

Abstract
Supertetrahedral clusters have been reported in two generally different types so far: one type possessing an organic ligand shell, no or low charges, and high solubility, while the other cluster type is ligand‐free with usually high charges and low or no solubility in common solvents. The latter is a tremendous disadvantage regarding further use of the clusters in solution. However, as organic substituents usually broaden the HOMO–LUMO gaps, which cannot be overcompensated by the (limited) cluster sizes, a full organic shielding comes along with drawbacks regarding opto‐electronic properties. We therefore sought to find a way of generating soluble clusters with a minimum number of organic substituents. Here, we present the synthesis and full characterization of two salts of [Sn10O4S16(SBu)4]4− that are high soluble in CH2Cl2 or CH3CN, which includes first NMR and mass spectra obtained from solutions of such salts with mostly inorganic supertetrahedral clusters. The optical absorption properties of this new class of compounds indicates nearly unaffected band gaps. The synthetic approach and the spectroscopic findings were rationalized and explained by means of high‐level quantum chemical studies.
Keywords: 119Sn-NMR spectroscopy, DFT calculations, ionothermal syntheses, supertetrahedral organotin chalcogenide clusters, X-ray diffraction
Selective access to partially butylated supertetrahedral clusters [Sn10O4S16(SBu)4]4− was achieved by employing a di‐butylated ionic liquid as reaction medium and alkylation agent. In contrast to purely inorganic [Sn10O4S20]8−, the butylated analogs are soluble in solvents like CH2Cl2 or CH3CN, without losing the typical opto‐electronic properties. Solution‐NMR data were assigned with the help of newly developed all‐electron relativistic DFT methods.
Introduction
The unique properties of materials based on supertetrahedral clusters, which are related to zeolites their general composition, have been documented in many reports in the past.[1, 2, 3, 4, 5, 6, 7, 8] In some cases, the desired properties require linkage of the clusters into networks—for instance, if certain pore sizes are needed for ion selectivity like in [H3O]3[Heta]4.2[H2dpp]0.3[In4Sn16O10S32]⋅33 H2O.[5] For photocatalytic properties, heterogeneous mixtures can be employed, for which the large diversity of open network structures like Na14[In17Cu3S35]⋅x H2O[6] or HAEM1.6[Ge3.2Zn0.8S8] (HAEM=N‐(2‐aminoethyl)‐morpholine‐H+)[7] are wonderful sources of tunable materials. Similar properties can also be achieved with salts of separate cluster units, as reported for the excellent photocatalytic activity of (C4C1C1Im)9[Cd3In17S31Cl4] (C4C1C1Im=1‐butyl‐2,3‐dimethylimidazolium).[8] However, all of the named materials need to be used as suspensions, as the relatively high charge of the anionic cluster substructures or the networks formed from them leads to an intrinsically low solubility in any common solvent. Consequently, also further derivatization or other chemical treatment has proven unsuccessful. This applies also to a few compounds possessing solubility in highly polar solvents: It was shown that a replacement of S atoms with Se atoms in (C4C1C1Im)9[Cd3In17S31Cl4] allows solubility in DMSO (dielectric constant ϵ≈41),[8] and compounds with supertetrahedral clusters of lower charges, like salts of [M4Sn4E17]10− (M=Co, Fe, Mn, Zn; Cd, Hg; E=S, Se, Te),[9] dissolve in highly polar solvents like water (ϵ≈80) or formamide (ϵ=109). In turn, clusters that are soluble in less polar solvents through shielding by organic group at bridging chalcogenolate ligands do usually show reduced opto‐electronic features owing to significantly enhanced optical gaps,[10, 11] unless the cluster sizes are getting huge, such as [Cd54S32(SPh)48(H2O)4]4−,[12] so that surface effects are overcompensated by a sufficiently large cluster volume.
The logical way of overcoming the named limitations would be the design and formation of compounds with molecular supertetrahedral clusters that exhibit a small degree of organic shielding only‐sufficient to solubilize the compounds in common organic solvents like CH2Cl2 (ϵ=9.1) or CH3CN (ϵ=37.5), yet without affecting the HOMO–LUMO gaps, similar to recent reports in the field of polygermanide anions forming highly soluble derivatives [Ge9{Si(SiMe3)3}]−,[13] [Ge9{Si(SiMe3)3}3{SnPh3}],[14] or [RGe9‐CH=CH−CH=CH‐Ge9R]4− (R=(2Z,4E)‐7‐amino‐5‐aza‐hepta‐2,4‐dien‐2‐yl).[15] The only problem to be solved is thus finding a corresponding synthetic approach for such target compounds. So far, all attempts to alkylate chalcogenido metalate clusters in a post‐synthetic way failed‐either due to the incompatibility of the alkylation reagents, like MeI or Meerwein salt, with the solvents from which the clusters were (irreversibly) obtained in crystalline form, or owing to the inherent insolubility of the cluster compounds in (organic) solvents appropriate for alkylation reactions. The extremely low nucleophilicity of chalcogenido metalate clusters in general is another important aspect that prohibited success in this direction so far.
Recently, we reported first promising results by presenting a method that allowed to form salts of partially methylated chalcogenido metalate clusters, which were heretofore only known as purely inorganic and highly charged molecules.[16, 17] The methylation was achieved by ionothermal syntheses[18] in imidazolium‐based ionic liquids (ClCmCnIm)X (Cl, Cm, Cn=alkyl chains with l, m, or n carbon atoms in 1‐, 2‐, or 3‐position of the imidazolium ring Im; X=Cl, Br, [BF4]).[19] With l or n being 1, thus ionic liquids with methyl substituents at one of the nitrogen atoms, the methyl group was selectively transferred to the terminal chalcogenide ligands of the investigated clusters, which served to reduce the negative charge of the anion by the number of alkyl substituents—for instance from −8 in [Sn10O4S20]8− [20, 21, 22] to −4 in [Sn10O4S16(SMe)4]4−.[16] Interestingly, the use of ionic liquids with a combination of any other alkyl groups and methyl substituents at the nitrogen atoms of the imidazolium ring also led to the selective transfer of the methyl group in all tested cases. However, as the methylation did not afford the desired increase in solubility, we continued to seek for a way of selectively transferring alkyl groups with larger chain lengths in order to ultimately from readily soluble supertetrahedral clusters.
To avoid a methyl transfer, the new studies started out with the synthesis of a symmetrically butyl‐substituted ionic liquid, (C4C1C4Im)Br (A), and its use in the cluster formation reaction. Indeed, a selective butylation of the discrete supertetrahedral clusters was achieved this way, as the methyl group at the C atom in 2‐position of the imidazolium ring is not transferred at all. This result is not only remarkable with respect to the synthetic approach that allows for a butylation of an extremely weak nucleophile, but finally allowed access to first salts of mainly inorganic supertetrahedral clusters that show high solubility in common organic solvents like CH2Cl2 and CH3CN. Hence, we do not only present the structural characterization of the products, but also show first NMR and mass spectra obtained from solutions of compounds with supertetrahedral cluster anions exhibiting purely inorganic bridging ligands.
Results and Discussion
Syntheses and Quantum Chemical Studies Regarding the Formation
The target compound 1 was obtained in two different salts with the sum formulas (C4C1C4Im)4+x [Sn10O4S16(SBu)4]Brx with x=3 (1 a) and x=2 (1 b). 1 a was prepared by in situ butylation during the cluster formation reaction, which was carried out by treatment of K4[SnS4]⋅4 H2O[23] in (C4C1C4Im)Br (A)[24] in the presence of 2,6‐dimethylmorpholine (DMMP) at 180 °C. Crystals of 1 b form upon dissolving crystals of compound 1 a in CH3CN and subsequently removing the solvent under reduced pressure. We attribute this finding to the high tendency of the ionic liquids to remain in solution, such that the crystals of 1 a lose one formula unit of it under these conditions. Scheme 1 summarizes the synthetic steps towards compounds 1 a and 1 b.
Scheme 1.
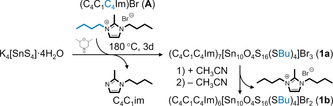
Survey of the synthesis of compounds 1 a and 1 b by ionothermal treatment of K4[SnS4]⋅4 H2O in (C4C1C4Im)Br (A) and further work‐up.
The structures of 1 a and 1 b were determined by means of single‐crystal X‐ray diffraction.[25] The presence of butyl substituents and the ionic liquid cations were confirmed in the solid state by means of Raman spectroscopy, and in solution by mass spectrometry, which provided us with the first mass spectrum of this cluster type at all.
In order to better understand why these reactions take place so readily and selectively, quantum chemical calculations were performed using density functional theory (DFT) methods (for details, see the Supporting Information).[26, 27, 28, 29, 30, 31, 32] We calculated reaction energies for the following reactions yielding methylated or butylated supertetrahedral clusters, respectively (Scheme 2).
Scheme 2.
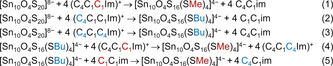
Reactions studied by means of DFT calculations.
Corresponding reaction energies in kJ mol−1 for reactions (1)–(3) are −617.5 (1), −586.5 (2), −592.5 (3). All values point to exoenergetic processes. A comparison of the energies of reactions (1) and (2) furthermore indicates a clear preference of a methylation (and not a butylation) to take place if (C4C1C1Im)+ is used. Finally, the butylation becomes more exoenergetic with the use of (C4C1C4Im)+ as butylation agent than with (C4C1C1Im)+. All of the calculated energies are thus in full agreement with our experimental findings.
Reaction energies that are calculated for reactions that include differently high charges of reactants and products can be afflicted with systematic shifts from COSMO. Although these should be similar for the analogous reactions (1) to (3), thus rendering the trends reliable, we also considered (hypothetical) exchange reactions (4)=(1)–(2) and (5)=(1)–(3), in which the charges of the involved species are the same on both sides of the reaction arrow with reaction energies in kJ mol−1 of −25.0 (4) and −31.0 (5). These numbers confirm that a methylation in general is preferred over a butylation. This rationalizes that the usage of an ionic liquid in which the nitrogen atoms of the imidazolium ring bear exclusively butyl substituents is essential for the formation of butylated clusters. We note in passing that corresponding calculations with Meerwein salt as methylation agent also yield (even more) negative reaction energies; yet such reactions cannot be practically carried out, as there is no solvent that is compatible to both the starting materials and Meerwein salt.
Crystal Structures
Compound 1 a crystallizes in the triclinic space group type P , with four formula units per unit cell. The butyl chains of both the anions and the cations are highly disordered, thus could not be fully localized on the difference Fourier map during the refinement. However, as outlined below, other analytical techniques, the analysis of the void space, and the comparison with compound 1 b allowed to confirm the composition. Compound 1 b crystallizes in the same space group type, but with two formula units only in the unit cell. The crystal structures are illustrated in Figure 1.
Figure 1.
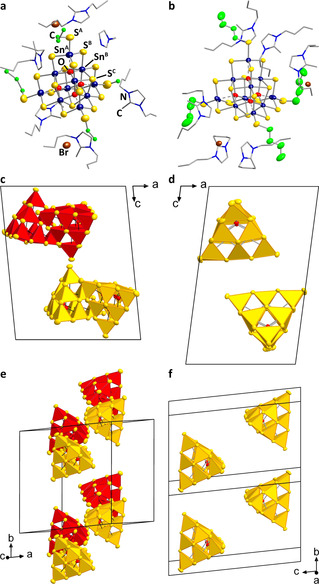
Crystal structure of compounds 1 a (a, c, e) and 1 b (b, d, f). Asymmetric units (a, b), with cluster atoms (excluding H atoms) and Br atoms given as displacement ellipsoids (50 % probability), imidazolium cations shown in wire mode, and H atoms are omitted for clarity. Simplified view of the packing of the anions in the crystal structures (c–f; Sn/O/S units shown only), with {SnS4} subunits given in polyhedral representation and the neighboring pairs of anions in 1 a indicated as pairs of clusters with yellow or red polyhedra, respectively. Structure diagrams with full labelling Scheme are given in the Supporting Information (Figures S6 and S7).
Besides the fact that the crystal structure of 1 b is only slightly affected by disorder, as the removal of one equivalent of (C4C1C4Im)Br obviously increased the lattice energy and thus crystallinity of the compound, the cluster molecules in both compounds are very similar (Figure 1 a,b; for a comprehensive list of structural parameters, see Table S4). Sn−Sterminal distances, which are indicative of an alkylation of the terminal S ligands, amount to 2.427(4)–2.448(4) Å in 1 a and 2.429(4)–2.500(7) Å in 1 b, while they were reported to be significantly shorter (2.355–2.374 Å) in the purely inorganic cousins.[20, 21, 22] Yet, the observed values are in good agreement with the Sn−Sterminal distances found for the methylated clusters (2.419(17)–2.511(11) Å).[16]
A notable difference between the two compounds, however, is the number x of co‐crystallizing formula units of the ionic liquid. In 1 b, x is unambiguously determined to be 2, while for 1 a the corresponding number had to be derived from the refinement of atoms plus an analysis of the differences in void spaces. By using the SQUEEZE tool[33] in OLEX2,[25] the void space volumes V VS were obtained by subtraction of the calculated volumes of the cluster units per unit cell from the unit cell volumes. The difference in V VS between the two compounds, ΔV VS=V VS(1 a)−V VS(1 b), amounts to 494 Å3, which is in perfect agreement with the calculated volume of one formula unit of (C4C1C4Im)Br (497 Å3) obtained with SQUEEZE from the structure of 1 b. This comparison also supports the correctness of the entire formula of 1 a, in which the number of counterions is anticipated to be seven—in agreement with the number of cluster anions and Br atoms found on the difference Fourier map, while not all of the imidazolium cations could be localized this way. Obviously, the different number of co‐crystallizing (C4C1C4Im)Br units additionally affects the overall packing of anions and cations in the crystal (Figure 1 c–f, and Figures S6,S7). The cluster anions are evenly distributed in the crystal structure of 1 b with center⋅⋅⋅center distances of 15.7714(2)–19.1377(3) Å. The relative orientation of the cluster units is a bit different in 1 a. Here, two clusters show a closer proximity, hence forming pairs (center⋅⋅⋅center distance: 12.0893(1) Å), while the other center⋅⋅⋅center distances are in the range of 15.7675(2) to 22.1975(3) Å.
Spectroscopy, Spectrometry, and Quantum Chemical studies of the Spectroscopic Findings
Single crystals of 1 a and 1 b dissolve well in organic solvents, which discriminates them from all reported salts of supertetrahedral chalcogenido metalate anions. Solutions in CH3CN‐d3 were thus analyzed by 1H‐, 13C‐, and 119Sn‐NMR spectroscopy (Figure 2 and Figure S8). The predominant peaks that are highlighted in the 1H‐NMR spectrum (Figure 2 a) confirm the presence of the (C4C1C4Im)+ cations (blue highlight). As the single crystals were covered by residues of the reaction medium that possesses a honey‐like consistence, we can also demonstrate the fate of the ionic liquid cation that served for the butylation: the resulting species could be identified as minor component C4C1im in the spectrum (im=imidazole; red highlight in Figure 2 a). The butyl signals of the alkylated cluster anions could not be assigned due to the very similar chemical shift of the other butyl substituents.
Figure 2.
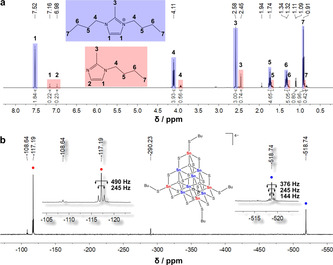
NMR spectra of a solution of single crystals of 1 b in CH3CN‐d3 with residues of attached reaction medium (causing minor signals). a) 1H‐NMR spectrum. b) 119Sn‐NMR spectrum with zoom into the signals assigned to the two different Sn atomic sites (cf. Figure 1 a: SnA, red; SnB, blue). Note that the relative intensities do not reflect the relative abundance owing to the measurement technique.
The 119Sn‐NMR spectra (Figure 2 b) show two predominant signals for the butylated supertetrahedral cluster, one at −117.19 ppm (SnA) and one at −518.74 ppm (SnB). Both signals exhibit satellites that we assign to 2 J(Sn−O/S−Sn) couplings: The satellites of the first show coupling constants of 490 and 245 Hz, while the values for the latter are 376, 245, and 144 Hz. The observed 119Sn chemical shifts are in accordance with literature data for corresponding coordination numbers at the SnIV atoms:[34, 35, 36, 37] SnA atoms are situated in between a tetrahedral and a trigonal bipyramidal ligand environment, {SnS4⋅⋅⋅O} (Sn⋅⋅⋅O 2.477(5)–2.550(4) Å), SnB atoms exhibit a pseudo‐octahedral coordination, {SnS4O2} (Sn−O 2.077(5)–2.116(6) Å). Furthermore, the coupling constants are in good agreement with data reported for [(PhCH2Sn)12(μ3‐O)14(μ‐OH)6], for which 2 J values were found to be between 483 and 115 Hz.[38]
As it is difficult to judge about the relative intensities with the naked eye (and as integration of the peaks is afflicted with high inaccuracies owing to different relaxation times in different chemical environments of the Sn atoms), we corroborated our assignment of the 119Sn data by means of DFT calculations. We note that the applied methodology for the calculation of coupling constants was implemented in the turbomole program suite only recently, and applied here for the first time: the NMR shifts were calculated within the quasi‐relativistic all‐electron exact two‐component (X2C) method[39] in its scalar‐relativistic one‐component variant,[40] coupling constants with two‐component X2C.[41] The finite nucleus model[42] and the diagonal local approximation for the unitary decoupling transformation (DLU)[43] were used, and further triple‐ζ basis sets[44] and integration grids optimized for all‐electron calculations (size 4a).[45] Calculated chemical shifts (w.r.t. SnMe4) are found at −5.8 ppm for SnA and −411.4 ppm for SnB. Both are thus systematically shifted by ≈115 ppm relative to the experimental values, but exhibit an excellent agreement of the shift difference Δδ≈406 ppm with respect to the experimental spectrum (Δδ≈401 ppm), which confirms our assignment. Measurements of indirect spin‐spin coupling constants show a coupling constant of 245 Hz that is present in both Sn peaks (see Figure 2 b), and thus most probably originates from the coupling 2 J(SnA−SnB). For the “inner” SnB atoms, this is accompanied by a second coupling constant that is lower by 101 Hz, and probably originates from the 2 J(SnB−SnB) coupling. Calculated numbers overall are higher [mean values of 363.3 Hz for 2 J(SnA−SnB) and 337.3 Hz for 2 J(SnB−SnB)], but again, 2 J(SnA−SnB) is significantly larger than 2 J(SnB−SnB). We note that in view of the large range of values for Sn coupling constants, the comparably large shift in absolute number is not unexpected, similar as for the chemical shielding constants. The weak satellites in the experimental spectrum at 499 Hz for SnA and 376 Hz for SnB cannot be attributed to 119Sn coupling constants.
As an additional prove of the identity of the cluster units in 1 a and 1 b, Raman spectra were recorded on single crystals of both compounds and compared (Figure S9). Both spectra exhibit the characteristic signal group of the Sn/O/S framework between 100 and 400 cm−1, while the signals at around 700 cm−1 and around 2900 cm−1 are clearly different from the signals observed for single crystals of the methylated anions. This confirms that the cluster anions in 1 a are also butylated.
Another peculiarity of the butylated supertetrahedra and their significant solubility is the possibility to transfer them into the gas phase without decomposition. By means of electrospray‐ionization mass spectrometry (ESI‐MS) in negative ion mode, we were able to detect the butylated anion‐as a mono‐anionic aggregate along with three (C4C1C4Im)+ cations, {(C4C1C4Im)3[Sn10O4S16(SBu)4]}−. The overview spectrum and the high‐resolution of the molecular peak is shown in Figure 3. The full version of the inset with all m/z values of the individual peaks is provided in the Supporting Information (Figure S10). We would like to emphasize that this is the first spectrum of a supertetrahedral cluster that possesses such a small number of organic substituents.
Figure 3.
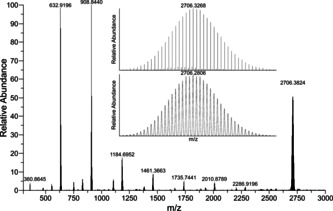
Overview ESI(−) mass spectrum of a fresh solution of single crystals of compound 1 b in CH3CN. The high‐resolution mass peak of the cluster anion in 1 b, detected along with two counterions as the mono‐anionic aggregate {(C4C1C4Im)3[Sn10O4S16(SBu)4]}−, is shown in the inset. Note that the slight deviation of decimal places in the high‐resolution spectrum versus the overview spectrum is normal and due to the differences in data processing. Further details, including the m/z values for all individual peaks, are found in the Supporting Information (Figure S10).
To probe to which extent the four organic groups would affect the electronic properties of the clusters, we recorded optical absorption spectra of the solid compounds as well as their solutions, and compared them to the spectrum of the methylated and the ligand‐free analogs. As illustrated in Figures 4 a and b, both solid compounds exhibit very similar values for their onset of absorption at around 3.5 eV in accordance with their colorless appearance. The salt of the methylated cluster[16] exhibits a very similar excitation energy (3.35 eV). Corresponding values of the salts of the purely inorganic cluster anion [Sn10O4S20]8− were not reported, but for compounds with a two‐dimensional linkage of such clusters, the value was determined to be 2.98 eV, which we take as a lower limit of the value of the separated cluster molecules.[46] This suggests that the partial alkylation only slightly affects the electronic structure of the cluster, and that the chosen alkyl group makes no difference in this regard. As expected, the spectrum recorded in solution (Figure 4 c) indicates a slightly blue‐shifted onset of absorption for the individual clusters—by ≈0.1 eV to 3.62 eV.
Figure 4.
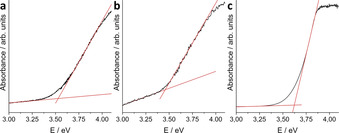
Optical absorption spectra recorded on single crystals of 1 a (a) and 1 b (b). Spectrum of a solution (c=2 mmol L−1) of single crystals of 1 b in CH3CN (c).
Time‐dependent density functional theory (TD‐DFT) calculations[26, 28, 29, 30, 31, 32, 47] served to explain and visualize the absorption event. The calculated spectra (line spectra and superimposed Gaussians) of calculated species [Sn10O4S20]8−, [Sn10O4S16(SMe)4]4−, and [Sn10O4S16(SBu)4]4−, are shown in Figure 5 along with the difference of densities of excited and ground states of the first absorption band, calculated as described in Ref. [48].
Figure 5.
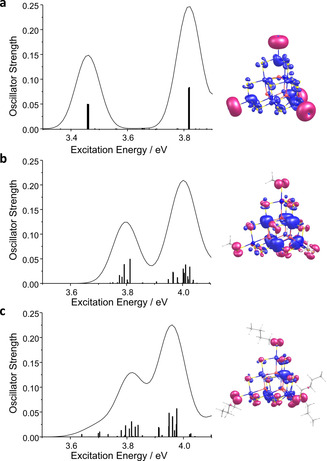
TD‐DFT excitation spectra of [Sn10O4S20]8− (a), [Sn10O4S16(SMe)4]4− (b), and [Sn10O4S16(SBu)4]4− (c), with superimposed Gaussians (left). Difference of densities the first absorption band (right; contours of densities plotted at 0.0008 a.u.); red contours indicate a surplus of electron density for the ground state, blue contours indicate a surplus of electron density for the excited state.
The calculated absorption energies (and respective onsets of absorption) are in excellent agreement with the data measured on the individual clusters in solution, shifted by only ≈0.1 eV to higher energies. Within the calculated spectra, the alkylated clusters show a slight blue‐shift of ≈0.35⋅⋅⋅0.4 eV with respect to the purely inorganic cluster, hence in perfect agreement with the blue‐shift of ≈0.35⋅⋅⋅0.5 eV observed in the experimental spectra. This blue‐shift can be understood upon inspection of the involved orbitals. The lowest energy transition is dominated by charge transfer from S(3p) orbitals of the corner S ligands (red contours in Figure 5) into orbitals of the cluster core (blue contours in Figure 5). As one of these S(3p) orbitals per S atom is involved in the S−C bond to the alkyl group, thus lowered in energy, its excitation requires more energy than in the purely inorganic case. This explains, why the overall effect is notably smaller than for fully protected clusters, where all surface chalcogenide ligands are involved in chalcogen−ligand bonds. Hence, while we significantly vary the solubility of the super‐tetrahedra, we keep their opto‐electronic properties comparably constant (with a slight tendency towards smaller energy differences between the first and second excitation bands) when going from ligand‐free clusters to methylated, and finally, butylated versions.
Conclusion
Reactions of K4[SnS4]⋅4 H2O in and with the ionic liquid 1,3‐dibutyl‐2‐methylimidazolium bromide, (C4C1C4Im)Br, allowed for the straight‐forward synthesis of salts of partially butylated supertetrahedral cluster anions [Sn10O4S16(SBu)4]4−. In contrast to methylated or purely inorganic analogs, the compounds are fully soluble in common organic solvents CH2Cl2 and CH3CN. We could thus record first NMR and mass spectra of solutions of this cluster type, which are presented in this work. Remarkably, the partial and selective alkylation of the supertetrahedra's terminal S ligands does only slightly affect the electronic structures of the compounds, which possess only slightly blue‐shifted excitation energies in regard to the purely inorganic salts. DFT calculations confirmed that the methylation in general is preferred over the butylation, and they allowed for the assignment of shifts and coupling constants to inner/outer Sn atoms. Time‐dependent DFT calculations showed that the lowest electronic excitation band is dominated by charge transfer from 3p orbitals of the corner S ligands into orbitals of the cluster core. Overall, the findings provided us with a unique perspective of a rich follow‐up chemistry and, ultimately, use of soluble supertetrahedral clusters for opto‐electronic applications in the future.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG) in the framework of the Priority Programme SPP1708. Open access funding enabled and organized by Projekt DEAL.
B. Peters, G. Stuhrmann, F. Mack, F. Weigend, S. Dehnen, Angew. Chem. Int. Ed. 2021, 60, 17622.
Dedicated to Professor Ulrich Siemeling on the occasion of his 60th birthday
Contributor Information
Prof. Dr. Florian Weigend, Email: florian.weigend@chemie.uni-marburg.de.
Prof. Dr. Stefanie Dehnen, Email: dehnen@chemie.uni-marburg.de.
References
- 1.
- 1a.Wu T., Bu X., Liao P., Wang L., Zheng S.-T., Ma R., Feng P., J. Am. Chem. Soc. 2012, 134, 3619–3622; [DOI] [PubMed] [Google Scholar]
- 1b.Xu X., Wang W., Liu D., Hu D., Wu T., Bu X., Feng P., J. Am. Chem. Soc. 2018, 140, 888–891. [DOI] [PubMed] [Google Scholar]
- 2.Xiong W.-W., Li J.-R., Hu B., Tan B., Li R.-F., Huang X.-Y., Chem. Sci. 2012, 3, 1200–1204. [Google Scholar]
- 3.Wang Y., Zhu Z., Sun Z., Hu Q., Li J., Jiang J., Huang X.-Y., Chem. Eur. J. 2020, 26, 1624–1632. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.Manos M. J., Iyer R. G., Quarez E., Liao J. H., Kanatzidis M. G., Angew. Chem. Int. Ed. 2005, 44, 3552–3555; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3618–3621; [Google Scholar]
- 4b.Manos M. J., Malliakas C. D., Kanatzidis M. G., Chem. Eur. J. 2007, 13, 51–58. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X.-M., Sarma D., Wu Y.-Q., Wang L., Ning Z.-X., Zhang F.-Q., Kanatzidis M. G., J. Am. Chem. Soc. 2016, 138, 5543–5546. [DOI] [PubMed] [Google Scholar]
- 6.Zheng N., Bu X., Vu H., Feng P., Angew. Chem. Int. Ed. 2005, 44, 5299–5303; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5433–5437. [Google Scholar]
- 7.Sasan K., Lin Q., Mao C., Feng P., Nanoscale 2016, 8, 10913–10916. [DOI] [PubMed] [Google Scholar]
- 8.Hao M., Hu Q., Zhang Y., Luo M., Wang Y., Hu B., Li J., Huang X.-Y., Inorg. Chem. 2019, 58, 5126–5133. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a.Palchik O., Iyer R. G., Liao J. H., Kanatzidis M. G., Inorg. Chem. 2003, 42, 5052–5054; [DOI] [PubMed] [Google Scholar]
- 9b.Zimmermann C., Melullis M., Dehnen S., Angew. Chem. Int. Ed. 2002, 41, 4269–4272; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 4444–4447; [Google Scholar]
- 9c.Dehnen S., Brandmayer M. K., J. Am. Chem. Soc. 2003, 125, 6618–6619; [DOI] [PubMed] [Google Scholar]
- 9d.Ruzin E., Fuchs A., Dehnen S., Chem. Commun. 2006, 4796–4798. [DOI] [PubMed] [Google Scholar]
- 10.Soloviev V., Eichhöfer A., Fenske D., Banin U., J. Am. Chem. Soc. 2000, 122, 2673–2674. [DOI] [PubMed] [Google Scholar]
- 11.Soloviev V., Eichhöfer A., Fenske D., Banin U., J. Am. Chem. Soc. 2001, 123, 2354–2364. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a.Zheng N., Bu X., Lu H., Zhang Q., Feng P., J. Am. Chem. Soc. 2005, 127, 11963–11965; [DOI] [PubMed] [Google Scholar]
- 12b.Nguyen K. A., Pachter R., Day P. N., Su H., J. Chem. Phys. 2015, 142, 234305. [DOI] [PubMed] [Google Scholar]
- 13.Li F., Muñoz-Castro A., Sevov S. C., Angew. Chem. Int. Ed. 2012, 51, 8581–8584; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8709–8712. [Google Scholar]
- 14.Li F., Sevov S. C., J. Am. Chem. Soc. 2014, 136, 12056–12063. [DOI] [PubMed] [Google Scholar]
- 15.Bentlohner M. M., Klein W., Fard Z. H., Jantke L.-A., Fässler T. F., Angew. Chem. Int. Ed. 2015, 54, 3748–3753; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3819–3824. [Google Scholar]
- 16.
- 16a.Peters B., Santner S., Donsbach C., Vöpel P., Smarsly B., Dehnen S., Chem. Sci. 2019, 10, 5211–5217; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b.Peters B., Reith S., Dehnen S., Z. Anorg. Allg. Chem. 2020, 646, 964–967. [Google Scholar]
- 17.Peters B., Lichtenberger N., Dornsiepen E., Dehnen S., Chem. Sci. 2020, 11, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a.Wasserscheid P., Welton T., Ionic Liquids in Synthesis, 2nd ed., Wiley-VCH, Weinheim, 2008; [Google Scholar]
- 18b.Morris R. E., Chem. Commun. 2009, 2990–2998. [DOI] [PubMed] [Google Scholar]
- 19.Niedermeyer H., Hallett J. P., Villar-Garcia I. J., Hunt P. A., Welton T., Chem. Soc. Rev. 2012, 41, 7780–7802. [DOI] [PubMed] [Google Scholar]
- 20.Schiwy W., Krebs B., Angew. Chem. Int. Ed. Engl. 1975, 14, 436; [Google Scholar]; Angew. Chem. 1975, 87, 451. [Google Scholar]
- 21.Parise J. B., Ko Y., Chem. Mater. 1994, 6, 718–720. [Google Scholar]
- 22.Kaib T., Kapitein M., Dehnen S., Z. Anorg. Allg. Chem. 2011, 637, 1683–1686. [Google Scholar]
- 23.Ruzin E., Jakobi S., Dehnen S., Z. Anorg. Allg. Chem. 2008, 634, 995–1001. [Google Scholar]
- 24.Boruah K., Borah R., ChemistrySelect 2019, 4, 3479–3485. [Google Scholar]
- 25.
- 25a.Sheldrick G. M., Acta Crystallogr. Sect. A 2015, 71, 3–8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b.Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 71, 3–8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c.Dolomanov O. V., Bourhis L. J., Gildea R. J., Howard J. A. K., Puschmann H., J. Appl. Crystallogr. 2009, 42, 339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25d.Depo- sition Numbers 2070230, 2070231, and 2070232 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 26.TURBOMOLE V7.5 2020, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH since 2007; available from https://www.turbomole.org.
- 27.Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
- 28.Weigend F., Baldes A., J. Chem. Phys. 2010, 133, 174102. [DOI] [PubMed] [Google Scholar]
- 29.Weigend F., Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 30.Metz B., Stoll H., Dolg M., J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar]
- 31.Klamt A., Schüürmann G., J. Chem. Soc. Perkin Trans. 2 1993, 2, 799–805. [Google Scholar]
- 32.Perdew J. P., Ernzerhof M., Burke K., J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar]
- 33.Spek A. L., Acta Crystallogr. Sect. C 2015, 71, 9–18. [DOI] [PubMed] [Google Scholar]
- 34.Jiang T., Ozin G. A., Bedard R. L., Adv. Mater. 1994, 6, 860–865. [Google Scholar]
- 35.Rangan K. K., Trikalitis P. N., Canlas C., Bakas T., Weliky D. P., Kanatzidis M. G., Nano Lett. 2002, 2, 513–517. [Google Scholar]
- 36.Multinuclear NMR (Ed.: Kennedy J. D., McFarlane W., Mason J.), Plenum, New York, 1987, pp. 305–334. [Google Scholar]
- 37.Gielen M., Davies A. G., Pannell K., Tiekink E., Tin Chemistry: Fundamentals, Frontiers, and Applications, Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 38.Plasseraud L., Cattey H., Richard P., Z. Naturforsch. 2011, 66b, 262–268. [Google Scholar]
- 39.Kutzelnigg W., Liu W., J. Chem. Phys. 2005, 123, 241102. [DOI] [PubMed] [Google Scholar]
- 40.Franzke Y. J., Weigend F., J. Chem. Theory Comput. 2019, 15, 1028–1043. [DOI] [PubMed] [Google Scholar]
- 41.Franzke Y. J., Mack F., Weigend F., J. Chem. Theory Comput. 2021, 10.1021/acs.jctc.1c00167. [DOI] [PubMed] [Google Scholar]
- 42.Visscher L., Dyall K. G., At. Data Nucl. Data Tables 1997, 67, 207–224. [Google Scholar]
- 43.Peng D., Middendorf N., Weigend F., Reiher M., J. Chem. Phys. 2013, 138, 184105. [DOI] [PubMed] [Google Scholar]
- 44.Pollak P., Weigend F., J. Chem. Theory Comput. 2017, 13, 3696–3705. [DOI] [PubMed] [Google Scholar]
- 45.Franzke Y. J., Tress R., Pazdera T. M., Weigend F., Phys. Chem. Chem. Phys. 2019, 21, 16658–16664. [DOI] [PubMed] [Google Scholar]
- 46.Huang S.-L., He L., Chen E.-X., Lai H.-D., Zhang J., Lin Q., Chem. Commun. 2019, 55, 11083–11086. [DOI] [PubMed] [Google Scholar]
- 47.Bauernschmitt R., Ahlrichs R., Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar]
- 48.Kühn M., Weigend F., J. Chem. Phys. 2014, 141, 224302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information