

Abstract
The increasing number of available genomes, in combination with advanced genome mining techniques, unveiled a plethora of biosynthetic gene clusters (BGCs) coding for ribosomally synthesized and post‐translationally modified peptides (RiPPs). The products of these BGCs often represent an enormous resource for new and bioactive compounds, but frequently, they cannot be readily isolated and remain cryptic. Here, we describe a tunable metabologenomic approach that recruits a synergism of bioinformatics in tandem with isotope‐ and NMR‐guided platform to identify the product of an orphan RiPP gene cluster in the genomes of Nocardia terpenica IFM 0406 and 0706T. The application of this tactic resulted in the discovery of nocathioamides family as a founder of a new class of chimeric lanthipeptides I.
Keywords: chimeric lanthipeptide, macrocyclic imide, metabologenomic approach, Nocardia terpenica, RiPPs
Chimeric Lanthipeptides: Employing a tunable metabologenomic approach, nocathioamides A–C were isolated and elucidated. They inaugurate a new RiPP family, characterized by a unique and hyper degree of modifications, including an intramolecular imide bridge.
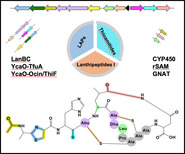
Introduction
RiPPs outline a significantly expanding category of peptide‐based natural products with multiple classes covering a vast chemical space. Such a broad scope of structural modifications translates into different biological activities, ranging from antibacterial, antifungal to antiviral potencies,[1] which can be therapeutically exploited. However, intriguing ecological and physiological roles are also reported for RiPPs.[2] Lanthipeptides, the most prevalent and exhaustively mined class of RiPPs, are structurally branded by their unifying installation of the thioether‐bridged amino acids lanthionine (Lan) and methyllanthionine (MeLan) as one of the frequent post‐translational modifications (PTMs) of RiPPs.[3] The typical introduction of sulfur and oxygen heterocycles within the core peptide comprising thazol(in)e and (methyl)oxazol(in)e moieties has also been perceived in several RiPP variants, such as linear azoline scaffolds (LAPs), head‐to‐tail monocyclic skeletons (cyanobactins), and multicyclic frameworks with a nitrogen‐containing heterocycle (pyritides).[4] Among the rare alterations of RiPPs, thioamitides arise in which thioamide(s) can be exceptionally incorporated into the peptide backbone to leverage their structural diversification and bioactive potential.[5] With the explosion of affordable sequencing technologies in orchestration with improved bioinformatics algorithms for biosynthetic gene cluster (BGC) identification and defined motif annotation,[6] the quest has been paved towards the discovery of a hidden arsenal of novel RiPP‐based molecules across various bacterial (meta)genomes. Nevertheless, connecting the RiPPs genetic locus of interest to the cognate metabolite(s) is yet a challenging task[7] due to the production of complex metabolite mixtures, the silence of many BGCs under laboratory cultivation conditions or if the source organism is considered uncultivable. The latter two hurdles are nowadays overcome by the application of heterologous expression and synthetic biology, while analytical problems are foremost addressed by the integration of mass spectrometry (MS)‐based metabolomics techniques.[8]
Bacteria from the genus Nocardia are foremost known in a medical context since they cause severe infections to humans and animals via nocardiosis. However, they have emerged over the last two decades also as talented producer strains of structurally diverse and highly bioactive natural products.[9] Beside nonribosomal peptides and polyketides, Nocardia spp. are also privileged producers of RiPPs, such as nocathiacins and nocardithiocin as thiopeptides[9] besides nocavionin[10] as lipolanthines. Within the course of our genome‐driven investigations of Nocardia strains,[11] we noted that the highly similar genomes of Nocardia terpenica IFM 406 and 706T both contain, beside nocavionin,[10] a further RiPP‐BGC which codes for a core ribosomal peptide co‐localized with a unique combination of post‐translational enzymes that have not been observed so far for lanthipeptides. Here, we report a genome‐guided identification, isolation and characterization of three structurally novel RiPPs, designated as nocathioamides A–C, from these strains, which represent the first members of a new class of chimeric lanthipeptides.
Results and Discussion
Genome Mining of the Nocathioamide Biosynthetic Gene Cluster in Nocardia terpenica IFM 0406 and 0706
Mining the genomes of N. terpenica IFM 406 and 706, employing the bioinformatic tools antiSMASH 5.1[6a] and RODEO[6b, 12] readily revealed the presence of a putative lanthipeptide BGC (Table S1). The comparatively large nta gene cluster (17.1 kb) consists of 14 open reading frames (ORFs), annotated as ntaA‐ntaN (Figure 1). NtaA‐ntaJ are transcribed in one direction, while ntaK‐ntaN are translated in the opposite orientation. The gene ntaA encodes a 51‐aa precursor peptide which showed no sequence similarity to any known RiPPs. The leader peptide possessed the conserved FDLN motif and towards its C‐terminal end two proline residues (Figure 1), which typically occurs in prepeptides of class I lantibiotics and suggest, with P commonly located at position −2, two putative cleavage sites.[13] To complicate matters further, NtaA contains in addition one characteristic tandem alanine “AA” and two “PA” cleavage sites[14] which give rise to multiple possibilities for dividing the precursor peptide into an N‐terminal leader peptide and a C‐terminal core peptide. Application of the automated web‐based tool RIPPMiner[15] corroborated these findings and predicted the formation of a 12 or 14mer. At this stage of the study, we assumed that the cleavage site is located within the “PAAPAC” sequence (residues −4 to +2) of the NtaA precursor peptide, which culminates in 12 to 15mer. We additionally expected that the dehydratases NtaF (Protein family PF04 738) and NtaG (PF14028) catalyze up to three dehydrations (on Thr4, Ser6 and Ser7), enabling thereby, in conjunction with the LanC‐like enzyme NtaB, the formation of up to three (methyl)lanthionine bridges (with Cys2, Cys10 and Cys11). Moreover, the nta cluster contained two YcaO‐encoding genes (ntaJ and ntaL) which would lead, depending on their type and partner protein, either to (methyl)azoline(s) formation or conversion of an amide(s) into a backbone thioamide(s).[12] The partner proteins were readily identified employing the RODEO[12] algorithm: the set of ntaK/ntaL was predicted to code for a pair of TfuA/YcaO proteins (PF07812/PF02624),[16] and ntaI/ntaJ to encode the tandem Ocin‐ThiF/YcaO (TIGR03693/ PF02624). Thus, we hypothesized that at the resultant RiPP would be thioamidated by NtaK/NtaL and that at least one Ser/Thr/Cys residue is converted into a (methyl)azoline catalyzed by NtaI/NtaJ. Notably, the application of RRE‐Finder implemented in antiSMASH 6.0[17] unveiled that the RiPP recognition element (RRE), which binds specifically to the precursor peptide (NtaA) guiding the post‐translationally modifying enzymes to their substrates, is fused to NtaI (Table S1). Taking into account the presence of a SagB‐dehydrogenase‐encoding gene[18] (ntaE, PF00881), it is conceivable that (methyl)azoline would be oxidized to (methyl)azole during the maturation of the RiPP. Consequently, the presence of a (methyl)azole ring system reduces, in turn, the maximum number of possible (methyl)lanthionine‐bridges from three to two. The analysis of the remaining ORFs of the nta BGC revealed genes for two GNAT‐N‐acetyltransferases (ntaH, PF13527 and ntaN, PF00583), a CYP450 oxygenase (ntaM, PF00067), a hydrolase (ntaD, PF01738) and a methyltransferase (ntaC, PF01135). Thus, we anticipated that the final peptide to be twice N‐acetylated and harbors an additional hydroxyl or keto‐group. Since methyltransferases catalyze a diverse spectrum of chemical reactions ranging from actual methyl transfer over epimerization to C−C crosslinking reactions,[1b, 19] no reliable prediction can be currently performed. In summary, the bioinformatic analysis suggested the production of a RiPP, formed by 12–15 amino acids featuring one to two (methyl)lanthionine bridges, thioamide bond(s), (methyl)azole ring(s) and possibly oxygenated in addition to acetylated amino acid residues. Due to the unprecedented peptide sequence and the predicted modifications of the resultant peptide, we embarked on the screening for the product(s) of the nta BGC.
Figure 1.
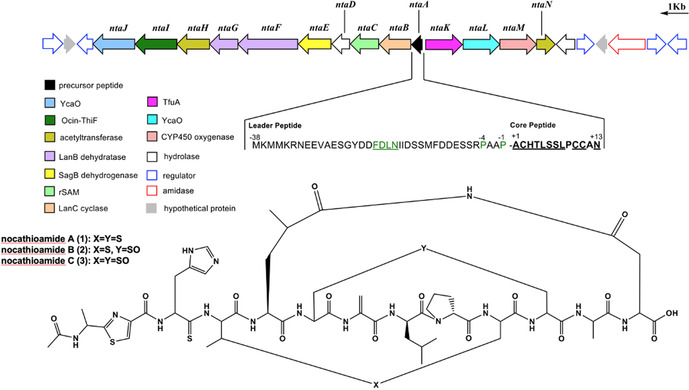
Top: Biosynthetic gene cluster coding for nocathioamides A–C. Bottom: Chemical structures of nocathioamides.
Targeted Identification and Isolation of Nocathioamides using a Configured Metabologenomic Approach
Counting on the bioinformatically predicted structural features and the limitation of formulating only a very broad mass range of the mature peptide, a three‐layered metabolomic workflow was designed to track down the resultant RiPP using OSMAC‐mass spectrometry (OSMAC‐MS), stable isotope labeling and 1H‐13C HMBC NMR (Figure S1). A panel of extracts, prepared under different culture conditions (30 different culture recipes, while temperature and fermentation periods were kept constant), was screened by liquid chromatography‐high resolution (LC‐HR)‐MS/MS with particular attention given to nitrogen‐containing metabolites with a mass higher than 1000 Da. The pharma media‐based extracts offered a reproducible candidate set of structurally related ions (m/z 1315, 1331 and 1347) with a peptidic nature inferred via their molecular formula predictions, and the intense formation of doubly charged species (Figures S2,S2A). Initial attempts to interpret the peptides sequence using the MS/MS technique were not successful, which were attributed by then to the processing degree of the substrates under investigation.
To corroborate the candidate compounds, three feeding experiments, employing isotopically labeled (15NH4)2SO4, [2H10] L‐leucine and [2H7] L‐ proline, respectively, were carried out on a small scale with strain IFM 0406. As postulated, the 15N‐labelling experiment led to uniformly 15N‐labeled peptides. In agreement with our hypothesis, each of the three candidate compounds showed in their corresponding LC/MS spectra a characteristic 15N‐isotope‐envelope pattern that pointed to the presence of 12 to 13mer (Figures 2 A and S3,S4A). In order to prove that the targeted peptides are indeed the product of the nta gene cluster, [2H10] L‐leucine was fed, which was expected to be incorporated twice and to produce a+9/10 and a+18/19/20 Da pattern. Strikingly, mass shifts of +6, +9, and +15 Da were observed for all candidate compounds’ pseudomolecular ions (Figures 2 A and S5,S6A), which readily hinted on the crude level prior to any isolation steps that one Leu residue must have undergone an unusual modification. Within the same context and driven by the numerous predicted possible cleavage sites, the labeled substrate [2H7] L‐proline was supplemented into IFM 0406 cultures to validate more the features in question and the probable hydrolytic site within the prepeptide. Notably, the proline experiment exhibited an extra 7 Da as a clear indication of a single encoded proline residue within the peptides under investigation (Figures 2 A and S7,S8A), which on the one hand validated that we are tracking down the products of interest and on the other hand automatically shrank the cleavage position from the “PAAPAC” to the “AC” sequence (residues +1 to +2) which corroborates, in turn, the formation of a 12 to 13mer.
Figure 2.
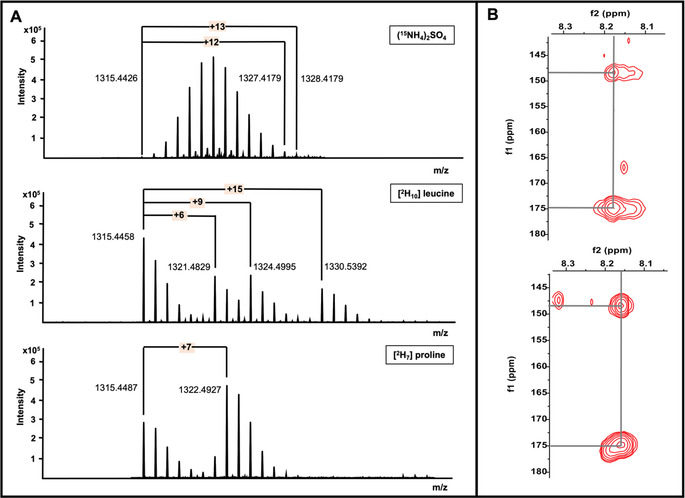
Details of the labeling experiments and decisive NMR key correlations, vital for the metabologenomic approach. A) HR‐MS profiles of labeled nocathioamide A [M+H]+; B) 1H‐13C HMBC screening of azole(s) on (sub)fractions.
At this point and motivated by the suggested molecular formula which showed no fit with any known compound derived from a natural resource using SciFinder® (Figures S9–S11), IFM 0406 was fermented on a large scale (22L). Upon C18‐vacuum liquid chromatography (RP‐VLC), we took advantage of the in silico predicted rare thioamide and common (methyl)azole moieties, which produce distinctive downfield resonances in NMR spectra.
Guided by LC‐MS of RP‐VLC fractions, a series of 1H‐13C HMBC NMR experiments were complementarily executed on the (sub)fractions exhibiting masses > m/z 1300 (Figure S23). Cross correlations, typical for oxazole and/or thiazole entities (δ H/C 8.18/148.32 and δ H/C 8.18/174.91) were exclusively observed in the VLC‐fraction, eluting with 70 % MeOH which was, beside the known secondary metabolite brasilicardin A, mainly enriched with the unknown peptides in pursuit (Figures 2 B and S23A). In contrast, pronounced downfield 1H‐13C HMBC cross‐peaks (δ C>200 ppm) were not detectable at all in any fractions, thusly crippling the envisioned thioamide‐based NMR screening scheme. However, upon further purification and having this molecular family in hand, the thioamide motif was found to reveal weaker resonances under the applied standard NMR conditions. Using Sephadex LH‐20 column chromatography, and followed by sequential rounds of RP‐HPLC (Figures S13–S15), pure nocathioamides A (1), B (2) and traces of C (3) were obtained.
Structure Elucidation of Nocathioamides A–C
In combination with extensive NMR analyses, the suggested elemental composition of 1, possessing m/z 1315 [M+H]+ and 658 [M+2 H]2+, was deduced to be C54H74N16O15S4 with 26 degrees of unsaturation (RDB) (Figure S9). The observation of a doubly charged species in the mass spectrum with a large number of exchangeable amide NH protons (δ H 6.5–10.5 ppm) and carbonyl carbons (δ C 160–185 ppm) from 1D NMR assured the peptidic nature of 1 (Figures S34,S35). The 13C‐NMR spectra in the differently applied solvents (d4 ‐, d3 ‐CH3OH) showed the presence of two major conformers (ratio: 3:1) (Figures S25 and S49). Although a high degree of spectral overlap existed, 11 spin systems were assembled from the HSQC, COSY, TOCSY, and HSQC‐TOCSY data. Briefly, among the deduced partial structures were the amino acids 2×Ala, 1×His, 1×α‐aminobutyric acid (Abu), 1×Leu, 1×4‐methylglutamate (4‐Meglu), 3×AlaS, 1×Pro and 1×Asn. 1H‐15N and 1H‐13C‐based HMBC cross correlations between the side chain hydrogens with the remaining nitrogen and carbon atoms completed each amino acid moiety. Notably, one low‐relaxing quarternary atom δ C 207.99 ppm), indicative for a thiocarbonyl functional group, showed cross‐peaks with H2‐β (His) in the HMBC spectrum, which allowed the definition of thiocarbonyl‐histidine (Figure 3 A). This fact was unequivocally apparent from a significant downfield shift of the nitrogen of Abu, involved in the thioamide bond formation (Figure 3 B). Moreover, HMBC resonances from a carbonyl group to H2‐β, H‐γ and H3‐δ of a Leu residue proved it to be oxidized at the remaining C‐δ in the form of 4‐Meglu. Further interpretation of the 1H‐15N‐ and 1H‐13C HMBC data also revealed the presence of a thiazole (Thz) and a dehydroalanine (Dha) residues and acetylation of the N‐terminal Ala moiety (Ala‐1). The defined subunits were step‐wise connected using a combination of foremost 1H‐13C HMBC correlations from α‐hydrogen resonances to the (thio)amide carbonyls of the adjacent amino acid and the observed Hα,β(i)→ HN(i+1) cross‐peaks in the 1H‐1H NOESY spectra to give the primary linear sequence of 1 (Figure 4). The so far delineated structure accounted only for 23 out of the 26 RDBs, indicating that the final three degrees of unsaturation must arise from three cyclization events. Analysis of 1H‐1H NOESY and 1H‐13C HMBC spectra, recorded either in d 3‐ or d 4‐MeOH and d 6‐DMSO, proved that residues 4–10 (Abu–AlaS‐10) and 6–11 (AlaS‐6–AlaS‐11) were connected via β‐thioether bonds while 5–13 (4‐Meglu–Asn) were interlinked with an imide functional group (Figures 4, and S16–S21). The finding of the unprecedented imide fragment was essentially gleaned from 1H‐15N HSQC spectrum of 1 (Figures 3 B, and S97), which provoked the reinspection of 1H‐15N HMBC couplings, disclosing that H2‐β (Asn) were correlated to a nitrogen atom at 172.8 ppm, consistent with reported 15N shift values for imides (Figures 3 C, and S114A).[20] Since literature about 15N‐NMR shift values is scarce, and the listed values strongly depend on the referencing method, we furthermore validated the 15N shift readings by the measurement of the commercially available glutarimide, as an imide model compound, whose imide nitrogen resonated at 172.3 ppm (Figure S100), thereby corroborating our conclusion and completing the 2D structure of nocathioamide A (1). Using the results from the initial labeling studies, we were also able to define the absolute configuration of three amino acids. Thus, the interpretation of the MS‐shift patterns showed that both leucine residues are D‐configurated, while the proline unit possesses the L‐configuration.[21]
Figure 3.
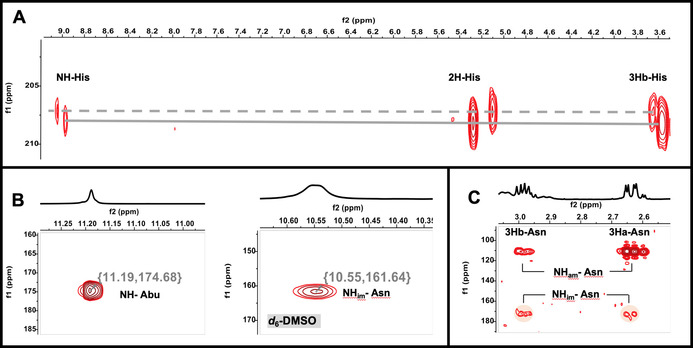
Key NMR correlations: A) 1H‐13C HMBC NMR spectrum highlighting cross‐peaks between C=S and 2 H, 3Hb and the amidic hydrogen of His; B) 1H‐15N HSQC NMR spectra highlighting the chemical shifts of the NH of Abu connected to C=S and the macrocyclic imide NH of Asn; C) 1H‐15N HMBC NMR spectrum showing the key cross‐peaks between 3Ha, and 3Hb of Asn with the amidic and imidic NH of Asn.
Figure 4.
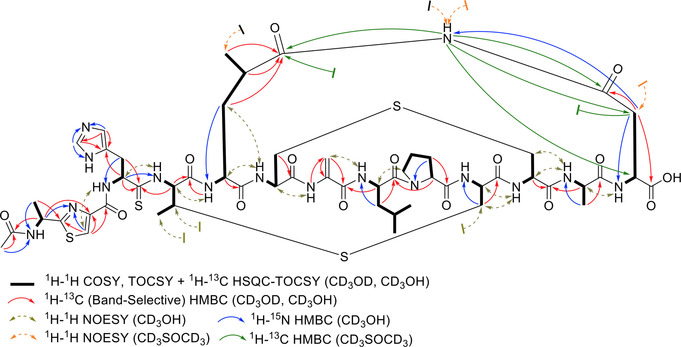
NMR key correlations of nocathioamide A (1).
Nocathioamide B (2) gave a prominent doubly charged [M+2 H]2+ peak by HRESIMS for C54H74N16O16S4 (Figure S10), with an additional 16 mass units translated into an extra oxygen‐atom relative to nocathioamide A (1). Furthermore, the NMR spectra of 2 were nearly identical with those of 1 (Figures S51, and S83), except that the α‐ and β‐carbons of the lanthionine‐bridge forming residues 6 and 11 (AlaSO‐6, and AlaSO‐11) showed a stark change in their chemical shifts where the α‐carbons shifted upfield whilst the β‐carbons resonated strongly downfield while the hydrogen values were merely not affected (Tables S3 and S6). Since such a diagnostic change in chemical shifts typically occurs upon the oxidation of the thioether functionality,[22] 2 was deduced to be the S‐monooxidized product of 1. Unlike 1 and 2, nocathiamide C (3) was solely analyzed by HR‐ESI‐MS for C54H74N16O17S4 (Figure S11), exhibiting 32 additional Da (2×O) in comparison with 1. Thus, this difference could be readily accounted for two S‐oxide groups instead of the typical thioether bridges present in 1.
To expedite further the molecular family, the recruitment of a Global Natural Products Social (GNPS)‐based molecular networking analysis seamlessly unveiled an additional suite of nocathioamide variants (Figure S124 and Table S8).[23]
Evaluation of the Biological Activity
In antibacterial, antifungal and cytotoxicity assays compounds 1 and 2 were both found to be inactive up to the highest concentration tested (MIC >32 μg mL−1, IC50 >64 μg mL−1, Table S9). Since 1 and 2 did not exhibit the classic biological activity of RiPPs, we assume that nocathioamides might possess an ecological or physiological function.[2] The identification of this function will be the subject of further studies.
Biosynthetic Considerations and Classification of Nocathioamides
The performed bioinformatic prediction of the final product was largely correct but was, owing to the novelty of the involved genes cocktail, not able to predict a) the exact cleavage site, and therefore the length of the peptide, b) the imide ring closure, c) the catalytic activity of the methyltransferase NtaC and d) that only one acetylation reaction will take place despite the presence of two acetyltransferase encoding genes (ntaH and ntaN). Regarding the latter issue, only one acetyltransferase is apparently contributing in the biosynthesis. We hypothesize that NtaH is the decisive enzyme for the N‐acetylation of the Ala‐1 residue due to the analysis of the very recently sequenced genome of Longimycelium tulufanense CGMCC 4.5737[24] (NZ_BMMK01000000) which also contained the nta cluster (Table S1). Strikingly, all annotated nta genes exhibited a high identity and similarity score, except for NtaN which showed no significant similarity with all proteins of strain CGMCC 4.5737. Since the nocathioamides A–C (1–3) bear no C‐ or N‐methylated groups, but instead carries two D‐configured leucine residues, the methyltransferase, NtaC, was hypothesized to be involved in the amino acid epimerization process, which is well established in a RiPP biosynthetic context.[25]
From a PTM point of view, nocathioamides represent a hypermodified RiPPs. 11 out of its 13 residues underwent different and specific enzymatic modifications to deliver unique chemical alterations [(Me)Lan, thioamidation, azole formation, N‐acetylation, (C and S) oxidation, dehydration, epimerisation, and macroimidation]. Most intriguing is the unusual oxidation of the δ‐carbon atom of Leu5 to deliver 4‐Meglu, possibly mediated by the action of the cytochrome P450 enzyme (CYP450) NtaM, which prepares the molecule for a macrocylic imide ring closure. To the best of our knowledge, such a regio‐specific CYP450‐based oxidation reaction of Leu was formerly reported so far for only seven NRPS‐derived natural products, such as echinocandins, nostopeptolide, polyoxypeptin B, griselimycin, perthamide C, monamycin D and ilamycin,[26] but never in a RiPP context (Figure S123). Moreover, the involved oxidative reactions never led to an imide macrocyclization, but rather to acyclic 4‐methylglutamate or five‐ and six‐membered heterocycles such as 4‐methylproline or cyclic hemiaminal substructures, respectively. Imide moieties themselves exist in several secondary metabolites, however either as succinimide or glutarimide units and not as a key element for ring closures of macrocycles, highlighting the uniqueness of nocathioamides again.
Another feature of the nocathioamide family is the single and double sulfoxidation of Lan and MeLan bridges. Sulfoxidated variants are described still in only a few RiPPs[22, 27] and the process is either enzymatically mediated by a clustered monooxygenase[22a] or occurs spontaneously under alkaline conditions.[22c]
For the classification of nocathioamides, according to the rules of the RiPP community,[1b] we consulted the nta BGC. Concerning the precursor peptide we found a hitherto unknown new AP cleavage site. Furthermore, we observed the presence of a split lanB‐like dehydratase (ntaF/ntaG) beside a lanC like cyclase (ntaB), which clearly categorized the nocathioamides as a member of the class I lanthipeptides. Notably, the co‐occurrence of a split lanB with lanC was singly reported for class I lantibiotics of the antifungal pinensins despite its frequent existence in LAPs and thiopeptides.[4, 28] It is also noteworthy to mention the thioamidation, which exclusively occurs at the His residue. In contrast to the few so far discovered thioamide containing RIPPs, recent genome mining efforts commenced to leverage and expand this chemical space with new thioamidated candidates either as thioviridamide‐similar entities[29] or as thiopeptides, exemplified by saalfelduracin and thiopeptin.[12] Interestingly, the associations of lanthionine synthetases with either F‐dependent and/or TfuA‐paired YcaOs were bioinformatically projected before by the teams of Mitchell and van der Donk to chart novel multi‐biosynthetic systems[30] which were followed up by their engineering efforts through combinatorial biosynthesis to mix and match new‐to‐nature hybrid RiPPs as thiazolinyl‐lanthipeptides (I, II).[31] Along the same lines, the nocathioamide family elegantly symbolises the first‐in‐nature combinatorial tribrid RiPPs hovering over three different class‐defining biosynthetic machineries of lanthipeptides, LAPs and thioamitides, decorated with an additional unique PTM.
Conclusion
In this study, we demonstrated the utility of a newly optimized metabologenomic approach through its application to an orphan gene cluster found in the genomes of N. terpenica IFM 0406 and 0706T. This approach led to the isolation of three novel peptides, nocathioamides A–C (1–3), which would have been overlooked by regular LC/MS screening due to peak overlapping and/or the appearance as a minor peak (Figure S12). We achieved the isolation by the bioinformatic prediction of the resulting peptide and used this information, after a round of media optimization, to screen for specific mass shifts of some amino acid residues upon labeling besides particular NMR features after fractionation. The benefit of the isotopic labeling experiments was threefold: They early connected the suspected chemotypes with the BGC of interest, narrowed down the cleavage site and allowed the determination of the absolute configuration of the involved amino acids. Such a developed hybrid MS/NMR tactic represents a valuable complement to the existing genome mining strategies, particularly for RiPPs lacking predictable bioactivity or mass ranges. Albeit the adopted approach applied in this study resulted in characterizing a thiazole‐containing RiPP, it still could be modulated to target other rare functional groups of RiPPs. For example, the integration of a 1H‐15N‐HSQC NMR screening component can enable to delve more RiPPs featuring imide moieties or unusual nitrogen crosslinks. Furthermore, with a refocused 1H‐13C‐HMBC sequence that takes into account the low‐relaxing quarternary atoms of thioamide carbonyl groups, it could be implemented to unearth further thioamidated RiPPs.
While the discovery of nocathioamides brings up several firsts into RiPPs, the nta BGC sets the stage for studying the interplay of the involved enzymes to elucidate the currently enigmatic biosynthetic events. This will possibly allow to expand the biobricks toolbox in synthetic biology towards engineering customized hybrid RiPPs exploiting the rare PTMs and the exceptional combinations of the different biogenesis machineries offered by nocathioamides chimeric gene cluster.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. D. Wistuba and her team (Mass Spectrometry Department, Institute for Organic Chemistry, University of Tübingen, Germany) for HR‐MS measurements and T. Wommer for expert technical assistance. We are also very thankful to Prof. S. Peter (Institute of Medical Microbiology and Hygiene, University of Tübingen, Tübingen, Germany) for providing fungal test strains. H.S. gratefully acknowledges the Ministry of Higher Education of Egypt (MOHE) for funding. S.A. gratefully acknowledge the funding received from BMBF German‐Indonesian Cooperation project NAbaUnAk and for a Ph.D scholarship from the German Academic Exchange Service (DAAD). H.B.‐O. acknowledges funding by the German Center for Infection Research (DZIF, TTU 09‐818). The authors acknowledge infrastructural support by the Cluster of Excellence EXC 2124 Controlling Microbes to Fight Infection (CMFI), project ID 390838134, funded by the Deutsche Forschungsgemeinschaft (DFG). Open access funding enabled and organized by Projekt DEAL.
H. Saad, S. Aziz, M. Gehringer, M. Kramer, J. Straetener, A. Berscheid, H. Brötz-Oesterhelt, H. Gross, Angew. Chem. Int. Ed. 2021, 60, 16472.
References
- 1.
- 1a.Ortega M. A., van der Donk W. A., Cell Chem. Biol. 2016, 23, 31–44; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b.Montalbán-López M., Scott T. A., Ramesh S., Rahman I. R., van Heel A. J., Viel J. H., Bandarian V., Dittmann E., Genilloud O., Goto Y., Grande Burgos M. J., Hill C., Kim S., Koehnke J., Latham J. A., Link A. J., Martinez B., Nair S. K., Nicolet Y., Rebuffat S., Sahl H.-G., Sareen D., Schmidt E. W., Schmitt L., Severinov K., Süssmuth R. D., Truman A. W., Wang H., Weng J.-K., van Wezel G. P., Zhang Q., Zhong J., Piel J., Mitchell D. A., Kuipers O. P., van der Donk W. A., Nat. Prod. Rep. 2021, 38, 130–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y., Rebuffat S., J. Biol. Chem. 2020, 295, 34–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Repka L. M., Chekan J. R., Nair S. K., van der Donk W. A., Chem. Rev. 2017, 117, 5457–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a.Burkhart B. J., Schwalen C. J., Mann G., Naismith J. H., Mitchell D. A., Chem. Rev. 2017, 117, 5389–5456; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b.Hudson G. A., Hooper A. R., DiCaprio A. J., Sarlah D., Mitchell D. A., Org. Lett. 2021, 23, 253–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahanta N., Szantai-Kis D. M., Petersson E. J., Mitchell D. A., ACS Chem. Biol. 2019, 14, 142–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a.Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S. Y., Medema M. H., Weber T., Nucleic Acids Res. 2019, 47, W81–W87; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b.Tietz J. I., Schwalen C. J., Patel P. S., Maxson T., Blair P. M., Tai H.-C., Zakai U. I., Mitchell D. A., Nat. Chem. Biol. 2017, 13, 470–478; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c.Santos-Aberturas J., Chandra G., Frattaruolo L., Lacret R., Pham T. H., Vior N. M., Eyles T. H., Truman A. W., Nucleic Acids Res. 2019, 47, 4624–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong Z., He B., Li J., Li Y.-X., Synth. Syst. Biotechnol. 2020, 5, 155–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a.Cox C. L., Tietz J. I., Sokolowski K., Melby J. O., Doroghazi J. R., Mitchell D. A., ACS Chem. Biol. 2014, 9, 2014–2022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b.Mohimani H., Kersten R. D., Liu W. T., Wang M., Purvine S. O., Wu S., Brewer H. M., Pasa-Tolic L., Bandeira N., Moore B. S., Pevzner P. A., Dorrestein P. C., ACS Chem. Biol. 2014, 9, 1545–1551; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c.Cao L., Gurevich A., Alexander K. L., Naman C. B., Leao T., Glukhov E., Luzzatto-Knaan T., Vargas F., Quinn R., Bouslimani A., Nothias L. F., Singh N. K., Sanders J. G., Benitez R. A. S., Thompson L. R., Hamid M.-N., Morton J. T., Mikheenko A., Shlemov A., Korobeynikov A., Friedberg I., Knight R., Venkateswaran K., Gerwick W. H., Gerwick L., Dorrestein P. C., Pevzner P. A., Mohimani H., Cell Syst. 2019, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhakal D., Rayamajhi V., Mishra R., Sohng J. K., J. Ind. Microbiol. Biotechnol. 2019, 46, 385–407. [DOI] [PubMed] [Google Scholar]
- 10.Wiebach V., Mainz A., Siegert M.-A. J., Jungmann N. A., Lesquame G., Tirat S., Dreux-Zigha A., Aszodi J., Le Beller D., Süssmuth R. D., Nat. Chem. Biol. 2018, 14, 652–654. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a.Buchmann A., Eitel M., Koch P., Schwarz P. N., Stegmann E., Wohlleben W., Wolański M., Krawiec M., Zakrzewska-Czerwinska J., Méndez C., Botas A., Núñez L. E., Morís F., Cortés J., Gross H., Genome Announc. 2016, 4, e01391; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b.Chen J., Frediansyah A., Männle D., Straetener J., Brötz-Oesterhelt H., Ziemert N., Kaysser L., Gross H., ChemBioChem 2020, 21, 2205–2213; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c.Buchmann A., Gross H., Microbiol. Resour. Announc. 2020, 9, e00689-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwalen C. J., Hudson G. A., Kille B., Mitchell D. A., J. Am. Chem. Soc. 2018, 140, 9494–9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Angelopoulou A., Warda A. K., O'Connor P. M., Stockdale S. R., Shkoporov A. N., Field D., Draper L. A., Stanton C., Hill C., Ross R. P., Front. Microbiol. 2020, 11, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asaduzzaman S. M., Sonomoto K., J. Biosci. Bioeng. 2009, 107, 475–487. [DOI] [PubMed] [Google Scholar]
- 15.Agrawal P., Khater S., Gupta M., Sain N., Mohanty D., Nucleic Acids Res. 2017, 45, W80–W88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a.Mahanta N., Liu A., Dong S., Nair A. K., Mitchell D. A., Proc. Natl. Acad. Sci. USA 2018, 115, 3030–3035; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b.Liu J., Lin Z., Li Y., Zheng Q., Chen D., Liu W., Org. Biomol. Chem. 2019, 17, 3727–3731. [DOI] [PubMed] [Google Scholar]
- 17.Kloosterman A. M., Shelton K. E., van Wezel G. P., Medema M. H., Mitchell D. A., mSystems 2020, 5, e00267-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S. W., Mitchell D. A., Markley A. L., Hensler M. E., Gonzalez D., Wohlrab A., Dorrestein P. C., Nizet V., Dixon J. E., Proc. Natl. Acad. Sci. USA 2008, 105, 5879–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a.Mahanta N., Hudson G. A., Mitchell D. A., Biochemistry 2017, 56, 5229–5244; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b.Gibson M. I., Fornerisa C. C., Seyedsayamdost M. R. in Cyclic Peptides (Eds.: Koehnke J., Naismith J., van der Donk W. A.), Royal Society of Chemistry, London, 2017, pp. 141–163; [Google Scholar]
- 19c.Lu J., Li Y., Bai Z., Lv H., Wang H., Nat. Prod. Rep. 2021, 10.1039/D0NP00044B. [DOI] [PubMed] [Google Scholar]
- 20.Levy G. C., Lichter R. L. in Nitrogen-15 Nuclear Magnetic Resonance Spectroscopy, Wiley, New York, 1979. [Google Scholar]
- 21.Bode H. B., Reimer D., Fuchs S. W., Kirchner F., Dauth C., Kegler C., Lorenzen W., Brachmann A. O., Grün P., Chem. Eur. J. 2012, 18, 2342–2348. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a.Somma S., Merati W., Parenti F., Antimicrob. Agents Chemother. 1977, 11, 396–401; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b.Vertesy L., Aretz W., Bonnefoy A., Ehlers E., Kurz M., Markus A., Schiell M., Vogel M., Wink J., Kogler H., J. Antibiot. 1999, 52, 730–741; [DOI] [PubMed] [Google Scholar]
- 22c.Maffioli S. I., Iorio M., Sosio M., Monciardini P., Gaspari E., Donadio S., J. Nat. Prod. 2014, 77, 79–84. [DOI] [PubMed] [Google Scholar]
- 23.Wang M., Carver J. J., Phelan V. V., Sanchez L. M., Garg N., Peng Y., Nguyen D. D., Watrous J., Kapono C. A., Luzzatto-Knaan T., Porto C., Bouslimani A., Melnik A. V., Meehan M. J., Liu W.-T., Crüsemann M., Boudreau P. D., Esquenazi E., Sandoval-Calderon M., Kersten R. D., Pace L. A., Quinn R. A., Duncan K. R., Hsu C.-C., Floros D. J., Gavilan R. G., Kleigrewe K., Northen T., Dutton R. J., Parrot D., Carlson E. E., Aigle B., Michelsen C. F., Jelsbak L., Sohlenkamp C., Pevzner P., Edlund A., McLean J., Piel J., Murphy B. T., Gerwick L., Liaw C.-C., Yang Y.-L., Humpf H.-U., Maansson M., Keyzers R. A., Sims A. C., Johnson A. R., Sidebottom A. M., Sedio B. E., Klitgaard A., Larson C. B., Boya P C. A., Torres-Mendoza D., Gonzalez D. J., Silva D. B., Marques L. M., Demarque D. P., Pociute E., O'Neill E. C., Briand E., Helfrich E. J. N., Granatosky E. A., Glukhov E., Ryffel F., Houson H., Mohimani H., Kharbush J. J., Zeng Y., Vorholt J. A., Kurita K. L., Charusanti P., McPhail K. L., Nielsen K. F., Vuong L., Elfeki M., Traxler M. F., Engene N., Koyama N., Vining O. B., Baric R., Silva R. R., Mascuch S. J., Tomasi S., Jenkins S., Macherla V., Hoffman T., Agarwal V., Williams P. G., Dai J., Neupane R., Gurr J., Rodriguez A. M. C., Lamsa A., Zhang C., Dorrestein K., Duggan B. M., Almaliti J., Allard P.-M., Phapale P., Nothias L.-F., Alexandrov T., Litaudon M., Wolfender J.-L., Kyle J. E., Metz T. O., Peryea T., Nguyen D.-T., VanLeer D., Shinn P., Jadhav A., Müller R., Waters K. M., Shi W., Liu X., Zhang L., Knight R., Jensen P. R., Palsson B. O., Pogliano K., Linington R. G., Gutierrez M., Lopes N. P., Gerwick W. H., Moore B. S., Dorrestein P. C., Bandeira N., Nat. Biotechnol. 2016, 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linhuan W., Juncai M., Int. J. Syst. Evol. Microbiol. 2019, 69, 895–898. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a.Bhushan A., Egli P. J., Peters E. E., Freeman M. F., Piel J., Nat. Chem. 2019, 11, 931–939; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b.Korneli M., Fuchs S. W., Felder K., Ernst C., Zinsli L. V., Piel J., ACS Synth. Biol. 2021, 10, 236–242. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a.Ma J., Huang H., Xie Y., Liu Z., Zhao J., Zhang C., Jia Y., Zhang Y., Zhang H., Zhang T., Ju J., Nat. Commun. 2017, 8, 391; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b.Tang M.-C., Zou Y., Watanabe K., Walsh C. T., Tang Y., Chem. Rev. 2017, 117, 5226–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simone M., Monciardini P., Gaspari E., Donadio S., Maffiolo S. I., J. Antibiot. 2013, 66, 73–78. [DOI] [PubMed] [Google Scholar]
- 28.Mohr K. I., Volz C., Jansen R., Wray V., Hoffmann J., Bernecker S., Wink J., Gerth K., Stadler M., Müller R., Angew. Chem. Int. Ed. 2015, 54, 11254–11258; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11406–11410. [Google Scholar]
- 29.
- 29a.Frattaruolo L., Lacret R., Cappello A. R., Truman A. W., ACS Chem. Biol. 2017, 12, 2815–2822; [DOI] [PubMed] [Google Scholar]
- 29b.Kawahara T., Izumikawa M., Kozone I., Hashimoto J., Kagaya N., Koiwai H., Komatsu M., Fujie M., Sato N., Ikeda H., Shin-ya K., J. Nat. Prod. 2018, 81, 264–269. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a.Zhang Q., Doroghazi J. R., Zhao X., Walker M. C., van der Donk W. A., Appl. Environ. Microbiol. 2015, 81, 4339–4350; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b.Walker M. C., Eslami S. M., Hetrick K. J., Ackenhusen S. E., Mitchell D. A., van der Donk W. A., BMC Genomics 2020, 21, 387; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30c.Wu C., van der Donk W. A., Curr. Opin. Biotechnol. 2021, 69, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burkhart B. J., Kakkar N., Hudson G. A., van der Donk W. A., Mitchell D. A., ACS Cent. Sci. 2017, 3, 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary