

Abstract
Ir‐catalyzed asymmetric alkene hydrogenation is presented as the strategy par excellence to prepare saturated isoprenoids and mycoketides. This highly stereoselective synthesis approach is combined with an established 13C‐NMR method to determine the enantioselectivity of each methyl‐branched stereocenter. It is shown that this analysis is fit for purpose and the combination allows the synthesis of the title compounds with a significant increase in efficiency.
Keywords: Archaea, asymmetric hydrogenation, lipids, membrane spanning, metathesis
Cyclo‐archaeol, iso‐caldarchaeol, caldarchaeol, and mycoketide are prepared by employing asymmetric hydrogenation to generate the multiple stereocenters.

Introduction
Long‐chain syn‐1,5 methyl‐branched lipids are biologically highly relevant compounds from either isoprenoid or polyketide origin. The architecture is found in a range of natural products such as tocopherols (vitamin E),[1] a plethora of insect pheromones,[2] lipids[3] and ketides[7] (Figure 1). Archaeal membrane phospholipids, are produced via an isoprenoid pathway. Archaea are able to thrive under harsh conditions such as high salinity, acidity and temperature and the methyl‐branched lipids are essential for membrane fluidity and decreased ion permeability.[3] Next to bilayer forming lipids, archaea produce membrane‐spanning lipids, of which caldarchaeol is the most well‐known. The stereochemistry of the methyl substituents was established by the Heathcock group, who synthesized the C40‐lipid chain.[4] This C40‐unit was later also synthesized by Czeskis.[5] Still much is unknown about the biosynthesis of these macrocyclic archaeal lipids. It is unclear how the macrocycle is formed, which enzymes are involved in this process or even the exact structures of the precursors.[6]
Figure 1.

Natural products bearing a large number of syn‐1,5 methyl‐branches.
Evolutionary unrelated, syn‐1,5 methyl‐branched lipid chains are also part of phosphomycoketides; long‐chain lipids of polyketide origin found in Mycobacterium tuberculosis. These compounds have received considerable attention as antigens, conferred by the antigen presenting protein CD1c.[7, 8] Co‐crystallisation of mannosyl‐phosphomycoketide with CD1c demonstrated that the stereochemistry of the five methyl branches in mannosyl phosphomycoketide (“all‐S”) is critical for binding.[9, 10]
The synthesis of syn‐1,5 methyl‐branched chains is challenging as the compounds are largely devoid of functional groups and the stereocenters are mutually independent so chirality‐transfer cannot be used. Therefore, all stereocenters have to be introduced individually by either chiral pool‐based approaches or asymmetric synthesis. Two successful strategies for the synthesis of these long chain syn‐1,5 methyl‐branched natural products have been reported. Starting from Roche ester (5) (Figure 2 a); the group of Kakinuma synthesized intermediate 7 by first extending 5, and subsequently coupling the methyl bearing intermediates with a sulfone‐halide coupling.[11, 12, 13]
Figure 2.
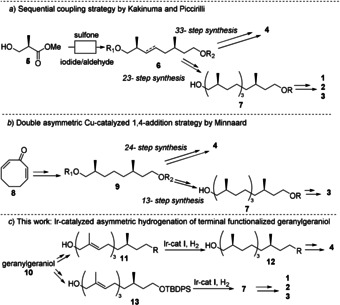
Overview of the strategies for the synthesis of long chain syn‐1,5 methyl‐branched lipids, by Kakinuma,[10, 11, 12] Piccirilli,[13, 14] and Minnaard.[15, 16]
Using 7 as the basis of their synthesis of archaeal lipids; they reported the first total synthesis of cyclo‐archaeol 3 in 1994[11] in 28 steps. In 1998, also using 7, the first total synthesis of both isomers of the 72‐membered macrocycle 1 and 2 were reported, both in a 45‐step synthesis.[13] Piccirilli et al. employed a similar strategy, using Julia‐Kocienski olefinations to extend the 1,5 methyl‐branched chain.[14] En route to mannose phosphomycoketide, they prepared mycoketide 4 in 33 steps.[15]
The second strategy relies on the construction of the stereocenters via asymmetric synthesis. Our group used a Cu‐catalyzed asymmetric conjugate addition of dimethylzinc to introduce two (syn) methyl‐branched centers starting from cycloocta‐2,7‐dienone (Figure 2 b).[16] This strategy allowed the synthesis of cyclo‐archaeol 3 in 22‐steps via intermediate 7,[17] and the first asymmetric synthesis of mycoketide 4 in 24‐steps.[18] Clearly, the synthesis of syn‐1,5 methyl‐branched lipids is laborious due to the large number of steps. In particular for mycoketide, an attractable synthesis of 4 is in need as mannose phosphomycoketide is studied as a biomarker and TB‐vaccine candidate.[19, 20] Mixtures of archaeal lipids can be isolated from cultures, but the pure compounds are not available and this holds especially for caldarchaeol/iso‐caldarchaeol as these cannot be separated.
In 2006, the Pfaltz group reported on iridium‐catalyzed asymmetric hydrogenation of unfunctionalized alkenes,[22] showcased with a concise and highly enantioselective synthesis of vitamin E. This approach allows the direct stereoselective conversion of isoprene units into a methyl‐branched stereocenter. In a subsequent publication, Pfaltz and co‐workers reported the asymmetric hydrogenation of farnesol, employing iridium catalyst I.[24] We realized that asymmetric hydrogenation of more functionalized isoprenoid systems would provide a very efficient entry into all‐syn 1,5‐methyl systems, provided that the starting material would be readily available. In addition, the scope of the hydrogenation reaction should be sufficiently broad, and the enantiopurity of each methyl‐branched center could be determined a posteriori. As a proof of the efficiency of such an approach, we projected the synthesis of the 36‐membered macrocycle cyclo‐archaeol 3, the two 72‐membered macrocycles caldarchaeol 1 and iso‐caldarchaeol 2, and mycoketide 4. It turns out that all these compounds can be produced with a considerable decrease in step‐count compared to the existing routes, which ultimately affords the mycoketides as a readily available group of antigens for immunological research on tuberculosis.
Results and Discussion
Construction of the syn‐1,5 Methyl Array
For the synthesis of macrocyclic archaeal lipids 1, 2 and 3 we developed a strategy based on intermediate 21 (Scheme 1). Subsequently, appropriately functionalized derivatives of 21 can be connected to either the secondary or primary hydroxyl group of the glycerol scaffold to prepare the different variants of macrocycles. In addition, 19 serves as the basis for the preparation of the mycoketide. Building block 21 would have to be constructed via the Ir‐catalyzed asymmetric hydrogenation of geranylgeraniol derivative 20. To make the synthesis as efficient as possible we planned the installation of a hydroxyl moiety on the unfunctionalized terminus of geranylgeraniol, which already has the full carbon skeleton in place.
Scheme 1.

Synthesis of building block 21: a) Ru{(S)‐Tol‐BINAP}(OAc)2, H2 (50 bar), MeOH, rt, 16 h, 94 %; b) Imidazole, TBDPSCl, CH2Cl2, 0 °C, 4 h, 95 %; c) NBS, 2:1 THF/H2O, 0 °C, 5 h; d) KOt‐Bu, THF, 0 °C, 30 min, 49 % over 2‐steps; e) H5IO6, THF, 0 °C, 1 h; f) (carbethoxyethylidene)triphenylphosphorane, THF, reflux, 16 h, 72 % over 2‐steps; g) DIBAL‐H, CH2Cl2, −78 °C, 30 min, 98 %; h) 1 mol % Cat I, H2 (50 bar), CH2Cl2, 0 °C, 16 h, 94 %.
To obtain multigram quantities of all‐E‐geranylgeraniol, commercially available annatto seeds (Bixa orellana) were extracted with heptane followed by fractional distillation and column chromatography of the extract. This provided 35 g of pure all‐E‐geranylgeraniol from 10 kg of seeds. Exploratory hydrogenation reactions revealed, congruent with observations of Pfaltz and co‐workers,[23, 24, 25] that the hydroxy group caused a small but distinct decrease in the stereoselectivity of the hydrogenation of the proximal alkene. Therefore, to obtain 21 in the maximum diastereoselectivity (d.r.), we started the synthesis with a Noyori asymmetric hydrogenation of the allylic alcohol (Scheme 1),[24, 26] with excellent enantioselectivity, followed by TBDPS protection. To functionalize the terminus, we initially opted for a SeO2 catalyzed olefin oxidation to install a hydroxyl function.[27] However, in the consecutive Ir‐catalyzed hydrogenation to the fully saturated system, we observed a disappointing d.r. of the terminal C4‐Me. Although a 5–10 % decrease in enantioselectivity has been observed in Ir‐catalyzed hydrogenation of allylic alcohols, the current reaction faced a 30–35 % loss of selectivity at this position. Transformation of 20 to the aldehyde revealed, finally, that the Riley oxidation to 20 had produced a 1:3 cis‐trans mixture. This is problematic as the cis‐isomer is transformed to the anti‐diastereomer in the hydrogenation. Due to the low‐yielding Riley oxidation (20 % for the E‐isomer) and the problematic separation of the isomeric mixture, a change in strategy was necessary. We chose for a high yielding epoxidation/oxidation/olefination sequence to install the desired trans alcohol, yielding 20 in 35 % yield over 5‐steps.[28]
Triene 20 was hydrogenated with iridium catalyst I and 50 bar of hydrogen, to yield the desired all‐syn methyl‐branched chain.
Both diastereomeric Mosher's esters revealed a 94 % selectivity on the terminal C4‐Me branch. In order to determine the d.r. of the “internal” methyl branches, the 13C‐NMR‐based method of Curran et al. was applied.[29] Based on a series of model compounds, this method predicts the chemical shifts of syn and anti‐stereoisomers in 1,5‐methyl branched systems with high fidelity. The signals of the “anti‐isomers” of the C3‐Me and C2‐Me branches could be located (Figure 3), but due to signal overlap the syn/anti ratio could only be determined by approximation. The signal of anti C4‐Me, on the contrary, is isolated and integrates as 6 % of the syn C4‐Me. The integral is the cumulation of S,R and R,S, as in both systems C4‐Me has an anti‐relation with the neighboring methyl‐branch. As the Mosher's ester method revealed a selectivity on C21 of 94 %, it is safe to state that there is an absolute minimum amount of anti C3‐Me. This means 21 is obtained with a minimum over‐all d.r. of 89 %, with the hydrogenation protocol developed by Pfaltz and introducing three stereocenters in one reaction.[24] 21 is prepared in 8‐steps, 31 % yield, a considerable improvement over the 13‐step synthesis, 13 % yield previously reported.[17] Furthermore, just four column purifications are required for the synthesis of 21, which ultimately enables a large‐scale synthesis.
Figure 3.
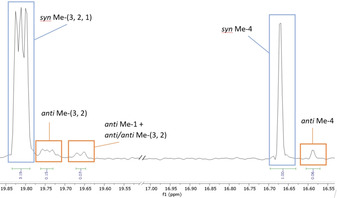
13C‐NMR analysis of compound 21 (note: cut‐out 17.05–19.50 ppm). Resolution enhanced with TRAF apodization.
The Synthesis of Cyclo‐Archaeol 3
With building block 21 in hand, we commenced with the synthesis of cyclo‐archaeol 3. Previously, our group reported the synthesis of 3 using ring‐closing metathesis.[17] The terminal alkenes were installed at a late stage via a Wittig olefination. We opted for a similar approach, but decreasing the step‐count by installing the terminal alkenes at an early stage (Scheme 2). In this way, 21 is used for the alkylation of both the secondary and the primary position of the glycerol backbone. The oxidation, olefination, deprotection sequence gave terminal alkene 24 in 68 % yield with only one column purification required.
Scheme 2.

Synthesis of cyclo‐archaeol: a) (COCl)2, DMSO, Et3N, CH2Cl2, −60 °C, 1 h; b) Methyltriphenylphosphonium iodide, BuLi, THF, −30 °C; c) TBAF, THF, rt, 4 h, 68 % over 3‐steps; d) (S)‐benzylglycidylether, (S,S)‐(salen)CoIII⋅OTs, O2‐atmosphere, rt, neat, 16 h, 92 %; e) MsCl, Et3N, CH2Cl2, 0 °C, 2 h, 96 %; f) NaH, 15‐crown‐5, THF, 16 h, rt, 76 %; g) Grubbs 2nd gen catalyst, CH2Cl2, rt, 48 h, 83 %; h) Pt/C, H2, 2:1 MeOH/CH2Cl2, rt, 16 h; i) Pd/C, H2, EtOAc, 6 h, 74 % over 2‐steps.
To obtain di‐ether 27 as the precursor for the ring‐closing metathesis, a Co‐salen catalyst was employed to ring‐open (S)‐benzylglycidylether with 24 as the nucleophile. While this cobalt catalyst is most well‐known for the kinetic resolution of terminal epoxides with water, according to Jacobsen,[30] we have shown that the catalyst also enables the synthesis of glycerol‐based ether lipids.[17] We opted for this strategy due to the low yields observed in the dialkylation of mono‐protected glycerol with mesylate 26. The combination of this Co‐catalyst and a terminal alkene is unprecedented as cobalt is known to facilitate oxidation.[31] We were therefore pleased to isolate the desired mono‐ether 25 in 92 % yield after 16 h reaction time. The secondary hydroxy group was etherified in a base‐mediated reaction with 26. Initial studies of this reaction gave mediocre yields (35–50 %), as under alkaline conditions the mesylate is prone to elimination. Aspinall et al. speculated that an α‐ether oxygen coordinates to the formed sodium alkoxide, leading to a stable chelate.[32] It turned out that the addition of crown ether significantly increased the yield of the etherification to 76 %, likely by dissociating the sodium ion. The application of this protocol to the one‐step di‐ether formation, however, led to a yield of 40 %.
The 36‐membered macrocycle was closed with Grubbs II catalyst in a high‐yielding ring‐closing metathesis giving 28 in 83 % yield. The internal alkene was hydrogenated with Pt/C, as palladium is known to racemize α‐stereocenters via alkene isomerization. Subsequent Pd/C mediated hydrogenolysis of the benzyl group furnished the desired 3 in 74 % yield over 2‐steps. Cyclo‐archaeol 3 was made in 11 % yield over 17‐steps, which is a significant improvement compared to the previous 22‐step synthesis by our group.[17] In addition, just nine column purifications are required in the entire synthesis.
The Synthesis of iso‐Caldarchaeol
With the 36‐membered macrocycle in‐hand we were optimistic that we could construct both isomers of the 72‐membered macrocycle with a similar approach. The main challenge here is the coupling of the two di‐ether fragments. We were able to construct the C40 chain via the dimerization of 25, employing a Ru‐catalyzed metathesis. The highest conversion towards the dimer of 25 was observed using 20 mol % Hoveyda‐Grubbs 2nd generation catalyst (Scheme 3). Notably, the cis and trans isomers differed significantly in R f, and the cis‐isomer was co‐polar with the starting material. After Pt/C catalyzed hydrogenation, pure saturated 29 was isolated in 65 % yield over 2‐steps.
Scheme 3.

Synthesis of iso‐caldarchaeol: a) Grubbs‐Hoveyda 2nd gen catalyst, CH2Cl2, rt, 24 h; b) Pt/C, 2:1 MeOH/CH2Cl2, rt, 16 h, 65 % over 2‐steps; c) NaH, 26, 15‐crown‐5, THF, 50 °C, 16 h, 55 %; d) Grubbs 2nd gen catalyst, CH2Cl2, rt, 48 h; e) Pt/C, H2, 2:1 MeOH/CH2Cl2, rt, 16 h; f) Pd/C, H2, EtOAc, rt, 16 h, 30 % over 3‐steps.
We proceeded with the alkylation of both secondary hydroxy groups in 29 with mesylate 26. The conditions previously used in the alkylation of 25 led to a mediocre yield of 35 %, however, increasing the reaction temperature to 50 °C increased the yield to 55 %. Exposing 30 to ring‐closing metathesis furnished the 72‐membered macrocycle 31, although because starting material and product are both exceptionally apolar, we were not able to recover the remaining starting material from the mixture. Subsequent hydrogenation and hydrogenolysis by Pt/C and Pd/C, respectively, provided 2 in 30 % yield over the final 3‐steps.
Iso‐caldarchaeol 2 was prepared in an overall step‐count of 20 and 3 % yield starting from geranygeraniol, which is substantially more efficient than the previously reported 45‐step synthesis.[13] Furthermore, just 9‐column purifications are required for the synthesis of 2.
The Synthesis of Caldarchaeol
Caldarchaeol is considerably more complicated to synthesize than iso‐caldarchaeol as it lacks the inherent symmetry of the latter. The methyl‐branched chains are connected crosswise to the glycerol head groups. Hence it requires the synthesis of two orthogonally protected fragments. We commenced with PMB (p‐methoxybenzyl)‐protection and desilylation of 21 (Scheme 4). One part of the resulting alcohol 34 was converted to mesylate 41 in 90 % yield, whereas the other part was subjected to CoIII‐salen catalyzed ring‐opening of (S)‐benzylglycidylether, giving the corresponding mono‐substituted benzyl‐glycerol in 83 % yield. The secondary hydroxy group was subsequently etherified with mesylate 26 to give di‐ether 36 in 60 % yield. PMB‐mesylate 41 was used to alkylate mono‐substituted benzylglycerol 25 to obtain 42, the counterpart of 36. We noted a consistently lower yield in alkylation reactions with compounds possessing a PMB group compared to alkylations leading to di‐alkene 27 (60 % vs. 75 %). Stacking of the PMB group with the benzyl‐protecting group possibly increases the steric hindrance around the secondary hydroxy group, hampering the alkylation.
Scheme 4.

Synthesis of caldarchaeol: a) NaH, 15‐crown‐5, PMBCl, rt, 16 h, 88 %; b) TBAF, THF, rt, 5 h, 95 %; c) (S)‐benzylglycidylether, (S,S)‐(salen)CoIII⋅OTs, O2‐atmosphere, rt, neat, 16 h, 83 %; d) NaH then 26, 15‐crown‐5, THF, 16 h, rt, 60 %; e) DDQ, 1:4 H2O/THF, 0 °C, 1 h, 81 %; f) MsCl, Et3N, THF, 0 °C, 1 h; g) thiophenol, K2CO3, DMF, rt, 16 h; h) (NH4)2MoO4, H2O2, n‐BuOH, THF, rt, 16 h, 69 % over 3‐steps; i) MsCl, Et3N, CH2Cl2, rt, 16 h, 90 % j) 25, NaH, 15‐crown‐5, THF, 16 h, rt, 60 %; k) DDQ, 1:4 H2O/THF, 0 °C, 1 h, 84 %; l) (COCl)2, DMSO, Et3N, CH2Cl2, −60 °C, 1 h, 98 %; m) BuLi, THF, −25 °C, 30 min, 55 %; n) Ac2O, DMAP, pyridine, rt, 16 h, 92 %; o) Grubbs 2nd gen catalyst, CH2Cl2, rt, 48 h, 52 % (73 % based on recovered sm); p) SmI2, HMPA, THF, rt, 1.5 h, 71 %; q) Pt/C, H2, 2:1 MeOH/CH2Cl2, rt, 16 h; r) Pd/C, H2, EtOAc, rt, 16 h, 76 % over 2‐steps.
To couple the two lipid fragments we decided to use a Julia olefination. Compared to its more advanced Kocienski‐modification, the Julia reaction is advantageous in this synthesis as the double bond is generated in a separate step. It has been shown that the presence of an internal alkene gives a mixture of 72 and 36‐membered rings in ring‐closing metathesis.[33] The hydroxy‐sulfone formed in the Julia reaction avoids this complication. The PMB‐ether in 42 was converted into the corresponding aldehyde via oxidative deprotection followed by a Swern oxidation to obtain 44 in excellent yield. Counterpart 36 was converted into the corresponding phenyl sulfone via oxidative deprotection (81 % yield).
The two lipid fragments 40 and 44 were connected in an n‐BuLi mediated Julia reaction to provide the desired hydroxy‐sulfone 45 in 55 % yield, which is consistent with previous reports.[13] Initially we planned to acetylate the hydroxy group of 45 after the ring‐closing metathesis, but it turned out that acetylation of the macrocycle was surprisingly more difficult than the acetylation of 45, which proceeded in excellent yield. Formation of the 72‐membered macrocycle proceeded smoothly using Grubbs 2nd generation catalyst, and provided 47 in 52 % yield (73 % yield brsm). Exposure of 47 to SmI2 [34] gave the desired alkene 48 in 71 % yield, and subsequent hydrogenation by Pt/C and hydrogenolysis of the benzyl functions gave 1 in 76 % yield over the final 2‐steps. Caldarchaeol 1 was synthesized in 28‐steps, a dramatic improvement compared to the previous 45‐step synthesis.[13]
The Synthesis of Mycoketide
Also mycoketide 4 can be efficiently prepared from geranylgeraniol, via intermediate 19. For this, we opted to do a late‐stage asymmetric hydrogenation including the remnant double bond from the olefination. After removal of the TBDPS group in 19, (Scheme 5 a) the hydroxy moiety was subjected to a Swern oxidation, and the resulting corresponding aldehyde was immediately used in a Julia‐Kocienski olefination with sulfone 59, prepared from commercially available materials. We were pleased to observe that the reaction was fully chemoselective and left the ethyl ester untouched. Compound 50 was isolated in 82 % yield over 2‐steps. Subsequent reduction with DIBAL afforded allylic alcohol 51 in 95 % yield. Ir‐catalysed asymmetric hydrogenation produced the fully saturated product 52 in quantitative yield. The stereoselectivity of the hydrogenation reaction was analyzed in the same way as for 21. Comparing the 13C chemical shifts with those described by Buter et al.,[35] we could extrapolate a 94 % d.r. for C1‐Me in 52 (for a detailed analysis see Supporting Information). The signal for C1‐Me in an anti‐relationship to its neighboring C2‐Me is separated from the adjacent peaks. Although the anti‐signal of C2‐Me is overlapping with the anti‐signals of C5‐Me and C4‐Me, the combined integration showed an identical selectivity as in compound 21 (i.e. 98 % selectivity). The e.r. of C4‐Me and C5‐Me had already been determined. All in all, 52 had been prepared with 87 % diastereomeric purity.
Scheme 5.

A) Synthesis of mycoketide: a) TBAF, THF, rt, 4 h, 95 %; b) (COCl)2, DMSO, Et3N, CH2Cl2, −60 °C, 1 h; c) 59, LHMDS, THF, −40 °C, 30 min, 82 % over 2‐steps; d) DIBAL‐H, CH2Cl2, −78 °C, 30 min, 95 %; e) 1 mol % Cat I, H2 (50 bar), CH2Cl2, 0 °C, 16 h; f) (COCl)2, DMSO, Et3N, CH2Cl2, −60 °C, 1 h; g) 60, KHMDS, THF, 30 min, −40 °C, 80 % over 3‐steps; h) 20 mol % 5‐ethyl riboflavin, N2H4⋅H2O, n‐BuOH, EtOH, rt, 48 h; i) Pd/C, H2, EtOAc, rt, 16 h, 88 % over 2‐steps. Scheme 5 B) synthesis of 59; a) 0.5 mol % Cat I, H2 (50 bar), CH2Cl2, 0 °C, 16 h, 97 %, 97 % e.r.; b) 1‐phenyl‐1H‐tetrazole‐5‐thiol, PPh3, DIAD, THF, 0 °C, 2 h; c) m‐CPBA, CH2Cl2, rt, 24 h, 73 % over 2‐steps.
Alcohol 52 was converted to the corresponding aldehyde and reacted in a Julia‐Kocienski olefination with 60, which in turn had been readily prepared from ((bromoethoxy)methyl)benzene. The group of Markó has reported a small but significant increase in yield using KHMDS, instead of LHMDS as the base in an olefination with a structurally related sulfone.[36] This translated to the current system, and with KHMDS, 54 was isolated in 80 % yield over 3‐steps.
Alkene 54 was reduced with a flavine catalyzed diimide reduction to avoid racemization of the adjacent stereocenter.[37] Final hydrogenolysis of the benzyl ether afforded 4 in 80 % yield. Mycoketide 4 was synthesized in a 15‐step longest linear sequence in 16 % overall yield, also for this compound a significant improvement compared to the previously reported 17‐steps and 8 % overall yield.[18]
Conclusion
The Ir‐catalyzed asymmetric alkene hydrogenation developed by Pfaltz et al. has been used as the basis of a highly efficient synthesis of saturated isoprenoids and mycoketides. An essential part of this approach is the ability to efficiently determine the stereoselectivity of the hydrogenation, as multiple mutually independent stereocenters are formed in the hydrogenation reaction. The 13C‐NMR based method proposed by Curran et al. proved to be extremely versatile in this respect, allowing a fast, streamlined synthesis of the archaeal membrane lipids caldarchaeol, iso‐caldarchaeol, and cyclo‐archaeol. These molecules will be of invariable use in resolving the remaining enzymatic steps in archaeal phospholipid biosynthesis. In addition, mycoketide has been prepared in a much more efficiently than previously reported. This is important as mannose phosphomycoketide, prepared from mycoketide, is considered an important antigen of M. tuberculosis and is under study as a biomarker of TB and as a TB‐vaccine component. Current immunological research is hampered by the availability of this compound, adding to the value of a concise synthesis of ketide 4.[38]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank Prof. A. Pfaltz for his kind donation of catalyst I in an early stage of the project. Prof. G. Appendino is kindly acknowledged for discussions on the isolation of geranylgeraniol.
R. L. H. Andringa, N. A. W. de Kok, A. J. M. Driessen, A. J. Minnaard, Angew. Chem. Int. Ed. 2021, 60, 17497.
References
- 1.Dowd P., Hershline R., Ham S. W., Naganathan S., Nat. Prod. Rep. 1994, 11, 251–264. [DOI] [PubMed] [Google Scholar]
- 2.Ando T., Yamakawa R., Nat. Prod. Rep. 2015, 32, 1007–1041. [DOI] [PubMed] [Google Scholar]
- 3.Jacquemet A., Barbeau J., Lemiègre L., Benvegnu T., Biochimie 2009, 91, 711–717. [DOI] [PubMed] [Google Scholar]
- 4.Heathcock C., Finkelstein B., Aoki T., Poulter C., Science 1985, 229, 862–864. [DOI] [PubMed] [Google Scholar]
- 5.Czeskis B. A., Alexeev I. G., Moiseenkov A. M., Russ. Chem. Bull. 1993, 42, 1246–1253. [Google Scholar]
- 6.Jain S., Caforio A., Driessen A. J. M., Front. Microbiol. 2014, 5, 641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moody D. B., Ulrichs T., Mühlecker W., Young D. C., Gurcha S. S., Grant E., Rosat J. P., Brenner M. B., Costello C. E., Besra, et al., Nature 2000, 404, 884–888. [DOI] [PubMed] [Google Scholar]
- 8.Matsunaga I., Sugita M., Clin. Dev. Immunol. 2012, 2012, 981821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsunaga I., Bhatt A., Young D. C., Cheng T. Y., Eyles S. J., Besra G. S., Briken V., Porcelli S. A., Costello C. E., Jacobs W. R., et al., J. Exp. Med. 2004, 200, 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scharf L., Li N. S., Hawk A. J., Garzón D., Zhang T., Fox L. M., Kazen A. R., Shah S., Haddadian E. J., Gumperz J. E., Saghatelian A., Faraldo-Gómez J. D., Meredith S. C., Piccirilli J. A., Adams E. J., Immunity 2010, 33, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eguchi T., Terachi T., Kakinuma K., J. Chem. Soc. Chem. Commun. 1994, 5, 137–138. [Google Scholar]
- 12.Eguchi T., Arakawa K., Terachi T., Kakinuma K., J. Org. Chem. 1997, 62, 1924–1933. [DOI] [PubMed] [Google Scholar]
- 13.Eguchi T., Ibaragi K., Kakinuma K., J. Org. Chem. 1998, 63, 2689–2698. [DOI] [PubMed] [Google Scholar]
- 14.Li N. S., Piccirilli J. A., Tetrahedron 2013, 69, 9633–9641. [Google Scholar]
- 15.Li N. S., Scharf L., Adams E. J., Piccirilli J. A., J. Org. Chem. 2013, 78, 5970–5986. [DOI] [PubMed] [Google Scholar]
- 16.Van Summeren R. P., Reijmer S. J. W., Feringa B. L., Minnaard A. J., Chem. Commun. 2005, 1387–1389. [DOI] [PubMed] [Google Scholar]
- 17.Ferrer C., Fodran P., Barroso S., Gibson R., Hopmans E. C., Damsté J. S., Schouten S., Minnaard A. J., Org. Biomol. Chem. 2013, 11, 2482–2492. [DOI] [PubMed] [Google Scholar]
- 18.van Summeren R. P., Moody D. B., Feringa B. L., Minnaard A. J., J. Am. Chem. Soc. 2006, 128, 4546–4547. [DOI] [PubMed] [Google Scholar]
- 19.Joosten S. A., Ottenhoff T. H. M., Lewinsohn D. M., Hoft D. F., Moody D. B., Seshadri C., Vaccine 2019, 37, 3022–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.James C. A., Seshadri C., Front. Immunol. 2020, 11, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crich D., Dudkin V., J. Am. Chem. Soc. 2002, 124, 2263–2266. [DOI] [PubMed] [Google Scholar]
- 22.Bell S., Wüstenberg B., Kaiser S., Menges F., Netscher T., Pfaltz A., Science 2006, 311, 642–644. [DOI] [PubMed] [Google Scholar]
- 23.Wang A., Fraga R. P. A., Hörmann E., Pfaltz A., Chem. Asian J. 2011, 6, 599–606. [DOI] [PubMed] [Google Scholar]
- 24.Wang A., Wüstenberg B., Pfaltz A., Angew. Chem. Int. Ed. 2008, 47, 2298–2300; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2330–2332. [Google Scholar]
- 25.Roseblade S. J., Pfaltz A., Acc. Chem. Res. 2007, 40, 1402–1411. [DOI] [PubMed] [Google Scholar]
- 26.Sita L. R., J. Org. Chem. 1993, 58, 5285–5287. [Google Scholar]
- 27.Umbreit M. A., Sharpless K. B., J. Am. Chem. Soc. 1977, 99, 5526–5528. [Google Scholar]
- 28.Handa S., Nair P. S., Pattenden G., Helv. Chim. Acta 2000, 83, 2629–2643. [Google Scholar]
- 29.Yeh E. A. H., Kumli E., Damodaran K., Curran D. P., J. Am. Chem. Soc. 2013, 135, 1577–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaus S. E., Brandes B. D., Larrow J. F., Tokunaga M., Hansen K. B., Gould A. E., Furrow M. E., Jacobsen E. N., J. Am. Chem. Soc. 2002, 124, 1307–1315. [DOI] [PubMed] [Google Scholar]
- 31.Reetz M. T., Töllner K., Tetrahedron Lett. 1995, 36, 9461–9464. [Google Scholar]
- 32.Aspinall H. C., Greeves N., Lee W. M., McIver E. G., Smith P. M., Tetrahedron Lett. 1997, 38, 4679–4682. [Google Scholar]
- 33.Arakawa K., Eguchi T., Kakinuma K., J. Org. Chem. 1998, 63, 4741–4745. [Google Scholar]
- 34.Keck G. E., Savin K. A., Weglarz M. A., J. Org. Chem. 1995, 60, 3194–3204. [Google Scholar]
- 35.Buter J., Yeh E. A. H., Budavich O. W., Damodaran K., Minnaard A. J., Curran D. P., J. Org. Chem. 2013, 78, 4913–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pospíšil J., Markó I. E., Org. Lett. 2006, 8, 5983–5986. [DOI] [PubMed] [Google Scholar]
- 37.Smit C., Fraaije M. W., Minnaard A. J., J. Org. Chem. 2008, 73, 9482–9485. [DOI] [PubMed] [Google Scholar]
- 38.Since acceptance of this manuscript, a related study was published: Falk I. D., Gál B., Bhattacharya A., Wei J. H., Welander P. V., Boxer S. G., Burns N. Z., Angew. Chem. Int. Ed. 2021, 10.1002/anie.202104051; [DOI] [Google Scholar]; Angew. Chem. 2021, 10.1002/ange.202104051. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information