

Abstract
Reductive transformations of easily available oxidized matter are at the heart of synthetic manipulation and chemical valorization. The applications of catalytic hydrofunctionalization benefit from the use of liquid reducing agents and operationally facile setups. Metal‐catalyzed hydroborations provide a highly prolific platform for reductive valorizations of stable C=X electrophiles. Here, we report an especially facile, broad‐scope reduction of various functions including carbonyls, carboxylates, pyridines, carbodiimides, and carbonates under very mild conditions with the inexpensive pre‐catalyst Mn(hmds)2. The reaction could be successfully applied to depolymerizations.
Keywords: amides, carbon dioxide, carbonates, depolymerization, hydroboration, manganese
Mn(hmds)2 enables clean hydroborations of C=X electrophiles (nitriles, amides, esters, pyridines, carbonates, diimides) at 20–80 °C. The protocol was applied to polymer degradations (polyesters, polycarbonates) and CO2 reduction. Mechanistic studies indicate the catalytic role of MnII hydrides, for example, the isolated cluster [MnH(hmds)]6.

Introduction
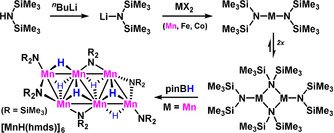
The availability of selective reduction reactions is at the heart of chemical transformations, derivatizations, and valorizations. Combinations of inexpensive and non‐toxic catalysts, easily available reductants, and mild reaction conditions may provide especially sustainable synthesis routes toward valuable chemical entities. Over the past decades, metal‐catalyzed hydrogenations and hydrofunctionalizations have emerged as powerful reduction tools with wide applicability to bulk chemicals, fine chemicals, pharmaceuticals, agrochemicals, and materials.[1, 2] Hydroboration reactions have taken a prominent position by virtue of the wide availability of boranes, their low toxicity, the high reactivity toward various unsaturated molecules, and the high levels of selectivity control. Metal‐catalyzed hydroborations operate at relatively mild conditions in the absence of hazardous reagents such as volatile gases, pyrophoric metals or reactive metal hydrides.[3, 4] Many Lewis acidic main group metal catalysts and mostly late‐transition metal catalysts were success‐fully employed. Earth‐abundant metal complexes (e.g. Fe, Co, Ni, Cu, Zn, Mg, Al) exhibited good activities in hydroborations of various C=C, C=O, C=N, and other unsaturated bonds.[5] Our group employed the commercially available metal amide LiN(SiMe3)2 (lithium hexamethyldisilazide, Li(hmds)) to catalytic hydroborations of nitriles to amines.[6] The catalyst was especially active toward aromatic nitriles while aliphatic nitriles, esters, and other carbonyl derivatives showed low conversions. Related reactions were reported with other alkali/alkaline earth metal complexes, albeit the protocols were mostly limited to activated electrophiles. A common feature of all reports of catalytic hydro‐borations of polar C=X bond motifs is the employment of catalysts that bear a strongly basic, mostly anionic ligand that appears to be crucial to the activation of the poorly nucleophilic borane. Selected examples include amides, amidinates, iminates, alkyls, hydrides, and alkoxides.[7, 8] Simple binary metal amide salts were recently reported as efficient hydroboration catalysts (M=Li, Mg, Fe, La and Y).[6, 7] Manganese, being the 3rd most abundant transition metal and non‐toxic in most compounds, is rather under‐utilized as hydrofunctionalization catalyst.[9, 10] There is a handful of reports on Mn‐catalyzed hydroborations of alkenes, carbonyls, and carboxylate derivatives that employ sophisticated pincer‐type ligands and/or require the addition of co‐catalytic bases or reductants to assure high catalyst activity and selectivity (Scheme 1).[10] We surmised that the combination of the rich coordination chemistry of transition metal ions with the stereo‐electronic and physical properties of the hexamethyldisilazide may enable an effective yet operationally simple hydroboration protocol based on the following conditions: i) the catalyst can be prepared by anion metathesis from commercial Li(hmds) and an inexpensive, non‐toxic 3d metal salt; ii) the bulky amide may form active borate complexes, create a lipophilic catalyst periphery that enhances solubilization, and prohibit catalyst aggregation under the reducing conditions; iii) the catalytic hydroboration operates under mild conditions without sophisticated additives/ligands. Herein, we document the benefits of using a most user‐friendly protocol for broad‐scope hydroborations of various unsaturated C=X bonds. The simple pre‐catalyst Mn(hmds)2 enabled clean reductions of diverse sets of carbonyls, carboxylates, pyridines, carbodiimides, carbonates, and polymer degradations but fully tolerated halides, arenes, and alkenes (Scheme 1, bottom right).
Scheme 1.

Top: Catalytic hydroborations of polar substrates as a key strategy of reduction and valorization. Center: Recently reported Mn catalysts for C=X hydroborations. Bottom: Lipophilic and basic metal‐hmds salts as inexpensive catalysts for C=X hydroborations.
Results and Discussion
We initiated our study of hydroboration reactions of a small set of electrophilic substrates with pinacolborane, pinBH, in the presence of simple metal salt catalysts (Table 1). In extension of previously reported hydroborations of nitriles with the inexpensive catalyst Li(hmds), we employed alternative metal salts bearing the same lipophilic, bulky, and Lewis basic ligand hmds. Transition metal‐hmds complexes can easily be prepared by anion metathesis of the corresponding metal halides with Li(hmds) and precipitation of insoluble lithium halides (Scheme 2). We have prepared the 3d‐metal complexes M(hmds)2 (M=Mn, Fe, Co) which exist as dimer or higher aggregates in solution.[11] Further, we have employed the recently reported hexanuclear cluster MnH6(hmds)6 that bears the key component of a hydroboration reaction: a metal ion for substrate coordination, an active hydride ligand, and the amide for borane activation. Again, the synthesis can be easily accomplished by a formal anion metathesis of Mn(hmds)2 with pinacolborane (pinBH).[12] We investigated a set of diverse substrates in metal‐catalyzed hydro‐borations at 20–50 °C (Table 1): Benzonitrile (1), ethyl benzoate (3), di‐n‐butyl carbonate (5), and 4‐methylpyridine (7). While the Li(hmds)‐catalyzed reaction was not effective for pyridines, FeII and CoII bis(hexamethyldisilazides) exhibited significantly lower reactivity with benzonitrile and the carbonate (entries 4, 5). Clean and high‐yielding formation of the respective alkoxyboronate and amidoboronate hydroboration products 2, 4, 6, and 8, respectively, under mild conditions was only observed with Mn catalysts (entries 6, 9). Other boranes (i.e. H3N⋅BH3, Me2NH⋅BH3, catechol‐borane) gave significantly lower conversion. Importantly, the combination MnBr2/Li(hmds) did not allow pyridine conversion while being effective for hydroborations of 1, 3, and 5 (entry 8). The hexanuclear cluster MnH6(hmds)6 (0.83 mol %), bearing a basic amido ligand and an active hydride per MnII, afforded excellent yields of all four reduction products (entry 9). These observations bear special significance as the very few literature reports of Mn catalysts involve sophisticated ligand design for achieving high catalytic activity.[10] The optimized conditions were applied to diverse substrates under air‐ and moisture‐free conditions (Scheme 3). The hydroboration of aromatic and aliphatic nitriles afforded very high yields of the desired primary amines (Scheme 3, top), with tolerance of I, Br, Cl, F, CF3, thio‐ether, cyclopropanes, and heterocycles. Ortho‐substituted benzo‐nitriles required slightly elevated temperatures. This Mn(hmds)2 catalysis exhibited enhanced conversions of aliphatic nitriles over the previous Li(hmds)‐protocol. Bifunctional substrates with nitrile and ketone, ester, pyridine functions, respectively, underwent clean reduction of all functional groups.
Table 1.
Survey of metal/amide catalysts for carbonyl group hydroborations.[a]

|
Entry |
Catalyst (mol %) |
Yield of hydroboration [%][b] |
|||
|---|---|---|---|---|---|
|
|
|
2 a |
4 a |
6 a |
8 a |
|
1 |
– |
0 |
0 |
0 |
0 |
|
2 |
HN(SiMe3)2 (10) |
0 |
0 |
0 |
0 |
|
3 |
LiN(SiMe3)2 (10) |
95 |
61 |
79 |
<5 |
|
4 |
Fe[N(SiMe3)2]2 (5) |
48 |
68 |
<5 |
45[c] |
|
5 |
Co[N(SiMe3)2]2 (5) |
15 |
12 |
<5 |
55[c,d] |
|
6 |
Mn[N(SiMe3)2]2 (5) |
99 |
99 |
99 |
93[c] |
|
7 |
MnBr2 (10) |
0 |
0 |
0 |
0 |
|
8 |
MnBr2 (5) + LiN(SiMe3)2 (10) |
87 |
64 |
77 |
<5 |
|
9 |
Mn6H6[N(SiMe3)2]6 (0.83) |
98 |
99 |
99 |
91[c] |
[a] Conditions: 0.2 mmol substrate, 2.0–3.1 equiv HBpin, catalyst, 0.6 mL [D6]benzene. [b] 1H‐NMR yields vs. internal hexamethylbenzene. [c] Ratios of regioisomers 8 a:8 a′ >90/10 (by 1H NMR). [d] Further unidentified products.
Scheme 2.
Synthesis of various metal hexamethyldisilazides, M(hmds)1–2.[11a, 12a]
Scheme 3.

Mn(hmds)2‐catalyzed hydroboration of nitriles, esters, amides, and pyridines. 1H‐NMR yields vs. internal hexamethylbenzene. [a] 10 mol % Li(hmds) instead of [Mn]. [b] 50 °C, 20 h. For details, see the ESI.
Reactions of chiral nitriles proceeded with full stereoretention. (S)‐(+)‐2‐Methyl butyronitrile was reduced at 50 °C to the β‐chiral butylamine in near‐quantitative yield ( =6.2°, CH3OH).[12b] Catalytic hydroborations of carboxylates have recently been reported with transition metal catalysts,[13] bulky organo‐magnesium,[14, 15] amidomagnesium,[7a, 16] and rare earth metal catalysts.[7d, 7e, 17, 18] Catalytic Mn(hmds)2 enabled clean ester reduction with HBpin at room temperature (Scheme 3, center). Alkanoates and benzoates were reactive; halides, CF3, and heteroaryl substituents were tolerated. The Li(hmds)‐catalyzed reaction gave significantly lower conversion. Reductions of chiral esters proceeded with full stereoretention: (L)‐(−)‐lactide gave (S)‐(−)‐1,2‐propanediol in quantitative yield ( =29.2°, CHCl3).[19] The Mn‐catalyzed hydroboration could also be applied to the depolymerization of polyesters (Scheme 4). The reduction of carboxamides typically requires harsher conditions due to the higher resonance stabilization. Chemoselectivity issues may arise from the reaction mechanism that may lead to aldehyde or amine products. The catalytic deoxygenative hydroboration of amides to amines is a safe alternative to hydrogenations or the use of LiAlH4. Rare earth metal catalysts were recently reported;[7f, 7g, 17] an abnormal N‐heterocyclic carbene‐potassium catalyst was active at 40 °C;[20] the combination KOtBu/BEt3 enabled amide reductions at 25–60 °C with pinacolborane;[21] 2,6‐di‐tert‐butyl‐phenolate lithium‐THF was catalytically active at 60 °C.[22] The simple pre‐catalyst Mn(hmds)2 proved active in the hydroboration of alkyl and aryl carboxamides with pinacolborane to primary amines in very good yields at 50 °C (Scheme 3, center). The reaction does not involve aldehyde or imine intermediates as no scrambling of the alkylamine group was observed.
Scheme 4.

Mn(hmds)2 catalyzed hydroboration of carbodiimides, cyclic and linear carbonates, depolymerization of polyesters and polycarbonates, and carbon dioxide. 1H‐NMR yields vs. internal hexamethylbenzene. [a] 10 mol % Li(hmds) instead of [Mn], [b] 80 °C, 20 h, [c] 80 °C, 72 h in [D8]THF. Details are given in the ESI.
Catalytic reductions of pyridines and related N‐heteroarenes constitute an attractive synthetic strategy of dearomatization toward the formation of six‐membered nitrogen‐containing heterocycles, a structural motif of utmost importance to bioactive molecules. Regioselective hydroborations of pyridines with pinacolborane toward 1,4‐dihydropyridines operate with Ni, Ru, and Mg catalysts; La, Rh, Th, Zn, Fe and Ni were employed for regio‐selective 1,2‐reductions.[23, 24] Typically, the presence of Brønsted basic or phosphine ligand have enabled high 1,2‐regio‐selectivities and yields. When subjecting the simple Mn complex Mn(hmds)2 to catalytic hydroborations of pyridines, excellent yields and high regiocontrol was observed at 50 °C (Scheme 3, bottom). The reaction tolerated Cl, Br, CF3, benzylic H groups and could be extended to quinolines, isoquinolines, phenanthridines and benzo[d]imidazoles. Regioselective 1,2‐hydroborations were highly favoured with 4‐substituted N‐heterocycles, electron‐with‐drawing substituents, or benzanellated N‐heterocycles. Having explored the efficacy of Mn(hmds)2‐catalyzed hydroborations of carboxyl derivatives and pyridines, we turned our attention to reductions of organic molecules bearing carbon in its highest oxidation state +IV. Carbodiimides are the nitrogen‐homologues of carbon dioxide and find applications as precursors to amidines, guanidines, and heterocycles and as dehydration reagents.[25, 26] The hydroboration of carbodiimides leads to formamidines, another hydroboration event gives aminals. The selective mono‐hydroboration of carbodiimides is challenging and only few examples were reported. The reaction with 9‐borabicyclo[3.3.1]‐nonane at high temperature gives mixtures of mono‐ and dihydroboration products.[27] Catalytic hydroboration with strongly basic organometallic complexes (Mg, K) and amido complexes (Hf, actinides) selectively gave the formamidine products.[8d, 28] Subjection of carbodiimides to the Mn(hmds)2‐catalyzed hydro‐boration conditions led to selective mono‐reduction to the desired formamidines at room temperature (Scheme 4, top). Bulky carbodimides (tBu, 2,6‐diisopropylphenyl) were cleanly converted at elevated temperature. In no case was the double reduction product observed, even with excess amounts of HBpin. The reaction showed perfect regiocontrol with unsymmetrical carbo‐diimides: N‐tert‐butyl‐N′‐ethylcarbodiimide was exclusively converted to the ethylaminoboronate isomer. Organic carbonates are large‐scale technical products of carbon dioxide fixation. The reduction of carbonates delivers methanol as the formal reduction product of CO2. The thermodynamic stability of carbonates renders them applicable as organic solvents, even in catalytic hydrogenations. Very few catalysts have been reported for hydrogenations of organic carbonates at elevated pressure and temperature.[29] The polar mechanisms of catalytic hydroboration reactions provide a more prolific scenario for carbonate reduction. Recent literature documented the efficacy of transition, main group, and lanthanide metal catalysts. Leitner and co‐workers employed Mn catalysts with PNP‐pincer ligands.[10c] High activity was only attained by addition of base (NaOtBu) and at high temperature (90 °C). Rueping et al. reported the dibutyl‐magnesium‐catalyzed hydroboration of carbonates at 65 °C in toluene;[30] a MgI ketiminate complex catalyzed the hydro‐boration of carbonates at room temperature;[16] lanthanum amide catalysts were recently employed by Xue et al.[7d] Following our initial observation of the efficient di‐n‐butyl carbonate reduction to methanol and the n‐butyl borate (Table 1), we further explored the scope of this transformation in the presence of 5 mol % Mn(hmds)2 at room temperature. Acyclic and cyclic carbonates were reduced to methanol (and higher alcohols/diols) in near‐quantitative yields at room temperature with no excess of the reducing reagent (Scheme 4, center). Chloride, alkene, and allyl ether functions were fully tolerated. No loss of stereoinformation was observed with stereomerically pure carbonates. Mn‐catalyzed hydro‐boration of cyclic cis‐ and trans‐cyclohexene carbonates afforded afforded quantitative yields of methanol and the cis‐ and trans‐cyclohexane‐1,2‐diols, respectively, with full preservation of stereochemistry. The general conditions of this Mn catalysis were also applied to the reductive depolymerization of polyesters and polycarbonates (Scheme 4, center). The degradation of end‐of‐life polycarbonates/polyesters into the valuable alcohol building blocks is a viable process for the valorization of waste. Hydrogenative depolymerizations may require harsh conditions (54 bar H2, 160 °C).[31] Recently, Cantat et al. reported catalytic hydrosilylations with the Lewis acid catalyst B(C6F5)3 and Ir catalysts, respectively.[32, 33]
With catalytic Mn(hmds)2, polyethylene terephthalate (PET) was successfully converted to the corresponding monomers para‐xylylene glycol and ethylene glycol in 71 % yield. Higher yields were obtained in THF at 80 °C. This is the first example of a Mn‐catalyzed hydroboration of PET. Poly‐ϵ‐caprolactone (PCL) underwent facile reductive depolymerization in the presence of catalytic Mn(hmds)2 (5 mol % per ester unit) and HBpin. PCL samples with average molecular weights of 14 000 and 80 000, respectively, were reduced to the 1,6‐hexandiol derivative in quantitative yields at 20 °C. Under identical conditions, polypropylene carbonate (PPC), the co‐polymer of CO2 and propylene oxide, was cleanly reduced. The propylene glycol and methanol derivatives were obtained in 87 % yield with no excess of the reducing reagent. Finally, we have extended the general conditions to the hydroboration of carbon dioxide (CO2). There is a growing number of homogeneous ligand‐metal catalysts that enable hydroborations of CO2 with available boranes (e.g. pinBH, catBH, BH3⋅SMe2, 9‐BBN).[5h, 33] The resultant C1‐hydrocarbons (formaldehyde, formic acid or methanol) constitute valuable chemical building blocks. Upon employment of Mn(hmds)2, CO2 was reduced by HBpin with 100 % selectivity to methanol at ≈1 bar, 80 °C (21 % yield vs. borane, 1H NMR). The hexanuclear cluster [MnH(hmds)]6 afforded similar conversion and selectivity under identical conditions (Scheme 4, bottom right). Efforts to prevent rapid catalyst decomposition in the absence of suitable donor molecules (in THF, with additives L‐lactide or propylene carbonate) showed no change of selectivity.
Mechanistic Studies
We have performed combined synthetic, kinetic, and spectroscopic experiments to elucidate the operating reaction mechanism, including the role of reaction components, kinetic studies and isotope effects, catalyst poisoning, and analyses of side products (Scheme 5). The initial optimizations of the general procedure (see Table 1) documented the high activity of manganese hydride clusters that were superior to the related Li, Fe and Co catalysts. A key to efficient hydroboration activity of catalyst species certainly resides in the ability of the metal center to act as Lewis acid. The role of Mn(hmds)2 in the coordinative activation of polar C=X bond substrates was corroborated by the isolation of an adduct upon reaction of Mn(hmds)2 with PhCN in hexane. The resultant [Mn(hmds)2(NCPh)2] was characterized by single crystal structure analysis (Scheme 5, top).[34] This observation is in stark contrast to the related Li(hmds)‐catalyzed reaction where the higher Lewis acidity of the Li ion mediated nitrile insertion into the Li‐N(TMS)2 bond and subsequent 1,3‐TMS transfer to give amidinatolithium complexes.[6] Our efforts into the identification of complexes formed from the reaction of [Mn(hmds)2(NCPh)2] with HBpin were unsuccessful, but proved the catalytic efficacy of the Lewis adduct by clean formation of PhCH 2N(Bpin)2 and hmds‐Bpin (1H‐NMR). The rapid hydride transfer from pinBH to transition metal‐hmds complexes suggests that Mn‐hydride species act as active catalysts. We observed the characteristic immediate colour change to dark brown upon addition of HBpin to Mn(hmds)2 in various solvents (benzene, toluene, hexane). The hexanuclear complex [Mn6H6(hmds)6] was isolated from such reactions as dark brown crystals.[12a] The identical catalytic activity of the mixture Mn(hmds)2/borane and [Mn6H6(hmds)6]—the latter formed from the stoichiometric reaction of HBpin with Mn(hmds)2—is in full accord with the postulated catalytic role of this (or related) Mn clusters. This hypothesis is supported by the observation of a strong dependence of hydroboration reactivity on the sequence of substrate addition. The hydroboration of pyridines only proceeded when HBpin was added to the solution of Mn(hmds)2 in benzene prior to the pyridine (nitrile and ester reduction proved independent on the order of substrate additions). We therefore postulate the formation of [Mn6H6(hmds)6] and other homologous oligonuclear manganese hydride species from the reaction of HBpin with Mn(hmds)2. This is supported by the isolation of crystalline [Mn6H6(hmds)6] and the concomitant formation of an insoluble white precipitate as major product, which both exhibited the same catalytic activity as the mixture of Mn(hmds)2/borane in the hydroboration (4‐methoxybenzonitrile, dibutyl carbonate). Attempts to grow single crystals from the THF‐soluble white precipitate were unsuccessful; the molecular mass was beyond the detection range of LIDFI‐MS (>2000 m/z). ICP‐OES (23.7 % Mn), elemental analysis (N: 6.37; H: 8.83; C: 33.84), the absence of boron, and the presence of equimolar active hydrides in its molecular structure are in full accord with a higher aggregate of the formula [MnH(hmds)]n (calcd: Mn: 25.4; N: 6.47; H: 8.85; C: 33.31). The latter was corroborated by the identical yields of benzylalcohol from reactions of [MnH(hmds)]6 and the postulated higher cluster [MnH(hmds)]n, respectively, with benzaldehyde (Scheme 5, center). If the isolated cluster complex [MnH(hmds)]6 constitutes a competent model of the true catalyst species, direct hydroboration should occur when adding suitable substrates. The reaction mixture of Mn6H6(hmds)6 and 6 equiv benzonitrile displayed a broad signal at 9.4 ppm in the 1H NMR which can be assigned to [Mn]‐N=CHPh species. Indeed, aqueous work‐up documented 84 % conversion of PhCN and clean formation of the imine HN=CHPh. A similar reaction with addition of 6 equiv HBpin (i.e. equimolar Mn:PhCN:HBpin) showed clean formation of the doubly reduced primary benzyl‐amine. The intermediate imine PhCH=NBpin was not detected by 1H NMR of the crude reaction. The same observations were made on hydroborations in the presence of Mn(hmds)2: Equimolar reactions of Mn(hmds)2, HBpin, and PhCN afforded the same [Mn]‐N=CHPh species and hmds‐Bpin, and upon work‐up the imine. With excess amounts of borane (3 equiv), benzylamine was cleanly formed. The Mn(hmds)2‐catalyzed hydroboration of 4‐methoxybenzonitrile with HBpin followed a pseudo‐1st order rate of nitrile consumption over the period of full conversion (Scheme 5, center). The individual reaction orders in catalyst and borane at low conversions (<25 %) were 1, respectively; the nitrile exhibited an order of zero. Nearly identical reaction orders in [Mn] were observed for Mn(hmds)2 and [Mn6H6(hmds)6] (0.97 and 0.83) so that mononuclear Mn‐hydride complexes very likely constitute the most active catalyst species. The determination of kinetic isotopic effects (KIE) from competing reactions with equimolar H‐Bpin and D‐Bpin resulted in a k H/k D ratio of ≈2.1 (nitrile, carbodiimide) and ≈1.6 (ester) (It is important to note that these values must be determined at low conversions and constitute lower limits of the real KIE due to the more rapid depletion of the faster reacting HBpin).[35] This (presumably) 1° KIE argues for a rate‐determining hydride transfer in the overall mechanism (B‐H to Mn or Mn‐H to C=X). The clear distinction between homotopic and heterotopic catalysis mechanisms is not trivial, as sensitive and insightful analytical tools that operate under the reaction conditions are rare. A first indication may be derived from selective catalyst poisoning studies.[36] The reductive conditions of the hydroboration reactions with basic pre‐catalyst Mn(hmds)2 and the active hydride reagent HBpin may facilitate reduction events at the Mn catalyst to produce Mn0 species in the absence of strong ligands. However, the operation of pre‐catalyst reduction was not observed (no H2 evolution, no diborane formation, no pinacol formation with aldehydes). Furthermore, no inhibition of catalyst activity by a potential Mn0‐amalgam phase was observed upon addition of 200 equiv Hg (per Mn) to the hydroboration of 4‐methoxybenzonitrile.[37] The same interpretation can be derived from the unaltered reaction rate when adding trimethylphosphine (PMe3)—a strong ligand for reduced Mn species. No “hidden” BH3 catalysis was operating (11B NMR).[38] Based on the collected synthetic, kinetic, and spectroscopic data, we postulate a catalytic mechanism that is initiated with the formation of [MnH(hmds)]x complexes from reaction of Mn(hmds)2 and HBpin (Scheme 5, bottom). In the presence of suitable donor‐functions in the substrates, deaggregation yields the mononuclear active catalyst species. The hydride transfer from HBpin to the C=X moiety or the Mn catalyst is rate‐limiting. The basic hmds and C‐X ligands are most likely involved in the activation of the borane to facilitate hydride transfer by intramolecular σ‐bond metathesis.
Scheme 5.

Key experiments and postulated mechanism (NR2=N(SiMe3)2).
Conclusion
In summary, we have reported an operationally facile protocol for reductions of a wide array of functional molecules by utilization of simple pre‐catalysts comprising of the transition metal Mn and the Lewis base hmds. The catalytic hydroboration reaction operates with the inexpensive liquid reductant HBpin under very mild conditions. Reductive valorizations of stable C=X electrophiles such as carbonyls, carboxylates, pyridines, carbo‐diimides, and carbonates were accomplished in high selectivities. The reaction conditions tolerated various functional groups and neighbouring stereocenters and may prove useful in chemo‐selective reductions of multifunctional molecules (reactivity trend: amide > ester > nitrile > carbonate > pyridine ≫ CO2 >≫ alkene). The same protocol was applied to polymer degradations of poly‐esters and polycarbonates. CO2 was reduced with moderate activity. Mechanistic studies indicate the catalytic role of inter‐mediate MnII‐hydride complexes, the absence of Mn0 species, and a rate‐limiting hydride transfer from the borane. The identification of the pre‐catalyst Mn(hmds)2 and the observation of active [MnH(hmds)]x intermediates complement the growing arsenal of metal catalysts bearing sophisticated (pincer) ligands and/or basic co‐catalysts. Without elaborate ligand design, the cooperative effect of an early transition metal and a Lewis basic ligand may serve as a generic platform for further applications to reactions of polar functional groups with soft reagents.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the European Research Council (CoG 683150). We thank Dr. Uttam Chakraborty for XRD support. Open access funding enabled and organized by Projekt DEAL.
P. Ghosh, A. Jacobi von Wangelin, Angew. Chem. Int. Ed. 2021, 60, 16035.
In memory of Kilian Muñiz
References
- 1.
- 1a.The Handbook of Homogeneous Hydrogenation (Eds.: de Vries J. G., Elsevier C. J.), Wiley-VCH, Weinheim, 2007; [Google Scholar]
- 1b.Nishimura S., Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001; [Google Scholar]
- 1c.Catalysis without Precious Metals (Ed.: Bullock R. M.), Wiley-VCH, Weinheim, 2010; [Google Scholar]
- 1d.Non-Noble Metal Catalysis: Molecular Approaches and Reactions (Ed.: Klein Gebbink R. J. M., Moret M.-E.), Wiley-VCH, Weinheim, 2019; [Google Scholar]
- 1e.Alig L., Fritz M., Schneider S., Chem. Rev. 2019, 119, 2681–2751; [DOI] [PubMed] [Google Scholar]
- 1f.Garduño J. A., García J. J., ACS Catal. 2020, 10, 8012–8022. [Google Scholar]
- 2.
- 2a.Hydrofunctionalization (Eds.: Ananikov V. P., Tanaka M.), Springer, Berlin, 2011; [Google Scholar]
- 2b.Magano J., Dunetz J. R., Org. Process Res. Dev. 2012, 16, 1156–1184; [Google Scholar]
- 2c.Modern Reduction Methods (Eds.: Andersson P. G., Munslow I. J.), Wiley-VCH, Weinheim, 2008; [Google Scholar]
- 2d.Brown H. C., Krishnamurthy S., Tetrahedron 1979, 35, 567–607; [Google Scholar]
- 2e.Seyden-Penne J., Reductions by Alumino and Borohydrides in Organic Synthesis 2nd ed., Wiley-VCH, New York, 1997. [Google Scholar]
- 3.
- 3a.Elangovan S., Garbe M., Jiao H., Spannenberg A., Junge K., Beller M., Angew. Chem. Int. Ed. 2016, 55, 15364–15368; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15590–15594; [Google Scholar]
- 3b.Mukherjee A., Srimani D., Chakraborty S., Ben-David Y., Milstein D., J. Am. Chem. Soc. 2015, 137, 8888–8891; [DOI] [PubMed] [Google Scholar]
- 3c.Chakraborty S., Leitus G., Milstein D., Chem. Commun. 2016, 52, 1812–1815, and references therein. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.Gribble G. W., Chem. Soc. Rev. 1998, 27, 395–404; [Google Scholar]
- 4b.Lu Z., Williams T. J., Chem. Commun. 2014, 50, 5391–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For reviews on transition metal-catalyzed hydroboration:
- 5a.Beletskaya I. P., Pelter A., Tetrahedron 1997, 53, 4957–5026; [Google Scholar]
- 5b.Miyaura N. in Catalytic Heterofunctionalization (Eds.: Togni A., Grützmacher H.), Wiley-VCH, Weinheim, 2001, chap. 1, pp. 1–45; [Google Scholar]
- 5c.Vogels C. M., Westcott S. W., Curr. Org. Chem. 2005, 9, 687–699; [Google Scholar]
- 5d.Park S., Chang S., Angew. Chem. Int. Ed. 2017, 56, 7720–7738; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7828–7847; [Google Scholar]
- 5e.Hayrapetyan D., Khalimon A. Y., Chem. Asian J. 2020, 15, 2575–2587; [DOI] [PubMed] [Google Scholar]
- 5f.Obligacion J. V., Chirik P. J., Nat. Rev. Chem. 2018, 2, 15–34; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5g.Shegavi M. L., Bose S. K., Catal. Sci. Technol. 2019, 9, 3307–3336; [Google Scholar]
- 5h.Chong C. C., Kinjo R., ACS Catal. 2015, 5, 3238–3259; [Google Scholar]
- 5i.Park S., ChemCatChem 2020, 12, 3170–3185; [Google Scholar]
- 5j.Chatterjee B., Gunanathan C., J. Chem. Sci. 2019, 131, 118. [Google Scholar]
- 6.Ghosh P., Jacobi von Wangelin A., Org. Chem. Front. 2020, 7, 960–966. [Google Scholar]
- 7.
- 7a.Barman M. K., Baishya A., Nembenna S., Dalton Trans. 2017, 46, 4152–4156; [DOI] [PubMed] [Google Scholar]
- 7b.Weidner V. L., Barger C. J., Delferro M., Lohr T. L., Marks T. J., ACS Catal. 2017, 7, 1244–1247; [Google Scholar]
- 7c.Baishya A., Baruah S., Geetharani K., Dalton Trans. 2018, 47, 9231–9236; [DOI] [PubMed] [Google Scholar]
- 7d.Xu X., Kang Z., Yan D., Xue M., Chin. J. Chem. 2019, 37, 1142–1146; [Google Scholar]
- 7e.Barger C. J., Motta A., Weidner V. L., Lohr T. L., Marks T. J., ACS Catal. 2019, 9, 9015–9024; [Google Scholar]
- 7f.Barger C. J., Dicken R. D., Weidner V. L., Motta A., Lohr T. L., Marks T. J., J. Am. Chem. Soc. 2020, 142, 8019–8028; [DOI] [PubMed] [Google Scholar]
- 7g.Ye P., Shao Y., Ye X., Zhang F., Li R., Sun J., Xu B., Chen J., Org. Lett. 2020, 22, 1306–1310. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a.Bisai M. K., Das T., Vanka K., Sen S. S., Chem. Commun. 2018, 54, 6843; [DOI] [PubMed] [Google Scholar]
- 8b.Yadav S., Pahar S., Sen S. S., Chem. Commun. 2017, 53, 4562–4564; [DOI] [PubMed] [Google Scholar]
- 8c.Kim H., Lee J. H., Hwang H., An D. K., New J. Chem. 2020, 44, 11330–11335; [Google Scholar]
- 8d.Panda T. K., Banerjee I., Sagar S., Appl. Organomet. Chem. 2020, 34, e5765; [Google Scholar]
- 8e.Yan B., He X., Ni C., Yang Z., Ma X., ChemCatChem 2021, 13, 851–854; [Google Scholar]
- 8f.Bedi D., Brara A., Findlater M., Green Chem. 2020, 22, 1125–1128; [Google Scholar]
- 8g.Saha S., Eisen M. S., ACS Catal. 2019, 9, 5947–5956; [Google Scholar]
- 8h.Weetman C., Anker M. D., Arrowsmith M., Hill M. S., Kociok-Köhn G., Liptrota D. J., Mahon M. F., Chem. Sci. 2016, 7, 628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a.Mukhopadhyay T. K., Flores M., Groy T. L., Trovitch R. J., J. Am. Chem. Soc. 2014, 136, 882–885; [DOI] [PubMed] [Google Scholar]
- 9b.Carney J. R., Dillon B. R., Campbell L., Thomas S. P., Angew. Chem. Int. Ed. 2018, 57, 10620–10624; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10780–10784; [Google Scholar]
- 9c.Trovitch R. J., Acc. Chem. Res. 2017, 50, 2842–2852; [DOI] [PubMed] [Google Scholar]
- 9d.Yang X., Wang C., Chem. Asian J. 2018, 13, 2307–2315, and references therein; [DOI] [PubMed] [Google Scholar]
- 9e.Bhunia M., Sreejyothi P., Mandal S. K., Coord. Chem. Rev. 2020, 405, 213110; [Google Scholar]
- 9f.Behera R. R., Ghosh R., Panda S., Khamari S., Bagh B., Org. Lett. 2020, 22, 3642–3648; [DOI] [PubMed] [Google Scholar]
- 9g.Martínez-Ferraté O., Chatterjee B., Werlé C., Leitner W., Catal. Sci. Technol. 2019, 9, 6370–6378; [Google Scholar]
- 9h.Sousa S. C. A., Realista S., Royo B., Adv. Synth. Catal. 2020, 362, 2437–2443; [Google Scholar]
- 9i.Kelly C. M., McDonald R., Sydora O. L., Stradiotto M., Turculet L., Angew. Chem. Int. Ed. 2017, 56, 15901–15904; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16117–16120; [Google Scholar]
- 9j.Mukhopadhyay T. K., Ghosh C., Flores M., Groy T. L., Trovitch R. J., Organometallics 2017, 36, 3477–3483; [Google Scholar]
- 9k.Ma X., Zuo Z., Liu G., Huang Z., ACS Omega 2017, 2, 4688–4692; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9l.Wei D., Buhaibeh R., Canac Y., Sortais J.-B., Chem. Commun. 2020, 56, 11617–11620; [DOI] [PubMed] [Google Scholar]
- 9m.Ganguli K., Mandal A., Sarkar B., Kundu S., Tetrahedron 2020, 76, 131439; [Google Scholar]
- 9n.Royo B., Adv. Organomet. Chem. 2019, 72, 59–102, and references therein; [Google Scholar]
- 9o.Dong J., Yuan X.-A., Yan Z., Mu L., Ma J., Zhu C., Xie J., Nat. Chem. 2021, 13, 182–190. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a.Zhang G., Zeng H., Wu J., Yin Z., Zheng S., Fettinger J. C., Angew. Chem. Int. Ed. 2016, 55, 14369–14372; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14581–14584; [Google Scholar]
- 10b.Vasilenko V., Blasius C. K., Wadepohl H., Gade L. H., Angew. Chem. Int. Ed. 2017, 56, 8393–8397; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8513–8517; [Google Scholar]
- 10c.Erken C., Kaithal A., Sen S., Weyhermüller T., Hölscher M., Werlé C., Leitner W., Nat. Commun. 2018, 9, 4521–4528; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d.Vasilenko V., Blasius C. K., Gade L. H., J. Am. Chem. Soc. 2018, 140, 9244–9254; [DOI] [PubMed] [Google Scholar]
- 10e.Barman M. K., Das K., Maji B., J. Org. Chem. 2019, 84, 1570–1579; [DOI] [PubMed] [Google Scholar]
- 10f.Kostera S., Peruzzini M., Kirchner K., Gonsalvi L., ChemCatChem 2020, 12, 4625–4631; [Google Scholar]
- 10g.Zhang G., Zeng H., Li S., Johnson J., Mo Z., Neary M. C., Zheng S., Dalton Trans. 2020, 49, 2610–2615; [DOI] [PubMed] [Google Scholar]
- 10h.Nguyen T. T., Kim J.-H., Kim S., Oh C., Flores M., Groy T. L., Baik M. H., Trovitch R. J., Chem. Commun. 2020, 56, 3959–3962; [DOI] [PubMed] [Google Scholar]
- 10i.Brzozowska A., Zubar V., Ganardi R. C., Rueping M., Org. Lett. 2020, 22, 3765–3769; [DOI] [PubMed] [Google Scholar]
- 10j.Vijjamarri S., O'Denius T. M., Yao B., Kubátová A., Du G., Organometallics 2020, 39, 3375–3383; [Google Scholar]
- 10k.Garhwal S., Kroeger A. A., Thenarukandiyil R., Fridman N., Karton A., de Ruiter G., Inorg. Chem. 2021, 60, 494–504; [DOI] [PubMed] [Google Scholar]
- 10l.Thenarukandiyil R., Satheesh V., Shimon L. J. W., de Ruiter G., Chem. Asian J. 2021, 16, 999–1006. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a.Andersen R. A., K.Faegri, Jr. , Green J. C., Haaland A., Lappert M. F., Leung W. P., Rypdal K., Inorg. Chem. 1988, 27, 1782–1786; [Google Scholar]
- 11b.Bradley D. C., Hursthouse M. B., Malik K. M. A., Möseler R., Transition Met. Chem. 1978, 3, 253–254. [Google Scholar]
- 12.
- 12a.Chakraborty U., Reyes-Rodriguez E., Demeshko S., Meyer F., Jacobi von Wangelin A., Angew. Chem. Int. Ed. 2018, 57, 4970–4975; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5064–5069; [Google Scholar]
- 12b.Brunner H., Wachter J., Schmidbauer J., Sheldrick G. M., Jones P. G., Organometallics 1986, 5, 2212–2219. [Google Scholar]
- 13.
- 13a.Khalimon A. Y., Farha P., Kuzmina L. G., Nikonov G. I., Chem. Commun. 2012, 48, 455–457; [DOI] [PubMed] [Google Scholar]
- 13b.Zhang G., Wu J., Zheng S., Neary M. C., Mao J., Flores M., Trovitch R. J., Dub P. A., J. Am. Chem. Soc. 2019, 141, 15230–15239. [DOI] [PubMed] [Google Scholar]
- 14.Mukherjee D., llern A., Sadow A. D., Chem. Sci. 2014, 5, 959–964. [Google Scholar]
- 15.Mukherjee D., Shirase S., Spaniol T. P., Mashima K., Okuda J., Chem. Commun. 2016, 52, 13155–13158. [DOI] [PubMed] [Google Scholar]
- 16.Cao X., Wang W., Lu K., Yao W., Xue F., Ma M., Dalton Trans. 2020, 49, 2776–2780. [DOI] [PubMed] [Google Scholar]
- 17.Tamang S. R., Singh A., Bedi D., Bazkiaei A. R., Warner A. A., Glogau K., McDonald C., Unruh D. K., Findlater M., Nat. Catal. 2020, 3, 154–162. [Google Scholar]
- 18.Patnaik S., Sadow A. D., Angew. Chem. Int. Ed. 2019, 58, 2505–2509; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 2527–2531. [Google Scholar]
- 19.Petursson S., Tetrahedron: Asymmetry 2009, 20, 887–891. [Google Scholar]
- 20.Bhunia M., Sahoo S. R., Das A., Ahmed J., Sreejyothi P., Mandal S. K., Chem. Sci. 2020, 11, 1848–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao W., Wang J., Zhong A., Wang S., Shao Y., Org. Chem. Front. 2020, 7, 3515–3520. [Google Scholar]
- 22.Bisai M. K., Gour K., Das T., Vanka K., Sen S. S., Dalton Trans. 2021, 50, 2354–2358. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a.Arrowsmith M., Hill M. S., Hadlington T., Kociok-Köhn G., Weetman C., Organometallics 2011, 30, 5556–5559; [Google Scholar]
- 23b.Tamang S. R., Singh A., Unruh D. K., Findlater M., ACS Catal. 2018, 8, 6186–6191; [Google Scholar]
- 23c.Kaithal A., Chatterjee B., Gunanathan C., Org. Lett. 2016, 18, 3402–3405; [DOI] [PubMed] [Google Scholar]
- 23d.Yu H.-C., Islam S. M., Mankad N. P., ACS Catal. 2020, 10, 3670–3675. [Google Scholar]
- 24.
- 24a.Dudnik A. S., Weidner V. L., Motta A., Delferro M., Marks T. J., Nat. Chem. 2014, 6, 1100–1107; [DOI] [PubMed] [Google Scholar]
- 24b.Oshima K., Ohmura T., Suginome M., J. Am. Chem. Soc. 2012, 134, 3699–3702; [DOI] [PubMed] [Google Scholar]
- 24c.Liu H., Khononov M., Eisen M. S., ACS Catal. 2018, 8, 3673–3677; [Google Scholar]
- 24d.Lortie J. L., Dudding T., Gabidullin B. M., Nikonov G. I., ACS Catal. 2017, 7, 8454–8459; [Google Scholar]
- 24e.Zhang F., Song H., Zhuang X., Tung C.-H., Wang W., J. Am. Chem. Soc. 2017, 139, 17775–17778; [DOI] [PubMed] [Google Scholar]
- 24f.Liu J., Chen J.-Y., Jia M., Ming B., Jia J., Liao R.-Z., Tung C.-H., Wang W., ACS Catal. 2019, 9, 3849–3857; [Google Scholar]
- 24g.Wang X., Zhang Y., Yuan D., Yao Y., Org. Lett. 2020, 22, 5695–5700; [DOI] [PubMed] [Google Scholar]
- 24h.Liu X., Li B., Hua X., Cui D., Org. Lett. 2020, 22, 4960–4965. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a.Zhang W., Nishiura M., Hou Z., Chem. Commun. 2006, 3812–3814; [DOI] [PubMed] [Google Scholar]
- 25b.Alonso-Moreno C., Antinolo A., Carrillo-Hermosilla F., Otero A., Chem. Soc. Rev. 2014, 43, 3406–3425; [DOI] [PubMed] [Google Scholar]
- 25c.Ong T.-G., Yap G. P. A., Richeson D. S., J. Am. Chem. Soc. 2003, 125, 8100–8101; [DOI] [PubMed] [Google Scholar]
- 25d.Ishikawa T., Kumamoto T., Synthesis 2006, 737–752. [Google Scholar]
- 26.
- 26a.Edelmann F. T., Chem. Soc. Rev. 2012, 41, 7657–7672; [DOI] [PubMed] [Google Scholar]
- 26b.Werner D., Deacon G. B., Junk P. C., Anwander R., Chem. Eur. J. 2014, 20, 4426–4438; [DOI] [PubMed] [Google Scholar]
- 26c.Karmel I. S. R., Fridman N., Eisen M. S., Organometallics 2015, 34, 636–643; [Google Scholar]
- 26d.Kumar R. K., Punniyamurthy T., RSC Adv. 2012, 2, 4616–4619. [Google Scholar]
- 27.Boese R., Köster R., Yalpani M., Z. Naturforsch. B 1994, 49, 1453–1458. [Google Scholar]
- 28.
- 28a.Weetman C., Hill M. S., Mahon M. F., Chem. Eur. J. 2016, 22, 7158–7162; [DOI] [PubMed] [Google Scholar]
- 28b.Rauch M., Ruccolo S., Parkin G., J. Am. Chem. Soc. 2017, 139, 13264–13267; [DOI] [PubMed] [Google Scholar]
- 28c.Liu H., Kulbitski K., Tamm M., Eisen M. S., Chem. Eur. J. 2018, 24, 5738–5742; [DOI] [PubMed] [Google Scholar]
- 28d.Shen Q., Ma X., Li W., Liu W., Ding Y., Yang Z., Roesky H. W., Chem. Eur. J. 2019, 25, 11918–11923; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28e.Khononov M., Fridman N., Tamm M., Eisen M. S., Eur. J. Org. Chem. 2020, 3153–3160. [Google Scholar]
- 29.
- 29a.Balaraman E., Gunanathan C., Zhang J., Shimon L. J. W., Milstein D., Nat. Chem. 2011, 3, 609–614; [DOI] [PubMed] [Google Scholar]
- 29b.Han Z., Rong L., Wu J., Zhang L., Wang Z., Ding K., Angew. Chem. Int. Ed. 2012, 51, 13041–13045; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 13218–13222; [Google Scholar]
- 29c.Kumar A., Janes T., Espinosa-Jalapa N. A., Milstein D., Angew. Chem. Int. Ed. 2018, 57, 12076–12080; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12252–12256; [Google Scholar]
- 29d.Kaithal A., Hölscher M., Leitner W., Angew. Chem. Int. Ed. 2018, 57, 13449–13453; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13637–13641; [Google Scholar]
- 29e.Zubar V., Lebedev Y., Azofra L. M., Cavallo L., El-Sepelgy O., Rueping M., Angew. Chem. Int. Ed. 2018, 57, 13439–13443; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13627–13631; [Google Scholar]
- 29f.Ferretti F., Scharnagl F. K., Dall'Anese A., Jackstell R., Dastgir S., Beller M., Catal. Sci. Technol. 2019, 9, 3548–3553; [Google Scholar]
- 29g.Dahiya P., Gangwar M. K., Sundararaju B., ChemCatChem 2021, 13, 934–939; [Google Scholar]
- 29h.vom Stein T., Meuresch M., Limper D., Schmitz M., Hölscher M., Coetzee J., Cole-Hamilton D. J., Klankermayer J., Leitner W., J. Am. Chem. Soc. 2014, 136, 13217–13225. [DOI] [PubMed] [Google Scholar]
- 30.Szewczyk M., Magre M., Zubar V., Rueping M., ACS Catal. 2019, 9, 11634–11639. [Google Scholar]
- 31.Krall E. M., Klein T. W., Andersen R. J., Nett A. J., Glasgow R. W., Reader D. S., Dauphinais B. C., Mc Ilrath S. P., Fischer A. A., Carney M. J., Hudson D. J., Robertson N. J., Chem. Commun. 2014, 50, 4884–4887. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a.Feghali E., Cantat T., ChemSusChem 2015, 8, 980–984; [DOI] [PubMed] [Google Scholar]
- 32b.Monsigny L., Berthet J.-C., Cantat T., ACS Sustainable Chem. Eng. 2018, 6, 10481–10488. [Google Scholar]
- 33.Kostera S., Peruzzini M., Gonsalvi L., Catalysts 2021, 11, 58–83, and references therein. [Google Scholar]
- 34.Deposition Number 2002499 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 35.Gómez-Gallego M., Sierra M. A., Chem. Rev. 2011, 111, 4857–4963. [DOI] [PubMed] [Google Scholar]
- 36.Gärtner D., Sandl S., Jacobi von Wangelin A., Catal. Sci. Technol. 2020, 10, 3502–3514. [Google Scholar]
- 37.Rogers E. I., Šljukić B., Hardacre C., Compton R. G., Electroanalysis 2008, 20, 2603–2607. [Google Scholar]
- 38.Bage A. D., Nicholson K., Hunt T. A., Langer T., Thomas S. P., ACS Catal. 2020, 10, 13479–13486. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary