

Abstract
Introduction
One of the hallmarks of injured skeletal muscle is the appearance of elevated skeletal muscle proteins in circulation. Human skeletal muscle generally consists of a mosaic of slow (type I) and fast (type IIa, IIx/d) fibers, defined by their myosin isoform expression. Recently, measurement of circulating fiber‐type specific isoforms of troponin I has been used as a biomarker to suggest that muscle injury in healthy volunteers (HV) results in the appearance of muscle proteins from fast but not slow fibers. We sought to understand if this is also the case in severe myopathy patients with Becker and Duchenne muscular dystrophy (BMD, DMD).
Methods
An enzyme‐linked immunosorbent assay (ELISA) that selectively measures fast and slow skeletal troponin I (TNNI2 and TNNI1) was used to measure a cross‐section of patient plasma samples from HV (N = 50), BMD (N = 49), and DMD (N = 132) patients. Creatine kinase (CK) activity was also measured from the same samples for comparison.
Results
TNNI2 was elevated in BMD and DMD and correlated with the injury biomarker, CK. In contrast, TNNI1 levels were indistinguishable from levels in HV. There was an inverse relationship between CK and TNNI2 levels and age, but no relationship for TNNI1.
Discussion
We define a surprising discrepancy between TNNI1 and TNNI2 in patient plasma that may have implications for the interpretation of elevated muscle protein levels in dystrophinopathies.
Keywords: biomarker, creatine kinase, muscle injury, muscular dystrophy, troponin
Short abstract
Abbreviations
- BMD
Becker muscular dystrophy
- CK
creatine kinase
- DMD
Duchenne muscular dystrophy
- EDTA
ethylenediaminetetraacetic acid
- ELISA
enzyme‐linked immunosorbent assay
- eMHC
embryonic myosin heavy chain
- HRP
horseradish peroxidase
- HV
healthy volunteer
- IgG
immunoglobulin G
- IR
infrared
- IRB
Institutional Research Board
- MLPA
Multiplex ligation‐dependent probe amplification
- NADH
nicotinamide adenine dinucleotide
- NADP
nicotinamide adenine dinucleotide phosphate
- PBS
phosphate‐buffered saline
- PBS‐T
phosphate‐buffered saline containing 0.1% Tween‐20
- REC
Research Ethics Committee
- TNNI
troponin inhibitory subunit
- U
unit
1. INTRODUCTION
Human skeletal muscle consists of both fast (type IIa, IIx) and slow (type I) myofibers defined by the expression of their myosin isoform, a sarcomere contractile protein, required for force development. Human skeletal muscle consists of approximately 50% slow and 50% fast fibers,1 although individual muscles can vary in their slow/fast proportion.2 Each fiber type has distinctive properties with respect to contractile speed, substrate utilization, and fatigue resistance, and the body uses these different properties to efficiently coordinate movement.
When healthy muscle is subjected to excessive, unaccustomed exercise, it develops soreness and sustained reductions in strength and range of motion. Proteins also leak from injured muscle fibers into circulation, including creatine kinase (CK), lactate dehydrogenase, and myoglobin. These biomarkers are not unique to either fast or slow fibers and so do not provide detail regarding differences in fiber responses to injury. Troponin I (TNNI) is a component of the troponin complex that controls initiation of contraction of muscle by calcium. It is distinct in that there is a different isoform for each type of striated muscle: TNNI1 in slow skeletal muscle, TNNI2 in fast skeletal muscle, and TNNI3 in cardiac muscle. Selective enzyme‐linked immunosorbent assays (ELISAs) have been used to demonstrate that TNNI2 but not TNNI1 is elevated in circulation after injurious exercise,3 even under extreme conditions.4
Duschene muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are caused by an absence (DMD) or truncation (BMD) of the dystrophin protein.5 Dystrophin provides a structural link between the actin cytoskeleton and the basement membrane through the dystrophin‐glycoprotein complex.6 When dystrophin is absent or truncated, contraction of muscle leads to heightened muscle stress and injury with normal use.
While the sensitivity to injury is much higher in DMD muscle than in BMD or healthy muscle, fast fibers still appear to be more susceptible than slow fibers, with young DMD patients exhibiting histological evidence of disruption in fast fibers7 and early loss of type IIx fibers.8 We sought to explore the relative susceptibility of these fibers to leak muscle contents.
2. METHODS
2.1. Patient samples
Healthy volunteer (HV) frozen plasma samples were purchased from BioIVT (Westbury, NY) and are confirmed to have been collected with institutional review board (IRB) approval and with appropriate informed consent. Plasma and serum for affected individuals were received from the Newcastle MRC Centre Biobank for Rare and Neuromuscular Diseases (DMD), and a Becker muscular dystrophy biomarker study at Binghamton University – SUNY (BMD). Upon receipt, all samples were aliquoted into working volumes of 50–100 μL and stored at −80°C to minimize freeze–thaw damage. For the BMD biomarker study at Binghamton University – SUNY, recruitment was through foundation social media posts, with centralized remote consent of subjects via telephone (IRB approval by Binghamton University). Patients had centrally curated DMD gene testing reports with mutation data consistent with a BMD diagnosis (in‐frame mutations, or other mutations consistent with partial dystrophin expression). For patients with a clinical diagnosis but without a prior genetic report, genetic confirmation was made via multiplex ligation‐dependent probe amplification (MLPA) genetic analysis (Fullerton Genetics Laboratory, Asheville, NC).
A mobile phlebotomist was sent to the subject's home for sample collection. Red top serum vacutainer tubes, containing silica act clot activator, were used for the blood collection. If a subject required MLPA testing, an ethylenediaminetetraacetic acid (EDTA) tube would be added for those collections, but was not used for any other analysis. After the serum tubes were left to clot for 30 min, they were processed in a centrifuge at 1000‐1300 × g for 10 min. The serum (top layer) fluid was then pipetted from the vacutainer tube and transferred into cryovials and immediately frozen on dry ice for shipment and later storage at −80°C. Serum samples were sent frozen on dry ice to Binghamton University and stored at −80°C. Samples were collected from 2017 to 2019 and analyzed in 2019.
Plasma samples from the Newcastle MRC Centre Biobank were collected from patients attending clinics at The John Walton Muscular Dystrophy Research Centre. DMD donors were genetically diagnosed via the NHS Northern Genetics Service Laboratory prior to providing blood samples to the MRC Centre Biobank for Rare & Neuromuscular Diseases. A multidisciplinary clinical team from the John Walton Muscular Dystrophy Research Centre were responsible for the clinical observations of this cohort. Collection of samples for storage and subsequent release for research was ethically approved by North East ‐ Newcastle & North Tyneside 1 Research Ethics Committee (REC reference: 19/NE/0028). Informed consent was obtained from donors using REC approved consent forms. Purple top BD EDTA Vacutainers were used to collect peripheral blood from the DMD cohort. Blood was drawn into vacutainers, gently inverted 5–10 times to ensure adequate mixing of blood with EDTA and then centrifuged at 1500 × g for 10 min. The upper plasma fraction was transferred via pipette into cryovials and immediately stored at −80°C. Samples were collected over a period of 9 y (2010–2019) and stored at −80°C prior to release to Edgewise Therapeutics for analysis.
2.2. CK assay
Blood plasma CK activity was assayed using a coupled‐reaction kit purchased from Pointe Scientific (Canton, MI). Plasma was diluted 25‐fold with phosphate‐buffered saline (PBS), of which 2 μL was added to the 384‐well plate. The CK assay reagent (70 μL, 4:1 kit Buffer A:Buffer B) was added using the Multidrop Combi (ThermoFisher, Inc., Waltham, MA) and the reaction progress monitored by absorbance at 340 nm for 30 min with the SpectraMax M3 plate reader (Molecular Devices, San Jose, CA) over approximately 20–30 min. Following the termination of the reaction, pathlength correction values were measured with near‐infrared (IR) absorbance at 900 and 975 nm. The raw absorbance data were processed in Microsoft Excel to exclude points with A340 > 2.5 and to correct for pathlength using a system‐specific K‐Factor of 0.168, which was measured and applied as described.9 The corrected absorbance data vs time was fit to a linear model in GraphPad Prism (GraphPad Software, San Diego, CA) to yield reaction slopes, which were compared to a standard curve of nicotinamide adenine dinucleotide (NADH; 5–100 μM) to yield enzyme rates in U/L, where U is defined as the amount of enzyme that results in the reduction of 1 μmol·L−1·min−1 nicotinamide adenine dinucleotide phosphate (NADP).
3. TNNI ELISAS
Plasma concentrations of TNNI isoforms for slow and fast muscle were measured by capture ELISA. The slow isoform (TNNI1) was measured using a commercially available test kit (LS‐F7068, LifeSpan Biosciences, Inc, Seattle, WA) and was performed according to the manufacturer's instructions. The fast isoform (TNNI2) was assayed as described previously.4 Briefly, high‐binding ELISA plates were coated with α‐TNNI2 monoclonal antibody (Clone 7G2, OriGene, Inc., Rockville, MD) at a concentration 6.4 μg/mL overnight at 4°C. The wells were blocked with 1% w/v non‐fat dry milk in PBS for 30 min at 37°C., followed by incubation for 2 h at 37°C with the samples or recombinant human TNNI2 as a standard curve. The wells were washed with PBS containing 0.1% Tween‐20 (PBS‐T) and incubated with 1 μg/mL polyclonal α‐TNNI2 antibody (PA5‐76303, ThermoFisher, Inc.) for 90 min at 37°C. After washing with PBS‐T, the detection antibody (horseradish peroxidase [HRP]‐conjugated goat‐α‐rabbit IgG, 0.08 μg/mL, Pierce Biosciences) was added for 45 min at 37°C, and the HRP was visualized with Ultra‐TMB colorimetric reagent (ThermoFisher) followed by quenching with 2 N H2SO4 and measurement of the absorbance at 410 nm. Selectivity of these assays for fast vs slow TNNI has previously been confirmed using human muscle extracts.4
4. RESULTS
Demographics of subjects included in the study are described in Table 1. We observed a wide range of CK activities in all groups with higher average values in BMD patients compared to HV (678 ± 923 U/L and 355 ± 1743 U/L, respectively) and greater activity than both BMD and HV in DMD samples (3402 +/− 2969 U/L; P < .0001) (Figure 1A).
TABLE 1.
Demographics of muscular dystrophy patients and healthy controls
| Healthy (n = 50) | BMD (n = 49) | DMD (n = 132) | |
|---|---|---|---|
| Age (y) | 45.5 +/− 15.9 | 38.4 +/− 18.8 | 13.0 +/− 5.7 |
| Age range (y) | 19–75 | 6–73 | 2–33 |
| Non‐ambulant | – | 17 (35%) | 60 (45%) |
Note: Data are mean ± SD.
FIGURE 1.
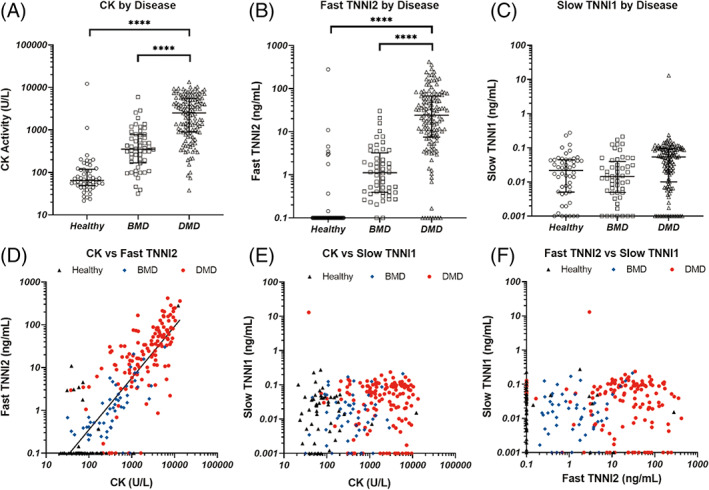
Plasma concentrations of CK enzymatic activity (A), TNNI1 (B), and TNNI2 (C) were measured in samples from BMD patients (squares) and DMD patients (triangles), with HV as controls (circles). In each panel, the error bars represent the median +/− the interquartile range. In panels (B) and (C), samples that exhibited no detectable TNNI concentration were assigned values equal to the assay's limit of detection (0.1 ng/mL and 0.001 ng/mL for fast and slow TNNI, respectively) When compared with each other, a significant correlation was found between CK and fast TNNI2 (D), with an R2 of 0.67. There was no significant correlation between CK and slow TNNI1 (E) nor between fast TNNI2 and slow TNNI1 (F) In panels (D) – (F), healthy samples are represented as black triangles, BMD as blue diamonds, and DMD red circles. ****: P < .0001. All other comparisons are non‐significant
We observed a similar pattern of elevated TNNI2 concentration (Figure 1B; HV: 6.0 +/− 38.9, BMD: 3.1 +/− 5.7, DMD: 51.0 +/− 71.6 ng/mL), although unlike CK, 83% of HV samples had TNNI2 concentrations below the level of detection for the assay (0.1 ng/mL). In contrast, all samples but one had concentrations of TNNI1 below 0.4 ng/mL (Figure 1C) with no clear relationship between disease group and concentration (HV: 0.036 +/− 0.052, BMD: 0.034 +/− 0.048, DMD: 0.16 +/− 1.13 ng/mL). A significant correlation was present between TNNI2 and CK (R2=0.68, P < .001) (Figure 1D) while no such correlation existed between TNNI1 and CK or TNNI1 and TNNI2 (Figure 1E, F).
CK activity was highest in younger DMD patients with a decline in activity in older patients (Figure 2A, T1/2 for the first‐order decay fit: 5.6 y). Similar results were obtained for TNNI2 (Figure 2B) but not TNNI1 (Figure 2C). In the BMD patients, CK and TNNI2 levels were higher in younger patients, but the relationship with age was less clear and flattened compared to DMD (Figure 2D, E ). As with DMD, TNNI1 levels in BMD were low and had no age relationship (Figure 2F).
FIGURE 2.
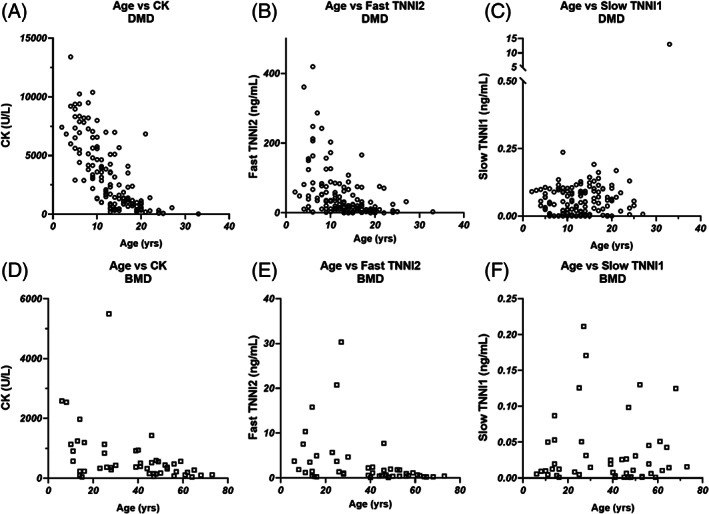
Concentration of CK enzymatic activity (A), TNNI2 (B), and TNNI1 (C) vs patient age in DMD patient samples. The same comparisons were made for BMD patients in panels (D, E, and F) for CK, TNNI2, and TNNI1, respectively
Non‐ambulant DMD patients had significantly lower CK activity and TNNI2 concentrations than ambulant patients (Figure 3A, B, CK: 798 +/− 955 U/L vs 3276 +/− 1704, TNNI2: 25.2 +/− 32.2 vs 82.6 +/− 86.9) but not lower TNNI1 (Figure 3C). Within the BMD patient population, the changes were not significant at the 95% confidence level (Figure 3D–F).
FIGURE 3.
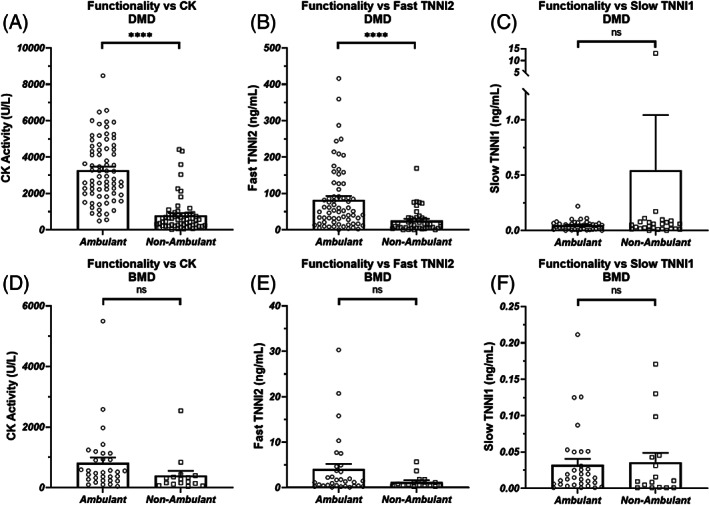
Ambulatory status for DMD was compared against plasma concentrations of CK enzymatic activity (A), TNNI2 (B), and TNNI1 (C). The same comparisons were made for BMD (D, E, and F). A patient was defined as “ambulatory” so long as the patient was not described as wholly dependent upon a wheelchair for mobility. Bars represent the mean +/− the standard error for the population. ****: P < .0001, ns: non‐significant
5. DISCUSSION
This cross‐sectional, retrospective study describes unexpected differences between fast and slow skeletal muscle biomarkers in DMD/BMD patient plasma. Although the data demonstrate a marked difference between fast and slow biomarkers between groups, it should be noted that a limitation of the current study is that these are not age matched, leaving the possibility of a different conclusion with closer age comparison. Also of note, aggregated CK activity levels in our BMD and DMD samples were lower than those measured in recent published datasets,10 possibly due to differences between assay systems. Finally, the mean CK of 355 U/L in the HV population was higher than is generally observed in healthy populations. This high value is driven by two individuals with much higher levels than the rest of the population (approximately 1000 and 10 000 U/L; Figure 1A). Unaccustomed exercise in healthy individuals can raise circulating CK above upper limits of normal and beyond 10 000 U/L.11 Therefore, the most likely explanation is that these individuals performed some form of strenuous exercise prior to sampling. The population median (65.0, shown in Figure 1A) is in line with normal ranges for males (22–33412).
The findings of differential troponin levels do, however, agree with a previous study that included two myopathy patients,13 although the specific disorders and absolute concentration of TNNI isoforms were not provided in that study. Our findings are also similar to studies in HV performing injurious eccentric exercise. In this setting, transient elevations in TNNI2 but not TNNI1 were observed after unilateral arm exercise3 or more extreme whole body exercise regimens4 and levels of TNNI2 were correlated with those of CK. Elevations in both TNNI1 and TNNI2 have been recorded in severe trauma and ischemia,13 suggesting that the release of TNNI1 into circulation may require muscle injury beyond that caused by contraction‐based injury, even in the context of dystrophin absence or mutation. Similar observations have also been made for cardiac TNNI3, which is only measurable in circulation after a severe ischemic event.14
Differences in disease susceptibility between fast and slow skeletal muscle fibers has been documented in both clinical and preclinical studies. Histological evidence of increased fast fiber disruption was first documented in DMD patients by examination of the fiber distribution of embryonic myosin heavy chain (eMHC), a contractile protein only expressed in regenerating fibers.7 Similar findings have also been documented in preclinical models of DMD, including dystrophic pigs and dogs.15, 16 Transgenic mdx mice that have a higher proportion of slow fibers also have milder disease progression.17, 18 Our observations provide additional detail regarding this relative susceptibility but also raise a number of questions.
As disease progresses in DMD, fibrosis and fatty tissue slowly replace muscle. Remaining skeletal muscle fibers also exhibit advanced pathophysiology including co‐expression of fast and slow myosin along with eMHC, which is observed in mixed, slow, and fast fibers.19 Rates of muscle turnover (measured with eMHC) are approximately constant with age (~30%),20 suggesting that changes in muscle composition do not affect rates of injury. In light of these changes, our results of low TNNI1, independent of age are surprising and suggest that slow or mixed fast/slow fiber injury or turnover occurs differently from fast fibers. One possible explanation is that membrane injury in DMD and BMD is considerably more severe in pure fast fibers compared to slow fibers. Fast myofibers develop greater relative force,21 and this would be expected to impart greater physical stresses on the dystrophin‐deficient membrane. Structural response to stress may also be different as fast fibers contain smaller integrating z‐discs22 that are more susceptibility to disruption.23 Additional studies will be required to determine why slow muscle fibers resist membrane rupture with contraction injury and may inform novel strategies to protect fast fibers from excessive disruption in BMD and DMD.
Our observations of decreasing CK and TNNI2 with age and ambulatory status in DMD are consistent to those seen with other studies.10, 24 This trajectory is usually assigned to decreases in muscle volume due to replacement through fibrosis. Our observations of differences between TNNI1 and TNNI2 levels with age suggest that the decreases in CK may be more nuanced, reflecting only the injury status of fast fibers.
CK is the most commonly measured circulating biomarker of muscle injury, and elevated levels are commonly used to diagnose pediatric skeletal muscle myopathies. Our cross‐sectional data reveal interesting differences between the distribution of CK and TNNI2. The majority of HV (83%) had TNNI2 levels below the lower level of detection of the ELISA (<0.1 ng/mL), while only 4% of BMD and 6% of DMD patients had non‐measurable levels of TNNI2. In contrast, CK activity was measurable in all samples tested with large overlap between HV and BMD activity. This difference is consistent with other cross‐sectional examinations of TNNI2 vs CK.10 A more sensitive TNNI2 assay will be required to properly define basal concentrations in HV and differences between healthy, BMD, and DMD patients. Greater understanding of these differences may provide a useful biomarker tool for the assessment of novel treatment effects in patients with relatively lower CK activity including BMD or older DMD patients.
CONFLICT OF INTEREST
Benjamin Barthel and Alan Russell are paid employees of Edgewise Therapeutics, Inc. The remaining authors have no conflicts of interest.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ACKNOWLEDGMENTS
Source of funding, private industry.
Barthel BL, Cox D, Barbieri M, et al. Elevation of fast but not slow troponin I in the circulation of patients with Becker and Duchenne muscular dystrophy. Muscle & Nerve. 2021;64:43–49. 10.1002/mus.27222
DATA AVAILABILITY STATEMENT
Data available on request from the authors
REFERENCES
- 1.Saltin B, Gollnick PD. Skeletal muscle adaptability: significance for metabolism and performance. In: Peachey LD, Adrian RH, Geiger SR, eds. Handbook of Physiology: A Critical, Comprehensive Presentation of Physiological Knowledge and Concepts. Bethesda, MD: American Physiological Society; 1983:555‐631. [Google Scholar]
- 2.Edgerton VR, Smith JL, Simpson DR. Muscle fibre type populations of human leg muscles. Histochem J. 1975;7(3):259‐266. [DOI] [PubMed] [Google Scholar]
- 3.Chapman DW, Simpson JA, Iscoe S, Robins T, Nosaka K. Changes in serum fast and slow skeletal troponin I concentration following maximal eccentric contractions. J Sci Med Sport. 2013;16(1):82‐85. [DOI] [PubMed] [Google Scholar]
- 4.Chen TC, Liu HW, Russell A, et al. Large increases in plasma fast skeletal muscle troponin I after whole‐body eccentric exercises. J Sci Med Sport. 2020;23:776‐781. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman EP, Fischbeck KH, Brown RH, et al. Characterization of dystrophin in muscle‐biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N Engl J Med. 1988;318(21):1363‐1368. [DOI] [PubMed] [Google Scholar]
- 6.Gao QQ, McNally EM. The Dystrophin complex: structure, function, and implications for therapy. Compr Physiol. 2015;5(3):1223‐1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webster C, Silberstein L, Hays AP, Blau HM. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell. 1988;52(4):503‐513. [DOI] [PubMed] [Google Scholar]
- 8.Pedemonte M, Sandri C, Schiaffino S, Minetti C. Early decrease of IIx myosin heavy chain transcripts in Duchenne muscular dystrophy. Biochem Biophys Res Commun. 1999;255(2):466‐469. [DOI] [PubMed] [Google Scholar]
- 9.Lampinen J, Raitio M, Perälä A, Oranen H, Harinen R. Correction method for photometric DNA quantification assay. Thermo Fisher Scientific Application Notes. 2012;1‐7. [Google Scholar]
- 10.Burch PM, Pogoryelova O, Goldstein R, et al. Muscle‐derived proteins as serum biomarkers for monitoring disease progression in three forms of muscular dystrophy. J Neuromuscul Dis. 2015;2(3):241‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen TC, Yang TJ, Huang MJ, et al. Damage and the repeated bout effect of arm, leg, and trunk muscles induced by eccentric resistance exercises. Scand J Med Sci Sports. 2019;29(5):725‐735. [DOI] [PubMed] [Google Scholar]
- 12.George MD, McGill NK, Baker JF. Creatine kinase in the U.S. population: impact of demographics, comorbidities, and body composition on the normal range. Medicine (Baltimore). 2016;95(33):e4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simpson JA, Labugger R, Collier C, Brison RJ, Iscoe S, Van Eyk JE. Fast and slow skeletal troponin I in serum from patients with various skeletal muscle disorders: a pilot study. Clin Chem. 2005;51(6):966‐972. [DOI] [PubMed] [Google Scholar]
- 14.Garg P, Morris P, Fazlanie AL, et al. Cardiac biomarkers of acute coronary syndrome: from history to high‐sensitivity cardiac troponin. Intern Emerg Med. 2017;12(2):147‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuasa K, Nakamura A, Hijikata T, Takeda S. Dystrophin deficiency in canine X‐linked muscular dystrophy in Japan (CXMDJ) alters myosin heavy chain expression profiles in the diaphragm more markedly than in the tibialis cranialis muscle. BMC Musculoskelet Disord. 2008;9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frohlich T, Kemter E, Flenkenthaler F, et al. Progressive muscle proteome changes in a clinically relevant pig model of Duchenne muscular dystrophy. Sci Rep. 2016;6:33362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selsby JT, Morine KJ, Pendrak K, Barton ER, Sweeney HL. Rescue of dystrophic skeletal muscle by PGC‐1alpha involves a fast to slow fiber type shift in the mdx mouse. PLoS One. 2012;7(1):e30063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chalkiadaki A, Igarashi M, Nasamu AS, Knezevic J, Guarente L. Muscle‐specific SIRT1 gain‐of‐function increases slow‐twitch fibers and ameliorates pathophysiology in a mouse model of duchenne muscular dystrophy. PLoS Genet. 2014;10(7):e1004490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marini JF, Pons F, Leger J, et al. Expression of myosin heavy chain isoforms in Duchenne muscular dystrophy patients and carriers. Neuromuscul Disord. 1991;1(6):397‐409. [DOI] [PubMed] [Google Scholar]
- 20.Janghra N, Morgan JE, Sewry CA, et al. Correlation of Utrophin levels with the dystrophin protein complex and muscle fibre regeneration in Duchenne and Becker muscular dystrophy muscle biopsies. PLoS One. 2016;11(3):e0150818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller MS, Bedrin NG, Ades PA, Palmer BM, Toth MJ. Molecular determinants of force production in human skeletal muscle fibers: effects of myosin isoform expression and cross‐sectional area. Am J Physiol Cell Physiol. 2015;308(6):C473‐C484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luther PK. The vertebrate muscle Z‐disc: sarcomere anchor for structure and signalling. J Muscle Res Cell Motil. 2009;30(5–6):171‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friden J, Sjostrom M, Ekblom B. Myofibrillar damage following intense eccentric exercise in man. Int J Sports Med. 1983;4(3):170‐176. [DOI] [PubMed] [Google Scholar]
- 24.Zatz M, Rapaport D, Vainzof M, et al. Serum creatine‐kinase (CK) and pyruvate‐kinase (PK) activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. J Neurol Sci. 1991;102(2):190‐196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request from the authors