

Abstract
Understanding the role of common polymorphisms in modulating the clinical phenotype when they co‐occur with a disease‐causing lesion is of critical importance in medical genetics. We explored the impact of apparently neutral common polymorphisms, using the gene encoding the urea cycle enzyme, ornithine transcarbamylase (OTC), as a model system. Distinct combinations of genetic backgrounds embracing two missense polymorphisms were created in cis with the pathogenic p.Arg40His replacement. In vitro enzymatic assays revealed that the polymorphic variants were able to modulate OTC activity both in the presence or absence of the pathogenic lesion. First, we found that the combination of the minor alleles of polymorphisms p.Lys46Arg and p.Gln270Arg significantly enhanced enzymatic activity in the wild‐type protein. Second, enzymatic assays revealed that the minor allele of the p.Gln270Arg polymorphism was capable of ameliorating OTC activity when combined in cis with the pathogenic p.Arg40His replacement. Structural analysis predicted that the minor allele of the p.Gln270Arg polymorphism would serve to stabilize the OTC wild‐type protein, thereby corroborating the results of the experimental assays. Our findings demonstrate the potential importance of cis‐interactions between common polymorphic variants and pathogenic missense mutations and illustrate how standing genetic variation can modulate protein function.
Keywords: enzymatic activity, genetic modifiers, OTC gene, pathogenic replacements, polymorphic variants, structural modeling
1. INTRODUCTION
The establishment of clear and unambiguous genotype‐phenotype correlations can be a challenging task when an array of clinical phenotypic outcomes are associated with different lesions in the same gene or even with the same pathogenic replacement. Such clinical/phenotypic diversity is a strong indication that other factors, whether genetic (e.g., genetic modifiers) or environmental (e.g., diet), are playing a role in modulating the phenotype‐genotype correlation (Domingo et al., 2019; Hartman & Tullman‐Ercek, 2019; Nadeau, 2001). In this study, we sought to explore the impact of common polymorphic variants as intragenic genetic modifiers using ornithine transcarbamylase (OTC) (E.C. 2.1.3.3) as a model system.
The concept of genetic modifiers was first proposed 80 years ago by J.B.S. Haldane (Haldane, 1941). Genetic modifiers may be defined as genes/variants (either coding or noncoding) that are capable of modifying the clinical/phenotypic effect of a pathogenic mutation in a given gene (Nadeau, 2001; Rahit & Tarailo‐Graovac, 2020; Weatherall, 2001). When such variants are located within regulatory regions, they may alter gene regulation and expression, whereas when present within coding regions, they may affect protein structure, function, stability, turnover, and protein‐protein interactions (Zhang et al., 2012). Thus, in principle, genetic modifiers may serve both to ameliorate or aggravate the phenotypic consequences of a given pathogenic mutation (Lehner, 2011). Previous studies have identified genetic modifiers within the regulatory regions of key genes involved in cystic fibrosis, sickle cell disease, retinitis pigmentosa, and OTC deficiency (Cruz et al., 2014; Darrah et al., 2010; Hillian et al., 2008; Jang et al., 2018; Li et al., 2020; Luksan et al., 2010; Steinberg & Sebastiani, 2012) among others. Missense polymorphic variants have also been identified as genetic modifiers (Chen et al., 2001; Cooper et al., 2013; Dakal et al., 2017; Li et al., 2018; Silva et al., 2004; van Leeuwen et al., 2016), an example of which is p.His558Arg in the SCN5A gene (NM_000335.5:c.1673A>G [p.His558Arg]) which, in combination with pathogenic SCN5A lesions, serves to modulate the severity of the Brugada syndrome phenotype (Makielski et al., 2003; Matsumura et al., 2017; Niu et al., 2006; Poelzing et al., 2006).
Understanding the contribution that apparently neutral polymorphic variation can make in modulating the clinical phenotype represents a key step towards the realization of personalized genomic medicine. To explore this issue, we focussed on the X‐linked OTC gene which encodes a urea cycle enzyme conserved in all mammalian species. OTC is a homotrimeric enzyme of the urea cycle that catalyses the formation of citrulline and inorganic phosphate from carbamoyl phosphate and ornithine (Shi et al., 2000; Smith & Garg, 2017). Deficiency of the enzyme generally leads to hyperammonemia, seizures, coma, and in some cases, premature death (Yamaguchi et al., 2006). More than 400 disease‐causing mutations have been identified in the OTC gene, most of them missense replacements (Caldovic et al., 2015; Stenson et al., 2020). The majority of these lesions are private or found in very few families (Yamaguchi et al., 2006). One exception is the p.Arg40His replacement (NM_000531.5:c.119G>A [p.Arg40His]) which has been reported in a number of distinct families as a consequence of both recurrent and identical‐by‐descent mutation (Hidaka et al., 2020; Koya et al., 2019; Matsuda et al., 1996; Nishiyori et al., 1997; Zhou, Huang, Ma & Zhu, 2020). OTC activity measured from the necropsied liver of five deceased patients with a confirmed p.Arg40His mutation has been found to be between 1.3% and 12% of the wild‐type OTC level (Matsuda et al., 1996) whereas the mutant (p.Arg40His‐harboring) OTC enzyme expressed in vitro in COS‐1 cells exhibited 10.2% (Matsuda et al., 1996) to 28% (Nishiyori et al., 1997) of wild‐type activity. The p.Arg40His mutation has been associated with both the early onset and the late‐onset phenotype in both females and males (Hidaka et al., 2020; Koya et al., 2019; Zhou et al., 2020). In a few cases, the sudden appearance of metabolic distress in otherwise apparently healthy carriers of the p.Arg40His mutation has been suggested to be associated with specific acute dietary changes (Cavicchi et al., 2014; Hidaka et al., 2020; Koya et al., 2019; Pinner et al., 2010).
In addition to the numerous individually rare pathogenic lesions, OTC also harbors two common polymorphic variants: p.Lys46Arg (NM_000531.5:c.137A>G [p.Lys46Arg]), and p.Gln270Arg (NM_000531.5:c.809A>C [p.Gln270Arg]) whose putative functional significance in terms of their influence on clinical/phenotypic heterogeneity has until now remained unaddressed. To investigate whether these two missense polymorphisms might act as intragenic modifiers of the OTC gene in concert with the p.Arg40His deleterious mutation, we recreated in vitro all the possible genetic backgrounds in which specific allelic combinations of the polymorphic and deleterious variants could co‐exist, and evaluated the resulting OTC enzymatic activity in each case.
2. MATERIALS AND METHODS
2.1. OTC allelic and haplotypic population frequencies
Population frequencies of OTC polymorphic variants p.Lys46Arg (rs1800321) and p.Gln270Arg (rs1800328) were obtained from the gnomAD browser V2.2.1 (Karczewski et al., 2020). Haplotype analysis was conducted using data from the 1000 Genomes Project (1KGP) Phase 3 available from the Ensembl Genome Browser release 101 which included the following populations: African (African Caribbean in Barbados, African ancestry in the Southwest US, Esan in Nigeria, Gambian in Western Division, Luhya in Webuye, Mende in Sierra Leone, Yoruba in Ibadan); American (Colombian in Medellin, Mexican ancestry in Los Angeles, Peruvian in Lima, Puerto Rican in Puerto Rico); East Asian (Chinese Dai in Xishuangbabba, Han Chinese in Beijing, Southern Han Chinese, Japanese in Tokyo, Kinh in Ho Chi Minh City); European (Utah residents with Northern and Western European ancestry, Finnish in Finland, British in England and Scotland, Iberian populations in Spain, Tuscany in Italy) and South Asian (Bengali in Bangladesh, Gujarati Indian in Houston, Indian Telugu in the UK, Punjabi in Lahore, Sri Lankan Tamils in the UK).
2.2. Isolation of the human OTC gene
Human liver RNA was purchased from Ambion™ (Thermo Fisher Scientific). One microgram liver RNA was used for complementary DNA (cDNA) synthesis with iScript™ cDNA Synthesis kit (Bio‐Rad) according to the manufacturer's recommendations. The human OTC full coding sequence (1065 base pair) was isolated from the cDNA by polymerase chain reaction (PCR) and cloned into the pHTP0 vector (NZYtech). PCR was performed with HotStarTaq® Master Mix Kit (Qiagen) using tailed primers (forward 5ʹ‐TCAGCAAGGGCTGAGGACCAGGGGACTTTGATAAG‐3ʹ and reverse 5ʹ‐TCAGCGGAAGCTGAGGTTTCCCCATAAACCAACTCA‐3ʹ) for cloning into the pHTP0 vector. PCR conditions were as follows: initial denaturation at 95°C for 15 min; 35 cycles of denaturation at 94°C for 30 s; annealing at 58°C for 30 s; elongation at 72°C for 1 min 30 s; with a final single 20 min elongation step. PCR products were analyzed on a 1% agarose gel. Bands of the expected size were excised, purified with NYZtech Gelpure (NZYtech), and ligated to pHTP0. pHTP0 plasmids were transformed into NZY5α competent cells (NZYtech). Ampicillin‐resistant clones were selected and grown overnight for plasmid purification with NZYtech Miniprep Kit (NZYtech). Plasmid sequences were confirmed by in‐house Sanger sequencing.
2.3. Site‐directed mutagenesis and cloning into the expression vector
pHTP0 plasmids containing the OTC gene were used for site‐directed mutagenesis to generate the requisite genetic backgrounds (Figure 1a). For each background, a specific set of primers was used (Figure 1b) and site‐directed mutagenesis PCR was performed using the NZYMutagenesis Kit (NZYtech) according to the manufacturer's instructions. PCR products were treated with DpnI and transformed into NZYStar competent cells (NZYtech). Positive colonies were selected and grown overnight for plasmid purification and sequenced as described above. To transfer OTC constructs from pHTP0 to the expression vector, PCR was performed using primers containing in‐built restriction sites EcoRI (forward: 5ʹ‐ CACCGGAATTCATGCTGTTTAATCTGAGGATCCT‐3ʹ) and SalI (reverse 5ʹ‐ ACGCGTCGACTCAAAATTTAGGCTTCTGGAGCT‐3ʹ). PCR was performed with 10 ng input plasmid DNA, 10 µM each primer, and Phusion Flash High Fidelity PCR Mix (Thermo Fisher Scientific). PCR conditions were as follows: initial denaturation at 98°C for 10 s, 35 cycles of denaturation at 98°C for 1 s, annealing at 63°C for 5 s and elongation at 72°C for 1 min with a single final elongation step at 72°C for 20 s. PCR products of the expected size on 1% agarose gel bands were excised and purified as described above. Next, purified PCR products and pIRES2‐AcGFP1 Vector (TAKARA Bio), were treated with the corresponding restriction enzymes and ligated using T4 DNA ligase (Invitrogen, Thermo Fisher Scientific). All purified plasmid sequences were confirmed by Sanger sequencing.
Figure 1.
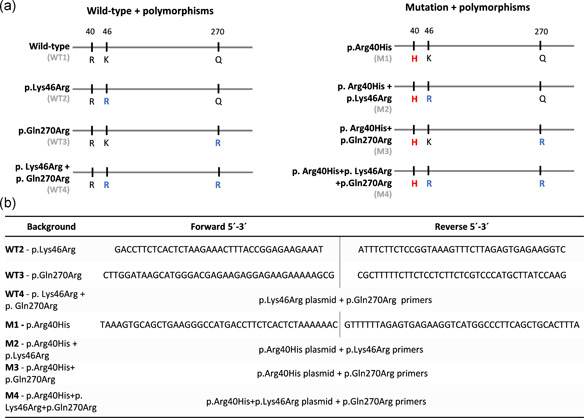
Genetic backgrounds and primer sets. (a) Schematic representation of the OTC genetic backgrounds analyzed in this study. Wild‐type backgrounds are termed WT1‐WT4, and the corresponding backgrounds containing the p.Arg40His mutation are termed M1‐M4 accordingly. Red indicates pathogenic amino acid replacement, blue indicates non‐pathogenic amino acid replacement. (b) Primer sets and strategies used to obtain each background sequence. OTC, ornithine transcarbamylase
2.4. Cell culture and expression of OTC protein
The expression of OTC protein was performed in HEK293T cells which lack endogenous OTC expression. HEK293T cells were maintained in Dulbecco's modified Eagle's medium high glucose, pyruvate, and supplemented with 10% fetal bovine serumand 5% Penicillin/Streptomycin. Cells were seeded at a density of 3 × 105 cells/well in 24‐well plates. After 24 h, cells were transfected using 1.5 µg selected OTC plasmid (pIRES‐AEGFP‐OTC) and Lipofectamine 2000 reagent (Invitrogen, Thermo Fisher Scientific) in Opti‐MEM reduced serum medium (Gibco, Thermo Fisher Scientific) according to the manufacturer's guidelines. 24 h after transfection, cells were inspected in the fluorescent cell imager ZOE™ (Bio‐RAD) to calculate transfection efficiency, by counting the number of cells expressing enhanced green fluorescent protein in relation to the total number of cells in the same field and no significant transfection efficiency differences were observed between backgrounds. Immediately after, cells were collected using passive lysis buffer 5X (Promega) supplemented with 10 µl protease inhibitor, and stored at −80°C. For each genetic background, three independent replicate assays were performed.
2.5. OTC activity assays
OTC activity assays were performed by adjusting the protocols described by Ceriotti, 1973; Pierson et al., 1977 and Suriano et al., 2007. Two chromogenic solutions were prepared:
Solution 1: antipyrine (4 g/L) + iron(III)sulfate (50 mg/L) in sulfuric acid 10% vol/vol
Solution 2: Brij®35 (0.3% wt/wt) + 2,3‐butanedione monoxime (5 g/L) in acetic acid 5% vol/vol
All reagents and kits used in these activity assays were purchased from Sigma‐Aldrich. Variable transfection efficiencies were standardized through total protein quantification of cell lysates using the Protein Quantification Kit‐Rapid. For each cell lysate sample, both basal and total citrulline levels were determined. All measurements were performed in 75 mM triethanolamine buffer pH 6.75 (the pH was adjusted with HCl), and incubations were performed in the dark. Total citrulline was determined by mixing 25 μl cell lysates containing 50 μg total protein, with 25 μl l‐ornithine hydrochloride 99% (7.5 mM) and 25 μl lithium carbamoylphosphate dibasic hydrate (75 mM). Basal citrulline levels were determined by mixing 25 μl cell lysates containing 50 μg total protein, with 50 μl TAE buffer. Citrulline standards were determined by mixing 25 μl citrulline solution (1 mM) with 50 μl TAE buffer. Samples were incubated for 30 min at 37°C; the enzymatic reaction was stopped by adding 1.5 ml Solution 1 and 1.5 ml Solution 2, and samples were further incubated for 20 min at 95°C. Next, 300 μl each sample was transferred in duplicate to a 96‐well plate and sample absorbance read at 460 nm in a BioTek Synergy™ Mx Microplate Reader. OTC activity in milliunits was calculated using the following formula:
Atot corresponds to the absorbance reading in samples where total citrulline was determined. Abas denotes the absorbance reading in samples where basal citrulline was determined. Astd refers to the absorbance reading in citrulline standards.
2.6. Statistical analysis
Statistical analyses were performed after checking data for normality (Shapiro–Wilk test), homogeneity of variances (Levene's test), and statistical outliers. The analysis of variance was performed by applying an analysis of variance test followed by multiple comparisons using either Tukey's test or the Dunn–Šidák correction. Significant differences were accepted for tests yielding p values <.05. The statistical software package used was GraphPad Prism 7.00 (San Diego).
2.7. Structural analysis
Comparative homology models of human OTC were constructed using the PDB 1C9Y human OTC crystal structure with 1.9 Å resolution (Shi et al., 2000) as a template. We created a total of eight homology models representing all possible genetic backgrounds, one for each missense variant haplotype identified in combination with the p.Arg40His mutation. Homology models were built using the SwissModel workspace (Waterhouse et al., 2018), and structural quality assessment of the models was performed by means of global model quality estimation and QMEAN scores (Benkert et al., 2010; Bertoni et al., 2017; Studer et al., 2019). Model visualization and structural analysis were performed using PyMOL™ molecular graphics system V.2.4.1 (Schrodinger, 2015). Structural stability analysis was run on the DUET server (Pires et al., 2014).
3. RESULTS
3.1. Haplotypic structure of p.Lys46Arg (rs1800321) and p.Gln270Arg (rs1800328) variants
Two polymorphic missense variants are known to occur in the human OTC gene (p.Lys46Arg and p.Gln270Arg) (Azevedo et al., 2002), for which the minor alleles attain frequencies of 0.19250 and 0.02912 (gnomAD v2.1.1), respectively. To assess the haplotype structure defined by these two variants in human populations, we included current data from the 1KGP (1000 Genomes Project Consortium et al., 2015), which presents information for a large number of populations. Out of the four possible haplotypes (Table 1), the most abundant was haplotype H1, defined by the Lys46‐Gln270 (wild‐type 1 [WT1]) combination, followed by H2 with the Arg46‐Gln270 (WT2) combination, H4 with both the alternative minor alleles Arg46‐Arg270 (WT4) and finally H3 with Lys46‐Arg270 (WT3).
Table 1.
Haplotype analysis of rs180321 and rs1800328 in various human populations
| Haplotype | rs1800321 p.Lys46Arg A>G | rs1800328 p.Gln270Arg A>G | All populations | African | American | East Asian | European | South Asian | Background |
|---|---|---|---|---|---|---|---|---|---|
| H1 | A | A | 0.851 | 0.614 | 0.840 | 0.997 | 0.721 | 0.962 | WT1 |
| H2 | G | A | 0.135 | 0.383 | 0.137 | 0.00131 | 0.225 | 0.0237 | WT2 |
| H3 | A | G | 0.00334 | ‐ | 0.00573 | ‐ | 0.0104 | 0.00696 | WT3 |
| H4 | G | G | 0.009 | ‐ | 0.0172 | 0.00131 | 0.0431 | 0.00557 | WT4 |
3.2. OTC activity assays
The amount of citrulline produced by cells transfected with plasmids encoding the full‐length wild‐type OTC protein (Lys46 + Gln270; WT1) was determined, as well as by cells transfected with plasmids encoding proteins harboring the p.Lys46Arg (WT2), p.Gln270Arg (WT3), and p.Lys46Arg + p.Gln270Arg (WT4) variants and negative controls (Mock) (Figure 2a). We also quantified the amount of citrulline produced in non‐transfected HEK293T cells, as a negative control. Cells transfected with Mock plasmids and non‐transfected cells were found to contain very low OTC activity, approximately 9.5 mU (3%) (Figure 2a,b), similar to what Suriano et al. observed in CHO cells expressing the Mock vector (Suriano et al., 2007). These control assays allowed us to demonstrate the virtual lack of endogenous citrulline in HEK293T cells but also to confirm that the pIRES‐AEGFP plasmid alone cannot drive citrulline production. The concentration of citrulline in cell lysates transfected with the plasmid encoding any of the wild‐type OTC configurations (WT1, WT2, WT3, and WT4) was significantly (p < .0001) higher in comparison to the negative controls, Mock and non‐transfected cells (Table S1).
Figure 2.
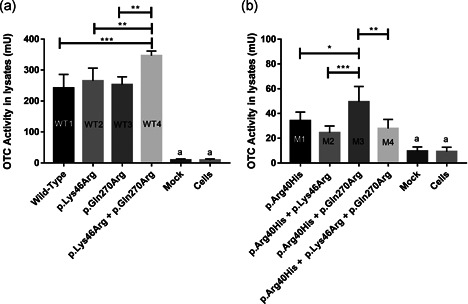
OTC enzymatic activity assays. (a) From left to right, OTC activity in cell lysates transfected with plasmids encoding wild‐type (WT) OTC, OTC harboring the p.Lys46Arg polymorphism, OTC carrying the p.Gln270Arg polymorphic variant, and OTC carrying both the p.Lys46Arg and p.Gln270Arg polymorphic variants. (b) From left to right, OTC activity in cell lysates transfected with plasmids encoding OTC harboring the p.Arg40His pathogenic mutation, OTC harboring the p.Arg40His mutation and the p.Lys46Arg polymorphic variant, OTC harboring the p.Arg40His mutation and the p.Gln270Arg polymorphic variant, OTC carrying the p.Arg40His mutation and both the p.Lys46Arg p.Gln270Arg polymorphisms. *p < .05, **p < .01, ***p < .0005. All tested conditions are significantly different from negative controls (a); see also Tables S1, S2, and S3 for detailed values. Mock: citrulline values (due to residual OTC activity) for cells transfected with an empty plasmid, Cells: citrulline values of non‐transfected cells. OTC, ornithine transcarbamylase
The enzymatic activity of WT1 OTC (239±52.9 mU) was similar to the wild‐type OTC enzymes harboring the polymorphic variants WT2 p.Lys46Arg (271 ± 48 mU) and WT3 p.Gln270Arg (253 ± 25.4 mU) singly. However, the OTC enzyme carrying the minor alleles of both polymorphic variants, WT4 (p.Lys46Arg + p.Gln270Arg), exhibited significantly higher activity (346 ± 15 mU) compared to the WT1 background (p = .002) and to the WT2 and WT3 backgrounds each of which carried only a single polymorphic variant (p < .005).
By contrast, significantly lower OTC activity was observed in those samples transfected with plasmids encoding OTC harboring the p.Arg40His mutation alone (M1) or combined with the polymorphic variants p.Lys46Arg and/or p.Gln270Arg (M2, M3, and M4) (Figure 2b) as compared with their wild‐type counterparts. The OTC protein carrying the p.Arg40His mutation yielded approximately 37±4.6 mU of OTC activity, corresponding to a reduction to about 15% of wild‐type activity, concordant with the results of previous studies (Matsuda et al., 1996; Nishiyori et al., 1997). Comparison of the enzymatic activities of wild‐type OTC protein (WT1) with those of OTC proteins harboring the p.Arg40His mutation (M1, M2, M3, and M4) showed a significant (p < .0001) reduction of enzymatic activity in all the latter (Table S2). OTC enzymes harboring the combinations p.Arg40His + p.Lys46Arg (M2), p.Arg40His + p.Gln270Arg (M3), and p.Arg40His + p.Lys46Arg + p.Gln270Arg (M4) exhibited lower OTC activity (26.8 ± 3.3, 51.6 ± 14, and 27.7 ± 8.9 mU, respectively) than the corresponding wild‐type enzymes. Although the OTC enzymes carrying the p.Arg40His mutation without either of the two polymorphic minor alleles showed low enzymatic activity, this activity was still significantly higher than the negative controls (p < .05) (Table S3). Pairwise comparison between the enzymatic activities of the OTC proteins harboring the p.Arg40His mutation alone or in combination with the minor alleles of the missense polymorphisms revealed that the M3 protein (p.Arg40His + p.Gln270Arg) displayed significantly increased enzymatic activity as compared to M1 (p.Arg40His), whereas M2 and M4 enzymes behaved similarly. These results suggest that the p.Gln270Arg variant may act as a genetic modifier of the p.Arg40His mutation in vitro.
3.3. Structural analysis of OTC
To explore the molecular mechanisms underlying the observed differences in enzymatic activity, we investigated the structural alterations in the OTC protein caused by the pathogenic mutation p.Arg40His and by the polymorphic variants p.Lys46Arg and p.Gln270Arg. We began by evaluating the three‐dimensional proximity and possible spatial interactions between the amino acid residue harboring the pathogenic mutation (amino acid residue 40) and the polymorphic missense variants (at amino acid residues 46 and 270), using 3D homology models constructed so as to recreate the eight possible protein backgrounds. Asall crystal structures of the human OTC protein available in the PDB database possess an arginine at position 270, we built the ancestral background (WT1) by changing the arginine to glutamine. A total of eight homology models were generated, corresponding to the same backgrounds that were tested in the enzymatic assays: that is, WT1, WT2, WT3, WT4, M1, M2, M3, and M4. Initial analysis showed that the three residues of interest, Arg40, Lys46, and Gln270, do not participate in any of the homotrimers' protein–protein interactions. We also measured the spatial distance between the three residues and found that Arg40 and Lys46 are located within 9.8 Å of each other (Site 1) whereas Gln270 is located on the opposite side of the monomer (Site 2) at distances of 20 and 30 Å from Lys46 and Arg40, respectively (Figure 3a).
Figure 3.
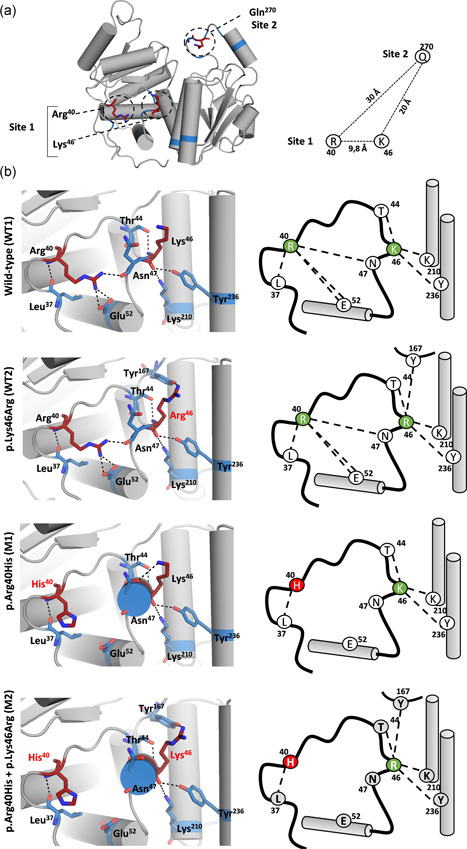
Structural analysis of OTC homology models. (a) Location of the three amino acid residues of interest (Arg40, Lys46, and Gln270) within the OTC monomer showing approximate distances between the target residues. (b) Detailed view of the hydrogen bond network alterations in Site 1 on various genetic backgrounds; each genetic background is indicated on the left. OTC, ornithine transcarbamylase
Structural analysis of Site 1 on the WT1 background revealed a network of hydrogen bonds that serve to link Arg40 and Lys46 to neighboring amino acid residues, but no direct interaction between Arg40 and Lys46 was observed (Figure 3b). In the WT2 model (p.Lys46Arg), we noted that the network of hydrogen bonds is preserved in comparison to the WT1 model, but that a new interchain bond is formed between Arg46 and Tyr167. The predicted conservation of the overall network of hydrogen bonds is in accord with p.Lys46Arg being a common variant with no significant structural or functional impact on the protein. By contrast, the pathogenic mutation, p.Arg40His (M1), was predicted to lead to the removal of three hydrogen bonds, two with Glu52 and one with Asn47 (Figure 3b). When we then considered the M2 model, which combines the His40 substitution with the minor allele Arg46 (p.Arg40His + p.Lys46Arg), we noted that the impact was additive in terms of the predicted alterations to the hydrogen bond network observed in the M1 (p.Arg40His) and WT2 (p.Lys46Arg) models, namely the loss of three hydrogen bonds in relation to p.Arg40His plus the gain of one interchain hydrogen bond with p.Lys46Arg.
In Site 2 (Figure 4), there is a network of four hydrogen bonds that links Gln270 to four neighboring residues (Arg277, Gln273, Gln271, and Ser267). In the WT3 model incorporating p.Gln270Arg, we still observe four hydrogen bonds, but these are connected to only three of the four neighboring amino acid residues. As in the case of the p.Lys46Arg variant, the conservation of the hydrogen bond network suggests that no major structural perturbations would be expected as a consequence of the replacement of Gln270 by Arg, as indeed is in accordance with the polymorphic frequency of this p.Gln270Arg variant in the general population.
Figure 4.
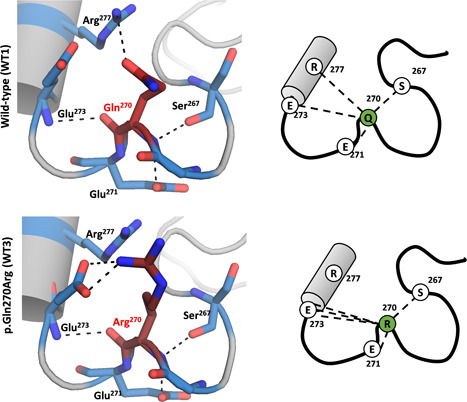
Structural analysis of Site 2 in OTC homology models. OTC, ornithine transcarbamylase
Structural analysis of the WT4, M3, and M4 models (p.Lys46Arg + p.Gln270Arg, p.Arg40His + p.Lys46Arg, and p.Arg40His + p.Lys46Arg + Gln270Arg, respectively) did not reveal any change in the network of predicted hydrogen bonds in comparison to the models previously analyzed (WT1, WT2, WT3, M1, and M2), possibly due to the considerable distance between Sites 1 and 2 in the OTC molecule.
As the loss of protein stability consequent to amino acid replacement is one of the main underlying causes of the disease phenotype (Yue et al., 2005), we next determined the impact of the variants under study on the overall structural stability of the OTC protein, by evaluating the change in Gibbs free energy (ΔΔG). We submitted the different homology models to the Duet server that aggregates two complementary tools, mCSM and SDM, to yield a consensus prediction of the change in protein stability upon a single amino acid replacement (Pires et al., 2014). By measuring and comparing the change of free energy, ΔΔG, between the folded and unfolded states of homology models harboring the pathogenic mutation and the polymorphic variants in relation to the native model, WT1, we aimed to infer the impact of each missense replacement on the structural stability of the protein (Kellogg et al., 2011; Magesh & George Priya Doss, 2014; Pires et al., 2014). This analysis predicted that the pathogenic mutation p.Arg40His was destabilizing. The polymorphic variant p.Lys46Arg was also predicted by mCSM and Duet to be destabilizing, but not to the same extent as p.Arg40His, whereas SDM predicted the p.Lys46Arg variant not to be destabilizing. By contrast, stability analysis by mCSM, Duet, and SDM of the model harboring the p.Gln270Arg variant produced a unanimous prediction that the Arg allele would be associated with an increase in stability.
4. DISCUSSION
The OTC protein is an evolutionarily conserved enzyme that plays a key role in the urea cycle, as evidenced by the hyperammonemia (which may be lethal) caused by impairment of OTC activity. Although a considerable number of disease‐causing mutations have been reported in the OTC gene, only two missense variants are known to occur in human populations at polymorphic frequencies: p.Lys46Arg (rs1800321) and p.Gln270Arg (rs1800328). These variants have generally been considered to be standing variation without any significant functional impact on the wild‐type protein. In this study, we first posed the question as to whether these protein variants could serve to modify OTC enzymatic activity in vitro. Second, we sought to explore the possibility that this standing polymorphic variation can modulate enzymatic activity in the specific context of the p.Arg40His pathogenic mutation.
To assess the potential impact of the two polymorphic OTC variants in cis with the p.Arg40His substitution, we combined data from in vitro activity assays with structural information from homology modeling for several different OTC proteins carrying distinct combinations of the polymorphic variants and the pathogenic p.Arg40His mutation.
Although the structural analysis revealed that both polymorphic variants alter the native network of hydrogen bonds observed in OTC (WT1) as well as alter the overall structural stability of the protein, enzymatic assays disclose that all wild‐type OTC enzymes (WT1, WT2, WT3, and WT4), irrespective of the polymorphic alleles present, displayed high level of enzymatic activity (Figure 2a), an expected finding as all these isoforms are present in the general population (Table 1). We however noted significantly enhanced enzymatic activity in association with the WT4 enzyme that combined the minor alleles of both polymorphisms in cis, in comparison with the other wild‐type enzymes, a strong indication that these two alleles in combination have the potential to function as a genetic modifier of OTC activity in the wild type background.
With respect to the various alternative versions of OTC harboring the p.Arg40His deleterious replacement, in vitro assays showed residual enzymatic activity corresponding to approximately 15% of WT1 activity (Figure 2b). This concurs with literature records reporting similar levels of enzymatic activity in liver samples obtained post mortem from patients carrying the p.Arg40His mutation (Matsuda et al., 1996) and with previous studies that characterized this mutation in vitro (Matsuda et al., 1996; Nishiyori et al., 1997). Although significantly reduced by comparison with wild‐type, the level of enzymatic activity associated with the various versions of OTC harboring the p.Arg40His mutation may well be enough to sustain quasi‐normal metabolic status as several OTC deficiency patients have been reported in the literature as presenting symptoms at distinct ages (Cavicchi et al., 2014; Hidaka et al., 2020; Koya et al., 2019; Matsuda et al., 1996; Pinner et al., 2010; Ploechl et al., 2001; Tuchman et al., 1994; Yoshino et al., 1990; Zhou et al., 2020).
The functional characterization of OTC protein carrying the p.Arg40His replacement in cis to the p.Lys46Arg and/or p.Gln270Arg polymorphic variants revealed that the combination of Arg46 with the pathogenic mutation p.Arg40His alone (M2), or in combination also with Arg270 (M4), exhibited reduced enzymatic activity, although this decrease was not significantly different from the activity observed in association with the M1 background. By contrast, functional characterization of OTC protein carrying the p.Arg40His in cis with the p.Gln270Arg polymorphic variant (M3) showed that the Arg270 allele contributed to a 37.8% increase in terms of OTC activity when compared with the major Gln270 allele (M1). This would therefore appear to be an example of what has been termed “antagonistic epistasis” (Lehner, 2011) by which a missense variant at a physically distant residue contributes to protein stability (Table 2) and partial rescue of enzymatic activity when in cis with a pathological missense mutation. Even though a significant change in protein activity was evident in vitro, we should be wary of extrapolating in relation to the clinical phenotype of individuals harboring the p.Gln270Arg polymorphic variant in cis to the p.Arg40His mutation because the in vivo context will inevitability exhibit much greater complexity. Moreover, in the specific setting of OTC deficiency, the incremental increase in enzymatic activity afforded by the p.Gln270Arg variant may be insufficient to maintain normal metabolic status, thereby preventing the development of clinical symptoms caused by insufficient enzymatic activity to maintain normal metabolic status.
Table 2.
Structural stability analysis of models harboring the variants p.Arg40His, p.Lys46Arg, and p.Gln270Arg in comparison to wild‐type (ΔΔG = 0)
| p.Arg40His | p.Lys46Arg | p.Gln270Arg | |
|---|---|---|---|
| mCSM | −1.457 | −0.678 | 0.044 |
| DUET | −1.551 | −0.303 | 0.382 |
| SDM | −0.13 | 0.26 | 0.38 |
Note: Values correspond to changes in Gibbs free energy (ΔΔG Kcal/mol). Negative values of ΔΔG indicate a protein destabilizing replacement, whereas positive values of ΔΔG indicate a stabilizing replacement.
The interplay between rare disease‐causing mutations and common polymorphic variants has been documented for only a very limited number of genes/proteins involved in human genetic disease (Chan et al., 2006; Cheng et al., 2011; Cooper et al., 2010; Gonzalez & Ostermeier, 2019; Lage et al., 2014; Li et al., 2018; Matsumura et al., 2017; Niu et al., 2006; Poelzing et al., 2006; Raef et al., 2008; Silva et al., 2004; Zhang et al., 2008). For instance, in the SCN5A gene, the well‐known p.His558Arg polymorphic variant (NM_000335.5:c.1673A>G [p.His558Arg]) has been demonstrated to modulate the effect of several pathogenic mutations in the cardiac channel SCN5A (Makielski et al., 2003; Niu et al., 2006). Moreover, in the context of the POLG gene, the p.Glu1143Gly (NM_002693.2:c.3428A>G [p.Glu1143Gly]) polymorphic variant was found to ameliorate the effect of the pathogenic p.Trp748Ser (NM_002693.2:c.2243G>C [p.Trp748Ser]) mutation in the DNA polymerase gamma of an individual with ataxia‐neuropathy (Chan et al., 2006). Another interesting example is the case of the p.Val141Met [NM_006763.3:c.421G>A (p.Val141Met)] pathogenic mutation in the BTG2‐encoded protein which can be suppressed by either one of two polymorphic variants in cis (NM_006763.2:c.376G>T [p.Ala126Ser] and NM_006763.2:c.434G>A [pp.Arg145Gln]) (Jordan et al., 2015).
Here we show that a specific polymorphic OTC variant, p.Gln270Arg, acts as a modifier of an OTC mutation in vitro, possibly by buffering the destabilizing effect of the p.Arg40His mutation. Previous work has shown that the reduction in OTC activity consequent to the p.Arg40His substitution was due to degradation of the preprotein in the cytosol during mitochondrial import (Augustin et al., 2000; Mavinakere et al., 2001). It is thus likely that the observed spectrum of phenotypic severity depends on OTC enzymatic degradation before mitochondrial import, and so the presence of p.Gln270Arg, which appears to contribute to the stabilization of the protein, might delay its early degradation. As our experiments did not include any investigation of the import and processing of OTC expressed in cells, further reasons to explain the precise mechanism by which amino acid position 270 may contribute to protein stabilization cannot be advanced.
Although our results appear promising in terms of increasing our understanding of the intragenic molecular interactions that play a role in mediating protein stability, they cannot at this stage be used to predict the clinical severity of individuals harboring the OTC p.Arg40His variant. Indeed, we still do not know whether the sub‐group of patients who also possess the p.Gln270Arg variant manifest less severe clinical symptoms (or experience a later age of onset) than individuals with different combinations of genetic variants. Moreover, the effect of the p.Arg40His mutation on the stability of the OTC protein may prevail over that of the allelic combination of polymorphic amino acid residues in relation to the phenotypic outcome. This represents an important future research question to be addressed. It should, however, be appreciated that the critical impact of nongenetic factors, such as diet may render any definitive conclusion difficult to achieve.
In summary, we show in this study that putatively neutral missense polymorphisms in the OTC protein have the potential to play a significant role as genetic modifiers of a specific pathological mutation. It is likely that many other similar instances of genetic modifiers of inherited human conditions will eventually be discovered. They will await detailed in vivo and in vitro analyses that should ultimately illuminate our understanding of the interplay between rare pathogenic mutations and common apparently neutral variants. Thus, Haldane's (1941) words are as true today as they were 80 years ago: “a great deal more work will be required before we can judge whether the presence of modifiers….is common or rare in the human species.”
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
WEB RESOURCES
gnomAD browser V2.2.1: https://gnomad.broadinstitute.org/. 1000 Genomes Project (1KGP) Phase 3 available at the Ensembl Genome Browser release 101: https://www.ensembl.org/index.html. DUET Server: http://biosig.unimelb.edu.au/duet/. SwissModel: https://swissmodel.expasy.org/.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This study was supported by FEDER—Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020—Operational Program for Competitiveness and Internationalization (POCI), Portugal 2020, and by Portuguese funding through FCT ‐ Fundação para a Ciência e a Tecnologia, within the framework of the Project POCI‐01–0145‐FEDER‐007274 to i3S and by FCT research Project POCI‐01–0145‐FEDER‐29723. CS holds an FCT PhD Fellowship (SFRH/BD/137925/2018). ARC holds an FCT PhD Fellowship (SFRH/BD/141702/2018). The funders had no role in the design, collection, analysis or interpretation of the data, or in the writing of the manuscript.
Lopes‐Marques, M., Pacheco, A. R., Peixoto, M. J., Cardoso, A. R., Serrano, C., Amorim, A., Prata, M. J., Cooper, D. N., & Azevedo, L. (2021). Common polymorphic OTC variants can act as genetic modifiers of enzymatic activity. Human Mutation, 42, 978–989. 10.1002/humu.24221
Contributor Information
Mónica Lopes‐Marques, Email: monicaslm@hotmail.com.
Luísa Azevedo, Email: lazevedo@ipatimup.pt.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding authors upon request.
REFERENCES
- 1000 Genomes Project Consortium , Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., & Abecasis, G. R. (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin, L., Mavinakere, M., Morizono, H., & Tuchman, M. (2000). Expression of wild‐type and mutant human ornithine transcarbamylase genes in Chinese hamster ovary cells and lack of dominant negative effect of R141Q and R40H mutants. Pediatric Research, 48(6), 842–846. 10.1203/00006450-200012000-00023 [DOI] [PubMed] [Google Scholar]
- Azevedo, L., Calafell, F., Vilarinho, L., & Amorim, A. (2002). Haplotype analysis and phylogeny of ornithine transcarbamylase polymorphisms. Annals of Human Genetics, 66(5‐6), 379–385. 10.1046/j.1469-1809.2002.00129.x [DOI] [PubMed] [Google Scholar]
- Benkert, P., Biasini, M., & Schwede, T. (2010). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics, 27(3), 343–350. 10.1093/bioinformatics/btq662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoni, M., Kiefer, F., Biasini, M., Bordoli, L., & Schwede, T. (2017). Modeling protein quaternary structure of homo‐ and hetero‐oligomers beyond binary interactions by homology. Scientific Reports, 7(1), 10480. 10.1038/s41598-017-09654-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldovic, L., Abdikarim, I., Narain, S., Tuchman, M., & Morizono, H. (2015). Genotype‐phenotype correlations in ornithine transcarbamylase deficiency: A mutation update. Journal of Genetics and Genomics, 42(5), 181–194. 10.1016/j.jgg.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavicchi, C., Donati, M., Parini, R., Rigoldi, M., Bernardi, M., Orfei, F., Gentiloni Silveri, N., Colasante, A., Funghini, S., Catarzi, S., Pasquini, E., la Marca, G., Mooney, S., Guerrini, R., & Morrone, A. (2014). Sudden unexpected fatal encephalopathy in adults with OTC gene mutations—clues for early diagnosis and timely treatment. Orphanet Journal of Rare Diseases, 9(1), 105. 10.1186/s13023-014-0105-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceriotti, G. (1973). Optimal conditions for ornithine carbamyl transferase determination. A simple micromethod without deproteinization. Clinica Chimica Acta, 47(1), 97–105. 10.1016/0009-8981(73)90065-x [DOI] [PubMed] [Google Scholar]
- Chan, S. S. L., Longley, M. J., & Copeland, W. C. (2006). Modulation of the W748S mutation in DNA polymerase gamma by the E1143G polymorphism in mitochondrial disorders. Human Molecular Genetics, 15(23), 3473–3483. 10.1093/hmg/ddl424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H., Jawahar, S., Qian, Y., Duong, Q., Chan, G., Parker, A., Meyer, J. M., Moore, K. J., Chayen, S., Gross, D. J., Glaser, B., Permutt, M. A., & Fricker, L. D. (2001). Missense polymorphism in the human carboxypeptidase E gene alters enzymatic activity. Human Mutation, 18(2), 120–131. 10.1002/humu.1161 [DOI] [PubMed] [Google Scholar]
- Cheng, J., Norstrand, D. W., Medeiros‐Domingo, A., Tester, D. J., Valdivia, C. R., Tan, B. H., Vatta, M., Makielski, J. C., & Ackerman, M. J. (2011). LQTS‐associated mutation A257G in α1‐syntrophin interacts with the intragenic variant P74L to modify its biophysical phenotype. Cardiogenetics, 1(1). 10.4081/cardiogenetics.2011.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, D. N., Chen, J. M., Ball, E. V., Howells, K., Mort, M., Phillips, A. D., Chuzhanova, N., Krawczak, M., Kehrer‐Sawatzki, H., & Stenson, P. D. (2010). Genes, mutations and human inherited disease at the dawn of the age of personalized genomics. Human Mutation, 31(6), 631–655. 10.1002/humu.21260 [DOI] [PubMed] [Google Scholar]
- Cooper, D. N., Krawczak, M., Polychronakos, C., Tyler‐Smith, C., & Kehrer‐Sawatzki, H. (2013). Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Human Genetics, 132(10), 1077–1130. 10.1007/s00439-013-1331-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz, N. M., Yuan, Y., Leehy, B. D., Baid, R., Kompella, U., DeAngelis, M. M., Escher, P., & Haider, N. B. (2014). Modifier genes as therapeutics: the nuclear hormone receptor Rev Erb alpha (Nr1d1) rescues Nr2e3 associated retinal disease. PLOS One, 9(1). e87942. 10.1371/journal.pone.0087942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakal, T. C., Kala, D., Dhiman, G., Yadav, V., Krokhotin, A., & Dokholyan, N. V. (2017). Predicting the functional consequences of non‐synonymous single nucleotide polymorphisms in IL8 gene. Scientific Reports, 7(1), 6525. 10.1038/s41598-017-06575-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah, R., McKone, E., O'Connor, C., Rodgers, C., Genatossio, A., McNamara, S., Gibson, R., Stuart Elborn, J., Ennis, M., Gallagher, C. G., Kalsheker, N., Aitken, M., Wiese, D., Dunn, J., Smith, P., Pace, R., Londono, D., Goddard, K. A., Knowles, M. R., & Drumm, M. L. (2010). EDNRA variants associate with smooth muscle mRNA levels, cell proliferation rates, and cystic fibrosis pulmonary disease severity. Physiological Genomics, 41(1), 71–77. 10.1152/physiolgenomics.00185.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo, J., Baeza‐Centurion, P., & Lehner, B. (2019). The causes and consequences of genetic interactions (epistasis). Annual Review of Genomics and Human Genetics, 20(1), 433–460. 10.1146/annurev-genom-083118-014857 [DOI] [PubMed] [Google Scholar]
- Gonzalez, C. E., & Ostermeier, M. (2019). Pervasive pairwise intragenic epistasis among sequential mutations in TEM‐1 β‐lactamase. Journal of Molecular Biology, 431(10), 1981–1992. 10.1016/j.jmb.2019.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane, J. (1941). The relative importance of principal and modifying genes in determining some human diseases. Journal of Genetics, 41(2‐3), 149–157. 10.1007/BF02983018 [DOI] [Google Scholar]
- Hartman, E. C., & Tullman‐Ercek, D. (2019). Learning from protein fitness landscapes: A review of mutability, epistasis, and evolution. Current Opinion in Systems Biology, 14, 25–31. 10.1016/j.coisb.2019.02.006 [DOI] [Google Scholar]
- Hidaka, M., Higashi, E., Uwatoko, T., Uwatoko, K., Urashima, M., Takashima, H., Watanabe, Y., Kitazono, T., & Sugimori, H. (2020). Late‐onset ornithine transcarbamylase deficiency: A rare cause of recurrent abnormal behavior in adults. Acute Medicine & Surgery, 7(1), e565. 10.1002/ams2.565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillian, A. D., Londono, D., Dunn, J. M., Goddard, K. A., Pace, R. G., Knowles, M. R., & CF Gene Modifier Study, G., Group, CF Gene Modifier Study Group . (2008). Modulation of cystic fibrosis lung disease by variants in interleukin‐8. Genes and Immunity, 9(6), 501–508. 10.1038/gene.2008.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, Y. J., LaBella, A. L., Feeney, T. P., Braverman, N., Tuchman, M., Morizono, H., Ah Mew, N., & Caldovic, L. (2018). Disease‐causing mutations in the promoter and enhancer of the ornithine transcarbamylase gene. Human Mutation, 39(4), 527–536. 10.1002/humu.23394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, D. M., Frangakis, S. G., Golzio, C., Cassa, C. A., Kurtzberg, J., Task Force for Neonatal, G., Davis, E. E., Sunyaev, S. R., & Katsanis, N. (2015). Identification of cis‐suppression of human disease mutations by comparative genomics. Nature, 524(7564), 225–229. 10.1038/nature14497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., Collins, R. L., Laricchia, K. M., Ganna, A., Birnbaum, D. P., Gauthier, L. D., Brand, H., Solomonson, M., Watts, N. A., Rhodes, D., Singer‐Berk, M., England, E. M., Seaby, E. G., Kosmicki, J. A., … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg, E. H., Leaver‐Fay, A., & Baker, D. (2011). Role of conformational sampling in computing mutation‐induced changes in protein structure and stability. Proteins, 79(3), 830–838. 10.1002/prot.22921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya, Y., Shibata, M., Senju, M., Honma, Y., Hiura, M., Ishii, M., Matsumoto, S., & Harada, M. (2019). Hyperammonemia in a woman with late‐onset ornithine transcarbamylase deficiency. Internal Medicine, 58(7), 937–942. 10.2169/internalmedicine.1851-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage, M. D., Pittman, A. M. C., Roncador, A., Cellini, B., & Tucker, C. L. (2014). Allele‐specific characterization of alanine: glyoxylate aminotransferase variants associated with primary hyperoxaluria. PLOS One, 9(4), e94338. 10.1371/journal.pone.0094338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner, B. (2011). Molecular mechanisms of epistasis within and between genes. Trends in Genetics, 27(8), 323–331. 10.1016/j.tig.2011.05.007 [DOI] [PubMed] [Google Scholar]
- Li, S., Datta, S., Brabbit, E., Love, Z., Woytowicz, V., Flattery, K., Capri, J., Yao, K., Wu, S., Imboden, M., Upadhyay, A., Arumugham, R., Thoreson, W. B., DeAngelis, M. M., & Haider, N. B. (2020). Nr2e3 is a genetic modifier that rescues retinal degeneration and promotes homeostasis in multiple models of retinitis pigmentosa. Gene Therapy. 10.1038/s41434-020-0134-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W., Yin, L., Shen, C., Hu, K., Ge, J., & Sun, A. (2018). SCN5A variants: Association with cardiac disorders. Frontiers in Physiology, 9, 1372–1372. 10.3389/fphys.2018.01372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luksan, O., Jirsa, M., Eberova, J., Minks, J., Treslova, H., Bouckova, M., Storkanova, G., Vlaskova, H., Hrebicek, M., & Dvorakova, L. (2010). Disruption of OTC promoter‐enhancer interaction in a patient with symptoms of ornithine carbamoyltransferase deficiency. Human Mutation, 31(4), E1294–E1303. 10.1002/humu.21215 [DOI] [PubMed] [Google Scholar]
- Magesh, R., & George Priya Doss, C. (2014). Computational pipeline to identify and characterize functional mutations in ornithine transcarbamylase deficiency. 3 Biotech, 4(6), 621–634. 10.1007/s13205-014-0216-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makielski, J. C., Ye, B., Valdivia, C. R., Pagel, M. D., Pu, J., Tester, D. J., & Ackerman, M. J. (2003). A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circulation Research, 93(9), 821–828. 10.1161/01.Res.0000096652.14509.96 [DOI] [PubMed] [Google Scholar]
- Matsuda, I., Matsuura, T., Nishiyori, A., Komaki, S., Hoshide, R., Matsumoto, T., Funakoshi, M., Kiwaki, K., Endo, F., Hata, A., Shimadzu, M., & Yoshino, M. (1996). Phenotypic variability in male patients carrying the mutant ornithine transcarbamylase (OTC) allele, Arg40His, ranging from a child with an unfavourable prognosis to an asymptomatic older adult. Journal of Medical Genetics, 33(8), 645–648. 10.1136/jmg.33.8.645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura, H., Nakano, Y., Ochi, H., Onohara, Y., Sairaku, A., Tokuyama, T., Tomomori, S., Motoda, C., Amioka, M., Hironobe, N., Toshishige, M., Takahashi, S., Imai, K., Sueda, T., Chayama, K., & Kihara, Y. (2017). H558R, a common SCN5A polymorphism, modifies the clinical phenotype of Brugada syndrome by modulating DNA methylation of SCN5A promoters. Journal of Biomedical Science, 24(1), 91. 10.1186/s12929-017-0397-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavinakere, M., Morizono, H., Shi, D., Allewell, N. M., & Tuchman, M. (2001). The clinically variable R40H mutant ornithine carbamoyltransferase shows cytosolic degradation of the precursor protein in CHO cells. Journal of Inherited Metabolic Disease, 24(6), 614–622. 10.1023/a:1012726207870 [DOI] [PubMed] [Google Scholar]
- Nadeau, J. H. (2001). Modifier genes in mice and humans. Nature Reviews Genetics, 2(3), 165–174. 10.1038/35056009 [DOI] [PubMed] [Google Scholar]
- Nishiyori, A., Yoshino, M., Kato, H., Matsuura, T., Hoshide, R., Matsuda, I., Kuno, T., Miyazaki, S., Hirose, S., Kuromaru, R., & Mori, M. (1997). The R40H mutation in a late onset type of human ornithine transcarbamylase deficiency in male patients. Human Genetics, 99(2), 171–176. 10.1007/s004390050333 [DOI] [PubMed] [Google Scholar]
- Niu, D.‐M., Hwang, B., Hwang, H.‐W., Wang, N. H., Wu, J.‐Y., Lee, P.‐C., Chien, J. C., Shieh, R. C., & Chen, Y. T. (2006). A common SCN5A polymorphism attenuates a severe cardiac phenotype caused by a nonsense SCN5A mutation in a Chinese family with an inherited cardiac conduction defect. Journal of Medical Genetics, 43(10), 817–821. 10.1136/jmg.2006.042192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson, D. L., Cox, S. L., & Gilbert, B. E. (1977). Human ornithine transcarbamylase. Purification and characterization of the enzyme from normal liver and the liver of a Reye's syndrome patient. Journal of Biological Chemistry, 252(18), 6464–6469. [PubMed] [Google Scholar]
- Pinner, J. R., Freckmann, M.‐L., Kirk, E. P., & Yoshino, M. (2010). Female heterozygotes for the hypomorphic R40H mutation can have ornithine transcarbamylase deficiency and present in early adolescence: a case report and review of the literature. Journal of Medical Case Reports, 4(1), 361. 10.1186/1752-1947-4-361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires, D. E. V., Ascher, D. B., & Blundell, T. L. (2014). DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Research, 42(W1), W314–W319. 10.1093/nar/gku411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploechl, E., Ploechl, W., Stoeckler‐Ipsiroglu, S., Pokorny, H., & Wermuth, B. (2001). Late‐onset ornithine transcarbamylase deficiency in two families with different mutations in the same codon. Clinical Genetics, 59(2), 111–114. 10.1034/j.1399-0004.2001.590208.x [DOI] [PubMed] [Google Scholar]
- Poelzing, S., Forleo, C., Samodell, M., Dudash, L., Sorrentino, S., Anaclerio, M., Troccoli, R., Iacoviello, M., Romito, R., Guida, P., Chahine, M., Pitzalis, M., & Deschênes, I. (2006). SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation, 114(5), 368–376. 10.1161/CIRCULATIONAHA.105.601294 [DOI] [PubMed] [Google Scholar]
- Raef, H., Baitei, E. Y., Zou, M., & Shi, Y. (2008). Genotype‐phenotype correlation in a family with primary cortisol resistance: possible modulating effect of the ER22/23EK polymorphism. European Journal of Endocrinology, 158(4), 577–582. 10.1530/eje-07-0629 [DOI] [PubMed] [Google Scholar]
- Rahit, K., & Tarailo‐Graovac, M. (2020). Genetic modifiers and rare mendelian disease. Genes, 11(3), 239. 10.3390/genes11030239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger, L. L. C. (2015). The PyMOL Molecular Graphics System, Version 1.7.4.5.
- Shi, D., Morizono, H., Aoyagi, M., Tuchman, M., & Allewell, N. M. (2000). Crystal structure of human ornithine transcarbamylase complexed with carbamoyl phosphate and L‐norvaline at 1.9 Å resolution. Proteins, 39(4), 271–277. [DOI] [PubMed] [Google Scholar]
- Silva, E., Dharmaraj, S., Li, Y. Y., Pina, A. L., Carter, R. C., Loyer, M., Traboulsi, E., Theodossiadis, G., Koenekoop, R., Sundin, O., & Maumenee, I. (2004). A missense mutation in GUCY2D acts as a genetic modifier in RPE65‐related Leber congenital amaurosis. Ophthalmic Genetics, 25(3), 205–217. 10.1080/13816810490513451 [DOI] [PubMed] [Google Scholar]
- Smith, L. D., & Garg, U. (2017). Chapter 5 ‐ Urea cycle and other disorders of hyperammonemia. In (Eds.) Garg, U. & Smith, L. D., Biomarkers in Inborn Errors of Metabolism (pp. 103–123). Elsevier. [Google Scholar]
- Steinberg, M. H., & Sebastiani, P. (2012). Genetic modifiers of sickle cell disease. American Journal of Hematology, 87(8), 795–803. 10.1002/ajh.23232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D., Mort, M., Ball, E. V., Chapman, M., Evans, K., Azevedo, L., Hayden, M., Heywood, S., Millar, D. S., Phillips, A. D., & Cooper, D. N. (2020). The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Human Genetics, 139(10), 1197–1207. 10.1007/s00439-020-02199-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer, G., Rempfer, C., Waterhouse, A. M., Gumienny, R., Haas, J., & Schwede, T. (2019). QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics, 36(6), 1765–1771. 10.1093/bioinformatics/btz828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suriano, G., Azevedo, L., Novais, M., Boscolo, B., Seruca, R., Amorim, A., & Ghibaudi, E. M. (2007). In vitro demonstration of intra‐locus compensation using the ornithine transcarbamylase protein as model. Human Molecular Genetics, 16(18), 2209–2214. 10.1093/hmg/ddm172 [DOI] [PubMed] [Google Scholar]
- Tuchman, M., Plante, R. J., McCann, M. T., & Qureshi, A. A. (1994). Seven new mutations in the human ornithine transcarbamylase gene. Human Mutation, 4(1), 57–60. 10.1002/humu.1380040109 [DOI] [PubMed] [Google Scholar]
- van Leeuwen, J., Pons, C., Mellor, J. C., Yamaguchi, T. N., Friesen, H., Koschwanez, J., Ušaj, M. M., Pechlaner, M., Takar, M., Ušaj, M., VanderSluis, B., Andrusiak, K., Bansal, P., Baryshnikova, A., Boone, C. E., Cao, J., Cote, A., Gebbia, M., Horecka, G, … Boone, C. (2016). Exploring genetic suppression interactions on a global scale. Science, 354(6312), aag0839. 10.1126/science.aag0839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F. T., de Beer, T., Rempfer, C., Bordoli, L., Lepore, R., & Schwede, T. (2018). SWISS‐MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296–W303. 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall, D. J. (2001). Phenotype‐genotype relationships in monogenic disease: lessons from the thalassaemias. Nature Reviews Genetics, 2(4), 245–255. 10.1038/35066048 [DOI] [PubMed] [Google Scholar]
- Yamaguchi, S., Brailey, L. L., Morizono, H., Bale, A. E., & Tuchman, M. (2006). Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Human Mutation, 27(7), 626–632. 10.1002/humu.20339 [DOI] [PubMed] [Google Scholar]
- Yoshino, M., Nishiyori, J., Yamashita, F., Kumashiro, R., Abe, H., Tanikawa, K., Ohno, T., Nakao, K., Kaku, N., & Fukushima, H. (1990). Ornithine transcarbamylase deficiency in male adolescence and adulthood. Enzyme, 43, 160–168. 10.1159/000468724 [DOI] [PubMed] [Google Scholar]
- Yue, P., Li, Z., & Moult, J. (2005). Loss of protein structure stability as a major causative factor in monogenic disease. Journal of Molecular Biology, 353(2), 459–473. 10.1016/j.jmb.2005.08.020 [DOI] [PubMed] [Google Scholar]
- Zhang, X., Chen, S., Zhang, L., Liu, M., Redfearn, S., Bryant, R. M., Oberti, C., Vincent, G. M., & Wang, Q. K. (2008). Protective effect of KCNH2 single nucleotide polymorphism K897T in LQTS families and identification of novel KCNQ1 and KCNH2 mutations. BMC Medical Genetics, 9(1), 87. 10.1186/1471-2350-9-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z., Miteva, M. A., Wang, L., & Alexov, E. (2012). Analyzing effects of naturally occurring missense mutations. Computational and Mathematical Methods in Medicine, 2012, 805827. 10.1155/2012/805827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Q., Huang, H., Ma, L., & Zhu, T. (2020). The application of next‐generation sequencing (NGS) in neonatal‐onset urea cycle disorders (UCDs): Clinical course, metabolomic profiling, and genetic findings in nine Chinese hyperammonemia patients. BioMed Research International, 2020, 5690915. 10.1155/2020/5690915 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon request.