

Abstract
Diethynyl phosphinates were developed as bisfunctional electrophiles for the site‐selective modification of peptides, proteins and antibodies. One of their electron‐deficient triple bonds reacts selectively with a thiol and positions an electrophilic moiety for a subsequent intra‐ or intermolecular reaction with another thiol. The obtained conjugates were found to be stable in human plasma and in the presence of small thiols. We further demonstrate that this method is suitable for the generation of functional protein conjugates for intracellular delivery. Finally, this reagent class was used to generate functional homogeneously rebridged antibodies that remain specific for their target. Their modular synthesis, thiol selectivity and conjugate stability make diethynyl phosphinates ideal candidates for protein conjugation for biological and pharmaceutical applications.
Keywords: antibody rebridging, bioconjugation, cysteine-selective modification, phosphorous-based electrophiles, protein double-modification
Diethynyl phosphonites were found to react specifically with two thiols in aqueous buffer to give constructs that are stable under physiological conditions. This enables the facile generation of protein (double)‐conjugates. Moreover, these molecules can be used to rebridge the interchain disulfides of therapeutic IgG antibodies.

Site‐selective modification of biomolecules with small chemical handles, such as fluorophores or bioactive compounds, enables a plethora of applications in life sciences and pharmacology.[1, 2] Most commonly, electrophilic reagents that target nucleophilic amino acids like lysine or cysteine (Cys) are employed.[1, 3, 4] Other than that, also most other nucleophilic amino acids can be selectively addressed, however these methods are less well established.[5, 6, 7, 8, 9] Additionally, the incorporation of non‐natural amino acids equipped with azides, trans‐cyclooctenes or tetrazines allows the biorthogonal modification of peptides and proteins.[10, 11]
Among the proteinogenic amino acids, Cys offers several advantages when it comes to protein modification. First, the enhanced nucleophilicity of the sulfhydryl group simplifies site‐selective modification. Moreover, its relatively low abundance on accessible protein surfaces often allows selective monofunctionalization of proteins.[3] Therefore, several approaches for selective labeling of Cys on proteins have been developed. Among them, thio‐Michael addition to maleimides remains the most widely used method. Although this reaction offers rapid kinetics at near neutral pH, the generated thiosuccinimide linkage is inherently unstable in the presence of external thiols, caused by retro‐Michael addition.[12]
In addition to the modification of single Cys residues with a specific payload, peptide or protein modifications using double reactive Cys‐specific reagents have been described in the literature.[13, 14, 15, 16, 17, 18] However, similar to normal maleimides, linkages generated with such reagents are often unstable towards reducing conditions or excess of small thiols.[13, 15] These bisfunctional reagents can be used for the intermolecular crosslinking of two Cys‐containing proteins.[15, 19, 20] Alternatively, in a process commonly referred to as disulfide rebridging, cystines can be converted following reduction and subsequent reaction with a biselectrophile, an approach that is particularly useful for antibody functionalization. In addition to the site‐selective modification with a defined stoichiometry, the covalent non‐reductive linkage between the antibody chains has been shown to increase the antibody's thermal stability.[21, 22] Recently, reagents based on functionalized dibromomaleimides,[23] divinylpyrimidines[19] and vinylsulfones[24] have been used to generate antibody–drug conjugates (ADCs) with a drug‐to‐antibody ratio of four.
Previously, our lab introduced unsaturated phosphonamidates and phosphonothioates as electrophilic reagents for chemoselective Cys modification, yielding highly stable conjugates.[14, 25, 26] These compounds can be generated from vinyl or alkynyl phosphonites and azides or electrophilic disulfides, respectively (Figure 1 a). Moreover, we have employed electrophilic PV‐reagents in peptide stapling,[26] protein–protein conjugation[14, 25] and in the production of efficacious ADCs.[27, 28]
Figure 1.

a) Electrophilic phosphonamidates and phosphonothiolates, generated from azides or electrophilic disulfides and vinyl or ethynyl phosphonites, react selectively with cysteine residues on proteins.[14, 25, 26] b) Development of substituted diethynyl phosphinates as reagents for selective thiol–thiol bioconjugation and rebridging of native disulfides, for example, in therapeutic antibodies.
Based on our previous findings, we envisioned that two ethynyl groups on the same phosphorous atom could be able to react subsequently with two sulfhydryl groups (Figure 1 b). Such molecules have been previously reported in the generation of P‐stereogenic heterocycles,[29] as intermediates in the synthesis of phosphascorpionate complexes[30] and as building blocks for macrocycles,[31] but to the best of our knowledge, they have not been used as reagents for cysteine modification. Using a small‐molecule‐based FRET‐system we show that thiol–thiol conjugates generated with diethynyl phosphinates are highly stable in human serum and in the presence of small thiols. Moreover, we used these to site‐selectively modify proteins for biological applications. Finally, diethynyl phosphinates were employed in the generation of functionalized, rebridged antibodies that were shown to remain selective for their antigen.
At the outset of our studies, we were aiming for the diethynyl equivalent to phosphonamidates: diethynyl phosphinamidates. Starting from the commercially available ethyl dichlorophosphite and ethynylmagnesium bromide, we obtained ethyl diethynyl phosphinite (I). Subsequently, we reacted the PIII‐compound with benzyl azide to obtain the PV biselectrophile via the Staudinger reaction. However, neither prolonged reaction times (>72 h) nor elevated temperatures (70 °C) led to the formation of the desired product. As an alternative route, diethyl phosphoramidous dichloride was substituted two times with ethynylmagnesium bromide and subsequently oxidized using hydrogen peroxide (II, 61 % yield). Initial testing of this compound on peptides and proteins revealed that it does react with thiols; however, the second addition could not be observed under physiological conditions. Moreover, we observed rapid hydrolysis of the P−N bond already under slightly acidic conditions (pH≤3), which limits the further application and analysis of II.
In light of these unfavorable results, our focus shifted towards the structurally related diethynyl phosphinates. Phosphinite I was successfully oxidized with hydrogen peroxide to afford ethyl diethynyl phosphinate (1) in 72 % yield. To test its reactivity towards thiols, compound 1 was mixed with an excess of ethanethiol (3 equiv) in DMF. Indeed, we observed full conversion of the starting material into the double thiol adduct after reacting it for 5 minutes at room temperature (rt, Scheme 1 a). Interestingly, a mixture of three different isomers was formed (as determined by UPLC) that could be separated by means of semi‐preparative HPLC (70 % combined isolated yield). Characterization of the obtained compounds with nuclear magnetic resonance (NMR) spectroscopy revealed that the observed ratio of 45:50:5 corresponds to the Z,Z, E,Z, and E,E isomer, respectively (Figure S1). However, we observed slow isomerization of the Z,Z and E,Z isomer to form the E,E product (Figure S2).
Scheme 1.
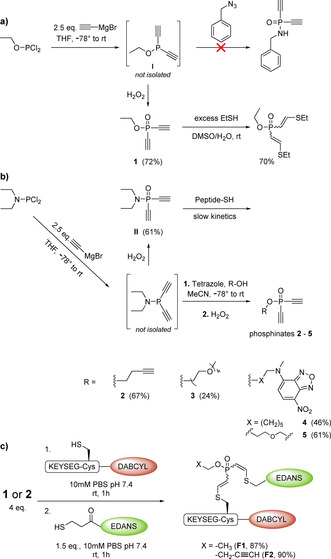
Synthesis of diethynyl phosphinates. a) Generation of phosphinate 1 and the formation of its thiol adducts. b) Synthetic route towards functional diethynyl phosphinates 2–5. c) Sequential thiol addition to diethynyl phosphinates allows to generate the quenched FRET pairs F1 and F2.
In order to access compounds with functional O‐substituents a one‐pot two‐step reaction starting from diethyl phosphoramidous dichloride was developed (Scheme 1 b). Using this route diethynyl phosphinates bearing alkynes as click handle (2), as well as tetraethylenglycol (3) and the fluorophore NBD (4 and 5) were synthesized (Scheme 1 b). In the course of this synthesis, we found diethynyl phosphinates to be very heat sensitive and only moderately stable on silica after they are oxidized. For this reason, we purified the compound as PIII‐derivative and oxidized as final step.
Stability of protein conjugates under physiological and biologically relevant conditions (e.g. in the presence of other thiols) is of utmost importance for their successful application. Especially, maleimide and electron‐deficient alkyne‐based thiol adducts have been reported to be susceptible towards exchange with other thiols as they are present inside of cells or in blood serum.[3, 32] To investigate the stability of phosphonamidate‐ and phosphonothiolate‐based thiol adducts, our group previously employed a fluorescence‐quenching assay.[14, 25] Using this assay, the stability of thiol conjugates generated from diethynyl phosphinate 1 as well as the stability of the P−O bond after thiol conjugation were investigated. Quenched products F1–F3 were synthesized from peptide P3, EDANS‐SH or EDANS‐N3 and the corresponding phosphinates 1 or 3 were used as bisreactive ethynyl phosphinates (Scheme 1 c and S3). For all constructs, excellent stability in physiological buffer, human serum and in the presence of excess free thiols was observed over the course of several days (Figure 2 a–c and Figure S4). Only under strong basic conditions did the conjugates degrade via β‐elimination at the linked Cys residue.
Figure 2.

a) FRET‐quenching assay to investigate the stability of the thiol conjugates. b),c) Observed EDANS fluorescence for constructs F1 and F2 in PBS, PBS supplemented with glutathione, human serum and in 0.1 m aq. NaOH over 72 h. d) General scheme for the site‐selective protein modification using diethynyl phosphinates and deconvoluted intact protein MS spectra of successfully labeled proteins. e) Conjugation of a cell‐penetrating R10‐peptide to mCherry‐5 allows delivery of mCherry into living cells with nucleolar localization and co‐localization of mCherry with NBD (scale bar 20 μm).
Having demonstrated that this compound class was highly thiol reactive and that the resulting conjugates were stable under various biologically relevant conditions, we started to test their applicability for protein modifications. At first phosphinate 1 was reacted with an eGFP mutant containing a single addressable Cys (eGFP C70M S147C). Using 10 equiv phosphinate 1 in PBS pH 7.4 containing 10 % DMSO as a co‐solvent, the reaction reached completion after 30 minutes (Figure 2 d). Analysis of the protein conjugate via CD and fluorescence spectroscopy showed that the secondary structure of eGFP is not altered upon modification (Figure S5). Tandem mass spectrometry (MS/MS) analysis of the tryptically digested protein verified that no amino acid other than Cys was modified by the phosphinate (Figure S5).
To prove the general applicability of Cys‐specific protein labeling using diethynyl phosphinates, various proteins of different size and nature (ubiquitin G76C, Histone H4 and recombinant human albumin) bearing one single addressable Cys residue were labeled. Incubation of the proteins (10–100 μm in PBS pH 7.4) with 10 equiv phosphinates 1–5 resulted in singly modified proteins after 10–60 minutes of reaction time (Figure 2 d). In contrast, reaction of 1 with an eGFP variant containing no addressable Cys (eGFP C70M) did not result in any protein labeling, further supporting excellent Cys selectivity.
Subsequently, we wanted to compare this new compound class with other Cys‐reactive electrophiles in terms of reaction kinetics. The estimation of reaction kinetics is particularly challenging for bisreactive reagents because the second thiol addition interferes with the analysis. However, we observed that on protein level the second thiol addition (i.e. the protein–protein cross‐linking) is significantly slower, probably because diffusion becomes a limiting factor. Consequently, we reacted 2 with one equivalent of eGFP in Tris buffer (pH 8.3) at a concentration of 90 μm at room temperature. The reaction progress was monitored by intact protein MS using eGFP C70M as internal standard and revealed a second‐order rate constant of approximately 0.47 m −1 s−1, which is comparable with previously reported PV‐electrophiles (Figure S6).[14, 25, 35]
Next, we wanted to explore whether we could make use of a two‐step labeling procedure with bisfunctional electrophiles to generate functional protein conjugates. Recently, our group described the use of linear polyarginine peptides (R10) to ensure cellular delivery of proteins into living cells with immediate bioavailability.[33] Following the two‐step labeling procedure, a Cys‐containing R10 was conjugated to eGFP via phosphinate 1 (Figure S7a). With the eGFP‐1‐R10 conjugate in hand, we then probed the cellular uptake into live cells following a previously developed protocol.[33] As expected, live‐cell imaging showed cellular uptake of the protein conjugate. Moreover, localization in the nucleoli was observed, which suggests that the eGFP‐R10‐conjugate shows stability inside living cells (Figure S7). To further validate the integrity of the whole phosphinate linkage, we used phosphinate 5 with NBD at the O‐substituent to generate an mCherry‐NBD‐R10 double conjugate. We found that this conjugate was also delivered into living cells and we observed co‐localization of mCherry with NBD. This further points towards the intracellular stability of the whole phosphinate linkage in our cargo conjugate (Figure 2 e, S8).
Motivated by these results, we next aimed for the rebridging of disulfides in IgG antibodies. Diethynyl phosphinates offer the potential to modify all four interchain disulfides, leading to a precise antibody‐to‐cargo ratio of four. Using the Her2‐targeting monoclonal IgG antibody Trastuzumab, we investigated the ability of diethynyl phosphinates for antibody rebridging (Figure 3 a).
Figure 3.

Reaction of Trastuzumab with diethynyl phosphinate 1 and subsequent analysis. a) General procedure for antibody rebridging using compound 1. b) Analysis of Trastuzumab before and after the reaction via SDS‐PAGE. c) Deconvoluted intact protein MS of the rebridged half antibody (2× modified with 1) after deglycosylation by PNGaseF.
Therefore, we slightly adapted a previously described protocol for antibody modification.[19] First, Trastuzumab (5 mg mL−1) was reduced using 10 equiv (2.2 equiv per disulfide) TCEP at 37 °C for half an hour. Subsequently, 5 equivalents of phosphinate 1 were added to the solution and the reaction was allowed to proceed at room temperature over night. SDS‐PAGE and intact protein MS analysis showed >95 % rebridging of the antibody, with the half antibody (covalently linked heavy and light chain) being the main product (Figure 3 b,c). In contrast, when unreduced Trastuzumab was reacted with 1, no rebridging or modification could be observed. Using a dedicated cross‐linking mass spectrometry search engine,[34] we were even able to validate the interchain cross‐link formed between the heavy and light chain of Trastuzumab (Figure S9).
Although we were not able to generate protein– or peptide–thiol adducts with phosphinamidate II, we tested its potential for antibody rebridging. Surprisingly, SDS gel analysis showed that II was able to cross‐link the antibodies' light and heavy chain, however only to approximately 33 % (Figure S10).
Encouraged by the excellent rebridging efficiency and site‐selectivity of diethynyl phosphinates, Trastuzumab was reacted with phosphinate 2 to allow for further functionalization of the antibody conjugate (Figure 4 a). Following the same procedure as described for 1, complete rebridging was observed after over‐night reaction. Again, the half antibody was formed as main product (Figure 4 b and S3.7). In the next step, the antibody was conjugated to fluorescein azide (FAM−N3) via copper‐aided azide–alkyne cycloaddition (CuAAC) to generate an antibody–fluorophore conjugate (AFC, Figure 4 a).
Figure 4.

Functional modification of Trastuzumab and its biological evaluation. a) Two‐step modification of the antibody with phosphinate 2, followed by on antibody CuAAC forming the fluorescein conjugate (half and full antibody). b) Analysis of the conjugate via SDS‐PAGE using coomassie staining (B) and in‐gel fluorescence (C). c) UV/Vis spectrum of the fluorescein‐conjugated antibody. d) Cell‐membrane labeling of Her2‐positive cells without any observed staining of Her2‐negative cells (scale bar 20 μm).
In‐gel fluorescence imaging and UV/Vis spectroscopy validated successful modification of the antibody (Figure 4 b,c). The fluorophore‐to‐antibody ratio was calculated to be 4.03, indicating quantitative conversion (SI 3.7).
The functionalized Trastuzumab clearly stained outer‐membrane‐bound Her2 on SKBR3 cells, while no fluorescence signal could be observed with the Her2‐negative cell line (Figure 4 d). This indicates that our rebridging and labeling strategy does not impede the antibodies' performance.
In conclusion, we herein present a novel technique for selective protein labeling and disulfide rebridging based on bisreactive unsaturated PV‐compounds. Diethynyl phosphinates show excellent reactivity and selectivity towards sulfhydryl groups in aqueous systems, which allows the easy and modular generation of protein conjugates with complex molecules. Moreover, the obtained constructs exhibit outstanding stability in the presence of an excess of small thiols, inside human serum and inside living cells. Antibodies rebridged with diethynyl phosphinates could be successfully modified with a fluorophore to obtain a FAR of four and retained their target selectivity. We believe that this simple and straightforward strategy for Cys‐selective protein and antibody modification will help to develop new tools for biological and biopharmaceutical applications.
Conflict of interest
The chemistry described in this manuscript is part of a patent application (Appl. Number: EP21170097.6).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. A. Schneider for valuable scientific discussion and for the expression of NLS‐mCherry, K. K.‐Hassanin and B. Kindt for excellent technical assistance and Dr. M.‐A. Kasper and Dr. P. Ochtrop for valuable input. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, SPP1623 HA 4468/10‐1 and RTG GRK2473), the Leibniz Society (SAW‐2018‐FMP‐4‐P5label, T18/2017) and the Einstein Foundation Berlin (Leibniz–Humboldt Professorship). C.E.S. is supported by a PhD fellowship of the Studienstiftung des Deutschen Volkes. Open access funding enabled and organized by Projekt DEAL.
C. E. Stieger, L. Franz, F. Körlin, C. P. R. Hackenberger, Angew. Chem. Int. Ed. 2021, 60, 15359.
In memory of Professor Rolf Huisgen
References
- 1.Hoyt E. A., Cal P. M. S. D., Oliveira B. L., Bernardes G. J. L., Nat. Rev. Chem. 2019, 3, 147–171. [Google Scholar]
- 2.Schumacher D., Hackenberger C. P. R., Curr. Opin. Chem. Biol. 2014, 22, 62–69. [DOI] [PubMed] [Google Scholar]
- 3.Gunnoo S. B., Madder A., ChemBioChem 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- 4.Ochtrop P., Hackenberger C. P. R., Curr. Opin. Chem. Biol. 2020, 58, 28–36. [DOI] [PubMed] [Google Scholar]
- 5.Jia S., He D., Chang C. J., J. Am. Chem. Soc. 2019, 141, 7294–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vantourout J. C., Rao Adusumalli S., Knouse K. W., Flood D. T., Ramirez A., Padial N. M., Istrate A., Maziarz K., deGruyter J. N., Merchant R. R., Qiao J. X., Schmidt M. A., Deery M. J., Eastgate M. D., Dawson P. E., Bernardes J. L., Baran P. S., J. Am. Chem. Soc. 2020, 142, 17236–17242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato S., Matsumura M., Kadonosono T., Abe S., Ueno T., Ueda H., Nakamura H., Bioconjugate Chem. 2020, 31, 1417–1424. [DOI] [PubMed] [Google Scholar]
- 8.Taylor M. T., Nelson J. E., Suero M. G., Gaunt M. J., Nature 2018, 562, 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang D., Nilchan N., Nanna A. R., Li X., Cameron M. D., Roush W. R., Park H. J., Rader C., Cell Chem. Biol. 2019, 26, 1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang K., Chin J. W., ACS Chem. Biol. 2014, 9, 16–20. [DOI] [PubMed] [Google Scholar]
- 11.Chen X., Wu Y. W., Org. Biomol. Chem. 2016, 14, 5417–5439. [DOI] [PubMed] [Google Scholar]
- 12.Shen B. Q., Xu K., Liu L., Raab H., Bhakta S., Kenrick M., Parsons-Reponte K. L., Tien J., Yu S. F., Mai E., Li D., Tibbitts J., Baudys J., Saad O. M., Scales S. J., McDonald P. J., Hass P. E., Eigenbrot C., Nguyen T., Solis W. A., Fuji R. N., Flagella K. M., Patel D., Spencer S. D., Khawli L. A., Ebens A., Wong W. L., Vandlen R., Kaur S., Sliwkowski M. X., Scheller R. H., Polakis P., Junutula J. R., Nat. Biotechnol. 2012, 30, 184–189. [DOI] [PubMed] [Google Scholar]
- 13.Smith M. E. B., Schumacher F. F., Ryan C. P., Tedaldi L. M., Papaioannou D., Waksman G., Caddick S., Baker J. R., J. Am. Chem. Soc. 2010, 132, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baumann A. L., Schwagerus S., Broi K., Kemnitz-Hassanin K., Stieger C. E., Trieloff N., Schmieder P., Hackenberger C. P. R., J. Am. Chem. Soc. 2020, 142, 9544–9552. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y., Zang C., An G., Shang M., Cui Z., Chen G., Xi Z., Zhou C., Nat. Commun. 2020, 11, 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canovas C., Moreau M., Bernhard C., Oudot A., Guillemin M., Denat F., Goncalves V., Angew. Chem. Int. Ed. 2018, 57, 10646–10650; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10806–10810. [Google Scholar]
- 17.Wang T., Riegger A., Lamla M., Wiese S., Oeckl P., Otto M., Wu Y., Fischer S., Barth H., Kuan S. L., Weil T., Chem. Sci. 2016, 7, 3234–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paterson D. L., Flanagan J. U., Shepherd P. R., Harris P. W. R., Brimble M. A., Chem. Eur. J. 2020, 26, 10826–10833. [DOI] [PubMed] [Google Scholar]
- 19.Walsh S. J., Omarjee S., Galloway W. R. J. D., Kwan T. T.-L., Sore H. F., Parker J. S., Hyvönen M., Carroll J. S., Spring D. R., Chem. Sci. 2019, 10, 694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith M. E. B., Schumacher F. F., Ryan C. P., Tedaldi L. M., Papaioannou D., Waksman G., Caddick S., Baker J. R., J. Am. Chem. Soc. 2010, 132, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun S., Akkapeddi P., Marques M. C., Martínez-Sáez N., Torres V. M., Cordeiro C., Boutureira O., Bernardes G. J. L., Org. Biomol. Chem. 2019, 17, 2005–2012. [DOI] [PubMed] [Google Scholar]
- 22.Bahou C., Love E. A., Leonard S., Spears R. J., Maruani A., Armour K., Baker J. R., Chudasama V., Bioconjugate Chem. 2019, 30, 1048–1054. [DOI] [PubMed] [Google Scholar]
- 23.Nunes J. P. M., Morais M., Vassileva V., Robinson E., Rajkumar V. S., Smith M. E. B., Pedley R. B., Caddick S., Baker J. R., Chudasama V., Chem. Commun. 2015, 51, 10624–10627. [DOI] [PubMed] [Google Scholar]
- 24.Badescu G., Bryant P., Bird M., Henseleit K., Swierkosz J., Parekh V., Tommasi R., Pawlisz E., Jurlewicz K., Farys M., Camper N., Sheng X., Fisher M., Grygorash R., Kyle A., Abhilash A., Frigerio M., Edwards J., Godwin A., Bioconjugate Chem. 2014, 25, 1124–1136. [DOI] [PubMed] [Google Scholar]
- 25.Kasper M., Glanz M., Stengl A., Penkert M., Klenk S., Sauer T., Schumacher D., Helma J., Krause E., Cardoso M. C., Leonhardt H., Hackenberger C. P. R., Angew. Chem. Int. Ed. 2019, 58, 11625–11630; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11751–11756. [Google Scholar]
- 26.Kasper M. A., Glanz M., Oder A., Schmieder P., Von Kries J. P., Hackenberger C. P. R., Chem. Sci. 2019, 10, 6322–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasper M., Stengl A., Ochtrop P., Gerlach M., Stoschek T., Schumacher D., Helma J., Penkert M., Krause E., Leonhardt H., Hackenberger C. P. R., Angew. Chem. Int. Ed. 2019, 58, 11631–11636; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11757–11762. [Google Scholar]
- 28.Kasper M., Gerlach M., Schneider A. F. L., Groneberg C., Ochtrop P., Boldt S., Schumacher D., Helma J., Leonhardt H., Christmann M., Hackenberger C. P. R., ChemBioChem 2020, 21, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harvey J. S., Giuffredi G. T., Gouverneur V., Org. Lett. 2010, 12, 1236–1239. [DOI] [PubMed] [Google Scholar]
- 30.Van Assema S. G. A., Tazelaar C. G. J., De Bas Jong G., Van Maarseveen J. H., Schakel M., Lutz M., Spek A. L., Slootweg J. C., Lammertsma K., Organometallics 2008, 27, 3210–3215. [Google Scholar]
- 31.van Assema S. G. A., de Jong G. B., Ehlers A. W., de Kanter F. J. J., Schakel M., Spek A. L., Lutz M., Lammertsma K., Eur. J. Org. Chem. 2007, 2405–2412. [Google Scholar]
- 32.Shiu H.-Y., Chan T.-C., Ho C.-M., Liu Y., Wong M.-K., Che C.-M., Chem. Eur. J. 2009, 15, 3839–3850. [DOI] [PubMed] [Google Scholar]
- 33.Schneider A. F. L., Kithil M., Cardoso M. C., Lehmann M., Hackenberger C. P. R., Nat. Chem. 2021, 10.1038/s41557-021-00661-x. [DOI] [PubMed] [Google Scholar]
- 34.Chen Z. L., Meng J. M., Cao Y., Yin J. L., Fang R. Q., Fan S. B., Liu C., Zeng W. F., Ding Y. H., Tan D., Wu L., Zhou W. J., Chi H., Sun R. X., Dong M. Q., He S. M., Nat. Commun. 2019, 10, 3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park Y., Baumann A. L., Moon H., Byrne S., Kasper M.-A., Hwang S., Sun H., Baik M.-H., Hackenberger C. P. R., Chem. Sci. 2021, 10.1039/d1sc01730f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary