

Abstract
Objective
Tofacitinib is an oral JAK inhibitor for the treatment of rheumatoid arthritis (RA), psoriatic arthritis, and ulcerative colitis, and has been previously investigated for psoriasis (PsO). This meta‐analysis of genome‐wide association studies (GWAS) was performed to identify genetic factors associated with increased risk/faster onset of herpes zoster (HZ) in subjects with RA or PsO receiving tofacitinib treatment, and to determine potential mechanisms that could be attributed to the varying rates of HZ across ethnicities.
Methods
In an ethnicity/indication‐specific, trans‐ethnic, trans‐population meta‐analysis of GWAS in subjects with RA or PsO from phase II, phase III, and long‐term extension studies of tofacitinib, 8 million genetic variants were evaluated for their potential association with time to an HZ event and incidence of an HZ event (case versus control) with tofacitinib treatment, using Cox proportional hazard and logistic regression analyses, respectively.
Results
In total, 5,246 subjects were included (3,168 with RA and 2,078 with PsO). After adjustment for age, baseline absolute lymphocyte count, genetically defined ethnicity, and concomitant methotrexate use (in RA subjects only), 4 loci were significantly associated with faster onset of HZ in European subjects (P < 5 × 10−8), including a single‐nucleotide polymorphism (SNP) near CD83 (frequency of risk allele ~2% in European subjects versus ~0.1% in East Asian subjects). In the trans‐ethnic, trans‐population meta‐analysis, the CD83 SNP remained significant. Four additional significant loci were identified in the meta‐analysis, among which a SNP near IL17RB was associated with faster onset of HZ (meta‐analysis hazard ratio 3.6 [95% confidence interval 2.40–5.44], P = 7.6 × 10−10; frequency of risk allele ~12% in East Asian subjects versus <0.2% in European subjects).
Conclusion
Genetic analysis of tofacitinib‐treated subjects with RA or PsO identified multiple loci associated with increased HZ risk. Prevalent variants near the immune‐relevant genes CD83 and IL17RB in European and East Asian populations, respectively, may contribute to risk of HZ in tofacitinib‐treated subjects.
INTRODUCTION
Tofacitinib is an oral JAK inhibitor for the treatment of rheumatoid arthritis (RA), psoriatic arthritis (PsA), and ulcerative colitis (UC), and has been previously investigated for psoriasis (PsO). Tofacitinib is an orally bioavailable small molecule whose inhibitory activity involves blockade of the ATP binding site (1). In cellular settings where the various JAKs signal in combination, tofacitinib preferentially inhibits signaling by heterodimeric receptors associated with JAK1 and/or JAK3, and has functional selectivity over JAK2 (1). The efficacy and safety of tofacitinib have been studied across multiple immune‐mediated inflammatory diseases, including RA (2, 3, 4, 5, 6, 7) and PsO (8, 9, 10, 11).
The safety profile of tofacitinib in subjects with RA or PsO is generally similar to that of tumor necrosis factor inhibitors and other biologic disease‐modifying antirheumatic drugs (bDMARDs), with the exception of herpes zoster (HZ) rates (12, 13, 14, 15). HZ risk is elevated in subjects with RA in comparison to the general population (16), and the risk is further increased in tofacitinib‐treated subjects (17), although multidermatomal or disseminated HZ cases have been infrequent (8% of HZ cases) in subjects receiving tofacitinib (13). This appears to be a class‐specific effect, because use of other JAK inhibitors targeting JAK1 or JAK1/JAK2 has resulted in an increased risk of HZ (18). HZ risk in tofacitinib‐treated subjects with RA increases with age, glucocorticoid use, tofacitinib dose, and enrollment within Asia (e.g., subjects from Japan and Korea have 2–3‐fold higher rates of HZ versus those from other regions) (19). Similarly, in tofacitinib‐treated subjects with PsO, HZ risk increases with age, tofacitinib dose, and Asian descent, and also prior bDMARD use (20). Subjects with UC and those with PsA receiving tofacitinib also experience higher rates of HZ when compared with subjects who have not been treated with tofacitinib (21, 22, 23). The higher HZ rates in Asian subjects observed in the RA and PsO studies (17, 20) could be attributable to multiple factors, including ascertainment bias, prevalence of a genetic clade of virus prone to reactivation, enhanced response to tofacitinib, or an interaction between JAK inhibition and a genetic polymorphism more common in Japan and Korea.
Genetic studies have identified variations in the HLA region as being associated with risk of HZ (24). We hypothesized that genetic factors may be associated with tofacitinib‐related HZ, and that the genetic variation across ethnicities may contribute to the variance in HZ rates. Identifying such genetic factors could help reveal the mechanisms of, and hence the risk of, varicella zoster virus (VZV) reactivation related to tofacitinib. We therefore conducted a genome‐wide trans‐ancestry meta‐analysis of HZ using DNA samples from RA and PsO subjects receiving treatment with tofacitinib in clinical studies. Furthermore, to understand the mechanism of an HZ‐associated variant near IL17RB, we correlated the allele count and the expression of candidate genes in immune cell types via an expression quantitative trait loci (eQTL) analysis.
PATIENTS AND METHODS
Population
This analysis included subjects with RA or PsO from phase II and phase III index tofacitinib studies (ClinicalTrials.gov identifiers NCT00413660, NCT00550446, NCT00603512, NCT00687193, NCT01059864, NCT00960440, NCT00847613, NCT00814307, NCT00856544, NCT00853385, NCT01039668, NCT00678210, NCT01276639, NCT01309737, NCT01241591, NCT01186744, and NCT01519089) and the corresponding long‐term extension (LTE) studies (ClinicalTrials.gov identifiers NCT00413699, NCT00661661, and NCT01163253) (for more details, see Supplementary Table 1 available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract). All subjects provided written informed consent.
Blood samples for genetic studies were genotyped, and passed sample quality control (QC). HZ events from both index and LTE studies were included in the analysis (data cutoff: April 2014).
Genotyping, imputation, and data QC
Germline DNA was extracted from peripheral blood. Single‐nucleotide polymorphism (SNP) data were generated using Illumina Human OmniExpress Plus Exome genome‐wide arrays, versions 1–4 (https://www.illumina.com/products/by‐type/microarray‐kits/infinium‐omni‐express‐exome.html). The genotype calls were conducted through GenomeStudio by Illumina. SNPs were imputed using IMPUTE2 (25), using reference panels from the 1000 Genomes Project phase I integrated variant set. Subjects who failed the sex match based on self‐reported sex or those who failed the heterozygosity check or relatedness test were excluded from the downstream analysis. Furthermore, SNPs that were estimated to have poor imputation performance (quality score <0.9) were removed from the analysis.
Up to 8 million autosomal SNPs were imputed. The allelic dosage of each genetic variant, ranging from 0 to 2 and calculated from posterior genotype probabilities from IMPUTE2, was used in each statistical model. As an additional QC step, allele frequencies of variants that produced the strongest association signals were compared with those reported in the gnomAD database (https://gnomad.broadinstitute.org/).
To determine the genetic ancestry of all subjects, we performed a principal components analysis (PCA) using EIGENSTRAT. Prior to PCA, the study data were combined with data from the 1000 Genomes Project. Independent autosomal SNPs across the genome were selected after pruning, and chromosomal regions known to be associated with ethnicity were also removed before running SmartPCA. Empiric ancestry groups were then determined based on the distribution over the first 2 principal components in each self‐reported population, using clinical data. Subjects who were more than 6 standard deviations from either of the 2 first principal components were removed in the final statistical analyses.
End points and analyses of associations
Two end points were evaluated: 1) time to HZ event with tofacitinib treatment, defined as the interval between the first tofacitinib treatment in either the index or the LTE studies and the earliest HZ event; and 2) numbers of HZ cases versus controls, in which HZ cases were subjects with investigator‐reported HZ, and controls were subjects who received tofacitinib in the index or LTE studies and did not develop HZ during the study observation period. Cox proportional hazard and logistic regression analyses were used for assessing associations with the time to HZ event and incidence of an HZ event (cases versus controls), respectively. R version 3.2 software was used for the statistical analyses.
Covariates
A set of baseline clinical variables that are known to, or could potentially, affect the rate of HZ were evaluated for inclusion as covariates in the analysis model. The covariates considered in RA studies included age (in years), sex, baseline weight, baseline rheumatoid factor status, baseline RA severity based on the Disease Activity Score in 28 joints (26), erythrocyte sedimentation rate, RA duration, baseline absolute lymphocyte count (ALC), baseline neutrophil count, glucocorticoid use, and concomitant methotrexate use. The covariates considered in PsO studies included age (in years), sex, baseline weight, baseline ALC, baseline neutrophil count, PsO duration, presence versus absence of PsA at baseline, and proportion of subjects achieving a 75% decrease in the Psoriasis Area and Severity Index (27) at week 12 or week 16 (depending on the trial). Tofacitinib dose was not included as a covariate because of the potential for dose switching in the LTE studies.
Covariates were selected via stepwise variable selection, using a P value cutoff of 0.05. In this analysis, age, baseline ALC, genetic population stratification, and concomitant methotrexate use (in RA subjects only) were included as covariates in the association test. Additionally, the first 3 principal components defined by genetic data within each ancestry subgroup were included in the analysis model.
Ancestry‐specific and trans‐ancestry genome‐wide association study (GWAS) meta‐analyses
Genetic ancestry subgroups of subjects (European, East Asian, South Asian, Hispanic, and Black) were defined as those clustering in principal component space, as estimated from genome‐wide genotype data in combination with self‐reported ethnicities. Ancestry‐specific GWAS were performed for European, East Asian, and Hispanic subgroups. The sample sizes for the Black and South Asian populations were small; these subgroups were therefore excluded from the GWAS.
Each SNP with a minor allele frequency (MAF) of >2% in each ethnicity subgroup within either the RA or PsO populations was tested for association, under an additive model with adjustment for covariates. A meta‐analysis across the ancestry subgroups and populations was conducted via a fixed effects model, with significant association defined as P values less than or equal to 5 × 10−8. The meta‐analysis included any SNPs with an MAF of >2% in at least 1 ethnicity subgroup; rarer alleles were not included, as the sample size would not be expected to provide adequate power for the risk estimate. Trans‐ethnicity allelic heterogeneity was assessed with Cochran’s Q test using the meta‐analysis random effects model, with a statistically significant level defined, using the conservative Bonferroni correction, as 0.005 (0.05 divided by 10). In an additional analysis, we restricted the meta‐analysis to variants with an MAF of >2% in all ethnic subgroups. Significant loci were labeled according to the gene nearest to the lead SNP, unless a compelling biologic candidate was mapped nearby. The overall design of the trans‐ancestry and trans‐population GWAS meta‐analysis is illustrated in Figure 1.
Figure 1.
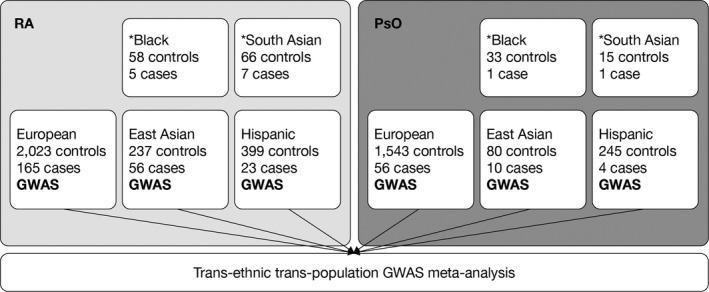
Overall design of the trans‐ancestry and trans‐population genome‐wide association study (GWAS) meta‐analysis in subjects with rheumatoid arthritis (RA) or psoriasis (PsO). *Black and South Asian subgroups were excluded from the GWAS meta‐analysis due to small sample sizes.
Assessment of associations between genetic variants and proportions of CD4+ T cell subtypes
To identify the role of genetic variants in regulating immune phenotypes, flow cytometry was performed on freshly isolated peripheral blood mononuclear cells (PBMCs) from 82 healthy Japanese individuals (see Supplementary Methods, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract).
Standardized human immunophenotyping was performed to classify CD4+ T cells into conventional Th1, Th2, Th17, and Treg cell types. Association of the genetic variants with the proportions of these CD4+ T cell subtypes was evaluated using an additive genetic model via linear regression analysis. In this analysis, only the association of the candidate SNP on CD4+ T cell subtypes was reported. Significance of the associations was defined as a P value cutoff of 0.05.
T cell subtype–specific eQTL analysis
Blood samples were collected from 29 healthy Japanese individuals. Naive CD4+ T cells from these individuals were collected via fluorescence‐activated cell sorting. These cells were cultured for 72 hours and differentiated into T cell subtypes via stimulation of CD3/CD28 (for Th0 cells), CD3/CD28 plus interferon‐γ (IFNγ) (for Th1 cells), CD3/CD28 plus interleukin‐4 (IL‐4) (for Th2 cells), CD3/CD28 plus IL‐1β plus IL‐6 plus IL‐23 plus transforming growth factor β (TGFβ) (for Th17 cells), or CD3/CD28 plus IL‐2 plus TGFβ plus all‐trans‐retinoic acid (for Treg cells). Gene expression of each cell type was measured using RNA sequencing with Illumina HiSeq 2000. Genotyping was conducted via Infinium OmniExpressExome BeadChips. Gene expression levels were quantified using Hisat2 (28) and HTSeq (29) using the GENCODE annotation (version 25), followed by normalization using probabilistic estimation of expression residuals (30, 31); the residuals were further treated by quantile normalization, and each gene expression value was then rank‐transformed to fit normal distribution. The association between variants and normalized expression values was analyzed using linear regression with an additive effects model. Within this analysis, only the eQTLs of the candidate SNP on the candidate gene are reported. Significant association was defined as a P value cutoff of 0.05.
The studies involving blood samples from healthy Japanese individuals were approved by the Ethics Committees of RIKEN and the University of Tokyo. Written informed consent was obtained from each volunteer.
RESULTS
Subjects
Overall, 9,640 subjects with RA or PsO were recruited in the 17 phase II, phase III, and LTE studies (Supplementary Table 1 [http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract]). DNA samples were collected from 5,605 subjects; 5,246 subjects (3,168 RA subjects and 2,078 PsO subjects) remained in the genetic studies following sample QC. A total of 5,027 subjects received ≥1 dose of tofacitinib in the index or LTE studies, and thus were retained in the analysis. The other 219 subjects were initially included in the placebo or comparator arms in the index studies and were not switched to tofacitinib in the LTE studies.
Of the tofacitinib‐treated subjects, 328 cases of HZ were reported (256 cases among RA subjects and 72 cases among PsO subjects). The numbers of subjects within each genetic ancestry subgroup were as follows: 3,787 European (75.3%), 671 Hispanic (13.3%), 383 East Asian (7.6%), 97 Black (1.9%), and 89 South Asian (1.8%) (Figure 1 and Supplementary Table 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract). The HZ rates and distribution of demographic and clinical characteristics of the subjects in this genotyped cohort were consistent with those in the overall trial populations (see details in Supplementary Tables 3 and 4, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract).
Identification of 4 genetic loci associated with increased HZ risk in European ancestry GWAS
Ancestry‐specific GWAS were performed in European, Hispanic, and East Asian ethnicity subgroups within the RA or PsO populations (Figure 1 and Supplementary Table 2 [http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract]). European ancestry–specific GWAS identified 4 loci (1 in RA subjects and 3 in PsO subjects) that were significantly associated with a faster time to an HZ event (P < 5 × 10−8) in tofacitinib‐treated subjects (Table 1 and Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract).
Table 1.
Genetic loci found to be associated with increased HZ risk in the European ancestry GWAS*
| Allele | Time to HZ event† |
Incidence of HZ (case versus control)‡ |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Disease, ethnicity | Locus | Chr |
Reference SNP ID |
Position |
Reference allele |
Alternative allele |
Alternative allele frequency |
HR (95% CI) | P | OR (95% CI) | P |
| RA | |||||||||||
| European | PMEPA1 | 20 | rs59967896 | 56216698 | T | TCAA | 0.03 | 3.75 (2.46–5.71) | 8.34 × 10−10 | 4.1 (2.51–6.69) | 2.34 × 10−7 |
| PsO | |||||||||||
| European | CD83 | 6 | rs112817503 | 14292820 | C | T | 0.03 | 5.74 (3.38–9.77) | 1.14 × 10−10 | 7.71 (4.02–14.8) | 6.25 × 10−8 |
| European | UGDH | 4 | rs150665541 |
39536523 |
C | T | 0.03 | 4.86 (2.79–8.45) | 2.10 × 10−8 | 5.46 (2.88–10.3) | 3.57 × 10−6 |
| European | VWF | 12 | rs200638456 |
6102826 |
T | TAC | 0.07 | 3.5 (2.25–5.46) | 2.94 × 10−8 | 4.04 (2.42–6.75) | 1.08 × 10−6 |
GWAS = genome‐wide association study; Chr = chromosome; SNP ID = single‐nucleotide polymorphism cluster identification; HR = hazard ratio; 95% CI = 95% confidence interval; OR = odds ratio; RA = rheumatoid arthritis; PsO = psoriasis.
Results of the Kaplan‐Meier analyses of time to herpes zoster (HZ) event are presented in Supplementary Figure 4 (available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract).
C‐statistics for the case versus control logistic regression model are presented in Supplementary Table 5 (available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract).
In the European RA population, the variant rs59967896, located on chromosome 20 within 6.7 kb 3′ of the prostate transmembrane protein androgen induced 1 (PMEPA1) locus, was significantly associated with time to an HZ event (hazard ratio [HR] 3.8, P = 8.3 × 10−10) and showed a marginal association (odds ratio [OR] 4.1, P = 2.3 × 10−7) in the HZ case versus control analysis. The variant rs59967896 had an alternative allele with a “CAA” insertion that, to our knowledge, had no reported functions (see Supplementary Figure 2A, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract). The PMEPA1 locus showed no associations with the HZ end points in the European PsO population, nor were there any associations evident in the Hispanic and East Asian populations of either RA or PsO subjects.
In the European PsO population, 3 genetic loci at CD83 (rs112817503), UGDH (rs150665541), and VWF (rs200638456) were associated with faster time to HZ (Supplementary Figures 2B–D [http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract]). SNP variant rs112817503 was associated with a faster time to onset of HZ (HR 5.7, P = 1.4 × 10−10) and increased risk of occurrence of an HZ event (OR 7.7, P = 6.3 × 10−8). CD83 was the closest coding gene to rs112817503, which was 155 kb away. Variant rs150665541 was associated with a faster time to onset of HZ (HR 4.9, P = 2.1 × 10−8) and increased risk of occurrence of an HZ event (OR 5.5, P = 3.6 × 10−6); it was located in the second intron of UGDH. Variant rs200638456 was associated with a faster time to onset of HZ (HR 3.5, P = 2.9 × 10−8) and increased risk of occurrence of an HZ event (OR 4.0, P = 1.1 × 10−6). The rs200638456 variant was located within an intronic region of VWF, with a repeated sequence of the dinucleotide “AC.” The alternative allele of rs200638456 had an additional insertion of the dinucleotide “AC.”
In the Hispanic and East Asian ancestry subgroups of RA and PsO subjects, GWAS analysis did not reveal any significant results (at the threshold of P < 5 × 10−8), likely because the sample sizes were modest. Other meta‐analyses across ethnicity subgroups and populations could reveal additional loci, especially those with consistent effects across these subgroups.
Identification of 4 additional genetic loci associated with increased HZ risk in trans‐ancestry and trans‐population GWAS meta‐analyses
A meta‐analysis of the ancestry‐ and population‐specific GWAS identified SNPs at 5 loci achieving genome‐wide significance (combined meta‐analysis P < 5 × 10−8) in the HZ case versus control analysis and/or in the time to HZ event analysis (Table 2 and Figure 2; see also Supplementary Figure 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract). These 5 loci included IL17RB, CD83, GPR141, TOX3, and ACSF3/CDH15. The strength of the genetic association with time to an HZ event and incidence of an HZ event (case versus control) for these loci and the ancestry/population‐specific effects of the top variants in the loci are presented in Supplementary Table 6 (available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract).
Table 2.
Genetic loci achieving genome‐wide significance in either the time to HZ event or HZ case versus control trans‐ancestry and trans‐population GWAS meta‐analyses*
| Allele | Time to HZ event |
Incidence of HZ (case versus control) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Genetic variant | Chr | Position |
Reference allele |
Alternative allele |
Allele frequency† |
HR (95% CI) | P | OR (95% CI) | P |
| IL17RB | rs58861611 | 3 | 54118714 | T | C | 0.093 | 3.6 (2.40–5.44) | 7.6 × 10−10 | 3.8 (2.17–6.67) | 3.0 × 10−6 |
| CD83 | rs112817503 | 6 | 14292820 | T | C | 0.022 | 3.6 (2.43–5.32) | 1.5 × 10−10 | 3.7 (2.35–5.87) | 2.1 × 10−8 |
| GPR141 | rs56114331 | 7 | 37802731 | C | T | 0.019 | 3.7 (2.37–5.73) | 6.3 × 10−9 | 3.4 (2.05–5.49) | 1.5 × 10−6 |
| TOX3 | rs79025327 | 16 | 52505167 | G | A | 0.072 | 2.9 (2.04–4.22) | 6.4 × 10−9 | 3.8 (2.28–6.23) | 2.2 × 10−7 |
| ACSF3 | rs142820005 | 16 | 89211411 | T | A | 0.055 | 2.3 (1.72–3.09) | 2.3 × 10−8 | 2.5 (1.81–3.45) | 2.7 × 10−8 |
The threshold for genome‐wide significance was defined as P < 5 × 10−8. HZ = herpes zoster; GWAS = genome‐wide association study; Chr = chromosome; HR = hazard ratio; 95% CI = 95% confidence interval; OR = odds ratio.
Allele frequency was calculated based on weighted allele frequency in each ancestry group by the standard deviation of the association effects.
Figure 2.
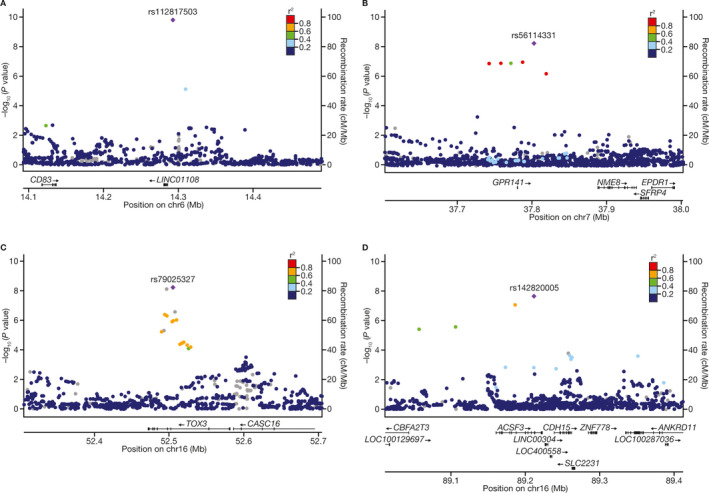
Regional association plots assessing the association of time to herpes zoster event with 4 genetic loci, at CD83 (A), GPR141 (B), TOX3 (C), and ACSF3 (D), in subjects with rheumatoid arthritis or psoriasis.
The association of CD83 was driven by the significant association observed in the European subgroup, as reported above. The frequency of the CD83 locus variant was lower in Hispanic subjects (~1%) than in European subjects (~2%), and was much rarer in East Asian subjects (~0.1%). The genetic effects in East Asian subjects could not be accurately estimated, due to the extremely low variant frequency. The trans‐ethnic and trans‐population meta‐analysis did not improve the significance levels for the CD83 variant in the European PsO population. Top variant rs56114331 in the GPR141 locus had a low allele frequency (1.7–2.1%) in Europeans, but was nevertheless higher than that in East Asian or Hispanic subjects (<1%). The significance of the GPR141 locus identified by meta‐analysis was mainly driven by the significant association in the European population of PsO subjects, although the sample size of the European population was not large enough to show significance in the European ancestry GWAS. Top variant rs79025327 in the TOX3 locus had a higher allele frequency in East Asian subjects (7–11%) compared with European or Hispanic subjects (~1–2%). The significant association of the TOX3 locus was mostly driven by the significant associations observed in the East Asian population of RA subjects. The ACSF3/CDH15 locus variant had the highest allele frequency in Europeans (~5.6%), and the significant association was mostly driven by European subjects with RA. The validity of these results requires further investigation, as many of them are associated with low‐frequency variants.
The robustness of the top associations was evaluated in several further analyses. We did not observe substantial deviations in allele frequencies for the top variants compared with those reported in the gnomAD database (Supplementary Table 7 [http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract]). In addition, no significant trans‐ethnic allelic heterogeneity effects were found after adjustment of the P values for multiple tests (see Supplementary Table 8, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract). However, when we restricted the meta‐analysis to variants with an MAF of >2% in all ethnic groups (5,685,609 SNPs), only 2 loci retained genome‐wide significance (CD83 and ACSF3), and 1 locus had suggestive genome‐wide significance (TOX3) (see Supplementary Table 9 at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract), suggesting that the findings presented herein are sensitive to the allele frequency threshold.
Association of IL17RB with a shorter time to HZ, suggesting a potential contributory role for Th2 shift
A SNP near IL17RB (rs58861611) was associated with faster time to HZ (meta‐analysis HR 3.6, P = 7.6 × 10−10) at the genome‐wide significance level, and was suggestively associated with HZ in the case versus control analysis (meta‐analysis OR 3.8, P = 3.0 × 10−6) (Table 2). Results from the Kaplan‐Meier analysis of time to HZ event are presented in Supplementary Figures 4A–F, and C‐statistics for the case versus control logistic regression model are presented in Supplementary Table 5 (http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract).
As shown in the detailed regional plots for the genetic association of the IL17RB locus and the ancestry/population‐specific effects of this SNP on HZ end points (Figure 3 and Supplementary Table 6 [http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract]), the association of IL17RB was driven by a risk allele common in East Asian subjects (~8–17%) but rare in European subjects (<0.2%). Within the ancestry‐ and population‐specific analyses, the most significant association with the HZ end points was seen in the East Asian subgroup of subjects with RA (HR 3.4, P = 3.2 × 10−7; OR 5.06, P = 2.4 × 10−6) (Supplementary Table 6).
Figure 3.
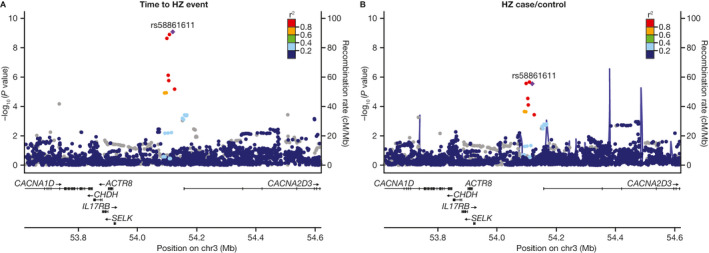
Regional association plots assessing the association of IL17RB with time to herpes zoster (HZ) event (A) and incidence of HZ (case versus control) (B) in subjects with rheumatoid arthritis or psoriasis. Each point represents a single‐nucleotide polymorphism (SNP) passing quality control in the trans‐ancestry meta‐analysis, plotted with its P value (on a −log10 scale) as a function of genomic position. The purple diamond indicates the lead SNP. Color coding of all other SNPs indicates linkage disequilibrium with the lead SNP (estimated using r2 values from East Asian populations in the 1000 Genomes Project database): red = r2 ≥ 0.8; gold = 0.6 ≤ r2 < 0.8; green = 0.4 ≤ r2 < 0.6; cyan = 0.2 ≤ r2 < 0.4; blue = r2 < 0.2; gray = r2 unknown.
Observation of altered T helper cells in rs58861611 carriers in healthy Japanese individuals
To elucidate the potential role of rs58861611 (IL17RB locus variant) in regulating immune phenotypes, flow cytometry was performed on freshly isolated PBMCs from 82 healthy Japanese individuals. Subjects were genotyped in parallel for the IL17RB rs58861611 SNP. Two subjects with the “CC” genotype were observed among these 82 healthy Japanese individuals, which is concurrent with the ~12% frequency of the “C” allele of rs58861611 in the overall Japanese population. As such, with the sample size being 82 subjects, the study had limited power to detect the variant impact on immune phenotypes.
The HZ risk allele of rs58861611 was significantly associated with lowered proportions of Th17 cells (P = 0.045) and Treg cells (P = 0.025) (Figures 4C and D). Similarly, a trend toward lowered proportions of Th1 cells was observed in rs58861611 variant carriers, although this was not statistically significant (Figure 4A). The effects of rs58861611 on Th2 cell proportions were also not significant (Figure 4B). These results suggest that rs58861611 may be associated with alterations in the proportions of T cell populations. However, due to the small sample size in this functional assessment, and the limited number of subjects with the IL17RB gene variant in the present study, this observation needs to be further evaluated in a larger cohort.
Figure 4.
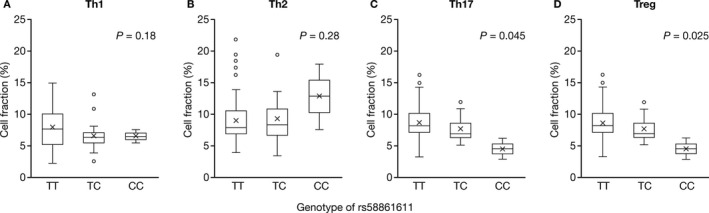
Correlation between genotype and cell fraction in peripheral blood CD4+ T cells from 82 healthy Japanese individuals. The test for significance of the data from the regression analyses of correlations between proportions of Th1 (A), Th2 (B), Th17 (C), and Treg cells (D) and genotype was performed using t‐statistics. The horizontal axis indicates the rs58861611 genotype groups. Data are shown as box plots, where each box represents the 25th to 75th percentiles, lines inside the boxes represent the median, the X indicates the mean, and lines outside the boxes represent the 10th and 90th percentiles. Circles indicate outliers.
Lack of association of rs58861611 with IL17RB gene expression in T helper cell–type specific eQTL analysis in a small cohort of healthy subjects
To further address the alterations in T cell proportions by the IL17RB variant, an eQTL analysis was performed to evaluate whether the rs58861611 variant impacts the gene expression of IL17RB or a nearby antisense sequence (AC012467.2) in T cell subpopulations. Th0, Th1, Th2, Th17, and Treg cells were induced from naive T cells from 29 healthy Japanese individuals. In this small cohort, there was only 1 “CC”‐homozygous subject, as expected. Low expression levels of IL17RB were observed in naive T cells, and its overall expression remained low in Th0, Th1, and Th17 cells, while the expression of IL17RB was increased in Th2 and Treg cells (see Supplementary Figures 5A–E, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41655/abstract), which has also been shown in other studies (32). Results of eQTL analysis did not reveal a significant association between the rs58861611 genotypes and IL17RB gene expression or the expression of AC012467.2, a potential antisense sequence, in any of the induced T cell subtypes. This result could be attributed to the low power of the analysis, since the sample size was small, or it is possible that rs58861611 may affect IL17RB expression in an untested cell type or through a mechanism unrelated to the messenger RNA expression of the IL17RB gene.
DISCUSSION
In this analysis, we sought to identify genetic factors contributing to the occurrence of HZ related to tofacitinib treatment. The GWAS identified numerous loci associated with an increased risk of VZV reactivation (i.e., faster time to HZ onset), including 5 loci identified in a meta‐analysis of the total pool, and loci identified in both ancestry‐ and population‐specific settings. These data indicate that 1 gene, IL17RB, may account for some of the HZ cases seen among East Asian subjects receiving tofacitinib (C‐statistic in the East Asian RA population = 0.78); however, no single gene accounts for all or the majority of cases of HZ in subjects receiving tofacitinib. Rather, the incidence of HZ in these populations is likely a result of interactions between many factors, including genetics and environmental factors.
In the ancestry‐ and population‐specific analyses, 4 genetic loci associated with faster development of HZ were identified in the European ancestry subgroup (1 in RA subjects, 3 in PsO subjects). CD83 represents a possible gene contributing to HZ risk, as a nearby variant, rs112817503, was significantly associated with HZ risk. CD83 is a marker of dendritic cell (DC) maturation; VZV infects mature monocyte‐derived DCs and impairs their functions by down‐regulating cell‐surface immune molecules, including CD83, CD80, and CD86 (33). Similarly, human cytomegalovirus (HCMV), a member of the herpesvirus family, can infect monocyte‐derived DCs. HCMV impairs the ability of the DCs to present antigens to T cells and thereby impairs the subsequent proliferation of T cells through multiple mechanisms, some of which involve release of soluble CD83 from DC membranes (34). Tofacitinib lowers CD80 and CD86 expression in DCs in vitro (35), suggesting that JAK inhibition could interact with a variant near CD83 to decrease presentation of virus in infected cells. The precise molecular mechanisms for these tofacitinib‐related effects are not known, but may be due to inhibition of IFNα. The CD83 association was driven by the data from the European subgroup, and remained significant in the trans‐ethnic GWAS meta‐analysis.
The PMEPA1 locus was associated with faster HZ development in European subjects with RA. The variant near PMEPA1 (i.e., TMEPA1) may also influence viral presentation, as HCMV reduces CD83 expression via TGFβ1 signaling (36), which is inhibited by PMEPA1 (37).
The VWF locus was associated with faster HZ development in European subjects with PsO. The VWF gene encodes the protein von Willebrand factor (vWF), which functions as both an antihemophilic factor carrier and a platelet‐vessel wall mediator in the blood coagulation system. The levels of vWF rise in multiple types of infections (38, 39). In a candidate gene study, VWF genetic variants were associated with human herpes simplex encephalitis, a rare complication following infection with herpes simplex virus type 1, which usually remains latent in neurons (40). Thus, the VWF gene may have roles in multiple infections; however, the mechanisms of vWF in HZ have not been directly studied. The VWF locus variant (rs200638456) is in moderate linkage disequilibrium (r2 = 0.64 in the European population) with an eQTL variant of CD9 (rs12099542) (41); thus, CD9 could also be a candidate causal gene for this association.
Within ancestry‐specific GWAS, no significant associations in the Hispanic and East Asian subgroups were identified, likely because the numbers of subjects in these ethnicity groups were small, and therefore this study had low power to detect differences.
It was hypothesized that combining ethnicity subgroups via meta‐analysis would increase the power to detect genetic factors for HZ risk; indeed, 4 additional loci were identified from the trans‐ancestry and trans‐population meta‐analysis, including a variant near the IL17RB gene prevalent in East Asian populations.
IL17RB encodes a cytokine receptor that specifically binds to IL‐25 (IL‐17E) and IL‐17B, in which IL‐17B is thought to be an antagonist of IL‐25 binding (32). IL‐25 induces Th2‐type cytokine production in IL17RB‐positive cells (32), and in a case report, genetic amplification of IL‐25 led to an overactive Th2 response with a phenotype of recurrent varicella (42). In this analysis, the IL17RB locus variant was also associated with lowered proportions of Th17/Treg cells in healthy Japanese individuals. Somewhat surprisingly, the IL17RB locus variant was not significantly associated with increased expression of IL17RB and did not show a significant effect on Th2 cell proportions, as might have been predicted from its known biologic effects. This may have been due to the small sample size and low power, or because we tested these effects in immune cells from healthy subjects and not under conditions of disease or tofacitinib exposure. These data suggest a potential mechanism by which the IL17RB variant contributes to HZ risk in Japanese individuals, as the imbalance of T cell subtypes may lead to a reduced threshold for VZV reactivation.
In addition, IL17RB is an expression marker that can be used to define invariant natural killer T (iNKT) cell heterogeneity (43). Studies have shown that iNKT cells produce Th1, Th2, or Th17 cytokines when challenged (43). The role of IL17RB and the JAK‐dependent cytokine IL‐15 in the development and ratio of iNKT cells has been characterized in mice: CD4+IL17RB+ iNKT cells produce large amounts of Th2 and moderate amounts of Th17 cytokines, whereas CD4+IL17RB− iNKT cells produce the anti‐viral Th1 cytokine IFNγ (43). CD4+IL17RB− iNKT cells express the IL‐15 receptor CD122, and require the presence of the JAK‐dependent cytokine IL‐15 for development (43). In mice hypomorphic for IL‐15 signaling, levels of Th1‐producing iNKT cells decrease, while levels of Th2‐producing iNKT cells increase (43). Deficient iNKT cells are characterized by low production of IFNγ; however, the functions of normal T cells and NK cells have been linked to disseminated HZ in response to vaccination in 2 case reports, despite an otherwise intact immune system (44, 45). These IL‐25 and iNKT studies and the association near the IL17RB gene suggest that the ratio of iNKT cell subsets at baseline may be important for HZ risk when combined with inhibition of IL‐15 signaling by tofacitinib.
Based on the significant loci identified in this analysis, we observed that genetic risks related to HZ are population‐ and ethnicity‐dependent. The CD83 variant was prevalent in European subjects; overall, its association with HZ was driven by the genetic effects observed in European subjects in the PsO population. The IL17RB variant was most prevalent in East Asian subjects; overall, its association with HZ was driven by the genetic effects observed in East Asian subjects in the RA population. This implies that genetic risk variants from different ethnicities may interact with disease conditions and tofacitinib exposure, jointly contributing to VZV reactivation. The IL17RB locus variant had an allele frequency of 8–17% in East Asian subjects, which was higher than the allele frequencies of all of the other HZ‐associated variants (<7% across populations) in this analysis. The common alleles from the IL17RB variant compared with other low‐frequency variants in other ethnicities may explain the higher HZ rate observed in tofacitinib‐treated East Asian individuals. Notably, the IL17RB variant did not show association with HZ in Asian subjects with PsO. This may have been because sample sizes were small or there were differences in HZ‐modifying risk factors between the PsO and RA populations. The functional mechanisms of the associations between the GPR141, TOX3, and ACSF3/CDH15 loci and HZ events also warrant further investigation.
This genetic and functional study is fundamentally limited by the relatively small sample sizes of the Hispanic and East Asian populations, as well as the assessment of multiple different subgroups (i.e., ethnicity and disease). Furthermore, we showed that the trans‐ancestry association findings are sensitive to the MAF threshold used. When we restricted the meta‐analysis to variants with an MAF of >2% in all ethnic groups, only 2 loci retained genome‐wide significance (CD83 and ACSF3), and 1 locus had suggestive genome‐wide significance (TOX3). These results highlight the importance of validating the current findings in large‐scale studies. An additional limitation is that data on prior HZ vaccination, which might have lowered the risk of VZV reactivation, were not collected in this study.
Overall, this analysis identified multiple genetic factors associated with HZ risk in tofacitinib‐treated subjects with RA or PsO. The findings provide novel insights into the molecular mechanisms contributing to VZV reactivation during tofacitinib treatment, which can be further validated in additional JAK inhibitor clinical studies or by genetic analysis of larger cohorts of East Asian subjects characterized by VZV response.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Bing had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Bing, Hirose, Kochi, Fujio, Valdez, Vincent, Clark.
Acquisition of data
Bing, Zhou, Tsuchida, Sumitomo, Zhang, Valdez.
Analysis and interpretation of data
Bing, Zhou, Chen, Hirose, Kochi, Tsuchida, Ishigaki, Zhang, Valdez, Martin, Clark.
ROLE OF THE STUDY SPONSOR
This study was sponsored by Pfizer Inc. Pfizer Inc. was involved in the study design, data collection, and data analysis. The studies of the CD4+ T cell subsets were supported by funding from Pfizer Inc. and Takeda Pharmaceutical Co., Ltd., and a grant from RIKEN. Medical writing support, under the guidance of the authors, was provided by Christina Viegelmann, PhD, and Sarah Piggott, MChem (CMC Connect, McCann Health Medical Communications) and was funded by Pfizer Inc. New York, in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med 2015;163:461–4). All authors, including those employed by the sponsor, were involved in data interpretation, revised the manuscript critically for intellectual content, and made the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Pfizer Inc.
Supporting information
Supplementary Material
Sponsored by Pfizer Inc. The studies of CD4+ T cell subsets were supported by funding from Pfizer Inc. and Takeda Pharmaceutical Co., Ltd., and by a grant from RIKEN.
Drs. Bing, Zhou, Zhang, and Clark were employees of Pfizer Inc. at the time of the analysis. Drs. Chen, Hirose, Valdez, Vincent, and Martin own stock or stock options in Pfizer Inc. Drs. Kochi, Tsuchida, Ishigaki, Sumitomo, and Fujio have received research funding from Chugai Pharmaceutical Co., Ltd., Pfizer Inc., and Takeda Pharmaceutical Co., Ltd.
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices 1) for indications that have been approved in the US and/or EU or 2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
References
- 1.Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, et al. The mechanism of action of tofacitinib: an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 2016;34:318–28. [PubMed] [Google Scholar]
- 2.Burmester GR, Blanco R, Charles‐Schoeman C, Wollenhaupt J, Zerbini C, Benda B, et al. Tofacitinib (CP‐690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet 2013;381:451–60. [DOI] [PubMed] [Google Scholar]
- 3.Fleischmann R, Kremer J, Cush J, Schulze‐Koops H, Connell CA, Bradley JD, et al. Placebo‐controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012;367:495–507. [DOI] [PubMed] [Google Scholar]
- 4.Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin‐Mola E, et al. Tofacitinib in combination with nonbiologic disease‐modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med 2013;159:253–61. [DOI] [PubMed] [Google Scholar]
- 5.Van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, et al. Tofacitinib (CP‐690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve‐month data from a twenty‐four–month phase III randomized radiographic study. Arthritis Rheum 2013;65:559–70. [DOI] [PubMed] [Google Scholar]
- 6.Van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 2012;367:508–19. [DOI] [PubMed] [Google Scholar]
- 7.Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley J, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 2014;370:2377–86. [DOI] [PubMed] [Google Scholar]
- 8.Bachelez H, van de Kerkhof PC, Strohal R, Kubanov A, Valenzuela F, Lee JH, et al. Tofacitinib versus etanercept or placebo in moderate‐to‐severe chronic plaque psoriasis: a phase 3 randomised non‐inferiority trial. Lancet 2015;386:552–61. [DOI] [PubMed] [Google Scholar]
- 9.Papp KA, Menter MA, Abe M, Elewski B, Feldman SR, Gottlieb AB, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two, randomized, placebo‐controlled, phase III trials. Br J Dermatol 2015;173:949–61. [DOI] [PubMed] [Google Scholar]
- 10.Bissonnette R, Iversen L, Sofen H, Griffiths CE, Foley P, Romiti R, et al. Tofacitinib withdrawal and retreatment in moderate‐to‐severe chronic plaque psoriasis: a randomized controlled trial. Br J Dermatol 2015;172:1395–406. [DOI] [PubMed] [Google Scholar]
- 11.Papp KA, Krueger JG, Feldman SR, Langley RG, Thaci D, Torii H, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: long‐term efficacy and safety results from 2 randomized phase‐III studies and 1 open‐label long‐term extension study. J Am Acad Dermatol 2016;74:841–50. [DOI] [PubMed] [Google Scholar]
- 12.Cohen S, Radominski SC, Gomez‐Reino JJ, Wang L, Krishnaswami S, Wood SP, et al. Analysis of infections and all‐cause mortality in phase II, phase III, and long‐term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol 2014;66:2924–37. [DOI] [PubMed] [Google Scholar]
- 13.Cohen SB, Tanaka Y, Mariette X, Curtis JR, Lee EB, Nash P, et al. Long‐term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheum Dis 2017;76:1253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curtis JR, Xie F, Yun H, Bernatsky S, Winthrop KL. Real‐world comparative risks of herpes virus infections in tofacitinib and biologic‐treated patients with rheumatoid arthritis. Ann Rheum Dis 2016;75:1843–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strober BE, Gottlieb AB, van de Kerkhof PCM, Puig L, Bachelez H, Chouela E, et al. Benefit‐risk profile of tofacitinib in patients with moderate‐to‐severe chronic plaque psoriasis: pooled analysis across six clinical trials. Br J Dermatol 2019;180:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smitten AL, Choi HK, Hochberg MC, Suissa S, Simon TA, Testa MA, et al. The risk of herpes zoster in patients with rheumatoid arthritis in the United States and the United Kingdom. Arthritis Rheum 2007;57:1431–8. [DOI] [PubMed] [Google Scholar]
- 17.Winthrop KL, Yamanaka H, Valdez H, Mortensen E, Chew R, Krishnaswami S, et al. Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol 2014;66:2675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol 2017;13:320. [DOI] [PubMed] [Google Scholar]
- 19.Winthrop KL, Curtis JR, Lindsey S, Tanaka Y, Yamaoka K, Valdez H, et al. Herpes zoster and tofacitinib: clinical outcomes and the risk of concomitant therapy. Arthritis Rheumatol 2017;69:1960–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winthrop KL, Lebwohl M, Cohen AD, Weinberg JM, Tyring SK, Rottinghaus ST, et al. Herpes zoster in psoriasis patients treated with tofacitinib. J Am Acad Dermatol 2017;77:302–9. [DOI] [PubMed] [Google Scholar]
- 21.Winthrop KL, Melmed GY, Vermeire S, Long MD, Chan G, Pedersen RD, et al. Herpes zoster infection in patients with ulcerative colitis receiving tofacitinib. Inflamm Bowel Dis 2018;24:2258–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mease P, Hall S, FitzGerald O, van der Heijde D, Merola JF, Avila‐Zapata F, et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med 2017;377:1537–50. [DOI] [PubMed] [Google Scholar]
- 23.Gladman D, Rigby W, Azevedo VF, Behrens F, Blanco R, Kaszuba A, et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med 2017;377:1525–36. [DOI] [PubMed] [Google Scholar]
- 24.Crosslin DR, Carrell DS, Burt A, Kim DS, Underwood JG, Hanna DS, et al. Genetic variation in the HLA region is associated with susceptibility to herpes zoster. Genes Immun 2015;16:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prevoo ML, van ’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight–joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 27.Fredriksson T, Pettersson U. Severe psoriasis—oral therapy with a new retinoid. Dermatologica 1978;157:238–44. [DOI] [PubMed] [Google Scholar]
- 28.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders S, Pyl PT, Huber W. HTSeq: a Python framework to work with high‐throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stegle O, Parts L, Durbin R, Winn J. A Bayesian framework to account for complex non‐genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol 2010;6:e1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parts L, Stegle O, Winn J, Durbin R. Joint genetic analysis of gene expression data with inferred cellular phenotypes. PLoS Genet 2011;7:e1001276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reynolds JM, Lee YH, Shi Y, Wang X, Angkasekwinai P, Nallaparaju KC, et al. Interleukin‐17B antagonizes interleukin‐25‐mediated mucosal inflammation. Immunity 2015;42:692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrow G, Slobedman B, Cunningham AL, Abendroth A. Varicella‐zoster virus productively infects mature dendritic cells and alters their immune function. J Virol 2003;77:4950–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gredmark‐Russ S, Söderberg‐Nauclér C. Dendritic cell biology in human cytomegalovirus infection and the clinical consequences for host immunity and pathology [review]. Virulence 2012;3:621–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kubo S, Yamaoka K, Kondo M, Yamagata K, Zhao J, Iwata S, et al. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte‐derived dendritic cells. Ann Rheum Dis 2014;73:2192–8. [DOI] [PubMed] [Google Scholar]
- 36.Arrode G, Boccaccio C, Abastado JP, Davrinche C. Cross‐presentation of human cytomegalovirus pp65 (UL83) to CD8+ T cells is regulated by virus‐induced, soluble‐mediator‐dependent maturation of dendritic cells. J Virol 2002;76:142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Itoh S, Itoh F. TMEPAI family: involvement in regulation of multiple signalling pathways. J Biochem 2018;164:195–204. [DOI] [PubMed] [Google Scholar]
- 38.McElroy AK, Erickson BR, Flietstra TD, Rollin PE, Towner JS, Nichol ST, et al. Von Willebrand factor is elevated in individuals infected with Sudan virus and is associated with adverse clinical outcomes. Viral Immunol 2015;28:71–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Regan N, Gegenbauer K, O’Sullivan JM, Maleki S, Brophy TM, Dalton N, et al. A novel role for von Willebrand factor in the pathogenesis of experimental cerebral malaria. Blood 2016;127:1192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdelmagid N, Bereczky‐Veress B, Atanur S, Musilová A, Zidek V, Saba L, et al. Von Willebrand factor gene variants associate with herpes simplex encephalitis. PLoS One 2016;11:e0155832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 2013;45:1238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green MR, Camilleri E, Gandhi MK, Peake J, Griffiths LR. A novel immunodeficiency disorder characterized by genetic amplification of interleukin 25. Genes Immun 2011;12:663–6. [DOI] [PubMed] [Google Scholar]
- 43.Watarai H, Sekine‐Kondo E, Shigeura T, Motomura Y, Yasuda T, Satoh R, et al. Development and function of invariant natural killer T cells producing T(h)2‐ and T(h)17‐cytokines. PLoS Biol 2012;10:e1001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levy O, Orange JS, Hibberd P, Steinberg S, LaRussa P, Weinberg A, et al. Disseminated varicella infection due to the vaccine strain of varicella‐zoster virus, in a patient with a novel deficiency in natural killer T cells. J Infect Dis 2003;188:948–53. [DOI] [PubMed] [Google Scholar]
- 45.Banovic T, Yanilla M, Simmons R, Robertson I, Schroder WA, Raffelt NC, et al. Disseminated varicella infection caused by varicella vaccine strain in a child with low invariant natural killer T cells and diminished CD1d expression. J Infect Dis 2011;204:1893–901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material