

Abstract
Objective
Identifying common genetic variants that confer genetic risk for cluster headache.
Methods
We conducted a case–control study in the Dutch Leiden University Cluster headache neuro‐Analysis program (LUCA) study population (n = 840) and unselected controls from the Netherlands Epidemiology of Obesity Study (NEO; n = 1,457). Replication was performed in a Norwegian sample of 144 cases from the Trondheim Cluster headache sample and 1,800 controls from the Nord‐Trøndelag Health Survey (HUNT). Gene set and tissue enrichment analyses, blood cell‐derived RNA‐sequencing of genes around the risk loci and linkage disequilibrium score regression were part of the downstream analyses.
Results
An association was found with cluster headache for 4 independent loci (r 2 < 0.1) with genomewide significance (p < 5 × 10−8), rs11579212 (odds ratio [OR] = 1.51, 95% confidence interval [CI] = 1.33–1.72 near RP11‐815 M8.1), rs6541998 (OR = 1.53, 95% CI = 1.37–1.74 near MERTK), rs10184573 (OR = 1.43, 95% CI = 1.26–1.61 near AC093590.1), and rs2499799 (OR = 0.62, 95% CI = 0.54–0.73 near UFL1/FHL5), collectively explaining 7.2% of the variance of cluster headache. SNPs rs11579212, rs10184573, and rs976357, as proxy SNP for rs2499799 (r 2 = 1.0), replicated in the Norwegian sample (p < 0.05). Gene‐based mapping yielded ASZ1 as possible fifth locus. RNA‐sequencing indicated differential expression of POLR1B and TMEM87B in cluster headache patients.
Interpretation
This genomewide association study (GWAS) identified and replicated genetic risk loci for cluster headache with effect sizes larger than those typically seen in complex genetic disorders. ANN NEUROL 2021;90:203–216
Cluster headache (CH) is a primary headache disorder characterized by attacks of intense unilateral orbital, supraorbital and/or temporal pain that last for 15 to 180 minutes and are associated with ipsilateral facial autonomic symptoms and/or restlessness. The majority of patients have episodic CH, with periods of attacks of weeks to months, alternating with attack‐free periods of at least 3 months. In 10 to 15% of patients, cluster periods never remit for longer than 3 months for at least 1 year, classifying them as chronic CH. The male‐to‐female ratio is 2:1.1 Smoking and psychiatric comorbidities are prevalent.2 Current treatment strategies include aborting acute attacks and aim to reduce attack frequency with preventive treatment.3 CH shows some phenotypic overlap with other trigeminal neuralgias, but also with migraine, for example, in that some patients with migraine may also report autonomic features. Certain similar pathophysiological pathways are hypothesized to be involved in both CH and migraine.4 Although these disorders share prominent features, they are clinically well distinguishable.5
The pathophysiology of CH is poorly understood, although vasomotor changes, inflammation, hypothalamic dysfunction, and dysregulation of the autonomic nervous system have been implicated as potential disease mechanisms.6 Twin and family studies have highlighted the involvement of genetic factors in CH.7 Thus far, most genetic studies used a hypothesis‐driven approach and have examined a limited number of variants in genes linked to presumed pathways in CH. Most studied are variants in HCRTR2, which encodes the hypocretin (orexin) type 2 receptor that binds neuropeptides hypocretin‐1 and ‐2 in the central nervous system. Still, initially positive genetic findings for HCRTR2 associations8, 9, 10 were not replicated in better‐powered studies.11, 12 Finally, the first, although very small hypothesis‐free, Italian genomewide association study (GWAS) investigating 99 patients with CH reported suggestive associations with genetic variants in ADCYAP1R1 and MME,13 but these findings were not replicated in a larger Swedish sample.14
To detect genetic variants for CH, we conducted a GWAS in a Dutch sample of 840 patients with CH and 1,457 controls from the same geographical region. Results were replicated in a Norwegian sample. Downstream analyses further assessed genes and mechanisms contributing to the pathogenesis of CH.
Methods
Patient Recruitment and Sample Collection
The Dutch cluster headache study included 862 Dutch patients with CH from the clinic‐based Leiden University Cluster headache neuro‐Analysis program (LUCA) that were recruited between 2010 and 2015 via the project's website. Patients with CH aged 18 years or older were included. Participants fulfilling the screening criteria were asked to complete an extended questionnaire that focused on signs and symptoms of CH as outlined in the International Classification of Headache Disorders (ICHD‐II or ICHD‐III) criteria for CH.15 , 16 Individual diagnoses were made upon visiting the outpatient clinic or using a validated algorithm (positive predictive value: ∼ 92%) based on ICHD criteria.17 CH cases were diagnosed in specialized headache centers to minimize misclassification. Controls (n = 1,671) were obtained from the Netherlands Epidemiology of Obesity Study (NEO) study,18 a population‐based sample that includes individuals aged 45 to 65 years living in a nearby municipality (Leiderdorp, The Netherlands) recruited between 2008 and 2012. Most cases and all controls originated from the same geographical region, in the Western part of the Netherlands. All participants were unrelated and of European ancestry. The local ethics committees approved the study. Written informed consent was obtained from all participants.
Genotyping, Quality Control, and Imputation in the Discovery Stage
Genomic DNA was extracted from peripheral blood leukocytes according to standard protocols and genotyping of both cases and controls was performed using the Illumina Infinium CoreExome‐24 version 1.1 array according to the protocol from the manufacturer. Cases were genotyped at the Genomics‐Core Facility at the Norwegian University of Science and Technology (Trondheim, Norway) and controls at the Centre National de Génotypage (Paris, France). For the cases, variant calling was performed with Genome Studio 2.0 following a standard quality protocol,19 and the CHARGE best practice calling of the HumanExome Bead chip.20 For the controls, calling was performed using the GenCall algorithm using standard settings as provided by Illumina. Quality control was performed according to standard procedures.21 Markers with high missingness rates (≥ 2%), monomorphic variants and those failing the Hardy–Weinberg equilibrium were excluded. Individuals were excluded if they had a high proportion of missing genotype data (≥ 2%), inconsistent sex information, were related (PI‐HAT ≥ 0.2), or were heterozygosity outliers. Principal component analysis (PCA) was performed on the pruned data set (with a 50‐kb sliding window, r 2 > 0.2) using PLINK and population outliers were excluded. No overt population substructure between cases and controls was observed (data not shown). After combining the genotyped single nucleotide polymorphism (SNP) information from LUCA and NEO, imputation was performed on the Michigan Imputation Server (https://imputationserver.sph.umich.edu/) using Haplotype Reference Consortium (HRC version 1.1 2016) as a reference panel after phasing by Eagle (version 2.3),22 using the default parameters.
In total, 345,064 SNPs from 2,297 individuals (840 cases and 1,457 controls) were available for imputation. Prior to analyses, variants with minor allele frequency (MAF) ≤ 0.01 or imputation INFO score ≤ 0.6 were excluded, resulting in 7,578,399 SNPs.
Statistical Analysis in the Discovery Stage
Case–control SNP association analysis was performed using a logistic regression model implemented in SNPTEST (version 2.5.2) for autosomal variants, with case–control status as outcome and assuming additive allelic effects. The model was adjusted for sex. In addition, the model was adjusted for the first 4 principal components to minimize effects of confounding and population stratification. A Manhattan and a quantile‐quantile (QQ) plot for test statistics were generated using R version 3.6.1 (R Foundation for Statistical Computing, Vienna, Austria). We determined lead SNPs that were independent from each other at r 2 < 0.1 and further apart than 500 kb, with association p < 5 × 10−8. Positional gene mapping and fine mapping of significant loci was performed using Functional Mapping and Annotation version 1.3.6 (FUMA), Probabilistic Identification of Causal SNPs (PICS), and Locuszoom.23, 24, 25 The proportion of variance explained by a given SNP was calculated using Nagelkerke pseudo R2.
Patient Recruitment and Data Generation in the Replication Stage
Cases were recruited at the Norwegian Advisory Unit on Headaches, St Olav's Hospital, Trondheim (Norway) between 2005 and 2016, with the inclusion criterion being the definite diagnosis of CH according to ICHD‐II or ICHD‐III,15 , 16 made by a neurologist with special competence in headache disorders to minimize misclassification. As controls, we used a random subset of 1,800 adult participants from the Nord‐Trøndelag Health Study (HUNT) who did not have CH defined by the International Classification of Disease (ICD)‐10 diagnosis G44.0 (Cluster headache syndrome) or the ICD‐9 diagnosis 346.2 (Migraine variants, including cluster headache).26
A sample of 159 CH cases were genotyped with the Illumina Infinium CoreExome‐24 version 1.1. Calling was performed with Genome Studio 2.0, using the cluster file from the largest batch of 58,996 HUNT All‐in controls (see below). The analysis followed the Genome Studio quality protocol,19 and the CHARGE best practice calling of the HumanExome Bead chip.20 The HUNT control samples were genotyped on 3 different Illumina HumanCoreExome arrays (HumanCoreExome12 version 1.0, HumanCoreExome12 version 1.1, and UM HUNT Biobank version 1.0), and called as described elsewhere.27 Markers with high missingness rates (≥ 2%), monomorphic variants and those failing the Hardy–Weinberg equilibrium were excluded. Individuals with high missingness rates (≥ 2%) or whose inferred sex contradicted with reported sex were excluded. A second round of quality control was performed after merging cases and all HUNT controls, excluding variants that were monomorphic, deviated from Hardy–Weinberg equilibrium, or had different genotype rate between cases and controls. Individuals were excluded if they had missingness ≥ 2%, outlying heterozygosity rate or were duplicates. Population outliers and non‐European samples were excluded. No overt population substructure between cases and controls was observed (data not shown). A total of 69,440 individuals passed quality control, including 144 cases. A dataset including the 144 cases and 1,800 randomly selected controls was imputed using Minimac3 (version 2.0.1) and the Hapmap r22 CEU panel. Variants with minor allele count < 3 or with imputation quality r 2 < 0.3 were excluded, resulting in 2,363,678 well‐imputed variants for 144 cases (38 women and 106 men) and 1,800 controls (952 women and 848 men). The study was approved by the local ethics committees. Written informed consent was obtained from all participants.
Replication Analysis
Association analysis was performed using a mixed logistic regression model implemented in SAIGE (version 0.35.8.3), where CH was modeled as the dependent variable, and the genetic variants as the independent variable. Sex and the first 8 principal components were included as covariates. From each independent significant locus (p < 5 × 10−8) in the discovery sample, the lead SNP, or a proxy SNP, was selected for replication. To correct for multiple testing, Bonferroni correction was applied for the number of loci tested (n = 4).
Sex Stratified Analysis
Analyses stratified for men and women were performed in SNPTEST to examine possible sex‐specific genetic effects. Both models were adjusted for the first 4 principal components.
Previously Reported Cluster Headache Loci
The 9 SNPs previously significantly associated with CH8, 9, 10, 13, 28, 29, 30, 31, 32 were tested for association in our discovery analysis. The p values were adjusted for multiple‐testing using Bonferroni correction.
Univariate Linkage Disequilibrium Score Regression
Linkage Disequilibrium Score Regression (LDSC version 1.0.1) was used to estimate the proportion of a true polygenic signal versus confounding factors, such as population stratification, and to calculate SNP‐based heritability.31 Variants present in the HapMap 3 reference set were used, after excluding variants (1) with large‐effect, explaining > 1% of phenotype variation, or variants in liquid disequilibrium (LD) with such; (2) with MAF ≤ 0.01 or imputation INFO score ≤ 0.9; and (3) in the HLA region. Heritability estimates were converted to the liability scale assuming a population prevalence of CH of 0.1%.6
Colocalization Analysis
To test whether the association signals for CH and migraine, on chromosome 6 near UFL1/FHL5, are consistent with a shared causal variant, we used a Bayesian colocalization procedure using the R package “coloc” with default settings.34 This test generates posterior probabilities for each locus weighting the evidence for 5 competing hypotheses regarding the sharing of causal variants, namely H0 (no causal variant for either trait); H1 or H2 (a causal variant only for trait 1 or 2); H3 (distinct causal variants, for each trait); and H4 (a single causal variant common to both traits). The analysis assumes a single causal SNP for each trait. For CH, we used the summary statistics from the discovery cohort and for migraine we used the summary statistics from Gormley et al35 without 23andMe (30,465 migraine cases and 143,147 controls); both populations are of European ancestry. Colocalization was tested for the region between the 2 nearest recombination hotspots.
Genetic Correlation
LDSC was also used to calculate genetic correlation between CH and migraine.33 For migraine, we used summary statistics from Gormley et al35 without 23andMe (30,465 migraine cases and 143,147 controls), excluding variants with MAF ≤ 0.01, INFO score ≤ 0.6, large‐effect variants or variants in an HLA region. In addition, the 38 genomewide significant migraine loci were tested for association with CH.35 Using the cor.test function in R, the correlation of the effect size (beta) between migraine and CH (current study) was calculated.
Gene‐Based Analysis
We performed the MAGMA gene‐based association analysis implemented in FUMA, using default settings to identify genes associated with CH.24 This calculates a gene test‐statistic (p value) based on all SNPs located within genes. SNPs were assigned to the genes obtained from Ensembl build 85 (only protein‐coding genes).
Tissue Specificity Analyses
To further test the relationship between tissue‐specific expression and genetic associations to CH, we examined all SNPs and their respective effect on the expression of genes up to 1 Mb away (cis‐expression quantitative trait locus [eQTL]), using FUMA quantitative trait locus (eQTL) mapping (https://fuma.gtlab.nl/tutorial#eQTLs); all SNPs were mapped based on each of the tissues in the Genotype Tissue Expression (GTEx) version 8 dataset using default setting.24 Additionally, we performed tissue expression analysis based on the MAGMA gene property in FUMA.24 This analysis tests for positive relationships between tissue‐specific gene expression in 30 general tissue types and 54 specific tissue types in the GTEx version 8 RNA‐seq data and gene‐based p values from the gene‐based analysis described above.
RNA‐Sequencing of Patients With CH and Controls
The genes identified by eQTL mapping with FUMA (see above) were further interrogated using existing RNA sequencing (RNA‐seq) data generated from peripheral venous blood samples from 39 patients with CH and 20 controls matched for age, sex, and smoking habits. Data generation and quality control is described in detail elsewhere.36 In short, RNA was extracted, using the PAXgene Blood miRNA kit, and sequenced using Illumina Hiseq4000. RNA‐seq reads were aligned and processed using the in‐house transcriptome analysis pipeline Gentrap (version 0.3.1). Within this pipeline, sequencing reads were aligned to the human genome reference GRCh38 using TopHat (version 2.0.13) and counted per gene using Htseq (version 0.6.1p1). The data were normalized for between‐sample variation and for within‐sample variation, using the Limma voom transformation. Differential expression analysis was performed in Limma, fitting a linear model correcting for age, gender, current smoking status, and leukocyte counts. The p values were adjusted for multiple‐testing using Bonferroni correction.
To determine the specificity of differential expression results obtained for CH, we examined the (nominally) significant genes from the CH RNA‐seq analysis in RNA‐seq data obtained from 26 patients with migraine and 20 age‐ and sex‐matched controls. Data generation and quality control have been previously described.37 In short, peripheral venous blood samples were drawn when the patients with migraine were migraine‐free for at least 5 days and headache‐free for 24 hours. RNA was extracted using PAXgene Blood RNA kit, sequenced (using Illumina Novaseq) and aligned. RNA‐seq reads were, after quality control, aligned to the human reference transcriptome, using kallisto (version 0.42.5). Resulting count matrices were corrected for library size and gene length, and normalized using the R package DESeq2. Differential expression was performed using the R package DESeq2 by fitting a generalized linear model, correcting for age.
Results
Study Participants
The clinical characteristics of cases and controls of the discovery sample are summarized in Table 1. There was a higher proportion of men (69% vs 44%) and smokers (52% vs 14%) among the cases compared to controls. Most patients had episodic CH (69%). A total of 13% of cases had migraine.
TABLE 1.
Clinical Characteristics of the Study Samplea
| Characteristics | Discovery sample | ||
|---|---|---|---|
| Patients with CH (n = 840) | Controls (n = 1,457) | p valueb | |
| Men | 579 (68.9) | 636 (43.7) | < 0.001 |
| Current daily smoking | 440 (52.4) | 202 (13.9) | < 0.001 |
| Episodic cluster headache | 577 (68.7) | ‐ | ‐ |
| Chronic cluster headache | 233 (27.7) | ‐ | ‐ |
| Migraine comorbidity | 106 (12.6) | ‐ | ‐ |
Data are expressed as numbers (percentages) unless otherwise stated.
Numbers and proportions may not add up to total of 100 due to rounding or missing values.
The p values of chi‐square test for categorical variables.
CH = cluster headache.
Association Analysis
Overall association results are shown in the Manhattan plot (Fig 1A) and the QQ plot (Fig 2). In total, 4 independent loci showed genomewide significant (p < 5 × 10−8) associations with CH (Fig 1B–E) with a combined explained variance of 7.2%. More specifically, we identified rs11579212 (odds ratio ]OR = 1.51, 95% confidence interval [CI] = 1.33–1.72 near RP11‐815 M8.1), rs6541998 (OR = 1.53, 95% CI = 1.37–1.74 near MERTK), rs10184573 (OR = 1.43, 95% CI = 1.26–1.61 near AC093590.1), and rs2499799 (OR = 0.62, 95% CI = 0.54–0.73 near UFL1/FHL5; Table 2). These lead SNPs had either a call rate or imputation metric close to 100%. Three of the 4 lead SNPs were present in the replication sample (rs11579212, rs6541998, and rs10184573), whereas for the SNP on chromosome 6 (rs2499799) we selected a proxy SNP (rs976357, r 2 = 1.0, D′ = 1). Lead SNPs of loci rs11579212 on chromosome 1, (OR = 1.58, 95% CI = 1.16–2.15) rs10184573 on chromosome 2 (OR = 1.74, 95% CI = 1.29–2.34) and rs976357 on chromosome 6 (OR = 0.44, 95% CI = 0.30–0.64) replicated after Bonferroni correction (Table 3).
FIGURE 1.
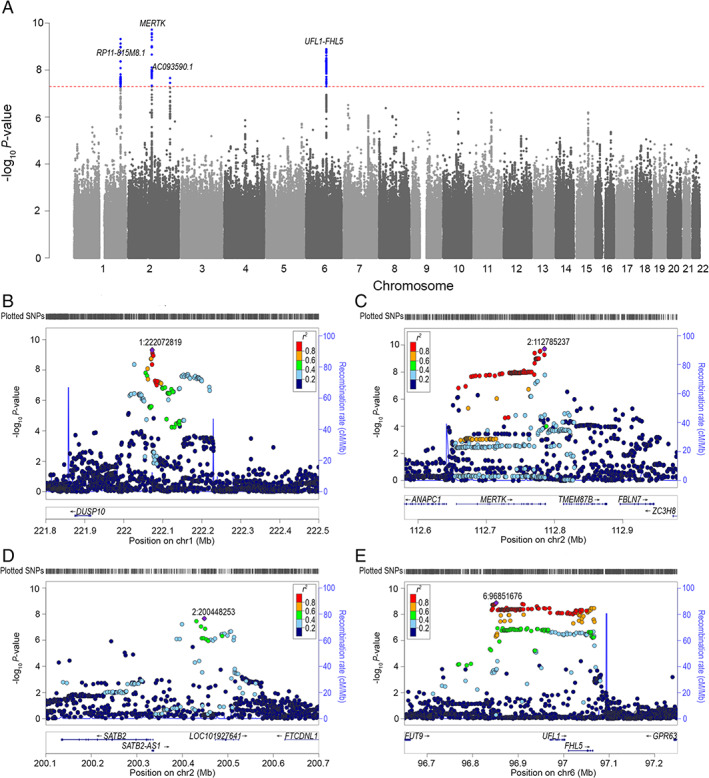
Manhattan plot and reginal plots for the discovery analysis. (A) Manhattan plot showing the ‐log10 p value for each SNP. Each marker was tested for association using an additive genetic model by logistic regression. The horizontal axis shows the chromosomal position and the vertical axis shows the significance of tested markers from logistic regression. The threshold for genome wide significance (p < 5 × 10−8) is indicated by a red dotted line. Markers that reach genomewide significance are shown in blue. (B–E) Regional Manhattan plots of the 4 genomewide significant cluster headache loci, with +/− 600 kb‐window. Each dot represents an SNP, the horizontal axis gives the genomic coordinate and the vertical axis the significance level (−log10 p value). The index SNP for each locus is marked with a purple diamond and annotated with its corresponding location number (CRCh37/hg19). SNPs are colored based on their correlation (r 2) with the labeled lead SNP according to the legend. The solid blue line shows the recombination rate from 1000 Genomes (EUR) data (right vertical axis). Gencode genes are shown. Figures were obtained from LocusZoom.24 (B) Locus: rs11579212 and 1:222072819. (C) Locus: rs6541998 and 2:112785237. (D) Locus: rs10184573 and 2:200448253. (E) Locus: rs2499799 and 6:96851676. SNP = single nucleotide polymorphisms. [Color figure can be viewed at www.annalsofneurology.org]
FIGURE 2.
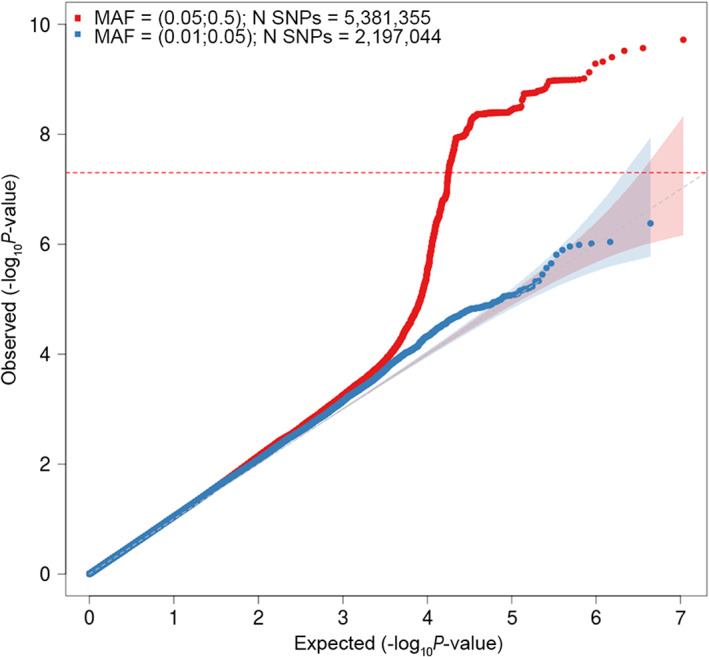
Quantile‐quantile (QQ) plot for association with cluster headache (CH). The horizontal axis shows ‐log10 p values expected under the null distribution. The vertical axis shows observed ‐log10 p values. Red = common SNPs (MAF ≥0.05), blue = low frequency SNPs (MAF = 0.005–0.05). Genomic inflation factor (λ) = 1.069. MAF = minor allele frequency; SNPs = single nucleotide polymorphisms. [Color figure can be viewed at www.annalsofneurology.org]
TABLE 2.
SNPs at the Four Loci Associated With Cluster Headache Discovery Sample
| SNP | Chr:Pos EA:NEAa | EAF | OR [95% CI] | p valueb | Nearest genec | eQTL mapped genesd |
|---|---|---|---|---|---|---|
| rs11579212 | 1:222072819 C:A | 0.34 | 1.51 [1.33–1.72] | 4.78 × 10−10 | RP11‐815 M8.1 | DUSP10 |
| rs6541998 | 2:112785237 C:T | 0.63 | 1.53 [1.37–1.74] | 1.91 × 10−10 | MERTK | TTL POLR1B FBLN7 ZC3H8 MERTK TMEM87B RGPD8 ZC3H6 |
| rs10184573 | 2:200448253 T:G | 0.44 | 1.43 [1.26–1.61] | 2.20 × 10−8 | AC093590.1 | SATB2 FTCDNL1 |
| rs2499799 | 6:96851676 C:T | 0.81 | 0.62 [0.54–0.73] | 1.29 × 10−9 | UFL1/FHL5 | UFL1 FHL5 GPR63 MMS22L FUT9 |
Chromosomal positions in GRCh37/hg19 coordinates.
Significant result (p < 5 × 10−8).
The nearest gene is based on ANNOVAR annotations with Ensembl build version 85.
The eQTL mapping was done in FUMA based on GTEx version 8.
Chr = chromosome; CI = confidence interval; EA = effect allele; EAF = effect allele frequency; NEA = non effect allele; OR = odds ratio; Pos = position; SNP = single nucleotide polymorphism.
TABLE 3.
Replication of the Significant Loci in an Independent Sample
| L | Chr | SNP discovery sample | SNP replication sample | Posa | OR [95% CI] | p valueb | Directionc |
|---|---|---|---|---|---|---|---|
| 1 | 1 | rs11579212 | rs11579212 | 222072819 | 1.58 [1.16–2.15] | 3.50 × 10−3 | + |
| 2 | 2 | rs6541998 | rs6541998 | 112785237 | 1.04 [0.78–1.40] | 0.78 | + |
| 3 | 2 | rs10184573 | rs10184573 | 200448253 | 1.74 [1.29–2.34] | 2.78 × 10−4 | + |
| 4 | 6 | rs2499799 | rs976357 (r 2 = 1.0) | 96849679 | 0.44 [0.30–0.64] | 2.76 × 10−5 | + |
Chromosomal positions in GRCh37/hg19 coordinates for the replication SNP.
Significant result (p < 0.05/4).
Direction; Same (+) or opposite (−) direction of association for discovery and replication analyses.
Chr = chromosome; CI = confidence interval; L = locus number; OR = odds ratio; Pos = chromosomal position; SNP = single nucleotide polymorphism.
The genomic inflation factor (λ) was 1.069 in the discovery analysis, whereas the LD score regression intercept was 1.044 (SE 0.0077), indicating moderate inflation due to factors other than polygenic architecture. We estimated the SNP‐based heritability (h 2) of CH at 30.3% (SE = 19.4%) on the observed scale. Assuming a population prevalence of 0.1% for CH this corresponds to a h 2 of 11.5% (SE = 7.4%) on the liability scale.
Fine mapping with PICS identified two variants with causal probability larger than 0.2, at rs11579212 (PICS probability = 0.40) and rs10184573 (PICS probability = 1.0), respectively.
Genetic Correlation of Cluster Headache with Migraine
The observed h 2 for migraine was 17.1% (SE = 1.56%). The genetic correlation between CH and migraine was 0.33 (SE = 0.021, p = 0.12). Next, we examined the 38 migraine‐associated loci reported in Gormley et al.35 Of the 37 migraine loci that were represented in our data set directly or by variants in high LD, one, located on chromosome 6, was associated with CH rs2971606, a proxy (r 2 = 1.0) for the migraine index variant rs67338227, in FHL5, p = 1.39 × 10−8 (Bonferroni corrected p corr = 0.5 × 10−6). The association had the same effect direction for migraine and CH. There was also moderate LD between the lead SNP for migraine and CH (r 2 = 0.64 in data from 1000 Genomes Project Phase 3 CEU). Still, colocalization analysis revealed a 67.5% posterior probability for the hypothesis (H3) that the causal variants for CH and migraine are distinct, higher than the 32.5% posterior probability for hypothesis (H4) that CH and migraine share a causal variant in this region.
The other 36 migraine loci were not associated with CH (data not shown), with the second strongest association seen for rs10786156 in PLCE1 (p = 2.82 × 10−3, p corr = 0.10). The migraine locus near MED14 on chromosome X (rs12845494) was not represented in our dataset. The effect sizes for the 37 loci combined correlated with those of CH, Pearson's r(35) = 0.59 (p = 1.36 × 10−4), even disregarding the overlapping FHL5 locus (Pearson's r(34) = 0.58, p = 2.18 × 10−4).
Sex‐Stratified Analyses
The low number of female cases gave limited power for the women‐only analysis. Rs6541998 was genomewide significant in men using sex‐stratified analyses; all other loci were nominally significant (p < 1 × 10−3) for both men and women with effects in the same direction. Using the method suggested by Clogg et al,38 we found no significant differences for the regression coefficients between men and women at the 4 lead SNPs (p value 0.54 for rs11579212; p value 0.62 for rs6541998; p value 0.57 for rs10184573; and p value 0.59 for rs2499799).
Previously Reported Cluster Headache Loci
Of the 9 different SNPs previously associated with CH, one replicated, rs1800759 in ADH4 (p = 0.00039, Bonferroni corrected p corr = 0.0035; Table 4). In contrast, none of the previously reported associations in HCRTR2, ADCYAP1R1, CLOCK, CHRNA3‐CHRNA5, and MME were replicated in our sample (see Table 4).
TABLE 4.
The Association of Previously Reported Cluster Headache Loci in Our Discovery Cluster Headache Sample
| Previously reported CH loci | Association with CH discovery sample | ||||||
|---|---|---|---|---|---|---|---|
| Nearest coding gene | Index SNP | EA | OR [95%CI] | p value | Ref | OR [95%CI] | pcorr‐valuea |
| ADCYAP1R1 | rs12668955 | G | 0.48 [0.34–0.07] | 9.1 × 10−6 | 13 | 1.05 [0.91–1.21] | 1 |
| ADH4 | rs1126671 | A | 2.33 [1.25–4.37] | 0.006b | 29 | 0.87 [0.76–0.99] | 0.36 |
| ADH4 | rs1126671 | A | ‐ | 0.03 | 30 | 0.87 [0.76–0.99] | 0.36 |
| ADH4 | rs1800759 | A | ‐ | 0.03 | 30 | 0.80 [0.70–0.90] | 0.0035 |
| CLOCK | rs12649507 | A | 1.29 [1.08–1.54] | 0.02 | 28 | 0.92 [0.80–1.05] | 1 |
| CHRNA3‐CHRNA5 | rs578776 | A | ‐ | 0.038 | 26 | 0.95 [0.83–1.09] | 1 |
| HCRTR2 | rs2653349 | G | 6.79 [2.25–22.99] | < 0.0002 | 8 | 1.08 [0.93–1.26] | 1 |
| HCRTR2 | rs2653349 | G | 1.97 [1.32–2.92] | 0.0007 | 10 | 1.08 [0.93–1.26] | 1 |
| HCRTR2 | rs3122156 | G | 0.82 [0.68–0.99] | 0.0421 (P corr 0.126) | 9 | 0.92 [0.80–1.06] | 1 |
| HCRTR2 | rs10498801 | G | 0.69 [0.49–0.97] | 0.030 | 27 | 1.03 [0.88–1.21] | 1 |
| MME | rs147564881 | C | ‐ | 0.019 | 13 | 0.24 [0.42–41.51] | 1 |
The SNPs previously reported to be associated with cluster headache and the corresponding OR (based on the same EA) and p values for these SNPs in the discovery sample.
The p values were Bonferroni corrected for 9 tests.
The p value is based on the carriers with homozygous AA genotype compared with GG/GA genotypes.
CH = cluster headache; CI = confidence interval; EA = effect allele; OR = odds ratio; SNP = single nucleotide polymorphism.
Downstream Bioinformatic Analysis
Using FUMA gene‐based eQTL mapping, 16 genes were mapped to the 4 loci (see Table 2). Additionally, gene‐mapping with MAGMA identified 5 genes whose expression was significantly influenced by the CH loci, TMEM87B, MERTK, FHL5, UFL1, and ASZ1 (Fig 3). Finally, we performed a MAGMA tissue expression analysis, which did not render any significant results (data not shown).
FIGURE 3.
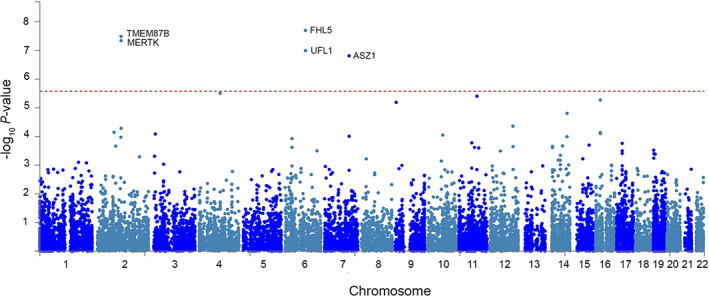
Gene‐based Manhattan plot. Input SNPs were mapped to 18,795 protein coding genes. The horizontal axis shows the chromosomal position and the vertical axis shows the significance of tested markers. The threshold for genome wide significance (p = 0.05/18,795 = 2.66 × 10−6) is indicated by a dotted line. SNP = single nucleotide polymorphism. [Color figure can be viewed at www.annalsofneurology.org]
RNA‐Seq Analyses
Using RNA‐seq data from white blood cells of 39 patients with CH and 20 controls, we assessed the 16 eQTL‐mapped genes derived from FUMA. Eleven genes were expressed in the samples, of which one was differentially lower expressed (POLR1B, p = 7.50 × 10−5, p corr = 8.3 × 10−4) and one was nominally differentially lower expressed (TMEM87B, p = 0.014, p corr = 0.15) in CH cases than in controls, both genes representing the rs6541998 locus (Table 5). The 2 genes were not differentially expressed in RNA‐seq data when comparing 26 patients with migraine with 20 controls (p = 0.50 and 0.45, respectively; see Table 5).
TABLE 5.
RNA‐seq Expression Data
| Gene | Expression in blood | Locus number | CH p value | CH p corr valuea | M p value |
|---|---|---|---|---|---|
| POLR1B | Yes | 2 | 7.50 × 10−5 | 8.3 × 10−4 | 0.50 |
| TMEM87B | Yes | 2 | 0.014 | 0.15 | 0.45 |
| ZC3H8 | Yes | 2 | 0.084 | 0.92 | ‐ |
| DUSP10 | Yes | 1 | 0.725 | 1 | ‐ |
| MERTK | Yes | 2 | 0.527 | 1 | ‐ |
| TTL | Yes | 2 | 0.465 | 1 | ‐ |
| MMS22L | Yes | 4 | 0.424 | 1 | ‐ |
| FBLN7 | Yes | 2 | 0.361 | 1 | ‐ |
| ZC3H6 | Yes | 2 | 0.285 | 1 | ‐ |
| FTCDNL1 | Yes | 3 | 0.123 | 1 | ‐ |
| UFL1 | Yes | 4 | 0.123 | 1 | ‐ |
| FHL5 | No | 4 | ‐ | ‐ | ‐ |
| FUT9 | No | 4 | ‐ | ‐ | ‐ |
| GPR63 | No | 4 | ‐ | ‐ | ‐ |
| RGPD8 | No | 2 | ‐ | ‐ | ‐ |
| SATB2 | No | 3 | ‐ | ‐ | ‐ |
Genes were selected based in the eQTL mapping in FUMA.22
The p values were Bonferroni corrected for 11 tests.
CH = cluster headache; eQTL = expression quantitative trait locus; FUMA = Functional Mapping and Annotation; M = migraine.
Discussion
We performed a GWAS in CH and identified 4 independent genetic risk loci, of which 3 replicated in an independent sample. The association effect sizes, with ORs around 1.5, are high compared to those usually observed in GWAS (https://www.ebi.ac.uk/gwas/).39 Whereas this may indicate that the risk for CH is driven by a limited number of loci with strong associations with CH, it is likely to be expected that follow‐up studies with larger sample sizes also will identify loci with smaller effect sizes. Except for the MERTK locus (rs6541998), all loci replicated in our replication sample, suggesting that the signals are genuine. Gene‐based mapping additionally found that expression of the ASZ1 gene may be influenced by one or more CH loci, providing a possible additional locus. RNA‐seq results show altered expression in patients with CH of POLR1B and TMEM87B, suggesting their involvement in CH. Although there seems to be a considerable SNP‐based heritability for CH, a robust estimation of SNP‐based heritability is not possible given the small sample size, hence heritability estimates should be interpreted with caution.
The main limitations of our study are that, (1) although we identified and replicated genomewide significant loci, the relatively small number of cases in the discovery sample will leave loci with smaller effect sizes or lower allele frequencies hidden; (2) it is unclear to what extent the present results can be extrapolated to ancestries other than European ancestry; and (3) although cases and controls were genotyped using the same platform, genotyping was performed in different laboratories possibly introducing batch effects. Therefore, we made significant efforts to circumvent possible problems arising from our design by rigorous quality control. Overall, our case sample was representative of the general CH population with an ~ 2:1 male–female ratio, chronic CH of ~ 30% and without any familial confounder, as familial cases were removed in the quality control steps.1 The difference in the percentage of men and women for cases and controls was corrected for in the statistical analysis.
Among previously suggested loci to be involved in CH, we found evidence for significant association to the alcohol dehydrogenase 4 gene (ADH4), although the effect identified is opposite to what was previously reported and at the genome wide level it was not significant.32 In previous studies, ADH4 was investigated mainly because alcohol is considered both a trigger and possible risk factor for transformation from episodic to chronic CH.31, 32 Of note, we did not find evidence for an association of HCRTR2, as reported previously,8, 9, 10 nor for any of the other previously reported loci in CH.
A remarkable finding in our study was that one of the leading loci, represented by rs2499799, which covers both FHL5 and UFL1, has previously been identified as a migraine risk locus.35 FHL5 encodes a transcription factor that regulates cAMP‐responsive elements CREB6 and CREM, which play a role in synaptic plasticity and memory formation.40 UFL1 codes for the ubiquitin‐fold modifier 1 (UFM1)‐specific ligase 1, an ubiquitin‐like protein that allows UFL1 to conjugate to its substrates.41 The ubiquitin protease system (UPS) has been associated as a pathway in neuropsychiatric and neurodegenerative disorders.42 In the latest migraine GWAS meta‐analysis, the UFL1/FHL5 locus had an OR of 1.09 (1.08–1.11) based on the primary signal (rs67338227).35 The direction of the effect in the UFL1/FHL5 locus in our dataset was the same in both migraine and CH and, the lead SNPs for migraine and CH were in LD (r 2 = 0.64). Our colocalization analysis suggested that CH and migraine are more likely caused by distinct variants at this locus. Admittedly, this finding could also be a result of different LD patterns in the samples that were compared in the colocalization analysis. The other 36 independent loci implicated in migraine showed no association with CH. Our results suggest though that the UFL1/FHL5 locus is “specific” for CH and that the association is not due to the mere presence of comorbid migraine among patients with CH. This is further supported by the similar prevalence of migraine among cases in our discovery sample (13%) and the expected population prevalence (10–17%), although the number of migraine cases in controls was not collected.43 While no other migraine locus reached significance in our study individually, there was a moderate correlation between association effect sizes of CH and migraine for the 37 examined migraine loci. This may reflect a shared genetic architecture underlying both disorders, which is not surprising given that they share pathophysiological features, such as the involvement of the trigeminovascular system and efficacy of calcitonin gene‐related peptide (CGRP) monoclonal antibodies and triptans.4 It is possible that future studies with larger sample sizes may identify the involvement of more migraine loci in CH.
With respect to the other replicated loci, rs11579212 and rs10184573, which mapped to RP11‐815 M8.1 and AC093590.1, respectively, they have not previously been related to disease, and their role in CH pathogenesis remains unclear. Although rs6541998 did not replicate in the small replication sample, 2 genes (POLR1B and TMEM87B) in the locus showed differential expression in CH compared to controls in RNA‐seq analyses, whereas no such effect was seen in migraine data. POLR1B, encoding DNA‐directed RNA polymerase I subunit RPA2, has been associated with Treacher Collins and TMEM87B, encoding transmembrane protein 87B may be involved in restrictive cardiomyopathy.44, 45 MERTK, the nearest gene, encodes a receptor tyrosine kinase of the TAM (Tyro3, Axl, and MERTK) family, is among other tissues expressed in oligodendrocytes, astrocytes, and microglia in the brain and has an effect on the immune response.46 Unfortunately, the number of associated genes with CH is not large enough to perform meaningful further downstream pathway analyses. Based on the regression coefficients, we found no evidence for a different effect for the lead SNPs between men and women.
In conclusion, this GWAS of CH reveals 4 genetic risk loci for CH with unusually high effect sizes for a complex disorder, of which 3 replicated in an independent sample. One of the loci has previously been identified as a migraine risk locus. Our results suggest several genes to be involved in the pathogenesis of CH and offer a starting point for future research to elucidate the molecular mechanisms of this severe disease.
POST‐SCRIPT PARAGRAPH
Two parallel manuscripts (Harder et al and O'Connor et al), submitted to the journal, report the first replicated genomic loci associated with CH. Whereas Harder et al investigated Dutch CH cases (n = 840) and controls (n = 1,457) and Norwegian CH cases (n = 144) and controls (n = 1,800), O'Connor et al investigated UK cases (n = 852) and controls (n = 5,614) as well as Swedish cases (n = 591) and controls n = 1,134). The 4 loci reported by Harder et al correspond to 4 loci reported by O'Connor et al, with the index variants reported in the 2 studies being in linkage disequilibrium with each other (D′ = 0.86 and r 2 = 0.36 for rs11579212 and rs12121134; D′ = 0.98 and r 2 = 0.95 for rs6541998 and rs4519530; D′ = 0.95 and r 2 = 0.34 for rs10184573 and rs113658130; and D′ = 0.93 and r 2 = 0.38 for rs2499799 and rs11153082, in the 1000 Genomes data for European populations). The independent discovery of the 4 loci in the 2 studies provides additional support that they represent genuine risk loci for cluster headache.
Next, we combined the summary statistics from the four studies (Dutch, Norwegian, United Kingdom, and Swedish) using inverse‐variance weighted meta‐analysis as implemented in METAL (with the “STDERR” option), after harmonizing the datasets using EasyQC.47, 48 In total, 8,039,373 variants were analyzed. The association to CH remained significant for all 8 index variants (in the 4 loci) reported in the 2 papers: rs11579212 (effect allele, EA: C), OR = 1.31 (95% CI = 1.21–1.41), p value 8.98 × 10−13; rs12121134 (EA: T), OR = 1.40 (95% CI = 1.29–1.53), p value 9.18 × 10−15; rs6541998 (EA: C), OR = 1.40 (95% CI = 1.30–1.51), p value 2.37 × 10−19; rs4519530 (EA: C), OR = 1.41 (95% CI = 1.31–1.52), p value 4.18 × 10−29; rs10184573 (EA: T), OR = 1.38 (95% CI = 1.28–1.50), p value 3.35 × 10−16; rs113658130 (EA: C), OR = 1.54 (95% CI = 1.41–1.69), p value 1.28 × 10−21; rs2499799 (EA: C), OR = 0.77 (95% CI = 0.70–0.84), p value 2.73 × 10−8; rs11153082 (EA: G), OR = 1.33 (95% CI = 1.23–1.43), p value 2.98 × 10−14. The 8 index variants in the overlapping loci showed a consistent effect direction across the 2 studies. Colocalization analysis, to determine whether the reported loci of both papers represent the same causal variants, identified a high posterior probability for 3 loci (those on chromosomes 1 and 2) to likely represent the same causal variant.34 Rs12121134 and rs11579212 have a posterior probability that the causal variants are the same (H4) of 80.4%, for rs4519530 and rs6541998 H4 is 87.4% and for rs113658130 and rs10184573 H4 is 96.9%. For the locus on chromosome 6, the colocalization analysis shows a higher probability that the loci in the 2 studies represent distinct causal variants (H3: 78.7%) rather than the same causal variant (H4: 21.2%).
Finally, the meta‐analysis resulted in three additional loci becoming genomewide significant: (1) a locus on chromosome 7 with 31 significant (p value <5 × 10−8) variants with index variant rs6966836 (chr7:117002998, EA: C), OR = 1.25 (95% CI = 1.16–1.35), p value 2.06 × 10−9; (2) a locus on chromosome 10 with 2 significant variants with index variant rs10786156 (chr10:96014622, EA: C), OR = 1.24 (95% CI = 1.15–1.33), p value 7.61 × 10−9; and (3) a locus on chromosome 19 with 2 significant variants with index variant rs60690598 (chr19:55052198, EA: T), OR = 1.87 (95% CI = 1.51–2.33), p value 1.70 × 10−8.
Author Contributions
A.V.E.H., B.S.W., R.N., E.T., T.F.H., R.H.J., M.D.F., J.‐A.Z., G.M.T., and A.M.J.M.v.d.M. contributed to the conception and design of the study. A.V.E.H., B.S.W., R.N., L.S.V., S.B., L.J.A.K., I.d.B., F.R.R., K.W.v.D., E.O.C., C.F., E.S.K., L.F.T., R.F., and P.P.‐R. contributed to the acquisition and analysis of data. A.V.E.H., B.S.W., and R.N. contributed to drafting the text and preparing the figures.
Members of “Cluster Headache Genetics Working Group”
All members of the “Cluster Headache Genetics Working Group” were actively involved in patient collection. The members of the Cluster Headache Genetics Working group include Roemer B. Brandt, MD, Ilse F. de Coo, MD, and Patty G.G. Doesborg, MD, all at the Leiden University Medical Center (LUMC), Leiden, The Netherlands; Roser Corominas, PhD, from the University of Barcelona, Barcelona, Spain; Institut de Biomedicina de la Universitat de Barcelona (IBUB), Barcelona, Spain; Instituto de Salud Carlos III, Spain; Institut de Recerca Sant Joan de Déu (IR‐SJD), Esplugues de Llobregat, Spain; Victor J. Gallardo, MSc, at the Headache Research Group, Vall d'Hebron Institute of Research (VHIR), Universitat Autònoma de Barcelona, Barcelona, Spain; Nunu Lund, MD, PhD, from the Danish Headache Center, Rigshospitalet Glostrup, Glostrup, Denmark; Paavo Häppölä, MSc, at University of Helsinki, Helsinki, Finland.
Potential Conflicts of Interest
The authors declared no conflict of interest.
Acknowledgments
The authors thank the patients for their participation in this project. The Nord‐Trøndelag Health Study (The HUNT Study) is a collaboration between HUNT Research Centre (Faculty of Medicine and Health Sciences, NTNU, Norwegian University of Science and Technology), Trøndelag County Council, Central Norway Regional Health Authority, and the Norwegian Institute of Public Health. The genotyping was financed by the National Institute of health (NIH), University of Michigan, The Norwegian Research council, and Central Norway Regional Health Authority and the Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU). The genotype quality control and imputation has been conducted by the K.G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU). We express our gratitude to all individuals who participated in the Netherlands Epidemiology in Obesity study. We are grateful to all participating general practitioners for inviting eligible participants. We also thank P. van Beelen and all research nurses for collecting the data and P. Noordijk and her team for sample handling and storage and I. de Jonge, MSc for data management of the NEO study. This work was supported by the South‐Eastern Norway Regional Health Authority (grant no. 2015089 to J.A.Z.), and grants of the Netherlands Organization for Scientific Research, that is the Center of Medical System Biology established by the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research (to A.M.J.M.v.d.M.), Spinoza 2009 (to M.D.F.) and EU‐funded FP7 “EUROHEADPAIN” (grant no. 6026337 to M.D.F., G.M.T, and A.M.J.M.v.d.M.). The genotyping of Dutch and Norwegian case samples was provided by the Genomics Core Facility (GCF), Norwegian University of Science and Technology (NTNU). The GCF is funded by the Faculty of Medicine and Health Sciences at NTNU and Central Norway Regional Health Authority. The NEO study is supported by the participating Departments, the Division and the Board of Directors of the Leiden University Medical Centre, and by the Leiden University, Research Profile Area “Vascular and Regenerative Medicine.” The funding organizations had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
References
- 1.Lund N, Barloese M, Petersen A, et al. Chronobiology differs between men and women with cluster headache, clinical phenotype does not. Neurology 2017;88:1069–1076. [DOI] [PubMed] [Google Scholar]
- 2.Lund N, Petersen A, Snoer A, et al. Cluster headache is associated with unhealthy lifestyle and lifestyle‐related comorbid diseases: results from the Danish cluster headache survey. Cephalalgia 2019;39:254–263. [DOI] [PubMed] [Google Scholar]
- 3.Brandt RB, Doesborg PGG, Haan J, et al. Pharmacotherapy for cluster headache. CNS Drugs 2020;34:171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vollesen AL, Benemei S, Cortese F, et al. Migraine and cluster headache ‐ the common link. J Headache Pain 2018;19:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Headache classification Committee of the International Headache Society (IHS) the international classification of headache disorders, 3rd edition. Cephalalgia 2018;38:1–211. [DOI] [PubMed] [Google Scholar]
- 6.May A, Schwedt TJ, Magis D, et al. Cluster headache. Nat Rev Dis Primers 2018;4:18006. [DOI] [PubMed] [Google Scholar]
- 7.Sjaastad O, Shen JM, Stovner LJ, et al. Cluster headache in identical twins. Headache 1993;33:214–217. [DOI] [PubMed] [Google Scholar]
- 8.Rainero I, Gallone S, Valfrè W, et al. A polymorphism of the hypocretin receptor 2 gene is associated with cluster headache. Neurology 2004;63:1286–1288. [DOI] [PubMed] [Google Scholar]
- 9.Fourier C, Ran C, Steinberg A, et al. Analysis of HCRTR2 gene variants and cluster headache in Sweden. Headache 2019;59:410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schürks M, Kurth T, Geissler I, et al. Cluster headache is associated with the G1246A polymorphism in the hypocretin receptor 2 gene. Neurology 2006;66:1917–1919. [DOI] [PubMed] [Google Scholar]
- 11.Weller CM, Wilbrink LA, Houwing‐Duistermaat JJ, et al. Cluster headache and the hypocretin receptor 2 reconsidered: a genetic association study and meta‐analysis. Cephalalgia 2015;35:741–747. [DOI] [PubMed] [Google Scholar]
- 12.Gibson KF, Santos AD, Lund N, et al. Genetics of cluster headache. Cephalalgia 2019;39:1298–1312. [DOI] [PubMed] [Google Scholar]
- 13.Bacchelli E, Cainazzo MM, Cameli C, et al. A genome‐wide analysis in cluster headache points to neprilysin and PACAP receptor gene variants. J Headache Pain 2016;17:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ran C, Fourier C, Michalska JM, et al. Screening of genetic variants in ADCYAP1R1, MME and 14q21 in a Swedish cluster headache cohort. J Headache Pain 2017;18:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelzer N, Haan J, Stam AH, et al. Clinical spectrum of hemiplegic migraine and chances of finding a pathogenic mutation. Neurology 2018;90:e575–e582. [DOI] [PubMed] [Google Scholar]
- 16.Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 2nd edition. Cephalalgia. 2004;24 Suppl 1:9–160. [DOI] [PubMed] [Google Scholar]
- 17.Wilbrink LA, Weller CM, Cheung C, et al. Stepwise web‐based questionnaires for diagnosing cluster headache: LUCA and QATCH. Cephalalgia 2013;33:924–931. [DOI] [PubMed] [Google Scholar]
- 18.de Mutsert R, den Heijer M, Rabelink TJ, et al. The Netherlands epidemiology of obesity (NEO) study: study design and data collection. Eur J Epidemiol 2013;28:513–523. [DOI] [PubMed] [Google Scholar]
- 19.Guo Y, He J, Zhao S, et al. Illumina human exome genotyping array clustering and quality control. Nat Protoc 2014;9:2643–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grove ML, Yu B, Cochran BJ, et al. Best practices and joint calling of the HumanExome BeadChip: the CHARGE consortium. PLoS One 2013;8:e68095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson CA, Pettersson FH, Clarke GM, et al. Data quality control in genetic case‐control association studies. Nat Protoc 2010;5:1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loh PR, Danecek P, Palamara PF, et al. Reference‐based phasing using the haplotype reference consortium panel. Nat Genet 2016;48:1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watanabe K, Taskesen E, van Bochoven A, et al. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome‐wide association scan results. Bioinformatics 2010;26:2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krokstad S, Langhammer A, Hveem K, et al. Cohort profile: the HUNT study, Norway. Int J Epidemiol 2013;42:968–977. [DOI] [PubMed] [Google Scholar]
- 27.Ferreira MA, Vonk JM, Baurecht H, et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat Genet 2017;49:1752–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cainazzo MM, Tiraferri I, Ciccarese M, et al. O015. Evaluation of the genetic polymorphism of the α3 (CHRNA3) and α5 (CHRNA5) nicotinic receptor subunits, in patients with cluster headache. J Headache Pain 2015;16:A88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan Z, Hou L, Wan D, et al. Genetic association of HCRTR2, ADH4 and CLOCK genes with cluster headache: a Chinese population‐based case‐control study. J Headache Pain 2018;19:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fourier C, Ran C, Zinnegger M, et al. A genetic CLOCK variant associated with cluster headache causing increased mRNA levels. Cephalalgia 2018;38:496–502. [DOI] [PubMed] [Google Scholar]
- 31.Rainero I, Rubino E, Gallone S, et al. Cluster headache is associated with the alcohol dehydrogenase 4 (ADH4) gene. Headache 2010;50:92–98. [DOI] [PubMed] [Google Scholar]
- 32.Zarrilli F, Tomaiuolo R, Ceglia C, et al. Molecular analysis of cluster headache. Clin J Pain 2015;31:52–57. [DOI] [PubMed] [Google Scholar]
- 33.Bulik‐Sullivan BK, Loh PR, Finucane HK, et al. LD score regression distinguishes confounding from polygenicity in genome‐wide association studies. Nat Genet 2015;47:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gormley P, Anttila V, Winsvold BS, et al. Meta‐analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 2016;48:856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eising E, Pelzer N, Vijfhuizen LS, et al. Identifying a gene expression signature of cluster headache in blood. Sci Rep 2017;7:40218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kogelman LJ, Falkenberg K, Halldorsson GH, et al. Comparing migraine with and without aura to healthy controls using RNA sequencing. Cephalalgia 2019;39:1435–1444. [DOI] [PubMed] [Google Scholar]
- 38.Clogg CC, Petkova E, Haritou A. Statistical methods for comparing regression coefficients between models. Am J Sociol 1995;100:1261–1293. [Google Scholar]
- 39.Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet 2017;101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anttila V, Winsvold BS, Gormley P, et al. Genome‐wide meta‐analysis identifies new susceptibility loci for migraine. Nat Genet 2013;45:912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komatsu M, Chiba T, Tatsumi K, et al. A novel protein‐conjugating system for Ufm1, a ubiquitin‐fold modifier. EMBO J 2004;23:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheon S, Dean M, Chahrour M. The ubiquitin proteasome pathway in neuropsychiatric disorders. Neurobiol Learn Mem 2019;165:106791. [DOI] [PubMed] [Google Scholar]
- 43.Lipton RB, Stewart WF. Prevalence and impact of migraine. Neurol Clin 1997;15:1–13. [DOI] [PubMed] [Google Scholar]
- 44.Sanchez E, Laplace‐Builhe B, Mau‐Them FT, et al. POLR1B and neural crest cell anomalies in Treacher Collins syndrome type 4. Genet Med 2020;22:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu HC, Coughlin CR, Geiger EA, et al. Discovery of a potentially deleterious variant in TMEM87B in a patient with a hemizygous 2q13 microdeletion suggests a recessive condition characterized by congenital heart disease and restrictive cardiomyopathy. Cold Spring Harb Mol Case Stud 2016;2:a000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tondo G, Perani D, Comi C. TAM receptor pathways at the crossroads of Neuroinflammation and neurodegeneration. Dis Markers 2019;2019:2387614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winkler TW, Day FR, Croteau‐Chonka DC, et al. Quality control and conduct of genome‐wide association meta‐analyses. Nat Protoc 2014;9:1192–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]