

Abstract
Exploiting cooperative effects between Na and FeII centres present in tris(amide) ferrate complexes has led to the chemoselective ferration of pentafluorobenzene, benzene, toluene, anisole, and pyridine being realised at room temperature. The importance of this bimetallic partnership is demonstrated by neither the relevant sodium amide (NaHMDS or NaTMP) nor the FeII amide Fe(HMDS)2 efficiently metallating these substrates under the conditions of this study. By combining NMR studies with the isolation of key intermediates and DFT calculations, we offer a possible mechanism for how these reactions take place, uncovering a surprising reaction pathway in which the metals cooperate in a synchronised manner. Although the isolated products are formally the result of Fe‐H exchange, theoretical calculations indicate that the aromatic substrates undergo Na‐H exchange followed by fast intramolecular transmetallation to Fe, thus stabilizing the newly generated aryl fragment.
Keywords: cooperative effects, DFT calculations, iron, metallation, sodium
Combining iron with sodium in tris(amido) complexes has led to the selective ferration of non‐activated arenes being accomplished. A new cooperative mechanism is disclosed, whereby the sodium performs the metal‐H exchange and this is followed by a fast intramolecular trans‐metal trapping of the aryl anion by the iron centre.
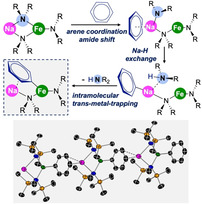
Heterobimetallic amide bases containing a complementary combination of an alkali metal with a less electropositive metal, such as Mg or Zn, have emerged as a powerful class of reagents for selectively deprotonating functionalised arenes.[1] Profiting from the cooperation between the two metals, these bimetallic strategies can offer greater functional group tolerance and special regioselectivities that conventional single‐metal bases cannot match.[2] Through the isolation and characterisation of informative organometallic intermediates from these reactions, Mulvey and co‐workers coined the concept of alkali‐metal‐mediated metallation (AMMM),[3] which enables the direct magnesiation (M=Mg) or zincation (M=Zn) of a wide range of aromatic substrates. For these processes, cooperative effects between the metals permit, in the most spectacular cases, the classical directed‐ortho‐metallation (DoM)[4] regioselectivities to be overriden.[1] These studies suggest that the aromatic substrate coordinates to the alkali metal, thereby entropically favoring the reaction and fixing the regioselectivity of the metalation, which is completed by the kinetically activated magnesiate (or zincate) moiety.[5] By expanding the scope of AMMM beyond main‐group chemistry, we recently reported that pairing the iron(II) amide Fe(HMDS)2 (HMDS=N{SiMe3}2) with NaHMDS in the same coordination compound allows regioselective ferration (Fe‐H exchange) of a wide range of fluoroarenes.[6] Since sodium is mandatory for the metallation to occur, these reactions can be described as examples of alkali‐metal‐mediated ferration (AMMFe) processes. This reactivity is surprising considering the lower polarity of Fe−N vs. Na−N bonds, which is why applications of iron amides in metallation are rare.[7]
Herein, we take a closer look into the mechanism involved in AMMFe reactions, assessing the role that the amide groups and the donor solvents play for the success of the Fe‐H exchange process. We also provide the first theoretical insights on how the bimetallic cooperation between Na and Fe is enabled. Furthermore, building on this new‐found knowledge, we introduce a more powerful bimetallic system that is capable of accomplishing the ferration of aromatic substrates that are significantly less activated than fluoroarenes.
We began our studies by investigating the reactivity of sodium tris(amido)ferrate [NaFe(HMDS)3] (1)[8] with pentafluorobenzene (C6F5H). This substrate was chosen due to its high degree of fluorination, since C−F functionalisation can compete with C−H bond metallation.[9] The reaction led to the isolation of [Na(HMDS)2Fe(C6F5)]∞ (2) as a yellow crystalline solid in 89 % yield as a result of the metallation of C6F5H (Figure 1 i). The reaction was monitored by 1H NMR spectroscopy and showed the concomitant formation of HMDS(H).
Figure 1.
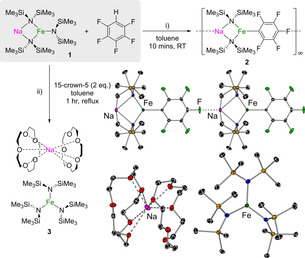
Reactivity of [NaFe(HMDS)3] (1) with C6F5H in: i) toluene and ii) in toluene containing 2 equiv 15‐crown‐5 to give 2 and 3, respectively.
X‐ray crystallographic studies confirmed the ferrated nature of the product (Figure 1), with the C−H bond replaced by a Fe−C σ‐bond. Whereas the pentafluoroaryl group binds terminally to the Fe centre, the F atom located in the sterically optimal C4‐position binds to the Na centre of a neighbouring unit, thereby giving rise to a polymeric arrangement. In each monomeric unit, the Na and Fe centres connect via two amide N bridges, with Na achieving further coordinative saturation by interacting anagostically with a Me group from each HMDS ligand (Figure 1).
The observed C−H chemoselectivity in the reaction of 1 with C6F5H is most intriguing considering previous reports involving homometallic FeII and Fe0 complexes, which react preferentially with this substrate at one of its C−F bonds.[10] This C−F bond preference has also been found for other transition metal complexes, especially in reactions with substrates having a high degree of fluorination.[9] Unlike these examples, here the reaction seems to be selective for the seemingly more activated H atom in C6F5H (in terms of the pK a value), although it should be noted that Fe(HMDS)2 on its own fails to react with this substrate even under refluxing conditions, thus confirming its poor metallating ability. On the other hand, NaHMDS does react with C6F5H, but in an unselective manner, generating a mixture of decomposition products including the salt NaF. Thus, although these findings demonstrate that the formation of 2 is cooperative in origin and can be categorised as an AMMFe process, the way that the Na/Fe cooperativity works remains unclear.
To shed some light on the possible mechanism of this Fe‐H exchange reaction, we next carried out the reaction of 1 with C6F5H in the presence of 2 equiv of the chelating crown ether 15‐crown‐5, which has a high sequestering ability for sodium cations. Under these conditions, no ferration was observed and instead, the crown‐separated sodium ferrate [{Na(15‐crown‐5)2}+{Fe(HMDS)3}−] (3) was obtained in 98 % yield (Figure 1 ii). Increasing the temperature to 110 °C for 1 h led to the same product, which suggests that the proximity of the two metals and the coordinative unsaturation of the sodium cation in 1 play an essential role. These findings also demonstrate that the anionic activation of the iron centre in the ferrate {Fe(HMDS)3}− is not sufficient on its own to promote the ferration of C6F5H.
Intrigued by the above findings, we next investigated the Fe‐H exchange mechanism using density functional theory calculations (see the Supporting Information for details). First, we modelled the co‐complexation of [{NaHMDS}3][11] with 3 equiv of Fe(HMDS)2 in toluene to give 3 equiv of sodium ferrate 1, a reaction that is exergonic by −4.5 kcal mol−1. Calculations indicated that this reaction becomes more favourable when toluene is coordinated to Na, leading to [(toluene)NaFe(HMDS)3] with a Gibbs energy in solution of −17.1 kcal mol−1. Building upon previous studies on cooperative bimetallic complexes,[1] we envisaged the ferration of C6F5H taking place by an initial coordination of the fluoroarene to the sodium centre through Na⋅⋅⋅F dative interactions (as shown in Figure 2 for I2) followed by cleavage of one Na−Namide bridging bond to facilitate Fe‐H exchange by the {Fe(HMDS)3}− anion. However, all attempts to model this reaction pathway led to sodiation of C6F5H as a result of the bulkiness of the HMDS ligand (see the Supporting Information). The lowest transition state found for this process (TS4 in Figure 2), after considering various structures with and without the presence of toluene coordinated to the Na centre, has an overall energy barrier of 34.7 kcal mol−1. This high activation energy seems incompatible with the experimental formation of 2 at room temperature, thus casting doubt on the viability of this pathway.
Figure 2.
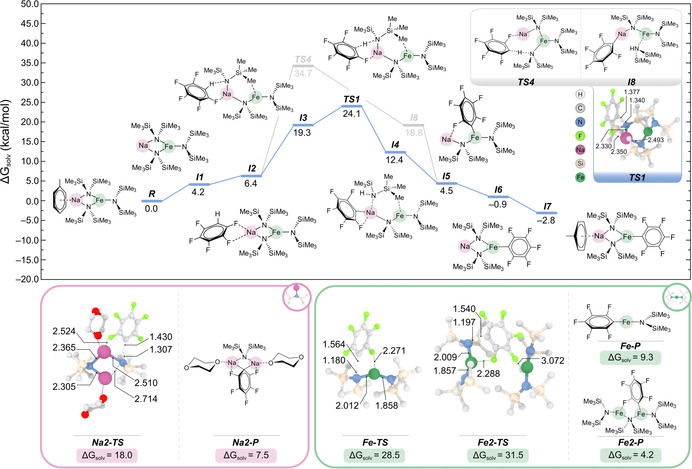
Top panel: Gibbs energy profile calculated in toluene at 298 K and 1 atm for the reaction of 1 with C6F5H using [(toluene)NaFe(HMDS)3] and [NaFe(HMDS)3] as active species. Bottom panel: Optimized structures of the transition states and reaction products involved in the metallation of C6F5H using the homometallic complexes [{(dioxane)Na(HMDS)}2] (red rectangle), [{Fe(HMDS)2}2], and Fe(HMDS)2 (green rectangle). Relevant bond distances [in Å] are also shown.
An alternative path with a much lower energy barrier was found after the dissociation of one bridging Fe−Namide bond in I2 to give I3, where the metal centres are connected by only one HMDS bridge. This pits the Lewis acidity of Fe against the unsaturated coordination of Na, and clearly Na wins out. In this intermediate, the amide bound terminally to Na is in prime position to perform the metallation of C6F5H, which affords I4 in a process that is exergonic by 6.9 kcal mol−1. This step occurs via TS1 with an activation energy of 24.1 kcal mol−1, which is in line with the mild experimental conditions for the synthesis of 2. Whereas this metallation is a Na‐H exchange, the intramolecular transmetallation of the fluoroaryl group by Fe was seen in a barrierless process during the geometry optimisation of I4 after removal of the protonated HMDS(H). This resulted in the formation of I5, wherein the fragile pentafluoroaryl anion is trapped and stabilised through formation of a more covalent Fe−C bond and a Na⋅⋅⋅F dative bond. In fact, I5 could be isolated experimentally (and structurally characterised) when the reaction was carried out in the presence of one equivalent of the donor 1,4‐dioxane (see 2‐diox in the Supporting Information). However, in neat toluene, the isomerization of I5 to I6, where the fluoroaryl group occupies a terminal site on Fe (as in 2), is energetically favoured by 7.4 kcal mol−1. Finally, coordination of toluene to Na gives I7, which drives the thermodynamics of the overall reaction forward, thereby making the ferration of C6F5H exergonic by −2.8 kcal mol−1.
The above mechanism explains the experimental isolation and characterisation of 2, as well as the deactivation seen on adding 15‐crown‐5 (Figure 1). The latter can be rationalised with Na being coordinatively saturated, which prevents coordination of the substrate and the terminal bonding of HMDS, as shown in I3. Hence, our DFT studies uncover two distinct but equally important metal roles for the successful outcome of the ferration reaction. In particular, sodium takes a central role in promoting the initial metallation, whereas Fe is crucial for stabilising the newly formed aryl anion. This scenario is reminiscent of the one previously described by Mulvey and co‐workers, as well as by us, in trans‐metal‐trapping (TMT) processes when LiTMP is mixed with a Group 13 complex, for example, Ga(CH2SiMe3)3.[12] However, these combinations fail to form a bimetallic base because of the steric incompatibility of the homometallic components, although they can still cooperate in a stepwise manner to deliver low‐polarity metallation. In this case, the lithium amide deprotonates the substrate, and the relevant aryllithium can then undergo co‐complexation with the Ga‐alkyl group to give a lithium gallate. An important difference between that system and the one reported herein is that dissociation between NaHMDS and Fe(HMDS)2 is not observed.[13] Instead, it is the cleavage of one Fe−N bridge in I2 which facilitates the formation of I3 and triggers the subsequent sodiation of the substrate. During this process, the Na and Fe centres remain connected by an amide bridge, which favours the transmetallation step. Furthermore, I3 can be envisaged as a pseudo‐monomeric sodium amide, in the sense that the Na binds to a terminal HMDS. Thus, I3 can be expected to be significantly more reactive in toluene than neat NaHMDS, which is a disolvated dimer.[14]
For comparison, the deprotonation of C6F5H was also modelled using the homometallic systems [{(dioxane)Na(HMDS)}2], which in the crystal exhibits a dimeric motif (see the Supporting Information for X‐ray crystallographic details), and Fe(HMDS)2. Iron amide Fe(HMDS)2 is also dimeric in the solid state, and in solution is in equilibrium with its monomeric variant.[15] Interestingly, all these reactions turn out to be endergonic by more than 4 kcal mol−1 (i.e. 7.5 kcal mol−1 for Na2‐P, 9.3 and 4.2 kcal mol−1 for Fe‐P and Fe2‐P, respectively), as seen in Figure 2 (see also Figures S3 and S4). Furthermore, the ferration reactions involved significantly higher energy transition states (28.0 and 31.5 kcal mol−1) than that calculated using bimetallic complex 1, whereas the sodiation reaction was found to have an activation barrier of only 18.0 kcal mol−1 (Na2‐TS), thereby proving the high reactivity of sodium amides. These findings highlight the important role of the Na amide in the ferration process, which involves the activation of the C−H bond after forming NaFe(HMDS)3 and the dissociation of the Fe−Namide bond.
We next pondered the effect of replacing NaHMDS by the more basic and more sterically encumbered NaTMP.[11] Thus, we first studied the co‐complexation of NaTMP with Fe(HMDS)2 in toluene, using the same approach as for the synthesis of 1. Mixing both homometallic components led to the instantaneous formation of a deep‐red solution, which deposited yellow crystals of the benzyl complex [Na(HMDS)2Fe(CH2Ph)]∞ (4) in 44 % yield, as a result of the lateral metallation of toluene (Figure 3 iii). Furthermore, switching to benzene, with its less‐activated hydrogen atoms, resulted in the formation of the phenyl ferrate [Na(HMDS)2Fe(Ph)]∞ (5) in 63 % yield (Figure 3 iv).
Figure 3.
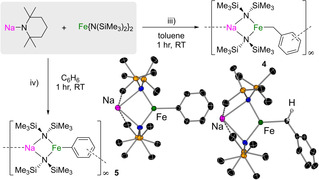
Ferration of: iii) toluene and iv) benzene using an equimolar cooperative NaTMP and Fe(HMDS)2 combination to yield 4 and 5.
Both 4 and 5 exhibit 1D polymeric structures in the solid state (see the Supporting Information for details), made up by monomeric four‐atom {NaNFeN} rings to which the metallated fragment (benzyl or Ph anion) coordinates terminally to the Fe centre. Polymerisation takes place through π‐coordination between the Na centre and the aromatic ring of an adjacent molecular unit.
Although the metallating power of the NaTMP/Fe(HMDS)2 partnership has been demonstrated, it should be noted that NaTMP on its own is inert towards the metallation of the above arenes. Of further significance, sodium ferrate 1 does not metallate these two solvents even under refluxing conditions. These findings can be rationalised by the initial formation of heteroleptic [NaFe(TMP)(HMDS)2] (I) by co‐complexation of the homometallic components, a process that DFT calculations predict to be exergonic by −5.1 kcal mol−1. In solution, this complex may exist as [(toluene)NaFe(TMP)(HMDS)2] with an even more favourable co‐complexation energy of −21.0 kcal mol−1 (see Figure S5 and Scheme S1). In contrast, previous studies found that heteroleptic [(TMEDA)NaFe(TMP)R2] (R=CH2SiMe3) fails to deprotonate benzene.[16]
A similar mechanism as that proposed for 1 can also be proposed for [NaFe(TMP)(HMDS)2] (I),[17] with coordination of the arene to the Na centre to facilitate its sodiation followed by fast intramolecular transmetallation to Fe to give the ferration product. However, the heteroleptic nature of this complex adds another level of complexity, as ligand scrambling may give rise to different active species in solution. Consequently, all possible complexes which might form in situ had to be considered in the modelling of the reaction mechanism. Among these species, [NaFe(TMP)3] (II) exhibits the lowest overall barrier of 24.0 kcal mol−1 (see the Supporting Information for mechanistic details, including ligand redistribution processes involving mixed Na/Fe species, Figure S9).
Next, we extended the study to anisole and pyridine. The former is a benchmark substrate in DoM chemistry,[4, 18] whereas pyridine is a more challenging substrate to metallate. This is mainly due to the lack of stability of its metalllated intermediates, even at extremely low temperatures. In addition, it is difficult to control the regioselectivity of the metallation process in the absence of other directing groups.[19] Pleasingly, both substrates undergo formal Fe‐H exchange at room temperature in hexane to give heterobimetallic [{(TMPH)Na2Fe(C6H4OMe)(TMP)2}+{Fe(HMDS)3}−] (6) and [(TMEDA)2Na(3‐C6H4N)Fe(HMDS)2] (7; Scheme 1 and Figure 4).
Scheme 1.
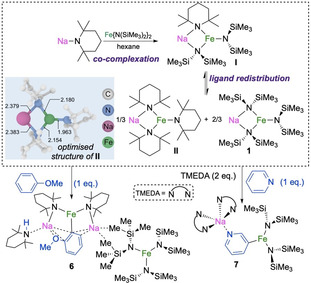
Ligand redistribution process for [NaFe(TMP)(HMDS)2] (I), modelled structure of [NaFe(TMP)3] (II), and ortho‐ and C3‐ferration of anisole and pyridine in hexane to produce 6 and 7, respectively.
Figure 4.
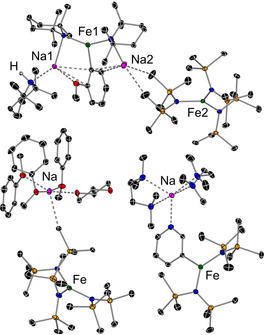
Molecular structures of 6 (top), 7 (bottom right), and 8 (bottom left) with displacement ellipsoids at 30 % probability; all H atoms are omitted.
X‐ray crystallographic studies confirmed the ortho‐ferration of anisole in 6, revealing a complex pseudo‐solvent‐separated ion‐pair structure. Its cation comprises an FeII centre bound to two TMP ligands and the ortho‐metallated anisyl fragment through its carbon atom, occupying the position vacated by a H atom (Figure 4 a). Each TMP acts as a bridge between the Fe and Na centres, with Na1 coordinating also to the OMe group and the anisyl‐Cipso atom as well as to TMP(H). In contrast, Na2 engages with two C atoms of the anisyl ring through π‐interactions and forms two electrostatic anagostic interactions with two Me groups of different HMDS ligands belonging to the {Fe(HMDS)3}− moiety of 6.
The constitution of 6 suggests that anisole is metallated by a putative [NaFe(TMP)3] (II) species, as predicted by DFT calculations with an overall barrier of 22.0 kcal mol−1 (Figure S11), similar to the aforementioned toluene reaction. The formation of 6 can be attributed to a possible ligand redistribution of [NaFe(TMP)(HMDS)2] (I) to give homoleptic sodium ferrates II and 1, as shown in Scheme 1 (for DFT details, see Schemes S2–S4 and S6). Furthermore, as II deprotonates anisole, the one equivalent of TMP(H) released acts as a donor for the Na cation to generate [{(TMPH)NaFe(C6H4OMe)(TMP)2], which in turn undergoes co‐complexation with 1 to give 6 (Scheme 1), a stable species which does not undergo any further ligand exchange (see the Supporting Information). Since 6 is stable and does not undergo ligand redistribution to regenerate the active [NaFe(TMP)3] base, only 0.33 equiv of anisole undergo ferration. This is consistent with the yield of isolated 6 (16 %, see the Supporting Information). The key role of the sodium amide in these ferration processes is further reinforced by the outcome of the reaction of [(dioxane)0.5NaFe(HMDS)3] (1‐diox) with anisole, which produces the coordination adduct [(PhOMe)3Na(diox)0.5}+{Fe(HMDS)3}−]2 (8), wherein anisole acts as a Lewis donor and its H atoms are intact, thereby forming the ion‐pair structure in Figure 4.
Despite the common presence of the pyridine ring in synthetically relevant molecules, the deprotonation of bare pyridine still remains a difficult task.[19] By combining BuLi with a lithium alkoxide at −78 °C using non‐polar hexane as a solvent, Caubère and co‐workers have reported the selective C2 metallation of pyridine in 90 % yield.[19a] Interestingly, here, reacting equimolar amounts of pyridine, NaTMP, and Fe(HMDS)2 at room temperature permitted isolation of sodium ferrate 7 in 72 % yield. Its structure shows a unique C3‐ferration of the heterocycle (Scheme 1 and Figure 4).
XRD studies showed that Fe binds to C3 of the newly generated pyridyl anion, whereas Na is attached to the N atoms and solvated by two chelating TMEDA ligands, which were added to aid crystallization. Unlike 4 and 5, both HMDS groups in 7 terminally bind to the Fe centre. In contrast with the known thermal instability of lithiated pyridine intermediates, which are usually generated at extremely low temperatures, 7 is stable in solution at room temperature. To assess the regioselectivity of the ferration process, pyridine was treated with an equimolar mixture of NaTMP and Fe(HMDS)2, and the mixture was quenched with D2O. Full conversion in terms of pyridine metalation was observed, and a 2.2:1 mixture of C3‐ and C4‐deuteropyridine was obtained. This preference for C3 not only contrasts with the regioselectivities previously reported (see above),[19] but also with those predicted for the metallation of pyridine in the gas phase (70–80 % deprotonation at the C4‐position and 20–30 % at C3).[20] Note that tris(HMDS) sodium ferrate is inert towards pyridine metallation and, instead, disproportionates into its single metal components [(py)(NaHMDS)]2 and [(py)2Fe(HMDS)2] (see the Supporting Information).
To conclude, a previously unconsidered mechanism for the ferration of aromatic substrates, using sodium ferrates as bases, has been posited. This bimetallic approach extends beyond fluoroarenes to less‐activated aromatic substrates, thereby demonstrating the power of these Na/Fe partnerships to promote chemical transformations which neither Na nor FeII amides are capable of achieving on their own. Future work will determine whether the mechanisms involved in AMMM depend subtly on the identity of the alkali metal and the metal centre, or whether the new mechanism posited here has more general applicability.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Robert Mulvey (University of Strathclyde) for his insightful comments on the work. Thanks also to the University of Bern and the SNF (188573 and 206021_177033) for the generous sponsorship of this research. The DJEI/DES/SFI/HEA Irish Centre for High‐End Computing (ICHEC) is acknowledged for generous provision of computational facilities and support. We also thank Martin Albrecht and Albert Farré (University of Bern) for their help with UV/Vis reaction monitoring studies.
L. C. H. Maddock, M. Mu, A. R. Kennedy, M. García-Melchor, E. Hevia, Angew. Chem. Int. Ed. 2021, 60, 15296.
In memory of Victor Riera
Contributor Information
Prof. Max García‐Melchor, Email: garciamm@tcd.ie.
Prof. Dr. Eva Hevia, Email: eva.hevia@dcb.unibe.ch.
References
- 1.Robertson S. D., Uzelac M., Mulvey R. E., Chem. Rev. 2019, 119, 8332. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a.Harrison-Marchand A., Mongin F., Chem. Rev. 2013, 113, 7470; [DOI] [PubMed] [Google Scholar]
- 2b.Mongin F., Harrison-Marchand A., Chem. Rev. 2013, 113, 7563. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a.Armstrong D. R., Clegg W., Dale S. H., Hevia E., Hogg L. M., Honeyman G. W., Mulvey R. E., Angew. Chem. Int. Ed. 2006, 45, 3775; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 3859; [Google Scholar]
- 3b.Martinez-Martinez A. J., Kennedy A. R., Mulvey R. E., O'Hara C. T., Science 2014, 346, 834. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.Beak P., Meyers A. I., Acc. Chem. Res. 1986, 19, 356; [Google Scholar]
- 4b.“The Directed ortho Metalation Reaction. A Point of Departure for New Synthetic Aromatic Chemistry”: Hartung C. G., Snieckus V. in Modern Arene Chemistry (Ed.: Astruc D.), Wiley-VCH, New York, 2002, pp. 330–367. [Google Scholar]
- 5.For recent reviews in s-block bimetallic compounds, see
- 5a.Gil-Negrete J. M., Hevia E., Chem. Sci. 2021, 12, 1982; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b.Gentner T. X., Mulvey R. E., Angew. Chem. Int. Ed. 2021, 60, 9247; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 9331. [Google Scholar]
- 6.Maddock L. C. H., Nixon T., Kennedy A. R., Probert M. R., Clegg W., Hevia E., Angew. Chem. Int. Ed. 2018, 57, 187; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 193. [Google Scholar]
- 7.Wunderlich S. H., Knochel P., Angew. Chem. Int. Ed. 2009, 48, 9717; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9897. [Google Scholar]
- 8.
- 8a.Maddock L. C. H., Cadenbach T., Kennedy A. R., Borilovic I., Aromi G., Hevia E., Inorg. Chem. 2015, 54, 9201; [DOI] [PubMed] [Google Scholar]
- 8b.Maddock L. C. H., Borilovic I., McIntyre J., Kennedy A. R., Aromi G., Hevia E., Dalton Trans. 2017, 46, 6683. [DOI] [PubMed] [Google Scholar]
- 9.Eisenstein O., Milani J., Perutz R. N., Chem. Rev. 2017, 117, 8710. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a.Vela J., Smith J. M., Yu Y., Ketterer N. A., Flaschenriem C. J., Lachicotte R. J., Holland P. L., J. Am. Chem. Soc. 2005, 127, 7857; [DOI] [PubMed] [Google Scholar]
- 10b.Xu X., Sun H., Shi Y., Jia J., Li X., Dalton Trans. 2011, 40, 7866. [DOI] [PubMed] [Google Scholar]
- 11.Mulvey R. E., Robertson S. D., Angew. Chem. Int. Ed. 2013, 52, 11470; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11682. [Google Scholar]
- 12.
- 12a.Uzelac M., Mulvey R. E., Chem. Eur. J. 2018, 24, 7786; [DOI] [PubMed] [Google Scholar]
- 12b.Uzelac M., Kennedy A. R., Hevia E., Inorg. Chem. 2017, 56, 8615; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c.Uzelac M., Kennedy A. R., Hevia E., Mulvey R. E., Angew. Chem. Int. Ed. 2016, 55, 13147; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13341; [Google Scholar]
- 12d.McLellan R., Uzelac M., Kennedy A. R., Hevia E., Mulvey R. E., Angew. Chem. Int. Ed. 2017, 56, 9566; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9694. [Google Scholar]
- 13.Variable 1H NMR studies of 1-diox in d8-toluene showed that the bimetallic constitution of the ferrate is retained even at −50 °C.
- 14.Woltornist R. A., Collum D. B., J. Am. Chem. Soc. 2020, 142, 6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olmstead M. M., Power P. P., Shoner S. C., Inorg. Chem. 1991, 30, 2547. [Google Scholar]
- 16.Supporting the key role of the NaTMP, diferration of benzene was achieved using a 2:1:1 mixture of NaTMP, [Fe(CH2SiMe3)2(TMEDA)], and TMP(H): Alborés P., Carrella L. M., Clegg W., García-Álvarez P., Kennedy A. R., Klett J., Mulvey R. E., Rentschler E., Russo L., Angew. Chem. Int. Ed. 2009, 48, 3317; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3367. [Google Scholar]
- 17.The NaTMP/Fe(HMDS)2 mixture also promotes the quantitative ferration of C6F5H generating 2 and TMP(H), see the Supporting Information for details.
- 18.Chadwick S. T., Rennels R. A., Rutherford J. L., Collum D. B., J. Am. Chem. Soc. 2000, 122, 421. [Google Scholar]
- 19.
- 19a.Gross P., Fort Y., Caubère P., J. Chem. Soc. Perkin Trans. 1 1997, 3071; [Google Scholar]
- 19b.“Reactions of Pyridines, Benzopyridines, and Azapyridines with Organomagnesiums and Organolithiums”: Schlosser M., Metalation of Azines and Diazines. Topics in Heterocyclic Chemistry, Vol. 31 (Eds.: Schnürch M., Mihovilovic M.), Springer, Berlin, 2013. [Google Scholar]
- 20.Schafman B. S., Wenthold P. G., J. Org. Chem. 2007, 72, 1645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary