

Abstract
CeCl3(thf) reacts at low temperatures with MeLi, t‐BuLi, and n‐BuLi to isolable organocerium complexes. Solvent‐dependent extensive n‐BuLi dissociation is revealed by 7Li NMR spectroscopy, suggesting “Ce(n‐Bu)3(thf)x” or solvent‐separated ion pairs like “[Li(thf)4][Ce(n‐Bu)4(thf)y]” as the dominant species of the Imamoto reagent. The stability of complexes Li3Ln(n‐Bu)6(thf)4 increases markedly with decreasing LnIII size. Closer inspection of the solution behavior of crystalline Li3Lu(n‐Bu)6(thf)4 and mixtures of LuCl3(thf)2/n‐BuLi in THF indicates occurring n‐BuLi dissociation only at molar ratios of <1:3. n‐BuLi‐depleted complex LiLu(n‐Bu)3Cl(tmeda)2 was obtained by treatment of Li2Lu(n‐Bu)5(tmeda)2 with ClSiMe3, at the expense of LiCl incorporation. Imamoto's ketone/tertiary alcohol transformation was examined with 1,3‐diphenylpropan‐2‐one, affording 99 % of alcohol.
Keywords: 7Li NMR spectroscopy, carbonyl alkylation, cerium, lithium, n-butyl
Seamless low‐temperature syntheses, crystallization and spectroscopies provide structural details of the heterobimetallic reagent routinely used for regioselective carbon−carbon bond forming reactions with carbonyl compounds. 7Li NMR spectroscopy takes on an important role here, tracking down n‐BuLi dissociation.

Introduction
The redox reagents Ceric Ammonium Nitrate (CAN=(NH4)2Ce(NO3)6) and SmI2(thf)2 as well as the binary alkylating agents CeCl3/LiR (R=alkyl like CH3 or n‐C4H9) constitute the most commonly employed rare‐earth‐metal reagents in organic transformations (including natural product synthesis).[1, 2, 3, 4] The cause of reactivity of the redox‐active compounds is well understood,[2, 3] and their crystal structures were revealed by X‐ray diffraction (XRD) analyses.[5, 6] In the solid state, CAN exhibits a three‐dimensional network of 12‐coordinate hexanitratocerate anions and ammonium cations interconnected by hydrogen bonding.[5] On the other hand, samarium diiodide crystallizes as a monomeric pentasolvate, SmI2(thf)5, from THF solution.[6] Due to the extreme air and moisture sensitivity and combined thermal instability, a detailed structural investigation of organocerium reagents has remained elusive.
In 1984, Tsuneo Imamoto et al. described the use of binary mixtures CeI3/RLi (R=Me, Et, n‐Bu, secBu, Ph) and CeCl3/RLi (R=n‐Bu, tBu) as effective reagents for regioselective carbon−carbon‐bond forming with various carbonyl compounds.[7] The best results were obtained when employing equimolar mixtures at −78 °C to −65 °C. Like for the Luche reagent (CeCl3(H2O)7/NaBH4),[8] cerium was launched as the least expensive rare‐earth metal while a greater part of transformations was performed with n‐BuLi as an easy‐to‐handle (as well as the cheapest) organolithium derivative.[9] Figure 1 depicts characteristic features of the nucleophilic addition of such organocerium reagents to carbonyl compounds, including smooth and selective 1,2‐addition in case of α,β‐unsaturated or easily enolizable substrate molecules,[7] functional group tolerance[10] as well as diastereocontrol via chelate coordination.[11]
Figure 1.

CeIII−carbonyl coordination directs highly selective nucleophilic addition reactions of binary CeX3/RLi (X=halogenido; R=alkyl).
The much improved selectivity compared to organolithium or Grignard reagents was assigned to a changed basicity of the organocerium reagent and enhanced hardness of the carbonyl carbon atom.[4] The latter originates from the strong oxophilicity of the trivalent cerium. Although Imamoto's seminal organocerium reagents embarked on a new and prosperous branch of organolanthanide chemistry, very few studies exist that target the structural elucidation of such bimetallic mixtures.[12] In sharp contrast, heterobimetallic main‐group organometallic reagents have been given much greater attention, and entitled modern ate chemistry.[13]
Imamoto‐type rare‐earth‐metal reagents, more recently employed for halogen‐rare‐earth‐metal exchange reactions[14] or Zweifel olefinations,[15] are as a rule generated in situ and have been designated “n‐Bu2LaCl⋅4 LiCl”,[16] “n‐Bu3Sm⋅5 LiCl”,[17] or simply “n‐Bu3Ce”.[15] The formulas were derived from X‐ray absorption fine structure (EXAFS) studies[18] or Raman spectroscopy.[15] Previous enlightening studies on the CeCl3/RLi binary system focused mainly on the composition and activation of the cerous chloride precursor[12, 19, 20] as well as the effect/effectiveness of reagent stoichiometry.[21] It was revealed that the generally applied thermal activation of the commercially available heptahydrate CeCl3(H2O)7 not only generates a material of composition [CeCl3(H2O)]n [12c] but also benefits from sonication.[20] Importantly, organocerium addition to hydrazones was found most effective and selective for a 1:1 stoichiometry of CeCl3/MeLi, but the active reagent formed at different stoichiometries was proposed to be a trimethylcerium species (supported by unreacted CeCl3).[21] Although this latter investigation “precluded firm conclusions”, the results “do point out the fallacy of ascribing reagent composition on the basis of mixing stoichiometry especially at low loadings of alkyllithium”.[21]
Herein we describe the successful isolation and structural characterization of rare‐earth‐metal n‐butyl complexes formed in LnCl3/n‐BuLi systems devoid of ancillary ligands. NMR spectroscopic studies involving the 7Li nucleus provide valuable insights into the solution behavior of such binary mixtures, pointing to the true organocerium species of the Imamoto reagent.
Results and Discussion
Synthesis and Solid‐State Structure of the Organocerium Derivatives Li3Ce(CH3)6(tmeda)3 and [Li(thf)4][CetBu4]. For assessing the CeCl3/LiR salt‐metathesis protocol we initially probed the methyl derivative, since this would rule out β‐H elimination as a potential decomposition pathway. Complexes of the type Li3LnMe6(tmeda)3 featuring the entire lanthanide series except for promethium and europium were accessed by Schumann et al. as early as 1978, via mixtures LnCl3/LiMe/OEt2/TMEDA (TMEDA=tetramethylethylenediamine).[22] Solid‐state structures applying XRD analysis were described for the rare‐earth metals erbium[22b] and holmium.[22c] Moreover, both the stabilizing effect of chelating tmeda, teeda (=tetraethylethylenediamine), and dme (=dimethoxyethylene) coligands and the enhanced instability of derivatives of the “lighter” and larger‐sized rare‐earth metals have been emphasized.[23] This is in accord with more recent findings by Okuda et al. on the stability of Li3LnMe6(thf)x (isolable for Ln smaller than Eu), which form the pentametallic ate complexes Li3Sc2Me9(thf)2(OEt2)3 and Li3Ln2Me9(thf)3(OEt2)2 (Ln=Y, Tb), when crystallized from diethyl ether solutions.[24] Such a LnIII‐size dependency on thermal stability is commonly observed in rare‐earth‐metal alkyl chemistry[25] and showcased for derivatives Ln(CH2SiMe3)3(thf)x,[26] [Li(dme)3][LntBu4] (see below),[27] and solvent‐free [LnMe3]n.[28] Since the cerium derivative Li3Ce(CH3)6(tmeda)3 (1) was only mentioned briefly as an impure product (δ CH3=−6.4 ppm),[22, 23] we re‐visited its synthesis applying a slightly modified version of the Schumann protocol. Accordingly, cerous methyl complex 1 could be synthesized at −10 °C in good yield (83 %), and was obtained in analytically pure, single‐crystalline form. The solid‐state structure of complex 1 turned out to be isostructural to the derivatives of the considerably smaller‐sized erbium and holmium (Figure 2).[22]
Figure 2.

Crystal structure of Li3Ce(CH3)6(tmeda)3 (1).[59] Atomic displacement ellipsoids set at 50 % probability. Hydrogen atoms omitted for clarity. Selected interatomic distances [Å] and angles [°]: Ce1–C1 2.6795(19), Li1–C1 2.205(3), Li1–N1 2.106(3); C1‐Ce1‐C1′ 88.05(9).
The 1H NMR spectrum revealed the methyl signal at δ=−4.08 ppm, reflecting a significant paramagnetic shift induced by CeIII (Supporting Information, Figure S4). As expected, the Ce−C distance of 2.6795(19) Å in 1 is considerably longer than those in the holmium (2.563(18) Å)[22b] and erbium congeners (2.57(2) Å).[22c] For further comparison, the Ce−C distances in 6‐coordinate Ce(CH2Ph)3(thf)3 and Ce(AlMe4)3 fall in the range 2.600(2)–2.614(2) Å[29] and 2.620(7)–2.646(8) Å,[30] respectively, while those in formally 3‐coordinate complexes Ce[CH(SiMe3)2]3 and Ce[C(SiHMe2)3]3 were detected at 2.475(7) Å[31] and 2.651(2)/2.659(2)/2.672(2) Å, respectively.[32]
Homoleptic anionic tert‐butyl complexes were previously reported for [Li(thf)x][Ln(t‐Bu)4] (Ln=Sm, Er: x=4; Y: x=3),[27a] [Li(OEt2)4][Er(t‐Bu)4],[22c] [Li(tmeda)2][Ln(t‐Bu)4] (Ln=Tb, Lu),[22c, 27b] and [Li(dme)3][Ln(t‐Bu)4] (Ln=Tb, Er).[27c] Crystal structures were obtained for [Li(tmeda)2][Lu(t‐Bu)4][27b] and [Li(dme)3][Er(t‐Bu)4],[27c] whereas the enhanced thermal instability of derivatives of the “lighter” rare‐earth metals was pointed out. In order to test our low‐temperature set‐up for organocerium derivatives prone to β‐H elimination,[33] we targeted the anionic fragment [Ce(t‐Bu)4]. Although the mixture CeCl3(thf)/t‐BuLi/THF gave access to complex [Li(thf)4][Ce(t‐Bu)4] (2) at −40 °C, single crystals could be obtained only from very concentrated, oily residues, not allowing for decent elemental analysis (1H NMR spectrum: δ tBu=2.39 ppm, Figure S7). Notwithstanding an XRD analysis revealed a 4‐coordinate cerium center (Figure 3), being isostructural to the previously reported erbium[27c] and lutetium derivatives.[27b] The Ce−C distances range from 2.501(11) to 2.544(11) Å and are considerably shorter than those in 6‐coordinate 1 (2.6795(19) Å), but match those of [Li(tmeda)2][Lu(t‐Bu)4] (2.32(2)–2.43(2) Å)[27b] and [Li(dme)3][Er(t‐Bu)4] (2.352(6)–2.395(6) Å),[27c] when taking into account the LnIII ion size.
Figure 3.

Crystal structure of [Li(thf)4][Ce(t‐Bu)4] (2).[59] Atomic displacement ellipsoids set at 30 % probability. Hydrogen atoms omitted for clarity. Selected interatomic distances [Å] and angles [°]: Ce1–C1 2.524(9), Ce1–C5 2.513(8), Ce1–C9 2.50(2), Ce1–C13 2.54(2); C‐Ce1‐C (range) 105.9(10)–111.8(8).
Synthesis and Solid‐State Structures ofn‐Butyl Derivatives Li3Ln(n‐Bu)6(thf)4 and Li2Ln(n‐Bu)5(tmeda)2 . Having proven the tamable thermal instability of organocerium complexes 1 and 2 we next tackled the feasibility of the respective n‐butyl derivatives. Initially, we attempted to isolate a crystalline cerium‐containing compound from reactions of cerium chloride (thf adduct) with various amounts of n‐butyllithium in tetrahydrofuran at low temperatures (−40 °C). Not quite unexpectedly, these endeavors proved to be unsuccessful in the first place. Having in mind the presumably enhanced stability of derivatives of the smaller‐sized rare‐earth metals and to better follow the metathesis reactions via NMR spectroscopy we quickly began to focus on lutetium. Indeed, ate complex Li3Lu(n‐Bu)6(thf)4 (3Lu , Figure 4) could be isolated from the reaction of LuCl3(thf)2 with 3.3 equivalents of n‐BuLi in n‐hexane. However, in order to accomplish complex 3Lu six equivalents of n‐BuLi must have reacted and associated per LuCl3(thf)2. It is also notable that four thf molecules have been accommodated in the complex despite the presence of only two in the lutetium chloride precursor. Therefore, THF appeared to be the limiting factor for this reaction. Optimization of the reaction conditions gave the so far best results when anhydrous rare‐earth‐metal chlorides LnCl3(thf)x (Ln=Sc, Y, La, Ce and Lu; covering the entire LnIII size range) were suspended in a mixture of n‐hexane and THF and cooled to −40 °C prior to the addition of n‐BuLi. Removal of the volatiles after 30 minutes under reduced pressure, extracting the remaining solid with n‐hexane, and concentrating the obtained solution gave crystalline Li3Ln(n‐Bu)6(thf)4 (3Ln , Scheme 1).
Figure 4.

Crystal structures of Li3Lu(n‐Bu)6(thf)4 (3Lu , left) Li2Lu(n‐Bu)5(tmeda)2 (4Lu , middle), and Li2Ce(n‐Bu)5(tmeda)2 (4Ce , right)[59] with atomic displacement ellipsoids set at 30 % probability. Hydrogen atoms, disorders and CH atoms of the THF molecules (3Lu ), and disorders of the n‐butyl groups (4Lu , 4Ce ) are omitted for clarity. Selected interatomic distances [Å] and angles [°] for 3Lu : Lu1–C1 2.553(17), Lu1–C5 2.559(18), Lu1–C9 2.46(2), Lu1–C13 2.550(19), Lu1–C17 2.44(2), Lu1–C21 2.58(3), Li1–C1 2.1555(3), Li1–C13 2.3324(3), Li2–C5 2.1928(3), Li2–C17 2.4854(3), Li2–C21 2.4623(3), Li3–C5 2.1307(3), Li3–C9 2.5339(4), Li3–C21 2.3886(3); C1‐Lu1‐C21 176.68(1), C5‐Lu1‐C13 177.55(1), C9‐Lu1‐C17 176.32(1), C1‐Lu1‐C5 88.63(1). 4Lu : Lu1–C7 2.468(2), Lu1–C11 2.5293(18), Lu1–C15 2.3797(19), Lu1–C19 2.4620(18), Lu1–C23 2.522(2), Li1–C7 2.217(4), Li1–C11 2.195(4), Li2–C19 2.224(4), Li2–C23 2.191(4); C7‐Li1‐C11 108.74(1), C19‐Li2‐C23 105.61(1). 4Ce : Ce1–C7 2.657(3), Ce1–C11 2.700(5), Ce1–C15 2.674(3), Ce1–C25 2.664(4), Ce1–C29 2.549(3), Li1–C7 2.185(6), Li1–C11 2.222(8), Li2–C15 2.183(6), Li2–C25 2.205(6); C7‐Li1‐C11 112.1(3), C15‐Li2‐C25 114.3(3).
Scheme 1.

Synthesis of n‐butyl complexes 3Ln and 4Ln .
Despite our best efforts, we could obtain crystals suitable for XRD analysis only for the lutetium derivative 3Lu . The crystalline material formed for the other “larger” rare‐earth‐metal centers displayed only very poor diffraction behavior. Complex 3Lu features a lutetium center surrounded by six n‐butyl ligands, which show distinct linkages to the lithium atoms. Two hydrocarbyl ligands are unsymmetrically bridged by one alkali metal, while the two remaining lithium atoms are unsymmetrically linked to three n‐butyl ligands each. The coordination sphere of the three lithium atoms is completed by four THF molecules.
The Lu−C distances range from 2.44(2) to 2.58(3) Å, matching those in Li3Lu(CH3)6(dme)3 (2.48(4)–2.57(4) Å),[23a] Lu(AlMe4)3 (2.455(2)–2.471(2) Å),[34] and Lu(GaMe4)3 (2.465(2)–2.493(2) Å).[35] All complexes 3Ln were characterized by NMR spectroscopy and elemental analysis. A 45Sc NMR experiment for 3Sc showed a broad signal pattern with a main peak centered at +502 ppm, clearly indicating σ‐bonded alkyl species in solution (Figure S17).[36, 37] For comparison, the 45Sc chemical shift of ScMe3(thf)x was detected at +601.7 ppm.[36] Similarly, the 1H‐89Y HSQC NMR spectrum of 3Y revealed one low‐field‐shifted 89Y resonance at +771 ppm (Figure S22), in accordance with a single organoyttrium species in solution (Y(CH2SiMe3)3(thf)2: δ(89Y)=882.7 ppm).[38] To further corroborate a similar composition of complexes 3Ln , we probed the stabilizing effect of TMEDA for the metal centers cerium and lutetium. Much to our delight, addition of stoichiometric amounts of three equivalents TMEDA prior to the crystallization of the reaction mixtures containing complexes 3Ln afforded complexes Li2Ln(n‐Bu)5(tmeda)2 (4Ln ) (Ln=Ce and Lu, Figure 4).
Surprisingly, besides the expected thf/tmeda donor ligand exchange, displacement of one n‐BuLi(tmeda) entity took place. Consequently, the rare‐earth‐metal centers in isostructural complexes 4Ce and 4Lu feature only five n‐butyl ligands. One of these n‐butyl ligands is terminal while of the remaining ones two are bridged by a lithium atom each, which are stabilized by one tmeda donor each. Such n‐BuLi(tmeda) displacement is in contrast to the Schumann methyl variants Li3Ln(CH3)6(tmeda)3 including complex 1. A plausible explanation for this is the increased steric bulk/basicity of the n‐butyl versus methyl ligands. Complexes 4Ce and 4Lu exhibit distinct Ln−C distances for the terminal and bridging n‐butyl ligands (Ce: 2.549(3) Å and 2.657(3)–2.700(5) Å; Lu: 2.3797(19) Å and 2.4620(18)–2.5293(18) Å). Overall, only very limited structural data are available on rare‐earth‐metal n‐butyl complexes likely due to the propensity for β‐H elimination. Cyclopentadienyl‐supported derivatives include constrained‐geometry complexes [(η5:η1‐C5Me4SiMe2NtBu)Y(μ‐n‐Bu)]2 (Y−C: 2.542(2)/2.544(2) Å) and terminal (η5:η1‐C5Me4SiMe2NtBu)Y(n‐Bu)(dme) (Y−C: 2.435(5) Å)[39] as well as metallocenes [(C5H4Me)2Ln(n‐Bu)]2 (Ln=Y: 2.551(8)/2.556(11)/2.587(13) Å, Dy: 2.536(18)/2.591(18) Å).[40] Because cerium and lutetium form the same type of complex for 4Ln , and based on other analytical data, monolanthanide derivatives of general formula Li3Ln(n‐Bu)6(thf)4 are also proposed for the remaining complexes 3Ln .
At this point we were once more challenged by the question why these reactions would lead to the isolation of ate complexes Li3Ln(n‐Bu)6(thf)4 rather than the envisaged “Ln(n‐Bu)xCl3−x (thf)y” (x=1–3). When starting from LnCl3(thf)x, which is very poorly soluble in THF, the simplest explanation would be that treatment of Lewis‐acidic rare‐earth‐metal chloride species with increasing amounts of the strong nucleophile n‐BuLi enhances its solubility, and is therefore more likely to react faster in consecutive reactions with n‐BuLi. The formation of isolable ate complexes Li3Ln(n‐Bu)6(thf)4 is further driven by the relatively small size of the n‐butyl ligand and by switching the solvent from coordinating (THF) to non‐coordinating (n‐hexane). Predominant ate complexation was also observed for Schumann's methyl complexes Li3LnMe6(thf)x [22] or diisopropylamido derivatives LiLn(NiPr2)4(thf)x.[41] Crucially, independent of the applied LuCl3(thf)x/n‐BuLi stoichiometry, complex 3Lu could be crystallized as the exclusive LuIII‐containing species, upon separation of the solution from unreacted rare‐earth‐metal halide, its evaporation to dryness, and extraction of the residue with n‐hexane (XRD unit‐cell check and 1H NMR spectroscopy indicated repeatedly formation of 3Lu ). This finding suggested that the typical reaction (preformation) conditions for a lutetium‐derived Imamoto reagent, LuCl3/n‐BuLi/THF/−78 °C/30 min should form Li3Lu(n‐Bu)6(thf)x as the dominant initial lutetium species in solution. Since any persisting equilibria in solution would impact the reactivity of the Imamoto alkylation reagent we next took a closer look at the solution behavior of bimetallic Li3Ln(n‐Bu)6(thf)4 and the binary system LnCl3(thf)x/n‐BuLi (Ln=Ce, Lu).
Solution Behavior ofn‐Butyl Complexes 3Ln and 4Ln Probed by NMR spectroscopy. All crystallized n‐butyl complexes are stable when stored as solids at −40 °C. However, when dissolved in any solvent, complexes 3La , 3Ce and 4Ce had fully decomposed after 24 h. In contrast, but not unexpectedly, the complexes of the smaller‐sized rare‐earth metals are stable in solution at −40 °C for up to one week. Warming complexes 3Ln and 4Ln to ambient temperature, decomposition was perceived visually within one hour (in both solution and solid state). To further determine the thermal stability of our complexes, variable‐temperature (VT) NMR spectra were measured (Figures S18, S23, S26, S36 and S42). Amazingly, β‐H elimination and 1‐butene formation was observed only from +30 °C onwards, being considerably more pronounced for 3La and 3Ce than for the respective complexes of the smaller‐sized rare‐earth metals. Because the heating was performed in 10 degree increments starting at −40 °C and held at each temperature for 15 minutes before measurement, it can be concluded that these complexes are relatively stable for a short amount of time even at ambient temperature.[33]
Crucially, both the 1H and 7Li NMR spectra of complexes 3Ln and 4Ln revealed that in solution a considerable portion of n‐BuLi gets displaced from the rare‐earth‐metal center. The degree of dissociation is highly dependent on the solvent and the rare‐earth metal. In general, n‐BuLi dissociation is more pronounced in THF than in toluene. Ate complexes 3Ln of the smaller‐sized rare‐earth metals yttrium, lutetium, and scandium are quite stable in toluene solution displaying minor n‐BuLi dissociation of ca. 1 %, 4 %, and 8 % respectively (Figures S14/S19/S37). On the other hand, n‐BuLi dissociation prevails for 3Ce and 3La (ca. 90 %). Interestingly, the presence of tmeda as a donor ligand in complexes 4Ln can either counteract or enforce n‐BuLi separation (3Ce /4Ce : >98 %/46 % versus 3Lu /4Lu : 4 %/20 %). For the organocerium(III) complexes, n‐BuLi ate complexation is easily detectable by paramagnetically shifted 7Li resonances. Figure 5 depicts the 7Li NMR spectra of complexes 1, 2, 3Ce , and 4Ce both in [D8]THF and [D8]toluene, clearly revealing a) the great stability of the hexamethylate complex 1, b) the persistence of ion‐separated tert‐butyl complex 2 also in solution, and c) the beneficial effect of tmeda (versus thf) donor ligands for intramolecular ate‐complex stabilization. The 7Li NMR spectrum of 4Ce suggests a clean separation into [LiCe(n‐Bu)4(tmeda)] and n‐BuLi (signal ratio 1:1).
Figure 5.

7Li NMR spectra (194.37 MHz, 233 K) of complexes 1, 2, 3Ce , and 4Ce , recorded in [D8]THF or [D8]toluene.
These findings have important implications for the composition of the “active” n‐BuLi‐derived Imamoto reagent. When used as a 1:1 mixture of CeCl3(thf) and n‐BuLi in THF at temperatures of −35 °C, the formation of ate complex Li3Ce(n‐Bu)6(thf)4 (3Ce ) seems highly unfavored. This can be concluded from the 7Li NMR spectrum of 3Ce , which does indicate only a small portion of lithium and paramagnetic cerium(III) in close proximity (δ Li=81.5 ppm). An equimolar mixture of CeCl3(thf) and n‐BuLi formed in situ in [D8]THF at −45 °C did not reveal any paramagnetically shifted 7Li NMR resonance (Figure S33). The latter seems to appear only at ratios <1:3. For comparison, the intramolecular ate complex LiCe[N(SiHMe2)2]4(thf) (in C6D6/1,2‐difluorobenzene)[42] and tmeda‐adduct 4Ce (in [D8]THF) display 7Li chemical shifts of 84.4 and 81.3 ppm. The 1H NMR spectrum in [D8]THF of 3La featuring the similarly sized lanthanum center lends further support to this assumption, as the n‐butyl resonances appear in an approximate 1:1 ratio (Figure S24). Moreover, 1:1 mixtures of CeCl3(thf) and n‐BuLi, obtained in THF at −45 °C contain a substantial amount (40–50 %) of unreacted CeCl3(thf) upon preformation for 30 minutes. Incomplete transmetalation in CeCl3/RLi mixtures has been pointed out previously at various occasions.[12a, 21] Therefore, unlike the smaller rare‐earth metals, which favor intramolecular ate complexation even in solution, the organocerium species prevailing under Imamoto conditions are most likely “Ce(n‐Bu)3(thf)x” or solvent‐separated ion pairs like “[Ce(n‐Bu)4(thf)y][Li(thf)4]”, similar to tert‐butyl complex 2. The formation of heteroleptic species “Ce(n‐Bu)xCly(thf)z” (x=1,2; x+y=3) seems very unlikely due to the persistence of ligand redistribution forming homoleptic complexes and/or the favorable occurrence of β‐H elimination. Non‐ate mixed hydrocarbyl/halide LnIII complexes have been structurally authenticated for phenyl and benzyl derivatives but have remained elusive for alkyl ligands capable of β‐H elimination. Representative examples include (C6H5)GdCl2(THF)4,[43] Ln(CH2Ph)2I(thf)3 (Ln=Y, Er),[44] and ion‐separated [YMeI(py)5][I].[24]
The dissociation behavior was further investigated in a series of NMR experiments (Figure 6), comparing complex 3Lu to the reactions of LuCl3(thf)2 with various amounts of n‐BuLi, and n‐BuLi itself. The 1H NMR spectrum of crystalline ate complex Li3Lu(n‐Bu)6(thf)4 (3Lu ) shows two signal sets for metal‐bonded CH2 groups (Figure 6, trace III/left). The Lu−CH2 moieties resonate at about −0.5 ppm, and hence are significantly shifted to lower field compared to the characteristic pattern of n‐BuLi at −1.0 to −1.5 ppm (cf. trace II/left).[45, 46] The two signal sets are clearly indicative of n‐BuLi dissociation in THF solution (Imamoto conditions). As in case of n‐BuLi, the two distinct signals for the lutetium‐bounded n‐Bu ligands might represent lutetium complexes of distinct aggregation “LixLu(n‐Bu)3+x (thf)y”(x=0–3). The dissociation of n‐BuLi in 3Lu is also corroborated by the 7Li NMR spectrum (trace III/right) showing the characteristic pattern of n‐BuLi[47, 48] and a very broad signal at 0 ppm, indicative of Lu‐n‐Bu‐Li moieties and rapid n‐BuLi exchange. Interestingly, when examining in situ formed solutions of LuCl3(thf)2/n‐BuLi (traces IV‐VII), free n‐BuLi was observed only when more than three equivalents of n‐BuLi were used per lutetium. This is supported by the respective 7Li NMR spectra, which also suggest the formation of free LiCl in case of <3 equivalents of n‐BuLi. For better comparison the 1H and 7Li spectra of n‐BuLi‐LiCl mixtures are shown (trace I). Moreover, addition of LuCl3(thf)2 to ate complex Li3Lu(n‐Bu)6(thf)4 (3Lu ) results in complete consumption of free n‐BuLi and formation of LiCl (trace VIII). This implies that dissociated n‐BuLi engages in “normal” ligand exchange with added LuCl3(thf)2. However, when the solvent is removed the in situ formed complexes scramble to the complex 3Lu , LuCl3 and LiCl (upon crystallization from n‐hexane).
Figure 6.

1H NMR (500.13 MHz, [D8]THF, 193 K) (left) and 7Li NMR spectra (194.37 MHz, [D8]THF, 193 K) (right) of LuCl3(thf)2 with x n‐BuLi (x=1, 3, 6, 12) compared to 3Lu , 3Lu +2 LuCl3(thf)2, n‐BuLi, and n‐BuLi+LiCl.
Based on these observations, we can hypothesize about the following scenario. The initial intermediate/transient product of the LuCl3(thf)2/x n‐BuLi reaction is certainly “Lu(n‐Bu)Cl2”. However, as soon as this heteroleptic complex is formed, its better solubility in THF (compared to LuCl3) will imply a more rapid reaction (compared to LuCl3) with the remaining n‐BuLi in solution. Thus, ate complexes of the type “LixLu(n‐Bu)3+x (thf)y”, and ultimately but not exclusively Li3Lu(n‐Bu)6(thf)4 (3Lu ), represent the dominant rare‐earth‐metal species in solution. As revealed by NMR spectroscopies, 3Lu is labile in solution and engages in a dissociation equilibrium with n‐BuLi; the displaced n‐BuLi should react further with LuCl3. Therefore, likely reaction products depending on the LuCl3(thf)2/n‐BuLi ratio are “Lu(n‐Bu)3” and “LixLu(n‐Bu)3+x (thf)y” with x≤2 for a ratio of 1:3 and smaller, and species “Lu(n‐Bu)3” and “LixLu(n‐Bu)3+x (thf)y” with x ranging from 1 to 3, for a ratio of larger than 1:3. A shortage of THF solvent via extraction of the reaction products into n‐hexane leads to ate complex Li3Lu(n‐Bu)6(thf)4 (3Lu ) as the only isolable (crystalline) species. Performing the reactions for longer time periods under otherwise identical conditions resulted in extensive decomposition.
Solution Behavior ofn‐Butyl Complexes 3Ln and 4Ln Probed by Derivatization Reactions. The preferred dissociation of n‐BuLi from ate complexes Li3Ln(n‐Bu)6(thf)4 (3Ln ) of the larger‐sized rare‐earth metals was further revealed by the reaction of crystalline 3Ce with LuCl3(thf)2 in THF, affording 3Lu in low crystalline yields of 23 % (XRD unit‐cell check). Interestingly, the dissociated n‐BuLi seems to exert a stabilizing effect on the organocerium species, since the solution turned brown rather quickly, upon addition of LuCl3(thf)2. In order to enforce the formation of n‐BuLi‐depleted “Ln(n‐Bu)3” we searched for reactions which would possibly convert any dissociated n‐BuLi selectively and ideally into products not affecting the isolation of putative “Ln(n‐Bu)3”. Luckily, such a reaction path could be observed for the treatment of 4Lu with trimethylsilyl chloride. The reaction was slow at −40 °C, but it produced a minor amount of crystalline LiLu(n‐Bu)3Cl(tmeda)2 (5, Scheme 2, Figure 7). On various other occasions, when trying to precipitate LiCl or extract “Ln(n‐Bu)3” from the LiCl‐containing residue, only progressive decomposition could be observed. Again, depending on the size of the rare‐earth‐metal center, complete decomposition took place in a few minutes (“Ce(n‐Bu)3”) or several hours (“Y(n‐Bu)3”). This behavior clearly shows the stabilizing effect of LiCl in these reactions making THF an ideal solvent.
Scheme 2.
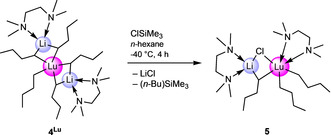
Reactivity of lutetium complex 4Lu with ClSiMe3: “lithium depletion”.
Figure 7.

Crystal structure of LiLu(n‐Bu)3Cl(tmeda)2 (5) with atomic displacement ellipsoids set at 50 % probability.[59] Hydrogen atoms and disorders of the n‐butyl groups are omitted for clarity. Selected interatomic distances [Å] and angles [°] for 5: Lu1–C1 2.421(3), Lu1–C5 2.367(3), Lu1–C18 2.447(3), Lu1–Cl1 2.7191(9), Lu1–N1 2.635(3), Lu1–N2 2.540(2), Li1–C18 2.287(6), Li1–Cl1 2.308(5); C18‐Lu1‐Cl1 89.26(9), C18‐Li1‐Cl1 104.6(2).
In the solid state, the lutetium atom of complex 5 adopts a distorted octahedral coordination geometry, involving three n‐butyl ligands, one chlorido ligand and a chelating tmeda molecule. The chlorido and one n‐butyl ligand bridge to the lithium atom forming a four‐membered ring. The coordination of the lithium atom is completed by the second tmeda molecule. Noteworthy, in the course of this reaction one tmeda ligand was transferred from lithium to lutetium. Striking are the distinct Lu−C distances of the terminal n‐butyl ligands of 2.367(3) and 2.421(3) Å. While the shorter distance approximately matches the terminal one of complex 4Lu (2.3797(19) Å), the longer distance is almost as long as that of the bridging n‐Bu ligand (2.447(3) Å). This might indicate a trans influence of the weakly coordinated chlorido ligand. In accordance with the crystal structure, the 1H NMR spectrum of 5 displays two distinct signal sets for the n‐butyl ligands in a 2:1 ratio (Figure S48).
Unsurprisingly, the n‐butyl ligands of complexes 3Ln get easily protonated in the presence of alcoholic substrates. As an example, treatment of Li3Ce(n‐Bu)6(thf)4 (3Ce ) with six equiv of neopentanol resulted in the crystallization of the heterobimetallic cluster Li3Ce2(OCH2 tBu)9(HOCH2 tBu)2(thf) (6, Scheme 3, Figure S1). On the basis of the crystal structure and Ce−O distances, the connectivity of 6 can be assigned as Li3Ce2(μ3‐OCH2 tBu)3(μ2‐OCH2 tBu)4(OCH2 tBu)2(HOCH2 tBu)2(thf). The μ2‐bridging neopentoxy ligands involve one lithium and cerium each, the μ3‐bridging ones connect two cerium atoms with one lithium, and the terminal ones are coordinated to one cerium center (Ce1). The coordination sphere of the lithium atoms is saturated with one thf and two alcohol donor molecules. The alcohol donors of 6 engage in hydrogen bonding with one terminal (O3) and one μ2‐bridging neopentoxy ligand (O10). Due to the distinct coordination modes, the Ce−O distances span a wide range of 2.196(2) to 2.608(2) Å, but match those of other cerous alkoxides.[49] Overall, the solid‐state structure of 6 features a completely asymmetric complex with all five metal centers displaying different coordination environments. A similar structure was reported for the yttrium neopentoxide Li3Y2(μ3‐OCH2 tBu)(μ3‐HOCH2 tBu)(μ2‐OCH2 tBu)5(OCH2 tBu)3(HOCH2 tBu)2.[50] The formation and structural characterization of Li3Ce2 complex 6 clearly reflects lithium depletion compared to the Li3Ce precursor 3Ce , and pictures the intricacy of 3Ce in solution. Complex 6 shows intricate solution behavior itself as evidenced by at least six signals in the 7Li NMR spectrum, including paramagnetically shifted ones (Figure S52).
Scheme 3.

Alcoholysis of cerium complex 3Ce with neopentanol.
Reactivity ofn‐Butyl Complexes 3Ln toward an Enolizable Ketone. In this study, 1,3‐diphenylpropan‐2‐one was employed as a test molecule (Table 1), since its selective conversion into the respective tertiary alcohol has been already shown in the original work by Imamoto.[7] Back then the following protocols were applied: [Ce, I2, THF, 0 °C (CeI3 formed in situ)/n‐BuLi (1 equiv), −65 °C, 30 min/ketone, −65 °C, 3 h] and [“CeCl3” (later determined as [CeCl3(H2O)]n), n‐BuLi (1 equiv), THF, −78 °C, 1 h/ketone, −65 °C, 3 h], yielding the alcohol in 98 % and 96 %, respectively.
Table 1.
Overview of the ketone reduction.

|
Entry[a] |
Compound |
Equiv ketone |
Solvent |
Additives |
Yield alcohol [%] |
Recovered ketone [%] |
|---|---|---|---|---|---|---|
|
1 |
CeCl3(thf) |
0.77 |
THF |
1 n‐BuLi |
99 |
0 |
|
2 |
CeCl3(thf) |
6 |
THF |
6 n‐BuLi |
77 |
20 |
|
3 |
3Ce |
6 |
THF |
– |
70 |
13 |
|
4 |
3Ce |
1 |
THF |
– |
88 |
3 |
|
5 |
3Ce |
6 |
Et2O |
– |
54[b] |
46[b] |
|
6 |
3Ce |
3 |
Et2O |
– |
78 |
15 |
|
7 |
3Ce |
1 |
Et2O |
– |
89 |
10 |
|
8 |
3Ce |
6 |
toluene |
– |
76 |
11 |
|
9 |
3Ce |
6 |
THF |
3 LiCl |
74 |
16 |
|
10 |
3Ce |
6 |
THF |
5 CeCl3(thf) |
89 |
8 |
|
11 |
3Ce |
6 |
THF |
5 “Ce turbo chloride” to ketone |
80 |
14 |
|
12 |
3Ce |
6 |
THF |
5 “Ce turbo chloride” after ketone |
90 |
1 |
|
13 |
3Ce |
6 |
THF |
5 Sc(OTf)3 after ketone |
62[b] |
38[b] |
|
14 |
3Ce |
6 |
THF |
5 AlCl3 after ketone |
79 |
21 |
|
15 |
3Ce |
6 |
THF |
3 TMEDA |
73 |
16 |
|
16 |
3Lu |
6 |
THF |
– |
74 |
23 |
|
17 |
3Lu |
6 |
THF |
3 TMEDA |
44 |
56 |
|
18 |
3Y |
6 |
THF |
– |
75 |
12 |
|
19[c] |
3Lu |
6 |
THF |
– |
62 |
38 |
|
20 |
– |
1 |
THF |
1 n‐BuLi |
50 |
50 |
[a] 0 °C. [b] Ratios determined by NMR spectroscopy. [c] Ambient temperature.
In our hands, the slightly modified version [CeCl3(thf), n‐BuLi (1 equivalent), THF, −35 °C, 30 min/ketone, −35 °C, 30 min] gave 99 % of alcohol (entry 1), clearly documenting the efficiency of the Imamoto transformation. The importance of using CeCl3/n‐BuLi in a 1:1 stoichiometry is revealed by the reactivity of crystalline or in situ formed complex 3Ce (entries 2 and 3). The yield of alcohol was significantly decreased at the expense of ketone enolization caused by dissociated n‐BuLi (for the exclusive reaction behavior of n‐BuLi, see entry 20). Conducting the reaction in diethyl ether also gave the best results when CeCl3(thf)/n‐BuLi was used in a 1:1 stoichiometry (entries 5–7). Surprisingly, complex 3Ce performed similarly in THF and toluene (entry 8).
To comprehend why the alcohol yield is lower in case of 3Ce than for CeCl3(thf)/n‐BuLi (ratio 1:1, entries 3 and 4 versus 1), various additives were tested. While the addition of LiCl did not affect the reaction outcome (entry 9), additional CeCl3(thf) did markedly increase the alcohol yield (entry 10). This effect was even significantly enhanced if “cerium turbo chloride” was added to the organocerium compound subsequently to the ketone (entry 12). “Cerium turbo chloride” is the combination of CeCl3 with two equivalents of LiCl and completely soluble in THF. It was prepared according to the method reported by the Knochel group.[51] As revealed by NMR spectroscopy in case of lutetium (Figure 6, trace VIII), dissociated n‐BuLi will react with added LnCl3 along with ligand scrambling and provide for the further supply of cerium‐bonded n‐butyl. Therefore the limited performance of complex 3Ce with 6 equivalents of ketone results from progressing n‐BuLi dissociation and its changed (reduced/non‐selective) reactivity toward the ketone. For ate complexes 3Ln of the smaller‐sized yttrium and lutetium, which are more stable in solution, the mediocre performance in the 6‐equivalent reaction might be attributable to the formation of sterically demanding alkoxy ligands in putative “Li3Ln[OC(CH2Ph)2 n‐Bu]6”. Overall, any effect of the rare‐earth‐metal size is not apparent since ate complexes 3Y and 3Lu showed a performance very similar to that of the cerium congener 3Ce (entries 16 and 18). The importance of performing the transformation in the presence of rare‐earth‐metal chlorides was also revealed when employing scandium triflate Sc(OTf)3 or AlCl3 as Lewis acids instead of additional CeCl3 (entry 13 and 14). Both reactions led to decreased alcohol yields and increased ketone recovery (via enolization). This and the distinct outcome of the ketone transformation when changing the order of ketone and “cerium turbo chloride” (entries 11 and 12) suggest an intramolecular ketone activation/alkylation scenario akin to a four‐membered transition state (1,2‐addition), rather than the participation of several metal complexes (Figure 8, A and B). Moreover, favorable ketone coordination/adduct formation[52, 53] and alkoxide formation[54] have been shown previously.
Figure 8.

Proposed “intramolecular” reaction involving a four‐membered transition state of the ketone reduction (A) rather than a multimolecular reaction path like B.
The impact of TMEDA was examined for in situ formed 4Ce and 4Lu (entries 15 and 17), indicating a pronounced n‐BuLi(tmeda) dissociation only in case of the lutetium reaction. Finally, performing the ketone transformation with 3Lu at ambient temperature resulted not unexpectedly in a significant drop of alcohol formation, but also documents the relative LnIII‐size‐dependent stability of the complexes at ambient temperature for a short time.
Finally, the isolation and crystallization of lithium alkoxide co‐products gave further insights into the Imamoto alkylation scenario. Prolonging the reaction time of the “incomplete” transformation of six equivalents of 1,3‐diphenylpropan‐2‐one with 3Ce (Table 1, entry 3) to three weeks led to the crystallization of the enolization product lithium 1,3‐diphenylprop‐1‐en‐2‐ate Li4[OC(=CHPh)CH2Ph]4(thf)4 (7, Figure S2). The dissociated, significantly more stable n‐BuLi (compared to 3Ce ) acts as a base, deprotonating unreacted ketone at the α‐position of the carbonyl moiety. This competitive reaction path reflects the main part of the recovered ketone, as listed in Table 1, since aqueous work‐up will involve keto–enol tautomerism. Complex 7 features a common structural motif in lithium alkoxide complexes[55, 56] with the lithium and alkoxy oxygen atoms occupying alternating positions of a cube. The lithium atoms are saturated with one THF molecule each.
Preliminary tests with CeCl3(thf)/n‐BuLi mixtures in the molar ratio 1:3 and acetone as the substrate led to the crystallization of the lithium alkoxide Li8[OCMe2(n‐Bu)]6Cl2(thf)6 (8, Figure S3). The incorporation of LiCl into the cluster core unambiguously documents that it is an integral part of the reagent solution. This is also revealed by the 7Li NMR spectra depicted in Figure 6 (right, traces VI‐VIII). Dissolved LiCl might also associate with the organocerium species (as detected for complex 5) thus exerting a stabilizing effect.
Proposed Formation and Reactivity of the Imamoto Organocerium Reagent (Scheme 4). Equimolar amounts of CeCl3 and n‐BuLi, when combined in THF at low temperatures (−40 °C), afford the cerous n‐butyl complex “Ce(n‐Bu)3(thf)x” or solvent‐separated ion pairs like “[Ce(n‐Bu)4(thf)y][Li(thf)4]”, as suggested by a) 1H and 7Li NMR spectroscopies and b) considerable amounts of unreacted cerium chloride. In particular, 7Li NMR spectroscopy indicates complete transmetalation (complete consumption of n‐BuLi) and the absence of adjacent cerium and lithium centers.
Scheme 4.

The Imamoto organocerium reagent in action.
Unfortunately, single‐crystalline products were not accessible. The formation of such species is further corroborated by the solution behavior of pre‐isolated crystalline ate complex Li3Ce(n‐Bu)6(thf)4 (3Ce ), displaying extensive n‐BuLi dissociation in THF solution. Note that the product obtained from the reaction of CeCl3⋅2 LiCl (cerium turbo chloride) with three equivalents of n‐BuLi in THF at −30 °C was previously analyzed as “n‐Bu3Ce” by Raman spectroscopy (absence of significant CeCl3 and n‐BuLi Raman lines).[15] Activation of the carbonylic substrate and its transformation (here reduction to the alkoxy moiety) takes place at the same cerium center, and can proceed three times. Such 1,2‐addition reactions have been proven for yttrium methyls [YMe3]n,[57] [YMe2(thf)5][BPh4], and [YMe(thf)6][BPh4]2,[24] as well as neosilyls [Y(CH2SiMe3)2(thf)4][A] (A=BPh4, Al(CH2SiMe3)4) and [Y(CH2SiMe3)(thf)5][BPh4]2 [58] employing fluorenone and benzophenone. Aqueous work‐up will lead to the alcoholic product and a recyclable inorganic cerium compound.
Conclusion
Seamless low‐temperature synthesis and crystallization techniques give access to isolable organocerium complexes Li3Ce(CH3)6(tmeda)3, [Li(thf)4][Ce(t‐Bu]4], Li3Ce(n‐Bu)6(thf)4, and Li2Ce(n‐Bu)5(tmeda)2. 1H/7Li NMR spectroscopic studies on in situ formed solutions of equimolar mixtures of CeCl3(thf) and n‐BuLi in THF at −35 °C suggest non‐isolable “Ce(n‐Bu)3(thf)x” or solvent‐separated ion pairs like “[Li(thf)4][Ce(n‐Bu)4(thf)y]” as effective organocerium species in respective Imamoto‐type alkylation reactions of carbonylic substrates. This hypothesis is corroborated by the solution behavior of ate complex Li3Ce(n‐Bu)6(thf)4, which displays extensive displacement of n‐BuLi. As revealed for the benchmark substrate molecule 1,3‐diphenylpropan‐2‐one, the prevailing dissociation of n‐BuLi results in decreased regioselectivity of the ketone/alcohol transformation. In contrast, ate complexes of the type Li3Ce(CH3)6(thf)x could be likely species in methyllithium derived Imamoto‐type reagents as revealed by the high stability of Li3Ce(CH3)6(tmeda)3 in THF solution. Not unsurprisingly, the effective composition of such CeCl3/RLi reagents is highly dependent on the rare‐earth metal, the hydrocarbyl group, and the solvent. Finally, the presence of THF‐soluble co‐product LiCl in in situ formed reagents is proposed to adopt an active role by exerting a stabilizing effect on the organocerium species. Compound LiLu(n‐Bu)3Cl(tmeda)2 features a structural snapshot of the likely involvement of LiCl in the soluble part of Imamoto‐type transformations.
It is a fact that a large number of organometallics‐promoted organic transformations (including the in situ formation of the multi‐component organometallic reagent) is routinely performed at low temperatures (−78→0 °C). It can be safely assumed that the application of state‐of‐the‐art cold‐chain techniques will continue to promote a better understanding of the reagent's formation, composition, and effectiveness, ultimately leading to both optimized reagents and conditions for substrate conversion.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Klaus Eichele for his help with the NMR measurements. Open access funding enabled and organized by Projekt DEAL.
T. Berger, J. Lebon, C. Maichle-Mössmer, R. Anwander, Angew. Chem. Int. Ed. 2021, 60, 15622.
Dedicated to Professor Karl W. Törnroos on the occasion of his 65th birthday
References
- 1.
- 1a.Kagan H. B., Namy J. L., Tetrahedron 1986, 42, 6573–6614; [Google Scholar]
- 1b.Trost B. M., Fleming I., Imamoto T. in Comprehensive organic synthesis, Pergamon, Oxford, 1991, Chapter 1.8; [Google Scholar]
- 1c.Molander G. A., Chem. Rev. 1992, 92, 29–68; [Google Scholar]
- 1d.Imamoto T., Lanthanides in organic synthesis, Academic Press, London, 1994. [Google Scholar]
- 2.
- 2a.Tse-Lok H., Synthesis 1973, 1973, 347–354; [Google Scholar]
- 2b.Nair V., Mathew J., Prabhakaran J., Chem. Soc. Rev. 1997, 26, 127–132; [Google Scholar]
- 2c.Das A. K., Coord. Chem. Rev. 2001, 213, 307–325; [Google Scholar]
- 2d.Nair V., Balagopal L., Rajan R., Mathew J., Acc. Chem. Res. 2004, 37, 21–30; [DOI] [PubMed] [Google Scholar]
- 2e.Nair V., Deepthi A., Tetrahedron 2009, 65, 10745–10755; [Google Scholar]
- 2f.Sridharan V., Menéndez J. C., Chem. Rev. 2010, 110, 3805–3849. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a.Namy J. L., Grirad P., Kagan H. B., Nou. J. Chim. 1977, 1, 5–7; [Google Scholar]
- 3b.Kagan H. B., J. Alloys Compd. 2006, 408–412, 421–426; [Google Scholar]
- 3c.Nicolaou K. C., Ellery S. P., Chen J. S., Angew. Chem. Int. Ed. 2009, 48, 7140–7165; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7276–7301; [Google Scholar]
- 3d.Szostak M., Procter D. J., Angew. Chem. Int. Ed. 2012, 51, 9238–9256; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9372–9390; [Google Scholar]
- 3e.Szostak M., Fazakerley N. J., Parmar D., Procter D. J., Chem. Rev. 2014, 114, 5959–6039; [DOI] [PubMed] [Google Scholar]
- 3f.Just-Baringo X., Procter D. J., Acc. Chem. Res. 2015, 48, 1263–1275; [DOI] [PubMed] [Google Scholar]
- 3g.Péter Á., Procter D. J., Chimia 2020, 74, 18–22; [DOI] [PubMed] [Google Scholar]
- 3h.Miller R. S., Sealy J. M., Shabangi M., Kuhlman M. L., Fuchs J. R., Flowers R. A., J. Am. Chem. Soc. 2000, 122, 7718–7722; [Google Scholar]
- 3i.Prasad E., Flowers R. A., J. Am. Chem. Soc. 2002, 124, 6895–6899; [DOI] [PubMed] [Google Scholar]
- 3j.Chopade P. R., Prasad E., Flowers R. A., J. Am. Chem. Soc. 2004, 126, 44–45; [DOI] [PubMed] [Google Scholar]
- 3k.J. A.Teprovich, Jr. , Balili M. N., Pintauer T., R. A.Flowers II , Angew. Chem. Int. Ed. 2007, 46, 8160–8163; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8308–8311; [Google Scholar]
- 3l.R. A.Flowers II , Synlett 2008, 2008, 1427–1439; [Google Scholar]
- 3m.Choquette K. A., Sadasivam D. V., Flowers R. A., J. Am. Chem. Soc. 2010, 132, 17396–17398; [DOI] [PubMed] [Google Scholar]
- 3n.Sadasivam D. V., Teprovich J. A., Procter D. J., Flowers R. A., Org. Lett. 2010, 12, 4140–4143; [DOI] [PubMed] [Google Scholar]
- 3o.Chciuk T. V., Anderson W. R., Flowers R. A., J. Am. Chem. Soc. 2016, 138, 8738–8741; [DOI] [PubMed] [Google Scholar]
- 3p.Maity S., Flowers R. A., J. Am. Chem. Soc. 2019, 141, 3207–3216. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.Imamoto T., Pure Appl. Chem. 1990, 62, 747–752; [Google Scholar]
- 4b.Imamoto T., Organocerium Reagents, Vol. 1, Chiba University, Japan, 1992; [Google Scholar]
- 4c.Liu H.-J., Shia K.-S., Shang X., Zhu B.-Y., Tetrahedron 1999, 55, 3803–3830; [Google Scholar]
- 4d.Bartoli G., Marcantoni E., Marcolini M., Sambri L., Chem. Rev. 2010, 110, 6104–6143; [DOI] [PubMed] [Google Scholar]
- 4e.Ho T.-L., in Fieser and Fieser's Reagents for Organic Synthesis, Wiley&Sons Inc, New York, 2017. [Google Scholar]
- 5.Beineke T. A., Delgaudio J., Inorg. Chem. 1968, 7, 715–721. [Google Scholar]
- 6.Evans W. J., Gummersheimer T. S., Ziller J. W., J. Am. Chem. Soc. 1995, 117, 8999–9002. [Google Scholar]
- 7.
- 7a.Imamoto T., Kusumoto T., Yokoyama M., J. Chem. Soc. Chem. Commun. 1982, 1042–1044; [Google Scholar]
- 7b.Imamoto T., Kusumoto T., Tawarayama Y., Sugiura Y., Mita T., Hatanaka Y., Yokoyama M., J. Org. Chem. 1984, 49, 3904–3912; [Google Scholar]
- 7c.Imamoto T., Sugiura Y., J. Organomet. Chem. 1985, 285, C21–C23; [Google Scholar]
- 7d.Imamoto T., Sugiura Y., Takiyama N., Tetrahedron Lett. 1984, 25, 4233–4236; [Google Scholar]
- 7e.Imamoto T., Takiyama N., Nakamura K., Hatajima T., Kamiya Y., J. Am. Chem. Soc. 1989, 111, 4392–4398. [Google Scholar]
- 8.
- 8a.Luche J. L., J. Am. Chem. Soc. 1978, 100, 2226–2227; [Google Scholar]
- 8b.Luche J.-L., Rodriguez-Hahn L., Crabbé P., J. Chem. Soc. Chem. Commun. 1978, 601–602; [Google Scholar]
- 8c.Gemal A. L., Luche J. L., J. Am. Chem. Soc. 1981, 103, 5454–5459. [Google Scholar]
- 9.
- 9a.Li H., Synlett 2012, 23, 1407–1408; [Google Scholar]
- 9b.Rathman T. L., Schwindeman J. A., Org. Process Res. Dev. 2014, 18, 1192–1210. [Google Scholar]
- 10.For examples, see:
- 10a.Sato T., Kato R., Gokyu K., Fujisawa T., Tetrahedron Lett. 1988, 29, 3955–3958; [Google Scholar]
- 10b.Bartoli G., Marcantoni E., Sambri L., Tamburini M., Angew. Chem. Int. Ed. Engl. 1995, 34, 2046–2048; [Google Scholar]; Angew. Chem. 1995, 107, 2163–2164. [Google Scholar]
- 11.For examples, see:
- 11a.Fujioka H., Fuji M., Okaichi Y., Yoshida T., Annoura H., Kita Y., Tamura Y., Chem. Pharm. Bull. 1989, 37, 602–605; [Google Scholar]
- 11b.Alcaraz C., Groth U., Angew. Chem. Int. Ed. Engl. 1997, 36, 2480–2482; [Google Scholar]; Angew. Chem. 1997, 109, 2590–2592. [Google Scholar]
- 12.
- 12a.Shakoor A., Jacob K., Thiele K.-H., Z. Anorg. Allg. Chem. 1985, 521, 57–60; [Google Scholar]
- 12b.Jacob K., Thiele K.-H., Z. Anorg. Allg. Chem. 1986, 536, 147–152; [Google Scholar]
- 12c.Evans W. J., Feldman J. D., Ziller J. W., J. Am. Chem. Soc. 1996, 118, 4581–4584; [Google Scholar]
- 12d.Evans W. J., Ansari M. A., Feldman J. D., Doedens R. J., Ziller J. W., J. Organomet. Chem. 1997, 545–546, 157–162; [Google Scholar]
- 12e.Conlon D. A., Kumke D., Moeder C., Hardiman M., Hutson G., Sailer L., Adv. Synth. Catal. 2004, 346, 1307–1315; [Google Scholar]
- 12f.Kim J. E., Weinberger D. S., Carroll P. J., Schelter E. J., Organometallics 2014, 33, 5948–5951; [Google Scholar]
- 12g.Kim J. E., Zabula A. V., Carroll P. J., Schelter E. J., Organometallics 2016, 35, 2086–2091. [Google Scholar]
- 13.
- 13a.Mulvey R. E., Organometallics 2006, 25, 1060–1075; [Google Scholar]
- 13b.Mulvey R. E., Mongin F., Uchiyama M., Kondo Y., Angew. Chem. Int. Ed. 2007, 46, 3802–3824; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3876–3899; [Google Scholar]
- 13c.Mulvey R. E., Acc. Chem. Res. 2009, 42, 743–755; [DOI] [PubMed] [Google Scholar]
- 13d.Gentner T. X., Mulvey R. E., Angew. Chem. Int. Ed. 2021, 60, 9247–9262; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 9331–9348. [Google Scholar]
- 14.Music A., Didier D., Synlett 2019, 30, 1843–1849. [Google Scholar]
- 15.Music A., Hoarau C., Hilgert N., Zischka F., Didier D., Angew. Chem. Int. Ed. 2019, 58, 1188–1192; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1200–1204. [Google Scholar]
- 16.Benischke A. D., Anthore-Dalion L., Berionni G., Knochel P., Angew. Chem. Int. Ed. 2017, 56, 16390–16394; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16608–16612. [Google Scholar]
- 17.Anthore-Dalion L., Benischke A. D., Wei B., Berionni G., Knochel P., Angew. Chem. Int. Ed. 2019, 58, 4046–4050; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4086–4090. [Google Scholar]
- 18.Wei B., Zhang D., Chen Y.-H., Lei A., Knochel P., Angew. Chem. Int. Ed. 2019, 58, 15631–15635; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 15777–15782. [Google Scholar]
- 19.Dimitrov V., Kostova K., Genov M., Tetrahedron Lett. 1996, 37, 6787–6790. [Google Scholar]
- 20.
- 20a.Greeves N., Lyford L., Tetrahedron Lett. 1992, 33, 4759–4760; [Google Scholar]
- 20b.Greeves N., Lyford L., Pease J. E., Tetrahedron Lett. 1994, 35, 285–288. [Google Scholar]
- 21.Denmark S. E., Edwards J. P., Nicaise O., J. Org. Chem. 1993, 58, 569–578. [Google Scholar]
- 22.
- 22a.Schumann H., Müller J., Angew. Chem. Int. Ed. Engl. 1978, 17, 276; [Google Scholar]; Angew. Chem. 1978, 90, 307; [Google Scholar]
- 22b.Schumann H., Pickardt J., Bruncks N., Angew. Chem. Int. Ed. Engl. 1981, 20, 120–121; [Google Scholar]; Angew. Chem. 1981, 93, 127–128; [Google Scholar]
- 22c.Schumann H., Mueller J., Bruncks N., Lauke H., Pickardt J., Schwarz H., Eckart K., Organometallics 1984, 3, 69–74. [Google Scholar]
- 23.
- 23a.Schumann H., Lauke H., Hahn E., Pickardt J., J. Organomet. Chem. 1984, 263, 29–35; [Google Scholar]
- 23b.Schumann H., J. Less-Common Met. 1985, 112, 327–341. [Google Scholar]
- 24.Kramer M. U., Robert D., Arndt S., Zeimentz P. M., Spaniol T. P., Yahia A., Maron L., Eisenstein O., Okuda J., Inorg. Chem. 2008, 47, 9265–9278. [DOI] [PubMed] [Google Scholar]
- 25.Zimmermann M., Anwander R., Chem. Rev. 2010, 110, 6194–6259. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a.Lappert M. F., Pearce R., J. Chem. Soc. Chem. Commun. 1973, 126; [Google Scholar]
- 26b.Bambirra S., Bouwkamp M. W., Meetsma A., Hessen B., J. Am. Chem. Soc. 2004, 126, 9182–9183; [DOI] [PubMed] [Google Scholar]
- 26c.Schumann H., Freckmann D. M. M., Dechert S., Z. Anorg. Allg. Chem. 2002, 628, 2422–2426. [Google Scholar]
- 27.
- 27a.Wayda A. L., Evans W. J., J. Am. Chem. Soc. 1978, 100, 7119–7121; [Google Scholar]
- 27b.Schumann H., Genthe W., Hahn E., Pickardt J., Schwarz H., Eckart K., J. Organomet. Chem. 1986, 306, 215–225; [Google Scholar]
- 27c.Noh W., Girolami G. S., Polyhedron 2007, 26, 3865–3870. [Google Scholar]
- 28.
- 28a.Dietrich H. M., Raudaschl-Sieber G., Anwander R., Angew. Chem. Int. Ed. 2005, 44, 5303–5306; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5437–5440; [Google Scholar]
- 28b.Gerber L. C. H., Le Roux E., Törnroos K. W., Anwander R., Chem. Eur. J. 2008, 14, 9555–9564. [DOI] [PubMed] [Google Scholar]
- 29.Wooles A. J., Mills D. P., Lewis W., Blake A. J., Liddle S. T., Dalton Trans. 2010, 39, 500–510. [DOI] [PubMed] [Google Scholar]
- 30.Occhipinti G., Meermann C., Dietrich H. M., Litlabø R., Auras F., Törnroos K. W., Maichle-Mössmer C., Jensen V. R., Anwander R., J. Am. Chem. Soc. 2011, 133, 6323–6337. [DOI] [PubMed] [Google Scholar]
- 31.Avent A. G., Caro C. F., Hitchcock P. B., Lappert M. F., Li Z., Wei X.-H., Dalton Trans. 2004, 1567–1577. [DOI] [PubMed] [Google Scholar]
- 32.Pindwal A., Patnaik S., Everett W. C., Ellern A., Windus T. L., Sadow A. D., Angew. Chem. Int. Ed. 2017, 56, 628–631; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 643–646. [Google Scholar]
- 33.Evans W. J., Meadows J. H., Wayda A. L., Hunter W. E., Atwood J. L., J. Am. Chem. Soc. 1982, 104, 2015–2017. [Google Scholar]
- 34.Zimmermann M., Frøystein N. Å., Fischbach A., Sirsch P., Dietrich H. M., Törnroos K. W., Herdtweck E., Anwander R., Chem. Eur. J. 2007, 13, 8784–8800. [DOI] [PubMed] [Google Scholar]
- 35.Zimmermann M., Litlabø R., Törnroos K. W., Anwander R., Organometallics 2009, 28, 6646–6649. [Google Scholar]
- 36.Barisic D., Diether D., Maichle-Mössmer C., Anwander R., J. Am. Chem. Soc. 2019, 141, 13931–13940. [DOI] [PubMed] [Google Scholar]
- 37.Mortis A., Barisic D., Eichele K., Maichle-Mössmer C., Anwander R., Dalton Trans. 2020, 49, 7829–7841. [DOI] [PubMed] [Google Scholar]
- 38.Elvidge B. R., Arndt S., Zeimentz P. M., Spaniol T. P., Okuda J., Inorg. Chem. 2005, 44, 6777–6788. [DOI] [PubMed] [Google Scholar]
- 39.Voth P., Arndt S., Spaniol T. P., Okuda J., Ackerman L. J., Green M. L. H., Organometallics 2003, 22, 65–76. [Google Scholar]
- 40.Grindell R., Day B. M., Guo F.-S., Pugh T., Layfield R. A., Chem. Commun. 2017, 53, 9990–9993. [DOI] [PubMed] [Google Scholar]
- 41.Spallek T., Heß O., Meermann-Zimmermann M., Meermann C., Klimpel M. G., Estler F., Schneider D., Scherer W., Tafipolsky M., Törnroos K. W., Maichle-Mössmer C., Sirsch P., Anwander R., Dalton Trans. 2016, 45, 13750–13765. [DOI] [PubMed] [Google Scholar]
- 42.Bayer U., Bock L., Maichle-Mössmer C., Anwander R., Eur. J. Inorg. Chem. 2020, 101–106. [Google Scholar]
- 43.Lin G., Jin Z., Zhang Y., Chen W., J. Organomet. Chem. 1990, 396, 307–313. [Google Scholar]
- 44.Mills D. P., Wooles A. J., McMaster J., Lewis W., Blake A. J., Liddle S. T., Organometallics 2009, 28, 6771–6776. [Google Scholar]
- 45.McGarrity J. F., Ogle C. A., J. Am. Chem. Soc. 1985, 107, 1805–1810. [Google Scholar]
- 46.Tai O., Hopson R., Williard P. G., Org. Lett. 2017, 19, 3966–3969. [DOI] [PubMed] [Google Scholar]
- 47.
- 47a.Fraenkel G., Fraenkel A. M., Geckle M. J., Schloss F., J. Am. Chem. Soc. 1979, 101, 4745–4747; [Google Scholar]
- 47b.Reed D., Barr D., Mulvey R. E., Snaith R., J. Chem. Soc. Dalton Trans. 1986, 557–564; [Google Scholar]
- 47c.Jones A. C., Sanders A. W., Bevan M. J., Reich H. J., J. Am. Chem. Soc. 2007, 129, 3492–3493. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a.Fraenkel G., Henrichs M., Hewitt M., Su B. M., J. Am. Chem. Soc. 1984, 106, 255–256; [Google Scholar]
- 48b.Qu B., Collum D. B., J. Am. Chem. Soc. 2006, 128, 9355–9360; [DOI] [PubMed] [Google Scholar]
- 48c.Barozzino-Consiglio G., Rouen M., Oulyadi H., Harrison-Marchand A., Maddaluno J., Dalton Trans. 2014, 43, 14219–14228. [DOI] [PubMed] [Google Scholar]
- 49.Boyle T. J., Tribby L. J., Bunge S. D., Eur. J. Inorg. Chem. 2006, 4553–4563. [Google Scholar]
- 50.Boyle T. J., Neville M. L., Sears J. M., Cramer R., ChemistrySelect 2016, 1, 473–481. [Google Scholar]
- 51.Krasovskiy A., Kopp F., Knochel P., Angew. Chem. Int. Ed. 2006, 45, 497–500; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 511–515. [Google Scholar]
- 52.Heeres H. J., Maters M., Teuben J. H., Helgesson G., Jagner S., Organometallics 1992, 11, 350–356. [Google Scholar]
- 53.Crozier A. R., Törnroos K. W., Maichle-Mössmer C., Anwander R., Eur. J. Inorg. Chem. 2013, 409–414. [Google Scholar]
- 54.Bayer U., Anwander R., Dalton Trans. 2020, 49, 17472–17493. [DOI] [PubMed] [Google Scholar]
- 55.Weiss E., Angew. Chem. Int. Ed. Engl. 1993, 32, 1501–1523; [Google Scholar]; Angew. Chem. 1993, 105, 1565–1587. [Google Scholar]
- 56.For recent examples of lithium enolates, see:
- 56a.Kolonko K. J., Wherritt D. J., Reich H. J., J. Am. Chem. Soc. 2011, 133, 16774–16777; [DOI] [PubMed] [Google Scholar]
- 56b.Guang J., Liu Q. P., Hopson R., Williard P. G., J. Am. Chem. Soc. 2015, 137, 7347–7356; [DOI] [PubMed] [Google Scholar]
- 56c.Jermaks J., Tallmadge E. H., Keresztes I., Collum D. B., J. Am. Chem. Soc. 2018, 140, 3077–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dietrich H. M., Meermann C., Törnroos K. W., Anwander R., Organometallics 2006, 25, 4316–4321. [Google Scholar]
- 58.Nakajima Y., Okuda J., Organometallics 2007, 26, 1270–1278. [Google Scholar]
- 59.Deposition Numbers 2069609 (1), 2069616 (2), 2069613 (3 Lu), 2069615 (4 Ce), 2069610 (4 Lu), 2069611 (5), 2069612 (6), 2069617 (7), and 2069614 (8) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary