

Abstract
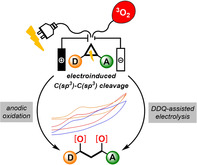
We describe the first electrochemical activation of D–A cyclopropanes and D–A cyclobutanes leading after C(sp3)−C(sp3) cleavage to the formation of highly reactive radical cations. This concept is utilized to formally insert molecular oxygen after direct or DDQ‐assisted anodic oxidation of the strained carbocycles, delivering β‐ and γ‐hydroxy ketones and 1,2‐dioxanes electrocatalytically. Furthermore, insights into the mechanism of the oxidative process, obtained experimentally and by additional quantum‐chemical calculations are presented. The synthetic potential of the reaction products is demonstrated by diverse derivatizations.
Keywords: C(sp3)−C(sp3) cleavage, cyclobutanes, cyclopropanes, donor–acceptor systems, synthetic electrochemistry
The first electrochemical activation of donor–acceptor (D–A) cyclopropanes and D–A cyclobutanes has been applied for the formal insertion of molecular oxygen into these compounds. C(sp3)−C(sp3) cleavage of the ring moieties was achieved either by direct anodic electrolysis or by utilizing a DDQ‐assisted electrochemical strategy. Quantum‐chemical calculations and supportive experiments provided mechanistic understanding of the electrocatalytic process.
Introduction
Donor–acceptor (D–A) cyclopropanes are strained[1] C3‐synthons that are valuable building blocks for synthetic organic chemistry. The vicinal substitution pattern of donor and acceptor substituents markedly increases the reactivity of these special cyclopropanes,[2] leading to facile C(sp3)−C(sp3) cleavage. Dating back to the late 1970s, when the versatile reactivity of these readily available carbocycles was first reported by Wenkert and Reissig,[3] D–A cyclopropanes have been widely used for a variety of organic transformations.[4] Over this span of four decades elegant methods for the activation of D–A cyclopropanes have been developed, paving the way for the formal generation of highly reactive intermediates by C(sp3)−C(sp3) cleavage. Of all reported approaches, Lewis acid catalysis is the most frequently used concept (Scheme 1 A).[5] By coordination of the acceptor moiety to a Lewis acid the polarization of the vicinally substituted C−C bond increases, allowing D–A cyclopropanes to take part in reactions in their formal zwitterionic form. Utilization of such 1,3‐dipoles has led to multiple (3+n)‐cycloadditions,[6] rearrangements[7] and ring‐opening reactions.[8] Another activation mode reported by Sparr and Gilmour[9] employed chiral iminium/enamine catalysis to react cyclopropyl carbaldehydes to 1,3‐dichlorides enantioselectively (Scheme 1 B).[10] This organocatalytic strategy allows the functionalization of a 1,3‐dipole concomitantly by electrophilic and nucleophilic chlorine sources. In contrast to this method, our group has developed a radical approach for the 1,3‐dichlorination of D–A cyclopropanes.[11] This concept makes use of a homolytic cleavage of the polarized C−C bond, leading to a radical intermediate that reacts further with chlorine radicals derived from iodobenzene dichloride (Scheme 1 C). Encouraged by these previously described activation modes and earlier work by Buriez on the ring‐opening of aminocyclopropanes via Shono‐type anodic oxidations,[12] we became interested in the electrochemical activation of D–A cyclopropanes by single electron transfer (SET) initiation (Scheme 1 D). According to the hypothesis, the single electron oxidation or reduction would result in the formation either of radical cationic or anionic species, both of which are unexplored reactive intermediates of D–A cyclopropanes. Considering the fact that synthetic electrochemistry is experiencing a renaissance steered by environmental and synthetic challenges,[13] an electroactivation of D–A cyclopropanes is highly desired. Herein, we report the electrocatalytic opening of D–A cyclopropanes by anodic oxidation (see Scheme 1 D top, a reductive pathway has not yet been realized), provide experimental and theoretically corroborated mechanistic insights and apply this concept to 1,3‐bisfunctionalization with triplet oxygen.
Scheme 1.
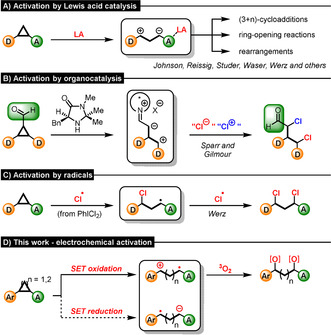
Overview of known strategies A)–C) for the activation of D–A cyclopropanes and D) our alternative electrochemical approach.
Results and Discussion
Our initial investigations of the anodic activation started with simple cyclic voltammetry experiments on various D–A cyclopropanes 1. The cyclic voltammograms of compounds bearing electron‐neutral (1 a, 1 f, Figure 1) and more electron‐rich (1 d, 1 j) arene donors gave clear indications of an irreversible redox process. Furthermore, the cyclopropanes followed the expected trend showing decreased oxidation potentials (from E 1/2=1.67 V for 1 f to E 1/2=1.37 V for 1 j, vs. Fc/Fc+ in HFIP) with increased electron richness.[14] Based on these results, the activation of such compounds to their radical cations by direct electrolysis at an anode is very likely. In order to establish a model reaction for this concept, we decided to use triplet oxygen as a reaction partner because of its abundance, cheapness and diradical character,[15] which leads to reactions with both radical cations and radical anions.[16]
Figure 1.
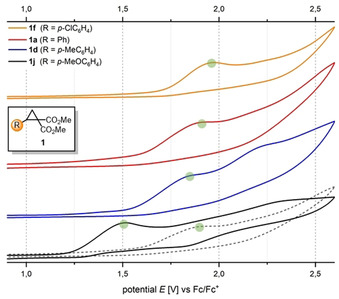
Cyclic voltammograms of D–A cyclopropanes 1 a, 1 d, 1 f and 1 j recorded in HFIP (solid line) and MeCN (dashed lines); NBu4BF4 (0.02 m) as electrolyte.
Our screening for the electrochemical incorporation of molecular oxygen started with the search for conditions that allow selective oxidation of the model substrate 1 a. For that purpose we decided to use the IKA ElectraSyn 2.0 apparatus and chose hexafluoroisopropanol (HFIP) as solvent, since it had been reported to be an excellent medium for electrochemical conversions by stabilizing redox intermediates.[17] When electrolyzing 1 a at a constant potential of +2.40 V at glassy carbon electrodes in an undivided cell, we obtained β‐hydroxy ketone 2 a in a yield of 88 % (Table 1, entry 1A). This provided evidence that a C(sp3)−C(sp3) cleavage in cyclopropane 1 a took place and that oxygen is a reasonable reaction partner for the model reaction. Efforts to use redox mediators such as DDQ (entry 2A) or CoTPP (entry 3A) only afforded traces of 2 a. Additionally, HFIP was found to be essential since other solvents such as MeCN or 2,2,2‐trifluoroethanol (TFE; entries 4A and 5A) led to poor reactivity. If the current was switched off (entry 6A), no conversion of the starting material 1 a was observed. Whereas cyclopropanes 1 d and 1 f were also successfully converted under standard conditions A, we encountered problems when applying these conditions to the electron‐rich cyclopropane 1 j (entry 1B). We assume that the anodic oxidation in HFIP would favor the formation of the radical cation at the oxygen of the para‐methoxy substituent by stabilizing interactions,[18] causing the cyclopropyl moiety to stay intact. While screening other solvents, the highest formation of the desired product 2 j using potentiostatic conditions was found to take place in aprotic MeCN albeit in a comparatively poor yield of 45 % (entry 2B). A higher yield of 60 % was obtained when using DDQ (entry 3B, standard conditions B) as a redox modulator at a constant potential of +1.30 V. Substoichiometric amounts of DDQ (entry 4B) led to lower yields, but gave evidence of DDQ acting catalytically. Potentiostatic electrolysis without a mediator (entry 5B, U=+1.30 V) or performing the reaction without current (entry 6B) left the starting material unchanged and confirmed that DDQ catalyzes the reaction only when current is applied. An even better yield was obtained when more expensive CoTPP was used for redox modulation (79 %, entry 7B).
Table 1.
Reaction optimizations.

|
Entry |
Deviation from standard conditions A |
Yield 2 a [%][a] |
|---|---|---|
|
1A |
none |
88 |
|
2A |
DDQ (100 mol %) at +1.30 V[b] |
traces |
|
3A |
CoTPP (2.5 mol %) at +2.00 V |
traces |
|
4A |
MeCN as solvent |
n.d. |
|
5A |
TFE as solvent |
traces |
|
6A |
no current |
n.d. |
|
Entry |
Deviation from standard conditions B |
Yield 2 j [%][a] |
|---|---|---|
|
1B |
standard conditions A |
n.d. |
|
2B |
no mediator at +2.40 V |
45 |
|
3B |
none |
60 |
|
4B |
DDQ (25 mol %) |
47 |
|
5B |
no mediator at +1.30 V |
n.d. |
|
6B |
no current |
n.d. |
|
7B |
CoTPP (2.5 mol %) at +2.00 V |
79 |
All reactions were stirred at rt, overnight and performed on scales of 150 μmol. No reference electrode was used to set the cell voltage. [a] Isolated yields, traces indicate minor conversion detected. [b] Performed in MeCN and HFIP, delivering traces of product 2 a in both reactions only. n.d.=not detected; GC=glassy carbon; DDQ=2,3‐dichloro‐5,6‐dicyano‐1,4‐benzoquinone; CoTPP=cobalt(II) tetraphenylporphyrin; TBA=tetra‐n‐butyl ammonium.
With optimized conditions in hand, we investigated the scope of the electrochemical insertion of oxygen by testing a variety of cyclopropanes under direct electrolysis conditions (Scheme 2, standard conditions A). Methyl substitution at the ortho‐, meta‐ and para‐positions of the aromatic donor led to the formation of the desired products 2 b–d in very good yields (81–85 %). para‐Halogenated compounds also delivered the β‐hydroxy ketones 2 e–g in high yields ranging from 80–86 %. When modifying the acceptor groups, the ethyl ester derivative 2 h (85 %) was obtained. Additionally, we successfully scaled up the reaction with model substrate 1 a (to 3.08 mmol, 83 %) and confirmed the structure of the product 2 a by X‐ray diffraction analysis. Next, we examined electron‐rich aromatic donors under indirect electrolysis conditions. Applying conditions B (DDQ, MeCN) afforded the thiophene‐containing product 2 i (60 %). For naphthyl 2 k (42 %, conditions C) and biphenyl 2 l (61 %) donors, DDQ modulation in HFIP facilitated selective product formation. In addition, the para‐acetoxy substituted product 2 m was isolated in 51 % yield. While efforts to convert 2 k–m by direct electrolysis led to complex product mixtures, no conversion was observed under conditions B.
Scheme 2.
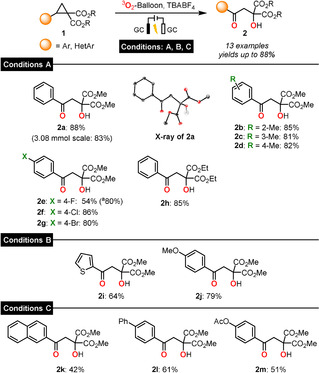
Scope of the electrosynthesis of β‐hydroxy ketones 2. [a] Yield when constant current electrolysis I=10 mA was applied. Conditions A=standard conditions A: cyclopropane (150 μmol), TBABF4 (0.02 m), HFIP (3 mL), O2‐atm., GC/GC, U=+2.40 V, rt; Conditions B=standard conditions B: cyclopropane (150 μmol, 1.0 equiv.), DDQ (1.0 equiv.), TBABF4 (0.02 m), MeCN (6 mL), O2‐atm., GC/GC, U=+1.30 V, rt; Conditions C: cyclopropane (150 μmol, 1.0 equiv.), DDQ (1.0 equiv.), TBABF4 (0.02 m), HFIP (3 mL), O2‐atm., GC/GC, U=+1.30 V, rt. No reference electrode was used to set the cell voltage.
Since D–A cyclobutanes have been reported to show similar reactivity to D–A cyclopropanes,[19] we wished to establish whether 4‐membered ring systems would also be converted when subjected to our established protocol. The direct electrolysis in HFIP of D–A cyclobutane 3 a (Scheme 3) bearing a phenyl donor proceeded smoothly to give the 1,2‐dioxane 4 a in an excellent yield of 99 %. Further investigations on ortho‐, meta‐ and para‐methyl‐substituted starting materials 3 b–d also led to the respective dioxanes (4 b–d: 75–86 %). In addition, reactions with para‐halogenated compounds afforded the desired products 4 e–g in good yields ranging from 68–74 %. We were surprised that cyclobutanes led to cyclic products while cyclopropanes converted to open‐chain molecules and therefore tested the feasibility of transforming 1,2‐dioxanes 4 into their acyclic isomers 5. This was indeed achieved under basic conditions using NEt3, enabling the formation of γ‐hydroxy ketones 5 a–g in up to quantitative yields via a Kornblum‐DeLaMare rearrangement.[20] The formation of 1,2‐dioxanes 4 and γ‐hydroxy ketones 5 was proved by X‐ray crystallography of 4 f and 5 a.
Scheme 3.
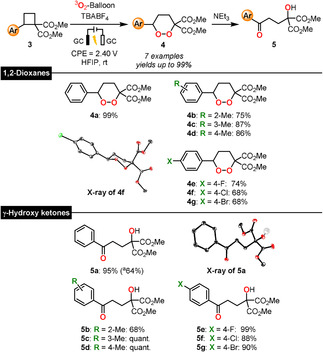
Scope of the electrosynthesis of 1,2‐dioxanes 4 and γ‐hydroxy ketones 5. [a] Yield for an one‐pot approach from 3 to 5. No reference electrode was used to set the cell voltage.
Based on a variety of mechanistic experiments and theoretical calculations, we propose the following plausible mechanism for the formation of oxidized products from electrochemically activated D–A cyclopropanes. Under direct electrolysis conditions (Scheme 4) the arene in 1 a is initially oxidized at the anode, leading to radical cation II by C(sp3)−C(sp3) cleavage of the cyclopropane moiety. Radical combination between II and triplet oxygen, followed by intramolecular 5‐exo‐trig cyclization, delivers intermediate IV. Oxidative activation of unreacted cyclopropane molecules 1 a via chain propagation furnishes the dioxolane 6 a as the final product of the electrocatalytic process. We propose analogous reaction intermediates bearing C4 chains for the electrosynthesis of 1,2‐dioxanes 4 from D–A cyclobutanes 3. Under assumed indirect conditions, the role of DDQ is not completely clear. Since we observed complex product mixtures resulting from overoxidation when applying direct electrolysis conditions to electron‐rich aryls, we assume that DDQ modulates the redox potential of such substrates (e.g. by π‐π stacking). Hence, a direct anodic oxidation at lower potential becomes possible, leading to more selective reactions. A pathway via cathodic reduction appears to be unlikely because of the low reducing power of anionic DDQ species and our following mechanistic investigation.
Scheme 4.
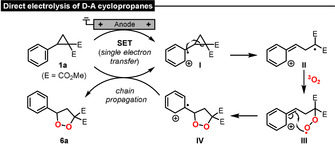
Mechanistic proposal for the direct anodic oxidation.
Our mechanistic investigations started with divided cell experiments (Scheme 5 A) in order to prove the postulated initial redox step. Conducting the reactions with 1 a/1 l in both the anodic and cathodic compartment under the previously described conditions A and C (see Scheme 2), evidence for an oxidative mechanism for the direct electrolysis and the DDQ‐assisted electrolysis was obtained. Furthermore, the radical cation species II was trapped in an electrophilic aromatic substitution reaction with toluene (Scheme 5 B). The isolated product 7 (63 %; p:o 8:1) confirms the formation of a benzyl cation within the proposed oxidative mechanism. Reactions with cyclopropanes 1 n/o (Scheme 5 C), which do not bear aromatic donors, led to no conversion when using direct electrolysis or the mediated strategy. These results indicate that the anodic oxidation at the aromatic system is crucial for success. Next, we examined the importance of the polarization of the activated C(sp3)−C(sp3) bond by reacting cyclopropanes bearing a dichloride 1 p and a dinitrile 1 q acceptor moiety. Since both substrates remained unreacted, the well‐established diester motif appears to be crucial for electrochemical ring cleavage. Additionally, when TEMPO (2.0 equiv.) was added under standard conditions A, the reaction was completely suppressed, suggesting that radical species are involved. By applying charge‐based constant voltage electrolysis, we investigated the time required for the starting material 1 a/1 j to fully convert (Scheme 5 D, for full data see Supporting Information). With respect to both strategies, full conversion of the starting materials 1 a/j was observed after providing only substoichiometric amounts (0.3 F mol−1) of electrons to the system. This unequivocally proves that radical chain propagation is involved. In order to explore the nature of the activating step (SET vs. HAT), we synthesized deuterated D–A cyclopropanes d‐1 a/j (Scheme 5 E). When converting these deuterated compounds under corresponding reaction conditions the deuterated dioxolanes (d‐6 a: 39 %, d‐6 j: 71 %) were exclusively obtained, confirming that no initial hydrogen atom transfer (HAT) takes place. Since DDQ is known to mediate hydrogen atom transfers[21] and could in principle transfer the abstracted deuterium atom back, H2O (10 equiv.) was additionally included to prevent such a pathway by H/D‐exchange. Because non‐deuterated dioxolanes are only observed as mixtures containing acyclic isomers, we became interested in the stability of the deuterated compounds d‐6 a/j. Quantum‐chemical investigations delivered a relatively high energy barrier (E a=51.3 kcal mol−1) for the spontaneous conversion of non‐deuterated dioxolane 6 a to β‐hydroxy ketone 2 a, which may indicate that tunneling plays a key role in this isomerization. Supplementary tunneling calculations (see Supporting Information) support this hypothesis. Electrochemical synthesis of compound 6 p (33 %) starting from the bisaryl‐substituted D–A cyclopropane 1 p suggested further that dioxolanes 6 are the final products of these electrocatalyic processes. Divided cell experiments, charge‐based electrolysis and conversion of deuterated material under direct electrolysis conditions using D–A cyclobutane 3 a or d‐3 a delivered analogous results.
Scheme 5.
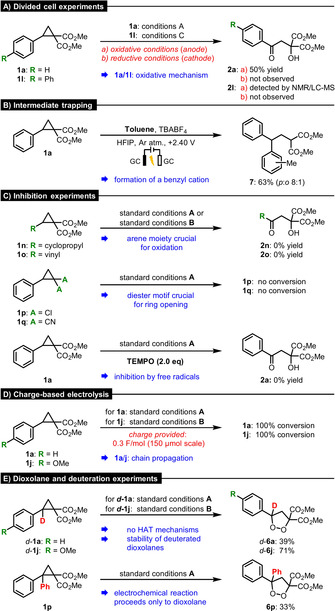
Experimental investigation of the mechanistic proposals.
The results of the quantum‐chemical calculations (DFT/TPSSh/def2‐TZVP, see Supporting Information for details) for the direct electrolysis of D–A cyclopropane 1 a (Scheme 6) are consistent with the proposed electrochemical activation mechanism. After the initiating SET, intermediate I is generated; the calculated Löwdin charges and spin densities confirm that the additional +1 charge and also the unpaired electron are mostly localized in the phenyl moiety as suggested above (see Scheme 4). In the following reaction steps, intermediates II, III and IV are formed in slightly exothermic reactions. The calculated spin densities agree with the expected radical positions according to Scheme 4 and the highest share of positive partial charge remains at the phenyl substituent. The calculated enthalpy differences in reaction steps 1 a → I (+147.3 kcal mol−1) and 4 → 6 a (−150.3 kcal mol−1) support an exothermic chain propagation process (net energy gain: −3.0 kcal mol−1) via SET.
Scheme 6.
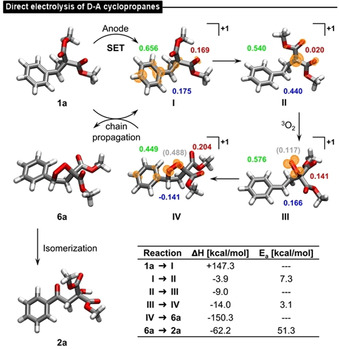
Results of quantum‐chemical calculations (DFT/TPSSh/def2‐TZVP) for the direct electrolysis of 1 a. Optimized intermediates are shown according to the proposed mechanism (Scheme 4). Spin densities are represented in orange isosurfaces. The contribution to the total charge deviation compared to 1 a is shown for the phenyl (green), cyclopropyl (blue) and the methyl ester (red) parts (contribution of added dioxygen shown in gray). Change in enthalpy and activation energies for each reaction step is given in the table.
Finally, we explored some follow‐up chemistry of the oxidized products derived from the electrocatalytic reactions. Starting with β‐hydroxy ketone 2 a (Scheme 7), a full reduction of the ketone to 8 was achieved by Pd/C‐catalyzed hydrogenolysis (90 %). In a simple condensation reaction between hydrazine and 2 a, the biologically interesting pyridazinone 9 was accessed in 81 % yield.[22] The structure of 9 was confirmed by X‐ray crystallography.[23] A three‐step one‐pot sequence (elimination, Diels–Alder reaction and cyclization/nitrogen insertion) via acetal 10 led to the formation of dihydropyridine 11 in 22 % yield. Further, we converted 1,2‐dioxane 4 a to diol 12 (68 %) by hydrogenolysis and synthesized oxime 13 (81 %) from γ‐hydroxy ketone 5 a. Reduction of 13 to its primary amine delivered the lactam 14, which contains a quaternary stereocenter, by spontaneous cyclization in 80 % yield.
Scheme 7.
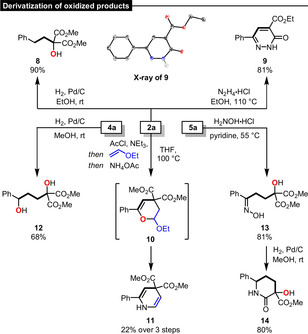
Derivatization of oxidized products 2 a, 4 a and 5 a.
Conclusion
We have developed strategies for the efficient insertion of molecular oxygen into D–A cyclopropanes and D–A cyclobutanes, which capitalize on the first electrocatalytic activation of these molecules. A diverse scope was established for the synthesis of β‐ and γ‐hydroxy ketones and 1,2‐dioxanes, containing aromatic donor groups. Mechanistic insights were gained by a variety of designed labelling, trapping, inhibition and half‐cell experiments. Additional DFT calculations, including tunneling studies, gave clear indications that radical cations can be accessed by electrochemical C(sp3)−C(sp3) cleavage. Finally, derivatization of the reaction products led to the simple formation of various nitrogen‐containing heterocycles. Our current efforts are focusing on further functionalizations of D–A cyclopropanes by electrochemical activation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the European Research Council (ERC Consolidator Grant “GAINBYSTRAIN” to D.B.W.). We thank Prof. Dr. Siegfried R. Waldvogel from University of Mainz for helpful advice on our initial electrochemical experiments. Open access funding enabled and organized by Projekt DEAL.
S. Kolb, M. Petzold, F. Brandt, P. G. Jones, C. R. Jacob, D. B. Werz, Angew. Chem. Int. Ed. 2021, 60, 15928.
Contributor Information
Simon Kolb, https://www.werzlab.de.
Prof. Dr. Christoph R. Jacob, Email: c.jacob@tu-braunschweig.de.
Prof. Dr. Daniel B. Werz, Email: d.werz@tu-braunschweig.de.
References
- 1.Gordon M. S., J. Am. Chem. Soc. 1980, 102, 7419. [Google Scholar]
- 2.
- 2a.Reissig H.-U., Zimmer R., Chem. Rev. 2003, 103, 1151; [DOI] [PubMed] [Google Scholar]
- 2b.Novikov R. A., Potapov K. V., Chistikov D. N., Tarasova A. V., Grigoriev M. S., Timofeev V. P., Tomilov Y. V., Organometallics 2015, 34, 4238; [Google Scholar]
- 2c.Werz D. B., Biju A. T., Angew. Chem. Int. Ed. 2020, 59, 3385; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3410. [Google Scholar]
- 3.
- 3a.Wenkert E., Alonso M. E., Buckwalter B. L., Chou K. J., J. Am. Chem. Soc. 1977, 99, 4778; [Google Scholar]
- 3b.Piers E., Reissig H.-U., Angew. Chem. Int. Ed. Engl. 1979, 18, 791; [Google Scholar]; Angew. Chem. 1979, 91, 857; [Google Scholar]
- 3c.Reissig H.-U., Hirsch E., Angew. Chem. Int. Ed. Engl. 1980, 19, 813; [Google Scholar]; Angew. Chem. 1980, 92, 839; [Google Scholar]
- 3d.Brückner C., Reissig H.-U., Angew. Chem. Int. Ed. Engl. 1985, 24, 588; [Google Scholar]; Angew. Chem. 1985, 97, 578. [Google Scholar]
- 4.
- 4a.Yu M., Pagenkopf B. L., Tetrahedron 2005, 61, 321; [Google Scholar]
- 4b.Schneider T. F., Kaschel J., Werz D. B., Angew. Chem. Int. Ed. 2014, 53, 5504; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5608; [Google Scholar]
- 4c.Carson C. A., Kerr M. A., Chem. Soc. Rev. 2009, 38, 3051; [DOI] [PubMed] [Google Scholar]
- 4d.Grover H. K., Emmett M. R., Kerr M. A., Org. Biomol. Chem. 2015, 13, 655; [DOI] [PubMed] [Google Scholar]
- 4e.O'Connor N. R., Wood J. L., Stoltz B. M., Isr. J. Chem. 2016, 56, 431; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4f.Singh P., Varshnaya R. K., Dey R., Banerjee P., Adv. Synth. Catal. 2020, 362, 1447; [Google Scholar]
- 4g.Cavitt M. A., Phun L. H., France S., Chem. Soc. Rev. 2014, 43, 804. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a.Lewis G. N., Valency and Structure of Atoms and Molecules, Wiley, New York, 1923; [Google Scholar]
- 5b.Corma A., García H., Chem. Rev. 2003, 103, 4307. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a.Benfatti F., de Nanteuil F., Waser J., Chem. Eur. J. 2012, 18, 4844; [DOI] [PubMed] [Google Scholar]
- 6b.Goldberg A. F. G., O'Connor N. R., Craig R. A., Stoltz B. M., Org. Lett. 2012, 14, 5314; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c.Zhu W., Fang J., Liu Y., Ren J., Wang Z., Angew. Chem. Int. Ed. 2013, 52, 2032; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2086; [Google Scholar]
- 6d.Mackay W. D., Fistikci M., Carris R. M., Johnson J. S., Org. Lett. 2014, 16, 1626; [DOI] [PubMed] [Google Scholar]
- 6e.Xu H., Hu J.-L., Wang L., Liao S., Tang Y., J. Am. Chem. Soc. 2015, 137, 8006; [DOI] [PubMed] [Google Scholar]
- 6f.Garve L. K. B., Petzold M., Jones P. G., Werz D. B., Org. Lett. 2016, 18, 564; [DOI] [PubMed] [Google Scholar]
- 6g.Racine S., Hegedüs B., Scopelliti R., Waser J., Chem. Eur. J. 2016, 22, 11997; [DOI] [PubMed] [Google Scholar]
- 6h.Kaicharla T., Roy T., Thangaraj M., Gonnade R. G., Biju A. T., Angew. Chem. Int. Ed. 2016, 55, 10061; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10215; [Google Scholar]
- 6i.Sabbatani J., Maulide N., Angew. Chem. Int. Ed. 2016, 55, 6780; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6892; [Google Scholar]
- 6j.Chidley T., Vemula N., Carson C. A., Kerr M. A., Pagenkopf B. L., Org. Lett. 2016, 18, 2922; [DOI] [PubMed] [Google Scholar]
- 6k.Dey R., Banerjee P., Org. Lett. 2017, 19, 304; [DOI] [PubMed] [Google Scholar]
- 6l.Chagarovskiy A. O., Vasin V. S., Kuznetsov V. V., Ivanova O. A., Rybakov V. B., Shumsky A. N., Makhova N. N., Trushkov I. V., Angew. Chem. Int. Ed. 2018, 57, 10338; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10495; [Google Scholar]
- 6m.Kreft A., Jones P. G., Werz D. B., Org. Lett. 2018, 20, 2059; [DOI] [PubMed] [Google Scholar]
- 6n.Augustin A. U., Busse M., Jones P. G., Werz D. B., Org. Lett. 2018, 20, 820; [DOI] [PubMed] [Google Scholar]
- 6o.Augustin A. U., Merz J. L., Jones P. G., Mlostoń G., Werz D. B., Org. Lett. 2019, 21, 9405; [DOI] [PubMed] [Google Scholar]
- 6p.Petzold M., Jones P. G., Werz D. B., Angew. Chem. Int. Ed. 2019, 58, 6225; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6291; [Google Scholar]
- 6q.Lücht A., Kreft A., Jones P. G., Werz D. B., Eur. J. Org. Chem. 2020, 2560; [Google Scholar]
- 6r.Ahlburg N. L., Jones P. G., Werz D. B., Org. Lett. 2020, 22, 6404; [DOI] [PubMed] [Google Scholar]
- 6s.Jacob A., Jones P. G., Werz D. B., Org. Lett. 2020, 22, 8720. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a.Schneider T. F., Kaschel J., Awan S. I., Dittrich B., Werz D. B., Chem. Eur. J. 2010, 16, 11276; [DOI] [PubMed] [Google Scholar]
- 7b.Kaschel J., Schmidt C. D., Mumby M., Kratzert D., Stalke D., Werz D. B., Chem. Commun. 2013, 49, 4403; [DOI] [PubMed] [Google Scholar]
- 7c.Ivanova O. A., Chagarovskiy A. O., Shumsky A. N., Krasnobrov V. D., Levina I. I., Trushkov I. V., J. Org. Chem. 2018, 83, 543; [DOI] [PubMed] [Google Scholar]
- 7d.Shim S. Y., Choi Y., Ryu D. H., J. Am. Chem. Soc. 2018, 140, 11184. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a.Shimada S., Hashimoto Y., Sudo A., Hasegawa M., Saigo K., J. Org. Chem. 1992, 57, 7126; [Google Scholar]
- 8b.Lifchits O., Charette A. B., Org. Lett. 2008, 10, 2809; [DOI] [PubMed] [Google Scholar]
- 8c.Goudreau S. R., Marcoux D., Charette A. B., J. Org. Chem. 2009, 74, 470; [DOI] [PubMed] [Google Scholar]
- 8d.de Nanteuil F., Loup J., Waser J., Org. Lett. 2013, 15, 3738; [DOI] [PubMed] [Google Scholar]
- 8e.Wales S. M., Walker M. M., Johnson J. S., Org. Lett. 2013, 15, 2558; [DOI] [PubMed] [Google Scholar]
- 8f.Xia Y., Lin L., Chang F., Fu X., Liu X., Feng X., Angew. Chem. Int. Ed. 2015, 54, 13748; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13952; [Google Scholar]
- 8g.Ivanov K. L., Villemson E. V., Budynina E. M., Ivanova O. A., Trushkov I. V., Melnikov M. Y., Chem. Eur. J. 2015, 21, 4975; [DOI] [PubMed] [Google Scholar]
- 8h.Karmakar R., Suneja A., Singh V. K., Org. Lett. 2016, 18, 2636; [DOI] [PubMed] [Google Scholar]
- 8i.Das S., Daniliuc C. G., Studer A., Org. Lett. 2016, 18, 5576; [DOI] [PubMed] [Google Scholar]
- 8j.Lücht A., Patalag L. J., Augustin A. U., Jones P. G., Werz D. B., Angew. Chem. Int. Ed. 2017, 56, 10587; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10723; [Google Scholar]
- 8k.Das S., Daniliuc C. G., Studer A., Angew. Chem. Int. Ed. 2017, 56, 11554; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11712; [Google Scholar]
- 8l.Richmond E., Vuković V. D., Moran J., Org. Lett. 2018, 20, 574; [DOI] [PubMed] [Google Scholar]
- 8m.Singh K., Bera T., Jaiswal V., Biswas S., Mondal B., Das D., Saha J., J. Org. Chem. 2019, 84, 710; [DOI] [PubMed] [Google Scholar]
- 8n.Das S., Daniliuc C. G., Studer A., Angew. Chem. Int. Ed. 2018, 57, 4053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4117; [Google Scholar]
- 8o.Lücht A., Sobottka S., Patalag L. J., Jones P. G., Reissig H.-U., Sarkar B., Werz D. B., Chem. Eur. J. 2019, 25, 10359; [DOI] [PubMed] [Google Scholar]
- 8p.Augustin A. U., Jones P. G., Werz D. B., Chem. Eur. J. 2019, 25, 11620; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8q.Guin A., Rathod T., Gaykar R. N., Roy T., Biju A. T., Org. Lett. 2020, 22, 2276. [DOI] [PubMed] [Google Scholar]
- 9.Sparr C., Gilmour R., Angew. Chem. Int. Ed. 2011, 50, 8391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8541. [Google Scholar]
- 10.
- 10a.Dickmeiss G., de Sio V., Udmark J., Poulsen T. B., Marcos V., Jørgensen K. A., Angew. Chem. Int. Ed. 2009, 48, 6650; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6778; [Google Scholar]
- 10b.Halskov K. S., Kniep F., Lauridsen V. H., Iversen E. H., Donslund B. S., Jørgensen K. A., J. Am. Chem. Soc. 2015, 137, 1685; [DOI] [PubMed] [Google Scholar]
- 10c.Sanchez-Diez E., Vesga D. L., Reyes E., Uria U., Carrillo L., Vicario J. L., Org. Lett. 2016, 18, 1270; [DOI] [PubMed] [Google Scholar]
- 10d.Halskov K. S., Næsborg L., Tur F., Jørgensen K. A., Org. Lett. 2016, 18, 2220; [DOI] [PubMed] [Google Scholar]
- 10e.Levens A., Ametovski A., Lupton D. W., Angew. Chem. Int. Ed. 2016, 55, 16136; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16370; [Google Scholar]
- 10f.Wallbaum J., Garve L. K. B., Jones P. G., Werz D. B., Chem. Eur. J. 2016, 22, 18756; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10g.Blom J., Vidal-Albalat A., Jørgensen J., Barløse C. L., Jessen K. S., Iversen M. V., Jørgensen K. A., Angew. Chem. Int. Ed. 2017, 56, 11831; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11993. [Google Scholar]
- 11.Garve L. K. B., Barkawitz P., Jones P. G., Werz D. B., Org. Lett. 2014, 16, 5804. [DOI] [PubMed] [Google Scholar]
- 12.Madelaine C., Six Y., Buriez O., Angew. Chem. Int. Ed. 2007, 46, 8046; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8192. [Google Scholar]
- 13.
- 13a.Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b.Wiebe A., Gieshoff T., Möhle S., Rodrigo E., Zirbes M., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 5594; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5694; [Google Scholar]
- 13c.Hilt G., ChemElectroChem 2020, 7, 395; [Google Scholar]
- 13d.Shatskiy A., Lundberg H., Kärkäs M. D., ChemElectroChem 2019, 6, 4067; [Google Scholar]
- 13e.Francke R., Little R. D., ChemElectroChem 2019, 6, 4373; [Google Scholar]
- 13f.Kawamata Y., Baran P. S., Joule 2020, 4, 701; [Google Scholar]
- 13g.Oyanagi S., Ishii H., Inagi S., Fuchigami T., J. Electrochem. Soc. 2020, 167, 155511; [Google Scholar]
- 13h.Wang F., Gerken J. B., Bates D. M., Kim Y. J., Stahl S. S., J. Am. Chem. Soc. 2020, 142, 12349; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13i.Pollok D., Waldvogel S. R., Chem. Sci. 2020, 11, 12386; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13j.Arndt S., Weis D., Donsbach K., Waldvogel S. R., Angew. Chem. Int. Ed. 2020, 59, 8036; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 8112; [Google Scholar]
- 13k.Dhawa U., Tian C., Wdowik T., Oliveira J. C. A., Hao J., Ackermann L., Angew. Chem. Int. Ed. 2020, 59, 13451; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 13553; [Google Scholar]
- 13l.Meyer T. H., Choi I., Tian C., Ackermann L., Chem 2020, 6, 2484; [Google Scholar]
- 13m.Chen H., Dong F., Minteer S. D., Nat. Catal. 2020, 3, 225. [Google Scholar]
- 14.For kinetic studies on D–A cyclopropanes see: Kreft A., Lücht A., Grunenberg J., Jones P. G., Werz D. B., Angew. Chem. Int. Ed. 2019, 58, 1955; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1975. [Google Scholar]
- 15.Borden W. T., Hoffmann R., Stuyver T., Chen B., J. Am. Chem. Soc. 2017, 139, 9010. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a.Feldman K. S., Parvez M., J. Am. Chem. Soc. 1986, 108, 1328; [Google Scholar]
- 16b.Mizuno K., Kamiyama N., Otsuji Y., Chem. Lett. 1983, 12, 477; [Google Scholar]
- 16c.Feldman K. S., Simpson R. E., Tetrahedron Lett. 1989, 30, 6985; [Google Scholar]
- 16d.Kulinkovich O., Astashko D., Tyvorskii V., Ilyina N., Synthesis 2001, 1453; [Google Scholar]
- 16e.Zhao Q., Wong H. N. C., Tetrahedron 2007, 63, 6296; [Google Scholar]
- 16f.Lu Z., Parrish J. D., Yoon T. P., Tetrahedron 2014, 70, 4270; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16g.Ge L., Wang D.-X., Xing R. I., Ma D., Walsh P. J., Feng C., Nat. Commun. 2019, 10, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16h.Xiong C., Cheng K., Wang J., Yang F., Lu J., Zhou Q., J. Org. Chem. 2020, 85, 9386; [DOI] [PubMed] [Google Scholar]
- 16i.Budde S., Goerdeler F., Floß J., Kreitmeier P., Hicks E. F., Moscovitz O., Seeberger P. H., Davies H. M. L., Reiser O., Org. Chem. Front. 2020, 7, 1789. [Google Scholar]
- 17.
- 17a.Francke R., Cericola D., Kötz R., Weingarth D., Waldvogel S. R., Electrochim. Acta 2012, 62, 372; [Google Scholar]
- 17b.Ayata S., Stefanova A., Ernst S., Baltruschat H., J. Electroanal. Chem. 2013, 701, 1; [Google Scholar]
- 17c.Imada Y., Röckl J. L., Wiebe A., Gieshoff T., Schollmeyer D., Chiba K., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 12136; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12312; [Google Scholar]
- 17d.Röckl J. L., Dörr M., Waldvogel S. R., ChemElectroChem 2020, 7, 3686. [Google Scholar]
- 18.
- 18a.Ammer J., Mayr H., J. Phys. Org. Chem. 2013, 26, 59; [Google Scholar]
- 18b.Colomer I., Chamberlain A. E. R., Haughey M. B., Donohoe T. J., Nat. Rev. Chem. 2017, 1, 0088. [Google Scholar]
- 19.
- 19a.Shimada S., Saigo K., Nakamura H., Hasegawa M., Chem. Lett. 1991, 20, 1149; [Google Scholar]
- 19b.Parsons A. T., Johnson J. S., J. Am. Chem. Soc. 2009, 131, 14202; [DOI] [PubMed] [Google Scholar]
- 19c.Allart E. A., Christie S. D. R., Pritchard G. J., Elsegood M. R. J., Chem. Commun. 2009, 7339; [DOI] [PubMed] [Google Scholar]
- 19d.Moustafa M. M. A. R., Stevens A. C., Machin B. P., Pagenkopf B. L., Org. Lett. 2010, 12, 4736; [DOI] [PubMed] [Google Scholar]
- 19e.Shenje R., Martin M. C., France S., Angew. Chem. Int. Ed. 2014, 53, 13907; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14127; [Google Scholar]
- 19f.Vemula N., Stevens A. C., Schon T. B., Pagenkopf B. L., Chem. Commun. 2014, 50, 1668; [DOI] [PubMed] [Google Scholar]
- 19g.Perrotta D., Racine S., Vuilleumier J., de Nanteuil F., Waser J., Org. Lett. 2015, 17, 1030; [DOI] [PubMed] [Google Scholar]
- 19h.Hu J.-L., Wang L., Xu H., Xie Z., Tang Y., Org. Lett. 2015, 17, 2680; [DOI] [PubMed] [Google Scholar]
- 19i.Reissig H.-U., Zimmer R., Angew. Chem. Int. Ed. 2015, 54, 5009; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5093; [Google Scholar]
- 19j.Feng L.-W., Ren H., Xiong H., Wang P., Wang L., Tang Y., Angew. Chem. Int. Ed. 2017, 56, 3055; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3101; [Google Scholar]
- 19k.Ahmed A., Christie S. D. R., Pritchard G. J., Tetrahedron Lett. 2017, 58, 3028; [Google Scholar]
- 19l.Kreft A., Ehlers S., Jones P. G., Werz D. B., Org. Lett. 2019, 21, 6315. [DOI] [PubMed] [Google Scholar]
- 20.Kornblum N., DeLaMare H. E., J. Am. Chem. Soc. 1951, 73, 880. [Google Scholar]
- 21.For studies of DDQ on redox reactions see:
- 21a.Chiba K., Arakawa T., Tada M., J. Chem. Soc. Perkin Trans. 1 1998, 2939; [Google Scholar]
- 21b.Hubig S. M., Rathore R., Kochi J. K., J. Am. Chem. Soc. 1999, 121, 617; [Google Scholar]
- 21c.Utley J. H. P., Rozenberg G. G., J. Appl. Electrochem. 2003, 33, 525; [Google Scholar]
- 21d.Francke R., Little R. D., Chem. Soc. Rev. 2014, 43, 2492; For cyclic voltammetry measurements of DDQ see: [DOI] [PubMed] [Google Scholar]
- 21e.Memarian H. R., Ghazaie M., Mehneh S. K., Z. Naturforsch. 2009, 64b, 1187; [Google Scholar]
- 21f.Röse P., Emge S., König C. A., Hilt G., Adv. Synth. Catal. 2017, 359, 1359; For interactions of DDQ with electron-rich arenes see: [Google Scholar]
- 21g.Das S., Natarajan P., König B., Chem. Eur. J. 2017, 23, 18161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ragusa G., Gómez-Cañas M., Morales P., Rodríguez-Cueto C., Pazos M. R., Asproni B., Cichero E., Fossa P., Pinna G. A., Jagerovic N., Fernández-Ruiz J., Murineddu G., Eur. J. Med. Chem. 2017, 127, 398. [DOI] [PubMed] [Google Scholar]
- 23.Deposition Numbers 2058833 (for 2a), 2058834 (for 4f), 2058835 (for 5a), and 2058836 (for 9) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary