

Abstract
Treatment of solid cancers with chimeric antigen receptor (CAR) T cells is plagued by the lack of ideal target antigens that are both absolutely tumor specific and homogeneously expressed. We show that multi-antigen prime-and-kill recognition circuits provide flexibility and precision to overcome these challenges in the context of glioblastoma. A synNotch receptor that recognizes a specific priming antigen, such as the heterogeneous but tumor-specific glioblastoma neoantigen epidermal growth factor receptor splice variant III (EGFRvIII) or the central nervous system (CNS) tissue-specific antigen myelin oligodendrocyte glycoprotein (MOG), can be used to locally induce expression of a CAR. This enables thorough but controlled tumor cell killing by targeting antigens that are homogeneous but not absolutely tumor specific. Moreover, synNotch-regulated CAR expression averts tonic signaling and exhaustion, maintaining a higher fraction of the T cells in a naïve/stem cell memory state. In immunodeficient mice bearing intracerebral patient-derived xenografts (PDXs) with heterogeneous expression of EGFRvIII, a single intravenous infusion of EGFRvIII synNotch-CAR T cells demonstrated higher antitumor efficacy and T cell durability than conventional constitutively expressed CAR T cells, without off-tumor killing. T cells transduced with a synNotch-CAR circuit primed by the CNS-specific antigen MOG also exhibited precise and potent control of intracerebral PDX without evidence of priming outside of the brain. In summary, by using circuits that integrate recognition of multiple imperfect but complementary antigens, we improve the specificity, completeness, and persistence of T cells directed against glioblastoma, providing a general recognition strategy applicable to other solid tumors.
INTRODUCTION
Although chimeric antigen receptor (CAR) T cells have demonstrated remarkable outcomes in treating hematologic malignancies (1), development of effective CAR T therapies for solid cancers remains a challenge, due in large part to the difficulty in identifying optimal target surface antigens. Very few antigens are truly tumor specific, and the resulting on-target/off-tumor cross-reaction of the engineered T cells with normal tissues can cause lethal toxicities (2–5). Conversely, even if antigens with high tumor specificity are identified, these targets are often heterogeneously expressed, and selective CAR targeting allows for escape of antigen-negative tumor cells (6). Thus, there is a general need for new tumor recognition strategies that can navigate the concurrent challenges of specificity and heterogeneity to increase the therapeutic benefit of CAR T cells against solid cancers.
A concrete example of this dual challenge is found in glioblastoma (GBM). The epidermal growth factor receptor splice variant III (EGFRvIII) is a highly GBM-specific neoantigen found in a subset of patients (7–10). Although this might seem like an ideal tumor-specific antigen target, previous clinical studies targeting GBM with an anti-EGFRvIII CAR showed consistent tumor recurrence. EGFRvIII expression in the tumors is highly heterogeneous, and EGFRvIII− tumor cells can escape and grow, despite effective killing of EGFRvIII+ cells by the CAR T cells (6, 11, 12). In contrast, alternative glioma-associated target antigens, including ephrin type A receptor 2 (EphA2) and interleukin 13 receptor α2 (IL13Rα2), are expressed on the surface of the vast majority of GBM cells (13–15), but have imperfect specificity. Although they are not expressed in normal central nervous system (CNS) tissues, they are expressed in some normal, non-CNS tissues such as the liver, kidney, esophagus, and genital organs (www.humanproteomemap.org). Thus, it is challenging to find a single ideal surface GBM antigen that is both specific and homogeneous enough to limit the dual challenges of off-target toxicity and incomplete killing.
We therefore proposed that T cells that recognize multi-antigen combinations provide a possible solution to the conundrum of simultaneously optimizing specificity of tumor recognition and completeness of killing. We previously developed “prime-and-kill” circuits in which a synNotch receptor [an engineered receptor that activates a transcriptional output when it recognizes its cognate antigen (16)] is primed to induce expression of a CAR directed against a killing antigen (17, 18). These synNotch-CAR circuits function as Boolean AND gates, requiring the recognition of both priming (synNotch) and killing (CAR) antigens. These circuits can also be considered “IF-THEN” circuits, as they execute CAR-directed killing only if first primed by the synNotch ligand. We hypothesized that, by carefully choosing the priming and killing antigens, such multi-antigen circuits could yield hybrid recognition behaviors that could navigate trade-offs between specificity and heterogeneity.
Here, we pursue two such multi-antigen targeting strategies for GBM: either priming with a tumor-specific but heterogeneous neoantigen (EGFRvIII) or priming with a normal CNS-specific antigen, myelin oligodendrocyte glycoprotein (MOG). SynNotch receptor priming by these antigens was used to locally induce expression of a tandem CAR that recognizes the more homogeneous GBM antigens, EphA2 and IL13Rα2 (13, 19). The expression of EphA2 and IL13Rα2 in other normal tissues makes them nonideal antigen targets for conventional, single-target CAR T cells. These antigens, however, could serve as effective killing targets, if higher selectivity was restricted by the required priming antigen. Thus, we hypothesized that T cells engineered with prime-and-kill circuits might induce CAR-driven cytotoxicity that was spatially restricted only to the vicinity of priming cells, thereby avoiding off-tumor killing in distant normal tissues that express the killing antigen but that lack the priming antigen.
RESULTS
Prime-and-kill circuit in T cells can overcome antigen heterogeneity by executing trans-killing
Figure 1 and fig. S1 (A and B) illustrate our rationale for designing a prime-and-kill circuit in T cells: Engineered T cells are first primed by a synNotch receptor that recognizes a cancer-specific but heterogeneous antigen, EGFRvIII, and then induce the expression of a CAR that kills by recognizing a homogeneous though imperfectly tumor-specific antigens, such as EphA2 or IL13Rα2. To achieve homogeneous killing and to further reduce the possibility of tumor escape, we used a tandem CAR that simultaneously targets the two killing antigens, EphA2 OR IL13Rα2 (13, 19). The tandem CAR, which functions as an OR-gate, has an extracellular region containing an α-EphA2 single-chain antibody and an IL13 mutein, a variant of IL13 ligand that binds with higher affinity to IL13Rα2 over IL13Rα1 (fig. S1C) (19, 20). The general strategy in this circuit design was to take advantage of the specificity of the priming antigen combined with the homogeneity of the killing antigens (Fig. 1A), in principle yielding specific and complete tumor killing.
Fig. 1. Multi-antigen prime-and-kill circuits in T cells provide a general strategy to overcome antigen heterogeneity while still maintaining high tumor specificity.
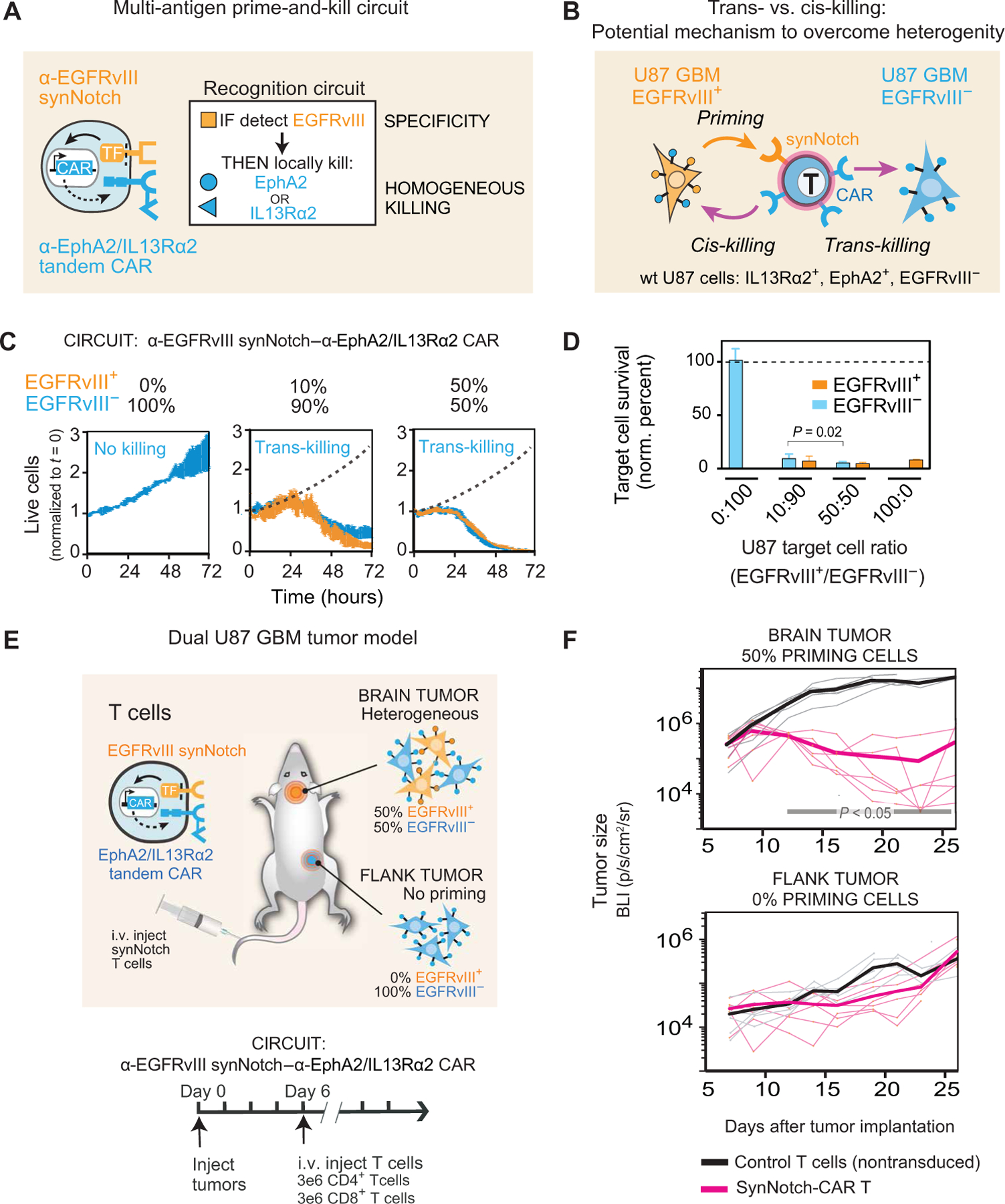
(A) Design of synNotch-CAR circuit primed by EGFRvIII neoantigen: α-EGFRvIII synNotch receptor induces expression of tandem α-EphA2/IL13Rα2 CAR (TF, transcription factor). These cells should be activated to kill EphA2+ or IL13Rα2+ target cells only if exposed to EGFRvIII+ cells. (B) Such T cells could overcome priming antigen heterogeneity if they can execute trans-killing, where priming and killing antigens are expressed on different but neighboring cells. (C) Real-time killing assays using heterogeneous mixtures of EGFRvIII+ and EGFRvIII− target cells show efficient trans-killing. Primary CD8+ human T cells transduced with α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR circuit were cultured with indicated ratios of EGFRvIII+ versus EGFRVIII− U87 cells at an E:T ratio of 5:1 and imaged over 3 days using IncuCyte. The EGFRvIII+ cell population (priming cells) is shown in yellow, and the EGFRvIII− cell population (target cells) is shown in blue. The presence of as low as 10% priming cells yielded strong killing of EGFRvIII− target cells, although killing was slightly slower compared to that observed with 50% priming cells (P = 0.0149 and P = 0.0218, t test at 48 and 72 hours, respectively). The dotted black line shows the growth of 100% target cells as a reference (n = 3, error bars denote SEM). See movie S1. (D) Relative survival of each cell type in experiments from (C) (at 72 hours). Killing of target cells is efficiently primed by as low as 10% priming cells (EGFRvIII+). No killing is observed in the absence of priming cells (n = 3, error bars denote SD). A t test was used for statistical comparison. (E) NCG mice were simultaneously implanted with two GBM tumors: a heterogeneous tumor comprising EGFRvIII+ and EGFRvIII− U87 cells (1:1 ratio) in the brain, and a homogeneous EGFRvIII− U87 tumor implanted subcutaneously in the flank. Mice were treated 6 days after tumor implantation with intravenous (i.v.) infusion of 3 million CD4+ and CD8+ synNotch-CAR T cells (n = 6) or control nontransduced T cells (n = 6). (F) Tumor size was measured by luciferase bioluminescence imaging (BLI) over time as the number of photons per second per square centimeter per steradian (p/s/cm2/sr). Tumor size curves for individual mice treated with synNotch-CAR T cells are shown in light pink; curves for mice treated with nontransduced T cells are shown in gray. Thicker lines correspond to geometric means. P < 0.05 by Mann-Whitney test on day 12 and onward, whereas the flank tumor grew at the same rate as in the mice treated with nontransduced T cells.
We postulated that priming based on a heterogeneous antigen could yield complete tumor killing, if a T cell primed by one cell could kill a different target cell bearing the killing antigen but lacking the priming antigen, a process we define as trans-killing (Fig. 1B). In contrast, cis-killing would describe priming and killing based on antigens presented on the same cell. To test whether synNotch-CAR T cells could execute trans-killing, we first engineered U87 GBM cells to constitutively express the priming antigen EGFRvIII. Native U87 cells do not express EGFRvIII, but they do express both EphA2 and IL13Rα2 killing antigens (11, 12, 21, 22), as demonstrated by the effective killing of U87 cells by T cells transduced with constitutive CAR targeting EphA2 or IL13Rα2 (fig. S1D). Thus, we could coculture U87 EGFRvIII+ cells with U87 EGFRvIII− cells and test whether the presence of EGFRvIII+ priming cells was sufficient to induce T cell killing of the EGFRvIII− target cells. We mixed the U87-EGFRvIII+ and U87-EGFRvIII− cells in indicated ratios to recapitulate different degrees of heterogeneity observed in patients with GBM and then tested whether target cell killing by our synNotch-CAR T cells was induced by the presence of priming cells.
We found that CD8+ T cells engineered with the α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR circuit could effectively kill EGFRvIII− target cells in vitro, if and only if there were also EGFRvIII+ priming cells present (Fig. 1, C and D, and fig. S1, E and F). Priming cells did not need to be present at very high proportions; the presence of as low as 10% priming cells yielded strong killing of EGFRvIII− target cells, although killing was slightly slower compared to that observed with 50% priming cells (P = 0.0149 and P = 0.0218 at 48 and 72 hours, respectively). In contrast, no killing of the EGFRvIII− target cells was observed in the absence of priming cells. In these assays, we independently tracked the kinetics of killing of the two different tumor cell populations using two different fluorescent labels over 72 hours (movie S1). We also validated effective trans-killing using target cells expressing the model priming and killing antigens, green fluorescent protein (GFP) and CD19, respectively (fig. S2). Together, these in vitro studies show that synNotch-CAR T cells can effectively execute trans-killing.
EGFRvIII-triggered synNotch-CAR cells kill tumors in vivo only if they contain priming cells
On the basis of these in vitro data, we next evaluated antitumor activities of synNotch-CAR T cells in mice bearing GBM xenografts. First, we confirmed that EGFRvIII-primed T cells could perform trans-killing of EGFRvIII− GBM cells in vivo but only in tumors with EGFRvIII+ priming cells. As proof of principle, we implanted dual tumors into NOD (nonobese diabetic) CRISPR Prkdc Il2r gamma (NCG) immunodeficient mice. In the brain, we implanted a 1:1 mixture of EGFRvIII+ and EGFRvIII− U87 tumor cells. In the flank of the same animals, we implanted EGFRvIII− U87 tumor cells only (Fig. 1E). Here, the flank tumor represents a potentially cross-reactive normal tissue that expresses the killing antigens but not the priming antigen. In contrast, the brain tumor has both priming and killing antigens. On day 6 after the tumor implantation, the mice received intravenous administration of synNotch-CAR T cells or control nontransduced T cells (n = 6 per group). All mice treated with control T cells showed tumor growth at both sites and reached the study endpoint with a median survival of 25.5 days. The mice treated with synNotch-CAR T cells, in contrast, demonstrated significant suppression of the intracranial tumor growth compared with that in control mice (P < 0.05; Fig. 1F). However, the mice treated with the synNotch-CAR T cells did not show statistically significant suppression of the flank tumor compared with the control group (P = 0.4; Fig. 1F). The selective lack of killing in the nonpriming flank tumor suggests that the cytotoxic activity of the synNotch-CAR T cells is spatially confined to tumors expressing both priming and killing antigens.
We also performed a systematic comparison of killing of implanted tumors with 0, 50, and 100% EGFRvIII+ U87 cells (fig. S3). We found that the cohort of mice that received synNotch-CAR T cells did not show any clearance of the 0% EGFRvIII+ tumors but showed equally effective tumor clearance of both the 50 and 100% EGFRvIII+ tumors (P < 0.001 and P < 0.001, respectively; fig. S3B). Thus, in this context, the efficacy of synNotch-CAR T cells is not negatively affected by EGFRvIII antigen heterogeneity.
EGFRvIII-triggered synNotch-CAR T cells efficiently and durably clear heterogeneous GBM6 PDX tumors better than constitutive CAR T cells
We then sought to evaluate the efficacy of synNotch-CAR T cells in a tumor model that exhibits naturally occurring heterogeneity of EGFRvIII expression. We identified the GBM6 patient-derived xenograft (PDX) tumor as an aggressive GBM model that shows intrinsic EGFRvIII heterogeneity (Fig. 2A) (23). We also confirmed the ability of T cells bearing the α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR circuit to detect and kill GBM6 cells in vitro (fig. S4, A and B). We also confirmed that there was a subpopulation of GBM6 cells with undetectable EGFRvIII antigen that were resistant to killing by the α-EGFRvIII CAR (fig. S4, C and D). Thus, GBM6 represents an ideal tumor model in which to identify circuits that could overcome problems caused by antigen heterogeneity.
Fig. 2. SynNotch-CAR T cells show improved efficacy and durability compared to individual parental constitutive CARs in clearing heterogeneous GBM6 PDX tumors.
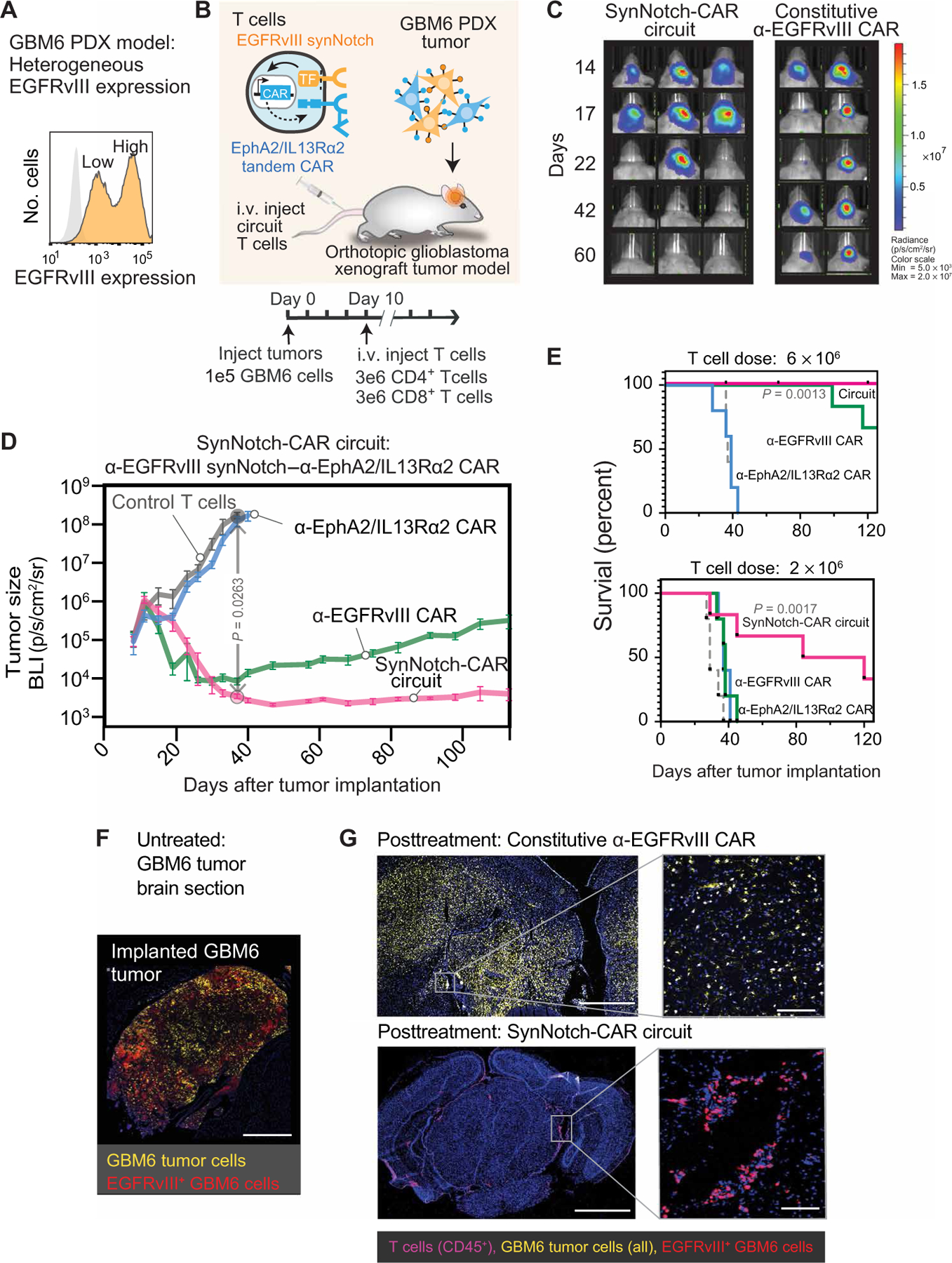
(A) Flow cytometry analysis of a patient-derived xenograft (PDX) GBM6 tumor model shows intrinsic heterogeneity of EGFRvIII expression. (B) Timeline for in vivo tumor experiments with GBM6 tumors. GBM6 tumors expressing mCherry and luciferase were orthotopically implanted in brains of NCG mice. Ten days after tumor implantation, mice were infused intravenously (i.v.) with 3 million each of CD4+ and CD8+ T cells expressing no construct (control) (n = 5), α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR circuit (n = 6), constitutively expressed α-EGFRvIII CAR (n = 5), or constitutively expressed α-EphA2/IL13Rα2 tandem CAR (n = 5). (C) Longitudinal bioluminescence imaging of GBM6 tumor–bearing mice treated with α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells and conventional α-EGFRvIII CAR T cells. Each column represents one mouse over time. (D) Time course of tumor size measured by bioluminescence. P = 0.0263, two-way ANOVA followed by a Dunnett’s test nontransduced versus synNotch-CAR T cells at day 37. Error bars represent means ± SEM of five to six individual mice from one experiment. (E) Kaplan-Meier survival curves for high-dose (6 × 106 cells) and low-dose (2 × 106 cells) treatments. Statistical significance was calculated using log-rank Mantel-Cox test. (F) Fluorescence microscopy of representative section of untreated GBM6 xenograft tumor, isolated 15 days after tumor implantation, shows heterogeneous expression of EGFRvIII. Scale bar, 500 µm. (G) Top: Representative fluorescence microscopy of a brain and tumor section isolated 107 days after treatment with conventional α-EGFRvIII CAR T cells shows the presence of tumor but loss of EGFRvIII expression. Bottom: Representative fluorescence microscopy of a brain section isolated 110 days after treatment with EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells reveals clearance of a GBM6 xenograft tumor and sustained presence of T cells. Scale bars, 1 mm (left) and 50 µm (right).
We implanted GBM6 tumors in the brains of immunodeficient NCG mice and treated them with T cells bearing the α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR circuit. As controls, we treated mice with nontransduced T cells or T cells constitutively expressing either the α-EGFRvIII CAR or α-EphA2/IL13Rα2 tandem CAR (Fig. 2, B to D). All of the mice receiving the untransduced T cells (n = 5) died of tumor progression by day 43 after tumor implantation (Fig. 2, C and D). Treatment with the constitutiveα-EphA2/IL13Rα2 tandem CAR was largely ineffective. Treatment with the constitutive α-EGFRvIII CAR T cells yielded initial tumor shrinkage but consistently resulted in recurrence of tumors in all mice (Fig. 2, C and D), recapitulating what has been previously observed in clinical trials deploying this CAR (6). In notable contrast, all of the mice treated with the synNotch-CAR T cells showed complete and long-term remission of the GBM6 tumors. This durable and complete tumor clearance was reproducible (fig. S4E) and was also reflected in significantly increased survival of mice treated with synNotch-CAR T cells (P = 0.0263 at day 37; Fig. 2D). The synNotch-CAR T cells were also significantly more effective than the constitutive α-EGFRvIII CAR T cells at a lower, suboptimal T cell dosage [P = 0.0017, log-rank (Mantel-cox) test; Fig. 2E].
To more carefully evaluate the outcome of the different T cell treatments, we performed postmortem immunofluorescence analysis of mice after 100-day treatment with either the α-EGFRvIII CAR T cells or the synNotch-CAR T cells. Brain slices isolated from 15-day-long tumor-bearing mice without T cell treatment are shown in Fig. 2F (GBM6 tumor cells, yellow; EGFRvIII antigen, red). The constitutive α-EGFRvIII CAR–treated brains showed persistence of GBM6 tumor cells but no remaining T cells (Fig. 2G, top). The remaining GBM6 cells, however, were all negative for EGFRvIII expression, consistent with tumor escape via selective growth of EGFRvIII− tumor cells. In contrast, the brain sections from mice treated with synNotch-CAR T cells showed complete elimination of GBM6 tumor cells and revealed persistent T cells in the brain parenchyma and meninges (Fig. 2G, bottom). In summary, PDX mouse experiments showed that the prime-and-kill circuit could achieve more specific and complete killing of tumor cells than constitutively expressed CARs targeted against a single antigen.
Immunofluorescence and intravital imaging show that T cell priming and killing is spatially restricted to GBM tumor
To track the mechanism of synNotch-CAR T cells in more detail, we sectioned brains isolated from GBM6 tumor–bearing mice only 6 days after T cell injection and evaluated them by immunofluorescence microscopy. In these experiments, GBM6 tumor cells were labeled with the fluorescent protein mCherry; primed T cells could be detected via a GFP-tag fused to the induced α-EphA2/IL13Rα2 CAR; apoptotic cells could be detected by staining for cleaved caspase 3 (Fig. 3A). We found that T cell priming and apoptosis spatially overlaid with the GBM6 tumor. Thus, T cell priming and killing were restricted only to the GBM6 tumor, and no collateral killing was observed, even in regions adjacent to the tumor. The same analysis with GBM6 tumor–bearing mice treated with nontransduced T cells showed no evidence of apoptosis in the tumor or elsewhere (Fig. 3B). Further imaging showed robust infiltration of transferred, hCD45+ T cells and priming selectively in GBM6 tumor tissue; primed T cells were absent in adjacent normal brain tissue (fig. S5A). Closer examination of these multicolor fluorescent images revealed cytolytic events in the tumor in which apoptotic GBM6 cells are surrounded by primed T cells (fig. S5B).
Fig. 3. Early time point imaging shows thatA priming and killing of synNotch-CAR T cells are precisely restricted to tumor region.

(A) Representative confocal fluorescent microscopy of brain slices from GBM6 tumor–bearing mice isolated at 6 days after infusion with α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells reveals primed T cells expressing CAR-GFP only within the tumor bed. Cleaved caspase 3 is also only observed within the tumor. Scale bars, 1 mm (left) and 100 µm (right). Overlay of tumor, cleaved caspase 3, and primed GFP T cell is shown in fig. S5B. (B) Representative confocal images of brain slices from GBM6 tumor–bearing mice isolated at 6 days after treatment with nontransduced T cells. Scale bar, 1 mm. (C) Intravital two-photon imaging of synNotch-CAR T cells shows priming within the GBM6 xenograft tumor. Tumors were implanted at a depth of 3 mm below the right frontal cortex, and cranial windows were implanted. Scale bar, 400 µm. See movie S2. (D) Half-life of CAR expression decay in synNotch-CAR T cells after removing priming cells. T cells were primed in vitro with GBM6 cells for 60 hours and then isolated by cell sorting. GFP-tagged CAR expression was observed over time. MFI, median fluorescence intensity. Decay experiment was performed in the absence (top) or presence (bottom) of target U87 cells lacking priming antigen. Exponential decay fit was used to determine the CAR half-life. Decay curves depict one representative of four different donors.
To more directly visualize T cell priming, we also performed intravital imaging 2 days after T cell injection (Fig. 3C and movie S2). These time-lapse movies showed a large number of primed T cells only within the tumor, as well as unprimed T cells that became primed as they approached the tumor. These movies showed that the primed T cells remained stably localized within the tumor over the full course of the 1-hour-long movie, presumably because the T cells were interacting with target cells. Although the primed T cells had dynamic cell processes reaching out to contact neighboring cells, they did not show any sign of rapid trafficking in or out of the tumor. This relatively stable residence of the primed T cells in the tumor may explain the highly specific and localized killing observed with these T cells.
Another feature that is likely to contribute to high spatial targeting specificity is that when the cells leave a microenvironment with priming antigens, the synNotch-induced CAR expression will stop, and the CAR protein will decay. We measured CAR protein decay using a GFP-tagged α-EphA2/IL13Rα2 CAR after removal of a long-term priming cell stimulus (Fig. 3D). We found that CAR expression decayed with a half-life of about 10 hours. This was slightly faster if CAR target cells, such as U87 tumor cells, were present, as these likely accelerated CAR endocytosis. The decay of CAR expression in the observed timeframe likely means that cells that leave the priming environment are not able to sustain a meaningful proliferative and killing response over the course of days to weeks.
SynNotch-induced CAR T cells show a more stem-like phenotype, reduced exhaustion, and improved in vivo persistence compared to constitutive CARs
One of the most surprising findings of these in vivo studies was the improved tumor clearance capacity of the synNotch-CAR T cells compared to the constitutive α-EphA2/IL13Rα2 CAR T cells, because both sets of T cells used the same CAR molecule. Moreover, this difference was only observed in vivo, as the constitutive tandem CAR T cells are highly effective at tumor killing in vitro (Fig. 4A). These puzzling observations suggested that there were additional features of synNotch-CAR circuits that yielded improved antitumor activity in vivo.
Fig. 4. SynNotch-CAR T cells show enhanced naïve/stem cell memory phenotype, reduced exhaustion, and improved persistence in vivo.
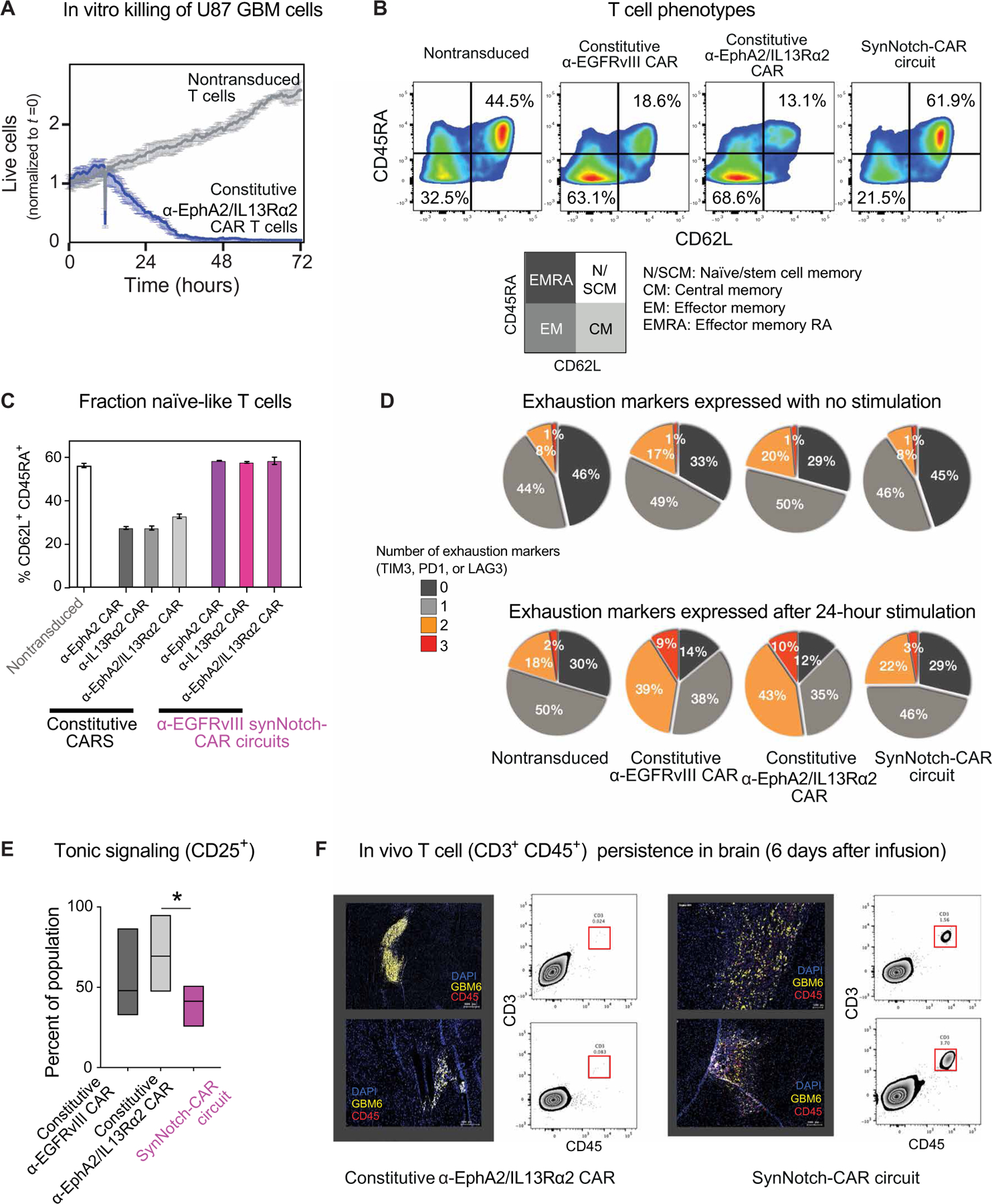
(A) In vitro killing by T cells constitutively expressing α-EphA2/IL13Rα2 tandem CAR (relative to nontransduced control). (B) Flow cytometry plots distinguishing naïve-like cells (CD45RA+CD62L+), central memory cells (CD45RA−CD62L+), effector memory cells (CD45RA−CD62L−), and effector memory RA cells (CD45RA+CD62L−) (representative of three experiments from different donors). Percentages of cells in the naïve/memory stem cell state and effector memory state are highlighted. T cells were rested for 10 days in vitro after transfection before phenotypic analysis. (C) Percentage of CD62L+CD45RA+ T cells in indicated CAR T cells and synNotch-CAR T cells (n = 3 per group). (D) Expression of exhaustion markers by indicated CAR and synNotch-CAR T cells, both without and with stimulation by target cells. Pie chart shows percentage of cells that express 0, 1, 2, or 3 exhaustion markers (PD1, LAG3, or TIM3), average of three different donors. (E) Tonic signaling in constitutive versus α-EGFRvIII-synNotch induced α-EphA2/IL13Rα2 CAR T cells, measured by percent CD25+ cells (one-way ANOVA followed by a Dunnett’s test, P < 0.05, n = 5 different donors). Boxes represent min to max with median center line. (F) SynNotch-CAR T cells show improved persistence in vivo compared to constitutive CAR T cells. GBM6 tumor–bearing mice were euthanized 6 days after infusion of either the constitutive tandem CAR T cells (left) or α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells (right). Representative confocal fluorescent microscopy of brain slices and single-cell suspensions of tumor-bearing brain show few T cells (CD3+CD45+) in constitutive CAR T cell–treated tumors. In contrast, mice treated with synNotch-CAR T cells reveal high number of T cells in tumors.
A general challenge in treating solid cancers with CAR T cells is exhaustion of the T cells, which prevents persistent antitumor activity. Recent studies have indicated that tonic signaling by constitutively expressed CARs can play a major role in increasing their susceptibility to exhaustion, leading to poor in vivo T cell persistence (24, 25). We therefore examined the differentiation state of our different types of engineered T cells by flow analysis and found that all of the α-EGFRvIII synNotch-CAR circuit T cells evaluated in this study contained a higher fraction of cells in a naïve-like state (CD62L+ CD45RA+, naïve or stem central memory), a phenotype associated with greater in vivo antitumor activity (26, 27), compared to the equivalent constitutive CAR T cells (Fig. 4B). We tested different EGFRvIII synNotch–primed circuits, inducing the α-EphA2 CAR, the α-IL13Rα2 CAR, or the tandem α-EphA2/IL13Rα2 CAR, and in all cases, the synNotch-induced version of the CAR yielded a much higher fraction of naïve-like T cells compared to the analogous constitutively expressed CAR (Fig. 4C). Furthermore, this phenotypic improvement seems to be shared by multiple synNotch-CAR circuits (fig. S6).
We evaluated the exhaustion state of the T cells using a panel of exhaustion makers [programmed cell death protein 1 (PD1), lymphocyte activation gene 3 (LAG3), and T cell immunoglobulin and mucin domain containing 3 (TIM3)] expressed on T cells, both without stimulation or 24 hours after stimulation with GBM target cells (Fig. 4D). In all cases, the constitutively expressed CAR T cells expressed a higher number of exhaustion markers compared to nontransduced T cells or synNotch-CAR T cells. Particularly, upon stimulation, T cells constitutively expressing CAR had significantly more populations expressing two or three exhaustion markers compared with the nontransduced T cells (P < 0.05), whereas synNotch-CAR T cells were not significantly different from the nontransduced T cells (P = 0.999, P = 0.42, P = 0.83, and P = 0.95, respectively, for 0, 1, 2, and 3 exhaustion markers).
Tonic signaling by constitutively expressed CARs has been shown to lead to increased T cell exhaustion (24, 25). Thus, we hypothesized that the synNotch-CAR circuits might show reduced exhaustion and increased stem-like phenotypes because of reduced tonic signaling. The synNotch-CAR T cells showed lower tonic signaling than constitutive CAR T cells as measured by CD25 expression in the absence of stimulation (P = 0.037; Fig. 4E). In addition, using mass cytometry analyses, synNotch-CAR T cells showed higher expression of the stemness marker transcription factor T cell factor 1 (TCF1) and lower expression of the exhaustion marker CD39 compared with constitutive CAR T cells (fig. S7). Thus, synNotch regulation of the CAR appears to prevent tonic signaling and subsequent T cell differentiation and exhaustion.
A more naïve-like and less exhausted phenotype is linked to stronger T cell proliferation and persistence in vivo. We directly investigated T cell persistence in vivo at 6 days after infusion into the tumor-bearing mice and found abundant synNotch-CAR T cells in the brain. In contrast, we found no surviving constitutive tandem CAR T cells in parallel experiments (Fig. 4F). Together, these findings are consistent with a model in which synNotch-CAR circuits prevent tonic signaling normally observed in constitutively expressed CARs, thereby allowing the T cells to maintain a more naïve, stem-like state less prone to differentiation and exhaustion. Thus, restricting CAR expression locally to the tumor (where priming signals are present) not only increases T cell targeting specificity but also yields a much more potent and persistent T cell state.
Identification of MOG as a CNS-specific antigen that could be used for tissue-specific priming of inducible CAR T cells
Given the robust trans-killing we observed with EGFRvIII-primed T cells, both in vitro and in vivo, we hypothesized that synNotch-CAR T cells might be capable of priming off of cells near a tumor, even if these priming cells lacked the killing antigen and were not malignant cells. This type of trans-killing could be very useful if, for example, a T cell could be primed by recognizing tissue-specific antigens expressed on nonmalignant cells closely associated in space with the tumor (Fig. 5A). For example, in the case of GBM, which rarely metastasizes outside of the CNS (28), we might design T cell circuits that are primed by a CNS-specific antigen that then triggers local killing by inducing expression of the tandem CAR against the GBM antigens EphA2 and IL13Rα2. Extensive previous studies indicate that these two killing antigens are not expressed in the normal CNS (15, 29, 30). Thus, creating a T cell that restricts its killing to targets that are both in the CNS and EphA2 or IL13Rα2 positive would potentially achieve both high tumor specificity and complete killing. A CNS antigen-primed circuit might thereby provide a solution for treating EGFRvIII-negative GBM.
Fig. 5. Design of brain antigen-primed synNotch-CAR circuit to target GBM.
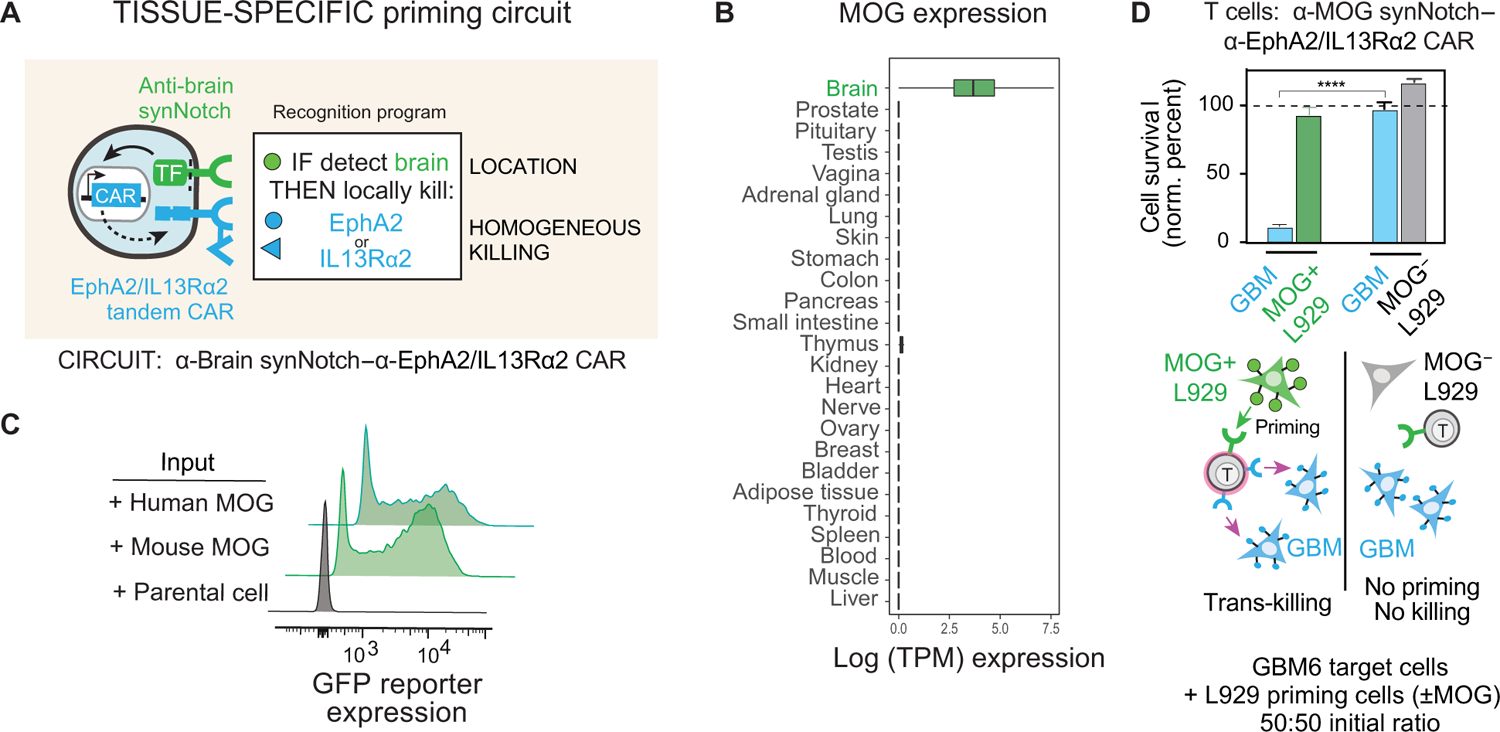
(A) “Tissue-specific priming” circuit is designed to restrict killing only to the brain, preventing damage to normal, nonbrain tissues that express killing antigens, EphA2 and IL13Rα2. In principle, this circuit should selectively identify GBM cells as the only brain-localized cells that are EphA2+ or IL13Rα2+. (B) Box and whisker plots showing tissue-specific expression of MOG across a subset of tissue samples in GTEx v7. Units shown are log scale–normalized RNA-sequencing counts (transcripts per million) taken from GTEx portal v7 (https://gtexportal.org/). (C) Primary CD8+ T cells expressing α-MOG synNotch and GFP reporter were cocultured with either parental K562 cells or K562 cells expressing mouse or human MOG. Flow cytometry histograms show induction of GFP reporter only in the presence of MOG+ K562 cells (representative of three experiments). (D) Primary CD8+ T cells transduced with α-MOG synNotch–α-EphA2/IL13Rα2 CAR circuit were cocultured with GBM6 target cells and L929 priming cells either expressing or not expressing mouse MOG. Relative cell survival of both target GBM and priming L929 cells was tracked over 72 hours (n = 3, error bars indicate SD). ****P < 0.0001, t test compared to those cocultured with MOG− L929 cells. The cell population ratio was 1:1:1, 1 × 104 cells each. No significant killing of the L929 priming cell population was observed in either experiment.
We bioinformatically identified two candidate brain cell surface proteins, cadherin 10 (CDH10), a CNS-specific cadherin, and MOG, a surface protein on the myelin sheath of neurons. The predicted tissue expression of these antigens is shown in Fig. 5B and fig. S8. For this study, we focused primarily on using MOG as a priming antigen, as we empirically found it to be more CNS specific (analysis of CDH10 priming antigen shown in fig. S8). We identified antibody sequences that bind MOG and used them to construct cognate synNotch receptors. We confirmed that these synNotch receptors could be activated by cells expressing either human or mouse MOG protein (Fig. 5C).
We engineered CD8+ T cells with the α-MOG synNotch–α-EphA2/IL13Rα2 CAR circuit and cocultured them with GBM6 target cells in the presence or absence of priming cells (L929 fibroblasts engineered to express MOG). We found that α-MOG synNotch–α-EphA2/IL13Rα2 CAR T cells could effectively kill the GBM6 cells, which do not express MOG, but only in the presence of MOG+ priming cells (Fig. 5D). We did not observe any off-target killing of the cocultured priming cells (which lack EphA2 or IL13Rα2 expression), showing that the synNotch-CAR T cells maintained specificity for only killing the CAR antigen–positive tumor cells in the mixture. These in vitro studies demonstrated that the synNotch-CAR T cells could be safely and efficiently primed for tumor killing by spatially associated nontumor cells expressing priming antigen.
To test whether the CNS-specific antigen-primed T cells could be effective in vivo, we treated NCG mice implanted with intracranial GBM6 PDX tumors with T cells bearing the α-MOG synNotch–α-EphA2/IL13Rα2 CAR circuit (Fig. 6A). The GBM6 tumor cells do not express MOG; therefore, in order for the T cells to kill tumor cells, they must be primed by MOG endogenously expressed in the host mouse CNS. The MOG-primed T cells proved to be highly effective at clearing the GBM6 tumor (P < 0.001, day 40; Fig. 6B) and increasing mouse survival (P = 0.005; Fig. 6C) relative to control mice treated with nontransduced T cells. In mice in which GBM6 tumors were implanted in both the flank and the brain, tumors in the flank were not cleared by the MOG-primed T cells (fig. S8G), consistent with a need for a localized CNS antigen to license the T cells for killing. Similarly, mice treated with T cells bearing the α-CDH10 synNotch–α-EphA2/IL13Rα2 CAR circuit showed effective priming (fig. S8B) and killing of GBM6 both in vitro and in vivo (fig. S8, C to F). However, these synNotch-CAR T cells showed poor discrimination in killing of brain versus flank tumors, suggesting that MOG is a superior CNS-specific priming antigen relative to CDH10 (fig. S8G).
Fig. 6. Tissue-specific priming of synNotch-CAR T cells by brain-specific antigen MOG induces effective killing of GBM6 brain tumors in vivo.
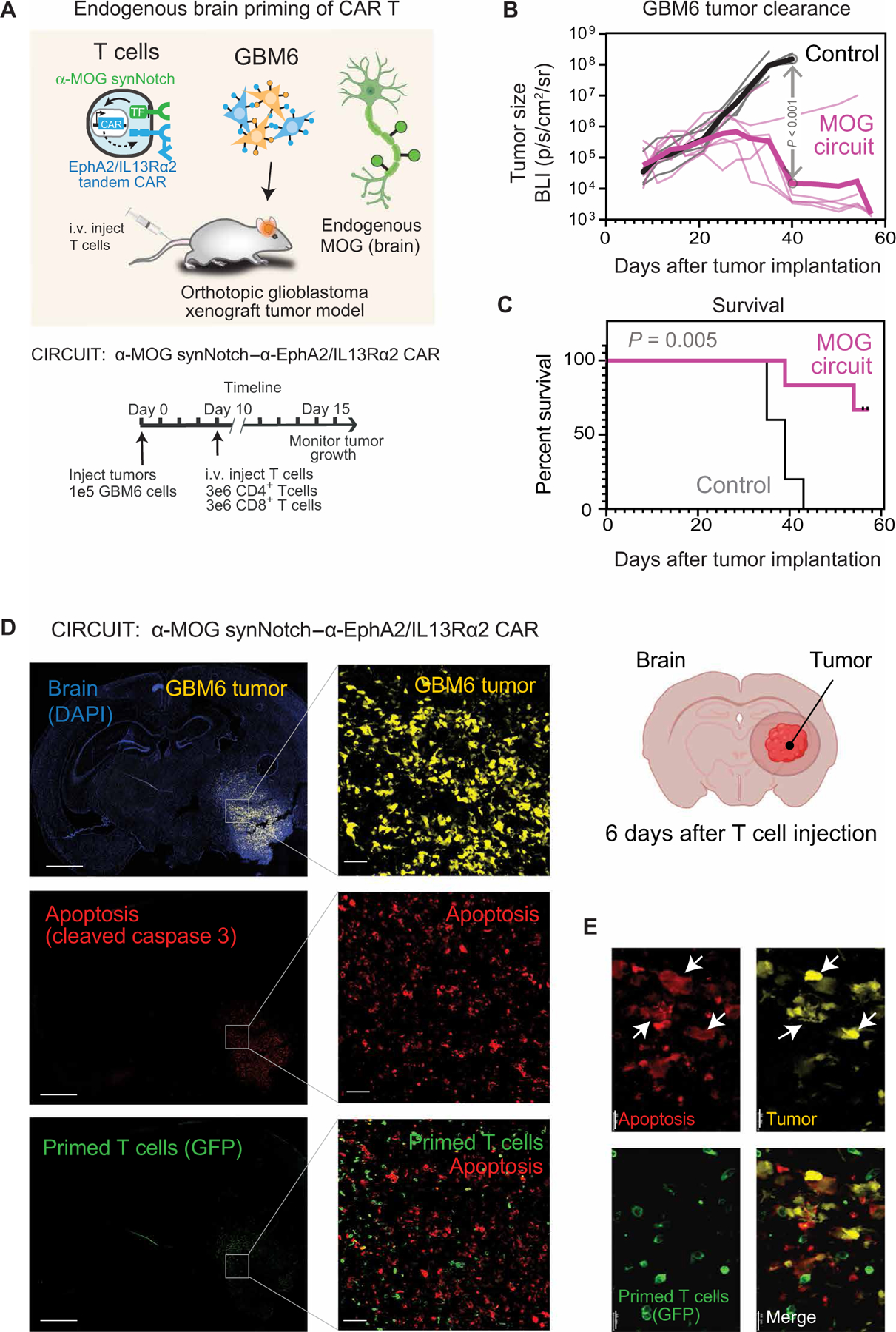
(A) GBM6 tumor cells were stereotactically implanted into brains of NCG mice. GBM6 cells were engineered to express mCherry and luciferase to allow for tracking of tumor size. Ten days after tumor implantation, mice were infused intravenously with 3 million each of CD4+ and CD8+ T cells. T cells expressed either no construct (nontransduced control) (n = 5) or α-MOG synNotch–α-EphA2/IL13Rα2 CAR circuit (n = 6). (B and C) Tumor size (B) and survival (C) were monitored over time. Tumor size was determined by bioluminescence imaging. Negative control treatment (nontransduced T cells) is shown in gray, and α-MOG SynNotch-CAR circuit treatment is shown in pink. P < 0.001 by t test with Holm-Sidak correction for multiple comparisons (on day 40). Thin lines show traces for individual animals; thick line shows geometric mean. See fig. S9G for analysis of off-target specificity (using non–brain-implanted tumor). Survival was analyzed over 60 days by log-rank (Mantel-Cox) test. P = 0.005. (D) GBM6 tumor–bearing mice were euthanized 6 days after α-MOG SynNotch-CAR T cell infusion. Representative confocal fluorescent microscopy of brain sections reveals that T cell–mediated killing (cleaved caspase 3 staining) is restricted to the tumor. Scale bars, 2 mm (left) and 50 µm (right). (E) Insets (single-stained images) are enlargements of outlined regions in main images. Expression of cleaved caspase 3 is confined to the tumor cells (yellow), as indicated by white arrows in the overlay image. Scale bar, 20 µm.
Postmortem immunofluorescence analysis of mice treated with the MOG-primed T cells revealed a high number of T cells in the tumor, with many of these in a primed, CAR-expressing state (Fig. 6D). In tumor-adjacent brain tissue, we observed fewer T cells, but these were also primed and expressing the CAR. In ontrast, although many transplanted T cells were found in the spleen, none of them was primed. Similar brain-specific priming of T cells was observed by flow cytometry analysis of brain and spleen samples (fig. S9). These results are consistent with T cell priming throughout the brain, presumably combined with more robust T cell expansion within the tumor as a result of CAR activation and local release of proliferative cytokines.
Given that these T cells are primed by nonmalignant cells in the CNS, we wanted to more carefully examine cell killing within the brains of treated animals. We sectioned brain tissue isolated from mice 6 days after T cell injection and evaluated them by immunofluorescent imaging for both tumor cells and cell apoptosis (Fig. 6, D and E). Cell killing was restricted only within the boundaries of the tumor and did not extend into the normal CNS tissue.
The effectiveness of this CNS-specific antigen-primed circuit represents an advance in engineering therapeutic cells. Not only does this circuit represent a possible way to treat EGFRvIII− GBM but also it represents a general strategy for targeting cell therapies to specific tissues or organs. Overall, we developed two successful multiple-antigen recognition circuits that rely on either a heterogenous cancer-specific antigen or a CNS tissue-specific antigen to overcome GBM heterogeneity, off-target toxicity, and T cell persistence (fig. S10).
DISCUSSION
These results show multiple ways to engineer synNotch-CAR circuits that synergistically combine recognition of complementary antigens that are otherwise imperfect as single-antigen targets (fig. S10). One can build circuits that are primed by a highly tumor-specific neoantigen, like EGFRvIII, or a tissue-specific antigen, like MOG. Critically, these priming antigens need not be homogeneously expressed on all tumor cells, or be present on any of the tumor cells at all, in the case of tissue-specific priming. Once primed, the T cells can then be programmed to execute complete tumor killing by inducing expression of CARs that target antigens homogenously expressed by tumors, even if these antigens have imperfect specificity individually. By integrating information from multiple antigens and multiple cells, these circuits essentially give improved capability for nuanced recognition of a tumor as a complex tissue and thus open up many new possibilities for how to recognize and attack tumors in safer and more specific ways. Other related strategies combine CARs and bispecific engagers to integrate multi-antigen combinations (31). The work shown here demonstrates that synNotch-CAR T cells have multiple facets that distinguish them from conventional constitutively expressed single-target CAR T cells, several of which may prove to be highly advantageous in treating other solid cancers beyond GBM (fig. S10).
An inherent dilemma constraining the design of any targeted cancer therapeutic is that increasing specificity to limit off-target toxicity concurrently increases the chance of tumor escape via intrinsic heterogeneity or loss of the molecular target. Here, we show that a prime-and-kill circuit that integrates two or more antigens can be used to navigate this dual optimization problem by relying on highly specific priming antigens, such as the GBM neoantigen EGFRvIII, to then induce the expression of CARs that kill based on a highly homogeneous antigen or set of antigens, such as EphA2 and IL13Rα2. Neither the priming nor killing antigens need to be perfect. EGFRvIII is specific but nonhomogeneous, whereas EphA2 and IL13Rα2 are more homogeneous but imperfectly tumor specific. Because their deficiencies are different, these types of antigens can be combined to yield a multi-antigen circuit that is both highly specific and capable of more complete killing. We empirically observe that the EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells are able to effectively and completely clear GBM6 PDX tumors without killing surrounding normal tissue or EphA2- or IL13Rα2-positive cells in other parts of the body that lack colocalized priming antigen.
The ability of these circuits to balance specificity and completeness of killing stems from the ability of the T cells to be primed by one type of cell and to then be induced to kill CAR antigen–positive target cells within the local environment (trans-killing). Once T cells move away from the environment with priming antigen, they lose CAR expression within hours and are thus unable to sustain a long-term proliferative and killing response, which takes days to weeks. Thus, the T cells are essentially capable of integrating over space to specifically launch potent, large-scale killing only in a microenvironment that contains the right combination of colocalized multiple antigens. Therefore, this targeting strategy is likely to be useful in preventing both toxicity and escape in many other solid tumors, presuming that similar complementary antigen combinations can be identified (32). This represents one of the first antitumor strategies that can overcome the intrinsic trade-off between specificity and limiting evasive escape.
An additional benefit of prime-and-kill circuit in T cells is that the simple act of placing CAR expression under regulated control maintains the T cells in a more naïve-like state that is more durable and less subject to exhaustion. Here, we find that T cells with the constitutive EphA2/IL13Ra2 CAR show no antitumor activity in vivo, whereas T cells with the synNotch-induced version durably clear the GBM tumor. Similar improved antitumor activity has been observed for synNotch-CAR T cells targeting other cancers beyond GBM (33), suggesting that these circuits may provide an effective general strategy for treating many solid cancers. These findings are consistent with recent findings that CAR T cell exhaustion can be limited by intermittent drug-induced repression of CAR signaling (34). In all of these cases, limiting tonic signaling appears to prevent the accumulation of exhausted, terminally differentiated cells. Restricting CAR expression until the T cells enter the tumor thus appears to be a logical strategy for optimizing cell potency and persistence.
Another new finding in this study is that tissue-specific antigens, such as MOG, whose expression is restricted to the CNS, can be harnessed as part of a multi-antigen targeted recognition signature for engineered T cell therapies. We find that T cells expressing an α-MOG synNotch receptor can be effectively and specifically primed in vivo in the CNS by endogenously expressed MOG. Moreover, if these cells are induced to express the α-EphA2/IL13Rα2 CAR, they only kill CAR antigen–expressing cells in the CNS and not those implanted outside of the CNS. This priming based on tissue-specific antigens is possible because the T cells can then execute trans-killing of tumor cells in the immediate proximity of the nonmalignant priming cells. This approach bypasses the need for a cancer-specific priming antigen and could be used for GBM that is EGFRvIII negative. This strategy works for the killing antigens EphA2 and IL13Rα2 because these antigens are not normally expressed in the brain, except in the case of tumors (15, 29, 30).
The ability to harness normal tissue antigens as part of a multi-antigen targeting signature adds a powerful new way to target cell-based therapies. For example, CNS-specific targeting could be used to more effectively target other CNS tumors or CNS metastases. CNS-targeted cells could also be used to treat other types of CNS diseases beyond cancer, including neuroinflammation or neuro-degeneration. Moreover, it is possible that identification of other highly tissue-specific antigens together with more sophisticated circuits (35) could lead to improved targeting of engineered cells to tissues and organs beyond the CNS.
We recognize several limitation of our present study. First, our strategy to design optimal multi-antigen recognition circuits is based on available gene expression data, for both tumors and normal tissues, and these data are not complete and limited in resolution, as not all tissues have been analyzed, most is bulk tissue expression rather than single cell, and most is focused on mRNA level rather than protein level. The process of choosing optimal tissue-specific priming antigens and the best killing antigens for any cancer will improve as better genomic data become available. Second, use of immunocompromised mice bearing human GBM and T cells limits our ability to fully characterize the interaction between synNotch-CAR T cells and the native GBM microenvironment. Syngeneic mouse models could help address this, although, ultimately, implementation of a carefully designed phase 1 clinical trial will be required to evaluate the safety and efficacy of our strategies in human patients.
Here, we have engineered combinatorial recognition T cells that harness existing CAR targets that have previously been clinically shown to be imperfect. We show that by linking these multiple imperfect targets into an integrated multi-antigen recognition T cell, we can generate next-generation T cell therapies that outperform traditional CAR T cells in preclinical models. We are hopeful that similar strategies can be applied to find new cell treatments for other challenging solid cancers.
MATERIALS AND METHODS
Study design
The objective of this study was to evaluate the therapeutic potential of synNotch-CAR T cells, in comparison with constitutively expressed CAR T cells, to overcome antigenic heterogeneity, off-target toxicity, and persistence in a mouse model bearing human PDX GBM. For in vivo experiments, 6- to 10-week-old NCG mice were used, with 5 to 10 mice per group to ensure statistical power. Animal size sample was determined by power analysis based on preliminary data. Before CAR T cell treatment, mice were randomized on the basis of bioluminescent imaging to ensure similar average tumor sizes across groups. There was no blinding. Survival was evaluated until predetermined Institutional Animal Care and Use Committee (IACUC)–approved endpoint was reached. The brain of each euthanized mouse was collected to confirm the absence of tumor. Before in vivo experiments, the T cell circuits were tested in vitro using coculture assays. Circuits were tested in T cells from multiple donors. Number of repeats is specified in each figure legend.
Construct design
SynNotch receptors were built by fusing α-EGFRvIII 139 scFv (11), α-MOG M26 scFv (36), or α-CDH10 scFv (Sidhu laboratory) to mouse Notch1 (NM_008714) minimal regulatory region (res.1427–1752) and Gal4 DBD VP64. All synNotch receptors contain N-terminal CD8a signal peptide (MALPVTALLLPLALLL HAARP) for membrane targeting and α-myc–tag (EQKLISEEDL) or flag-tag (DYKDDDDK) for detecting surface expression with α-myc A647 (Cell Signaling, no. 2233) or α-flag A647 (R&D Systems, no. IC8529R); see Morsut et al. (16) for synNotch sequence. Receptors were cloned into a modified pHR′SIN:CSW vector containing a phosphoglycerate kinase (PGK) orspleenfocus-formingvirus(SFFV) promoter. ThepHR′SIN:CSW vector was also used to make response element plasmids with five copies of the Gal4 DNA binding domain target sequence (GGAG-CACTGTCCTCC GAACG) upstream from a minimal cytomegalovirus (CMV) promoter. Response element plasmids also contain a PGK promoter that constitutively drives mCherry or blue fluorescent protein (BFP) expression to easily identify transduced T cells. CARs were built by fusing EphA2 scFv (37), IL13 mutein [E13K,K105R] (38), or IL13 mutein [E13K,K105R]-G4Sx4-EphA2 scFv (37, 38) to the hinge region of the human CD8α chain and transmembrane and cytoplasmic regions of the human 4–1BB, and CD3z signaling domains. Inducible CAR constructs were cloned into a Bam HI site 3′ to the Gal4 response elements. CARs were tagged C-terminally with GFP, BFP, myc-tag, or flag-tag to verify surface expression.
Primary human T cell isolation and culture
Primary CD4+ and CD8+ T cells were isolated from donor blood after apheresis by negative selection (STEMCELL Technologies, nos. 15062 and 15063). Blood was obtained from Blood Centers of the Pacific, as approved by the University of California San Francisco (UCSF) Institutional Review Board. T cells were cryopreserved in RPMI 1640 (UCSF cell culture core) with 20% human AB serum (Valley Biomedical, no. HP1022) and 10% dimethyl sulfoxide. After thawing, T cells were cultured in human T cell medium consisting of X-VIVO 15 (Lonza, no. 04–418Q), 5% human AB serum, and 10 mM neutralized N-acetyl L-cysteine (Sigma-Aldrich, no. A9165) supplemented with IL-2 (30 U/ml; National Cancer Institute Biological Resources Branch Preclinical Repository) for all experiments except for the IncuCyte experiments. IncuCyte experiments were cultured in RPMI 1640 with 5% human AB serum supplemented with IL-2 (30 U/ml).
Lentiviral transduction of human T cells
Pantropic vesicular stomatitis virus G (VSV-G)–pseudotyped lentivirus was produced via transfection of Lenti-X 293T cells (Clontech, no.11131D) with a pHR′SIN:CSW transgene expression vector and the viral packaging plasmids pCMVdR8.91 and pMD2.G using Fugene HD (Promega, no. E2312). Primary T cells were thawed the same day and, after 24 hours in culture, stimulated with Human T-Activator CD3/CD28 Dynabeads (Life Technologies, no. 11131D) at a 1:3 cell:bead ratio. At 48 hours, viral supernatant was harvested and, in some assays, concentrated using a Lenti-X concentrator (Clontech, no. 631231). Primary T cells were exposed to the lentivirus for 24 hours. At day 4 after T cell stimulation, Dynabeads were removed, and T cells were expanded until day 9, when they were rested and could be used in assays. T cells were sorted with the Beckton Dickinson (BD) FACSAria Fusion or Sony SH800S Cell Sorter. AND-gate T cells exhibiting basal CAR expression were gated out during sorting.
Human T cell phenotyping
T cell phenotypes were assessed using the following antibodies: phycoerythrin (PE) anti-CD25 (clone BC96, 302606, BioLegend) for T cell activation; PE-Cy7 anti-PD1 (clone EH12.1, 561272, BD Biosciences), BV785 anti-TIM3 (clone F38–2E2, 345031, BioLegend), and AF700 anti-LAG3 (clone 3DS223H, 56–2239-42, Thermo Fisher Scientific) for T cell exhaustion; BV605 anti-CD45RA (clone HI100, 304133, BioLegend) and allophycocyanin (APC)–Cy7 anti-CD62L (clone DREG-56, 304813, BioLegend) T cell differentiation state. Briefly, posttransfected T cells were sorted, expanded, and rested for about 10 days and then analyzed by flow cytometry. To assess influence of antigen-dependent activation on T cell phenotype, T cells were cocultured at an Effector:Target (E:T) ratio of 1:1 (50,000:50,000) for 24 hours with target GBM6 cells prestained with CellTrace Far Red (C34564, Thermo Fisher Scientific) per the manufacturer’s instructions to distinguish them from T cells. We used a 1:100 antibody dilution. A total volume of 50 µl per staining reaction was used in staining buffer [phosphate-buffered saline (PBS) with 2% fetal bovine serum]. Samples were incubated at 4°C for 15 min and washed with staining buffer. T cells were analyzed by flow cytometry.
Cell lines
Cell lines used were K562 myelogenous leukemia cells [American Type Culture Collection (ATCC), no. CCL-243], L929 mouse fibroblast cells (ATCC, no. CCL-1), U87 MG GBM cells (ATCC, no. HTB-14), and GBM6 PDX cells (gift of F. Furnari, Ludwig Institute and University of California San Diego). U87-EGFRvIII–negative luciferase (12) and U87 MG cells were lentivirally transduced to stably express GFP or mCherry, respectively, under control of the SFFV promoter. At 72 hours after transduction, cells were sorted on an Aria Fusion cell sorter (BD Biosciences) on the basis of GFP or mCherry expression and expanded. Cell lines were sorted for expression of the transgenes. U87-luciferase and U87-luciferase-mCherry cells were stably transduced with nonmutated EGFR using a retroviral construct (gift of M. Meyerson; Addgene, plasmid no. 11011) to generate an EGFRvIII-negative cell line that grows at a similar rate as the EGFRvIII-positive U87 cells. GBM6 cells were lentivirally transduced to stably express both mCherry and firefly luciferase. These cells were cultured in Dulbecco’s modified Eagle’s medium F12 medium, with supplements of EGF (20 µg/ml), fibroblast growth factors (20 µg/ml), and heparin (5 µg/ml). K562 cells were lentivirally transduced to stably express surface CDH10 (CDH10 extracellular membrane was fused to the platelet-derived growth factor transmembrane domain). K562s and L929 cells were lentivirally transduced to stably express full-length MOG.
In vitro stimulation of synNotch T cells
For in vitro synNotch-CAR T cell and tumor cell coculture experiments, 1 × 104 tumor cells (either U87 or GBM6) were cultured overnight in a flat-bottom 96-well tissue culture plate. The next morning, 1 × 104 to 5 × 104 T cells were added to the plate and cocultures were analyzed for 24 to 96 hours for activation and specific lysis of tumor cells. For in vitro synNotch T cell stimulations cocultured with three different cell populations, an additional 1 × 104 priming cells (either K562 or L929) were added to the initial overnight culture. Flow cytometry was performed using BD LSRII or Attune NxT Flow Cytometer; analysis was performed by FlowJo software (Tree Star).
Assessment of synNotch-CAR T cell cytotoxicity
CD8+ synNotch-CAR T cells were stimulated for 24 to 96 hours as described above with target cells expressing the indicated antigens. The degree of specific lysis of target cells was determined by comparing the fraction of target cells alive in the culture compared to treatment with nontransduced T cell controls, unless stated otherwise. Cell death was monitored by shift of target cells out of the side scatter and forward scatter region normally populated by the target cells. Alternatively, cell viability was analyzed using the IncuCyte Zoom system (Essen BioScience). Tumor cells were plated into a 96-well plate at a density of 1.0 × 104 cells per well in triplicate overnight. T cells were added at the indicated concentrations into each well next day at a final volume of 200 µl per well. Target cells and T cells were cocultured as described above. Two fields of view were taken per well every 15 min. Mean florescence intensity was calculated using IncuCyte Zoom software (Essen BioScience) to determine target cell survival. Data were summarized as means ± SEM.
Assessment of CAR expression decay in vitro
EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells were cocultured with excess of target GBM6 cells for 60 hours to induce steady-state CAR expression. Primed, CAR-expressing GFP-positive cells (α-EphA2/IL13Rα2 CAR is fused to GFP) were sorted and then cultured alone or with U87 tumor cells. GFP fusion reporter was monitored over time by flow analysis as a readout of CAR decay. Data were fit to exponential decay to estimate the half-life of CAR expression.
In vivo mouse experiments
All mouse experiments were conducted according to IACUC-approved protocols. For orthotopic heterogeneous model with U87 tumors, a mixture of 1.5 × 104 U87-luc-EGFRvIII–positive mCherry cells and 1.5 × 104 U87-luc-EGFRvIII–negative GFP cells was implanted intracranially into 6- to 10-week-old female NCG mice (Charles River), with 6 to 10 mice per group. For homogeneous U87-luc-GFP-EGFRvIII–positive model, 3 × 104 cells were injected into the brains of NCG mice. For orthotopic heterogeneous model with GBM6, 10 × 105 GBM6-luc-mCherry cells were intracranially implanted into 6- to 8-week-old female NCG mice with 5 to 10 mice per group. After anesthesia with 1.5% isoflurane, stereotactic surgery for tumor cell implantation (injection volume: 2 µl) was performed with the coordination of the injection site at 2 mm right and 1 mm anterior to the bregma and 3 mm into the brain. Before and for 3 days after surgery, mice were treated with an analgesic (meloxicam and buprenorphine) and monitored for adverse symptoms in accordance with the IACUC. In the subcutaneous model, NCG mice were injected with either 1.0 × 106 U87-luc-mCherry+ or 1.2 × 105 GBM6-luc-mCherry cells subcutaneously in 100 µl of Hanks’ balanced salt solution on day 0. Tumor progression was evaluated by luminescence emission on Xenogen IVIS Spectrum after intraperitoneal injection of 1.5 mg of D-luciferin (GoldBio; injection volume, 100 µl). Before treatment, mice were randomized such that initial tumor burden in control and treatment groups was equivalent. Mice were intravenously treated with 6.0 × 106 engineered or non-transduced T cells via tail vein in 100 µl of PBS. Survival was evaluated over time until predetermined IACUC-approved endpoint (hunching, neurological impairments such as circling, ataxia, paralysis, limping, head tilt, balance problems, and seizures) was reached (n = 6 to 10 mice per group).
Cranial window implantation
Eight-week-old NCG mice underwent tumor implantation with GBM6 xenograft as described above. Ten to 12 days after tumor injection, mice underwent implantation of a cranial window and custom-designed titanium headplate (UC Berkeley Physics Machine Shop; design by K. Poskanzer) according to the following procedure. The cranial window consisted of a No.1 4-mm glass coverslip (Warner Scientific) glued onto a No.1 3-mm glass coverslip with optical adhesive (Norland 71, Norland Products) and cured with 365-nm ultraviolet light for 15 min. Five hours before surgery, mice were intraperitoneally injected once with dexamethasone (2.8 mg/kg). At time of surgery, mice were anesthetized with 1.5 to 3% isoflurane and secured into a stereotactic frame with ear bars. The head was shaved, an ovular flap of skin was removed from the top of the head, and skin margins were glued down with biocompatible cyanoacrylate glue (VetBond, 3M). The titanium headplate was superglued (KrazyGlue) to the skull and secured with dental cement at the end of the surgery (C&B Metabond Kit, Parkell). A 3-mm-diameter craniotomy was performed using a Foredom micro-drill with 1-mm and 0.5-mm carbide drill bits (McMaster-Carr). The exposed brain was gently irrigated with ice-cold artificial cerebro-spinal fluid (150 mM NaCl, 2.5 mM KCl, 10 mM Hepes, pH 7.3), and the cranial window was secured with superglue and dental cement. Mice were provided with heat, fluids, and analgesics during post-surgical recovery in accordance with institutional IACUC regulations. T cell injections were given 15 days after tumor implantation, and imaging was performed at 1 and 2 days after adoptive transfer of T cells.
Two-photon in vivo microscopy
Intravital two-photon images were acquired with Zeiss LSM 780 NLO equipped with a Ti:Sapphire laser (MaiTai HP, Spectra Physics) tuned to 760 nm (for excitation of tagBFP+ synNotch-CAR T cells and mCherry+ tumor) and 900 nm (for excitation of GFP+ synNotch-CAR T cells), respectively, and focused through a Zeiss 20× water immersion objective (numerical aperture of 1.0). Before imaging, mice were anesthetized with isoflurane and the headplate was fixed into the head posts of a custom-made moving stage (Thorlabs, UC Berkeley Physics Machine Shop). Anesthesia was maintained at 1% isoflurane through a nose cone, and body temperature was kept stable via a temperature-controlled heating pad. Images of 598 µm × 598 µm areas of in vivo tumors were acquired at 512 pixel × 512 pixel resolution for standard images and 1024 pixel × 1024 pixel resolution for higher-resolution images. Volume images were acquired over a 30- to 200-µm Z range in 5- or 10-µm steps. Time-lapse datasets were acquired either in single planes over time periods up to 45 min or in combined time + Z series over a 598 × 598 area (X × Y) with variable Z ranges (Z = 5 to 100 µm) with 1- to 5-µm steps. Movies were processed in Zen software for three-dimensional reconstructions.
Immunofluorescence
Mice were euthanized before being transcardially perfused with cold PBS. Brains were then removed and fixed overnight in 4% paraformaldehyde–PBS before being transferred to 30% sucrose and were allowed to sink (1 to 2 days). Subsequently, brains were embedded in O.C.T. Compound (Tissue-Tek; 4583; Sakura Finetek). Serial 10-mm coronal sections were then cut on freezing microtome and stored at −20°C. Sections were later thawed, fixed with 10% formalin for 10 min followed by incubation in blocking buffer (PBS–5% normal donkey serum) for 40 min, and stained with primary antibodies overnight at 4°C. Primary antibodies used were the following: CD45 (D9M8I) XP Rabbit mAb (Cell Signaling Technology; 1:100), Anti-EGFRvIII, clone DH8.3 (MilliporeSigma; 1:100), EphA2 (D4A2) XP Rabbit mAb (Cell Signaling Technology; 1:100), Anti-IL13 receptor alpha 2 antibody (Abcam; 1:100), and Cleaved Caspase 3 (Asp175) Rabbit mAb (Cell Signaling Technology; 1:400).
Secondary antibodies raised in donkey and conjugated with Alexa Fluor 647 were used at 4°C for 2 hours to detect primary labeling. Sections were stained with nuclear dye DRAQ7 (Abcam) or 4′,6-diamidino-2-phenylindole. (DAPI) (Thermo Fisher Scientific). Images were acquired using either a Zeiss Axio Imager 2 microscope (×20 magnification) with TissueFAXS scanning software (TissueGnostics) or a Zeiss LSM 780 microscope (×20 magnification) with Zeiss Zen imaging software. Exposure times and thresholds were kept consistent across samples within imaging sessions. Single-color images were used to elucidate the regions for quantification. DAPI was used for nuclear segmentation. Colocalization of CD45 (AF647) and GFP signal with nuclear stain was done to obtain counts for GFP+ T cells (StrataQuest Software).
Assessment of engineered T cells in vivo
For all experiments involving phenotyping of adoptively transferred engineered T cells, brain and spleen were harvested after per-fusion with cold PBS. Brains were mechanically minced and treated at 37°C for 30 min with digestion mix consisting of collagenase D (30 mg/ml) and deoxyribonuclease (10 mg/ml) and soybean trypsin inhibitor (20 mg/ml). The resulting brain homogenate was resuspended in 70% Percoll (GE Healthcare), overlaid with 30% Percoll, and then centrifuged for 30 min at 650g. Enriched brain infiltrating T cells were recovered at the 70–30% interface and stained with fluorescently conjugated antibodies against CD3 (5 µl; catalog no. 555342, BD Biosciences) and CD45 (5 µl; catalog no. 564357, BD Biosciences) for 1 hour at 4°C. Before staining with antibodies, cells were stained with BD Horizon Fixability Viability Stain 780 (BD Biosciences) to discriminate live from dead cells. Data were collected on the Attune NxT Flow Cytometer, and the analysis was performed in FlowJo software (Tree Star).
Statistical analysis
Statistical significance was determined by specific tests and presented as means ± SEM or means ± SD as indicated in the figure legends. A Shapiro-Wilk test was used for data normalcy. To assess significant differences between single measurements of two groups of normally distributed data, unpaired or paired two-tailed Student’s t test was used; otherwise, Mann-Whitney U test was applied. To assess significant differences between more than two groups of normally distributed data, we performed one-way analysis of variance (ANOVA), followed by post hoc analyses: Comparison against a control group was performed using Dunnett’s multiple comparisons test, comparison of selected pairs of datasets was performed using Holm-Sidak’s multiple comparisons test, and comparison of all pairs of datasets was performed using Tukey’s multiple comparisons test. The Kaplan-Meier estimator was used to generate survival curves, and differences in survival distributions were assessed using the log-rank test. All P values are provided in the figures or their legends. All statistical analyses were performed with Prism software version 7.0 (GraphPad).
Supplementary Material
Movie S1. Real-time killing assays using different heterogeneous mixtures of EGFRvIII+ and EGFRvIII− target cells show efficient trans-killing.
Movie S2. Intravital imaging of circuit CAR T cells shows dynamic priming within the GBM6 xenograft tumor.
Data file S1. Raw data.
Fig. S1. Construction and testing of α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells against U87 GBM.
Fig. S2. Prime-and-kill circuit in T cells can overcome heterogeneity using a model antigen system in vitro.
Fig. S3. α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells mediate effective and localized antitumor response against U87 GBM that heterogeneously expresses EGFRvIII in the brain.
Fig. S4. α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells are effective against GBM6 tumor cells in vitro and in vivo.
Fig. S5. Fluorescence imaging of brain slices from NCG mice with implanted GBM6 tumors treated with α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells.
Fig. S6. SynNotch-CAR T cells show more naïve-like phenotype compared to comparable constitutively expressed CAR T cells.
Fig. S7. Mass cytometry shows that synNotch-induced circuits yield T cells with increased expression of stemness marker TCF1 and reduced expression of exhaustion marker CD39.
Fig. S8. Construction and testing of α-CDH10 synNotch–α-EphA2/IL13Rα2 CAR T cells.
Fig. S9. Brain-specific synNotch-CAR T cells mediate effective anti-GBM responses.
Fig. S10. Strategies for design of synNotch-CAR T cells to treat GBM.
Acknowledgments:
We thank J. Williams, R. Hernandez-Lopez, V. Tieu, N. Blizzard, J. Shon, H. Ogino, M. Shahin, and members of the W.A.L. laboratory and H.O. laboratory for assistance, advice, and helpful discussions.
Funding: This study was supported by HDFCCC Shared Resource Facilities, Laboratory for Cell Analysis and Preclinical Therapeutics Core, through NIH (P30CA082103). This work was supported by grants from the NIH—U54CA244438 (W.A.L.), R01 CA196277 (W.A.L.), and 1R35 NS105068 (H.O.); the Howard Hughes Medical Institute (W.A.L.); UCSF Glioblastoma Precision Medicine Program (W.A.L. and H.O.); and Parker Institute for Cancer Immunotherapy (W.A.L. and H.O.).
Footnotes
Competing interests: W.A.L. is on the Scientific Advisory Board for Allogene Therapeutics and is a shareholder of Gilead Sciences and Intellia Therapeutics. H.O. is on the Scientific Advisory Board for Neuvogen and Eureka Therapeutics. K.T.R. is a cofounder of Arsenal Biosciences. W.A.L., H.O., P.B.W., and J.H.C. are inventors on the following relevant patent/patent applications held/submitted by UCSF: Trans-antigen targeting in heterogeneous cancers and methods of use thereof (WO 2019/195576 A1) (2019); Methods of treating EGFRvIII-expressing glioblastomas (WO2019/195586 A1; US 62/654,012) (2019); Methods of treating glioblastomas (WO2019195596A1) (2019); W.A.L., H.O., P.B.W., J.H.C., and M.S.S. are inventors on the following relevant patent/patent applications held/submitted by UCSF: Use of MOG for priming a treatment for glioblastoma (US 62/980,882) (2020) and Use of brain-specific antigens to home, block and deliver cell-based treatments to the brain (US 62/980,885) (2020). H.O. is an inventor on patent Treatment of cancer using humanized anti-EGFRvIII chimeric antigen receptor US20190330356A1 (2019) held by University of Pennsylvania and University of Pittsburgh that covers EGFRvIII targeting.
Data and materials availability:
All data associated with this study are in the paper or the Supplementary Materials. All reagents will be made available to members of the research community after completion of a material transfer agreement. Reagent requests should be directed to W.A.L. (Wendell.lim@ucsf.edu) and copied to N. Blizzard (noleine.blizzard@ucsf.edu) and M. Broeker (Michael.Broeker@ucsf.edu).
REFERENCES AND NOTES
- 1.June CH, Sadelain M, Chimeric antigen receptor therapy. N. Engl. J. Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee C-CR, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA, Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan D-AN, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA, T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. J. Am. Soc. Gene Ther 19, 620–626 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, McMahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, Rosenberg SA, Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother 36, 133–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA, Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. J. Am. Soc. Gene Ther 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, Lacey SF, Navenot J-M, Zheng Z, Levine BL, Okada H, June CH, Brogdon JL, Maus MV, A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med 9, eaaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, Biegel JA, Hayes RL, Wong AJ, Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res 55, 5536–5539 (1995). [PubMed] [Google Scholar]
- 8.Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD, Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res 57, 4130–4140 (1997). [PubMed] [Google Scholar]
- 9.Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, Sawaya R, Aldape K, Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res 11, 1462–1466 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Thorne AH, Zanca C, Furnari F, Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro Oncol 18, 914–918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, Boesteanu AC, Cogdill AP, Chen T, Fraietta JA, Kloss CC, Posey AD Jr., Engels B, Singh R, Ezell T, Idamakanti N, Ramones MH, Li N, Zhou L, Plesa G, Seykora JT, Okada H, June CH, Brogdon JL, Maus MV, Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med 7, 275ra22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohno M, Ohkuri T, Kosaka A, Tanahashi K, June CH, Natsume A, Okada H, Expression of miR-17–92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J. Immunother. Cancer 1, 21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, Wu M-F, Orange JS, Sumazin P, Man T-K, Joseph SK, Hegde M, Ahmed N, Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol 20, 506–518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hegde M, Corder A, Chow KKH, Mukherjee M, Ashoori A, Kew Y, Zhang YJ, Baskin DS, Merchant FA, Brawley VS, Byrd TT, Krebs S, Wu MF, Liu H, Heslop HE, Gottschalk S, Gottachalk S, Yvon E, Ahmed N, Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther 21, 2087–2101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wykosky J, Gibo DM, Stanton C, Debinski W, EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol. Cancer Res 3, 541–551 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, Lim WA, Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 164, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA, Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, Smythe KS, Dudakov JA, Pierce RH, Rader C, Riddell SR, Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 35, 489–503.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zah E, Lin M-Y, Silva-Benedict A, Jensen MC, Chen YY, T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res 4, 498–508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kahlon KS, Brown C, Cooper LJN, Raubitschek A, Forman SJ, Jensen MC, Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res 64, 9160–9166 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Chow KKH, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, Rainusso N, Wu M-F, Liu H, Kew Y, Grossman RG, Powell S, Lee D, Ahmed N, Gottschalk S, T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther 21, 629–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krenciute G, Krebs S, Torres D, Wu M-F, Liu H, Dotti G, Li X-N, Lesniak MS, Balyasnikova IV, Gottschalk S, Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T cells to IL13Rα2-positive glioma. Mol. Ther. J. Am. Soc. Gene Ther 24, 354–363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, Paucar A, Yang H, Ohashi M, Zhu S, Wykosky J, Reed R, Nelson SF, Cloughesy TF, James CD, Rao PN, Kornblum HI, Heath JR, Cavenee WK, Furnari FB, Mischel PS, Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 343, 72–76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, Nagaraja S, Granja J, de Bourcy CFA, Majzner R, Satpathy AT, Quake SR, Monje M, Chang HY, Mackall CL, c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL, 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR, Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP, A human memory T cell subset with stem cell-like properties. Nat. Med 17, 1290–1297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Awan M, Liu S, Sahgal A, Das S, Chao ST, Chang EL, Knisely JPS, Redmond K, Sohn JW, Machtay M, Sloan AE, Mansur DB, Rogers LR, Lo SS, Extra-CNS metastasis from glioblastoma: A rare clinical entity. Expert Rev. Anticancer Ther 15, 545–552 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK, Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 1, 1253–1258 (1995). [PubMed] [Google Scholar]
- 30.Wykosky J, Gibo DM, Stanton C, Debinski W, Interleukin-13 receptor α2, EphA2, and Fos-related antigen 1 as molecular denominators of high-grade astrocytomas and specific targets for combinatorial therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 14, 199–208 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, Bailey SR, Boroughs AC, Frigault MJ, Leick MB, Scarfò I, Cetrulo CL, Demehri S, Nahed BV, Cahill DP, Wakimoto H, Curry WT, Carter BS, Maus MV, CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol 37, 1049–1058 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Dannenfelser R, Allen GM, VanderSluis B, Koegel AK, Levinson S, Stark SR, Yao V, Tadych A, Troyanskaya OG, Lim WA, Discriminatory power of combinatorial antigen recognition in cancer T cell therapies. Cell Syst 11, 215–228.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry ND, Montgomery G, Liu B, Roybal KT, SynNotch CAR circuits enhance solid tumor recognition and promote persistent anti-tumor activity in mouse models. Sci. Transl. Med 10.1126/scitranslmed.abd8836, (2021). [DOI] [PMC free article] [PubMed]
- 34.Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, Mackall CL, Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv 3, 711–717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams JZ, Allen GM, Shah D, Sterin IS, Kim KH, Garcia VP, Shavey GE, Yu W, Puig-Saus C, Tsoi J, Ribas A, Roybal KT, Lim WA, Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science 370, 1099–1104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Büdingen H-C, Hauser SL, Fuhrmann A, Nabavi CB, Lee JI, Genain CP, Molecular characterization of antibody specificities against myelin/oligodendrocyte glycoprotein in autoimmune demyelination. Proc. Natl. Acad. Sci. U.S.A 99, 8207–8212 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldgur Y, Susi P, Karelehto E, Sanmark H, Lamminmäki U, Oricchio E, Wendel H-G, Nikolov DB, Himanen JP, Generation and characterization of a single-chain anti-EphA2 antibody. Growth Factors 32, 214–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krebs S, Chow KKH, Yi Z, Rodriguez-Cruz T, Hegde M, Gerken C, Ahmed N, Gottschalk S, T cells redirected to interleukin-13Rα2 with interleukin-13 mutein–chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Rα1. Cytotherapy 16, 1121–1131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Real-time killing assays using different heterogeneous mixtures of EGFRvIII+ and EGFRvIII− target cells show efficient trans-killing.
Movie S2. Intravital imaging of circuit CAR T cells shows dynamic priming within the GBM6 xenograft tumor.
Data file S1. Raw data.
Fig. S1. Construction and testing of α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells against U87 GBM.
Fig. S2. Prime-and-kill circuit in T cells can overcome heterogeneity using a model antigen system in vitro.
Fig. S3. α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells mediate effective and localized antitumor response against U87 GBM that heterogeneously expresses EGFRvIII in the brain.
Fig. S4. α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells are effective against GBM6 tumor cells in vitro and in vivo.
Fig. S5. Fluorescence imaging of brain slices from NCG mice with implanted GBM6 tumors treated with α-EGFRvIII synNotch–α-EphA2/IL13Rα2 CAR T cells.
Fig. S6. SynNotch-CAR T cells show more naïve-like phenotype compared to comparable constitutively expressed CAR T cells.
Fig. S7. Mass cytometry shows that synNotch-induced circuits yield T cells with increased expression of stemness marker TCF1 and reduced expression of exhaustion marker CD39.
Fig. S8. Construction and testing of α-CDH10 synNotch–α-EphA2/IL13Rα2 CAR T cells.
Fig. S9. Brain-specific synNotch-CAR T cells mediate effective anti-GBM responses.
Fig. S10. Strategies for design of synNotch-CAR T cells to treat GBM.
Data Availability Statement
All data associated with this study are in the paper or the Supplementary Materials. All reagents will be made available to members of the research community after completion of a material transfer agreement. Reagent requests should be directed to W.A.L. (Wendell.lim@ucsf.edu) and copied to N. Blizzard (noleine.blizzard@ucsf.edu) and M. Broeker (Michael.Broeker@ucsf.edu).