

Abstract
A novel protocol for the transition metal‐free 1,2‐addition of polyfluoroaryl boronate esters to aldehydes and ketones is reported, which provides secondary alcohols, tertiary alcohols, and ketones. Control experiments and DFT calculations indicate that both the ortho‐F substituents on the polyfluorophenyl boronates and the counterion K+ in the carbonate base are critical. The distinguishing features of this procedure include the employment of commercially available starting materials and the broad scope of the reaction with a wide variety of carbonyl compounds giving moderate to excellent yields. Intriguing structural features involving O−H⋅⋅⋅O and O−H⋅⋅⋅N hydrogen bonding, as well as arene‐perfluoroarene interactions, in this series of racemic polyfluoroaryl carbinols have also been addressed.
Keywords: alcohol; 1,2-addition reaction; boronate esters; fluoroarene; transition metal-free
Herein we report a transition metal‐free procedure for the base‐promoted 1,2‐addition of polyfluorophenylboronates to aldehydes and ketones to generate organofluorine‐containing alcohols in yields up to 93 %.
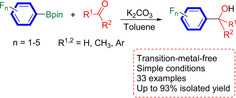
Introduction
Over the past few decades, the transition‐metal‐catalyzed 1,2‐addition of organometallic reagents to the C=O functionality of aldehydes and ketones has developed as a useful method for the synthesis of substituted secondary and tertiary alcohols.[1] Numerous reagents have been used for these reactions, including organomagnesium,[2] organozinc,[1, 3] organolithium,[4] organosilane,[5] organostannane,[6] organocerium,[7] and organoboron compounds.[8] In particular, organoboronate reagents offer significant advantages such as air and moisture stability, low toxicity, good functional group tolerance, and availability.[8] In 1998, Miyaura and co‐workers[9] first reported the addition of arylboronic acids to aldehydes using a Rh catalyst. In subsequent studies, other rhodium,[10] palladium,[11] platinum,[12] nickel,[13] copper,[14] iron,[15] cobalt,[16] and ruthenium[17] complexes have been developed as precatalysts for such reactions. However, transition metals can be expensive, toxic, and difficult to remove completely from the corresponding product. A transition metal‐free strategy would be highly desirable for these useful transformations. The reaction products for the addition of arylboronic acids to ketones, after hydrolysis, are tertiary alcohols, which are important building blocks for the synthesis of pharmaceuticals, agrochemical compounds, and natural products.[18] However, the nucleophilic addition of organometallic reagents to ketones can be challenging due to the inherent steric congestion around the carbonyl group, frequently resulting in the generation of products arising from side reactions such as reduction and aldol condensation.[19] Therefore, the development of an efficient, general, and convenient protocol for the synthesis of tertiary alcohols is of considerable interest.
Moreover, an ideal strategy to synthesize ketones, important and ubiquitous structural motifs,[20] lies in the transition metal‐catalyzed replacement of an aldehyde's C(O)‐H group with a carbon electrophile.[21] Recently, Zheng and co‐workers demonstrated the direct functionalization of aldehyde C−H bonds with aryl halides, using a precious metal palladium catalyst, which has proven to be a viable method to generate the corresponding ketone products.[22]
Polyfluoroarenes have gained extensive attention due to their important role in pharmaceutical, agrochemical, and advanced materials.[23] Thus, identifying practical and efficient concepts for the introduction of fluorine or fluorinated building blocks is highly desirable. Several studies have been reported regarding the polyfluorophenylation of aldehydes. For example, in 1999, Knochel and co‐workers[24] used fluorinated aryl bromides to perform pentafluorophenylation of aldehydes (Scheme 1 a). More recently, Lam and co‐workers[25] used a copper catalyst (Scheme 1 b) and Gu and co‐workers[26] (Scheme 1 b) used an N‐heterocyclic carbene (NHC) organocatalyst to obtain fluorinated aryl carbinols using polyfluorophenyl trimethylsilane as a nucleophile for the addition to aldehydes. In 2015, Huang and co‐workers[27] (Scheme 1 c) reported a Mg‐mediated polyfluoroaryl addition to aldehydes. Although some advancements in this field have been reported, these methods suffer from the requirement for highly flammable Grignard reagents, transition metals or NHC catalysts. Moreover, methods reported by Lam and co‐workers and Gu and co‐workers are limited to pentafluorophenyl trimethylsilane or 1,4‐bis (trimethylsilyl) tetrafluorobenzene as substrates.
Scheme 1.
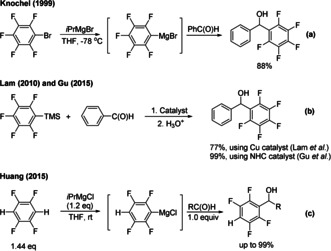
Approaches to access polyfluoroaryl carbinols via the addition to aldehydes.
Recently, we reported efficient methods to generate fluorinated arylboronic acid pinacol esters (ArF‐Bpin) via C‐F borylation of fluoroarenes using NHC‐ligated Ni complex[28a, 28b] and C‐Cl borylation of ArF‐Cl using Pd catalyst under base free condition.[28c] Likewise, we reported optimized conditions for the Suzuki–Miyaura cross‐coupling reaction of ArF‐Bpin compounds with ArX (X=Br, I) using a combination of CuI and 1,10‐phenanthroline as a catalyst precursor.[28d] Furthermore, we reported the palladium‐catalyzed homocoupling of fluorinated arylboronates,[28e] and the copper‐catalyzed oxidative cross‐coupling of electron‐deficient polyfluorophenyl boronate esters with terminal alkynes.[28f] We report herein the transition metal‐free polyfluorophenylation of ketones and aldehydes with fluorinated aryl boronates, which provides a convenient and novel strategy for the synthesis of alcohols and ketones.
Results and Discussion
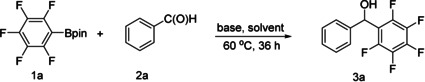
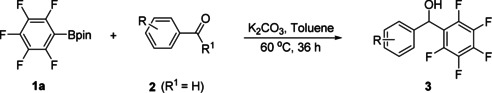
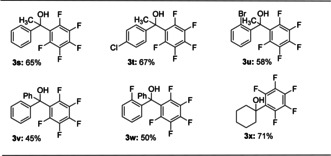
Addition of arylboronic acids to aldehydes using transition metal catalysts has been well developed. We expected that the use of more Lewis acidic pentafluorophenyl‐Bpin with a base would generate a nucleophilic intermediate in the absence of a transition metal. To verify our hypothesis, we initially examined the reaction of pentafluorophenyl‐Bpin (1 a) and benzaldehyde (2 a) as a model reaction. As shown in Table 1, secondary alcohol 3 a was observed as the addition product after hydrolysis when the mixture of 1 a and 2 a was heated in the presence of KOMe as the base (Table 1, entry 1). Encouraged by this first result, we screened the reaction parameters, including the base and the solvent, to improve the performance of the reaction. The employment of K2CO3 as the base dramatically increased the yield to 92 % (Table 1, entry 6). The experimental results revealed that heating is required as the room temperature reaction only afforded 3 a in trace amounts (Table 1, entry 7). Lower conversions were observed when reactions were conducted in coordinating solvents such as DMF, THF, and 1,4‐dioxane (Table 1, entries 8, 10, 11), and the lowest yield was obtained when CH3CN was used as the solvent (Table 1, entry 9). In addition, the reaction exhibited very poor performance under aerobic conditions (Table 1, entry 12). Interestingly, increasing the amount of K2CO3 to 3 equiv was not helpful (Table 1, entry 13). Decreasing the amount of K2CO3 (0.8 equiv) did not impact the performance of the reaction (Table 1, entry 14). No reaction took place when K2CO3 was absent (Table 1, entry 15), indicating that K2CO3 as the base is important for this reaction. Not surprisingly, adventitious water quenched the reaction (Table 1, entries 16, 17). However, under anhydrous conditions, the transition metal‐free polyfluorophenylation of benzaldehyde with pentafluorophenyl‐Bpin is feasible and leads to high yields of the desired product.
Table 1.
Optimization of the reaction conditions.[a]
|
Entry |
Base |
Solvent |
Yield [%][b] |
|---|---|---|---|
|
1 |
KOMe |
toluene |
20 |
|
2 |
KF |
toluene |
25 |
|
3 |
tBuOLi |
toluene |
52 |
|
4 |
Cs2CO3 |
toluene |
60 |
|
5 |
Na2CO3 |
toluene |
78 |
|
6 |
K2CO3 |
toluene |
92 |
|
7[c] |
K2CO3 |
toluene |
trace |
|
8 |
K2CO3 |
DMF |
50 |
|
9 |
K2CO3 |
CH3CN |
15 |
|
10 |
K2CO3 |
THF |
88 |
|
11 |
K2CO3 |
1,4‐dioxane |
79 |
|
12[d] |
K2CO3 |
toluene |
35 |
|
13[e] |
K2CO3 |
toluene |
83 |
|
14[f] |
K2CO3 |
toluene |
92 |
|
15 |
– |
toluene |
0 |
|
16[g] |
K2CO3 |
toluene |
66 |
|
17[h] |
K2CO3 |
toluene |
25 |
[a] Conditions: 1 a (0.44 mmol), 2 a (0.4 mmol), base (1.0 equiv), degassed and dried solvent (3 mL), 60 °C, 36 h, under argon. [b] Yields were determined by GC‐MS analysis vs. a calibrated internal standard and are averages of two runs. [c] Room temperature. [d] Under air. [e] K2CO3 (3 equiv). [f] K2CO3 (0.8 equiv). [g] K2CO3 (0.8 equiv), degassed wet toluene. [h] K2CO3 (0.8 equiv), wet toluene. Moisture and air are detrimental to the yield due to the instability of the fluorinated aryl boronate.[23g]
Using these optimized conditions, we evaluated the scope and the limitations of this reaction. As shown in Table 2, a series of aldehydes bearing electron‐withdrawing or ‐donating substituents at the para‐, meta‐, or ortho‐position all worked well with pentafluorophenyl‐Bpin to give the desired products (3 b–3 k). Notably, for reactions employing aldehydes bearing electron‐donating groups, increasing the reaction temperature to 80 °C for 48 hours was required to generate the corresponding products in acceptable yields. It should be noted that reactions using 4‐(diethoxymethyl)benzaldehyde resulted in cleavage of the diethoxymethyl group yielding 3 l. Furthermore, this methodology could be successfully extended to more complex aldehydes, such as those incorporating naphthyl and pyridyl groups (3 m and 3 n). The structures of compounds 3 f, 3 l, 3 m and 3 n were unambiguously confirmed via single crystal X‐ray analysis (vide infra). After a broad range of aromatic aldehydes were examined, reactions with aliphatic aldehydes were investigated using the optimized conditions. Gratifyingly, all reactions proceeded smoothly to afford the corresponding products (3 o–3 q). Importantly, aldehydes containing ester groups, which are well‐known to be sensitive towards Grignard reagents, also afforded the desired alcohols in excellent yield (3 r).
Table 2.
Scope of the reaction with respect to the different aldehyde substrates 2.[a,b]
|
|
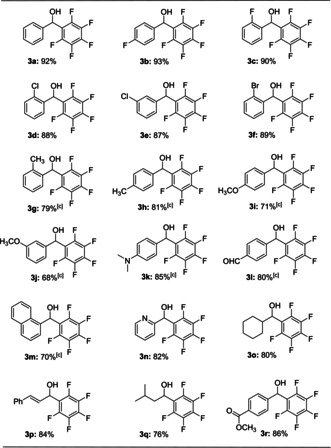
[a] Conditions: 1 a (0.44 mmol), 2 (0.4 mmol), K2CO3 (0.32 mmol), toluene (3 mL), 60 °C, 36 h, Ar. [b] Isolated yields are reported. [c] 80 °C, 48 h.
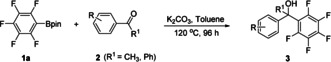
We then briefly investigated the scope using simple ketones (Table 3). When reactions were performed at 120 °C and for prolonged reaction times, the corresponding products were provided in moderate yields (3 s–3 u). Modest reaction yields were obtained when sterically hindered benzophenone and (2‐fluorophenyl)(phenyl)methanone were used (3 v–3 w). Importantly, cyclohexanone proceeded to give the desired products in good yield (3 x).
Table 3.
Scope of the reaction with respect to the different ketone substrates 2.[a,b]
|
|
[a] Conditions: 1 a (0.44 mmol), 2 (0.4 mmol), K2CO3 (0.32 mmol), toluene (3 mL), 120 °C, 96 h, Ar. [b] Isolated yields are reported.
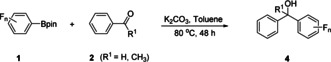
To explore further the utility of this reaction, we then examined the scope using other less substituted polyfluorophenyl boronate esters with benzaldehyde (Table 4). The compounds 2,3,5,6‐tetrafluorophenyl‐Bpin, 2,3,4,6‐tetrafluorophenyl‐Bpin, and 2,4,6‐trifluorophenyl‐Bpin also proved to be effective in these reactions and afforded the products in excellent yields (4 a–4 c). Furthermore, the reaction with 2,6‐difluorophenyl‐Bpin proceeded to give the desired product in 80 % yield (4 d). However, these reaction conditions were not suitable for the reaction of 2,5‐difluorophenyl‐Bpin and 2‐fluorophenyl‐Bpin with benzaldehyde. Surprisingly, reactions with these substrates resulted in the formation of ketones (4 e and 4 f) when a strong base was used. Tetrafluorophenyl‐Bpin reacted readily with acetophenone to yield product 4 g. Unfortunately, no reaction occurred when the aryl‐Bpin compound did not have an ortho‐fluorine substituent (4 h and 4 i), as 3‐fluorophenyl‐Bpin, phenyl‐Bpin, 4‐CH3‐phenyl‐Bpin and 4‐CN‐phenyl‐Bpin all failed to provide any product. These results demonstrate that the ortho‐fluorine group plays a key role in related conversions.
Table 4.
Scope of the reaction with respect to different polyfluorophenyl boronate substrates 1.[a,b]
|
|
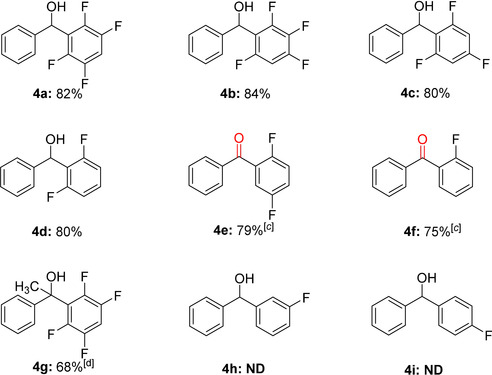
[a] Reaction conditions: 1 a (0.44 mmol), 2 (0.4 mmol), K2CO3 (0.32 mmol), toluene (3 mL), 80 °C, 48 h, Ar. [b] Isolated yields are reported. [c] t‐BuOLi (0.32 mmol). [d] 120 °C, 96 h.
To gain further insight into the aforementioned reactions, several mechanistic studies were conducted. The reaction of 2 a with pentafluorobenzene 5 under standard conditions was examined, yet 3 a was not formed in any detectable amounts (Scheme 2 a), indicating that the C‐Bpin moiety is essential and deprotonation of the fluoroarene or nucleophilic attack at the fluoroarene by the base is not a plausible pathway. Interestingly, for the standard reaction between 1 a and 2 a, the yield dropped dramatically if 18‐crown‐6 ether and K2CO3 were added (Scheme 2 b). This experimental result indicates that the presence of the potassium ion plays a crucial role for the outcome of the reaction. Furthermore, if the reaction of 1 a and 2 a was performed in the presence of only a catalytic amount of K2CO3 (20 mol %) (Scheme 2 c), reaction rates were reduced, and a week was required to produce 3 a in good, isolated yield. This finding again indicates that the potassium ion (or the base) plays an important role in the reaction. Substituting ortho‐fluorines by ortho‐chlorines, using either C6Cl5Bpin or 2,6‐dichlorophenyl‐1‐Bpin as substrates, did not yield any product as shown by in situ GCMS studies. Likewise, 2,3,4‐trifluorophenylBpin and 3,4,5‐trifluorophenylBpin substrates with only one or no ortho‐fluorine substituent also led to no detectable product formation. The presence of an ortho‐methoxy group on the aldehyde, however, did not inhibit the reaction.
Scheme 2.
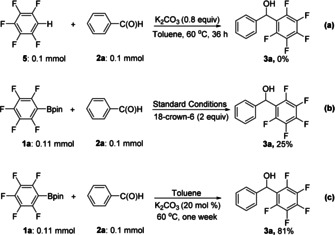
Preliminary mechanistic studies.
Based on previous studies[28, 29] and experimental observations, a mechanism for the 1,2‐addition of polyfluorophenylboronates to aryl aldehydes in the presence of K2CO3 as base is proposed, as shown in Scheme 3. K2CO3 interacts with the Lewis‐acidic Bpin moiety of substrate 1 to generate base adduct A, which weakens the carbon‐boron bond and ultimately cleaves the B−C bond along with attachment of a potassium cation to the aryl group. The resulting ArF − anion adduct B undergoes nucleophilic attack at the aldehyde carbon atom of substrate 2 to generate methanolate C. The methanolate oxygen atom then attacks the electrophilic Bpin group to obtain compound D. Transfer of K2CO3 from intermediate D to the boron atom of the more Lewis‐acidic polyfluorophenyl‐Bpin 1 finally closes the cycle and regenerates complex A. Thus, the primary reaction product is the O‐borylated addition product E, which was detected by HRMS and NMR spectroscopy for the perfluorinated derivative (Supporting Information, section VIII).
Scheme 3.
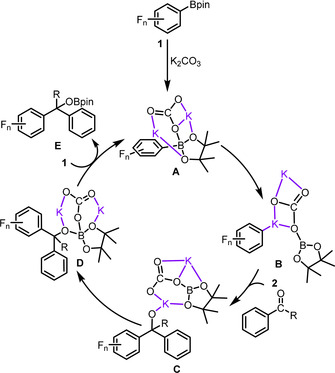
Proposed mechanism of the 1,2‐addition of polyfluorophenylboronates to aldehyde derivatives in the presence of K2CO3 as base.
To corroborate this mechanism, a detailed DFT study was performed on the model 1,2‐addition of 1 a to 2 a, the results of which are shown in Figure 1. In the initial step, K2CO3 coordinates to the Bpin moiety of 1 a and gives rise to the pentafluorophenyl‐Bpin‐base complex 6 with free energy decreasing by 27.2 kcal mol−1. The energy of compound 6 is set as the zero point of the energy profile. The pentafluorobenzene anion (ArF −) adduct 8 is formed endothermically by cleavage of the B‐C(ArF) bond via transition state 7‐ts with an energy barrier of 26.4 kcal mol−1. In the optimized structures of 7‐ts, K+ cations coordinate to C, O and F atoms, whereas there is only K‐O coordination in compound 6. Subsequent cleavage of the B‐C(ArF) bond can be facilitated by this pathway. The separated carbonate adduct and ArF − group in adduct 8 are connected and stabilized by K+ cations. Nucleophilic attack of ArF − at the aldehyde carbon atom via transition state 10‐ts occurs to achieve the coupling intermediate 11 with an energy of 17.6 kcal mol−1. This low activation energy barrier can be attributed to the coordination of K+ to the oxygen atom of the aldehyde, thus enhancing the electrophilicity of the aldehyde carbon atom. Subsequently, the methanolate oxygen atom attacks the Lewis‐acidic boron atom to give the corresponding compound 13 irreversibly via transition state 12‐ts. The overall energy barrier for this step is 16.2 kcal mol−1. Finally, K2CO3 in compound 13 coordinates to the boron of substrate 1 a via transition state 14‐ts, followed by cleavage of a B−O bond to give 16‐ts and eventually 17, regenerating the active species 6. As shown in Figure 1, the energy barriers for these two steps are very low, indicating that intermediate 13 transforms to product 17 swiftly. The step from pentafluorophenyl‐Bpin‐base compound 6 to product 17 is calculated to be exergonic by 14.3 kcal mol−1. The base‐assisted cleavage of Bpin and pentafluorophenyl (ArF) is calculated to be the rate determining step (RDS) with a free energy of activation of 26.4 kcal mol−1.
Figure 1.
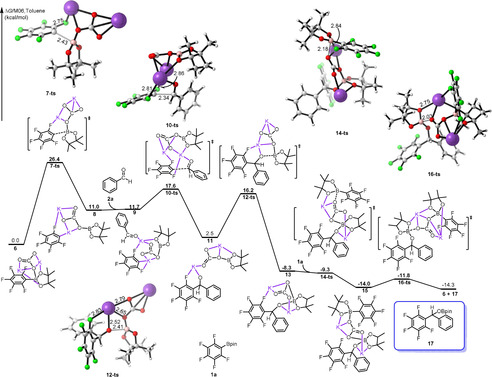
Free energy profile for the 1,2‐addition of pentafluorophenyl‐Bpin (1 a) and benzaldehyde (2 a) in the presence of K2CO3 as the base, calculated at the M06/(6–311++G(d, p), SMD)//B3LYP/(6–31+G(d)) level of theory. Relative free energies (ΔG) are given in kcal mol−1, and bond lengths are given in Å.
As shown in Figure 1, the cation K+ bonds with one or two F atoms in these intermediates and transition states, suggesting that the fluoride substituents possibly play an important role in the 1,2‐addition of polyfluorophenylboronates to aryl aldehydes. Therefore, we calculated the activation free energies of the RDS using polyfluorophenylboronates with different numbers and positions of fluorine substituents as the substrate. The results given in Figure 2 clearly show that the energy barrier rises with a reduction in the number of F substituents. The position of the fluorine atoms also affects the energy barrier, and ortho fluorine has a stronger effect on the barrier than F substituents at other positions. The barrier for 24, with an ortho‐F substituent, is higher than that of 22 by 2.6 kcal mol−1, whereas that of 26 with a para‐F substituent rises to 39.0 kcal mol−1. In fact, no reaction was observed under these conditions when 26 was used as the substrate, which is consistent with our calculated results. We conclude that the ortho‐F substituent is vital in this reaction for interaction with K+ along the reaction pathway, and that other F substituents also influence the reactivity for the 1,2‐addition of polyfluorophenylboronates to aryl aldehydes via their electron‐withdrawing effect. Thus, stronger electron‐withdrawing groups located at the para or meta carbons of polyfluorophenylboronates may promote this reaction.
Figure 2.
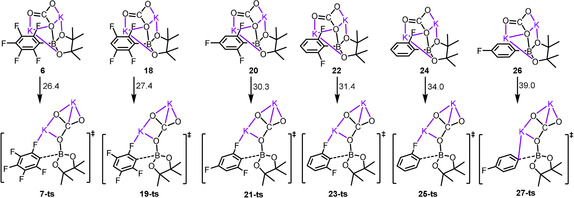
Free energies of activation of the cleavage of Bpin and ArF step calculated at the M06/(6–311++G(d, p), SMD)//B3LYP/(6–31+G(d)) level of theory. Relative free energies (ΔG) are given in kcal mol−1.
To ascertain the role of the K+ cation in these reactions, part of the free energy profile without the cation was also calculated at the same level of theory, and the results are given in Figure 3. Compared with the energy profile in Figure 1, in the absence of K+, the process of the methanolate oxygen anion 33 attack at the Lewis‐acidic boron in 30 becomes improbable, with an activation barrier of 41.4 kcal mol−1, although the initial cleavage of Bpin and pentafluorophenyl (ArF) step has a lower free energy of activation. Upon addition of 18‐crown‐6 to the reaction, the yields drop dramatically. As a counterion, K+ clearly regulates the nucleophilicity of CO3 2−, and promotes the reactivity by interaction with oxygen or fluorine atoms. Our DFT calculations indicate that both the ortho‐F substituents on the polyfluorophenylboronates and the counterion K+ are essential for the 1,2‐addition of polyfluorophenylboronates to aryl aldehydes.
Figure 3.
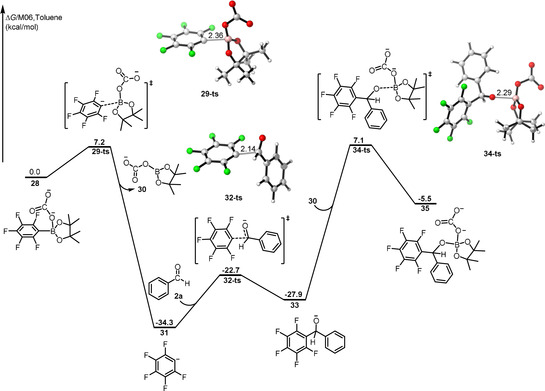
Free energy profile of 1,2‐addition of polyfluorophenylboronates with aryl aldehydes in the absence of K+ calculated by the M06/(6–311++G(d, p), SMD)//B3LYP/(6–31+G(d)) level of theory. Relative free energies (ΔG) are given in kcal mol−1, bond lengths are given in Å.
The structures of 3 f, 3 l, 3 m, 3 n, and 4 d were unambiguously confirmed by single crystal X‐ray diffraction. While the molecular structures are chiral (Figure 4), all the compounds represent racemic mixtures. Due to the presence of OH groups, the arrangement of the molecules in the crystal structures of all compounds is primarily determined by O−H⋅⋅⋅O or O−H⋅⋅⋅N hydrogen bonding (Supporting Information, Table S2). The presence of π⋅⋅⋅π stacking interactions between pentafluorophenyl and bromophenyl or naphthyl moieties (3 f and 3 m), respectively, is also observed in these examples (Figure 5, Table S3). Such an attractive interaction between arenes and perfluorinated arenes results from the different electronegativities of the hydrogen and fluorine atoms with respect to the carbon atoms of the aromatic rings and, hence, from opposite multipole moments of the aromatic groups. It is called the arene‐perfluoroarene interaction and can be applied as a supramolecular synthon in crystal engineering.[30] This was previously confirmed by Marder and co‐workers, who have shown that this type of interaction leads to the formation of highly ordered π‐stacks of alternating arene and perfluoroarene molecules in co‐crystals of arenes and perfluoroarenes.[30d, 31]
Figure 4.
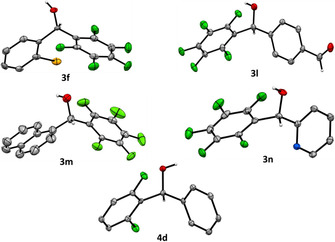
Molecular structures of compounds 3 f, 3 l, 3 m, 3 n and 4 d in the solid state at 100 K. Atomic displacement ellipsoids are drawn with 50 % probability. Only selected hydrogen atoms are shown for clarity. C gray, O red, N blue, Br orange, F green, H white.[36]
Figure 5.
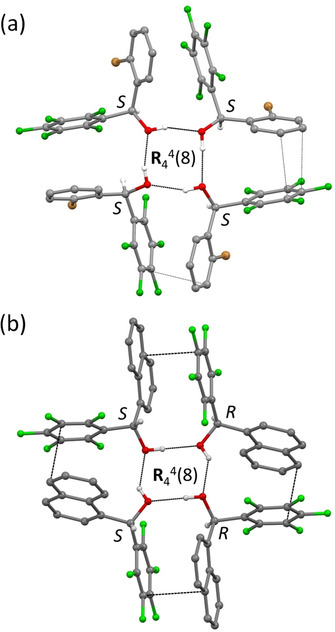
Compounds a) 3 f and b) 3 m self‐assemble to form tetramers via O−H⋅⋅⋅O hydrogen bonding and the corresponding graph set notation is R4 4(8).[31] π⋅⋅⋅π Stacking interactions between the bromophenyl or naphthyl and pentafluorophenyl groups, respectively, within the tetrameric unit are indicated by close C⋅⋅⋅C contacts (dashed lines). a) Each of the four symmetry‐independent tetramers of 3 f consists of molecules of the same chirality (RRRR or SSSS). Only one tetramer (SSSS) is shown here. b) In 3 m, the tetramer is centrosymmetric with (RRSS) chirality of the molecules.
In the crystal structures of compounds 3 f and 3 m, the combination of both O−H⋅⋅⋅O hydrogen bonding and arene‐perfluoroarene interaction leads to the intriguing formation of [O−H⋅⋅⋅]4 hydrogen‐bonded cyclic tetramers with graph set R 4 4(8) (Figure 5, Table S2).[32] The molecules of the tetramer interact via arene‐perfluoroarene π⋅⋅⋅π stacking between the bromophenyl or naphthyl and pentafluorophenyl moieties on the outside of the cyclic [O−H⋅⋅⋅]4 ring. The interplanar separations (3.281(7)–3.687(14) Å) are typical for π⋅⋅⋅π stacking interactions[30, 31] and the angles between the interacting planes are 4.96(19)–16.8(3)° (Table S3). In the higher symmetry compound 3 m (space group P21/c with Z′=2, where Z′ denotes the number of molecules in the asymmetric unit), arene‐perfluoroarene interactions are also present between the tetramers, in addition to C−H⋅⋅⋅π, C−H⋅⋅⋅F, and F⋅⋅⋅F interactions (Figure S6). Each tetramer of 3 m is centrosymmetric and, hence, contains molecules of opposite chirality (RRSS), leading to a racemic mixture (Figure 5 b). Tetramers are arranged in sheets parallel to the ‐plane (Figure S6). In contrast, compound 3 f crystallizes in the non‐centrosymmetric space group P1. There are 16 symmetry‐independent molecules in the asymmetric unit (Z′=16) of 3 f, which build up four symmetry‐independent hydrogen‐bonded cyclic tetramers (Figure S1). Each tetramer is constituted by molecules of the same chirality (RRRR or SSSS) (Figure 5 a). Thus, the chirality of the four tetramers in the asymmetric unit, i.e., (RRRR)(SSSS)(RRRR)(SSSS), leads to a racemic mixture, as shown in Figures 5 a, S1 and S3. Tetramers of mixed chirality are arranged in sheets parallel to the ‐plane with bromine atoms all pointing up or down within the sheet (Figures S2 and S3). Parallel sheets face each other either with the bromine atoms or without. In fact, crystals of 3 f represent one of the rare class of crystals for which Z′>1.[33, 34] While searching for a structure of higher symmetry, the cell parameters of 3 f were also determined at 200 K. As this resulted in a similar triclinic unit‐cell metric as was observed at 100 K, the occurrence of a phase transition at temperatures between 100 K and 200 K is unlikely.
Contrary to 3 f and 3 m, the dominance of hydrogen bonding and absence of arene‐perfluoroarene interactions in compounds 3 l (space group ), 3 n and 4 d (both space group C2/c) resulted in the formation of one‐dimensional hydrogen‐bonded chains (Figure 6). In 3 l and 3 n, the intermolecular O−H⋅⋅⋅O and O−H⋅⋅⋅N hydrogen bonding interaction takes place between the alcohol (O−H, donor) and the carboxaldehyde (O, acceptor) and pyridyl (N, acceptor) groups, respectively, the latter having a stronger hydrogen bond acceptor ability compared to the alcohol group (Table S2). Depending on the position of the acceptor atom in the molecule, hydrogen‐bonded chains are straight (3 l, Figure 6 a) or zig‐zag‐like (3 n, Figure 6 b). In 3 l, each one‐dimensional chain contains molecules of one particular chirality (either R or S), and chains of opposite chirality exhibit extensive π‐stacking interaction between the phenyl groups. In this way, double‐stranded linear chains projecting the C6F5 groups on both sides are formed, as shown in Figure 6 a. The C6F5 groups from neighboring strands undergo interdigitation and exhibit partial offset π⋅⋅⋅π interactions between fluorinated moieties and C−F⋅⋅⋅π interactions between phenyl and pentafluorophenyl groups (Figures S4 and S5, Table S3). In 3 n, one‐dimensional zig‐zag chains are formed by molecules of alternating chirality (RSRS…) (Figure 6 b). The pyridyl rings lie coplanar and the pentafluorophenyl groups interdigitate via partial offset π⋅⋅⋅π interactions to form a parallel ribbon‐like arrangement (Figure S7, Table S3). This structure exhibits a bilayer architecture as there are alternating hydrophobic and hydrophilic regions (Figures S7 and S8).[35] In 4 d, corrugated one‐dimensional chains are observed by the intermolecular O−H⋅⋅⋅O−H⋅⋅⋅hydrogen bonding interactions between the alcohol groups (Table S2), and molecules constituted of alternating pairs of same chirality (RRSSRRSS… as shown in Figure 6 c and Figure S9). Other intermolecular interactions observed in 4 d include C−H⋅⋅⋅F, C−H⋅⋅⋅π, and very weak, strongly offset π⋅⋅⋅π interactions (Table S3).
Figure 6.
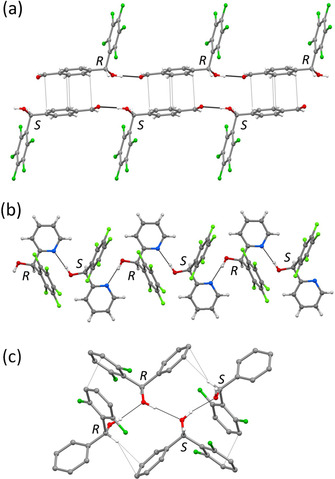
One‐dimensional hydrogen‐bonded chains are present in a) 3 l (O−H⋅⋅⋅O), b) 3 n (O−H⋅⋅⋅N), and c) 4 d (O−H⋅⋅⋅O). a) In 3 l, chains containing molecules of opposite chirality stack parallel via π⋅⋅⋅π interaction between the phenylcarboxaldehyde groups. b) A zig‐zag chain constituted by molecules of alternative chirality (RSRS…) is shown for compound 3 n. c) Compound 4 d exhibits corrugated chains with (RRSS…) chirality of the molecules. Additional weak interactions (C−H⋅⋅⋅π and partial π⋅⋅⋅π stacking) are shown.
Conclusion
We have demonstrated here the simple conditions for the 1,2‐addition of aldehydes and ketones with polyfluorophenylboronate compounds. This strategy has the following advantages: 1) transition metal‐free catalyst system; 2) a variety of aromatic and aliphatic aldehydes were found to be suitable substrates for this reaction using pentafluorophenyl‐Bpin in moderate to excellent yields; and 3) sterically hindered ketones also worked well to furnish the corresponding products. This method also introduces the use of polyfluorophenyl‐Bpin compounds instead of Grignard reagents for polyfluorophenylation of arylaldehyde and ketone substrates. Further studies of the synthesis and applications of polyfluorophenyl boronates are underway in our laboratory and will be reported in due course.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Z.L. and Y.M.T. are grateful to the China Scholarship Council for providing Ph.D. scholarships. X.L. is grateful to the China Scholarship Council for providing a sabbatical leave fellowship. G.K.K. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. Y.P.B. thanks the Universitas Padjadjaran for an Academic Leadership Grant (ALG). T.B.M. and U.R. thank the Julius‐Maximilians‐Universität Würzburg and the Deutsche Forschungsgemeinschaft (DFG) for support. S.A.W. acknowledges support from the Natural Science and Engineering Research Council of Canada. We thank AllyChem Co. Ltd. for a generous gift of B2pin2. Open access funding enabled and organized by Projekt DEAL.
Z. Liu, G. K. Kole, Y. P. Budiman, Y.-M. Tian, A. Friedrich, X. Luo, S. A. Westcott, U. Radius, T. B. Marder, Angew. Chem. Int. Ed. 2021, 60, 16529.
Contributor Information
Prof. Dr. Xiaoling Luo, Email: xiaolingluo@cqnu.edu.cn.
Prof. Dr. Udo Radius, Email: u.radius@uni-wuerzburg.de.
Prof. Dr. Todd B. Marder, Email: todd.marder@uni-wuerzburg.de.
References
- 1.
- 1a.Bolm C., Hildebrand J. P., Muniz K., Hermanns N., Angew. Chem. Int. Ed. 2001, 40, 3284–3308; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 3382–3407; [Google Scholar]
- 1b.Pu L., Yu H. B., Chem. Rev. 2001, 101, 757–824; [DOI] [PubMed] [Google Scholar]
- 1c.Fagnou K., Lautens M., Chem. Rev. 2003, 103, 169–196; [DOI] [PubMed] [Google Scholar]
- 1d.Hayashi T., Yamasaki K., Chem. Rev. 2003, 103, 2829–2844; [DOI] [PubMed] [Google Scholar]
- 1e.“9.04-Zinc and Cadmium”: Knochel P., Perrone S., Grenouillat N. in Comprehensive Organometallic Chemistry III, Vol. 9 (Eds.: Crabtree R. H., Mingos D. M. P., Knochel P.), Elsevier, Oxford, 2007, pp. 81–143. [Google Scholar]
- 2.
- 2a.Boudier A., Bromm L. O., Lotz M., Knochel P., Angew. Chem. Int. Ed. 2000, 39, 4414–4435; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 4584–4606; [Google Scholar]
- 2b.“9.03-Magnesium”: Knochel P., Gavryushin A., Krasovskiy A., Leuser H. in Comprehensive Organometallic Chemistry III, Vol. 9 (Eds.: Crabtree R. H., Mingos D. M. P., Knochel P.), Elsevier, Oxford, 2007, pp. 31–79; [Google Scholar]
- 2c.Muramatsu Y., Harada T., Chem. Eur. J. 2008, 14, 10560–10563. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a.Wu P.-Y., Wu H.-L., Uang B.-J., J. Org. Chem. 2006, 71, 833–835; [DOI] [PubMed] [Google Scholar]
- 3b.Shannon J., Bernier D., Rawson D., Woodward S., Chem. Commun. 2007, 3945–3947; [DOI] [PubMed] [Google Scholar]
- 3c.Lu G., Kwong F. Y., Ruan J. W., Li Y. M., Chan A. S. C., Chem. Eur. J. 2006, 12, 4115–4120; [DOI] [PubMed] [Google Scholar]
- 3d.Schmidt F., Stemmler R. T., Rudolph J., Bolm C., Chem. Soc. Rev. 2006, 35, 454–470; [DOI] [PubMed] [Google Scholar]
- 3e.Soai K., Niwa S., Chem. Rev. 1992, 92, 833–856. [Google Scholar]
- 4.“11.01-Lithium”: Gray M., Tinkl M., Snieckus V. in Comprehensive Organometallic Chemistry II, Vol. 11 (Eds.: Abel E. W., Stone F. G. A., Wilkinson G., McKillop A.), Elsevier, Oxford, 1995, pp. 1–92. [Google Scholar]
- 5.
- 5a.Hanamoto T., Yamada K., J. Org. Chem. 2009, 74, 7559–7561; [DOI] [PubMed] [Google Scholar]
- 5b.Oi S., Moro M., Inoue Y., Organometallics 2001, 20, 1036–1037; [Google Scholar]
- 5c.Fujii T., Koike T., Mori A., Osakada K., Synlett 2002, 298–300; [Google Scholar]
- 5d.Lerebours R., Wolf C., J. Am. Chem. Soc. 2006, 128, 13052–13053. [DOI] [PubMed] [Google Scholar]
- 6.Oi S., Moro M., Inoue Y., Chem. Commun. 1997, 1621–1622. [Google Scholar]
- 7.
- 7a.Imamoto T., Kusumoto T., Yokoyama M., J. Chem. Soc. Chem. Commun. 1982, 1042–1044; [Google Scholar]
- 7b.Liu H.-J., Shia K.-S., Shang X., Zhu B.-Y., Tetrahedron 1999, 55, 3803–3830; [Google Scholar]
- 7c.Bartoli G., Bosco M., Di Martino E., Marcantoni E., Sambri L., Eur. J. Org. Chem. 2001, 2901–2909; [Google Scholar]
- 7d.Bartoli G., Marcantoni E., Marcolini M., Sambri L., Chem. Rev. 2010, 110, 6104–6143; [DOI] [PubMed] [Google Scholar]
- 7e.Music A., Didier D., Synlett 2019, 30, 1843–1849; [Google Scholar]
- 7f.Music A., Hoarau C., Hilgert N., Zischka F., Didier D., Angew. Chem. Int. Ed. 2019, 58, 1188–1192; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1200–1204. [Google Scholar]
- 8.
- 8a.Yoshida K., Hayashi T., Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine (Ed.: Hall D. G.), Wiley-VCH, Weinheim, 2005; [Google Scholar]
- 8b.Suzuki A., Acc. Chem. Res. 1982, 15, 178–184; [Google Scholar]
- 8c.Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457–2483; For potassium organotrifluoroborates, see: [Google Scholar]
- 8d.Darses S., Genet J.-P., Chem. Rev. 2008, 108, 288–325; [DOI] [PubMed] [Google Scholar]
- 8e.Darses S., Genet J.-P., Eur. J. Org. Chem. 2003, 4313–4327; [Google Scholar]
- 8f.Pucheault M., Darses S., Genet J.-P., Chem. Commun. 2005, 4714–4716. [DOI] [PubMed] [Google Scholar]
- 9.Sakai M., Ueda M., Miyaura N., Angew. Chem. Int. Ed. 1998, 37, 3279–3281; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 3475–3477. [Google Scholar]
- 10.For selected references for Rh, see:
- 10a.Tajuddin H., Shukla L., Maxwell A. C., Marder T. B., Steel P. G., Org. Lett. 2010, 12, 5700–5703; [DOI] [PubMed] [Google Scholar]
- 10b.Focken T., Rudolph J., Bolm C., Synthesis 2005, 429–436; [Google Scholar]
- 10c.Ueda M., Miyaura N., J. Org. Chem. 2000, 65, 4450–4452; [DOI] [PubMed] [Google Scholar]
- 10d.Huang R., Shaughnessy K. H., Chem. Commun. 2005, 4484–4486; [DOI] [PubMed] [Google Scholar]
- 10e.Batey R. A., Thadani A. N., Smil D. V., Org. Lett. 1999, 1, 1683–1686; [Google Scholar]
- 10f.Gois P. M. P., Trindade A. F., Veiros L. F., André V., Duarte M. T., Afonso C. A. M., Caddick S., Cloke F. G. N., Angew. Chem. Int. Ed. 2007, 46, 5750–5753; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5852–5855; [Google Scholar]
- 10g.Marques C. S., Peixoto D., Burke A. J., RSC Adv. 2015, 5, 20108–20114; [Google Scholar]
- 10h.Jana R., Tunge J. A., J. Org. Chem. 2011, 76, 8376–8385; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10i.Morikawa S., Michigami K., Amii H., Org. Lett. 2010, 12, 2520–2523; [DOI] [PubMed] [Google Scholar]
- 10j.Trindade A. F., Andre V., Duarte M. T., Veiros L. F., Gois P. M. P., Afonso C. A. M., Tetrahedron 2010, 66, 8494–8502; [Google Scholar]
- 10k.White J. R., Price G. J., Plucinski P. K., Frost C. G., Tetrahedron Lett. 2009, 50, 7365–7368; [Google Scholar]
- 10l.Kuang Y., Wang Y., Eur. J. Org. Chem. 2014, 1163–1166. [Google Scholar]
- 11.For selected references for Pd, see:
- 11a.Yamamoto T., Ohta T., Ito Y., Org. Lett. 2005, 7, 4153–4155; [DOI] [PubMed] [Google Scholar]
- 11b.Lin S., Lu X., J. Org. Chem. 2007, 72, 9757–9760; [DOI] [PubMed] [Google Scholar]
- 11c.Kuriyama M., Shimazawa R., Shirai R., J. Org. Chem. 2008, 73, 1597–1600; [DOI] [PubMed] [Google Scholar]
- 11d.Zhao H., Cheng M., Zhang T., Cai M., J. Organomet. Chem. 2015, 777, 50–56; [Google Scholar]
- 11e.Yamamoto T., Furusawa T., Zhumagazin A., Yamakawa T., Oe Y., Ohta T., Tetrahedron 2015, 71, 19–26; [Google Scholar]
- 11f.Yamamoto T., Zhumagazin A., Furusawa T., Adv. Synth. Catal. 2014, 356, 3525–3529. [Google Scholar]
- 12.Liao Y. X., Xing C. H., He P., Hu Q. S., Org. Lett. 2008, 10, 2509–2512. [DOI] [PubMed] [Google Scholar]
- 13.For selected references for Ni, see:
- 13a.Takahashi G., Shirakawa E., Tsuchimoto T., Kawakami Y., Chem. Commun. 2005, 1459–1461; [DOI] [PubMed] [Google Scholar]
- 13b.Arao T., Kondo K., Aoyama T., Tetrahedron Lett. 2007, 48, 4115–4117; [Google Scholar]
- 13c.Bouffard J., Itami K., Org. Lett. 2009, 11, 4410–4413. [DOI] [PubMed] [Google Scholar]
- 14.For selected references for Cu, see:
- 14a.Zheng H., Zhang Q., Chen J., Liu M., Cheng S., Ding J., Wu H., Su W., J. Org. Chem. 2009, 74, 943–945; [DOI] [PubMed] [Google Scholar]
- 14b.Shintani R., Takatsu K., Hayashi T., Chem. Commun. 2010, 46, 6822–6824; [DOI] [PubMed] [Google Scholar]
- 14c.Zheng H., Ding J., Chen J., Liu M., Gao W., Wu H., Synlett 2011, 1626–1630. [Google Scholar]
- 15.Zou T., Pi S. S., Li J. H., Org. Lett. 2009, 11, 453–456. [DOI] [PubMed] [Google Scholar]
- 16.Karthikeyan J., Jeganmohan M., Cheng C. H., Chem. Eur. J. 2010, 16, 8989–8992. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a.Yamamoto Y., Kurihara K., Miyaura N., Angew. Chem. Int. Ed. 2009, 48, 4414–4416; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4478–4480; [Google Scholar]
- 17b.Li H., Xu Y., Shi E., Wei W., Suo X., Wan X., Chem. Commun. 2011, 47, 7880–7882. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a.Chen S. T., Fang J. M., J. Org. Chem. 1997, 62, 4349–4357; [DOI] [PubMed] [Google Scholar]
- 18b.Vu M. D., Das M., Liu X. W., ACS Catal. 2019, 9, 9009–9014; [Google Scholar]
- 18c.Liu Y.-L., Lin X.-Y., Adv. Synth. Catal. 2019, 361, 876–918. [Google Scholar]
- 19.Ishihara K., Hatano M., Synthesis 2008, 11, 1647–1675. [Google Scholar]
- 20.
- 20a.McDaniel R., Thamchaipenet A., Gustafsson C., Fu H., Betlach M., Betlach M., Ashley G., Proc. Natl. Acad. Sci. USA 1999, 96, 1846–1851; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b.Cuquerella M. C., Lhiaubet-Vallet V., Cadet J., Miranda M. A., Acc. Chem. Res. 2012, 45, 1558–1570; [DOI] [PubMed] [Google Scholar]
- 20c.Kamat P. V., Chem. Rev. 1993, 93, 267–300; [Google Scholar]
- 20d.Gooßen L. J., Rudolphi F., Oppel C., Rodríguez N., Angew. Chem. Int. Ed. 2008, 47, 3043–3045; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3085–3088; [Google Scholar]
- 20e.Gong W.-J., Liu D.-X., Li F.-L., Gao J., Li H.-X., Lang J.-P., Tetrahedron 2015, 71, 1269–1275; [Google Scholar]
- 20f.Wang J., Cary B. P., Beyer P. D., Gellman S. H., Weix D. J., Angew. Chem. Int. Ed. 2019, 58, 12081–12085; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 12209–12213; [Google Scholar]
- 20g.Lutter F. H., Grokenberger L., Hofmayer M. S., Knochel P., Chem. Sci. 2019, 10, 8241–8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a.Zhang X., MacMillan D. W. C., J. Am. Chem. Soc. 2017, 139, 11353–11356; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b.Swyka R. A., Zhang W., Richardson J., Ruble J. C., Krische M. J., J. Am. Chem. Soc. 2019, 141, 1828–1832; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c.Schwarz J. L., Schäfers F., Tlahuext-Aca A., Lückemeier L., Glorius F., J. Am. Chem. Soc. 2018, 140, 12705–12709; [DOI] [PubMed] [Google Scholar]
- 21d.Verheyen T., van Turnhout L., Vandavasi J. K., Isbrandt E. S., De Borggraeve W. M., Newman S. G., J. Am. Chem. Soc. 2019, 141, 6869–6874; [DOI] [PubMed] [Google Scholar]
- 21e.Swyka R. A., Shuler W. G., Spinello B. J., Zhang W., Lan C., Krische M. J., J. Am. Chem. Soc. 2019, 141, 6864–6868; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21f.Fan P., Lan Y., Zhang C., Wang C., J. Am. Chem. Soc. 2020, 142, 2180–2186; [DOI] [PubMed] [Google Scholar]
- 21g.Lei C., Zhu D., Tangcueco V. I. T., Zhou J. S., Org. Lett. 2019, 21, 5817–5822; [DOI] [PubMed] [Google Scholar]
- 21h.Wakaki T., Togo T., Yoshidome D., Kuninobu Y., Kanai M., ACS Catal. 2018, 8, 3123–3128. [Google Scholar]
- 22.Wang L., Wang T., Cheng G., Li X., Wei J., Guo B., Zheng C., Chen G., Ran C., Zheng C., ACS Catal. 2020, 10, 7543–7551. [Google Scholar]
- 23.
- 23a.Ahrens T., Kohlmann J., Ahrens M., Braun T., Chem. Rev. 2015, 115, 931–972; [DOI] [PubMed] [Google Scholar]
- 23b.Leroux F., Jeschke P., Schlosser M., Chem. Rev. 2005, 105, 827–856; [DOI] [PubMed] [Google Scholar]
- 23c.Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]
- 23d.Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320–330; [DOI] [PubMed] [Google Scholar]
- 23e.Sakamoto Y., Suzuki T., Kobayashi M., Gao Y., Fukai Y., Inoue Y., Sato F., Tokito S., J. Am. Chem. Soc. 2004, 126, 8138–8140; [DOI] [PubMed] [Google Scholar]
- 23f.Sakamoto Y., Suzuki T., Miura A., Fujikawa H., Tokito S., Taga Y., J. Am. Chem. Soc. 2000, 122, 1832–1833; [Google Scholar]
- 23g.Budiman Y. P., Westcott S. A., Radius U., Marder T. B., Adv. Synth. Catal. 2021, 363, 2224–2255. [Google Scholar]
- 24.Abarbri M., Dehmel F., Knochel P., Tetrahedron Lett. 1999, 40, 7449–7453. [Google Scholar]
- 25.Brogan S., Carter N. B., Lam H. W., Synlett 2010, 615–617. [Google Scholar]
- 26.Du G. F., Xing F., Gu C. Z., Dai B., He L., RSC Adv. 2015, 5, 35513–35517. [Google Scholar]
- 27.Jia X. J., Wang J., Ding X., Yang J., Huang Z. Y., J. Org. Chem. 2015, 80, 10874–10882. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a.Zhou J., Kuntze-Fechner M. W., Bertermann R., Paul U. S. D., Berthel J. H. J., Friedrich A., Du Z., Marder T. B., Radius U., J. Am. Chem. Soc. 2016, 138, 5250–5253; [DOI] [PubMed] [Google Scholar]
- 28b.Tian Y.-M., Guo X.-N., Kuntze-Fechner M. W., Krummenacher I., Braunschweig H., Radius U., Steffen A., Marder T. B., J. Am. Chem. Soc. 2018, 140, 17612–17623; [DOI] [PubMed] [Google Scholar]
- 28c.Budiman Y. P., Lorenzen S., Liu Z., Radius U., Marder T. B., Chem. Eur. J. 2021, 27, 3869–3874; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28d.Budiman Y. P., Friedrich A., Radius U., Marder T. B., ChemCatChem 2019, 11, 5387–5396; [Google Scholar]
- 28e.Budiman Y. P., Jayaraman A., Friedrich A., Kerner F., Radius U., Marder T. B., J. Am. Chem. Soc. 2020, 142, 6036–6050; [DOI] [PubMed] [Google Scholar]
- 28f.Liu Z., Budiman Y. P., Tian Y.-M., Friedrich A., Huang M., Westcott S. A., Radius U., Marder T. B., Chem. Eur. J. 2020, 26, 17267–17274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a.Pietsch S., Neeve E. C., Apperley D. C., Bertermann R., Mo F. Y., Qiu D., Cheung M. S., Dang L., Wang J. B., Radius U., Lin Z. Y., Kleeberg C., Marder T. B., Chem. Eur. J. 2015, 21, 7082–7098; [DOI] [PubMed] [Google Scholar]
- 29b.Eichhorn A. F., Fuchs S., Flock M., Marder T. B., Radius U., Angew. Chem. Int. Ed. 2017, 56, 10209–10213; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10343–10347. [Google Scholar]
- 30.
- 30a.Dahl T., Acta Chem. Scand. 1988, 42, 1–7; [Google Scholar]
- 30b.Williams J. H., Acc. Chem. Res. 1993, 26, 593–598; [Google Scholar]
- 30c.Coates G. W., Dunn A. R., Henling L. M., Dougherty D. A., Grubbs R. H., Angew. Chem. Int. Ed. Engl. 1997, 36, 248–251; [Google Scholar]; Angew. Chem. 1997, 109, 290–293; [Google Scholar]
- 30d.Collings J. C., Roscoe K. P., Thomas R. Ll., Batsanov A. S., Stimson L. M., Howard J. A. K., Marder T. B., New J. Chem. 2001, 25, 1410–1417. [Google Scholar]
- 31.
- 31a.Dai C., Nguyen P., Marder T. B., Scott A. J., Clegg W., Viney C., Chem. Commun. 1999, 2493–2494; [Google Scholar]
- 31b.Collings J. C., Batsanov A. S., Howard J. A. K., Marder T. B., Cryst. Eng. 2002, 5, 37–46; [Google Scholar]
- 31c.Collings J. C., Roscoe K. P., Robins E. G., Batsanov A. S., Stimson L. M., Howard J. A. K., Clark S. J., Marder T. B., New J. Chem. 2002, 26, 1740–1746; [Google Scholar]
- 31d.Smith C. E., Smith P. S., Thomas R. Ll., Robins E. G., Collings J. C., Dai C., Scott A. J., Borwick S., Batsanov A. S., Watt S. W., Clark S. J., Viney C., Howard J. A. K., Clegg W., Marder T. B., J. Mater. Chem. 2004, 14, 413–420; [Google Scholar]
- 31e.Collings J. C., Smith P. S., Yufit D. S., Batsanov A. S., Howard J. A. K., Marder T. B., CrystEngComm 2004, 6, 25–28; [Google Scholar]
- 31f.Watt S. W., Dai C., Scott A. J., Burke J. M., Thomas R. Ll., Collings J. C., Viney C., Clegg W., Marder T. B., Angew. Chem. Int. Ed. 2004, 43, 3061–3063; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3123–3125; [Google Scholar]
- 31g.Friedrich A., Collings I. E., Dziubek K. F., Fanetti S., Radacki K., Ruiz-Fuertes J., Pellicer-Porres J., Hanfland M., Sieh D., Bini R., Clark S. J., Marder T. B., J. Am. Chem. Soc. 2020, 142, 18907–18923. [DOI] [PubMed] [Google Scholar]
- 32.Etter M. C., Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar]
- 33.
- 33a.Steed J. W., CrystEngComm 2003, 5, 169–179; [Google Scholar]
- 33b.Anderson K. M., Goeta A. E., Hancock K. S. B., Steed J. W., Chem. Commun. 2006, 2138–2140; [DOI] [PubMed] [Google Scholar]
- 33c.Desiraju G. R., CrystEngComm 2007, 9, 91–92. [Google Scholar]
- 34.Brock C. P., Duncan L. L., Chem. Mater. 1994, 6, 1307–1312. [Google Scholar]
- 35.Moulton B., Zaworotko M. J., Chem. Rev. 2001, 101, 1629–1658. [DOI] [PubMed] [Google Scholar]
- 36.Deposition Numbers 2045652 (3f), 2045653 (3l), 2045654 (3m), 2045655 (3n), and 2045656 (4d) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary