and (iv) give rise to adducts bearing strategically positioned C=C unit(s) for further chemical diversification. Aimed at getting full advantage of these features, in recent years considerable efforts have been devoted to the development of catalytic enantioselective C−C and C−X bond‐forming reactions that proceed through either preformed (indirect methods)[6] or transiently generated (direct methods) di(poly)enolates and equivalents. As shown in Figure 1a, substrate‐catalyst interaction during the key bond‐forming step in direct methods may be covalent or noncovalent. The dienamine mediated approach relies on the activation of the unsaturated ketone or aldehyde substrate via covalent binding to the chiral primary or secondary amine catalyst.[7] Covalent substrate‐catalyst binding also operates when switterionic dienolate intermediates are involved, the latter generated in situ upon coupling of an unsaturated aldehyde, ketone or carboxylic acid derivative and a chiral nucleophilic catalyst, mainly N‐heterocyclic carbene (NHC) catalysts in the presence of base,[8] or trialkylphosphines.[9] In both covalent approaches main reaction control is intramolecular and the reactions usually proceed at the remote carbon center (vinylogous reactivity)[10] leading to (hetero)cyclic products through multistep bond‐forming cascades.
Figure 1.
(a) Activation modes for enolizable carbonyl compounds in asymmetric catalysis. (b) Direct C−H functionalization pathways via dienolate intermediates (E=electrophile).
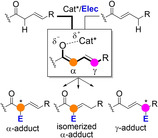
Complementing these covalent activation modes, enolizable unsaturated carbonyl compounds A and B may undergo deprotonation to afford linear dienolate species I in which the chiral (protonated) catalyst and the reactive center(s) on the enolate are not linked covalently, Figure 1b. Similarly, crossed dienolates II may be formed as noncovalent reactive complex from deprotonation of C. In the last couple of years, several applications involving these types of noncovalent intermediates have appeared in which chirality transfer to the product occurs with remarkable levels or site‐, regio‐ and stereocontrol. In addition, the reaction outcomes from intermediates like I and II are often divergent as compared to main pathways from extended enamines and azolium/phosphonium enolates, thus expanding the portfolio of direct enantioselective C−H functionalizations. This Minireview focuses on these noncovalent approaches and provides an overview of the major advances, challenges and opportunities within the field of asymmetric catalysis.
2. Early Studies with Preformed Metallic Di‐ and Trienolates
The interest in π‐extended enolates, dienolates in particular, can be traced back more than half a century. Early investigations already revealed that many factors, including the substrate substitution pattern (both steric and electronic effect), base/metal employed, temperature and solvent, have their impact on the reaction outcome. While mostly involving preformed dienolates and nonasymmetric variants, some of the underlined reactivity and selectivity issues are also relevant to modern asymmetric and catalytic approaches. Here, some selected contributions are highlighted.[11] Work in the late 50’s and 60’s in the context of alkali metal alkoxide‐promoted alkylation reactions of Δ4‐3‐keto steroids and related cyclic conjugate enones already revealed that the formed linear metal dienolate may follow diverting reaction pathways leading to α‐monoalkylated, α,α‐dialkylated, γ‐alkylated and isomerization products in variable amounts.[12] Subsequent studies addressed the issue of site‐selective deprotonation of related cyclic and acyclic α,β‐unsaturated ketones possessing two enolizable flanks. While crossed dienolates might be formed preferentially under kinetic conditions using bulky metal amides,[13] linear(extended) dienolates are favored under thermodynamic control using bases such as MOt‐Bu, MH or MNH2 (M=Na, K).[14] As a general trend, thus formed linear dienolates react mainly at Cα, occasionally at the O atom, but hardly at Cγ under kinetic conditions (Scheme 1).[15, 16] The observed regiochemistry can be rationalized primarily in terms of the polarity (charge control, “hardness”) of the participating reactants and their polarizability (frontier orbital coefficients, “softness”).[17] Substituent groups at the carbonyl α, β or γ position may also influence the observed regioselectivity. Wagner introduced the allopolarization concept as a refinement to help predicting the regioselectivity preferences in substituted dienolates and related systems.[18] By defining the “polarity index” as the ratio of the partial charges at two discrete reaction sites X and Y, it would follow that an increase (decrease) in polarity of the dienolate would enhance (diminish) charge control.
Scheme 1.
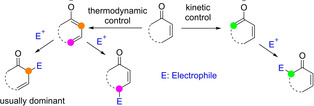
Metal dienolate formation and reactivity trends.
The reversibility of certain reactions is another factor that may alter the regioisomer distribution. For example, reactions potentially reversible, such as aldol, Mannich and Michael reactions of both Li[19] and Zn[20] dienolates, were reported to lead to the α‐addition adducts at low temperature, but to evolve to the thermodynamically more stable γ‐addition products under warming. Also additives may modify drastically some inherent trends. In this respect, elegant work by Yamamoto demonstrated that precomplexation of the carbonyl substrates to a bulky aluminium salt (tris(2,6‐diphenylphenoxide, ATPH 4) may override the innate α‐regioselectivity by steric shielding, leading to γ‐addition cross aldol products exclusively (Scheme 2).[14, 21]
Scheme 2.
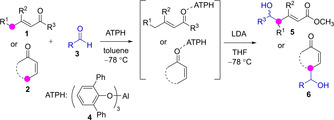
Substrate precomplexation to bulky Al‐phenoxides may override the innate α‐reactivity of metal dienolates leading to γ‐addition aldol products.
In comparison to dienolates, metal tri‐ and superior extended enolates present one or more additional nucleophilic sites, further complicating the reaction regioselectivity control. For example, among the few studies with these intermediates, work in the 80’s showed that aldol and Michael addition reactions of lithium trienolate dianion 7 afford mixtures of the three possible regioisomeric products 8/9, with α/γ/ϵ‐product ratios depending on the reaction conditions and the substitution pattern of the trienolate and the acceptor (Scheme 3).[22] One remarkable example of efficient regiocontrol with these challenging nucleophiles relies on the ATPH precomplexation strategy developed by Yamamoto. Thus, the cross‐aldol reactions of enolizable dienyl and trienyl aldehydes with both aliphatic and aliphatic aldehydes precomplexed to ATPH yield the ϵ‐adducts exclusively.[14]
Scheme 3.
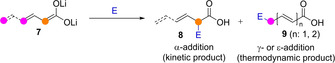
The regioselectivity issue in reactions involving dilithium di‐ and trienolate dianions by Mestres.
The development of Mukaiyama‐type addition reactions involving silyl di‐ and trienol ether derivatives have constituted a tremendous step forward in controlling the reactivity and regio‐ and stereoselectivity of multivalent enolate equivalents. The reader is referred to authoritative reviews on the subject.[6] Of interest, silyl di‐ and trienol ethers tend to react through the distal (γ‐ or ϵ‐carbon) rather than through the α‐carbon. This selectivity preference diverts from that of metallodienolates and nicely correlates with the calculated HOMO orbital coefficients and electrophilic susceptibilities at the different positions of silyl dienol ethers vs the corresponding lithium dienolates.[23]
One important aspect in the chemistry of dienolate intermediates refers to the starting carbonyl compound from which they are generated. In principle, enolizable both α,β‐ and β,γ‐unsaturated (namely, conjugate and skipped) carbonyl compounds may lead to the required dienolate species upon deprotonation with a base reagent or catalyst, but each substrate has their pros and cons. From the very beginning, it became clear that deprotonation of α,β‐unsaturated esters by some common metal amide bases may be troublesome due to competitive aza‐Michael addition.[24] The use of bulkier amide bases and hexamethylphosphoric triamide (HMPA) as an additive, at low temperature, allowed to overcome this shortcoming.[25] Another problem associated to conjugate enoyl systems is their low carbon acidity unless an additional activating group is positioned at Cγ,[26] which make them reluctant towards smooth enolization, even in the presence of stoichiometric base. In a recent example (Scheme 4), the treatment of conjugated enone 10 with a mixture of Cy2BCl and Et3N, followed by addition of aldehyde 13, failed to provide the desired aldol product 14, suggesting that the boron dienolate 12 was not produced. By contrast, applying the same aldolization conditions to the skipped enone 11 led to the aldol product 14 in 91 % yield.[27]
Scheme 4.
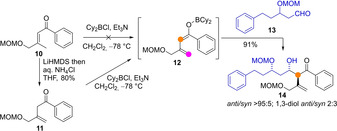
Recent example by Ito demonstrating that while deprotonation of skipped enones is suitable that of conjugate enones is reluctant.
As anticipated, deprotonation of β,γ‐unsaturated enoyl systems is comparatively easier. In recent years various catalytic approaches have been developed for the smooth, direct C−H functionalization of such systems via transiently generated di(tri)enolates with remarkable levels of chemo‐, site‐, regio‐ and stereoselectivity. As will be outlined in the following sections, one potential problem with these substrates is their tendency to isomerize to the thermodynamically favored conjugate systems.
3. Catalytic Methods
Key developments in the late 90’s and early 2000’s demonstrated the feasibility of direct, enantioselective transformations of unmodified carbonyl compounds mediated by their latent metal enolates generated in situ using soft enolization protocols.[28] These findings constituted a formidable step forward in synthetic methodology as both substrate activation and reaction stereoinduction were proven attainable under very convenient conditions. This approach resulted equally suitable to access latent metal dienolates, although the development of useful protocols relying on these latter intermediates elapsed owing in part to their attenuated reactivity and the need of additional regiochemistry control. Some years latter, purely organic chiral base catalysts were also demonstrated to be well suited for promoting the direct and asymmetric C−H functionalization of enolizable carbonyl compounds. Initially restricted to substrates bearing acidic CH units with two adjacent activating groups, organocatalytic reactions involving dienolate intermediates become available progressively.
In the following, methods belonging to both categories, namely those involving chiral metallic catalysts and chiral organocatalysts, are ordered according to the type of starting pronucleophilic carbonyl compound. It is important to note that methods for in‐ring functionalization of easily enolizable five, and to a lesser extent six, membered heterocyclic compounds, including oxazolones,[29] thiazolones,[30] imidazolones,[30] pyrazolones,[31] γ‐butenolides,[32] γ‐butyrolactams,[32a, 33] oxindoles,[34] and dihydropyrimidinones, Scheme 5, that involve aromatic enolate intermediates 15, constitute a differentiated group and are not considered here.[35]
Scheme 5.
In‐ring C−H functionalization of enolizable heterocycles involving aromatic di(poly‐)enolate intermediates are not covered in this work.
Stabilized allylic carbanions related to di‐ and trienolates may be derived from unsaturated compound classes other than carbonyl compounds, including sulfones, phoshonates, nitrocompounds or nitriles, particularly α,α‐dinitriles.[36] Due to their pioneering relevance, some key work on direct catalytic and asymmetric C−H functionalization of simple unsaturated nitriles is also described, as an exception.
3.1. Metal‐catalysis
3.1.1. Ester dienolates
In 2006 Kanai and Shibasaki reported the enantioselective intermolecular reductive aldol reactions of allenic esters 16 to methyl ketones 17 using chiral Cu(I) complexes and pinacolborane as the reducing reagent (Scheme 6).[37] The main reaction pathway of the reductively generated copper dienolate was shown to be highly dependent on the chiral phosphine ligand, allowing an effective control of the reaction regioselectivity. Whereas (R)‐DTBM‐SEGPHOS (L1) induced preferential formation of γ‐adduct 18 with perfect Z‐selectivity and high stereoselectivity, TANIAPHOS‐type ligands L2 favored exclusive formation of the α‐adduct as two epimers 19/20.
Scheme 6.
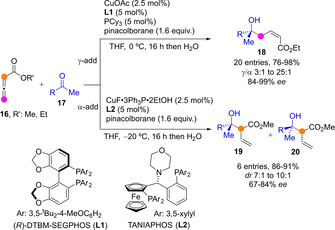
Cu(I)‐catalyzed reductive aldol between allenic esters and methyl ketones.
Soon after the same group documented the Mannich reaction between butenoate ester 21 and N‐phosphinoyl aldimines 22 to proceed smoothly in the presence of several metal aryloxides (Scheme 7).[38] The reaction scope was explored in its racemic version mainly, employing bis(p‐anisyl)barium complex as the catalyst. Under the basic reaction conditions the C=C double bond isomerized to the corresponding Morita‐Baylis‐Hillman (MBH) adduct 23 spontaneously, unless the ester γ‐position was disubstituted, which led to a diastereomeric mixture of α‐product 24. Three enantioselective examples using chiral aryloxy ligands L3 are reported, leading to enantioselectivities in the range 77 %‐80 % ee. Authors expand this catalytic strategy based on barium dienolate intermediates to the aldol addition reaction to aldehydes, which, again, proceeded through a α‐addition/isomerization path to produce conjugated aldols 25 in good yields and high enantioselectivity.[39] Isobutanal and n‐propanal, two readily enolizable aldehydes, were competent acceptors in this reaction although enantioselectivities were slightly lower than for other aldehydes. Control experiments showed that α‐aldol adduct 26 formed at low conversions, presented poor dr and ee values, and its formation is reversible, which led to the conclusion that the subsequent double bond isomerization took place under dynamic kinetic asymmetric conditions to afford the most stable conjugate aldol 25 in high ee.
Scheme 7.
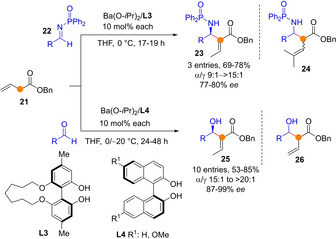
Ba(II)‐catalyzed Mannich and aldol additions of allylic esters with concomitant double bond isomerization by Shibasaki.
The system comprised of Cu(I)‐chiral diphosphine complex and Barton's base (2‐tert‐butyl‐1,1,3,3‐tetramethylguanidine) was shown by Yin to be able to promote the aldol addition of allylic esters 27 to aromatic aldehydes with high γ‐selectivity. Thus obtained adducts, upon subsequent one‐pot double bond E to Z isomerization and transesterification in the presence of a phosphine catalyst at 100 °C, led to the corresponding unsaturated δ‐lactone 28 in good yields and high enantioselectivity (Scheme 8).[40] Aliphatic aldehydes are also competent reaction partners yielding directly the lactone products 29 with similar efficiency and selectivity. Authors confirmed that under these catalytic conditions the parent conjugated (vinyl) esters 30 are unreactive, probably because of the inability of the catalytic system for deprotonating them to generate the required metal dienolate intermediate.
Scheme 8.
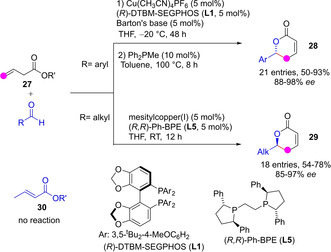
Cu‐catalyzed direct vinylogous aldol‐isomerization‐lactonization cascade to unsaturated δ‐lactones by Yin.
3.1.2. Amide dienolates
Reactions involving catalytically generated amide dienolate intermediates in the presence of a chiral metal complex and an amine base have also been developed. Yin described the asymmetric Mannich reaction between β,γ‐unsaturated N‐acyl pyrazoles 31 and N‐Boc aldimines 32 to smoothly proceed under chiral Cu(I)‐diphosphine complex/Et3N catalysis to afford the γ‐addition products 33 in high yields, essentially perfect regioselectivity and high enantioselectivities (Scheme 9).[41] According to the authors, increasing the steric hindrance of the metal dienolate by using bulky 3,5‐disubstituted pyrazole moiety in combination with bulky phosphine ligands (L1, L6) on the Cu(I) catalyst were critical design elements for cancelling the competitive α‐addition pathway. Remarkably, this catalytic approach was also productive with the doubly unsaturated N‐acyl pyrazole substrates 31 (n=1) which led to the bisvinylogous ω‐amino amide adducts 34 efficiently, provided that the catalyst loading is upgraded from 1 to 5 mol%. In this latter case, some adjustment of the reaction conditions and the chiral ligand was also necessary to attain high selectivity over the newly formed remote stereocenter.
Scheme 9.
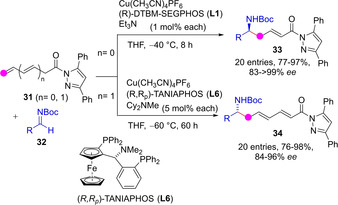
Cu(I)‐catalyzed vinylogous and bisvinylogous Mannich reaction involving pyrazole amide di‐ and tri‐enolates by Yin.
In subsequent studies, the same group found that the required metal dienolates can be generated from the parent conjugated amide systems provided that the Cγ position bears a F, Cl or Br substituent. The weakly electron‐withdrawing halogen atoms in 35 seem to acidify the Cγ‐H group without reducing significantly the nucleophilicity of this carbon in the formed dienolate. This way, vinylogous Mannich adducts 36 with contiguous halogen and amino functionality were produced in good yields, diastereomeric ratios from moderate to very high and generally high enantioselectivity (Scheme 10).[42]
Scheme 10.

Cu(I)‐catalyzed vinylogous Mannich reaction of in situ formed halogen‐substituted pyrazole amide dienolates.
Apart from C−C bond formation reactions, the catalytic nonasymmetric C−N bond formation involving metal dienolates as nucleophiles has been reported. Huang and Zhang described a metal‐catalyzed α‐amination of α,β‐unsaturated N‐acyl pyrazoles using azodicarboxylates as electrophilic reagents.[43] While the development of an enantioselective variant remains pending,[44] authors showed that preferential formation of α‐amination product is feasible by using Zn(OAc)2/DIPEA as the catalyst and γ‐amination product by using AgOAc/TMG, instead. A plausible explanation of this regiodivergent behavior of Zn(II) versus Ag(I) was provided based on the ability of the former to form chelate dienolate complexes.[45]
Shibasaki and coworkers developed a Cu(I)/diphosphine catalyzed direct asymmetric aldol addition reaction of α‐vinyl thioamides 37 and aliphatic aldehydes to give rise the α‐addition aldol products 38 in high yields, syn/anti ratios typically over 20 : 1 and high enantioselectivity (Scheme 11a). The use of a phenolic additive was crucial for attaining high stereoselectivity and the reaction is believed to proceed through a chair‐like transition state TS1.[46] In a related work, the same group developed a direct Cu(I)‐catalyzed aldol reaction of allylic 7‐azaindoline amides 39 with both aliphatic and aromatic/alkynyl aldehydes, which yielded the complementary syn (40) and anti (41) adducts, respectively (Scheme 11b).[47]
Scheme 11.
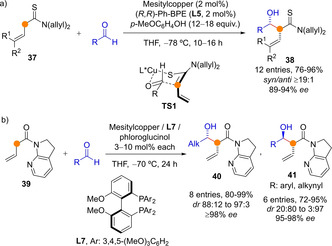
Copper‐catalyzed direct enantio‐ and diastereoselective aldol additions of α‐vinyl (thio)amides by Shibasaki.
3.1.3. Nitrile (aza)dienolates
In a series of papers Shibasaki's group has reported the Cu‐catalyzed C−H functionalization of allylic nitriles 42 with various electrophiles based on generation of an active carbon nucleophile by cooperative catalysis between a Lewis acid and a Brønsted base under mild reaction conditions.[48] Coordination to nitrile group of a Cu(I) salt (soft Lewis acid) facilitates concomitant Cα‐H deprotonation by a lithium aryloxide (hard base) to form the required aza‐dienolate intermediate.[49] In a first realization, the addition of these species to N‐phosphinoyl ketoimines 43 was reported to take place at Cα preferentially. Concomitant C=C double bond isomerization then affords the quaternary aza‐MBH adducts 44 with good yields, high Z/E ratios and high enantioselectivities using a chiral diphosphine ligand (Scheme 12).[48a] Unexpectedly, extension of this approach to ketones as acceptors followed a divergent reaction path and afforded the γ‐addition adducts 45 exclusively. The reaction needed polar solvents in order to provide practical yields of the tertiary carbinols, enantioselectivities from good to excellent and total Z selectivity.[48b] Refinement of this binary catalyst system by incorporating a hard Lewis base (phosphine oxide) as third component led to significant improvement on the isolated yields and enantioselectivities, even at reduced catalyst loading (down to 1 mol% or below).[48c]
Scheme 12.
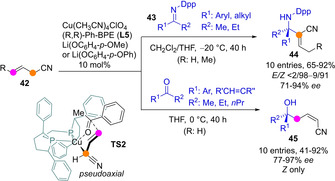
Cu(I)‐catalyzed asymmetric Cα‐Mannich‐ and Cγ‐aldol‐type reactions of allylic nitriles developed by Shibasaki's group. Dpp: diphenylphosphinoyl.
Surprisingly, applying the above catalytic activation of allylic nitriles to aldehydes as acceptors was nontrivial. The reaction outcomes were dependent on both the reaction times and the nature of the aldehyde (Scheme 13).[50] The reaction of acrylonitrile with α,β‐unsaturated aldehydes at −40 °C led to a time‐dependent mixture of α‐ and γ‐addition products, plus the isomerized MBH‐type adduct 47, suggesting the α‐addition reaction is reversible and the α‐adduct is not stable; with aromatic aldehydes, the γ‐addition product 46 predominated. In contrast, the reaction with aliphatic aldehydes afforded the α‐addition product 48 exclusively. The method was applied to a rapid enantioselective synthesis of δ‐valerolactone, which was implemented in the enantioselective synthesis of 49, a key intermediate for the preparation of the protein serine/threonine phosphatases as well as DNA topoisomerase II inhibitor fostriecin.
Scheme 13.
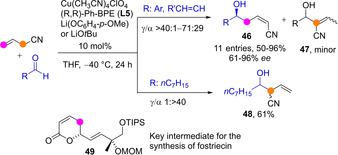
Acceptor‐dependent α vs γ regioselectivity in Cu‐catalyzed enantioselective aldol‐type additions of allyl nitriles to aldehydes by Shibasaki.
3.1.4. Ketone dienolates
Research on direct enantioselective C−C bond forming reactions involving metallic ketone dienolates remains less developed and are restricted to ketones with a single enolizable flank. Early examples with preformed metal dienolates established, once again, that the regioselectivity of alkylation reaction is multivariable. For instance, the favorable α‐reaction regioselectivity inherent to most alkali metal (e. g. Li+, Na+) dienolates alkylation may be reversed to favor γ‐reaction using Cu, Ge, Sn or Zn dienolates. The use of in situ generated Pd(II) dienolates as intermediates turned out to be a useful strategy to shift the regiochemistry of these reactions in favor of the γ site.[51] In this context, Buchwald developed a Pd‐catalyzed γ‐arylation(vinylation) of either α,β‐ or β,γ‐unsaturated ketones 50 with the corresponding aryl‐ or vinyl bromides (e. g. 51) in the presence of Cs2CO3 and a diphosphine ligand, the nature of which influenced both reaction chemo‐ and regioselectivity strongly (Scheme 14).[52] Using 4 mol% of 1,2‐bis(diphenylphosphino)ethane (dppe) in toluene at 100 °C, the γ‐arylation product was obtained exclusively. An enantioselective tandem γ‐arylation/aza‐Michael addition version using DTBM‐SEGPHOS (L1) was implemented to afford bicyclic indole derivatives 52.
Scheme 14.
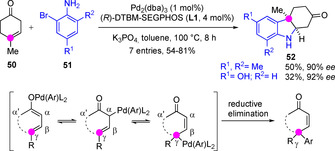
Pd‐catalyzed tandem γ‐arylation/aza‐Michael route to fused indoles from β,γ‐unsaturated cyclohexanones by Buchwald.
Liu and Feng reported that complexes comprised of ytterbium and a chiral diamide N,N’‐dioxide ligand are able to catalyze the tandem isomerization/α‐Michael addition of β,γ‐unsaturated 2‐acyl imidazoles 53 to deliver dimeric adducts 54/55 in the presence of Et3N as cocatalyst (Scheme 15).[53] The bidentate coordination ability of these particular ketones proved crucial for achieving good stereocontrol (ee up to >99 %; dr up to >19 : 1), but also to facilitate isomerization of the double bond, so that from a single starting material dual reactivity, nucleophilic and electrophilic, may be achieved under the same reaction conditions. Control experiments showed that Et3N cocatalyst does not play a significant role during the enolization process, but rather it facilitates double bond isomerization to occur with high E/Z selectivity. The α‐selective reactivity of these Yb dienolate intermediates generated in situ could also be applied against other acceptors, including Michael acceptors and ketoimines successfully, giving access to adducts 56–58.
Scheme 15.
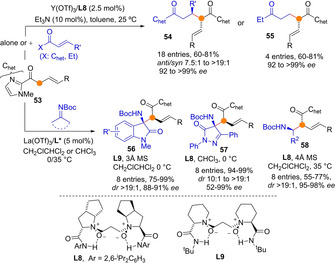
Rare earth/N,N‐dioxide catalysed α‐selective Michael and Mannich addition reactions of β,γ‐unsaturated 2‐acylimidazoles by Feng.
The above examples with not exception involve linear extended metal dienolates and the preferential α‐ or γ‐reactivity could be modulated by careful substrate design, including nature and position of substituents, and to some extent by the nature of the catalyst as well, including the metal. Such regioselectivity problems are not encountered in methods involving crossed metal dienolates, which generally lead to α‐addition adducts, although direct catalytic and asymmetric developments are less common. In a recent work, Trost demonstrated that the introduction of unsaturation adjacent to the carbonyl drastically improves the reactivity of α‐branched ketones in Zn‐catalyzed Mannich reactions with N‐carbamoyl imines (Scheme 16).[54] These reactions would proceed through transiently formed crossed dienolates, as shown in TS3. For example, the Zn(II) catalysed Mannich reactions of α‐fluoro enones (and ynones) 59 with N‐carbamoyl aldimines 60 to yield β‐tetrasubstituted β‐fluoamines 61 proceeded in good to excellent yields and enantioselectivity.[54a] Control experiments showed that while both α‐alkyl[54b] and α‐fluoro α‘‐unsaturated ketones reacted efficiently, the parent saturated ketones (simple enolate needs to be formed) were unreactive under the same catalytic conditions.
Scheme 16.
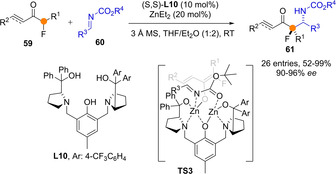
Direct, catalytic and α‐selective Mannich reactions of unsaturated α‐fluoroketones involving crossed Zn(II)‐dienolates by Trost.
3.2. Brønsted base catalysis
Most common chiral organic bases are weak, therefore unable to activate substrates with pKa values above 17.[55] For example, deprotonation of unfunctionalized enolizable carbonyl compounds is beyond the capability of the majority of chiral BB catalysts. Carbonyl compounds with a β,γ‐unsaturation would be slightly more acidic, increasing the chances to deliver the corresponding di‐(tri)enolate transiently. Also, the use of bifunctional BB/H‐bond catalysts or stronger BB catalysts may broaden the pool of enolizable substrates within reach. H‐Bond donor groups installed within the catalyst structure may synergistically coordinate to the carbonyl oxygen atom and assist during deprotonation. On the other hand, the potential of organic superbases is clear, as early work by Verkade's group using nonionic bases like C1 (measured pK a about 32[56]) nicely demonstrated. For example, the reaction between allylic esters and aldehydes took place within 3 hours at temperatures from −63 to −94 °C in the presence of C1 (20–40 mol%) to afford aldol products 62 from exclusive Cα addition (Scheme 17). When the allylic nitriles were employed instead of esters, the reaction also proceed through Cα but led to the corresponding MBH‐type adducts 63 as subsequent C=C bond isomerization occurred spontaneously.[57] In the following section main advances in this area are presented.
Scheme 17.
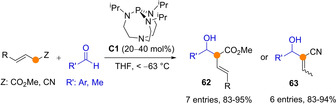
Organocatalytic aldol addition reactions of acrylic ester and nitrile using Verkade's strong bases.
3.2.1. Ester dienolates
The carbon acidity of simple carboxylic esters is low and so their activation by common chiral BB catalysts is not feasible. In contrast, β,γ‐unsaturated esters might be within reach, leading to dienolates as transient species. This was the case in the study by Zhao and co‐workers, who demonstrated that whereas phenyl 2‐phenylacetate 67 was not competent pronucleophile for the Mannich addition to N‐tosyl imines 65 promoted by tertiary amine/urea bifunctional catalyst C2, γ‐arylbutenoate 64 did work efficiently (Scheme 18).[55] Syn‐configured adducts 66, derived from reaction at the Cα of the transiently formed dienolate intermediate, were obtained in good yields and stereoselectivity. The nature of the O‐aryl group on esters has big impact on the outcome of the reaction, with best results attained using 2‐methyl naphth‐1‐yl group. In comparison, less sterically hindered aryl esters (OPh) provided poorer levels of stereocontrol, while highly demanding aryls, like 2,6‐dimethylphenyl esters, gave high enantio‐ and diastereoselectivity but a low reactivity (30 % of desired product was obtained).
Scheme 18.
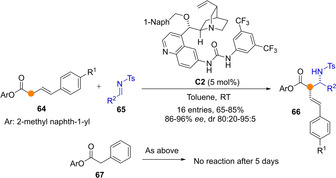
Direct urea/tertiary amine catalysed Mannich reaction of β,γ‐unsaturated aryl esters proceeding through Cα as reported by Zhao.
As compared to aryl esters, alkyl esters tend to be less reactive. Taking advantage of the cooperative activation by a second ester group in diester glutaconate, Quintard and co‐workers developed a bifunctional catalyst promoted direct addition of alkyl diesters 68 to nitroolefins 69.[58] The one‐pot treatment of the resulting adducts with Zn in acetic acid provided the corresponding cyclic trisubstituted pyrrolidines 70 in generally good yields and high enantio‐ and diastereoselectivity (Scheme 19). Given the symmetric structure of the transiently formed dienolate species from these particular diesters, the α/γ regioselectivity issue is irrelevant.
Scheme 19.
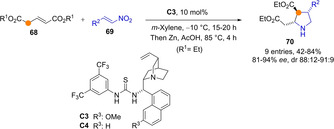
Tandem Michael addition/reduction/cyclisation involving symmetrical dienolates as key intermediates.
3.2.2. Thioester dienolates
In comparison to the parent oxoesters, carbon acidity of thioesters is slightly higher. Tang reported the base catalyzed γ‐selective amination of β,γ‐unsaturated 1‐naphthyl thioesters 71 with ditertbutyl diazoesters 72 (Scheme 20).[59] The reactions employing C 2 symmetric guanidine catalyst C5 proceeded at low temperature with high yields and enantioselectivity leading to product 73. The reaction was stereospecific and the Z‐configured thioester Z‐71 afforded the γ‐aminated products ent‐73 with opposite configuration at the newly formed C−N stereocenter. The effectiveness of these reactions is strongly influenced by the nature of the radical attached to sulfur: alkyl thioesters performed poorly, while aryl thioesters gave usually much better yields and selectivities. Density functional calculations favored a “side‐on” mechanism (TS4) with the catalyst working bifunctional rather than the alternative hetero‐Diels‐Alder (TS5) pathway. This catalytic γ‐amination process was also extendable to β,γ‐unsaturated N‐acyl oxazolidinones.
Scheme 20.
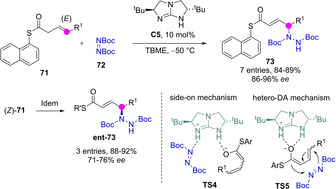
Stereospecific γ‐amination of β,γ‐unsaturated thioesters catalysed by chiral guanidine C5.
3.2.3. Amide dienolates
Since the introduction of pyrazoleamides as active pronucleophiles in asymmetric catalysis by Barbas,[60] these substrates turned out quite valuable for both metallic catalysis, vide supra, and organocatalysis. Dienolates transiently generated from deprotonation of allyl pyrazoleamides 74 by bifunctional base catalyst engaged in a tandem aldol/cyclisation process with isatins 75, with relief of pyrazole (Scheme 21). Using as low as 1 mol% Takemoto's catalyst C6, the corresponding spirocompounds 76 were obtained in very high yields and good enantioselectivity.[61]
Scheme 21.
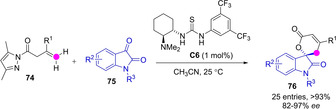
Asymmetric organocatalytic synthesis of spiroindoles through tandem γ‐selective aldol addition/lactonization by Wu.
Huang group reported the tandem intermolecular Michael/intramolecular oxa‐Michael addition between skipped N‐enoyl pyrazoles 77 and α,β‐unsaturated ketoesters 78 to afford dihydropyrans 79 (Scheme 22).[62] Authors proposed a cooperative coordination of the chiral catalyst to both dienolate and ketoester groups (TS6). The same group demonstrated that nitroolefins 69 are also well suited electrophiles leading to the γ‐addition Michael adducts 80 in good yields and very high stereoselectivity for all tested aromatic nitroolefins (R2=Ar). A low yield (28 %) was reported for an aliphatic nitroolefin. Subsequent treatment of thus obtained adducts with 20 mol% tetramethylguanidine (TMG) afforded the corresponding tetrasubstituted cyclobutanes 81 as single diastereomer.[63]
Scheme 22.
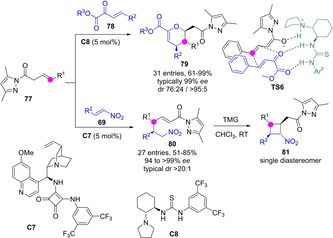
γ‐Selective Michael reactions of β,γ‐unsaturated N‐acyl pyrazoles triggered by bifunctional tertiary amine/H‐bond catalysts by Huang.
3.2.4. Ketone dienolates
Direct aldol addition reactions of allylic ketones to activated ketones as acceptors can be triggered by chiral tertiary amine/H‐bond bifunctional catalysts. These reactions proceed almost exclusively through the Cγ of the transiently generated ketone dienolate, thus providing an entry to remotely functionalized enone products. Huang, Jiang and coworkers have described the reaction between acyclic allyl ketones 82 and active ketones, both cyclic (e. g. isatins 75)[64] or acyclic (e. g. α‐CF3 ketones and α‐keto esters and phosphonates, 83),[65] to provide aldols 85 and 86 in high yields, good E/Z ratios, and enantioselectivities from good to excellent (Scheme 23). No examples involving ketones with two enolizable flanks were reported. More recently, Mukherjee extended the procedure to pyrazole‐2,3‐diones 84 as acceptors to produce pyrazolone derivatives 87 bearing a tertiary carbinol in good yields and enantioselectivities from moderate to high.[66]
Scheme 23.
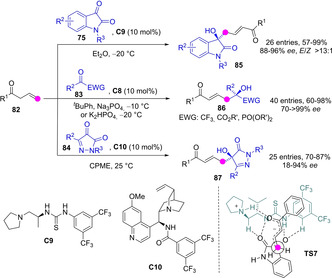
Organocatalytic γ‐selective aldol additions involving acyclic ketone dienolates and active ketones as acceptors by Jiang.
Transiently generated ketone dienolates also react through the Cγ against Michael acceptors to deliver cyclic products upon intramolecular trapping of the anionic intermediate. Hong and coworkers reported the direct reaction between allylic ketones 88 and nitroolefins 89 catalyzed by cinchona/squaramide bifunctional catalysts, to produce tetrasubstituted cyclobutanes 90 (Scheme 24).[67] The reaction is believed to occur via a tandem C−C/C−C bond forming process that commences with the Michael addition of the in situ formed dienolate to nitroolefin (stereomodel shown), followed by a subsequent intramolecular Michael cyclisation. The reaction was highly efficient with aromatic nitroalkenes, but the aliphatic ones led to diminished yields (54‐66 %). In a parallel development, Yang and Fang documented a Michael/heteroMichael tandem catalytic reaction between allylic ketones and unsaturated 1,2‐diketones 91 that afford tetrasubstituted dihydropyrans 92 under otherwise similar bifunctional tertiary amine/thiourea catalysis.[68] Once again, reactions involving either substrate with aliphatic substituents led to somewhat eroded yield or/and enantioselectivity. In another example, Albrecht described the addition/cyclisation reaction involving allyl ketones and cyclic unsaturated N‐tosyl imines 93 to yield benzofuran‐fused tetrahydropyridines 94 in good yields and high selectivities.[69]
Scheme 24.
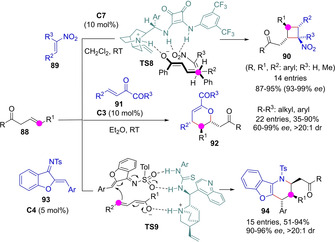
Organocatalytic tandem Michael/Michael processes initiated with γ‐addition of transiently formed ketone dienolates.
Alternatively, Palomo group described that in the presence of bifunctional BB/H‐bond catalysts like C11 reactions of transiently generated ketone dienolates, both cyclic and acyclic, with Michael acceptors proceeded through the Cα preferentially (Scheme 25).[70] For example, the reactions with (mainly aromatic) nitroolefins afforded adducts 95 with high regio‐ and stereoselectivity regardless the aromatic or aliphatic nature of the ketone R flank. As illustrated by the hydrogenation reaction leading to 96, simple reduction of the resulting adducts gives access to products derived from a formal enantioselective C−H functionalization of dissymmetrical alkyl alkyl ketones, a yet unconquered transformation using direct chemical approaches.[71] Among several subtypes of allyl ketones that successfully engaged in this catalytic reaction, α’‐hydroxyketones are worth of mention, since aftermath oxidation of the ketol moiety in adducts 97 smoothly affords the corresponding α‐branched carboxylic acids 98.
Scheme 25.
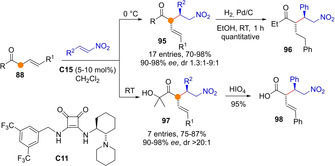
Organocatalytic asymmetric α‐selective Michael additions of transiently formed ketone dienolates by Palomo.
The behavior of alkynyl allyl ketones in the above reaction is tricky and strongly dependent on the substitution pattern of the allylic system. While unsubstituted allyl ketones 99 (R1, R2=H) invariably led to the MBH‐type products 101 via spontaneous double bond isomerization, adducts from β‐substituted ketones (R1≠H) showed to be resistant towards such isomerization, affording 100 as a mixture of diastereomers (Scheme 26). In addition, the reaction outcome of γ‐substituted ketones 99 (R1=H, R2≠H) was catalyst‐dependent: catalyst C11 led to mixtures of both 100 and 101, but the newly developed bulky catalyst C12 led to nonisomerized product 100 cleanly.[72]
Scheme 26.
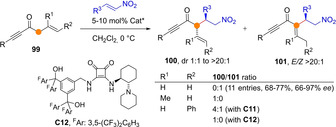
Catalyst‐ and substrate‐dependent reaction regioselectivity as reported by Palomo for transient dienolates from allyl alkynyl ketones.
Cα selective addition of transiently generated acyclic ketone dienolates is prevalent in reactions with activated ketimines, as described by Ma group.[73] Using nonsymmetrical thiourea catalysts C13, the Mannich reaction between aryl allyl ketones 82 and cyclic N‐sulfonyl iminoesters 102 afforded adducts 103 with high yields and selectivities (Scheme 27). When applied to alkyl allyl ketones diminished yields and both diastereo‐ and enantioselectivity were obtained. It was observed that the nature of the catalyst, specifically the stereochemical matching of the two chiral saccharide and diamine moieties, have a big influence not only on reaction stereoselectivity, but also on the α/γ ratio.
Scheme 27.
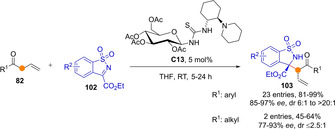
The glycosyl‐thiourea/amine catalysed α‐selective Mannich reaction between allyl ketones and activated ketimines by Ma.
The realization of highly enantioselective and α‐selective reactions involving α‐branched dienolate intermediates remains undeveloped; not only the attenuated reactivity and enantiofacial discrimination over a trisubstituted prochiral carbon are challenging,[74] but also the preference for the sterically less demanding γ‐reactivity vs the more demanding α‐reactivity needed to be addressed. Despite these difficulties, the α‐selective addition of α‐vinyl substituted cycloalkanones 104 (n=0,1,2 or 3) to several acceptors was feasible to carry out under the action of bifunctional tertiary amine/squaramide catalysis giving rise adducts in good yields and high enantioselectivity (Scheme 28).[75] Catalyst screening led to identify bulky catalysts C14 and C16 as best suited for the addition reaction to vinyl bis(sulfones) 105, while the 3,5‐dinitrophenyl analog C15 proved optimum for the addition to either nitroolefins or formaldehyde. Under the present conditions acyclic α‐branched dienolate precursors were ineffective substrates. Quantum calculations of purely intrinsic electronic parameters (e. g. charge distribution and Fukui nucleophilicity index) of the allegedly formed dienolate species failed in predicting the differences in reactivity of cyclic vs. acyclic enones. More relevant proved computing the transition state activation barriers of model reactions, which correlated well with the observed cyclic/acyclic reactivity difference. TS10 was identified as optimal transition state that correctly predicts the sense of α vs.γ site selectivity and reaction enantioselectivity.
Scheme 28.
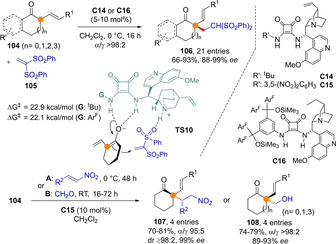
Organocatalytic α‐selective 1,2‐ and 1,4‐additions of α‐vinyl cycloalkanones leading to quaternary stereocenters developed by Palomo.
A reaction in which two transiently generated ketone dienolates, each reacting through Cα, are involved in consecutive steps, is reported by Li and Fang (Scheme 29).[76] The reaction involves aryl‐substituted allyl ketones 109 and alkynyl‐1,2‐diketones 110 as acceptor under PTC conditions to produce tetrasubstituted cyclopentenones 111 after an intermediate C→O acyl transfer. Attempts to render the process asymmetric by means of chiral ammonium salt C17⋅Br led to suboptimal enantiocontrol so far.
Scheme 29.
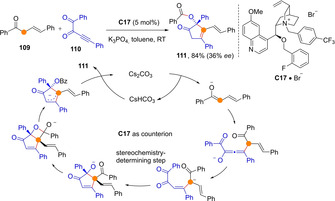
Sequential Cα/Cα coupling of intermediate ketone dienolates leading to tetrasubstituted cyclopentenones from acyclic precursors.
The isomerization of carbon‐carbon double bonds within otherwise untouched carbon skeletons may lead to formation of new stereocenters. Very recently, Paton and Dixon reported a bifunctional iminophosphorane C18‐catalyzed enantioselective synthesis of α,β‐unsaturated cyclohexanones 113 through a facially selective 1,3‐prototropic shift of β,γ‐unsaturated prochiral isomers 112 (Scheme 30).[77] The key and enantioselectivity‐determining step in this downstream derivatization process is the reprotonation at Cγ of a thiourea‐stabilized dienolate intermediate in which the catalyst basic site acts as a proton shuttle.
Scheme 30.
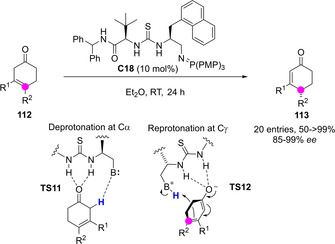
A thiourea/iminophosphorane catalytic approach to the enantioselective isomerization of .β,γ‐cyclohexenones by Paton and Dixon.
Relying on a dual covalent/noncovalent activation strategy, Xu and coworkers developed an enantioselective Michael addition of allylic ketones 82 to enals 114 that afford the γ‐addition adducts 115 in good yields and very high stereoselectivity (Scheme 31).[78] Authors found that concomitant activation of the enal via iminium ion formation and the nucleophile via H‐bond‐stabilized chiral dienolate was key to achieve both good conversion and efficient γ/α regioselectivity. Apparently, coordination of bis(sulfonamide) catalyst to dienolate anion causes shielding of the α position, thus favoring γ‐attack as in TS13. Starting from skipped cyclohexenones authors merged this process with a subsequent NHC‐catalyzed intramolecular Stetter reaction to provide a new entry to Hajos‐Wiecher‐type ketone skeleton enantioselectively.[79]
Scheme 31.
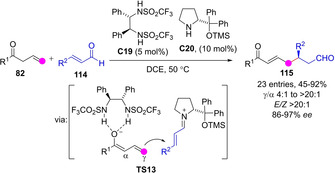
γ‐Selective Michael addition of transiently generated bis(sulfonamide)‐stabilized dienolates from allyl ketones by Xu.
3.2.5. Aldehyde dienolates
The low γ‐carbon acidity of the majority of α,β‐unsaturated carbonyl compounds makes deprotonation at this position by weak BB catalysts, such as tertiary amines, extremely difficult. Xu and coworkers conceived that the relatively active α‐aryl α,β‐unsaturated aldehydes may be an exception, particularly if reactive electrophiles are employed. Relying on a cinchona alkaloid/indanolamine‐derived bifunctional thiourea catalyst C21 they developed a tandem Michael/Henry reaction sequence between α‐aryl α,β‐unsaturated aldehydes 116 and aromatic nitroolefins for the construction of pentasubstituted cyclohexenes 117 (Scheme 32).[80] Although the reactions required from 4 to 8 days for completion and a 20 mol% catalyst loading, highly enantioenriched products with diastereoselectivities ranging from 3 : 1 to >20 : 1 were obtained. Interestingly, epimeric cycloadduct 118 could be obtained with similar efficiency by shifting to squaramide catalyst C22. The opposite nitroolefin face selectivity was explained by assuming two alternative approaching models TS14 and TS15, respectively.
Scheme 32.
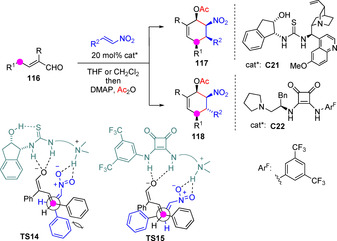
Diastereodivergent organocatalytic synthesis of substituted cyclohexenes involving aldehyde dienolates by Xu.
Variants of this reaction between aldehyde‐derived dienolates and nitroolefins were developed subsequently, which opened straightforward routes to substituted tetrahydro dibenzothiophenes 120/121 (X=S) and cyclohexenes 123 (Scheme 33).[81] In the latter development two mol‐equivalents of nitroolefin and two consecutive aldehyde dienolate‐mediated intermolecular addition steps, first one α‐selective and the second one γ‐selective (TS17), are involved.
Scheme 33.
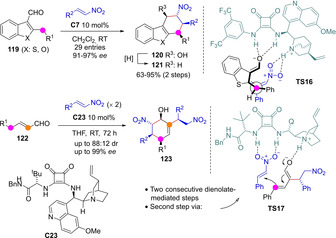
Tandem dienolate‐mediated addition/Henry reaction towards cyclohexene systems by Zhao.
3.3. “Out of the ring” dienolates from heterocyclic pronucleophiles
Some unsaturated heterocycles, particularly lactones and lactams, are prone to undergo “out of the ring” deprotonation to yield dienolate intermediate species of aromatic character. This activation strategy has been implemented successfully for the exocyclic asymmetric C−H functionalization of heterocyclic pronucleophiles using both metal and metal‐free catalytic approaches. In this context, two almost contemporary papers described the γ‐functionalization of α‐alkyliden‐2‐oxindoles via transiently generated dienolate intermediates. Qing and coworkers developed a substrate controlled Mannich reaction of α‐alkyliden‐2‐oxindoles with chiral N‐sulfinoyl aldimines mediated by titanium dienolates,[82] while Feng group reported the catalyst‐controlled direct Michael reaction between α‐alkyliden‐2‐oxindoles 124 and chalcones and related enones 125, using as catalysts Yb(III)‐N,N’‐dioxide chiral complexes (Scheme 34).[83] The reactions proceeded at the Cγ position of the intermediate dienolate affording Z‐configured products 126 preferentially with excellent enantiocontrol.
Scheme 34.
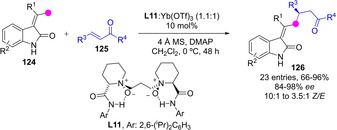
Ytterbium(III)‐catalyzed γ‐functionalization of 3‐alkylidene‐2‐oxindoles by Feng. DMAP: 4‐dimethylaminopyridine.
Complementing the metal‐catalyzed approach described above, metal‐free methods based on chiral base‐promoted deprotonation of several “enolizable” alkylidene‐heterocycles have appeared. Xu and coworkers described the catalytic vinylogous aldol‐type addition of alkylidene azlactones 127 to α‐keto esters 128 followed by cyclisation to deliver densely functionalized α,β‐unsaturated δ‐lactones (dihydropyranones) 129 featuring a quaternary stereocenter in good yields and excellent enantioselectivities (Scheme 35).[84] Later, Singh and coworkers developed a variant using aryl trifluoromethyl ketones 130 as acceptors.[85] The reaction is believed to proceed through intramolecular O‐acylation of the anionic aldolate with concomitant azlactone ring opening. Strikingly, TS models with opposite coordination geometries between the protonated catalyst and the Nu/Elec reactants were proposed in each work.[86]
Scheme 35.
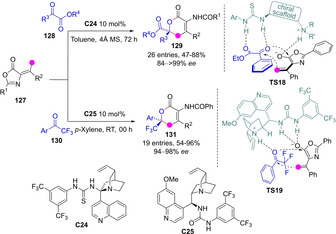
Dienolates from alkylidene oxazolones en route to unsaturated α‐amino γ‐lactones by Xu and Singh, independently.
Alternative activation approaches have been documented for the exocyclic functionalization of alkylidene oxazolones 132. Jiang and Chan described deprotonation in the presence of a tert‐leucine‐derived urea/quaternary ammonium salt C26 under PTC conditions and subsequent γ‐selective addition to 4‐nitro‐5‐styrylisoxazoles 133 to give rise enantioenriched α‐aminocyclohexenones 134 featuring two and three contiguous tertiary stereocenters in high stereoselectivity (Scheme 36).[87] On the other hand, Lian and Xu developed the γ‐selective allylic alkylation of this type of dienolates using catalytically activated MBH‐type carbonates 135.[88]
Scheme 36.
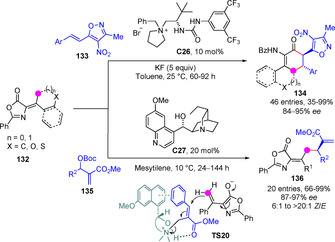
Asymmetric Michael additions and allylic alkylations of dienolates transiently generated from alkylideneoxazolones.
Structures containing the 2‐oxindole core are widespread among medicinally relevant compounds, sparking the development of asymmetric and catalytic syntheses of this class of compounds. In this context, organocatalytic methods have recently appeared that allow the direct C−H functionalization at the 2’ position of the oxindole side‐chain at C3 involving transiently generated dienolate species. Starting from enolizable 3‐alkyliden 2‐oxindoles 137, Curti, Rassu, Casiraghi and coworkers described the cinchona alkaloid/thiourea C28 catalyzed vinylogous Michael reaction of these pronucleophiles and nitroolefins to render compounds 138 (Scheme 37).[89] Products from dienolate γ‐addition were detected exclusively and, with the exception of aliphatic nitroolefins which turned to be less reactive and selective, adducts were obtained with high yields and essentially perfect enantio‐ and diastereocontrol. Expansion of this chemistry allowed to access chiral 2‐oxindole derivatives such as 139–142 with specific variations at the C3 side‐chain. For example, the complementary Z‐configured isomer 139,[90] products with a quaternary stereocenter (140, 141)[91] or adducts from a formal allylic alkylation reaction (142).[92] Addition to β,γ‐unsaturated α‐ketoesters has also been described.[93]
Scheme 37.
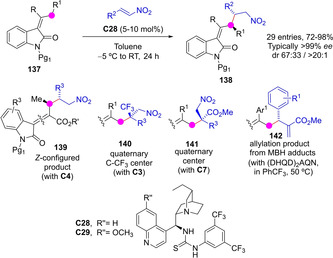
3‐Alkylidene‐2‐oxindoles as dienolate precursors and their enantioselective γ‐addition to different electrophiles.
In the above examples dienolates generated from alkyliden‐2‐oxindole have a single γ, or two equivalent γ and γ’, site(s) available, so site selectivity is not an issue. Bencivenni et al scrutinized alkyliden‐2‐oxindole substrates with two nonequivalent γ and γ’ reactive sites. They observed that, regardless the E/Z configuration of the substrate C=C double bond, the reaction catalyzed by C29 at room temperature afforded mixtures of addition products from both γ and γ’ sites, although isolated product always had Z configuration.[94] These observations were rationalized based on easy E/Z equilibration of starting material through σ bond rotation (s‐cis/s‐trans) in the intermediate dienolate. However, by performing the reactions at −20 °C the E/Z isomerization was suppressed and the reaction took place at the γ site exclusively, each geometrical isomer of starting material leading to different addition products stereospecifically. Kinetic studies that included isotopic and concentration effects indicated that deprotonation is the rate‐determining step of the reaction, and that the thiourea moiety of the catalyst interacts faster with nitrocompound than with oxindole. From these data authors propose dienolate, and not dienol, as the actual reaction intermediate (TS21, Figure 2) and justify the lowering of the E/Z isomerization rate as the concentration of nitroolefin reagent increases.
Figure 2.
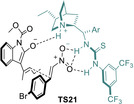
Model proposed by Bencivenni et al. for the above catalytic reactions involving 3‐alkylidene‐2‐oxindole‐derived dienolates as intermediates.
3.4. Aza‐dienolates from o‐allyl azaarenes
Nitrogen‐stabilized allylic anions (aza‐dienolates) may be formed via deprotonation of o‐allyl azaarenes. While this activation mechanism might constitute an approach for the catalytic side‐chain C−H functionalization of azaarenes, the realization of this idea in a direct and regio‐ and enantioselective manner has remained very much unexplored. Using ethereal solvents, 20 mol% LiH2PO4 and a newly developed bifunctional dipeptide‐based amine/urea catalyst C30, Jiang and coworkers reported the vinylogous (γ‐selective) direct aldol reaction of o‐allyl quinolones 143 and isatins 75 (R3=Me) to give 144 via intermediate aza‐dienolate (Scheme 38).[95] In this study authors found that extension of the method to acyclic trifluoromethyl ketones, e. g. 130, was not straightforward as yields dropped drastically. However, shifting to PTC conditions and using de novo developed ammonium salt C31 allowed the reaction with these latter ketones to work nicely.
Scheme 38.
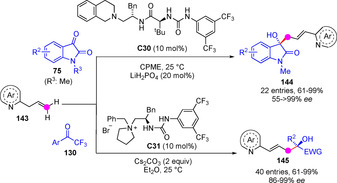
Enantioselective vinylogous aldol additions of azadienolates from o‐allyl azaarenes with activated ketones by Jiang.
Very recently, Yin reported the vinylogous (γ‐selective) aldol‐type addition of o‐allylazaarenes 143 to aromatic aldehydes in the presence of a chiral Cu‐bisphosphine catalyst and Barton's base as cocatalyst (Scheme 39).[96] It is interesting that by varying the chiral bisphophine ligand authors managed to control preferential formation of γ‐hydroxyl o‐vinylazaarenes with either E (146/147) or Z (148) configuration in yields from moderate to excellent and very high regio‐ and stereoselectivity.
Scheme 39.
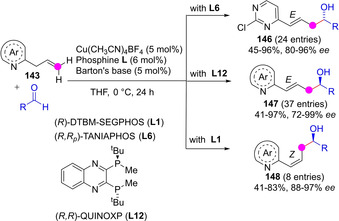
Ligand‐dependent azadienolate mediated vinylogous aldol‐type reactions of o‐allyl azaarenes by Yin.
3.5. Trienolates
Direct asymmetric and catalytic C−H functionalizations of enolizable substrates involving transiently generated trienolate species are essentially undeveloped. As compared with the parent enolates, extended trienolates are expected to present attenuated nucleophilicity, while regioselectivity, α vs. γ or ϵ, remains challenging. In the realm of metal catalysis, the work by Yin in bisvinylogous Mannich reaction involving copper trienolates derived from doubly unsaturated pyrazole amides, noted above (Scheme 12), is, as far as we know, the only single example. Within organocatalytic strategies, very recently Palomo's group reported the first example of Cα‐selective addition of transiently formed trienolates from doubly unsaturated thioester and ketones 149, using as acceptors nitroolefins (Scheme 40).[97] During initial screening of various Brønsted base catalysts, it was observed that, besides the α‐addition adduct 153, variable amounts of the isomerized starting material 151, the MBH‐type thioester/ketone product 154, and cyclohexene 150 were also obtained. Using cinchona‐derived squaramide catalyst C7 in dichloromethane at room temperature, the Michael addition adduct 153 could be obtained as the major product. Reaction optimization led to the discovery of a highly stereoselective domino process involving consecutive addition/isomerization events by combined action of chiral bifunctional catalyst C7 and MTBD as achiral stronger base cocatalyst. In this way tetrasubstituted cyclohexenes 150 were obtained in one‐pot from minimally functionalized starting materials.
Scheme 40.
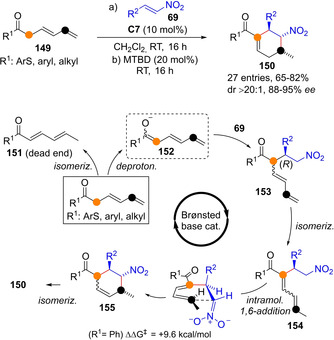
Trienolate‐mediated asymmetric one‐pot transformation of doubly unsaturated ketones/thioesters into substituted cyclohexenes by Palomo. MTBD: 7‐methyl‐1,5,7‐triazabicyclo[4.4.0]dec‐5‐ene.
3.6. Reactions involving electrophile‐catalyst chiral complexes
In the methods above the transiently generated dienolate forms a chiral noncovalent complex with the catalyst prior to reacting with a suitable electrophile. Methods based on a complementary strategy have also been developed in which an achiral dienolate species reacts with a covalently activated catalyst‐electrophile chiral complex. For example transiently generated dienolates of different nature are able to react with α,β‐unsaturated aldehydes, the latter activated in the form of chiral iminium species, to provide C−H functionalized carbonyl and related products enantioselectively. Thus, Albrecht developed an enantio‐ and diastereoselective entry to carboannulated naphthalene‐1(4H)‐one derivatives 159 through a tandem addition/intramolecular aldol condensation starting from naphthoquinone 156 (Scheme 41).[98] A similar strategy by Wu and Chi that ends up with an intramolecular Mannich addition reaction served to prepare tetrasubstituted cyclohexene carbaldehydes 160,[99] while Li, Cheng and coworkers described the enantioselective Michael addition reaction of dienolates from alkylidene oxindoles 137 to enals.[100] In this same context, Zanardi's group explored the reactivity of doubly unsaturated enolizable malononitriles against enals under dual activation of both nucleophile and electrophile. Indeed, they observed that the α,α‘‐dicyano dienes 158 may follow divergent deprotonation pathways: (i) deprotonation at the remote ϵ position (linear extended π‐system formation) with subsequent Michael addition leading to adducts 162, which could then be cyclisized via intramolecular Stetter reaction, or (ii) deprotonation at the γ’ site (crossed extended π‐system formation), followed by formal [4+2] cyclisation to adducts 163.[101]
Scheme 41.
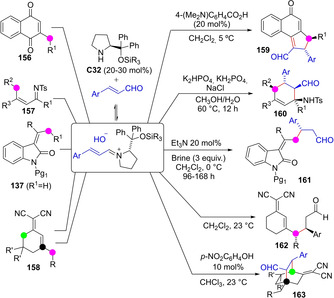
Direct addition reaction of various in situ generated dienolates with transiently generated chiral iminium ion from aldehydes.
On the other hand, addition of transiently generated dienolates to MBH carbonates was shown feasible upon activation of these latter by Lewis base (nucleophilic) catalysts. For instance, Huang and Jiang reported[102] the α‐selective addition of allyl aryl ketones 82 (R1=aryl) to MBH carbonates 164 in the presence of 10 mol% (DHQD)2PYR to afford adducts 165 in generally good yields and high selectivity (Scheme 42). The tBuO− anion detached upon coupling of the tertiary amine catalyst with the MBH adducts is postulated to act as a base for dienolate formation. Related developments using enolizable allyl sulfones[103] and 3,5‐dimethyl‐4‐nitroisoxazoles[104] as pronucleophiles have also been documented.
Scheme 42.
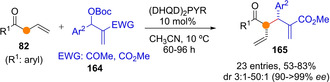
α‐Selective coupling reaction of in situ formed ketone dienolates with chiral amine‐activated MBH carbonates 164 by Huang and Jiang.
Yin and coworkers demonstrated the feasibility of in situ generation of achiral dienolates with concomitant activation of allylic electrophile reagent with a chiral metal complex catalyst for enantioselective C−H functionalization. These authors described the Ir‐catalyzed asymmetric vinylogous allylic alkylation of unsaturated lactones 167 and 168 with allylic carbonates 166 using TMG as the achiral base (Scheme 43).[105] The γ‐alkylation products are obtained with very high site‐ and stereoselectivities using phosphoramidite chiral ligands L13 and L14, respectively.
Scheme 43.
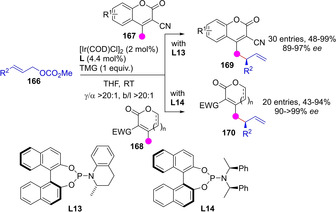
Ir‐catalyzed vinylogous allylic alkylation of β,γ‐unsaturated lactones by Yin.
4. Miscellaneous
Recently, an original strategy was devised by Du and Chen that uses transiently generated palladium(II) di‐ and trienolates as umpolung allylic alkylation reagents (Scheme 44). Under oxidative conditions (e. g. O2 atmosphere) these intermediates may evolve into electron‐deficient π‐allylpalladium complexes, which then react with substituted 2‐oxindoles 171 and related pronucleophiles through the remote Cγ (Cϵ in the case of doubly unsaturated ketones) site affording the corresponding quaternary stereocenters as in 172 with excellent enantioselectivity.[106] Just in the opposite direction, Zhao group reported the switch from alkoxide‐π‐allyl to metal dienolate reactivity by using Pd−Ti relay catalysis. Based on this umpolung strategy, vinylethylene carbonates 173 reacted with aurones 176 as special class of α,β‐unsaturated cyclic ketones to yield spiro‐heterocycles, such as 177, with three contiguous stereocenters in high diastereoselectivity, but moderate enantioselectivity.[107]
Scheme 44.
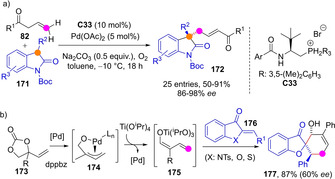
Umpolung strategies for the generation of ester and aldehyde metal dienolates in situ and their coupling with donor and acceptor reagents.
Alkynylogous enolates, the alkynyl analogs of metal dienolates, which might present the alkynyl and allenyl tautomeric forms 178 and 179, could be generated in situ from the corresponding alkynyl or allenyl esters. The reactivity of transiently formed alkynylogous enolates have been explored to a limited extent. Feng et al documented direct aldol reactions of allenyl esters 180 with isatin 181 via in situ formed chiral Au(I) allenyl enolates,[108] while Yin reported copper‐catalyzed direct aldol reactions of alkynyl esters 183 to various aromatic and aliphatic aldehydes via transiently generated Cu(I)/diphosphine alkynyl enolates (Scheme 45).[109] In both cases the reactions proceeded through Cγ, furnishing the corresponding allenyl carbinols (chiral 2,3‐allenols) 182 and 184/185 in good yields and excellent enantio‐ and diastereoselectivity.
Scheme 45.
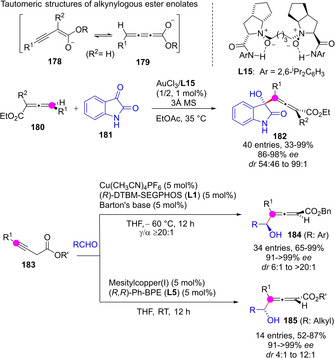
Alkynylogous enantioselective aldol reactions of homopropargylic esters under Au(III) and Cu(I) catalysis.
Johnson reported a dienolate mediated three‐component reductive coupling reaction between dimethyl phosphite 186, benzylidene pyruvates 78 and aldehydes catalyzed by bifunctional thiourea/iminophosphorane catalysts C34 to afford densely functionalized O‐phosphorylated aldol adducts 187 (Scheme 46).[110] Dienolates are generated in situ via base‐promoted Pudovik addition of dialkylphosphite to unsaturated keto ester and subsequent phospha‐Brook rearrangement, then reacting with aldehydes through the Cα exclusively and with good enantio‐ and diastereoselectivity.
Scheme 46.
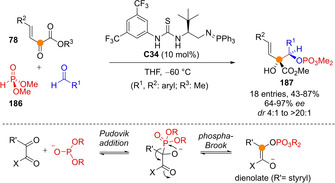
Asymmetric organocatalytic reducting coupling reactions between benzylidene pyruvates and aldehydes by Johnson.
Azadienolates 189 generated in situ from deprotonation of isatin‐derived nitrones 188 in the presence of chiral bifunctional BB catalysts may react with nitroolefins and activated ketimines 190 to afford the corresponding γ‐selective Michael addition adducts 191 or 1,3‐dipolar cycloadducts 192, respectively (Scheme 47).[111]
Scheme 47.
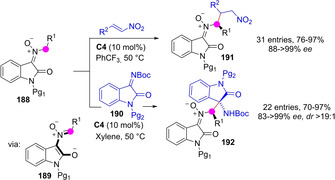
Catalytic asymmetric addition reactions involving nitrone‐derived azadienolates as intermediates.
Very recently, two enzymatic processes, the carbon‐carbon cyclization of oxepinone 193 to furnish fused bicyclic β‐lactone structure during vibralactone 194 biosynthesis,[112] and the decarboxylation of 3‐methylglutaconyl CoA to generate 3,3‐dimethylacrylyl‐CoA in an alternative biosynthetic pathway to isovaleryl‐coenzyme A,[113] have been proposed to proceed through enzyme‐coordinated dienolate intermediates (Scheme 48).
Scheme 48.
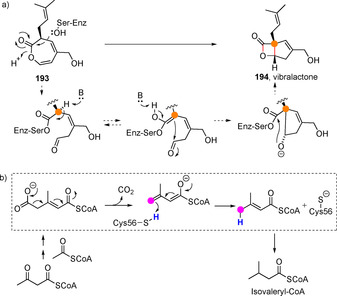
Recently reported enzymatic transformations involving dienolate intermediates.
5. Conclusions and Outlook
Catalytic, site‐ and enantioselective functionalization of β,γ‐unsaturated enolizable carbonyl compounds via transiently generated dienolates is nowadays feasible using both metallic and nonmetallic catalysts. Alone, or more often in combination with an organic base, metal complexes based on Cu(I), Ba(II), Zn(II), Yb(III), La(III), Ir(III), Pd(II) and Au(III), can trigger these reactions through tightly coordinated metal‐dienolate noncovalent intermediates. Although the reaction outcome in the majority of metal‐catalyzed reactions leads to adducts from the dienolate γ‐carbon attack (vinylogous reactivity), examples in which the complementary α‐addition product is obtained are also known. The γ vs. α reactivity preference is very much substrate‐dependent, but catalyst fine tuning is important to maximize or, in some instances, even reverse it. Likewise, purely organic chiral catalysts, particularly bifunctional Brønsted base/H‐bond catalysts, have been demonstrated to be useful in promoting highly enantioselective direct addition and coupling reactions of β,γ‐unsaturated enolizable compounds to electrophiles that proceed through intermediate dienolate species. Once again, the preferential α‐ or γ‐reaction pathway depends mainly on the substrate structure and substitution pattern, but organocatalysts structure has its impact. As compared to methods involving dienolate intermediates, examples dealing with trienolates remain very much unexplored yet and need further investigation. Similarly, most current protocols require nonconjugate (skipped) unsaturated substrates, and the conjugate analogs, while relatively more accessible and stable, remain challenging substrates because of lower carbon acidity. Development of stronger base catalysts may offer new opportunities in this regard. Although base‐promoted deprotonation is the most general access to di‐(tri)enolate intermediates, alternative approaches, including reductive variants starting from doubly unsaturated systems, may deserve some future attention. Finally, the methods covered here[114] constitute a nice complement to related activation approaches relying on covalent substrate‐catalyst coordination, such as in enamine‐ and acylammonium enolate mediated strategies.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Claudio Palomo studied Chemistry at the Instituto Químico de Sarria, in Barcelona, where he received his Chemical Engineering Degree in 1975. He obtained his Licenciatura in Chemistry in 1979 at the University of Barcelona and his Ph.D. in 1983 at the University of the Basque Country under the supervision of Prof. R. Mestres. In 1989 he was promoted to Professor and in 1991 he joined Prof. H. Rapoport at the University of California at Berkeley as a visiting professor. His current interests are in the area of asymmetric catalysis.

Biographical Information
Mikel Oiarbide obtained his PhD from the University of the Basque Country UPV/EHU (Spain) in 1991 working with Prof. C. Palomo and J.M. Aizpurua on tributyltin hydride mediated transformations. He then spent two years as a PDRA with Prof. H. Rapoport at UC Berkeley (USA) working on the synthesis of N‐glycosides pentostatin and coformycin. In 1994, he took up a tenure position at UPV/EHU and was promoted to Professor in 2003.

Acknowledgements
We thank the University of the Basque Country UPV/EHU (UFI QOSYC 11/22), the Basque Government (grant IT‐1236‐19) and Ministerio de Ciencia e Innovation (grant PID2019‐109633GB−C21), Spain, for their continuous financial support.
M. Oiarbide, C. Palomo, Chem. Eur. J. 2021, 27, 10226.
In memory of Prof. Kilian Muñiz.
Contributor Information
Prof. Mikel Oiarbide, Email: mikel.oiarbide@ehu.es.
Prof. Claudio Palomo, Email: claudio.palomo@ehu.es.
References
- 1.Braun M., Modern Enolate Chemistry: From Preparation to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, Germany, 2015. [Google Scholar]
- 2.
- 2a.Mukaiyama T., Narasaka K., Banno K., Chem. Lett. 1973, 1011–1014. First enantioselective version: [Google Scholar]
- 2b.Mukaiyama T., Kobayashi S., Uchiro H., Shiina I., Chem. Lett. 1990, 129–132. [Google Scholar]
- 3.For selected reviews on Mukaiyama aldol reaction, see:
- 3a.Matsuo J.-I., Murakami M., Angew. Chem. Int. Ed. 2013, 52, 9109–9118; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9280–9289; [Google Scholar]
- 3b.Yamashita Y., Yasukawa T., Yoo W.-J., Kitanosono T., Kobayashi S., Chem. Soc. Rev. 2018, 47, 4388–4480. [DOI] [PubMed] [Google Scholar]
- 4.Pioneering work:
- 4a.Yamada Y. M. A., Yoshikawa N., Sasai H., Shibasaki M., Angew. Chem. Int. Ed. Engl. 1997, 36, 1871–1873; [Google Scholar]; Angew. Chem. 1997, 109, 1942–1944; [Google Scholar]
- 4b.Yoshikawa N., Yamada Y. M. A., Das J., Sasai H., Shibasaki M., J. Am. Chem. Soc. 1999, 121, 4168–4178; [Google Scholar]
- 4c.Trost B. M., Ito H., J. Am. Chem. Soc. 2000, 122, 12003–12004. [Google Scholar]
- 5.Pioneering work using proline as aminocatalyst:
- 5a.Eder U., Sauer G., Wiechert R., Angew. Chem. Int. Ed. Engl. 1971, 10, 496–497; [Google Scholar]; Angew. Chem. 1971, 83, 492–493; [Google Scholar]
- 5b.Hajos Z. G., Parrish D. R., J. Org. Chem. 1974, 39, 1615–1621; [Google Scholar]
- 5c.List B., Lerner R. A., Barbas C. F., J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar]
- 6.For indirect methods based on preformed silyl enolates (vinylogous Mukaiyama reactions), see:
- 6a.Hosokawa (aldol) S., Tetrahedron Lett. 2018, 59, 77–88; [Google Scholar]
- 6b.Cordes M. H. C., Kalesse M., Org. React. 2019, 98, 173–773; [Google Scholar]
- 6c.Yin (Michael) Y., Jiang Z., ChemCatChem 2017, 9, 4306–4318; [Google Scholar]
- 6d.Schneider C., Abels F., Org. Biomol. Chem. 2014, 12, 3531–3543. [DOI] [PubMed] [Google Scholar]
- 7.Selected reviews:
- 7a.Jiang H., Albrecht Ł., Jørgensen K. A., Chem. Sci. 2013, 4, 2287–2300; [Google Scholar]
- 7b.Jurberg I. D., Chatterjee I., Tannerta R., Melchiorre P., Chem. Commun. 2013, 49, 4869–4883; [DOI] [PubMed] [Google Scholar]
- 7c.Marcos V., Alemán J., Chem. Soc. Rev. 2016, 45, 6812–6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For a review, see: Chen X.-Y., Liu Q., Chauhan P., Enders D., Angew. Chem. Int. Ed. 2018, 57, 3862–3873; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3924–3935. [Google Scholar]
- 9.For a review, see: Fan Y. C., Kwon O., Chem. Commun. 2013, 49, 11588–11619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For a recent review on the principle of vinylogy as applied to π-extended-type donors, see: Curti C., Battistini L., Sartori A., Zanardi F., Chem. Rev. 2020, 120, 2448–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For a review dealing with the regioselectivity of reactions involving heteroatom-stabilized allylic anions, see: Katritzky A. R., Piffl M., Lang H., Anders E., Chem. Rev. 1999, 99, 665–722. [DOI] [PubMed] [Google Scholar]
- 12.Ringold H. J., Malhotra S. K., J. Am. Chem. Soc. 1962, 84, 3402–3403. [Google Scholar]
- 13.Lee R. A., McAndrews C., Patel K. M., Reusch W., Tetrahedron Lett. 1973, 965–968. [Google Scholar]
- 14.Saito S., Shiozawa M., Ito M., Yamamoto H., J. Am. Chem. Soc. 1998, 120, 813–814. [Google Scholar]
- 15.
- 15a.Groenewegen P., Kallenberg H., van der Gen A.Tetrahedron Lett. 1978. 491–494; [Google Scholar]
- 15b.Vedejs E., Gapinski D. M., McElvain S. M.Tetrahedron Lett. 1981, 22, 4913–4916. [Google Scholar]
- 16.House H. O., Modern Synthetic Reactions, 2nd Ed. Benjamin, Menlo Park, Calif.1972, p. 492. [Google Scholar]
- 17.Pearson R. G., Hard and Soft Acids and Bases, Dowden, Hutchinson and Ross, Stroudsburg, Penn.1973. [Google Scholar]
- 18.For details, see: Gompper R., Wagner H. U., Angew. Chem. 1976, 88, 389–401; [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1976, 15, 321–333. [Google Scholar]
- 19.Examples involving: (ketone dienolates)
- 19a.Dugger R. W., Heathcock C. H., J. Org. Chem. 1980, 45, 1181–1185; (carboxylic acid-derived dianions); [Google Scholar]
- 19b.Casinos I., Mestres R., J. Chem. Soc. Perkin Trans. 1 1978, 1651–1655; [Google Scholar]
- 19c.Johnson P. R., White J. D., J. Org. Chem. 1984, 49, 4424–4429. [Google Scholar]
- 20.van Maanen H. L., Kleijn H., Jastrzebski J. T. B. H., Lakin M. T., Spek A. L., van Koten G., J. Org. Chem. 1994, 59, 7839–7848. [Google Scholar]
- 21.
- 21a.Saito S., Shiozawa M., Nagahara T., Nakadai M., Yamamoto H., J. Am. Chem. Soc. 2000, 122, 7847–7848; [DOI] [PubMed] [Google Scholar]
- 21b.Saito S., Nagahara T., Shiozawa M., Nakadai M., Yamamoto H., J. Am. Chem. Soc. 2003, 125, 6200–6210. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a.Ballester P., Costa A., García-Raso A., Gómez-Solivellas A., Mestres R., Tetrahedron Lett. 1985, 26, 3625–3628; [Google Scholar]
- 22b.Ballester P., Costa A., García-Raso A., Mestres R., J. Chem. Soc. Perkin Trans. 1 1988, 2797–2803. [Google Scholar]
- 23.In rationalizing site selectivity, steric effects cannot be underestimated. For further details, see: Denmark S. E., Heemstra J. R., Beutner G. L., Angew. Chem. Int. Ed. 2005, 44, 4682–4698; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 4760–4777. [Google Scholar]
- 24.Herrmann J. L., Kieczykowski G. R., Schlessinger R. H., Tetrahedron Lett. 1973, 14, 2433–2436. [Google Scholar]
- 25.Rathke M. W., Sullivan D., Tetrahedron Lett. 1972, 13, 4249–4252. [Google Scholar]
- 26.Mannich-type reactions of metal dienolates generated from γ-bromo-α,β-unsaturated esters, involving chiral N-sulfinyl imines:
- 26a.Chogii I., Njardarson J. T., Angew. Chem. Int. Ed. 2015, 54, 13706–13710; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13910–13914; [Google Scholar]
- 26b.Chogii I., Das P., Delost M. D., Crawford M. N., Njardarson J. T., Org. Lett. 2018, 20, 4942–4945. [DOI] [PubMed] [Google Scholar]
- 27.Kawamoto Y., Kobayashi T., Ito H., Org. Lett. 2019, 21, 5813–5816. [DOI] [PubMed] [Google Scholar]
- 28.For leading refences, see: (ketone enolates)
- 28a.Yamada Y. M. A., Yoshikawa N., Sasai H., Shibasaki M., Angew. Chem. Int. Ed. Engl. 1997, 36, 1871–1873; [Google Scholar]; Angew. Chem. 1997, 109, 1942–1944. (N-acylimide enolates); [Google Scholar]
- 28b.Evans D. A., Downey C. W., Hubbs J. L., J. Am. Chem. Soc. 2003, 125, 8706–8707. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a.Alba A.-N. R., Rios R., Chem. Asian J. 2011, 6, 720–734; [DOI] [PubMed] [Google Scholar]
- 29b.Fisk J. S., Mosey R. A., Tepe J. J., Chem. Soc. Rev. 2007, 36, 1432–1440; [DOI] [PubMed] [Google Scholar]
- 29c.Hewlett N. M., Hupp C. D., Tepe J. J., Synthesis 2009, 2825–2839; [Google Scholar]
- 29d.Mosey R. A., Fisk J. S., Tepe J. J., Tetrahedron: Asymmetry 2008, 19, 2755–2762. [Google Scholar]
- 30.Mielgo A., Palomo C., Beilstein J. Org. Chem. 2016, 12, 918–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu S., Bao X., Wang B., Chem. Commun. 2018, 54, 11515–11529. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a.Jusseau X., Chabaud L., Guillou C., Tetrahedron 2014, 70, 2595–2615; [Google Scholar]
- 32b.Mao B., Fañanás-Mastral M., Feringa B. L., Chem. Rev. 2017, 117, 10502–10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Montagnon T., Kalaitzakis D., Sofiadis M., Vassilikogiannakis G., Org. Biomol. Chem. 2020, 18, 180–190. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a.Zhou F., Liu Y.-L., Zhou J., Adv. Synth. Catal. 2010, 352, 1381–1470; [Google Scholar]
- 34b.Shen K., Liu X., Lin L., Feng X., Chem. Sci. 2012, 3, 327–334; [Google Scholar]
- 34c.Dou X. W., Lu Y. X., Chem. Eur. J. 2012, 18, 8315–8319; [DOI] [PubMed] [Google Scholar]
- 34d.Rios R., Chem. Soc. Rev. 2012, 41, 1060–1074; [DOI] [PubMed] [Google Scholar]
- 34e.Hong L., Wang R., Adv. Synth. Catal. 2013, 355, 1023–1052; [Google Scholar]
- 34f.Chauhan P., Chimni S. S., Tetrahedron: Asymmetry 2013, 24, 343–356; [Google Scholar]
- 34g.Cheng D., Ishihara Y., Tan B., C. F.Barbas, III, ACS Catal. 2014, 4, 743–762; [Google Scholar]
- 34h.Dalpozzo R., Org. Chem. Front. 2017, 4, 2063–2078. [Google Scholar]
- 35.Also, see ref 6c.
- 36.For work involving α,α-dicyanoolefins, see:
- 36a.Cuia H. L. and Chen Y. C., Chem. Commun. 2009, 4479–4486; [DOI] [PubMed] [Google Scholar]
- 36b.Kaur J., Chauhana P., Chimni S. S., Org. Biomol. Chem. 2016, 14, 7832–7847. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a.Zhao D., Oisaki K., Kanai M., Shibasaki M., Tetrahedron Lett. 2006, 47, 1403–1407; [Google Scholar]
- 37b.Zhao D., Oisaki K., Kanai M., Shibasaki M., J. Am. Chem. Soc. 2006, 128, 14440–14441. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi A., Aoyama N., Matsunaga S., Shibasaki M., Org. Lett. 2007, 9, 3387–3390. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi A., Matsunaga S., Shibasaki M., J. Am. Chem. Soc. 2009, 131, 10842–10843. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H.-J., Yin L., J. Am. Chem. Soc. 2018, 140, 12270–12279; [DOI] [PubMed] [Google Scholar]; CorrectionJ. Am. Chem. Soc. 2018, 140, 14526. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a.Zhang H.-J., Shi C.-Y., Zhong F., Yin L., J. Am. Chem. Soc. 2017, 139, 2196–2199; For a related catalytic Mannich-type reaction involving phosphonate and sulfonate-derived allylic anions, see: [DOI] [PubMed] [Google Scholar]
- 41b.Yue W.-J., Zhang C.-Y., Yin L., iScience 2019, 14, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong F., Yue W.-J., Zhang H.-J., Zhang C.-Y., Yin L., J. Am. Chem. Soc. 2018, 140, 15170–15175. [DOI] [PubMed] [Google Scholar]
- 43.Fu X., Bai H.-Y., Zhu G.-D., Huang Y., Zhang S.-Y., Org. Lett. 2018, 20, 3469–3472. [DOI] [PubMed] [Google Scholar]
- 44.A Co-catalyzed enantioselective γ-amination of β,γ-unsaturated N-acylpyrazoles appeared after preparation of this manuscript: Fu X., Hao Y., Bai H.-Y., Duan A., Zhang S.-Y., Org. Lett. 2021, 23, 25–30. [DOI] [PubMed] [Google Scholar]
- 45.Lin S., Lin Z., J. Org. Chem. 2019, 84, 12399–12407. [DOI] [PubMed] [Google Scholar]
- 46.Cui J., Ohtsuki A., Watanabe T., Kumagai N., Shibasaki M., Chem. Eur. J. 2018, 24, 2598–2601. [DOI] [PubMed] [Google Scholar]
- 47.
- 47a.Takeuchi T., Kumagai N., Shibasaki M., J. Org. Chem. 2018, 83, 5851–5858. For a review on the use of nonheteroaromatic amides in asymmetric catalysis, see:; [DOI] [PubMed] [Google Scholar]
- 47b.Kumagai N., Shibasaki M., Chem. Eur. J. 2016, 22, 15192–15200. [DOI] [PubMed] [Google Scholar]
- 48.(Mannich)
- 48a.Yazaki R., Nitabaru T., Kumagai N., Shibasaki M., J. Am. Chem. Soc. 2008, 130, 14477–14479; (Aldol); [DOI] [PubMed] [Google Scholar]
- 48b.Yazaki R., Kumagai N., Shibasaki M., J. Am. Chem. Soc. 2009, 131, 3195–3197; [DOI] [PubMed] [Google Scholar]
- 48c.Yazaki R., Kumagai N., Shibasaki M., J. Am. Chem. Soc. 2010, 132, 5522–5531. [DOI] [PubMed] [Google Scholar]
- 49.For a Pd-catalyzed α-addition of allylic nitriles to aldimines in the presence of NaHCO3 in THF to afford racemic Mannich-type products, see: Aydin J., Szabo K. J., Org. Lett. 2008, 10, 2881–2884. [DOI] [PubMed] [Google Scholar]
- 50.Otsuka Y., Takada H., Yasuda S., Kumagai N., Shibasaki M., Chem. Asian J. 2013, 8, 354–358. [DOI] [PubMed] [Google Scholar]
- 51.
- 51a.Yamamoto Y., Hatsuya S., Yamada J., J. Chem. Soc. Chem. Commun. 1988, 86; [Google Scholar]
- 51b.Yamamoto Y., Hatsuya S., Yamada J., J. Org. Chem. 1990, 55, 3118–3128; [Google Scholar]
- 51c.Terao Y., Satoh T., Miura M., Nomura M., Tetrahedron Lett. 1998, 39, 6203–6206; [Google Scholar]
- 51d.Terao Y., Satoh T., Miura M., Nomura M., Bull. Chem. Soc. Jpn. 1999, 72, 2345–2350; [Google Scholar]
- 51e.Terao Y., Kametani Y., Wakui H., Satoh T., Miura M., Nomura M., Tetrahedron 2001, 57, 5967–5974. [Google Scholar]
- 52.Hyde A. M., Buchwald S. L., Angew. Chem. Int. Ed. 2008, 47, 177–180; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 183–186. [Google Scholar]
- 53.Kang T., Hou L., Ruan S., Cao W., Liu X., Feng X., Nat. Commun. 2020, 11, 3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.
- 54a.Trost B. M., Tracy J. S., Yusoontorn T., Hung C.-I. J., Angew. Chem. Int. Ed. 2020, 59, 2370–2374; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 2390–2394; [Google Scholar]
- 54b.Trost B. M., Hung C.-J., Gnanamani E., ACS Catal. 2019, 9, 1549–1557; Also, see:; [Google Scholar]
- 54c.Wang W., Dai J., Yang Q., Deng Y.-H., Peng F., Shao Z., Org. Lett. 2021, 23, 920–924. [DOI] [PubMed] [Google Scholar]
- 55.
- 55a.Guang J., Rout S., Bihani M., Larson A. J., Arman H. D., Zhao J. C.-G., Org. Lett. 2016, 18, 2648–2651; [DOI] [PubMed] [Google Scholar]
- 55b.Alonso D. A., Kitagaki S., Utsumi N., C. F.Barbas, III, Angew. Chem. Int. Ed. 2008, 47, 4588–4591; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4664–4667. [Google Scholar]
- 56.Kisanga P. B., Verkade J. G., Schwesinger R., J. Org. Chem. 2000, 65, 5431–5432. [DOI] [PubMed] [Google Scholar]
- 57.Kisanga P. B., Verkade J. G., J. Org. Chem. 2002, 67, 426–430. [DOI] [PubMed] [Google Scholar]
- 58.Quintard A., Rodriguez J., Org. Lett. 2017, 19, 722–725. [DOI] [PubMed] [Google Scholar]
- 59.Wang J., Chen J., Kee C. W., Tan C.-H., Angew. Chem. Int. Ed. 2012, 51, 2382–2386; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2432–23436. [Google Scholar]
- 60.Tan B., Hernández-Torres G., Barbas C. F. III, Angew. Chem. Int. Ed. 2012, 51, 5381–5385; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5477–5481. [Google Scholar]
- 61.Li T.-Z., Jiang Y., Guan Y.-Q., Sha F., Wu X.-Y., Chem. Commun. 2014, 50, 10790–10792. [DOI] [PubMed] [Google Scholar]
- 62.Qin J., Zhang Y., Liu C., Zhou J., Zhan R., Chen W., Huang H., Org. Lett. 2019, 21, 7337–7341. [DOI] [PubMed] [Google Scholar]
- 63.Cen Y., Wang Y., Wang S., Ma Y.-Y., Zhao D.-G., Zhan R., Huang H., Org. Lett. 2020, 22, 7135–7140. [DOI] [PubMed] [Google Scholar]
- 64.Zhu B., Zhang W., Lee R., Han Z., Yang W., Tan D., Huang K.-W., Jiang Z., Angew. Chem. Int. Ed. 2013, 52, 6666–6670; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6798–6802. [Google Scholar]
- 65.Jing Z., Bai X., Chen W., Zhang G., Zhu B., Jiang Z., Org. Lett. 2016, 18, 260–263. [DOI] [PubMed] [Google Scholar]
- 66.Ray B., Mukherjee S., J. Org. Chem. 2018, 83, 10871–10880. [DOI] [PubMed] [Google Scholar]
- 67.Akula P. S., Hong B.-C., Lee G.-H., Org. Lett. 2018, 20, 7835–7839. [DOI] [PubMed] [Google Scholar]
- 68.
- 68a.Li X., Kong X., Yang S., Meng M., Zhan X., Zeng M., Fang X., Org. Lett. 2019, 21, 1979–1983. For a related inverse-electron-demand oxa-Diels-Alder reaction of allylic ketones with isatin-derived unsaturated keto esters, see;30865466 [Google Scholar]
- 68b.Lin Y., Hou X.-Q., Li B.-Y., Du D−M., Adv. Synth. Catal. 2020, 362, 5728–5735. [Google Scholar]
- 69.
- 69a.Frankowski S., Skrzyńska A., Sieroń L., Albrecht Ł., Adv. Synth. Catal. 2020, 362, 2658–2665; For a formal [6+2] cycloaddition involving dienolate initiated tandem addition/cyclisation between allylic ketones (1,4-naphthoquinones) and dicyanoheptafulvene that proceed with poor enantioselectivity, see: [Google Scholar]
- 69b.Romaniszyn M., Gronowska K., Albrecht Ł., J. Org. Chem. 2019, 84, 9929–9936. [DOI] [PubMed] [Google Scholar]
- 70.
- 70a.Iriarte I., Olaizola O., Vera S., Gamboa I., Oiarbide M., Palomo C., Angew. Chem. Int. Ed. Ed. 2017, 56, 8860–8864; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8986–8990; [Google Scholar]
- 70b.Olaizola I., Campano T. E., Iriarte I., Vera S., Mielgo A., García J. M., Odriozola J. M., Oiarbide M., Palomo C., Chem. Eur. J. 2018, 24, 3893–3901. [DOI] [PubMed] [Google Scholar]
- 71.Cano R., Zakarian A., McGlacken G. P., Angew. Chem. Int. Ed. 2017, 56, 9278–9290; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9406–9418. [Google Scholar]
- 72.Campano T. E., Iriarte I., Olaizola O., Etxabe J., Mielgo A., Ganboa I., Odriozola J. M., García J. M., Oiarbide M., Palomo C., Chem. Eur. J. 2019, 25, 4390–4397. [DOI] [PubMed] [Google Scholar]
- 73.Qiao B., Huang Y.-J., Nie J., Ma J.-A., Org. Lett. 2015, 17, 4608–4611. [DOI] [PubMed] [Google Scholar]
- 74.Urruzuno I., Mugica O., Oiarbide M., Palomo C., Angew. Chem. Int. Ed. 2017, 56, 2059–2063; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2091–2095. [Google Scholar]
- 75.Urruzuno I., Mugica O., Zanella G., Vera S., Gómez-Bengoa E., Oiarbide M., Palomo C., Chem. Eur. J. 2019, 25, 9701–9709. [DOI] [PubMed] [Google Scholar]
- 76.Kong X., Song J., Liu J., Meng M., Yang S., Zeng M., Zhan X., Li C., Fang X., Chem. Commun. 2018, 54, 4266–4269. [DOI] [PubMed] [Google Scholar]
- 77.Golec J. C., Carter E. M., Ward J. W., Whittingham W. G., Simón L., Paton R. S., Dixon D. J., Angew. Chem. Int. Ed. 2020, 59, 17417–17422; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 17570–17575. [Google Scholar]
- 78.Gu Y., Wang Y., Yu T.-Y., Liang Y.-M., Xu P.-F., Angew. Chem. Int. Ed. 2014, 53, 14128–14131; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14352–14355. [Google Scholar]
- 79.Jia Z.-L., Wang Y., Zhao C.-G., Zhang X.-H., Xu P.-F., Org. Lett. 2017, 19, 2130–2133. [DOI] [PubMed] [Google Scholar]
- 80.Xie J.-K., Wang Y., Lin J.-B., Ren X.-R., Xu P.-F., Chem. Eur. J. 2017, 23, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 81.
- 81a.Wang Y., Lin J.-B., Xie J.-K., Lu H., Hu X.-Q., Xu P.-F., Org. Lett. 2018, 20, 5835–5839; [DOI] [PubMed] [Google Scholar]
- 81b.Majee D., Jakkampudi S., Arman H. D., Zhao J. C.-G., Org. Lett. 2019, 21, 9166–9170. [DOI] [PubMed] [Google Scholar]
- 82.Liu Y., Yang Y., Huang Y., Xu X.-H., Qing F.-L., Synlett 2015, 26, 67–72. [Google Scholar]
- 83.Xiao X., Mei H., Chen Q., Zhao X., Lin L., Liu X., Feng X., Chem. Commun. 2015, 51, 580–583. [DOI] [PubMed] [Google Scholar]
- 84.Gao T. P., Liu D., Lin J.-B., Hu X.-Q., Wang Z.-Y., Xu P.-F., Org. Chem. Front. 2016, 3, 598–602. [Google Scholar]
- 85.Joshi H., Yadav A., Das A., Singh V. K., J. Org. Chem. 2020, 85, 3202–3212. [DOI] [PubMed] [Google Scholar]
- 86.The literature covering asymmetric transformations triggered by tertiary amine/H-bonding bifunctional catalysts is plagued of TS proposals that fit to either coordination geometry, often termed as Takemoto's and Pápai's models. Alternative ways of how the various H-bond donors in the catalyst interact with basic sites on the nucleophile and electrophile have also been reported. It is unfortunate that, too often, such proposals lack due theoretical or experimental support. For more detailed discussion, see: Grayson M. N., Houk K. N., J. Am. Chem. Soc. 2016, 138, 9041–9044. [Google Scholar]
- 87.Zhu B., Lu B., Zhang H., Xu X., Jiang Z., Chang J., Org. Lett. 2019, 21, 3271–3275. [DOI] [PubMed] [Google Scholar]
- 88.Zhao S., Zhao Y.-Y., Lin J.-B., Xie T., Liang Y.-M., Xu P.-F., Org. Lett. 2015, 17, 3206–3209. [DOI] [PubMed] [Google Scholar]
- 89.
- 89a.Curti C., Rassu G., Zambrano V., Pinna L., Pelosi G., Sartori A., Battistini L., Zanardi F., Casiraghi G., Angew. Chem. Int. Ed. 2012, 51, 6200–6204; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6304–6308; [Google Scholar]
- 89b.Rassu G., Zambrano V., Pinna L., Curti C., Battistini L., Sartori A., Pelosi G., Zanardi F., Casiraghi G., Adv. Synth. Catal. 2013, 355, 1881–1886. [Google Scholar]
- 90.Curti C., Battistini L., Sartori A., Rassu G., Pelosi G., Lombardo M., Zanardi F., Adv. Synth. Catal. 2018, 360, 711–721. [Google Scholar]
- 91.
- 91a.Chen Q., Wang G., Jiang X., Xu Z., Lin L., Wang R., Org. Lett. 2014, 16, 1394–1397; [DOI] [PubMed] [Google Scholar]
- 91b.Zhong Y., Ma S., Xu Z., Changa M., Wang R., RSC Adv. 2014, 4, 49930–49933. [Google Scholar]
- 92.Feng J., Li X., Cheng J.-P., Chem. Commun. 2015, 51, 14342–14345. [DOI] [PubMed] [Google Scholar]
- 93.Jaiswal M. K., Kumar R., Singh S., Jain S., Vanka K., Singh R. P., Eur. J. Org. Chem. 2020, 5690–5694. [Google Scholar]
- 94.Di Iorio N., Righi P., Ranieri S., Mazzanti A., Margutta R. G., Bencivenni G., J. Org. Chem. 2015, 80, 7158–7171. [DOI] [PubMed] [Google Scholar]
- 95.Bai X., Zeng G., Shao T., Jiang Z., Angew. Chem. Int. Ed. 2017, 56, 3684–3688; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3738–3742. [Google Scholar]
- 96.Wang S.-Q., Liu Z.-C., Yue W.-J., Yin L., Angew. Chem. Int. Ed. 2021, 60, 4604–4608; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 4654–4658. [Google Scholar]
- 97.Olaizola O., Iriarte I., Zanella G., Gómez-Bengoa E., Ganboa I., Oiarbide M., Palomo C., Angew. Chem. Int. Ed. 2019, 58, 14250–14254; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14388–14392. [Google Scholar]
- 98.Skrzyńska A., Romaniszyn M., Pomikło D., Albrecht Ł., J. Org. Chem. 2018, 83, 5019–5026. [DOI] [PubMed] [Google Scholar]
- 99.Luo G., Huang Z., Zhuo S., Mou C., Wu J., Jin Z., Chi Y. R., Angew. Chem. Int. Ed. 2019, 58, 17189–17193; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17349–17353. [Google Scholar]
- 100.Feng J., Li X., Cheng J.-P., J. Org. Chem. 2017, 82, 1412–1419. [DOI] [PubMed] [Google Scholar]
- 101.Dell'Amico L., Rassu G., Zambrano V., Sartori A., Curti C., Battistini L., Pelosi G., Casiraghi G., Zanardi F., J. Am. Chem. Soc. 2014, 136, 11107–11114. [DOI] [PubMed] [Google Scholar]
- 102.Tong G., Zhu B., Lee R., Yang W., Tan D., Yang C., Han Z., Yan L., Huang K.-W., Jiang Z., J. Org. Chem. 2013, 78, 5067–5072. [DOI] [PubMed] [Google Scholar]
- 103.Jiang L., Lei Q., Huang X., Cui H.-L., Zhou X., Chen Y.-C., Chem. Eur. J. 2011, 17, 9489–9493. [DOI] [PubMed] [Google Scholar]
- 104.Kowalczyk-Dworak D., Kwit M., Albrecht Ł., J. Org. Chem. 2020, 85, 2938–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shi C.-Y., Xiao J.-Z., Yin L., Chem. Commun. 2018, 54, 11957–11960. [DOI] [PubMed] [Google Scholar]
- 106.Ran G.-Y., Yang X.-X., Yue J.-F., Du W., Chen Y.-C., Angew. Chem. Int. Ed. 2019, 58, 9210–9214; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9308–9312. [Google Scholar]
- 107.Yang L.-C., Tan Z. Y., Rong Z.-Q., Liu R., Wang Y.-N., Zhao Y., Angew. Chem. Int. Ed. 2018, 57, 7860–7864; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7986–7990. [Google Scholar]
- 108.Wang G., Liu X., Chen Y., Yang J., Li J., Lin L., Feng X., ACS Catal. 2016, 6, 2482–2486. [Google Scholar]
- 109.Zhong F., Xue Q.-Y., Yin L., Angew. Chem. Int. Ed. 2020, 59, 1562–1566; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1578–1582. [Google Scholar]
- 110.Horwitz M. A., Zavesky B. P., Martinez-Alvarado J. I., Johnson J. S., Org. Lett. 2016, 18, 36–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.
- 111a.Zhan G., Shi M.-L., Lin W.-J., Ouyang Q., Du W., Chen Y.-C., Chem. Eur. J. 2017, 23, 6286–6289; [DOI] [PubMed] [Google Scholar]
- 111b.Shi M., Zhan G., Du W., Chen Y., Acta Chim. Sin. 2017, 75, 998–1002; [Google Scholar]
- 111c.Sun Q., Li X., Su J., Zhao L., Ma M., Zhu Y., Zhao Y., Zhu R., Yan W., Wang K., Wang R., Adv. Synth. Catal. 2015, 357, 3187–3196. [Google Scholar]
- 112.Feng K.-N., Yang Y.-L., Xu Y.-X., Zhang Y., Feng T., Huang S.-X., Liu J.-K., Zeng Y., Angew. Chem. Int. Ed. 2020, 59, 7209–7213; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7276–7280. [Google Scholar]
- 113.
- 113a.Li Y., Luxenburger E., Müller R., Angew. Chem. Int. Ed. 2013, 52, 1304–1308; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1342–1346; [Google Scholar]
- 113b.Sheng X., Himo F., Angew. Chem. Int. Ed. 2020, 59, 22973–22977; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2020, 132, 23173–23177. [Google Scholar]
- 114.Note added in proof. The following papers on asymmetric catalytic transformations involving dienol(ate) intermediates appeared after preparation of this manuscript: (Metal catalysis)
- 114a.Li D., Zhang M., Yang Y., Peng T., Yang D., Gao W., Wang R., Org. Lett. 2020, 22, 9229–9233; [DOI] [PubMed] [Google Scholar]
- 114b.Tian J.-J., Liu N., Liu Q.-F., Sun W., Wang X.-C., J. Am. Chem. Soc. 2021, 143, 3054–3059. (Organocatalysis); [DOI] [PubMed] [Google Scholar]
- 114c.Jaiswal M. K., Kumar R., Singh S., Jain S., Vanka K., Singh R. P., Eur. J. Org. Chem. 2020, 5690–5694; [Google Scholar]
- 114d.Hayashi Y., Suga Y., Umekubo N., Org. Lett. 2020, 22, 8603–8607; [DOI] [PubMed] [Google Scholar]
- 114e.Wittmann S., Martzel T., Truong C. T. P., Toffano M., Oudeyer S., Guillot R., Bournaud C., Gandon V., Brière J.-F., Vo-Thanh G., Angew. Chem. Int. Ed. 2021, 60, 11110–11114; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 11210–11214. [Google Scholar]