

Abstract
Background and Aims
The liver plays a central role in all metabolic processes in the body. However, precise characterization of liver metabolism is often obscured by its inherent complexity. Phosphorylated metabolites occupy a prominent position in all anabolic and catabolic pathways. Here, we develop a 31P nuclear magnetic resonance (NMR)–based method to study the liver “phosphorome” through the simultaneous identification and quantification of multiple hydrophilic and hydrophobic phosphorylated metabolites.
Approach and Results
We applied this technique to define the metabolic landscape in livers from a mouse model of the rare disease disorder congenital erythropoietic porphyria (CEP) as well as two well‐known murine models of nonalcoholic steatohepatitis: one genetic, methionine adenosyltransferase 1A knockout mice, and the other dietary, mice fed a high‐fat choline‐deficient diet. We report alterations in the concentrations of phosphorylated metabolites that are readouts of the balance between glycolysis, gluconeogenesis, the pentose phosphate pathway, the tricarboxylic acid cycle, and oxidative phosphorylation and of phospholipid metabolism and apoptosis. Moreover, these changes correlate with the main histological features: steatosis, apoptosis, iron deposits, and fibrosis. Strikingly, treatment with the repurposed drug ciclopirox improves the phosphoromic profile of CEP mice, an effect that was mirrored by the normalization of liver histology.
Conclusions
In conclusion, these findings indicate that NMR‐based phosphoromics may be used to unravel metabolic phenotypes of liver injury and to identify the mechanism of drug action.
Abbreviations
- 3PG
3‐phosphoglycerate
- 6PG
6‐phosphogluconate
- ADPG
adenosine 5′‐diphosphoglucose
- CDHF
choline‐deficient high‐fat
- CDP
cytidine 5′‐diphosphate
- CEP
congenital erythropoietic porphyria
- CH
choline
- CL
cardiolipin
- CPX
ciclopirox
- DPX
di‐n‐butyl phthalate in xylene
- FADH2
flavin adenine dinucleotide
- FBP
fructose 1,6‐bisphosphate
- G1P
glucose 1‐phosphate
- G6P
glucose 6‐phosphate
- GDPG
guanosine 5′‐diphosphoglucose
- LPC
lysophosphatidylcholine
- MAT1A‐KO
methionine adenosyltransferase 1A knockout
- NADPH
nicotinamide adenine dinucleotide phosphate reduced form
- NDP
nucleoside diphosphate
- NMR
nuclear magnetic resonance
- NTP
nucleoside triphosphate
- PARP
polyadenosine‐diphosphoribose polymerase
- PC
phosphatidylcholine
- PCr
phosphocreatine
- PE
phosphatidylethanolamine
- PEP
phosphoenolpyruvate
- PI
phosphatidylinositol
- PPP
pentose phosphate pathway
- PS
phosphatidylserine
- S7P
sedoheptulose 7‐phosphate
- SM
sphingomyelin
- TC
total cholesterol
- TCA
tricarboxylic acid
- TG
triglyceride
- UROIIIS
uroporphyrinogen III synthase
- WT
wild type
- X5P
xylulose 5‐phosphate
More than 18,000 enzymatic reactions, integrated in some 26,000 metabolic pathways with a continuous throughput of 100,000 metabolites, constitute the human metabolism.( 1 ) At first glance, the map of the metabolic pathways that configures the human metabolism (or that of any organism) shows an inextricable complexity (https://www.genome.jp/kegg/pathway/map/map01100.html).( 2 ) In order to acquire a holistic view of metabolism, it is essential to reduce such complexity at the minimum functional information expenses. In this context, it has been proposed that much of the logic of metabolism may be explained by focusing on a small group of transferring molecules that power metabolism (including ATP, NADPH, and acetyl‐CoA).( 3 ) Here we argue that metabolism can be quantitatively described by focusing on the phosphorylated fraction of the metabolism, the “phosphorome,” because phosphorylated metabolites occupy a prominent position in all catabolic (degradative) and anabolic (synthetic) pathways. In fact, the enzymes that regulate their synthesis and degradation act as “chemical relays” that limit the flow through virtually all metabolic pathways, such as glycolysis, gluconeogenesis, the tricarboxylic acid (TCA) cycle, oxidative phosphorylation, the pentose phosphate pathway (PPP), the urea cycle, and glycogenesis, or the synthesis of phospholipids, triglycerides (TG), and cholesterol.( 4 )
Because of the impossibility of monitoring metabolism directly from human liver samples, animal models are used instead, being of particular importance in the medical research field.( 5 ) Rodents are useful models because of their relatively fast reproductive period and resemblance to human organ distribution. Consequently, a plethora of mouse models (dietary and genetic) are currently available to investigate the pathophysiological basis of many diseases associated with the liver and/or the potential therapeutic effect of drugs on such pathologies. Albeit with certain limitations, many studies have validated the metabolic resemblance, and most of the extracted conclusions are applicable to human beings as well.( 6, 7 )
The detection and quantification of metabolites from the liver suffer from an instrumental limitation because of (i) the range of concentration in which the metabolites appear in the organism (from millimolar to picomolar concentration), (ii) the chemical nature of the compounds (polar, nonpolar, etc.), and (iii) the complexity of the mixture. Phosphorylated metabolites can be measured by various techniques including liquid chromatography mass spectrometry( 8 ) and nuclear magnetic resonance (NMR) spectroscopy.( 9 ) 1H‐NMR spectroscopy of liver extracts is very sensitive but difficult to interpret because it retains full complexity of the metabolism and the metabolites cannot be quantified because of widespread signal overlap and strong background signals from water (Supporting Fig. S4C,D). This makes it overly complicated to resolve relevant and quantifiable information from liver extracts by 1H‐NMR, even when selecting only the phosphorylated metabolite subset (via 31P filtering). 31P‐NMR offers many advantages: relatively high sensitivity and a wide signal dispersion that prevents signal overlap.( 10 ) Phosphorylated metabolites from liver extracts have been investigated by solution NMR spectroscopy( 11, 12 ) and in vivo using MRI.( 13, 14 ) However, such studies are normally not quantitative, only report a very reduced subset of metabolites, and/or suffer from limitations in the liver extraction protocol. So far, 31P‐NMR quantitative analysis has been limited to in vitro studies.( 15 )
Here, we present a robust method for the characterization and quantification of the phosphorome composition from mouse liver samples. To that end, we have optimized all experimental aspects in the procedure, including sample preparation and NMR determination to spectral processing. Our phosphoromic analysis reports the absolute concentration of up to 54 assigned metabolites under standardized operating procedures (SOPs). As a proof of concept, we have elucidated the liver phosphorome from a mouse model of the autosomal recessive disorder congenital erythropoietic porphyria (CEP). Patients with CEP suffer from a deficient biosynthesis of the heme prosthetic group, which often results in steatohepatitis that progresses to fibrosis and, ultimately, hepatic failure.( 16 ) Our phosphoromic analysis quantitatively defines the liver metabolic landscape of CEP. We identify metabolic alterations in the activities of glycolysis, glycogenesis, PPP, oxidative phosphorylation, phospholipid metabolism, and apoptosis. Strikingly, the phosphoromic profile of CEP mice liver largely improved after treatment with the repurposed drug ciclopirox (CPX),( 17 ) which correlates with the normalization of the liver histology. We also analyzed the phosphoromic patterns of two well‐known murine models of NASH: one genetic, methionine adenosyltransferase 1A knockout (MAT1A‐KO) mice,( 18 ) and the other dietary, mice fed a choline‐deficient high‐fat (CDHF) diet.( 19 ) These findings indicate that phosphoromics analysis is an excellent reporter of liver metabolism and may be used to unravel metabolic phenotypes of liver injury and to identify the mechanism of drug action.
Materials and Methods
Mouse Models
Transgenic CEP female mice (n = 14, of which 7 were treated with CPX) and wild‐type (WT) littermates (n = 6) were generated as reported.( 17 ) Ten‐month‐old male MAT1A‐KO mice (n = 9) and WT littermates (n = 6) were generated as shown elsewhere.( 20 ) CDHF male mice (n = 9; C57B1/6 background control mice, n = 6) were generated as indicated.( 21, 22 ) All experiments were approved by CIC bioGUNE’s Biosafety and Bioethics Committee and the Country Council of Bizkaia and all animals received human care according to the criteria outlined in the NIH Guide for the Care and Use of Laboratory Animals. At the end of the animal experiments, livers were collected (always in the morning, with all the animals belonging to the same experiment sacrificed at the same time), cryogenized in liquid nitrogen and stored at −80°C until further use. All experiments were repeated between six and nine times to ensure statistical significance.
Liver Histology
Every staining was done in mouse liver tissue. Liver tissues were fixed in neutral buffered 10% formalin solution (Sigma‐Aldrich, HT501128‐4L) embedded in paraffin and cut into 5‐μm sections with HistoCore MULTICUT microtome (Leica). Liver sections were deparaffinized with Histo‐Clear I solution (Electron Microscopy Sciences, 64110‐01) and hydrated through decreasing concentration of alcohol solutions. For the hematoxylin/eosin staining, samples were stained with Harris hematoxylin (Sigma‐Aldrich, Merck, HHS128‐4L), washed in running tap water for 5 minutes, and after 1 second in 0.5% hydrochloric acid, the sections were stained with aqueous eosin solution (Sigma‐Aldrich, Merck, HT1102128‐4L) for 15 minutes. Finally, the samples were washed in running tap water for 3 minutes, dehydrated in increasing concentrations of alcohol solutions until 100%, and mounted with di‐n‐butyl phthalate in xylene (DPX) mounting medium (Sigma‐Aldrich, Merck 06522). For the Prussian blue stain (Polysciences, 24199) tissue sections were stained 2 times with 4% hydrochloric acid and 4% potassium ferrocyanide, 10 minutes each. The slides were counterstained with 1% nuclear fast red solution and rinsed twice. Finally, the samples were dehydrated in ethanol, cleared in Histo‐Clear I solution and mounted with DPX. For the cleaved caspase‐3, sections were unmasked 5 minutes at 600 W in a microwave followed by 15 minutes at 800 W with EDTA 1 mM pH 8.0. Then, samples were blocked for unspecific binding with 3% hydrogen peroxide for 10 minutes followed by 30 minutes of incubation with 2.5% normal goat serum. Sections were then incubated 1 hour at 37°C with the primary antibodies (cleaved caspase‐3 ref: 9661S; cleaved polyadenosine‐diphosphoribose polymerase [PARP], 94885, Cell Signaling) followed by 30 minutes of incubation with ImmPRESS goat anti‐rabbit IgG (Vector Laboratories, MP‐7451). Samples were developed with Vector VIP purple substrate (SK‐4600) and sections were counterstained 1 minute with Mayer’s hematoxylin, dehydrated, cleared, and mounted with DPX. For the Masson’s trichromic staining, slides were deparaffinized and hydrated, preheated in Bouin’s solution at 56°C for 15 minutes, and cooled in water. After the stain with working Weigert’s iron hematoxylin solution (5 minutes), slides were washed and stained with Biebrich scarlet‐acid fucshin (5 minutes). Samples were stained with working phosphotungstic/phosphomolybdic acid solution and aniline blue solution (5 minutes each). Finally, slides were placed in acetic acid, 1%, for 2 minutes, rinsed, dehydrated with alcohol, cleared in Histo‐Clear I, and mounted with DPX. For the Oil Red O staining, frozen sections were cut at 4 µm, fixed in 10% Formalin solution and washed during 5 minutes. After the incubation with 60% isopropanol, tissues were stained with Oil Red O (Sigma‐Aldrich, O0625‐25G) for 15 minutes and washed with 60% isopropanol, lightly stained with Mayer’s hematoxylin, and mounted. All images were captured using a Leica DM750 optical microscopy with a Leica ICC50W digital color camera.
NAFLD Activity Score
Total NAFLD activity score (NAS), which represents the sum of scores for steatosis, lobular inflammation, and ballooning, ranging from 0 to 8, was calculated for WT, CEP, and CEP + CPX mice based on the analysis of the Masson trichrome staining and according to the criteria published by Kleiner et al.( 23 )
Extraction Method
Aliquots of liver (between 20 and 300 mg) were each distributed evenly into 3 different Eppendorf tubes. Each 100 mg of liver was lysed in 774 µL of ice‐cold chloroform/methanol mixture (38.8/61.2 vol/vol), and Sodium trimethylsilylpropanesulfonate was added to 7.72 mM concentration to monitor the extraction yield. Livers were homogenized in a Precellys tissue homogenizer at 4,000 g for 2 × 30 seconds and 554 µL of a chloroform/water mixture (45.8/54.2 vol/vol) were then added to the homogenate. After vortexing, the solution was left on ice for 15 minutes before being centrifugated for 15 minutes at 9,000 g, 4°C. The two obtained phases were then carefully separated and dried in a SpeedVac for 3 hours. Pellets were stored at −80°C for further use.
NMR Spectroscopy
The 20‐300 mg of dry liver extract was resuspended in 300 µL solution according to the extract fraction. For the hydrophilic E1 fraction, we used Tris‐d11‐DCl buffer in deuterium oxide (100 mM), pD 7, with added sodium azide (1 mM), tetramethyl phosphonium chloride (1 mM) as 31P reference compound, and gadobutrol (0.5 mM) as paramagnetic relaxation enhancer. For the lipophilic E2 fractions, we used DMSO‐d6 with added triphenyl phosphine oxide (4 mM) as 31P reference compound. The resuspended extracts were filled into 5 mm Shigemi NMR tubes.
All NMR experiments were recorded at 298 K on a Bruker 600 MHz (12 T) Avance III spectrometer equipped with a QXI (1H,13C,15N,31P) or a BBO (BB,1H) probehead. For each sample, three different experiments were collected: (i) a 1D 31P zgip spectrum with inverse gated 1H decoupling (3‐11.5 hours), (ii) a 1D 1H p3919gp with water signals suppression using a binomial 3‐9‐19 pulse with echo gradient pair (7 minutes), and (iii) a 2D 31P,1H COLOC spectrum with coherence selection by gradients (5‐13.5 hours). The 1D 31P experiment was recorded with long interscan delays d1 > 3∙T1,max(31P) for quantification, where the paramagnetic relaxation enhancer (gadobutrol) added only to the hydrophilic E1 fraction allowed to use a short d1 = 1 second, whereas the hydrophobic E2 fraction required d1 = 5 seconds. The same set of spectra were recorded for 80 pure phosphorylated standard compounds (listed in Supporting Table S2) to facilitate the identification of phosphorylated liver metabolites. Each of the 31P standards was prepared at increased 5 mM concentration in the appropriate solution corresponding to either E1 or E2 extract (see previous description) to allow faster NMR data acquisition.
The phosphorylated liver metabolites were identified by comparison with the 31P chemical shifts of the pure standard compounds compiled in Supporting Table S2. In some cases, spiking experiments with selected pure standard compounds were performed to confirm the metabolite assignment. Assigned phosphorylated metabolites were then quantified by referencing their 31P peak integral against the added internal reference compound. In case of signal overlap, we applied peak deconvolution to assign corresponding peak areas.
Results
Phosphoromic Analysis of Mice Liver Extracts
Our approach to study phosphorylated metabolites in the liver is based on the direct observation and quantification of the phosphorous signals (31P‐NMR). Metabolite extraction from liver tissues and the NMR analysis is described in detail in the Supporting Material (Protocol and Supporting Figs. S1‐S4). After the analysis of the 31P‐NMR spectra of liver extracts at different concentrations, we determined that the detection limit is around 30 pmol of 31P‐compound per milligram of liver, with a signal‐to‐noise ratio of 4 (Supporting Fig. S5). Thus, a minimum of 20 mg and an optimal value of 100 mg of liver tissue is required for a reliable metabolite quantification by this method. However, we reckon that an optimized NMR hardware set (i.e., a dedicated heteronuclear‐detection cryoprobe) would result in 2‐5–fold increase in sensitivity, equivalent to a demand of less than 10 mg of liver tissue. With average mouse liver weights of about 1 g, the sample amount obtained from one animal can thus be partitioned to enable different phosphoromic analyses of specific liver fractions (e.g., from a tumor resection) or complementary histological, transcriptomic, and proteomic analyses.
The processing of mouse livers includes the metabolite extraction from the tissue and the subsequent fractionation into the hydrophilic (E1) and the hydrophobic (E2) extracts (Supporting Fig. S1). The reported ternary mixtures of chloroform (CHCl3):methanol (MeOH): water (H2O)( 24, 25 ) cannot be readily used here because liver homogenization releases water molecules, which often compromise phase separation. To improve it, we analyzed the ternary phase diagram (Supporting Fig. S2) to identify an optimal stoichiometry at 55:25:20 (w/w/w of CHCl3:MeOH:H2O). This extraction protocol ensures excellent reproducibility and good yields (69% ± 5%) as determined with an added reference compound (DSS). Remarkably, the protocol also ensures complete separation among phases with no metabolites found in the two fractions.
For quantitative analyses, the 31P‐NMR signal intensity has to be proportional to its concentration for each metabolite. To that end, the E1 extract was doped with a commercially available contrast agent that induces fast relaxation of the signal without increasing line broadening.( 26 ) This strategy could not be applied to the E2 extract because of limited solubility, and longer recovery times were used instead. Quantification was then achieved by adding specific internal references to each of the fractions. Independent analysis of replicas (between six and nine repetitions) was used to ensure reproducibility and statistical significance.
A representative 31P‐spectrum of mouse liver E1 and E2 extracts is shown in Fig. 1A, illustrating the excellent peak dispersion obtained. Resolved peaks were assigned by comparison with an in‐house database of 31P chemical shifts obtained for 80 metabolites (Supporting Table S1) collected at the same experimental conditions. Peak assignments and quantification (Supporting Table S3) were complemented and corroborated by heteronuclear 2D 1H, 31P‐COLOC correlation spectra of the 13P(‐O‐C‐)1Hn moieties present in most phosphorylated metabolites. In many cases, we spiked the sample with suspected phosphorylated compounds to confirm their assignment.
FIG. 1.
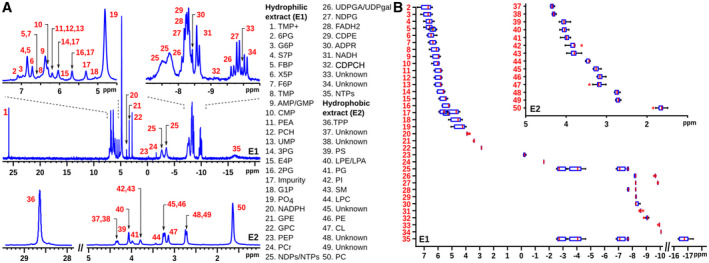
31P‐NMR spectroscopy of mouse liver extracts. E1 (E2) stands for the hydrophilic (lipophilic) extract. (A) A total of 50 different peaks are identified within the two extracts. For the abbreviation meanings, see Table 1. (B) Chemical shift variability of the different peaks was determined from a cohort of more than 50 spectra from liver samples. Abbreviation: NDPG, nucleoside diphosphate glucose.
31P‐NMR signal frequencies (i.e., chemical shifts) are very sensitive to temperature, pH, solvent, and metabolite concentrations. The chemical shift variance can be estimated for each metabolite from a comparison of several (>50) mouse liver spectra and is shown in Fig. 1B (and Supporting Table S4). Although some metabolites show significant chemical shift variance, these are not large enough to compromise the molecular assignment.
Although most metabolites are monophosphorylated and generate only one 31P‐NMR singlet signal, pyrophosphorylated metabolites generally show a corresponding number of distinct 31P signals with doublet (terminal phosphate groups) or triplet (central phosphate groups) fine structure. Nucleoside diphosphates (NDPs) and nucleoside triphosphates (NTPs), however, cannot be discriminated by their nucleobase and show broad 31P signals without fine structure but with partial overlap. Thus, the two signals numbered 25 in Fig. 1 correspond to the terminal α‐phosphates and β‐phosphates in NDPs and α‐phosphates and γ‐phosphates in NTPs, whereas signal 35 corresponds to the well separated central β‐phosphate in NTPs. Similar minor ambiguities hold for peaks 26 and 27 in Fig. 1. Interestingly, an ischemic tissue sample shows partial or complete absence of the NTP γ‐phosphate 31P NMR signal.
In case of the phospholipidic E2 extract, 31P signals derive from the polar head groups of phospholipids and are rather insensitive to the chemical composition of the distantly attached fatty acid side chains. As shown in Fig. 1A, the phosphatidylcholine signal (PC; peak 50 at 1.67 ppm) dominates the spectrum. In certain circumstances (such as in CEP mouse livers; vide infra), the E2 extract’s 31P spectrum also contains signals that correspond to lyso‐PC (LPC; peak 44 at 3.46 ppm).
Even though the present study mainly addressed liver tissue from mouse models, we have also validated the methodology with the analysis of human samples. Supporting Fig. S6 shows the 31P‐NMR spectrum of a biopsy from a patient with hepatocellular carcinoma, where all the metabolites under consideration have been identified and assigned.
Metabolic Significance of the Phosphorome
The 31P‐NMR platform contains 54 phosphorylated metabolites covering central metabolism (glycolysis, PPP, and the TCA cycle), glycogenesis, phospholipid, nucleotide, and nicotinamide metabolism, as well as 4 nonphosphorylated compounds (lactate, choline [CH], TG, and total cholesterol [TC]) (Fig. 2; Table 1). The metabolites that were identified and quantified in the liver are marked by red dots. The remaining phosphorylated metabolites were tested but could not be detected, presumably because their steady‐state concentration is below the detection limit (i.e., approximately 20 μM in the NMR tube with our NMR accessory). Overall, the phosphorome provides critical and comprehensive information on the entire liver metabolism: the hydrophilic E1 extract largely reports on the central carbon metabolism, whereas the hydrophobic E2 fraction yields information on the phospholipid composition. Thus, our 31P‐NMR–based approach provides quantitative information on the content of glucose 6‐phosphate (G6P), the first step in glucose catabolism, and its main catabolic products, uridine 5′‐diphosphoglucose (UDPG), 6‐phosphogluconate (6PG), and fructose 6‐phosphate, which are readouts of glycogenesis, the oxidative phase of the PPP, and glycolysis, respectively. The concentration of NTPs and the concentration of NADH, NADPH, and flavin adenine dinucleotide (FADH2) indicate the level of phosphoryl and electron‐transfer power that drives much of the anabolic pathways in the liver. The content of phosphocreatine (PCr), although not as important to maintain liver energy homeostasis as in the muscle, is also determined. Of greater importance to the liver is the quantification of 3‐phosphoglycerate (3PG), 2‐phosphoglycerate, phosphoenolpyruvate (PEP), and lactic acid, which yield information about glycolysis in relation to mitochondrial function (Warburg effect), and of xylulose 5‐phosphate (X5P), sedoheptulose 7‐phosphate (S7P), and erythrose 4‐phosphate, which indicate the activity through the reversible nonoxidative phase of the PPP that produces sugars for DNA and RNA synthesis. The information about the content of nucleosides triphosphates, diphosphates, and monophosphates (NTPs, NDPs, and NMPs) obtained by this phosphoromic platform is also critical, as the liver is the major site of nucleotide synthesis that is used for the replenishment and maintenance of the body pools. The content of adenosine 5′‐diphosphoribose (ADPR), a product of the activity of PARPs, also known as ADP‐ribosyltransferases, which is involved in multiple aspects of metabolic and cellular function, such as DNA damage,( 18 ) was also measured. In addition to ADPR and UDPG, other nucleotide sugars such as UDP‐galactose, UDP‐glucuronic acid, ADP‐glucose (ADPG), and guanosine 5′‐diphosphoglucose (GDPG), which make polysaccharides containing glucose, galactose, and glucuronic acid, were also quantified.
FIG. 2.
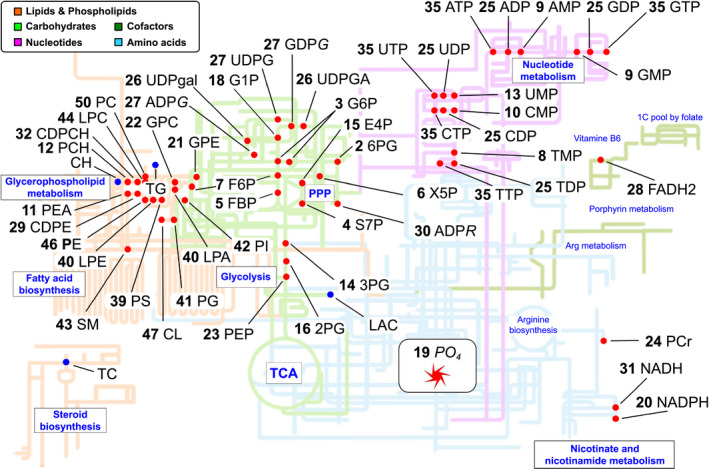
Map of the phosphorylated metabolism. Metabolites measured in liver extracts are represented as red circles when measured. Phosphorome gives information on central metabolism (glycolysis, PPP, and the TCA cycle), glycogenesis, phospholipid, nucleotide, nicotinamide, and energy metabolism. Arg, arginine.
TABLE 1.
Phosphorylated Metabolites Under Consideration
| Peak | Abbreviation | Compound | Extract |
|---|---|---|---|
| 16 | 2PG | 2‐Phosphoglycerate | E1 |
| 14 | 3PG | 3‐Phosphoglycerate | E1 |
| 2 | 6PG | 6‐Phosphogluconate | E1 |
| 25 | ADP | Adenosine 5′‐diphosphate | E1 |
| 27 | ADPG | Adenosine 5′‐diphosphoglucose | E1 |
| 30 | ADPR | Adenosine 5′‐diphosphoribose | E1 |
| 9 | AMP | Adenosine 5′‐monophosphate | E1 |
| 35 | ATP | Adenosine 5′‐triphosphate | E1 |
| 25 | CDP | Cytidine 5′‐diphosphate | E1 |
| 32 | CDPCH | Cytidine 5′‐diphosphocholine | E1 |
| 29 | CDPE | Cytidine 5′‐diphosphoethanolamine | E1 |
| CH | Choline | E1 (1H) | |
| 47 | CL | Cardiolipin | E2 |
| 10 | CMP | Cytidine 5′‐monophosphate | E1 |
| 35 | CTP | Cytidine 5′‐triphosphate | E1 |
| 15 | E4P | Erythrose 4‐phosphate | E1 |
| 7 | F6P | Fructose 6‐phosphate | E1 |
| 28 | FADH2 | Flavin adenine dinucleotide | E1 |
| 5 | FBP | Fructose 1,6‐bisphosphate | E1 |
| 18 | G1P | Glucose 1‐phosphate | E1 |
| 3 | G6P | Glucose 6‐phosphate | E1 |
| 25 | GDP | Guanosine 5′‐diphosphate | E1 |
| 27 | GDPG | Guanosine 5′‐diphosphoglucose | E1 |
| 9 | GMP | Guanosine 5′‐monophosphate | E1 |
| 22 | GPC | Glycerophosphocholine | E1 |
| 21 | GPE | Glycerophosphoethanolamine | E1 |
| 35 | GTP | Guanosine 5′‐triphosphate | E1 |
| LAC | Lactic acid | E1 (1H) | |
| 40 | LPA | Lysophosphatidic acid | E2 |
| 40 | LPE | Lysophosphatidylethanolamine | E2 |
| 44 | LPC | Lysophosphatidylcholine | E2 |
| 31 | NADH | β‐Nicotinamide adenine dinucleotide | E1 |
| 20 | NADPH | Nicotinamide adenine dinucleotide phosphate reduced form | E1 |
| 25 | NDP | Nucleoside diphosphate | E1 |
| 35 | NTP | Nucleoside triphosphate | E1 |
| 50 | PC | Phosphatidylcholine | E2 |
| 12 | PCH | Phosphocholine | E1 |
| 24 | PCr | Phosphocreatine | E1 |
| 46 | PE | Phosphatidylethanolamine | E2 |
| 11 | PEA | Phosphoethanolamine | E1 |
| 23 | PEP | Phosphoenolpyruvate | E1 |
| 41 | PG | Phosphatidylglycerol | E2 |
| 42 | PI | Phosphatidylinositol | E2 |
| 19 | PO4 | Inorganic phosphate | E1 |
| 39 | PS | Phosphatidylserine | E2 |
| 4 | S7P | Sedoheptulose 7‐phosphate | E1 |
| 43 | SM | Sphingomyelin | E2 |
| TC | Total cholesterol | E2 (1H) | |
| 25 | TDP | Thymidine 5′‐diphosphate | E1 |
| TG | Triglyceride | E2 (1H) | |
| 8 | TMP | Thymidine 5′‐monophosphate | E1 |
| 1 | TMP+ | Tetramethyl phosphonium chloride (REF) | E1 |
| 36 | TPP | Triphenyl phosphine oxide (REF) | E2 |
| 35 | TTP | Thymidine 5′‐triphosphate | E1 |
| 25 | UDP | Uridine 5′‐diphosphate | E1 |
| 27 | UDPG | Uridine 5′‐diphosphoglucose | E1 |
| 26 | UDPGA | Uridine 5′‐diphospho glucuronic acid | E1 |
| 26 | UDPgal | Uridine 5′‐diphosphogalactose | E1 |
| 13 | UMP | Uridine 5′‐monophosphate | E1 |
| 35 | UTP | Uridine 5′‐triphosphate | E1 |
| 6 | X5P | Xylulose 5‐phosphate | E1 |
E1 and E2 account for hydrophilic and hydrophobic extracts, respectively. CH, LAC, TC, and TG are obtained from 1D 1H‐NMR spectrum. Abbreviation: REF, added reference compounds.
PC, the most abundant cellular phospholipid, accounts for about 95% of the total CH cellular content. The remaining 5% includes CH, phosphocholine, cytidine 5′‐diphosphate (CDP), the rate‐limiting substrate in PC synthesis, glycerophosphocholine, the end‐product of PC catabolism, sphingomyelin (SM), and LPC. The 31P‐NMR‐based platform offers quantitative information of all these metabolites, except CH, which was measured by 1H‐NMR. Phosphoethanolamine, CDP‐ethanolamine, phosphatidylethanolamine (PE), and glycerophosphoethanolamine were also quantified by the 31P‐NMR–based platform. Other phospholipids determined by the platform include phosphatidylserine (PS), SM, phosphatidylinositol (PI), cardiolipin (CL), and phosphatidylglycerol (PG). TG and TC content was quantified by 1H‐NMR.
Liver Damage Caused by CEP
In this proof‐of‐concept study, we explored the potential value of the proposed phosphoromic approach by investigating a metabolic disease and the therapeutic effect of CPX, a drug for CEP treatment. CEP is a rare autosomal recessive disorder produced by a deficiency in the uroporphyrinogen III synthase (UROIIIS), the fourth enzyme in the heme biochemical pathway( 16, 19, 27 ) (Fig. 3). CEP results in a deficit in heme and the overproduction of uroporphyrin by‐products, especially type I uroporphyrin and coproporphyrin, which accumulate to toxic levels in different organs (Fig. 3). The disease is very severe, and it provokes hemolysis, severe anemia, splenomegaly, and phototoxic cutaneous lesions (blisters). As commonly observed in many porphyrias, the disease also generates hepatic damage.( 28 ) Histological analysis of livers from CEP mice (Fig. 4) shows steatosis with fibrosis, porphyrin deposits, and an accumulation of erythroid cells clustering around sinusoids.( 29, 30 ) This was further confirmed by the NAS (Supporting Table S5), showing values of moderate steatosis and fibrosis.
FIG. 3.
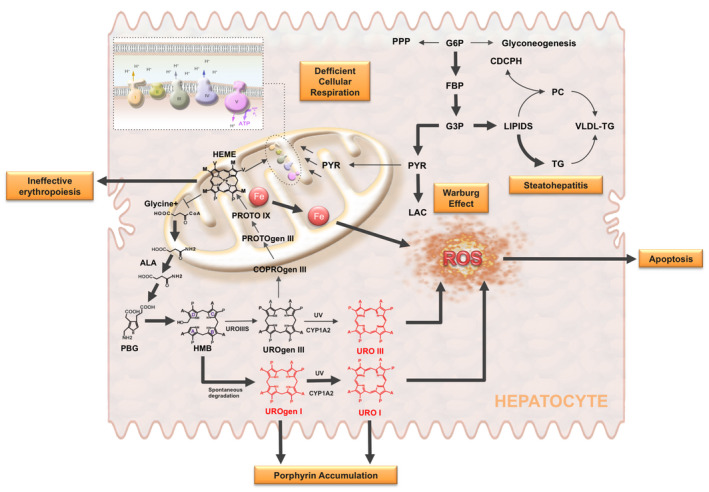
Porphyrin accumulation and oxidative stress in CEP. CEP is an autosomal recessive disorder of heme synthesis characterized by reduced activity of UROIIIS and the accumulation of nonphysiologic porphyrin metabolites (UROgen I and URO I). This results in ineffective erythropoiesis, iron deposition, mitochondrial dysfunction, and enhanced generation of reactive oxygen species (ROS), leading to the release of molecular danger signals triggering apoptosis in neighbor cells, probably erythroid cells. The diagram also shows the activation of glycolysis caused by mitochondrial dysfunction (Warburg effect) and how the excess of carbon units may be used for the various branching pathways that originate from glycolysis, like the de novo lipogenesis. This, together with the decrease of phosphatidylcholine (PC), which is required for VLDL synthesis and export, and the increased generation of ROS can explain the development of steatohepatitis in CEP mice. Abbreviations: ALA, aminolevulinate; COPROgen, coproporphyrinogen; CYP1A2, cytochrome P450 1A2; G3P, glyceraldehyde 3‐phosphate; HEME, heme group; HMB, hydroxymethylbilane; PGB, porphobilinogen; PROTO, protoporphyrin; PROTOgen, protoporphyrinogen; PYR, pyruvate; URO, uroporphyrin; UROgen, uroporphyrinogen; UV, ultraviolet radiation.
FIG. 4.
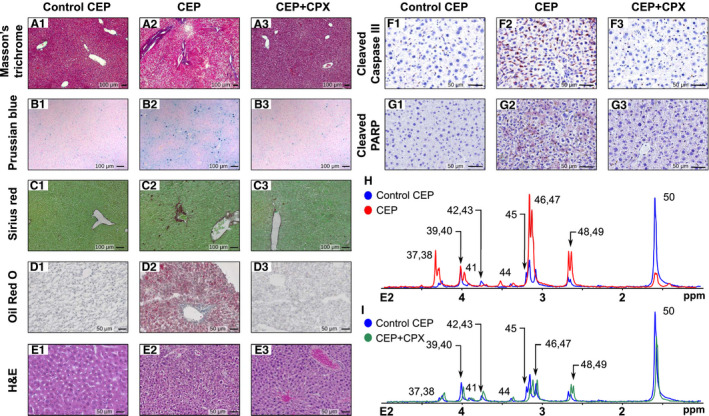
Impact of CEP condition in the phosphorome of mice liver and the recovery effect by CPX. (A‐G) Histology performed by (A) hematoxylin/eosin, (B) Prussian blue, (C) sirius red, (D) Oil Red O, (E) Masson's trichrome, (F) caspase‐3, or (G) PARP staining of control CEP (#.1) and CEP mice (#.2), with the latter also treated with CPX (#.3). CEP murine model (n = 7) shows steatosis with fibrosis, porphyrin deposits, and accumulation of erythroid cells clustering around sinusoids as well as apoptosis as compared with WT mice (n = 6). All these pathogenic features largely decrease after the administration of CPX (n = 7). (H,I) 31P spectra of lipophilic phase comparing WT (blue) peaks versuss CEP liver (red) and CEP liver treated with CPX (green). H&E, hematoxylin and eosin.
Liver samples from CEP mice were analyzed using the aforementioned phosphorome approach and compared with liver samples from WT mice to gain insight on the molecular basis of the CEP condition (Figs. 4, 5). Inspection of the E1 liver extract 31P‐NMR spectrum shows a reduction in G6P along with its downstream metabolites glucose 1‐phosphate (G1P) and UDPG/ADPG/GDPG (glycogenesis) and 6PG (PPP; Fig. 5A‐D). In line with this reduction in 6PG, the concentration of NADPH, the product of the oxidative phase of the PPP, is markedly reduced; so is the content of X5P and S7P, two representative metabolites of the nonoxidative phase of the PPP. Conversely, the content of fructose 1,6‐bisphosphate (FBP), the first metabolite connecting glucose metabolism into glycolysis, is increased (Fig. 5A.2). This suggests that glycogenesis and the PPP are slowed down in CEP mice to favor the flow of glucose through the glycolytic pathway. The finding that the liver content of 3PG and lactate, the glycolysis end‐product, are increased in CEP mice as compared with WT animals supports this hypothesis (Fig. 5A). This effect may be explained by the heme deficit, which is consubstantial to porphyria and results in mitochondrial respiration damage. This is consistent with the reduction in the respiratory cytochromes reported in cells isolated from peripheral blood of patients of different porphyries.( 31 ) This shift to anaerobic glycolysis (Warburg effect) in CEP liver associates with a decrease in NTPs, NADH, and FADH2 (Fig. 5E), wherein the latter is a redox factor produced during the TCA cycle and utilized in the last part of the electron transport chain, a raise in iron deposition, which produces liver injury by inflicting oxidative stress on hepatocytes (Fig. 4B) and an increase in ADP‐ribose (Fig. 5C), which is a molecular biomarker associated with cell injury and apoptosis.( 32, 33 )
FIG. 5.
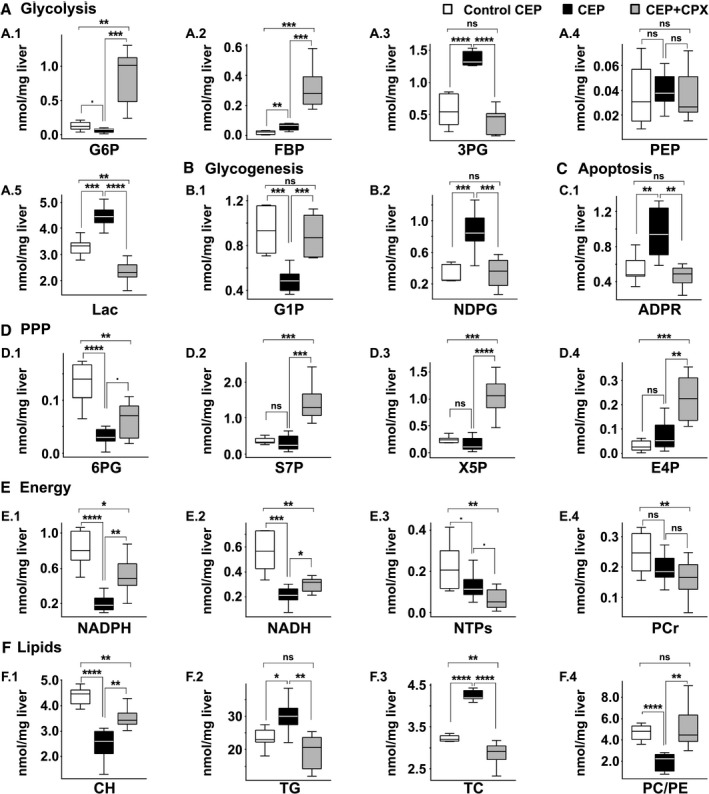
Metabolite quantification for the CEP model. Comparison of quantified metabolites (nmol/mg liver) between control CEP (white bars; n = 6), CEP (black bars; n = 7) and CEP treated with CPX (gray bars, n = 7). (A) Glycolysis, (B) Glycogenesis, (C) Apoptosis, (D) Pentose phosphate pathway, (E) Energy, (F) Lipids. P values of < 0.1, 0.05, 0.01, 0.001 and 0.0001 are represented by ·, *, **, *** and ****, respectively.
The analysis of the phospholipids in the E2 extract shows a pervasive accumulation of lipids in CEP liver as compared with WT mice, other than PC, which is reduced (Fig. 4H). The reduction of PC associates with a 50% reduction in the content of CDPCH, the rate‐limiting substrate in PC biosynthesis, and a greater content of LPC, a phospholipid mediator of hepatic lipotoxicity,( 34 ) TG, and TC (Fig. 5F), which are all common features of human NASH.( 35 ) Similarly, an accumulation of PG, PE, and CL has also been associated with mitochondrial malfunction.( 36 ) PS, PI, and SM levels remain unchanged, whereas the PC/PE and PC/(PE + SM) molar ratios are significantly reduced in CEP mice (Fig. 5F), which are indicators of impaired membrane integrity, liver failure, and steatohepatitis.( 37, 38 ) Of particular interest is the marked reduction in liver CH levels observed in CEP mice (Fig. 5F), which associates to the decrease in CDPCH and PC content. This condition has been shown to lead to impaired VLDL biosynthesis and export with a concomitant increase in intrahepatic TG.( 20, 39, 40 ) In agreement with these findings, liver histology of CEP mice also shows steatosis, inflammation, and fibrosis (Fig. 4A‐F; Supporting Fig. S7).
Altogether, we can conclude that mitochondrial dysfunction is the origin of an anaerobic glycolysis and lactate accumulation (Warburg effect) in CEP mice. In this scenario (Fig. 3), the excess of carbon units may be used for the various branching pathways that originate from glycolysis, like the de novo lipogenesis.( 41 ) This, together with the decrease of PC, which is required for VLDL synthesis and export, and the increase in iron deposition, which inflicts the generation of reactive oxidative substances, can explain the development of steatohepatitis in CEP mice (Fig. 3).
Because our phosphorome analysis disclosed a CEP‐induced steatohepatitis (Fig. 4D), we thought it would be instructive to compare its 31P‐fingerprint with that of two well‐known murine models of NASH: one genetic, MAT1A‐KO mice( 20 ) (Supporting Fig. S8), and the other dietary, mice fed a CDHF diet( 21, 22 ) (Supporting Fig. S9). The E1 liver extract from MAT1A‐KO mice liver showed an increase in hexose‐phosphates and triose‐phosphates (G6P, FBP, G1P, and 3PG) and a reduction in PEP, whereas lactic acid remained unchanged. ADP‐ribose was also elevated in MAT1A‐KO livers, whereas the content of 6PG, X5P, NADPH, NADH, and NTPs was significantly decreased (Fig. 6). The phosphoromic signature of mice fed a CDHF diet differed significatively from those in MAT1A‐KO (Fig. 6). Thus, although G6P, 3PG, and ADP‐ribose were also elevated in CDHF mice livers, FBP content was reduced, lactic acid increased, and the concentration of PEP, G1P, and 6PG remained unchanged (Fig. 6). The content of the ribose‐phosphates S7P and X5P were markedly reduced and the content of NTPs, NADH, and NADPH significantly increased in CDHF mice livers (Fig. 6). In turn, the phospholipid E2 extract highlights the general lipid disorder reported in MAT1A‐KO( 39 ) and CDHF mice,( 21 ) showing an accumulation of TG and a reduction in the PC/PE molar ratio (Fig. 6), a lipid pattern similar to that observed in CEP animals. As in the CEP model, the hepatic PC/PE and PC/(PE + SM) molar ratios are significantly reduced in MAT1A‐KO and drastically in CDHF mice (Fig. 6). Finally, the increase in hepatic CH observed in mice fed with a CDHF diet (Fig. 6F) represents less than 5% of the total hepatic CH. A small reduction in its transformation to CDPCH or betaine or an increase in PC hydrolysis (which accounts for about 95% of the total cellular CH) could explain this increase as an adaptative mechanism to compensate for the deficiency in CH.( 42 )
FIG. 6.
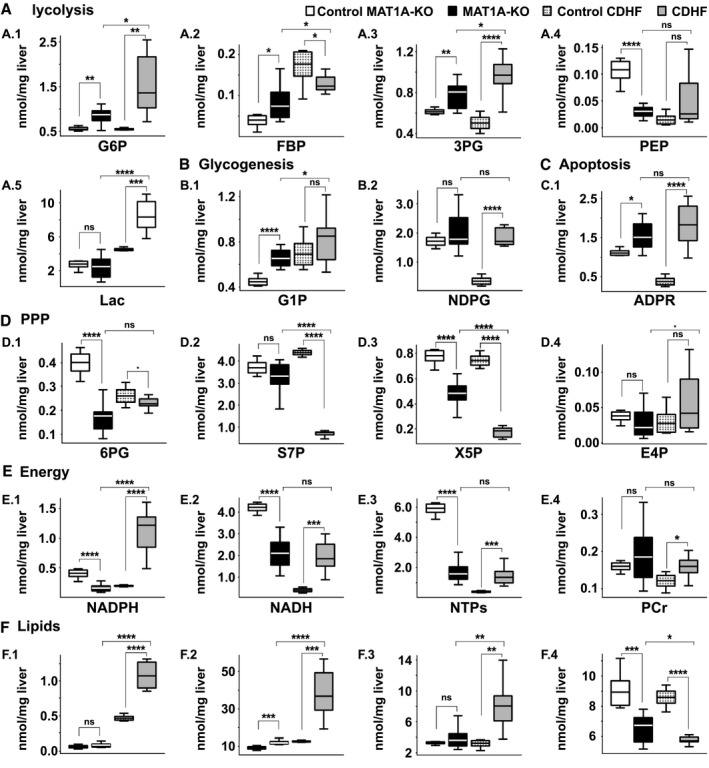
Metabolite quantification for validated NASH models. Comparison of quantified metabolites (nmol/mg liver) between control MAT1A‐KO (white bars; n = 6), MAT1A‐KO (black bars; n = 9), control CDHF (dotted gray bars; n = 6), and CDHF (gray bars; n = 9). (A) Glycolysis, (B) Glycogenesis, (C) Apoptosis, (D) Pentose phosphate pathway, (E) Energy, (F) Lipids. P values of < 0.1, 0.05, 0.01, 0.001 and 0.0001 are represented by ·, *, **, *** and ****, respectively.
In short, these results demonstrate that phosphoromics provide a global perspective of liver metabolism of a pathological state with respect to a “healthy” situation that may be used effectively to distinguish between different models of steatohepatitis( 20 ) and eventually to identify different pathogenic mechanisms.
Therapeutic Effect of CPX on CEP as Investigated by Phosphoromic Analysis of Liver Extracts
The bioactive conformation of UROIIIS is not stable thermodynamically, but its lifetime is long enough to exert its function.( 43 ) However, most of the inherited deleterious mutations in the UROIIIS gene accelerate protein aggregation and impair its homeostasis in the cell.( 44, 45, 46 ) Recently, we discovered that the antifungal CPX acts as a pharmacological chaperone by binding to UROIIIS and kinetically stabilizing the enzyme.( 17 ) A subtoxic concentration of CPX, administered to CEP mice for 45 days, is able to revert most of the pathological effects and to significantly reduce hemolysis, steatosis, and porphyrin deposition (Fig. 4).
Remarkably, the 31P‐NMR spectra from the E1 and E2 extracts indicate that the aberrant liver phosphorome of CEP mice was largely restored to normal on CPX administration (Figs. 4 and 5). Thus, the hepatic G6P level, which is markedly reduced in CEP mice liver, recovers even above the level of WT mice in CPX‐treated CEP mice (Fig. 5A). This therapeutic effect engenders an increase in the content of its downstream products G1P and UDPG/ADPG/GDPG (glycogenesis), and 6PG, X5P, and S7P (PPP), in some cases exceeding the value in WT animals (Fig. 5B,D). The content of lactate, which was increased in CEP mice liver, decreased significantly after CPX treatment until reaching normal values (Fig. 5A). Some of the energy‐associated metabolites, AMP/guanosine 5′‐monophosphate, cytidine 5′‐monophosphate and uridine 5′‐monophosphate, and NADPH also reached values comparable with WT after CPX treatment, whereas ADP‐ribose levels decreased (Fig. 5C,E). The later prompted us to immunostain liver sections of CEP mice for active caspase‐3, a readout of apoptosis known to be increased in NASH.( 47 ) Interestingly, CPX treatment inhibited caspase‐3 activation and PARP cleavage, which was increased in CEP mice (Fig. 4C; Supporting Fig. S5). All in all, these results underscore an improvement in central carbon metabolism and mitochondrial function in response to CPX treatment in CEP mice (Fig. 5; Supporting Table S4), which agrees with the histological findings( 17 ) (Fig. 4).
The therapeutic effect of CPX in CEP is best observed in the phospholipidic E2 liver extract (Fig. 4I), where the majority of the phospholipids as well as the PC/PE and PC/(PE + SM) molar ratios are restored to healthy liver values (Fig. 5F). These results from our phosphoromic analysis indicate that the stabilization of the UROIIIS and restoration of the heme level production are able to correct the membrane impairment and revert the liver injury, as observed in the histological analysis (Supporting Table S4).
Discussion
The human metabolome, as in other mammals, is a collection of more than 100,000 metabolites interconnected by more than 18,000 enzymatic reactions.( 1 ) These metabolites provide the building blocks and the energy to power all the physiological processes in the cell, but alterations in their concentration can lead to disease. Thus, a comprehensive readout of the human metabolome is a powerful way to monitor individual health or pathology and the biomolecular response to its therapy. However, this highly convoluted information may be overwhelmingly complex, and it is essential to reduce it with the minimum functional information losses possible. Phosphorylated metabolites are synthesized and catabolized by irreversible enzymes that operate as “chemical relays” limiting the flow through virtually all metabolic pathways (Fig. 2), creating a demand‐driven system that can “drag” a metabolite to or away from a specific metabolic pathway (catabolic or anabolic).
Here, we have shown that the liver phosphorome, that is, the set of liver’s phosphorylated metabolites, provides a snapshot of the organ’s metabolism in health, disease, and response to treatment, which correlates with liver histology and provides mechanistic information. Our 31P‐NMR–based phosphoromic platform was developed under strict SOPs for the extraction of metabolites, the precise assignment of metabolites by 31P‐NMR, and their quantification using a chemical standard in combination with relaxation accelerating paramagnetic adjuvants. This procedure worked both with murine and human liver samples and provides the absolute concentration of up to 54 phosphorylated metabolites, separated in 2 phases. The hydrophilic phase covers a wide array of pathways from glycolysis, glycogenesis, the PPP, TCA, and the phosphoryl‐transfer and electron‐transfer pathways that power much of the anabolic processes. The lipophilic phase provides information on the phospholipid metabolism (PE, PS, PI, SM, CL, and PC), which can be rationalized in terms of membrane integrity and composition. In addition, the platform provides quantitative information of four key nonphosphorylated metabolites: lactate, CH, TG, and TC. Although 100 mg liver was used in most of the present experiments, we show here that this amount can be lowered to a minimum of 20 mg without compromising the quantification of the phosphorylated metabolites by this method. As mentioned, we estimate that the sensitivity of the method may be improved with the available technology to push down the limit to 10 mg liver tissue, an amount that makes it feasible for use with liver needle biopsies.( 48 )
We have applied this phosphoromic platform to obtain interpretable connections between changes in phosphorylated metabolites and histological phenotypes. In the CEP model, we observed a reduction in energy harvesting and storage, as indicated by the decrease in NTPs, NADH, and FADH2, and an accumulation of liver lactate, a condition known as the Warburg effect. These findings were associated with histological evidence of heme accumulation, an indicator of cell injury, and apoptosis. We also observed in CEP mice a reduction in the PC/PE and PC/PE + SM ratios, which could be explained by the reduction of CDPCH, the rate‐limiting substrate in PC biosynthesis. Interestingly, changes in phosphorylated and nonphosphorylated metabolites (NADH, lactate, ADP‐ribose, PC/PE, TG, TC) coupled with the improvement of the histological phenotype in CEP mice treated with CPX. This successful proof of concept suggests broad and powerful use of the phosphoromics platform in drug development, e.g., to guide the preclinical selection of drug candidates for further clinical development.
We also applied this phosphoromic platform to identify individual metabolites and perturbations in metabolic pathways to two NASH mouse models: MAT1A‐KO and CDHF. MAT1A‐KO mice liver show an increase in hexose‐phosphates and triose‐phosphates (G6P, FBP, and 3PG) and a reduction in PEP, whereas lactate remained unchanged. This, together with the increase in G1P and the reduction of the products of the oxidative phase of the PPP (6PG and NADPH) and the reduction of NADH and NTPs, indicates an increase in gluconeogenesis and/or a decrease of oxidative metabolism in MAT1A‐KO. Work has shown a critical role of MAT1A in regulating mitochondrial function.( 49 ) Conversely, in CDHF mice, we observed an increase in energy harvesting and storing, as indicated by the increase in NADH, NADPH, NTPs, and PCr. These differences between both NASH models further demonstrate the power of this phosphoromic platform to provide mechanistic insights that may help to understand the heterogeneity of NAFLD and its progression to NASH as well as potentially important therapeutic target candidates. As expected, both NASH models showed an increase in hepatic TG and a decrease in the PC/PE molar ratio.
Author Contributions
G.B.‐S., S.C.L., J.M.M., and O.M. were responsible for conceptualization; G.B.‐S., J.B., B.M., M.F.F., J.A.‐F., T.D., and O.M. were responsible for methodology; G.B.‐S., J.B, D.F.‐R., F.L.‐O., M.B., and V.G.d.J. were responsible for formal analysis; G.B.‐S. and O.M. were responsible for writing (original draft presentation); G.B.‐S., S.C.L., R.N., J.M.M., and O.M. were responsible for writing (reviewing and editing). The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting information
Supplementary Material
Supported by the Department of Industry, Tourism and Trade of the Government of the Autonomous Community of the Basque Country (Elkartek BG2019) and the Severo Ochoa Excellence Accreditation from MCIU (SEV‐2016‐0644). O. M. acknowledges the Agencia Estatal de Investigación (Spain) for grants CTQ2015‐68756‐R and RTI2018‐101269‐B‐I00, and J. M. M. acknowledges the Agencia Estatal de Investigación (Spain) and CIBERehd for grant SAF2017‐88041‐R. S.C.L. acknowledges NIH for support (NIH grant R01DK123763).
Potential conflict of interest: Dr. Mato consults for, advises, and owns stock in Owl. He consults for and advises Abbott. He consults for Galmed.
Contributor Information
José M. Mato, Email: omillet@cicbiogune.es, Email: director@cicbiogune.es.
Oscar Millet, Email: omillet@cicbiogune.es.
References
Author names in bold designate shared co‐first authorship.
- 1.Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vázquez‐Fresno R, et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res 2018;46:D608‐D617. 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27‐30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh CT, Tu BP, Tang Y. Eight kinetically stable but thermodynamically activated molecules that power cell metabolism. Chem Rev 2018;118:1460‐1494. 10.1021/acs.chemrev.7b00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang ML, Yang SS. Metabolic signature of hepatic fibrosis: From individual pathways to systems biology. Cells 2019;8:1423. 10.3390/cells8111423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anstee QM, Goldin RD. Mouse models in non‐alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol 2006;87:1‐16. 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al‐awar A, Kupai K, Veszelka M, Szűcs G, Attieh Z, Murlasits Z, et al. Experimental diabetes mellitus in different animal models. J Diabetes Research 2016;2016:1‐12. 10.1155/2016/9051426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, et al. Mouse models of nonalcoholic steatohepatitis: Toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology 2019;69:2241‐2257. 10.1002/hep.30333. [DOI] [PubMed] [Google Scholar]
- 8.Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein post‐translational modifications with mass spectrometry. Nat Methods 2007;4:798‐806. 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 9.Kumar GS, Page R, Peti W. Preparation of phosphorylated proteins for NMR spectroscopy. Methods Enzymol 2019;614:187‐205. 10.1016/bs.mie.2018.07.004. [DOI] [PubMed] [Google Scholar]
- 10.Emwas AH, Roy R, McKay RT, Tenori L, Saccenti E, Gowda GAN, et al. NMR spectroscopy for metabolomics research. Metabolites 2019;9:123. 10.3390/metabo9070123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell JD, Cox IJ, Sargentoni J, Peden CJ, Menon DK, Foster CS, et al. A 31P and 1H‐NMR investigation in vitro of normal and abnormal human liver. BBA ‐ Mol Basis Dis 1993;1225:71‐77. 10.1016/0925-4439(93)90124-J. [DOI] [PubMed] [Google Scholar]
- 12.Harvey PJ, Gready JE, Hickey HM, Le Couteur DG, McLean AJ. 31P and1H NMR spectroscopic studies of liver extracts of carbon tetrachloride‐treated rats. NMR Biomed 1999;12:395‐401. https://doi.org/10.1002/(SICI)1099-1492(199910)12:6<395::AID-NBM568>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 13.Jeon MJ, Lee Y, Ahn S, Lee C, Kim OH, Oh BC, et al. High resolution in vivo 31P‐MRS of the liver: Potential advantages in the assessment of non‐alcoholic fatty liver disease. Acta Radiol 2015;56:1051‐1060. 10.1177/0284185114550850. [DOI] [PubMed] [Google Scholar]
- 14.Norén B, Lundberg P, Ressner M, Wirell S, Almer S, Smedby O. Absolute quantification of human liver metabolite concentrations by localized in vivo 31P NMR spectroscopy in diffuse liver disease. Eur Radiol 2005;15:148‐157. [DOI] [PubMed] [Google Scholar]
- 15.Bhinderwala F, Evans P, Jones K, Laws BR, Smith TG, Morton M, et al. Phosphorus NMR and its application to metabolomics. Anal Chem 2020;92:9536‐9545. 10.1021/acs.analchem.0c00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balwani M, Desnick RJ. The porphyrias: Advances in diagnosis and treatment. Blood 2012;120:4496‐4504. 10.1182/blood-2012-05-423186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urquiza P, Laín A, Sanz‐Parra A, Moreno J, Bernardo‐Seisdedos G, Dubus P, et al. Repurposing ciclopirox as a pharmacological chaperone in a model of congenital erythropoietic porphyria. Sci Transl Med 2018;10:eaat7467. 10.1126/scitranslmed.aat7467. [DOI] [PubMed] [Google Scholar]
- 18.Bock FJ, Chang P. New directions in poly(ADP‐ribose) polymerase biology. FEBS J 2016;283:4017‐4031. 10.1111/febs.13737. [DOI] [PubMed] [Google Scholar]
- 19.Christiansen AL, Aagaard L, Krag A, Rasmussen LM, Bygum A. Cutaneous porphyrias: Causes, symptoms, treatments and the danish incidence 1989‐2013. Acta Dermato‐Venereologica 2016;96:868‐872. 10.2340/00015555-2444. [DOI] [PubMed] [Google Scholar]
- 20.Alonso C, Fernández‐Ramos D, Varela‐Rey M, Martínez‐Arranz I, Navasa N, Van Liempd SM, et al. Metabolomic identification of subtypes of nonalcoholic steatohepatitis. Gastroenterology 2017;152:1449‐1461.e7. 10.1053/j.gastro.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolf M, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross‐talk with hepatocytes. Cancer Cell 2014;26:549‐564. 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Fondevila MF, Fernandez U, Gonzalez‐Rellan MJ, Da Silva Lima N, Buque X, Gonzalez‐Rodriguez A, et al. The L‐α‐lysophosphatidylinositol/GPR55 system induces the development of non‐alcoholic steatosis and steatohepatitis. Hepatology 2020. 10.1002/hep.31290. [Epub ahead of print] 73:2, 2021, 606‐624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 24.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959;37. https//doi.org/10.1139/o59‐099. [DOI] [PubMed] [Google Scholar]
- 25.Nagana Gowda GA, Raftery D. Analysis of coenzymes and antioxidants in tissue and blood using 1D 1 H NMR spectroscopy. Methods Mol Biol 2019;2037:97‐110. 10.1007/978-1-4939-9690-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mulder FAA, Tenori L, Luchinat C. Fast and quantitative NMR metabolite analysis afforded by a paramagnetic co‐solute. Angew Chemie ‐ Int Ed 2019;58:15283‐15286. 10.1002/anie.201908006. [DOI] [PubMed] [Google Scholar]
- 27.Erwin AL, Desnick RJ. Congenital erythropoietic porphyria: Recent advances. Mol Genet Metab 2019;128:288‐297. 10.1016/j.ymgme.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee WH, Tai WC, Wu PY. Congenital erythropoietic porphyria. Dermatologica Sin 2012;30:62‐65. 10.1016/j.dsi.2011.09.012. [DOI] [Google Scholar]
- 29.Bishop DF, Clavero S, Mohandas N, Desnick RJ. Congenital erythropoietic porphyria: Characterization of murine models of the severe common (C73R/C73R) and later‐onset genotypes. Mol Med 2011;17:748‐756. 10.2119/molmed.2010.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blouin JM, Duchartre Y, Costet P, Lalanne M, Ged C, Lain A, et al. Therapeutic potential of proteasome inhibitors in congenital erythropoietic porphyria. Proc Natl Acad Sci U. S. A. 2013;110:18238‐18243. 10.1073/pnas.1314177110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chacko B, Culp ML, Bloomer J, Phillips J, Kuo YF, Darley‐Usmar V, et al. Feasibility of cellular bioenergetics as a biomarker in porphyria patients. Mol Genet Metab Reports 2019;19:100451. 10.1016/j.ymgmr.2019.100451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, et al. Accumulation of free ADP‐ribose from mitochondria mediates oxidative stress‐induced gating of TRPM2 cation channels. J Biol Chem 2005;280:6138‐6148. 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 33.Sengupta A, Hon T, Zhang L. Heme deficiency suppresses the expression of key neuronal genes and causes neuronal cell death. Mol Brain Res 2005;137:23‐30. 10.1016/j.molbrainres.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, et al. Mechanisms of lysophosphatidylcholine‐induced hepatocyte lipoapoptosis. Am J Physiol ‐ Gastrointest Liver Physiol 2012;302:G77‐G84. 10.1152/ajpgi.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007;46:1081‐1090. 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 36.Lu YW, Claypool SM. Disorders of phospholipid metabolism: An emerging class of mitochondrial disease due to defects in nuclear genes. Front Genet 2015;6:3. 10.3389/fgene.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mato JM, Alonso C, Noureddin M, Lu SC. Biomarkers and subtypes of deranged lipid metabolism in non‐alcoholic fatty liver disease. World J. Gastroenterol 2019;25:3009‐3020. 10.3748/wjg.v25.i24.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z, Agellon LB, Allen TM, Umeda M, Jewell L, Mason A, et al. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab 2006;3:321‐331. 10.1016/j.cmet.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Iruarrizaga‐Lejarreta M, Varela‐Rey M, Fernández‐Ramos D, Martínez‐Arranz I, Delgado TC, Simon J, et al. Role of aramchol in steatohepatitis and fibrosis in mice. Hepatol Commun 2017;1:911‐927. https://doi:10.1002/hep4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vance DE. Physiological roles of phosphatidylethanolamine N‐methyltransferase. Biochim Biophys Acta, Mol Cell Biol Lipids 2013;1831:626‐632. 10.1016/j.bbalip.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 41.Liberti MV, Locasale JW. The warburg effect: How does it benefit cancer cells? Trends Biochem Sci 2016;41:211‐218. 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ueland PM. Choline and betaine in health and disease. J Inherit Metab Dis 2011;34:3‐15. 10.1007/s10545-010-9088-4. [DOI] [PubMed] [Google Scholar]
- 43.Fortian A, Castaño D, Ortega G, Laín A, Pons M, Millet O. Uroporphyrinogen III synthase mutations related to congenital erythropoietic porphyria identify a key helix for protein stability. Biochemistry 2009;48:454‐461. 10.1021/bi801731q. [DOI] [PubMed] [Google Scholar]
- 44.Fortian A, González E, Castaño D, Falcon‐Perez JM, Millet O. Intracellular rescue of the uroporphyrinogen III synthase activity in enzymes carrying the hotspot mutation C73R. J Biol Chem 2011;286:13127‐13133. 10.1074/jbc.M110.205849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blouin JM, Bernardo‐Seisdedos G, Sasso E, Esteve J, Ged C, Lalanne M, et al. Missense UROS mutations causing congenital erythropoietic porphyria reduce UROS homeostasis that can be rescued by proteasome inhibition. Hum Mol Genet 2017;26:1565‐1576. 10.1093/hmg/ddx067. [DOI] [PubMed] [Google Scholar]
- 46.ben Bdira F, González E, Pluta P, Laín A, Sanz‐Parra A, Falcon‐Perez JM, et al. Tuning intracellular homeostasis of human uroporphyrinogen III synthase by enzyme engineering at a single hotspot of congenital erythropoietic porphyria. Hum Mol Genet 2014;23:5805‐5813. 10.1093/hmg/ddu298. [DOI] [PubMed] [Google Scholar]
- 47.Wilson CH, Kumar S. Caspases in metabolic disease and their therapeutic potential. Cell Death Differ 2018;25:1010‐1024. 10.1038/s41418-018-0111-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sherlock S. Needle biopsy of the liver: A review. J Clin Pathol 1962;15:291‐304. 10.1136/jcp.15.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santamaria E, Avila MA, Latasa MU, Rubio A, Martin‐Duce A, Lu SC, et al. Functional proteomics of nonalcoholic steatohepatitis: Mitochondrial proteins as targets of S‐adenosylmethionine. Proc Natl Acad Sci U. S. A. 2003;100:3065‐3070. 10.1073/pnas.0536625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material