

Abstract
Single-cell motility is spatially heterogeneous and driven by metabolic energy. Direct linking cell mobility to cell metabolism is technically challenging but biologically important. Here we use singlecell metabolic imaging to measure glycolysis in individual endothelial cells with genetically-encoded biosensors capable of deciphering metabolic heterogeneity at subcellular resolution. We show that cellular glycolysis fuels endothelial activation, migration and contraction and that the high lactate production sites co-localize with active cytoskeletal remodeling within an endothelial cell. Mechanistically, RhoA induces endothelial glycolysis for the phosphorylation of cofilin and myosin light chain in order to reorganize the cytoskeleton and thus control cell mobility; RhoA activation triggers a glycolytic burst through the translocation of the glucose transporter SLC2A3/GLUT3 to fuel the cellular contractile machinery, as demonstrated across multiple endothelial cell types. Our data indicate that Rho-GTPase signaling coordinates energetic metabolism with cytoskeleton remodeling to regulate the motility of single endothelial cells.
Introduction
Endothelial cells exhibit motile behaviors in response to hemodynamics, during migration in angiogenesis, and during contractions in barrier failure; each of which involves drastic morphological changes that require energy production and consumption. Bulk measurements have revealed that 1) endothelial cells preferentially utilize glycolysis for fueling migration1, 2) endothelial cells are dynamically regulated by shear stress2–4, and 3) endothelial motilities are tightly regulated by the Rho family of small GTPases5. However, mechanistic coupling of motility to energy in typically heterogeneous cell populations requires simultaneous information at the single-cell level. Therefore, there is a critical need for developing single-cell metabolic measurements which can inform new mechanisms by linking cell motility phenotypes to energy production in the same cell. Furthermore, measuring energy production in spatially disparate subcellular structures of a single cell is expected to have mechanistic significance in cell biology since most cells dynamically regulate their metabolic programs as a result of nutrient availability, anabolic requirements, and energy demand.
By using single-cell imaging analysis, we were able to link glycolysis to motility in individual endothelial cells in order to understand spatially heterogeneous phenomena. This Förster resonance energy transfer (FRET) image-based approach uses deep learning6,7 to rapidly quantify the intracellular glycolytic metabolites including lactate, glucose, pyruvate, and ATP in single endothelial cells, thus linking metabolic states to key endothelial motile phenotypes including adaptation to shear stress, migration, and contraction. A strong correlation between morphology and glycolytic rate at the single-cell level was identified, which was further confirmed by the subcellular distribution of high glycolytic rates at sites of active actin turnover. Our single-cell metabolic measurements discovered that the activation of GTPase RhoA triggers a glycolytic burst essential for the endothelial contractile machinery. We further discovered that glucose transporter SLC2A3/GLUT3 controls the RhoA-induced endothelial glycolysis and subsequent contraction. These results highlight the significance of single-cell metabolic analyses in their ability to uncover a new paradigm of small GTPase-dependent coupling of energy production to cell mobility.
Results
Development of a single-cell metabolic imaging method
To study motile phenotypes of individual endothelial cells, we first devised a single-cell metabolism quantification method to monitor the lactate concentration and glycolytic rate of individual cells expressing a genetically encoded biosensor. Here we used Laconic, a Förster resonance energy transfer (FRET)-based lactate biosensor comprising of an mTFP/Venus FRET pair and a bacterial transcription factor LldR8. LldR binds lactate causing the FRET pair to change their intramolecular distance that leads to increased mTFP (acceptor)/Venus (donor) (or 1/FRET) signal. In this way, lactate concentrations are positively proportional to 1/FRET signals (Extended Data Fig. 1a). Laconic-expressing human endothelial cells were imaged, segmented using a semantic segmentation pipeline, and assembled into fluorescence traces under biochemical and/or biomechanical stimulations (Fig. 1a). This allowed us to monitor the terminal metabolite in glycolysis, lactate, in endothelial cells, thus directly revealing endothelial glycolytic reprogramming for energy production. A deep learning approach with U-nets7 using custom code in MATLAB rapidly and reliably performed pixel-level classification of images and segmentation of single cells with Boundary F1 score9 for identifying the cytoplasm > 90%, and class-specific cytoplasm accuracy, precision, recall, and Dice score > 80%, (Extended Data Fig. 1b, Extended Data Table 1). Single-cell fluorescence following segmentation was highly correlated with ground-truth fluorescence (Extended Data Fig. 1c).
Figure 1: Single-cell lactate production rate (LPR) distinguishes metabolically heterogeneous subpopulations.
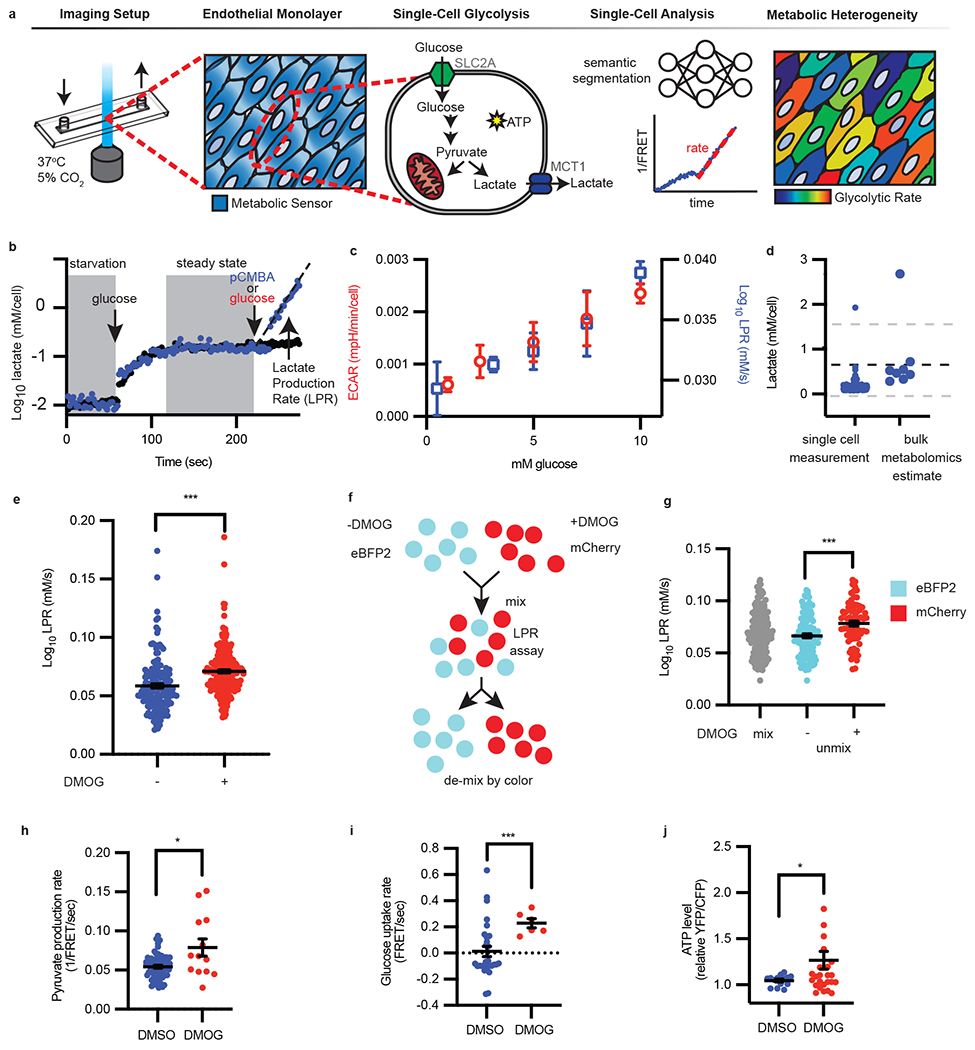
a, Schematic of single-cell metabolism quantification by deep learning. Endothelial cells were plated into imaging chambers capable of fluid exchange with controlled temperature and CO2. Once endothelial cells grew to form a monolayer, metabolite sensors were genetically introduced to measure glycolysis. Within each cell, glucose is converted to pyruvate and consumed by mitochondria or fermented into lactate and exported via MCT1, thus generating ATP. Deep learning-enabled semantic segmentation was used for linking cells into a 1/FRET time series for quantification of intracellular lactate over time to reveal metabolic heterogeneity. b, Following starvation, glucose was injected followed by glucose and MCT1 inhibitor pCMBA (blue dots) or glucose alone (red dots). The slope (black dotted line, eye guide) of each cell is the lactate production rate (LPR), filtered for fitting with R2 values ≥ 0.9. c, Single-cell LPR and per cell ECAR vs extracellular glucose concentration (n = 7, 226, 43, 200, 206 cells for LPR left to right, n= 4, 3, 4, 3, 3 wells for ECAR left to right; error bars are SEM). d, Comparison of steady-state lactate levels (n = 167 cells) with metabolomics (n = 8). The dotted black line is the mean metabolomics (n = 8) estimate of intracellular lactate measured by mass spectrometry. Gray dotted lines are standard deviation. e, LPR of vehicle DMSO (“−“) or DMOG (“+”) treated endothelial cells (n = 164 “−”, 256 “+”; error bars were SEM, p < 0.0001). f, eBFP2 or mCherry cells were treated with DMSO or DMOG, then mixed and imaged together; g, the LPR is then deconvoluted by color (n = 166 eBFP2, n = 79 mCherry; error bars are SEM, p < 0.0001). h-j, Pyruvate production rate (h, n = 67, 13, p = 0.048), glucose uptake rate (i, n = 29, 6, p = 0.0005), and ATP levels (j, n = 14, 29, p = 0.0323) in HAECs treated overnight with DMSO or DMOG measured using genetically encoded sensors. Error bars SEM. Statistical significance was determined by two-sided Welch’s t-test.
To reliably measure and calibrate physiological levels of intracellular lactate and glycolysis using laconic, we controlled intracellular lactate concentrations by permeabilizing human aortic endothelial cells (HAECs) and subjecting them to different concentrations of lactate. As shown in Extended Data Fig. 1a, the 1/FRET change is linear to 6 orders of magnitude of log lactate concentrations (R2 = 0.96). However, lactate export is a dynamic process in live cells that is regulated by the concentration gradient between the extra- and intra-cellular compartments10. Thus, in order to measure the glycolytic rate of the cell, lactate export was blocked through inhibition of monocarboxylate transporter 1 (MCT1), the major lactate transporter in endothelial cells11 (Fig. 1a). Upon MCT1 blockade by p-chloromercuribenzoic acid (pCMBA), we observed a continuous increase of lactate in individual cells, the slope of which we defined as the lactate production rate (LPR) (Fig. 1b), which reflects the overall glycolytic rate in a single endothelial cell. Furthermore, the single-cell LPR increased linearly, in agreement with bulk extracellular acidification measurements, as a function of extracellular glucose concentration (Fig. 1c). Employing a bootstrap algorithm simulating LPR, by drawing from ground truth fluorescence values and fitting slopes to the deep learning-measured fluorescence (Extended Data Fig. 1c), we estimated the absolute quantification error of LPR to be 5% (see Methods, Extended Data Fig. 1d).
Consistently, the steady-state intracellular concentration of lactate measured by single-cell FRET was within the range estimated by bulk mass spectrometry (~0-1.6 mM) (Fig. 1d). Corresponding to single-cell LPR, blockade of MCT1 with pCMBA10 also reduced extracellular acidification in a bulk bioenergetics flux assay (Extended Data Fig. 2a). Additionally, we were able to increase intracellular lactate at the single-cell level with glucose or oligomycin stimulation, and inhibit lactate production by 2-deoxyglucose, like those used in an extracellular flux glycolysis stress test at the bulk level (Extended Data Fig. 2b).
To test the LPR’s ability to distinguish heterogeneous glycolytic rates in cell populations, which is critical for differentiating subpopulations, we mimicked hypoxic conditions in endothelial cells by treating with dimethyloxaloylglycine (DMOG). DMOG inhibits prolyl hydroxylase, thereby promoting the stabilization of transcription factor hypoxia inducible factor-1α (HIF-1α)2. We found that the single-cell LPR was elevated in DMOG-treated cells (Fig. 1e), in agreement with an increase in ECAR measured in bulk extracellular flux (Extended Data Fig. 2c). We next forced metabolic heterogeneity by mixing vehicle (DMSO)- or DMOG-treated cells that were previously labeled with either eBFP2 or mCherry fluorescent proteins, respectively (Fig. 1f). By assessing the LPR of each cell and then de-mixing by color (Fig. 1g), our method distinguished authentic metabolic subpopulations.
In addition to lactate, we further employed other genetically encoded FRET-based sensors to measure key glycolytic metabolites including pyruvate12, glucose13, and ATP14 to cross-validate these findings with DMOG. Pyruvate production, glucose uptake, and ATP levels were all increased in DMOG-treated cells relative to DMSO control (Fig. 1h–j). Notably, pyruvate and glucose have alternative fates and ATP is not exclusively produced by glycolysis, whereas lactate production is a definitive surrogate for glycolysis. The results not only validated our primary results using the laconic sensor, but also laid a solid foundation for the general application of our single-cell metabolism method across different metabolites.
Glycolytic rates of single cells subjected to flow
We then assessed single-cell glycolytic heterogeneity in a spatial context by exploiting the hemodynamic flow-dependent activation of endothelial cells, which are generally subject to different shear stresses depending on the local flow conditions in the vasculature15. Disturbed flow-induced cell activation leads to atherosclerosis at arterial branch points15, driven by low shear stress-induced endothelial glycolysis2,3, and abrogated by high shear stress which is known to repress glycolysis4 through reducing the expression of a key glycolytic enzyme, PFKFB3, in a transcription factor KLF2-dependent manner4. However, the ability to study flow-induced metabolic changes has so far been limited to extracellular flux without spatial resolution2.
Using a cone and plate device to generate spatially heterogeneous shear forces over microscopic distances (Fig. 2a), we focused on an area at the flow boundary (indicated by endothelial elongation and alignment, Fig. 2b) to determine force-dependent metabolic rates. As shown in Fig. 2c, cells subjected to high shear stress had reduced LPR compared to those exposed to low shear stress, consistent with our previously published bulk data as measured by ECAR2. We next linked metabolism to a morphological phenotype (flow-induced endothelial cell elongation) at the single-cell level. By analyzing endothelial cells exposed to shear stress, we found a direct association between force-induced cell morphology and glycolytic rate. We plotted the LPR and shear stress as a function of cell length and found a negative correlation with LPR but a positive correlation with shear stress (Fig. 2d). At 20× magnification, cell length is strongly negatively correlated with LPR (Fig. 2e, f). This result was partially caused by the presence of external forces (shear stress), which could change metabolic rates independent of cell morphology, though shorter cells within the high shear stress region exhibit higher LPR (Fig. 2e).
Figure 2: Endothelial LPR in response to shear stress is heterogeneous.
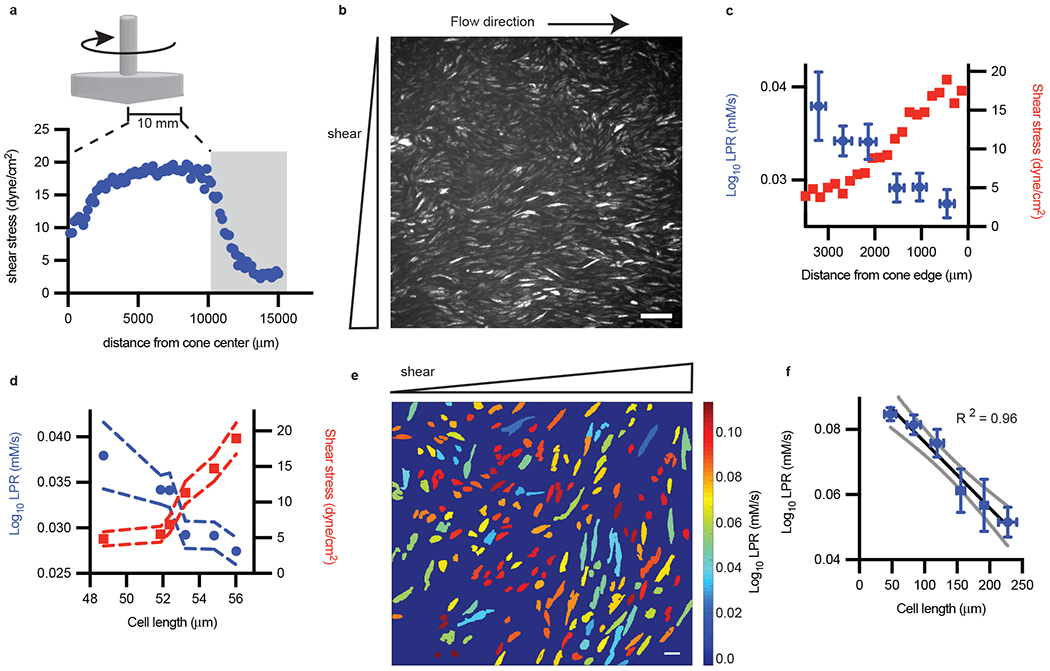
a, A cone and plate device was used to generate shear stress as measured by particle image velocimetry, with gray bar indicating area of interrogation. b, The corresponding laconic fluorescence image at the gray zone in a. Scale bar = 165 μm (representative of n = 3 biological replicates). c, LPR and shear stress vs distance from cone edge (n = 27, 47, 34, 34, 35, 33 left to right; error bars are SEM). d, Replot of c, with LPR and shear stress as a function of cell length, error bars are SEM. e, The LPR of segmented endothelial cells subjected to flow (color from cold to hot indicated the increase of LPR in each cell). Scale bar = 44 μm. f, Major axis cell length of endothelial cells (e) plotted against measured LPR (cell number n = 103, 47, 18, 8, 8, 4 low to high cell length; error bars are SEM). Standard deviation of regression coefficients for R2 for (f) is 0.074.
Glycolysis fuels the migration of single endothelial cells
Endothelial cells migrate during angiogenesis or wound closure. This process requires energy, involves dramatic changes in cell shape, and is dependent on RhoA/ROCK-1 remodeling of the actin cytoskeleton5. In typical wound assays, single-cell migrations are heterogeneous where some cells are motile while others remain stationary16. Previously, it has been shown that endothelial cell migration requires glucose, whereas endothelial cell growth requires glutamine usage in oxidative phosphorylation1,17. However, it is not known whether individual migrating cells are more glycolytic than stationary cells, which could elucidate whether metabolism is a driver of heterogeneous cell migration in a disrupted cell monolayer. We first demonstrated that glucose is necessary for migration whereas mitochondrial metabolism is dispensable for wound closure (treatment with Antimycin A, Fig. 3a). The results indicated that cell migration is driven by glycolysis. Consistently, HAECs treated with DMOG, which increases glycolytic capacity in a HIF-1α-dependent manner18, enhanced migration and are able to close wounds faster than control (Fig. 3b).
Figure 3: Single-cell metabolism links migratory cell phenotypes to glycolysis states.
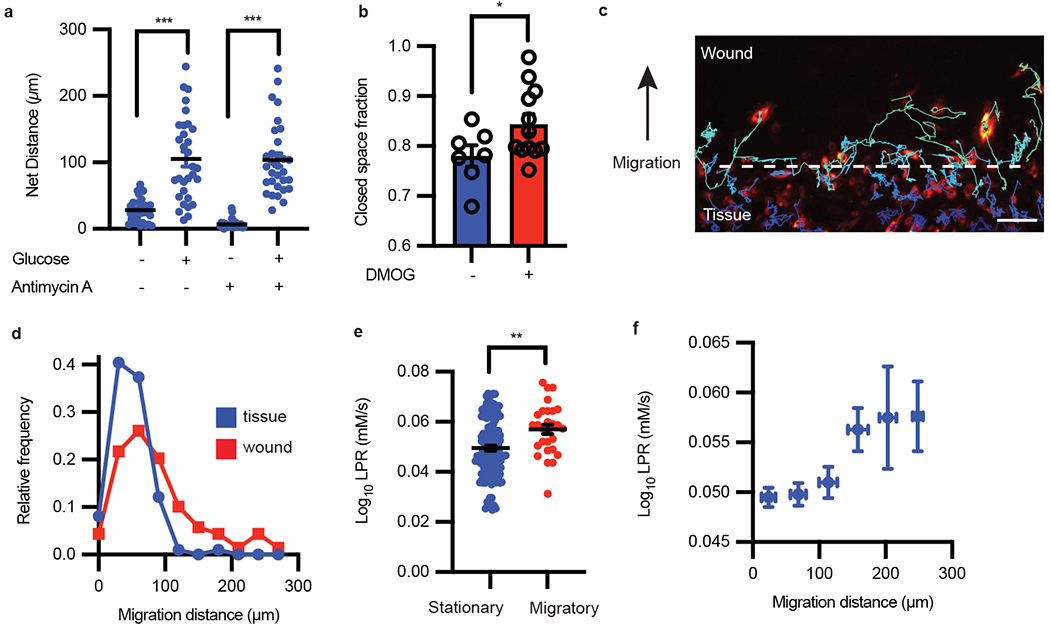
a, Migration distance of HAECs with or without glucose and with or without antimycin A (n = 31 cells each condition; error bars are SEM, p < 0.0001 for both pairs of conditions). b, Migration scratch assay quantification with and without DMOG treatment (n = 7 and 12, respectively; error bars are SEM, p = 0.0474). c, Migrating endothelial cells (false color) were tracked for 48 hours following wound formation. Scale bar = 150 μm. d, Frequency distribution of cell migration distance in the wound vs tissue. e, The LPR of cells that migrated less than 50 μm compared to cells that migrated more than 200 μm (cell number n = 141 and 28, respectively; error bars are SEM, p = 0.0012). f, The LPR change as a function of cell migrated distance (n = 114, 118, 42, 12, 8 from low to high migration distance; error bars are SEM). Statistical significance was determined by two-sided Welch’s t-test.
However, it remains unknown whether individual cells that migrated further were more glycolytic, as activation of HIF-1α by DMOG could turn on thousands of genes2. In order to elucidate whether cell glycolysis fuels individual cell motility, we applied single-cell metabolic assays to analyze migrating endothelial cells in a wound assay. By tracking individual cell migrations (Fig. 3c) over the course of 12 hours, we confirmed that cells in the more stationary “tissue” migrated less than cells in the more migratory “wound”, overall (Fig. 3d). We found that migratory cells, cells that traveled more than one cell length, were more glycolytic compared to stationary cells (Fig. 3e). We further found that cell migration distance is positively correlated with cell glycolysis (Fig. 3f), regardless of the final cell positions in the tissue or wound regions. These results suggest that the migration of endothelial cells is fueled by glycolysis, which is moreover proportional to work performed.
RhoA activation increases endothelial glycolysis
Having shown that glycolysis fuels cell migration, we next quantified the glycolytic rate during cell contraction, a key motile endothelial process that occurs over a short timespan (minutes) and is thus independent of transcriptional control. Because GTPase RhoA activation is central to induce the contractile apparatus of endothelial cells within minutes19, we hypothesized that controlled RhoA agonism would induce a glycolytic burst to power cell contractions. RhoA activation or inactivation in HAECs was achieved by overexpressing constitutively active RhoA (RhoA-Q63L) or dominant negative RhoA (RhoA-T19N). As shown in Figure 4a, RhoA-Q63L (CA) increased single cell LPR compared to RhoA-T19N (DN). To remove possible batch effects, we next conjugated fluorescent protein eBFB2 to RhoA-T19N and fluorescent protein mScarlet-i to RhoA-Q63L, followed by cell mixing and single-cell LPR measurements. Deconvolution by color showed significantly increased glycolysis in RhoA-activated (mScarlet-i positive) HAECs when compared to RhoA-inactivated (eBFP2 positive) cells (Fig. 4b). Switching fluorescent labeling of RhoA-Q63L and RhoA-T19N had no effect on the results (Extended Data Fig. 3a). We next treated HAECs with lysophosphatidic acid (LPA), another well-known RhoA agonist20, which significantly increased single-cell LPR compared to untreated control (Fig. 4c). Furthermore, treatment with thrombin, a serine protease that rapidly stimulates RhoA and subsequently RhoA kinase-1 (ROCK-1)21 to produce contractions and barrier breakdown, significantly increased LPR in 7 minutes, which was largely abrogated by the treatment of a specific ROCK-1 inhibitor, Y27632 (Fig. 4d). Consistently, glucose uptake and ATP levels were also elevated shortly after thrombin treatment (Fig. 4e). Thus, activation of GTPase RhoA causatively drives intracellular lactate production and glycolysis in single endothelial cells.
Figure 4: Single-cell imaging of lactate production reveals RhoA drives endothelial cell glycolysis, which is necessary for contractions.
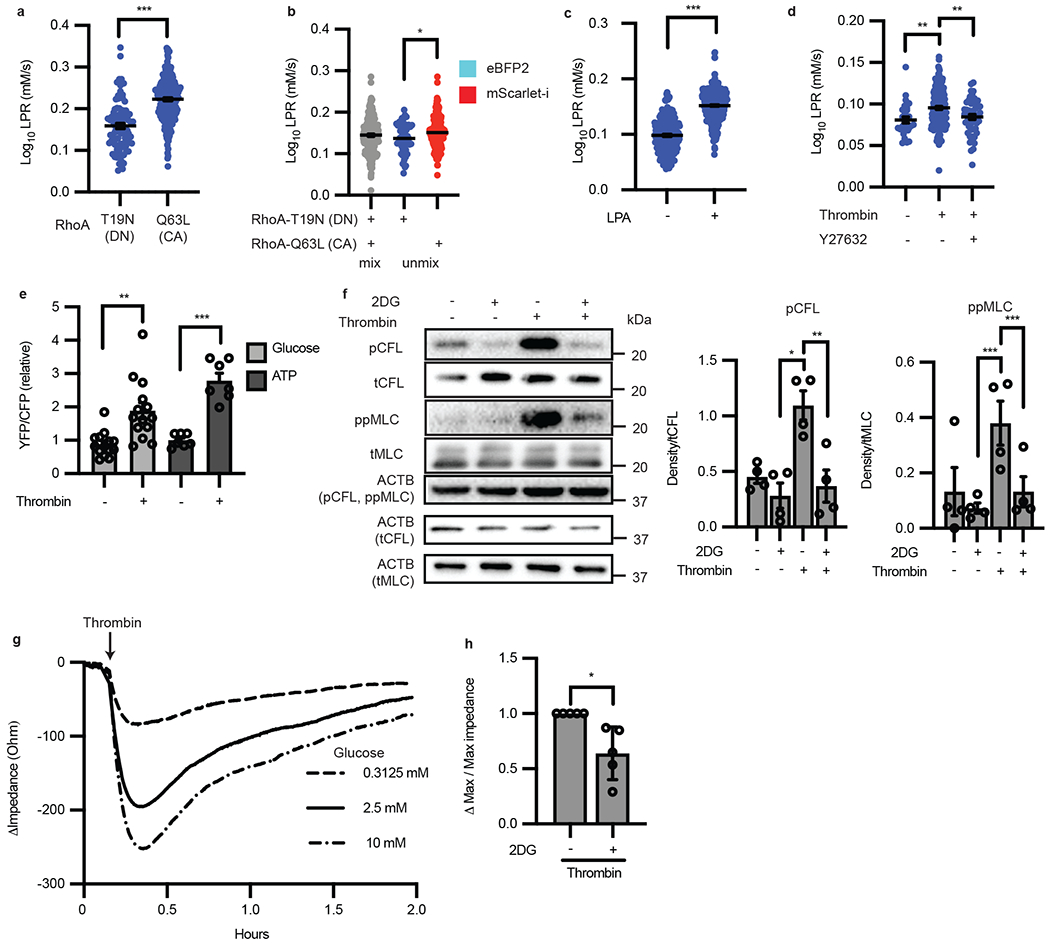
a, LPR of HAECs transfected with either RhoA-T19N (dominant negative “DN” form) or RhoA-Q63L (constitutively active “CA” form). (n = 91, 250 respectively, p < 0.0001). b, RhoA-Q63L-mScarlet-i transfected ECs have increased LPR compared to those transfected with RhoA-T19N-eBFP after unmixing by color (n = 52 and 174, respectively, p = 0.0193). c, LPR of HAECs treated with control (DMSO “−”) or 10 μm of lysophosphatidic acid for 30 min. (n = 244 and 423 respectively, p < 0.0001). d, Thrombin induced LPR (n = 171) compared to control (n = 28, p = 0.0028) which is suppressed with Y27632 (n = 51, p = 0.0042). e, Glucose and ATP levels before and after treatment with thrombin measured by FRET (n = 14 cells glucose, p = 0.0012; n = 7 cells ATP, p = 0.0001). f, Western blot of 2DG pre-treatment in the presence or absence of thrombin and phosphorylation of cofilin and MLC (n = 4 biological replicates. For pCFL, p = 0.008 for thrombin vs. 2DG and p = 0.0038 for thrombin vs thrombin + 2DG. For ppMLC, p < 0.0001 for both thrombin vs 2DG and thrombin vs thrombin + 2DG). Loading control (ACTB) each protein denoted in parentheticals. Original blot in Source Data a. g, TEER trace of HAECs after thrombin treatment in different extracellular glucose conditions. h, Inhibition of hexokinase with 2-DG reduces thrombin-induced permeability as measured by TEER (n = 5 each; biological replicates, p = 0.0283). Statistical significance determined by two-sided Welch’s t-test (a, b, c, e, h) or by one-way ANOVA followed by Bonferroni test (d, f). All error bars are SEM.
RhoA activates contractile machinery through glycolysis
Because the activity of RhoA-dependent kinases requires ATP, we hypothesized that the downstream functions of RhoA/ROCK-1 are dependent on glycolysis. RhoA/ROCK-1 activation causes rapid phosphorylation of cofilin (CFL) which enacts reorganization of the actin cytoskeleton from cortical actin to stress fibers19,22. RhoA/ROCK-1 activity also causes phosphorylation of myosin light chain (MLC) which actively pulls on stress fibers, causing cell contractions and intercellular gaps in endothelial monolayers (failure of the endothelial paracellular barrier) as well as the reduction in monolayer impedance23–25. Phosphorylation of CFL and MLC occurs in 2-30 minutes with relaxation of the cell to its basal state after 1 hour as the stimulus is depleted19,21. To test our hypothesis, we used hexokinase inhibitor 2-deoxyglucose (2-DG) to block glycolysis. Indeed, 2DG significantly reduced basal CFL and MLC phosphorylation in HAECs and moreover, completely blocked thrombin-induced phosphorylation of CFL and MLC (Fig. 4f), demonstrating that RhoA/ROCK1 downstream effects are dependent on active glycolysis. Consistently, the degree of contraction, or change in monolayer impedance, was dependent on the concentration of glucose in the media, as measured by transendothelial electrical resistance (TEER) (Fig. 4g). Concordantly, depleting endothelial glycolysis with 2-DG also prevented thrombin-induced barrier failure by TEER (Fig. 4h). Furthermore, inhibition of ROCK-1 with Y27632 followed by thrombin treatment phenocopied pre-treatment with 2DG (Extended Data Fig. 3b). These data collectively demonstrated that RhoA/ROCK1-dependent glycolysis is required for the contractile machinery of endothelial cells.
RhoA simulates contractions via glucose transporter SLC2A3
We next determined the molecular mechanisms by which RhoA induces glycolysis to stimulate endothelial contraction. Since the increase in glycolysis occurs over a short timespan (measured at 7 minutes) following RhoA activation (Fig. 4d) and availability of glucose determines the degree of downstream phosphorylation and contraction (Fig. 4f, g), we hypothesized that glucose uptake plays a critical role in RhoA-induced glycolysis. Previous studies have shown that SLC2A1/GLUT1 is a major glucose transporter in endothelial cells. SLC2A1 contributes to the basal endothelial glycolytic capacity in the blood brain barrier26 and pulmonary vasculature27, as well as to increased glycolysis stimulated by chronic (hours) exposure to hypoxia and disturbed flow in large arteries2. We therefore first tested the role of SLC2A1. Unexpectedly, small interfering RNA (siRNA)-mediated SLC2A1 knockdown did not reduce thrombin-induced phosphorylation of CFL and MLC in HAECs (Extended Data Fig. 4a). At the same time, qRT-PCR analysis identified that SLC2A3 is the most abundant glucose transporter transcript in HAECs (Extended Data Fig. 4b). We therefore tested the role of SLC2A3 in RhoA-induced glycolysis. Our data showed that SLC2A3 knockdown by siRNAs reduced basal phosphorylation of CFL and MLC and significantly abrogated thrombin-induced phosphorylation of CFL and MLC with three separate siRNAs (Fig. 5a, Extended Data Fig. 4c,d).
Figure 5: RhoA induces glucose transporter SLC2A3 translocation which drives subcellular glycolysis in thrombin stimulated contractile endothelial cells.
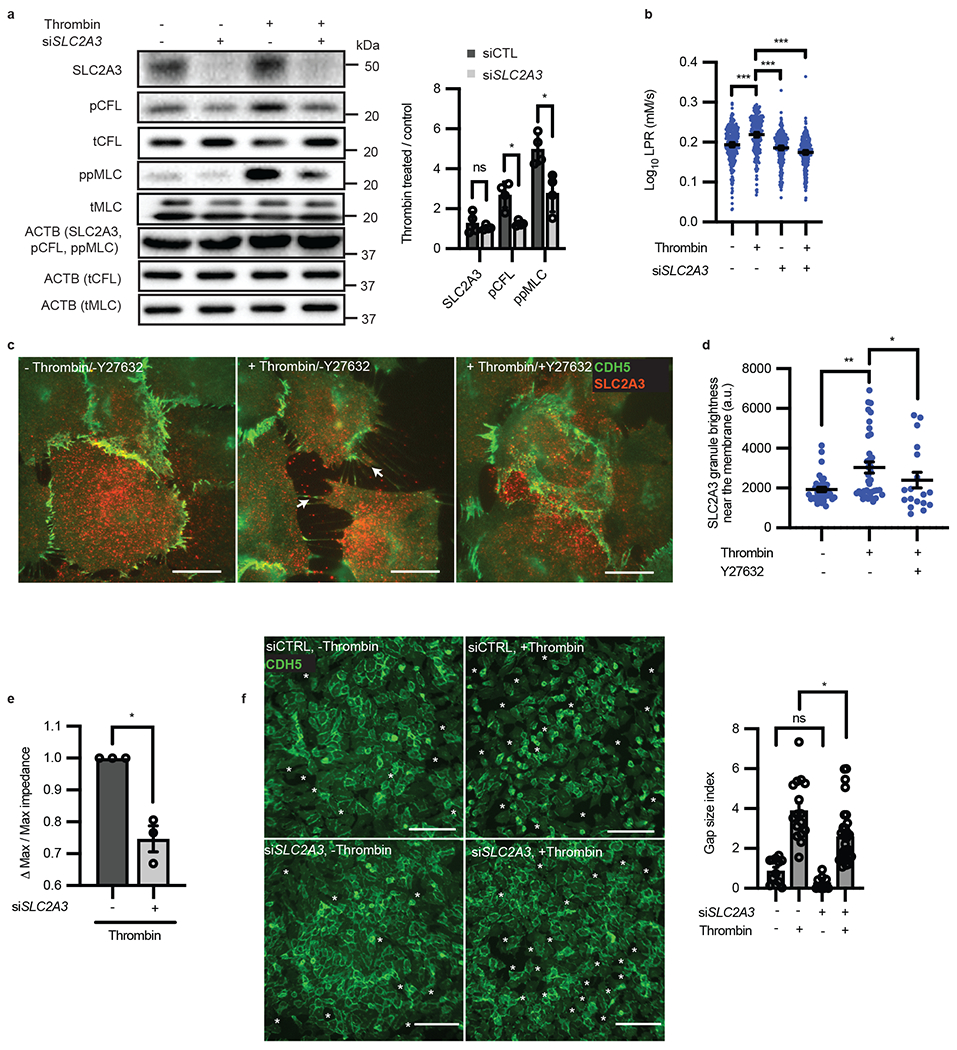
a, Western blot of phospho-cofilin and phospho-MLC in the presence or absence of siRNA targeted towards SLC2A3 and thrombin treatment (n = 4 all; biological replicates, p = 0.0140 for pCFL and p = 0.0233 for ppMLC). Loading control (ACTB) for each protein denoted in parentheticals. Original blot in Source Data b. b, LPR of siRNA targeted towards SLC2A3 or control with and without thrombin treatment (n = 277, 263, 275, 278 from left to right, p < 0.0001 for all comparisons). c, Representative immunofluorescence of membrane markers CDH5 and SLC2A3 in thrombin treated cells causes SLC2A3 granule localization to membrane spikes, which is inhibited by Y27632, by TIRF microscopy. Scale bar = 5 μm. d, Quantification of SLC2A3 granule brightness near the membrane of (c) (a.u. = arbitrary units, n = 36, 36, 17 images from left to right. p = 0.003 for thrombin vs. control, and p = 0.0499 for thrombin vs thrombin + Y27632). e, TEER measurement of thrombin-induced permeability changes in the presence or absence of siRNA targeted towards SLC2A3 (n = 4; biological replicates, p = 0.0248). f, Representative immunofluorescence of CDH5 and intercellular gaps, as quantified by gap size index, in HAECs treated with control or siRNA targeted towards SLC2A3 followed by thrombin treatment (n = 12, 16, 8, 32 images from left to right, p = 0.0184). Scale bar = 150 μm. Statistical significance determined by multiple unpaired two-tailed t-test with multiple comparisons adjustment by the Holm-Šídák method (a), two-sided Welch’s t-test (e, f) or by one-way ANOVA followed by Bonferroni test (b, d). All error bars are SEM.
Although SLC2A3 knockdown had no effect on basal endothelial glycolysis, it largely abolished the thrombin-induced glycolytic burst, as detected by single-cell LPR (Fig. 5b). In direct comparison, SLC2A1 knockdown led to a measurable decrease in LPR after thrombin stimulation but this decrease was far less than that of SLC2A3 knockdown (Extended Data Fig. 4e). Consistently, the thrombin-induced increase in glucose and ATP levels were abrogated by SLC2A3 knockdown (Extended Data Fig. 4f, g). Because the glycolytic burst could not be attributed to SLC2A3 transcriptional regulation given the acute response (measured within 7 minutes), we hypothesized that SLC2A3 is regulated post-transcriptionally by RhoA/ROCK-1. To test this hypothesis, we performed immunofluorescence imaging of SLC2A3 in thrombin-treated HAECs. Using total internal reflection fluorescence (TIRF) microscopy, which selectively illuminates membrane fluorophores close to the cover slip (within ~100 nm)28,29, we detected significantly increased intensity of SLC2A3 in granules near the plasma membrane of single HAECs following thrombin stimulation; such accumulation of SLC2A3 were abrogated by ROCK inhibitor Y27632, further suggesting the involvement of Rho/ROCK signaling pathway (Fig. 5c–d). In agreement with the SLC2A3-dependent glycolytic burst and phosphorylation of CFL and MLC, thrombin-induced endothelial contraction was significantly abrogated by SLC2A3 knockdown, demonstrated by increased monolayer impedance by TEER (Fig. 5e) and reduced intercellular gap size (gap size index, Fig. 5f), although the number of gaps was unchanged (gap index, Extended Data Fig. 4h)30. Furthermore, increasing the amount of extracellular glucose increased gap size and gap number, which was blunted by SLC2A3 knockdown (Extended Data Fig. 4i). Contraction (gap size index), but not gap number, was partially rescued under SLC2A3 knockdown by treatment with oligomycin to divert all glucose to be utilized via glycolysis (Extended Data Fig. 4j). Thrombin-induced phosphorylation of CFL and MLC were also significantly abrogated by SLC2A3 knockdown in human umbilical endothelial cells (HUVECs) (Extended Data Fig. 5a) and human lung microvascular cells (HMVECs) (Extended Data Fig. 5b). Thrombin-induced HUVEC contractions (as measured by cell size, Extended Data Fig. 5c) and HMVEC contractions (as measured by intercellular gaps, Extended Data Fig. 5d) were also significantly reduced by SLC2A3, indicating this pathway is conserved across multiple endothelial cell types.
The functional role of SLC2A3 in endothelial glycolysis and contraction is largely unknown. In order to explore the in vivo relevance of SLC2A3 in barrier regulation, we needed to first ascertain the expression profile of Slc2a1 and Slc2a3 in primary endothelial from different organs in mice. Gene Expression Omnibus datasets (GEOD) were downloaded from EndoDB, a database of curated endothelial transcriptomics datasets31. As shown in Extended Figure 6a, Slc2a3 has expression levels similar to Slc2a1 across multiple organs (normalized to Slc2a1). We next overexpressed SLC2A3 under the endothelial-specific CDH5/VE-cadherin promoter and evaluated vascular leak in mouse aortas. Mouse aortas, because to their intrinsic curvature, exhibit RhoA activation due to hemodynamic shear forces32. We employed a delivery system which integrates polyethylenimine (PEI) nanoparticles and plasmids with the endothelial-specific CDH5/VE-cadherin promoter. Intravenous injection of CDH5-driven, mScarlet-i-expressing plasmids resulted in mScarlet-i fluorescence in arterial endothelium, highlighted by co-localization with ve-cadh (which markes endothelium) adjacent to the lumen, but not the underlying media and adventitia (yellow signal in merge panel, Extended Fig. 6b), demonstrating endothelial-specific overexpression in mice. The same approach using CDH5-driven, SLC2A3-expresssing plasmids led to SLC2A3 overexpression in aortic endothelium but not in the media and adventitia in mice, measured by quantitative real-time PCR (Extended Data Fig. 6c). To demonstrate functional relevance of SLC2A3, mice were injected with PEI nanoparticles containing CDH5-SLC2A3 or CDH5-Ctrl plasmids 24 hours prior to sacrifice. 1 hour prior to sacrifice, Evans blue dye was injected retro-orbitally. Evans blue dye binds to albumin and transits the endothelial barrier in a paracellular fashion under conditions of diminished tight and adherent junctions and quantitatively measures the leakiness of mouse aortas33. Endothelial overexpression of SLC2A3 results in leakiness of aorta as quantified by Evans blue spectroscopy (OD 620), normalized to aorta weight after dye extraction (Extended Data Fig. 6d). This experiment suggests that SLC2A3 regulates the endothelial barrier in vivo. Collectively, these results demonstrated the importance of SLC2A3 on fueling RhoA/ROCK-1 downstream phosphorylation targets and its importance in both in vitro and in vivo barrier function.
Subcellular glycolysis and cytoskeletal remodeling overlap
After finding RhoA activation induces endothelial glycolysis and contraction within minutes at the single-cell scale (Fig. 4a–d), we sought to evaluate how glycolysis relates to morphological changes within individual endothelial cells with subcellular resolution. Reasoning that actin remodeling is dependent on high-energy phosphates, we hypothesized that localized changes in actin is correlated with glycolytic activity at the subcellular spatial scale. To test this hypothesis, we again stimulated actin remodeling with a RhoA agonist thrombin. By co-expressing laconic and Lifeact-RFP34 (an F-actin label) in endothelial cells, we measured LPR and actin remodeling in single endothelial cells with subcellular resolution at 40× magnification (Fig. 6a). Lifeact signal increases and shimmers at the edges of thrombin-induced contracting cells, indicating active actin remodeling. In order to visualize actin remodeling, we computed the maximum intensity projection of the difference in Lifeact signals in the 7 minutes following thrombin addition prior to performing the LPR assay (Fig. 6a, difference image). The LPR image was captured by performing pixel-wise calculation of the slope of the FRET signal after addition of pCMBA (Fig. 6a, LPR). As shown in the merged image, the LPR signal was spatially correlated with the Lifeact difference image (Fig. 6a, “LPR+difference” and “LPR+difference+nucleus”). The Lifeact difference and LPR images together demonstrated enhancement of signals on the contracting edge and center of the cell. To quantify the correlation, we calculated the degree of overlap using Manders’ overlap coefficient (MOC) and Pearson’s correlation coefficient (PCC). Thrombin-stimulated cells showed much higher levels of correlation between subcellular LPR and Lifeact than those of non-stimulated cells (Fig. 6b). This correlation was diminished by inhibition of ROCK (Y-27632), actin polymerization (Latrunculin B), and myosin II (para-amino-Blebbistatin) (Fig. 6b). Moreover, SLC2A3 knockdown reduced the correlation between Lifeact difference and LPR after thrombin treatment, indicating RhoA-dependent SLC2A3 activity is necessary for the co-occurrence of subcellular glycolysis and actin remodeling (Fig. 6c).
Figure 6: Subcellular distribution of LPR colocalizes with actin remodeling.
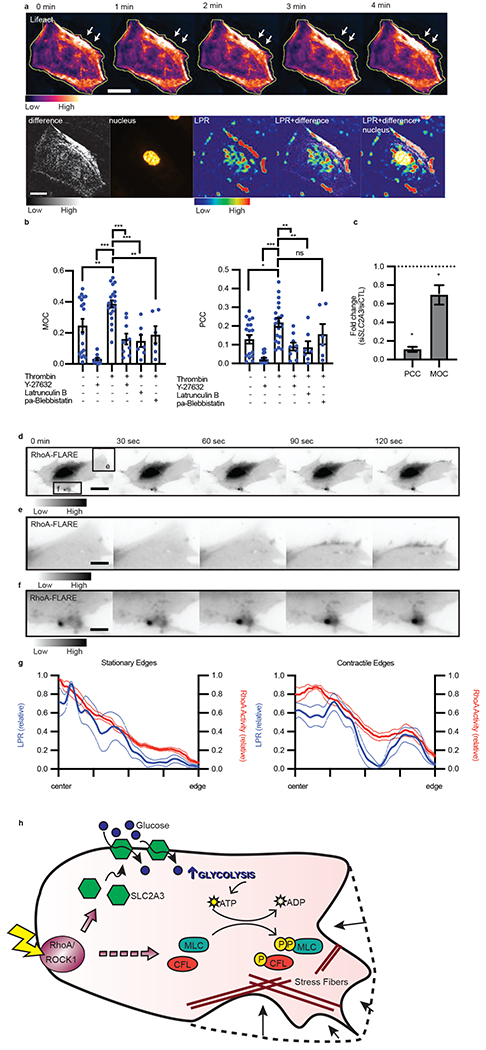
a, Representative montage of endothelial cell actin structure, as labeled with Lifeact-RFP, where the yellow line indicates original cell edge and white arrows point to contractile edge after treatment with thrombin (n = 14 biological replicates, 3 separate experiments). Maximum projection of change in actin structure (difference), nucleus, per pixel LPR map (LPR), and merges. Scale bar = 30 μm. b, MOC and PCC of unstimulated or stimulated cells in the presence of ROCK inhibitor (Y-27632), actin polymerization inhibitor (Latrunculin B), and myosin II inhibitor (paBlebbistatin: para-aminoblebbistatin), error bars are SEM. (n = 15, 14, 20, 9, 7, 6 from left to right. Thrombin vs. control, p = 0.0035; vs. Y27632, p < 0.0001; vs. thrombin + Y27632, p < 0.0001; vs. thrombin + latrunculin B. p = 0.0002; vs. thrombin + para-aminoblebbistatin, p = 0.0040). c, Fold change of PCC or MOC of thrombin treated cells under SLC2A3 knockdown compared to siCTRL, all error bars are SEM. (n = 14 for both, p = 0.0317 for PCC, p = 0.0151 for MOC). d, RhoA activity measured by RhoA-FLARE in a contracting endothelial cell (scale bar = 15 μm). e-f, Regions (e) and (f) from (d) demonstrate high RhoA activity in areas of active contraction (scale bar = 4.5 μm) (d-f representative of n = 9 cells, 3 biological replicates). g, Normalized LPR and RhoA activity distribution from cell center to edge of non-contractile/stationary edges and contractile/motile edges stimulated with thrombin. (LPR stationary, n = 4; RhoA stationary, n = 6; LPR contract, n = 8; RhoA contract, n = 9). Dotted lines are standard deviation. h, Proposed mechanism of RhoA regulating glycolysis and contraction. Agonism of RhoA/ROCK1 stimulates SLC2A3 translocation to the membrane to increase intracellular glycolysis, thereby providing the substrate for phosphorylation of downstream targets (MLC and CFL), resulting in stress fiber formation and, ultimately, contraction. Statistical significance was determined by one-way ANOVA followed by Bonferroni test (b) or by two-sided Welch’s t-test (c).
Similarly, we assessed subcellular glycolysis using Laconic in migrating endothelial cells. Here, we computed the Lifeact difference image in the 10 minutes prior to performing the LPR assay (Extended Data Fig. 7a, difference image). In migrating HAECs, we found that LPR and Lifeact difference were significantly correlated (Extended Data Fig. 7a), compared to control (performed by randomizing the LPR images 10,000 times and computing the MOC or PCC) (Extended Data Fig. 7b). These results indicate spatial cytoskeletal remodeling and glycolysis are correlated in multiple types of endothelial motilities.
To determine whether subcellular LPR was spatially related to RhoA, we next evaluated RhoA activity using a FRET-based biosensor RhoA-Flare35 in single contractile endothelial cells. We found enhanced RhoA activity and increased LPR co-located in similar areas following thrombin stimulation: the center of the cells (Fig 6d) as well as along the edges of the contractile aspects of the cell (Fig. 6d–f, and Extended Data Fig. 7c–l). In contrast, where the cell edge did not move, there was no FRET (RhoA activity) (Extended Data Fig. 7i). Edges with persistent contraction consistently demonstrated high RhoA-Flare FRET (Extended Data Fig. 7j). The similar distribution of RhoA activity and LPR was further confirmed by comparing stationary edges vs contractile edges across multiple cells, averaged together (Fig. 6g). This data revealed that the non-uniform localized distribution of metabolism correlates with cytoskeleton remodeling at the subcellular level. These results further showed co-localization between LPR and RhoA activities, suggesting that an endothelial cell generates energy using glycolysis to drive actin remodeling through GTPase RhoA.
Altogether, our single-cell metabolic assays revealed molecular mechanisms that RhoA/ROCK-1 activates SLC2A3 translocation to trigger a glycolytic burst to power the endothelial contractile apparatus across multiple endothelial types (Fig. 6h). Furthermore, subcellular analysis of LPR revealed the non-uniform, localized distribution of glycolysis at site of actin turnover and RhoA activity, indicating RhoA signaling coordinates energy production through glycolysis with energy consumption through cytoskeletal remodeling to control cell motility.
Discussion
Most studies measure cell metabolism using bulk assays. However, individual cells are inherently heterogeneous. Metabolic heterogeneity in endothelial cells dictates vascular activation and growth36,37, during T-cell differentiation controls cell function in infection38,39, within tumors affects responses to anticancer treatments40, and among yeast cells results in distinct subpopulations in response to sudden changes of substrate availability41, to name a few. Reprogrammed metabolic rates to support different cellular functions has rightfully gained attention but experimental methods to directly evaluate heterogeneous populations remain in infancy. To decipher the metabolic states of individual cells, we used a FRET-based metabolic imaging method to measure single-cell metabolism with subcellular resolution. Our single-cell metabolism method is complementary to existing ensemble metabolic assays and broadly applicable to different cell types. With this method, we were able to investigate the underlying molecular mechanisms of glycolysis in endothelial cells. This method enabled us to link heterogeneous glycolytic states to GTPase RhoA-activated phenotypes across variable time scales (low shear stress: days, migration: hours, and contraction: minutes) in individual endothelial cells. This method also allowed us to explore spatially heterogeneous metabolism and motility within individual cells. Given the feasibility, reproducibility, and robustness of this approach, it should have widespread applications to study metabolic support of single-cell functions in cell biology.
In addition to identifying single-cell metabolic heterogeneity, we found non-uniform distributions of glycolytic rates within single motile cells, specifically at sites of actin turnover and RhoA activity. RhoA-coupled glycolysis and contractile machinery demonstrated here also support an emerging paradigm that cellular metabolism and cytoskeleton architecture are intimately coordinated in an “energetic metabolon”. Our results also suggest that an endothelial cell is energetically frugal and samples the environment for energetic precursors only when necessary: here, the cell employs RhoA/ROCK-1 to couple energy production with acute energy-consuming responses such as cytoskeletal remodeling to enable contraction. This could be functionally essential, as the continued presence of high energy phosphates in a cell may make controlling enzymatic phosphorylation difficult. Thus, having a local, segregated pool of transmembrane receptors may enable digital-like spatial control of phosphorylation42. Co-localization of glycolytic enzymes with energetic demand could provide an efficient way for the cell to not only temporally determine when energy is produced, but spatially control where energy is consumed, preventing futile reactions, avoiding energy over-production, and overcoming diffusion limitation. “Glycolytic metabolons” have been suggested in endothelial cells, with increased ATP in in the perinuclear cytosol and lamellipodial ruffles36, and evidence that glycolytic enzymes bind to the actin cytoskeleton43. Here, using single-cell metabolic assays, we directly visualized the highly localized, enriched lactate production in subcellular endothelial edges and perinuclear sites, providing evidence to functionally support the “glycolytic metabolon” theory. Further studies linking the subcellular glycolytic rate and distribution of glycolytic machinery are needed to strengthen and validate this theory.
Integrating our single-cell metabolic method with classic molecular and functional analyses also allowed us to discover the causal role of RhoA in stimulating a SLC2A3-dependent glycolytic burst necessary for endothelial contraction. Although it is commonly recognized that SLC2A1/GLUT1 is a major glucose transporter in endothelial cells across diverse vascular beds and contributes to basal glycolysis under chronic transcriptional activation2,26,27,44, our results revealed the critical role of SLC2A3/GLUT3 in triggering a metabolic burst, defined by a quick increase in glucose uptake, glycolysis, and ATP level. ATP budget and nutrient availability plays an important role in cellular decision making, including certain endothelial behaviors17,37,45,46. Using this mechanism, a single endothelial cell can augment its glycolytic potential even further with SLC2A3 transmembrane recruitment during acute stimuli in cellular processes requiring an immediate energy burst. We also demonstrated that vascular leak was increased in an endothelial overexpression model of SLC2A3 in vivo. In highly-glycolytic platelets where SLC2A3 is enriched, thrombin (a potent platelet activator) can stimulate SLC2A3 plasma membrane translocation47, suggesting a conserved molecular function. Exactly how RhoA/ROCK-1 causes transmembrane expression of SLC2A3 requires further exploration. Investigation of SLC2A3’s role in regulating vascular pathophysiology could bolster the case that modulating signal transduction by interfering with cellular energetics could be a fruitful path to novel pharmacotherapeutics in diffuse vascular pathologies48. The physiological role of endothelial SLC2A3 in vascular homeostasis and disease in vivo deserves future investigation.
Importantly, we found lactate production to be highest at the cell edge and center of contractile cells. Though we attribute this to energy demand at locations of high RhoA activity, one could hypothesize that lactate could also be produced locally for usage for other cellular processes such as histone lactylation within the nucleus49. Perhaps the increase of local glycolysis could act as a signal for the cell to adapt to a changing environment, thus imprinting acute stimuli into cellular memory. Future investigation of subcellular endothelial glycolysis would benefit from tracing high energy phosphates from where they are produced, to sites where they are consumed, including consideration of nuclear lactate as it corresponds to gene regulation. For instance, FRET biosensors can be further engineered with a nuclear localization sequence or plasma membrane targeting.
In summary, we investigated spatially heterogeneous endothelial motilities by developing a single-cell imaging assay to measure glycolysis with subcellular resolution. We demonstrated that endothelial migration and contraction, critical endothelial phenotypes, were driven by glycolysis at the single-cell and subcellular levels. We found energy-demanding cytoskeletal remodeling to be conducted by RhoA through SLC2A3-mediated glucose uptake and glycolysis. Altogether, our single-cell metabolic assays and mechanistic findings demonstrated the critical interplay between signaling, metabolism, and motility in dictating non-uniform phenotypes of individual endothelial cells.
Methods
For additional methods, please see Supplementary Information.
Cell Culture and cloning
Human aortic endothelial cells (HAECs) (Lonza, CC-2535) and grown in endothelial growth media (EGM-2) supplemented with SingleQuots (Lonza, CC-3156 & CC-4176) and Antibiotic-Antimycotic (Gibco, 15240062) at 37°C with 5% CO2. HAECs were used from passage 6 to 8 for all experiments. Human pulmonary microvascular endothelial cells (HMVEC, Lonza, C12281) were maintained in EGM2-MV medium (Lonza, CC-3202) supplemented with 10% FBS. Human umbilical vein endothelial cells (HUVEC, Lonza C2519A) were maintained in EGM2-SingleQuot medium (2% FBS). HMVECs and HUVECs were used from passage 5 to 7 for all experiments. Cell lines were verified as mycoplasma free and that they are endothelial cells with CD31 verification by the vendor.
Laconic, LifeAct, ATeam, and FLIIP-glu700 expression
Laconic8, ATeam, or FLIIPglu700 containing plasmid (Addgene: 44238, 51958, and 17866 respectively) were cloned into Ad5 adenovirus by VectorLabs for construction of Adenovirus-Laconic (Ad-Lac), Adenovirus-ATeam (ad-ATeam), or adenovirus-FLIIPglu700 (ad-Glu). Ad-LifeAct-RFP (ad-LifeAct) was from Ibidi (60122). For transduction into endothelial cells, Ad-Lac at 50:1 multiplicity of infection (MOI), ad-ATeam at 100:1 MOI, and ad-Glu at 200:1 MOI in 1:1 EGM2 media:Opti-MEM (ThermoFisher, 11058021) and 3 μL GeneJammer (Agilent, 204130) per milliliter of final volume was added for 1 hour before replacement of media. For migration and contraction experiments, ad-Lac and ad-LifeAct were co-transduced each with MOI 50:1. Endothelial cells were used 48 hours post transduction to allow for cell recovery and maximal fluorescence.
T7-eBFP2, T7-mCherry, and RhoA overexpression
eBFP2 and mCherry constructs were generated from the Addgene plasmid #64323 and GeneCopoeia EX-C0726-M56-PFKFB3, respectively. For RhoA overexpression, pcDNA3-EGFP-RhoA-WT, pcDNA3-EGFP-RhoA-Q63L (Constituitively Active/CA), pcDNA3-EGFP-RhoA-T19N (Dominant Negative/DN) were purchased from Addgene (12965, 12968, 12967). EGFP was cloned out of each construct and replaced with either mScarlet-i (Addgene 85056 pLifeAct-mScarlet-i_N1) or eBFP2 using the vector and insert primers in Supplementary Table 1, followed by Gibson assembly (NEB, E2611). PCR products containing T7 promotors for in vitro transcription were generated using NEB Q5 polymerase (New England Biolabs, M0491) and the primers (Integrated DNA Technologies) listed in Supplementary Table 1.
mRNA transcripts were generated using mMESSAGE mMACHINE T7 Ultra Kit (ThermoFisher, AM1345). HAECs were transfected with 50 ng/cm2 of mRNA with Lipofectamine MessengerMAX (ThermoFisher, LMRNA003) overnight in 20% Opti-MEM:EGM2. On the day of the LPR assay, cells, each dually transfected with either A) mCherry and laconic or B) eBFP2 and laconic, were split into two groups – A and B by themselves or mixed together into an Ibidi chamber.
RhoA-Flare Expression
HAECs were plated 5 × 104 cells/well in 8-well glass bottom dish (Ibidi 80827) two days prior to the experiment. One day prior to the experiment, RhoA-Flare (Addgene, 12150) plasmid was incubated with fluorine-modified polyethylenimine (F-PEI) at a 4:1 plasmid:F-PEI mass ratio for 30 minutes in serum-free media (Opti-MEM) then incubated with cells for 4 hrs at 37 °C. The F-PEI were synthesized according to a previous method50. Briefly, heptafluorobutyric anhydride was added to PEI with a molar ratio of 7:1 then trimethylamine was added as 1.5-fold mole to heptafluorobutyric anhydride. This was stirred for 48 hours at room temperature then dialyzed with distilled water in a bag with a molecular weight cut off of 1 kDa. Media was exchanged after the 4 hr incubation to complete endothelial growth medium (EGM2) and cells were imaged the following day.
Thrombin, lysophosphatidic acid, Y27632, latrunculin B, para-amino-blebbistatin, or 2DG treatment
HAECs were first starved in EBM2 or Fluorobrite medium (Gibco, A1896701) for at least two hours prior to thrombin treatment for 10 minutes. Thrombin was used at 0.25 U/mL for all experiments except for Fig. 4d, which was 0.5 U/mL for 7-10 minutes prior to assay. Cells were treated with lysophosphatidic acid for 30 minutes prior to assay. Y27632, latrunculin B, para-amino blebbistatin, or 2DG containing samples were starved in EBM2 or Fluorobrite for one hour prior to thrombin treatment.
Metabolomics
Endothelial cells were plated at 500,000 cells/well using 6-well plates. Metabolites were extracted according to the manufacturer’s protocol for metabolomics (HMT Metabolomics). The average lactate concentration measured by capillary electrophoresis time-of-flight mass spectrometry was 37973.5 ± 29376.25 pmol per million of cells. Assuming an average length, height, and aspect ratio of 100 μm, 4 μm, and 1.12, respectively51, the estimated lactate concentration per cell was 0.77 ± 0.82 mM.
Laconic imaging
Imaging of laconic sensor was performed on an Olympus IX-71 microscope with a 10× objective. FRET filter sets were from Semrock: excitation 438/24, mTFP emission 483/32, Venus emission 542/27, and dichroic FF458-Di02. Hoechst and TRITC filter set for visualizing Hoechst stain, eBFP2, and mCherry were from Semrock: Hoescht excitation 337/50, Hoescht emission, 447/60, Hoescht dichroic FF409-Di03, TRITC excitation 556/20, TRITC emission 617/73, TRITC dichroic Di03-R488/561. FRET images were taken in rapid succession using a computer controlled Prior filter excitation and emission wheels and a Photometrics 95B camera. The microscope system was enclosed in a heated chamber maintained at 37 °C and 5% CO2 for all time-lapse microscopy experiments. The microscope system was controlled by MicroManager. Imaging flow chambers (Ibidi, 80176) were used according to the manufacturer’s instructions. Injections for exchanging buffers were performed manually through attached tubing.
Laconic calibration
Ad-laconic were transduced into HAECs and were replated into Ibidi chambers at approximately 100,000 cells / chamber 24 hours later, and then imaged 24 hours later. Cells were incubated in intracellular buffer (ICB) (10 mM NaCl, 130 mM KCl, 1.25 mM HEPES pH 7.2) with 10 μM nigericin and 2 μM of rotenone with varying concentrations of sodium lactate (0.0001 mM, 0.001 mM, 0.01 mM, 0.1 mM, 1 mM and 10 mM). mTFP and Venus fluorescence channel images were acquired every 2 seconds over 3 minutes per lactate concentration. The mTFP/Venus (donor/acceptor) intensity ratio per cell after background subtraction (hereafter, 1/FRET or FRET’ in shorthand) was obtained after segmentation (described below).
Lactate concentration and production rate (LPR) calculation
One day after Ad-Lac transduction, HAECs were re-plated into Ibidi chambers at approximately 100,000 cells / chamber. After waiting another 24 hours, they were starved in extracellular buffer (ECB) composed of (112 mM NaCl, 5 mM KCl, 1.25 mM CaCl2, 1.25 mM MgSO4, 10 mM HEPES, 24 mM NaHCO3, pH 7.4) or Fluorobrite MEM (Gibco, A1896701) for at least 1 hour. The first injection was 10 mM glucose in ECB, then after 3 minutes, 10 mM glucose and 500 μM of pCMBA in ECB. The fluorescence images of mTFP and Venus were obtained with exposures between 100-300 ms/frame every 2 seconds until completion of the assay. LPRs were calculated as follows: after pCMBA injection, the FRET’ rose (ΔFRET−1) as cells accumulated intracellular lactate. From the FRET’ vs lactate calibration curve, we define c as a constant in Equation 1 which we use in Equation 2 to finally define LPR in Equation 3.
| Eq. 1 |
| Eq. 2 |
| Eq. 3 |
Intracellular lactate was determined by first estimating the FRET’ signal at zero lactate, which was after starvation for 1 hour in extracellular buffer and perfusion with 10 mM pyruvate. Following 10 mM glucose perfusion, the FRET’ signal reached steady state after 2 minutes. At the end of each experiment, the FRET’ value at 10 mM lactate was measured such that each individual cell had its own two-point calibration curve. An average FRET’ value over 20 seconds during steady state was used to determine intracellular lactate.
DMOG stimulation of endothelial cells
HAECs were plated to confluency and transduced with Ad-Lac at 50 MOI two days prior to the LPR assay. One day prior to the LPR assay, cells were transfected with either T7-eBFP2 or T7-mCherry at 50 ng/cm2. The night before the assay, the cells were treated with 0.5 mM of DMOG or DMSO in EGM2. On the day of the LPR assay, cells, each dually labeled with either A) mCherry and laconic or B) eBFP2 and laconic, were split into two groups – A and B by themselves or mixed together into an Ibidi chamber.
RhoA-Q63L or RhoA-T19N expression in endothelial cells
HAECs were plated to confluency and transduced with Ad-Lac at 50 MOI two days prior to the LPR assay. One day prior to the LPR assay, cells were transfected with either T7-eBFP2-RhoA-Q63L, T7-mScarlet-i-RhoA-T19N, T7-eBFP2-RhoA-T19N, or T7-mScarlet-i-RhoA-Q63L at 25 ng/cm2. On the day of the LPR assay, cells, each dually labeled with either A) mScarlet-i and laconic or B) eBFP2 and laconic, were split into two groups – A and B by themselves or mixed together into an Ibidi chamber.
siRNA expression in endothelial cells with LPR measurements
Two days prior to the LPR assay, HAECs plated to confluency were transduced with with Ad-Lac at 50 MOI, Ad-ATeam at 100 MOI, or Ad-FLIIP-Glu700 at 200 MOI. 12 hours later the cells were transfected with either scrambled control or siRNA targeted towards SLC2A3 as above.
Deep learning enabled segmentation of cells
Sample generation for ground truth
Endothelial cells were transduced with Ad-Lac at 50 MOI as above. After 48 hours of transduction, cells were fixed for immunofluorescence as described above for VE-Cadh (CDH5) to mark cell boundaries and stained with Hoechst (1:1000). Samples were then stored at 4°C wrapped in parafilm to be imaged at a later time. Images of cells were acquired in the FRET channel and Hoechst channel at 10× magnification.
Ground truth labeling
Cellprofiler52 was used for ground truth labeling of 4 classes: background, border, cytoplasm, and nuclei. First, 1200×1200 pixel images in the FRET channel were background corrected with subtraction using a running average block size of 150 pixels across all images of the fixed samples. Next, using an adaptive threshold (Otsu) strategy, nuclei were identified after staining with Hoechst dye. Using nuclei as seeds, cell boundaries were found through the propagation algorithm. The cytoplasm was then defined as areas inside the cell boundaries (through immunofluorescence labeling with anti-CDH5 and secondary antibody anti-rabbit-Alexa 488), but not of the nuclei. Next, these images were recombined and labeled with a unique class identifier. Minor adjustments to the labeled images were made algorithmically and automatically in MATLAB, including making sure that a cell boundary encapsulates all cytoplasm and nuclei, and getting rid of cells that had no nuclei. Images of each label class were generated and visually inspected for accuracy. 432 images containing approximately 500 cells per image were used for training as described below. Of note, ground truth labeling required separate nuclei and boundary staining procedures which would have to be repeated on each separate dataset for precise determination of cell boundaries. This makes fast determination of LPR without manual boundary tracing impractical (we routinely collect hundreds of cells in each experiment). With deep learning, it became possible to avoid the two extra staining procedures and thereby significantly increases experimental throughput, and has the added benefit of being generalizable to other cell types and magnifications.
Deep-learning enabled semantic segmentation
The 432 image data set with labeled ground truth was randomized into training (70%), validation (15%), and test (15%) datasets. To enhance the dataset, patches were extracted and augmented with random reflection, rotation, and shear up to 10 degrees. Next, a U-net network7 (Extended Data Fig. 1b) was trained using conventional deep learning, with final layer pixel classification consisting of an inverse class weighting (based on class frequency) cross entropy loss function over 50 epochs. 128×128 image tile size was chosen, with 8 mini-batches, using stochastic gradient of descent with 0.95 momentum (~38,000 unique images prior to augmentation; Background pixels: 2.93×108; Border pixels: 2.81×107; Cell body pixels: 9.15×107; Nuclei pixels: 2.54×107). Initial learning rate was 0.001 and was dropped by a factor of 10 every 10 epochs. The training was performed in MATLAB using the Deep Learning Toolbox on 2 NVIDIA GeForce 1080 Ti and 1 NVIDIA Titan V GPU. Total training time was about 96 hours and was stopped after no further reduction of loss. In this manner, a semantic segmentation network capable of assigning each pixel a separate class was created. Measures of deep learning accuracy are listed in Extended Data Table 1. We found that inclusion of a nuclei class improved the classification of cell boundaries. Adding additional 2D convolutional layers to increase the receptive field did not improve the network accuracy but significantly reduced the learning rate. Convolutional layers had 3×3 kernel size. Skip connections were included in the U-net as depicted in Extended Data Fig. 1b.
Image Analysis
After feeding images into the semantic segmentation network, the output was further processed to remove cells below a pre-defined threshold, based on expected cell size. Next, FRET’ trajectories of each cell over time were generated. To accomplish this, individual cells were extracted from the labeled images: each cell was identified as an object surrounded by a boundary class and containing a nucleus. The mean position of the cell was matched to a cell with the closest position in a subsequent image, thus generating a position trajectory, and therefore FRET’ trajectory (computed as the average mTFP fluorescence intensity divided by the average Venus fluorescence intensity inside the boundary minus background signal) of every cell. The FRET’ trajectories were then subjected to a median filter of width 5 for denoising. To obtain the lactate production rate, pyruvate production rate, or glucose uptake rate, the slope of the FRET’ (or regular FRET for glucose sensor) signal was calculated over 40 second trajectories and fit against a linear model during the portion of the time the cells were exposed to MCT1 inhibitor. ATP levels were determined by the average FRET signal before and after addition of agonist. Only if the R2 value of the fit was greater than 0.9 was the data considered useable. Conversion of FRET’ to LPR was calculated as described above.
Quantification of image analysis error
We estimated the error in deep learning assisted semantic segmentation by first computing the error between ground truth fluorescence and segmented fluorescence. First, we used a test data set of cells expressing laconic for which we have ground truth and segmented these images with the deep learning algorithm. Then using the same image analysis pipeline, we plot the cell fluorescence as estimated from deep learning networks against the ground truth cell fluorescence (n ~ 20,000). The 95% confidence interval of the slope is 0.93-0.95, so there was consistently a small underestimation of the true fluorescence of each cell (Extended Data 1c).
To estimate how this error in fluorescence detection affects fitting of LPR data, we turn to Monte Carlo simulations of LPR (bootstrapping). Using the dataset (Extended Data 1c) which provides a real measure between ground truth and fluorescence, we generate a line x = y by randomly sampling the ground truth fluorescence, each of which corresponds to a deep-learning generated fluorescence, 100 times. The slope of this line is then calculated by performing linear regression. This random sampling of fluorescence values, followed by slope calculation, simulates LPR calculation in real samples. The distribution of these bootstrapped slopes is plotted after repeating this 10,000 times, resulting in a Gaussian distribution of slopes (Extended Data 1d). The red line is fit to a Gaussian. The mean of the fit Gaussian is 0.9, similar to the correlation between ground truth and deep learning fluorescence above (0.94). The standard deviation of the Gaussian fit is 0.05. Thus the error in estimating LPR, due to semantic segmentation, is 0.05/0.9 which is approximately 5%.
Per-pixel LPR
Cells were seeded into Ibidi chambers and the same protocol followed when imaging LPR as above, with the exception that a 40× plan-apochromat Olympus objective was used for imaging. After acquisition of the images, a tophat filter with width of 30 microns (in real space) was used to estimate the background. A Gaussian with width of 1 pixel was convolved with the original image to smooth noise artifact. To this smoothened image, the minimum of the estimated background image was subtracted. FRET’ was calculated by dividing the mTFP image from the YFP image. Following, the per pixel LPR was calculated after binning by 4 in an average sense in each image in the time series (after addition of pCMBA). The slope of each super pixel in the time series was then calculated; the value of the slope was replotted as the LPR image. LPR images were then resized to the original size.
Actin turnover and overlap calculations
Ten Lifeact-RFP images, taken 1 minute apart, were subtracted from each other in sequential order, and the maximum intensity of each of these difference images was projected as the final “difference” image. Both the Lifeact difference image and LPR images were normalized to the largest value in the respective image. To calculate the Manders’ Overlap Coefficient (MOC) or Pearson’s Correlation Coefficient (PCC) (complementary statistical measures of spatial correlation) between the Lifeact difference image and the per pixel LPR image, Lifeact difference images were first segmented by hand. The mask of the cell was then applied to the LPR per pixel image to avoid background effects. Next, the actin difference image was thresholded at 5% of maximum intensity. Finally, the MOC and PCC were calculated as in equations 4 and 5, respectively.
| Eq. 4 |
| Eq. 5 |
where x and y are Lifeact difference and LPR images with i rows and j columns, and 〈…〉 operator denotes mean. Simulated MOC or PCC was performed by randomizing the spatial location of the LPR pixel values within the masked area and calculating the MOC or PCC with the thresholded Lifeact difference image 10,000 times.
RhoA imaging, LPR vs RhoA activity
RhoA-Flare imaging was performed with FRET filter sets purchased from Semrock: excitation 438/24, mTFP emission 483/32, Venus emission 542/27, and dichroic FF458-Di02 on transfected cells. Integration times ranged from 100ms to 500ms with a Photometrics Prime95b camera using either a 40× or 100× plan-apochromat Olympus objective. Single cell LPR was performed on separate cells as described above. To calculate the LPR or RhoA activity as a function of distance from center, the cells were first segmented by hand in ImageJ. Line scan intensities of either RhoA activity (FRET) or LPR were performed from the geometric center of the cell to the edge of the contracting or non-contracting portions of the cell. These line scans were then normalized to maximum intensity and interpolated using cubic splines to the same number of data points (1000). The signals were then averaged to produce the final figure. Each line scan in the category of contractile or not is from a different cell.
Mouse studies
Gene Expression Omnibus datasets (GEOD) were downloaded from EndoDB (https://endotheliomics.shinyapps.io/endodb/), a database of curated endothelial transcriptomics datasets31. All mouse endothelial datasets are of isolated primary cells in the untreated (control) arms of the experiments.
All animal care and treatment procedures were approved by the University of Chicago Institutional Animal Care and Use Committee. Animals were handled according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and all experiments were compliant with ethical guidelines from The Guide for the Care and Use of Laboratory Animals. All experimental animals were assigned unique identifiers to blind experimenter to treatment. For the mouse model of vascular leak, C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME). All experimental animals were maintained in a pathogen-free environment with a humidity of 40-60%, a temperature of 20–24 °C, and a 12-h light/dark cycle at the University of Chicago. For in vivo human SLC2A3 or mScarlet-i transient overexpression, C57BL/6 mice, 10–12 wk old, were tail vein-injected with the solution containing 40 μg of endotoxin-free CDH5-SLC2A3 overexpression plasmid, CDH5-mScarlet-i, or control CDH5a containing plasmid DNA diluted with sterile 5% glucose and then mixed with polyethylenimine (PEI) nanoparticles (TurboFect in vivo Transfection Reagent) (Thermo Scientific) according to manual instructions. After 24 hours, mice were anesthetized with ketamine-xylazine (80 mg/kg:20 mg/kg) before harvest of aorta and intimal RNA isolation, Evans blue assay, or fixation, paraffin-embedding and sectioning for visualization of mScarlet-i.
Evans blue dye assay
After anesthesia, retro-orbital injection of 4 μL/g of 2% Evans blue dye was performed. 1 hour later, aortas were immediately cleaned and harvested, photographed, and weighed. Evans blue dye was extracted from the aortas by sonication in 50% trichloroacetic acid (4 μL/mg). Each sample was centrifuged, and the supernatant mixed with 100% ethanol in a 1:3 ratio. The OD of each sample was read at 620 nm on a Biotek-3 instrument and normalized to aorta weight.
Aorta intima isolation for qRT-PCR
After careful isolation, the aorta was quickly flushed with 350 μl of Qiazol lysis reagent (Qiagen) using a 29G insulin syringe and the elute was collected in a microfuge tube for intimal RNA isolation. The leftover aorta after flushing with Qiazol was homogenized with 700 μl of Qiazol for RNA isolation of media and adventitia. Reverse transcription and qRT-PCR were performed using the Maxima First Strand cDNA Synthesis Kit (ThermoFisher) according to manufacturer’s protocol. Mouse PCR primers are listed above.
Fluorescence of aortic intima
CDH5-mScarlet-i transfected aortas were fixed in 4% paraformaldehyde for 15 minutes and embedded in OCT after being immersed in OCT:sucrose=1:1 mixed solution for 24 hours. Frozen samples were sectioned in 5-8 μm. The sections were then mounted, permeablized, blocked, and incubated with antibody to CDH5, followed by fluorescent secondary antibody (as above). Samples were then incubated with ProLong Gold with DAPI (Thermo Fisher) and imaged with a confocal microscope (Olympus).
Statistics
The data were analyzed in Prism 9 (GraphPad Software Inc, La Jolla, CA). All data with error bars were shown as mean ± standard error of the mean (SEM) except for the metabolomics estimate which was the mean ± standard deviation (SD) (Fig. 1) to better highlight the distribution of intracellular lactate levels. Statistical significance was determined by unpaired Welch’s t-test for individual differences and one way ANOVA followed by Bonferroni’s test for multiple comparisons. All t-tests were two-tailed. All experiments were biological replicates except for extracellular flux experiments, which included technical replicates. All measurements were taking from distinct samples.
Extended Data
Extended Data Fig. 1. Single-cell metabolic assay utilizes deep learning-enabled segmentation.
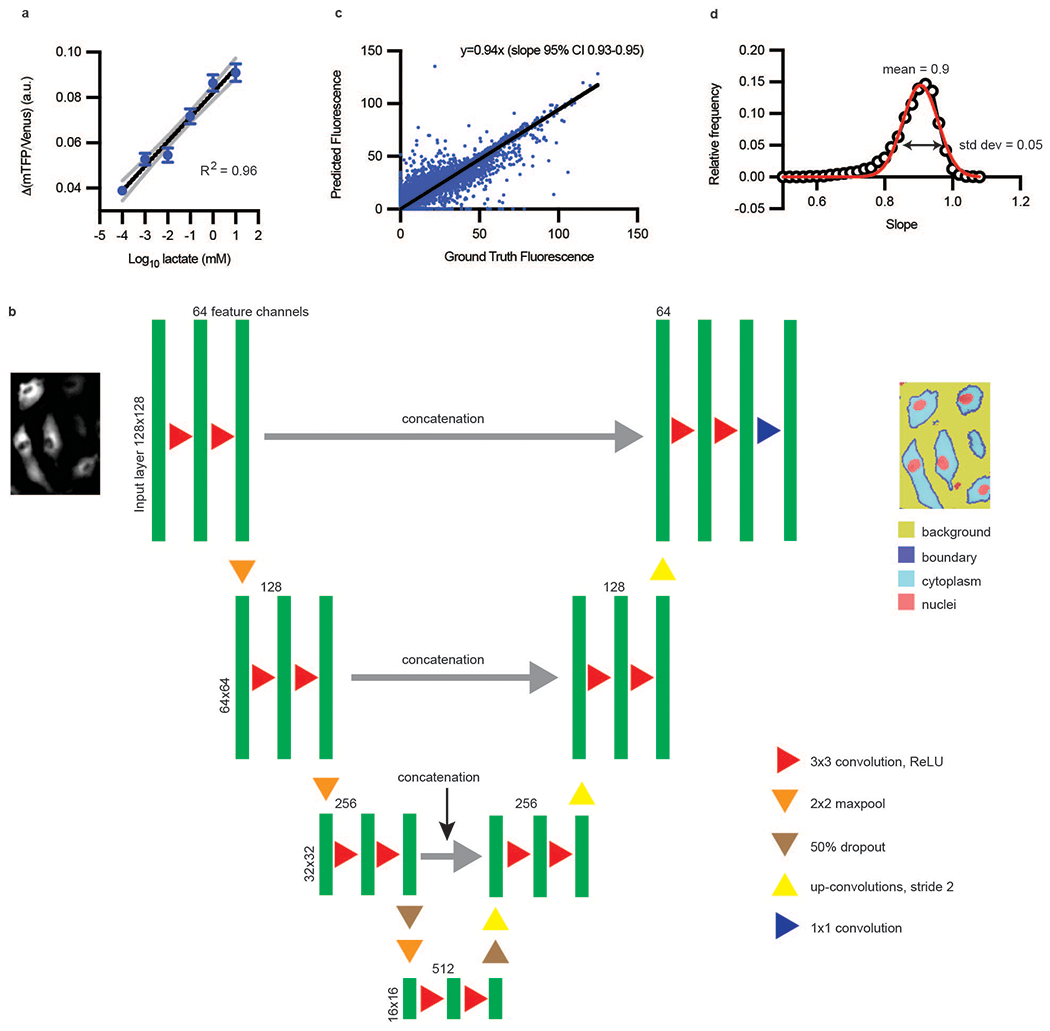
a, mTFP/Venus (or 1/FRET) change was linearly correlated over 6 orders of magnitude of log lactate (n = 287 cells; black line is semilog fit with R2 = 0.96, gray dotted lines are 95% confidence interval); error bars are SEM. Standard deviation of regression coefficients for R2 for (a) is 0.049. b, Deep learning architecture for semantic segmentation, modified from Ronneberger et al7. The U-Net consisted of a contracting path (encoder) and an expansive path (decoder). The contracting path consists of layers composed of two 3 × 3 convolutions, each followed by a rectified linear unit (ReLU) activation function. Each layer is followed by a 2 × 2 max pooling operation with a stride of two- which will double the number of feature channels being used. The convolutions start with 64 feature channels and 128 × 128 images and continue until they reach 512 channels and 16 × 16 images. In the expansive path, 2 × 2 up-convolutions (up sampling) of stride 2 to decrease by half the feature channels and increase the size of the image. The layers composed of 3 × 3 convolutions and ReLU are concatenated with the pair of layers in the contracting side, in order to reinclude the localization information. In the final layer, a 1 × 1 convolution layer is used to map the resulting 64-component feature channels vector to the 4 segmentation channels for background, boundary, cytoplasm, and nuclei. c, Ground Truth Fluorescence vs. Predicted Fluorescence, as evaluated with deep-learning-enabled semantic segmentation. (n ~ 20,000 cells) Ground truth fluorescence was defined as signal inside the cell boundary. d, Frequency distribution of fluorescence slopes. Data from (c) or are randomly sampled 100 times and fitted to a line. This process is repeated 10,000 times and the distribution of fitted slopes was plotted (open black circles). The red line is a Gaussian fit with mean 0.9 and standard deviation of 0.05. Thus, the error in estimating slopes of fluorescence is approximately 0.05/0.9, or approximately 5%.
Extended Data Fig. 2. Single-cell laconic accurately mimics bioenergetics assays.
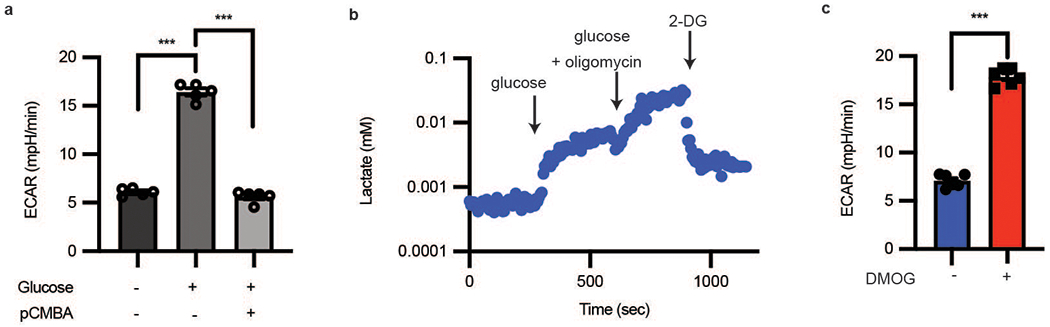
a, Extracellular flux assay of HAECs treated with glucose then pCMBA (n = 5) using Seahorse; biological replicates, error bars are SEM, p < 0.0001 for both comparisons. b, A single-cell glycolytic stress test using lactate FRET sensor. Cells were first starved in zero glucose buffer for 1 hour. Glucose addition increased intracellular lactate, which was further increased by adding oligomycin and glucose. Addition of 2-deoxyglucose (2-DG) blocked all glycolysis, causing intracellular lactate to drop. c, Overnight treatment of endothelial cells with 500 μM DMOG increases ECAR measured by extracellular flux relative to DMSO-treated cells using Seahorse. (n = 11 combined biological and technical replicates for each condition; error bars are SEM, p < 0.0001). Statistical significance determined by one-way ANOVA followed by Bonferroni test (a) or by two-sided Welch’s t-test (c).
Extended Data Fig. 3. RhoA activity induces contraction using glycolysis.
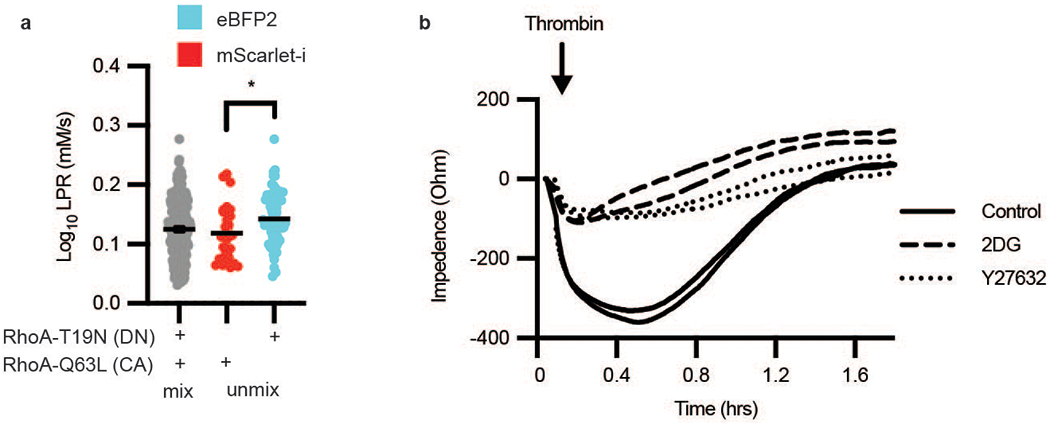
a, LPR of HAECs transfected with either RhoA-T19N-eBFP2 (dominant negative form, cyan) or RhoA-Q63L-mScarlet-i (constitutively active form, red), mixed, then deconvoluted by color (n = 174 “cyan” and 52 “red”, respectively). Error bars are SEM, p = 0.0193. b, TEER measurement of HAECs after thrombin treatment, either pre-treated with Y27632 or 2DG compared to control (no Y27632 or 2DG). Statistical significance determined by two-sided Welch’s t-test.
Extended Data Fig. 4. SLC2A3 but not SLC2A1 knockdown mitigates thrombin-induced phosphorylation of CFL and MLC and glycolysis.
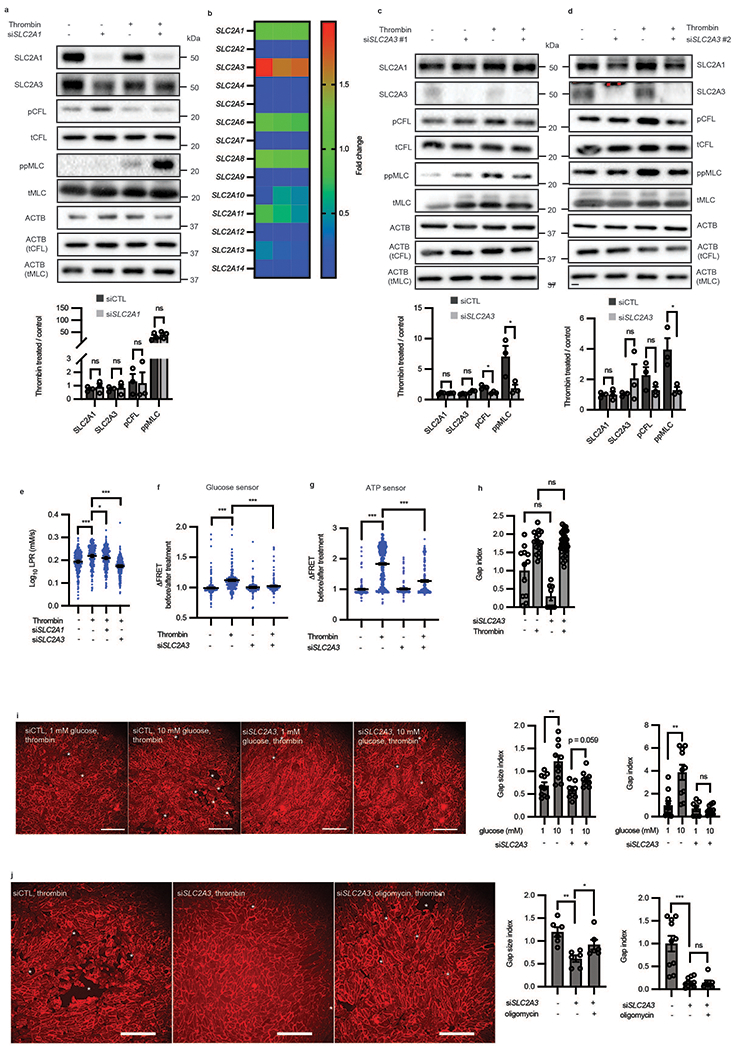
a, Western blot of SLC2A1, SLC2A3, phospho-cofilin and phospho-MLC in the presence or absence of siRNA targeted towards SLC2A1 and thrombin treatment (n = 3 biological replicates). Loading control for each protein is ACTB unless specifically denoted in parentheticals. Original blot in Source Data c. b, Heatmap of glucose transporter expression relative to SLC2A1 in HAECs captured by qRT-PCR (n = 3 biological replicates) c-d, Western blot of SLC2A1, SLC2A3, phospho-cofilin and phospho-MLC in the presence or absence of two distinct siRNA (separate from the one used in Figure 5a) (#1 in c and #2 in d) targeted towards SLC2A3 and thrombin treatment (n = 3 biological replicates. For c, p = 0.0190 for pCFL and 0.0490 for ppMLC. For d, p = 0.0272 for ppMLC.). Loading control for each protein is ACTB unless specifically denoted in parentheticals. Original blot in Source Data d-e. e, Single-cell LPR of HAECs treated with thrombin in the presence of siSLC2A1 or siSLC2A3 (n = 277 for thrombin vs. control, n = 281 for thrombin vs. thrombin + siSLC2A1, and n = 278 for thrombin vs. thrombin + siSLC2A3, p < 0.0001, p = 0.0384, p < 0.0001, respectively.). f-g, Glucose (f) and ATP levels (g) in single-cells in the presence or absence of SLC2A3 knockdown and thrombin treatment (f: n = 248, 248, 108, 190 from left to right, p < 0.0001 for all comparisons; g: 190, 598, 150, 287 from left to right, p < 0.0001 for all comparisons). h, Gap index of cells treated with or without thrombin in the presence or absence of SLC2A3 knockdown (n= 12, 16, 8, 32 from left to right). i-j, Representative images of HAECs treated with siSLC2A3 or control with varying concentrations of glucose (i) or with oligomycin (j) in the presence of thrombin. Red color is CDH5. Gap size index and gap index quantification of images (i, n = 10, 10, 7, 8 from left to right, p = 0.022 for gap size index and p = 0.0019; j, n = 6 for gap size index, p = 0.0013 for siSLC2A3 vs control and p = 0.040 for siSLC2A3 vs. siSLC2A3 + oligomycin, and n = 10, 11, 7 from left to right for gap index, p < 0.0001). White stars highlight gaps in the monolayer. Scale bars = 250 μm. Statistical significance determined by multiple unpaired two-tailed t-tests (a, c, d), one-way ANOVA followed by Bonferroni test (e-g, i) or by two-sided Welch’s t-test (h, j). All error bars are SEM.
Extended Data Fig. 5. SLC2A3 inhibition abrogates thrombin-induced phosphorylation of CFL and MLC and contraction across multiple endothelial cell types.
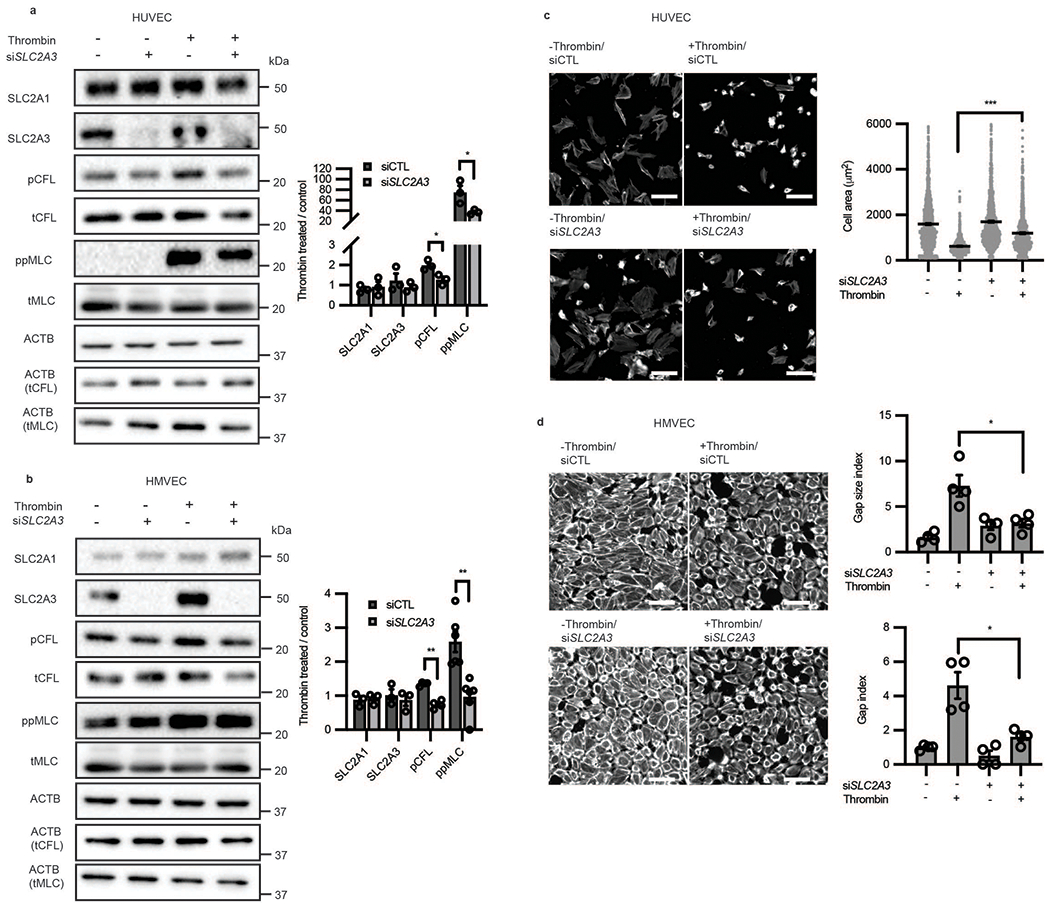
a-b, Western blot analysis of glucose transporter expression, phosphorylated CFL, and phosphorylated MLC, in human umbilical vein endothelial cells (HUVEC, a, n = 3; biological replicates, p = 0.0257 for pCFL and 0.0414 or ppMLC) and human microvascular endothelial cells (HMVEC, b, n = 3 for SLC2A1, SLC2A3, n = 4 for CFL and n = 5 for MLC; biological replicates, p < 0.001 for pCFL and p = 0.0014 for ppMLC). Loading control for each protein is ACTB unless specifically denoted in parentheticals. Original blot in Source Data f-g. c, Representative images and cell size of contractile HUVECs subjected to thrombin stimulation in the absence or presence of SLC2A3 knockdown. (n = 1375, 1279, 760, 1162 from left to right, p < 0.0001) d, Representative images, gap size index, and gap index of contractile HMVECs subjected to thrombin stimulation in the absence or presence of SLC2A3 knockdown. Representative images are phalloidin. (n = 4 biological replicates, p = 0.0171 for gap size index and p = 0.0262 for gap index) Statistical significance determined by two-sided Welch’s t-test. All error bars are SEM.
Extended Data Fig. 6. In vivo overexpression of SLC2A3 increased vascular leak.
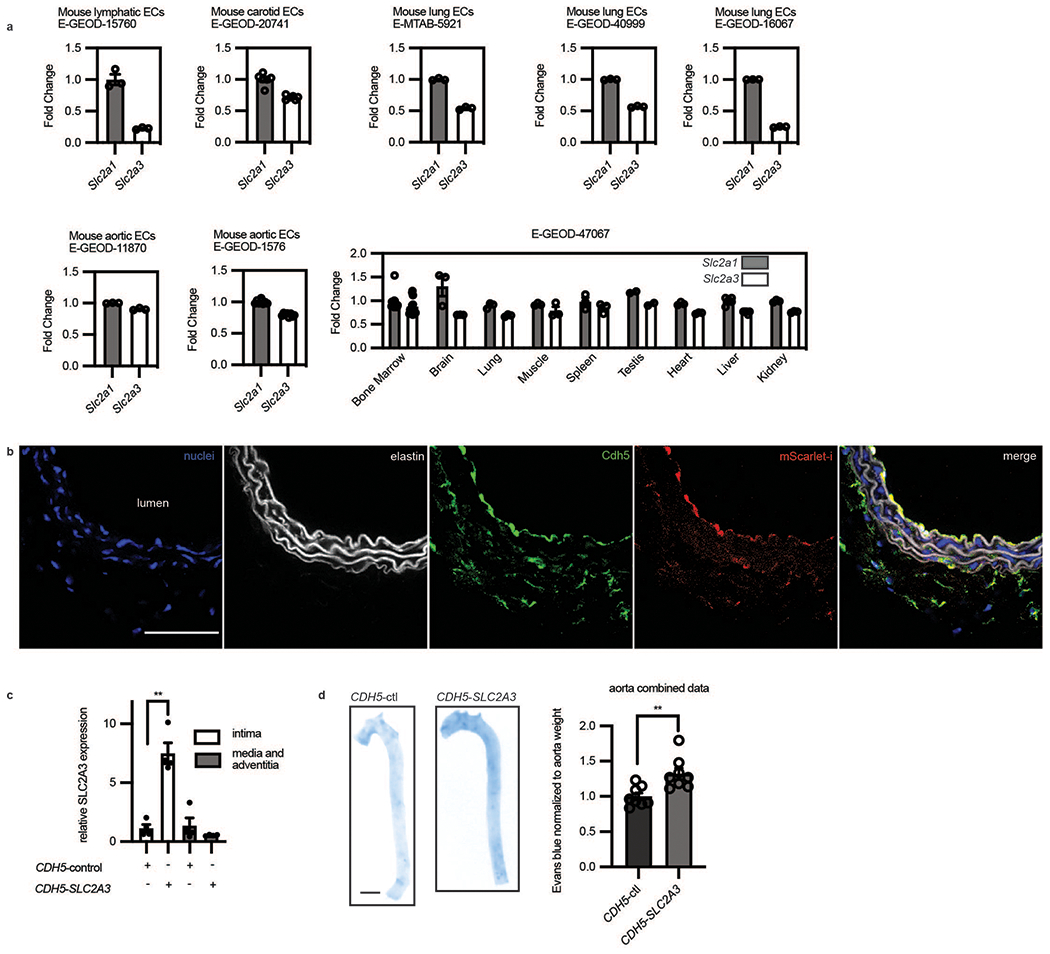
a, GEO datasets of primary endothelial mouse cells comparing Slc2a1 and Slc2a3 from different organs. Fold change indicates that data are normalized to Slc2a1 except for E-GEOD-47067 which was normalized to average of Slc2a1 in all tissues. Only controls of experiments in the GEO datasets were used in these analyses. (n = 3 independent biological replicates for each condition for E-GEOD-15760; n = 6 independent biological replicates for each condition for E-GEOD-20741; n = 3 independent biological replicates for each condition for E-MTAB-5921; n = 3 independent biological replicates for each condition for E-GEOD-40999; n = 3 independent biological replicates for each condition for E-GEOD-11870; n = 3 independent biological replicates for each condition for E-GEOD-1576; n = 3 independent biological replicates for condition in each organ for E-GEOD-47067) b, Immunofluorescence of an arterial section demonstrating enhanced mScarlet-i fluorescence (red) colocalized with ve-cadh (green) which marks the endothelium next to the lumen (yellow, merge) (Scale bar, 500 μm, representative of n = 4 mice). c, SLC2A3 expressing plasmid uses an endothelial specific CDH5 promoter. SLC2A3 is detected by qRT-PCR of the intima of the mouse aorta, and far less than in the media and adventitia. (n = 4 independent biological replicates, p = 0.0030) d, Evans blue (OD 620 nm) quantification of mouse aortas, normalized by weight. (n = 8 independent biological replicates for each condition, p = 0.0050). Statistical significance was determined by two-sided Welch’s t-test. All error bars are SEM.
Extended Data Fig. 7. Subcellular RhoA activity, actin turnover, and LPR distribution in single motile endothelial cells.
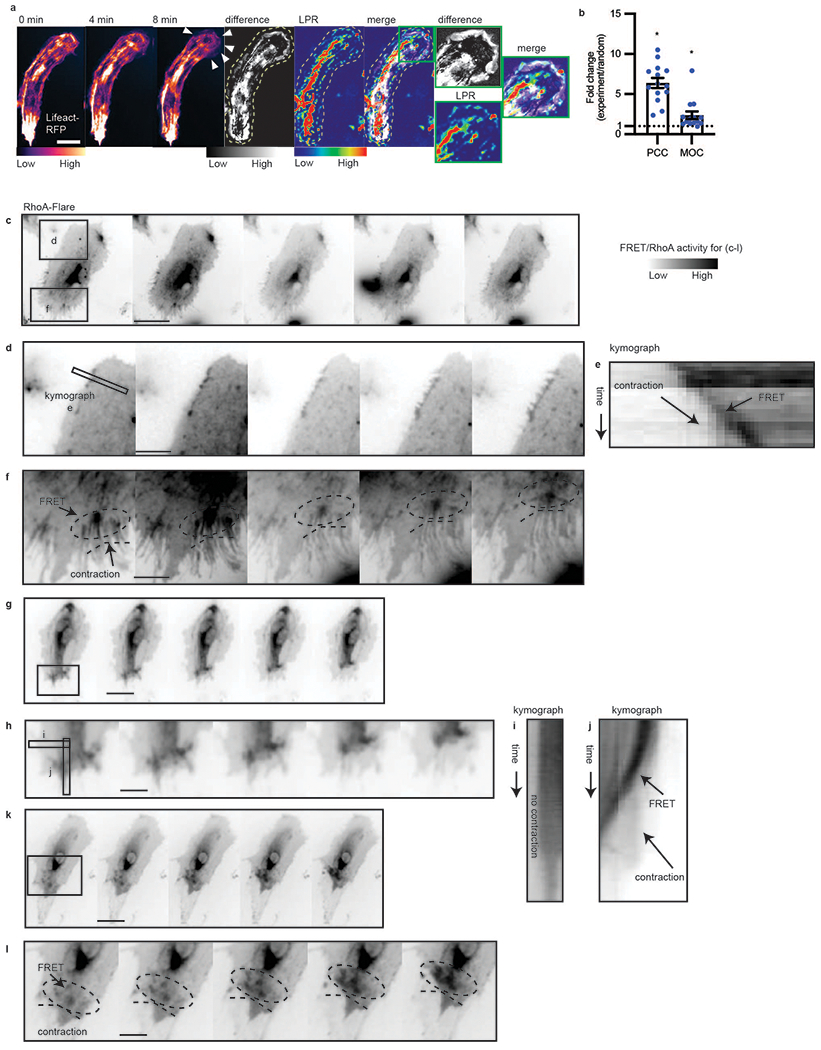
a, Montage of actin timelapse in a migratory endothelial cell; maximum projection of change in actin structure (Lifeact difference image, obtained by sequential subtraction of preceding 10 images, taken 1 minute apart, followed by maximum intensity projection of the absolute value of the differences), per pixel LPR map (LPR), Lifeact difference and LPR overlap (“difference+LPR”, merge), scale bar = 20 μm. White arrows in montage indicate cell spreading. Green outline shows enhanced LPR near the regions of cell spreading. Representative of n = 14 cells, 3 separate experiments. b, Pearson’s correlation coefficient (PCC) and Manders’ overlap coefficient (MOC) of Lifeact difference and LPR, compared to random. The random PCC/MOC is calculated by randomizing the LPR image and computing with the Lifeact difference image. Fold change of experimental PCC/MOC over randomized is shown. (n = 14). Error bars are SEM, p = 0.014 for PCC and p = 0.0489 for MOC. c-l, RhoA activity measured by RhoA-FLARE in contractile endothelial cells. Representative data of total n = 9 cells in 3 replicates. (Scale bar = 16 μm in c, g, k. Scale bar = 7 μm in d, f, h, l). d,f, regions of contractile cell from (c) showing RhoA membrane activity. Circle in (f) notes moving RhoA activity. e, kymograph showing contractile edge from (d) and increasing RhoA activity over time. h, region of contractile cell from (g) showing increased RhoA membrane activity. i-j, kymograph showing non-contractile edge (i) without RhoA increase and contractile edge (j) from (g) and increasing RhoA activity over time. l, region of contractile cell from (k) showing RhoA membrane activity. Dotted ellipses notes highlight increasing RhoA activity along with active contraction. Statistical significance determined by two-sided Welch’s t-test.
Extended Data Table 1:
Deep learning semantic segmentation metrics
| Matthew’s Correlation Coefficient | 0.85 | |||
|---|---|---|---|---|
| Global accuracy | 0.84 | |||
| Class | Background | Cytoplasm | Nuclei | Cell boundary |
| Class specific accuracy | 0.85 | 0.83 | 0.45 | 0.78 |
| Standard deviation of accuracy | 0.083 | 0.079 | 0.086 | 0.055 |
| Boundary F1 score | 0.96 | 0.95 | 0.75 | 0.94 |
| Standard deviation of Boundary F1 score | 0.05 | 0.047 | 0.079 | 0.071 |
| Precision | 0.85 | 0.83 | 0.45 | 0.78 |
| Recall | 0.97 | 0.84 | 0.31 | 0.38 |
| Dice score | 0.9 | 0.82 | 0.38 | 0.53 |
| Standard Deviation of Dice Score | 0.074 | 0.0659 | 0.064 | 0.064 |
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by NIH grants R00AI106941 (JH), R21AI120010 (JH), R01HL138223 (YF), and R01HL136765 (YF), NIH New Innovator Award DP2AI144245 (JH), T32EB009412 (DLH), T32HL007381 (DLH), F32HL134288 (DW), NIH Pathway to Independence Award K99HL145113 (DW), and R00HL145113 (DW). NVIDIA GPU Grant (DW), CSCTR Early Career Development Award (DW), and NSF Career Award 1653782 (JH) also supported this work.
Footnotes
Declaration of interests
There authors declare no competing interests.
Code Availability
The code generated during this study is available at https://github.com/wulab-code/laconic without restriction.
Data Availability
Gene Expression Omnibus datasets (GEOD) were downloaded from EndoDB (https://endotheliomics.shinyapps.io/endodb/): E-GEOD-15760, E-GEOD-20741, E-MTAB-5921, E-GEOD-40999, E-GEOD-16067, E-GEOD-11870, E-GEOD-1576, E-GEOD-47067. Original data that supports our findings in this study are available from the corresponding author upon reasonable request or at doi:10.5281/zenodo.4638059.
References
- 1.Kim B, Li J, Jang C & Arany Z Glutamine fuels proliferation but not migration of endothelial cells. EMBO J 36, 2321–2333 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu D, et al. HIF-1alpha is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng S, et al. Mechanical Activation of Hypoxia-Inducible Factor 1alpha Drives Endothelial Dysfunction at Atheroprone Sites. Arterioscler Thromb Vasc Biol 37, 2087–2101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doddaballapur A, et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler Thromb Vasc Biol 35, 137–145 (2015). [DOI] [PubMed] [Google Scholar]
- 5.van Nieuw Amerongen GP, Koolwijk P, Versteilen A & van Hinsbergh VW Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler Thromb Vasc Biol 23, 211–217 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Van Valen DA, et al. Deep Learning Automates the Quantitative Analysis of Individual Cells in Live-Cell Imaging Experiments. PLoS Comput Biol 12, e1005177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ronneberger O, Fischer P & Brox T U-Net: Convolutional Networks for Biomedical Image Segmentation. 234–241 (2015). [Google Scholar]
- 8.San Martin A, et al. A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS One 8, e57712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Csurka GL, Diane. What is a good evaluation measure for semantic segmentation? IEEE Trans. Pattern Anal. Mach. Intell. 26(2013/01/01). [Google Scholar]
- 10.Halestrap AP Monocarboxylic acid transport. Compr Physiol 3, 1611–1643 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Sonveaux P, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One 7, e33418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.San Martin A, et al. Imaging mitochondrial flux in single cells with a FRET sensor for pyruvate. PLoS One 9, e85780 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takanaga H, Chaudhuri B & Frommer WB GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim Biophys Acta 1778, 1091–1099 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imamura H, et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci U S A 106, 15651–15656 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies PF Flow-mediated endothelial mechanotransduction. Physiol Rev 75, 519–560 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayor R & Etienne-Manneville S The front and rear of collective cell migration. Nat Rev Mol Cell Biol 17, 97–109 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Diebold LP, et al. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nature Metabolism 1, 158–171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mole DR, et al. 2-Oxoglutarate analogue inhibitors of hif prolyl hydroxylase. Bioorganic & Medicinal Chemistry Letters 13, 2677–2680 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Fazal F, et al. Essential role of cofilin-1 in regulating thrombin-induced RelA/p65 nuclear translocation and intercellular adhesion molecule 1 (ICAM-1) expression in endothelial cells. J Biol Chem 284, 21047–21056 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Nieuw Amerongen GP, Vermeer MA & van Hinsbergh VW Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol 20, E127–133 (2000). [DOI] [PubMed] [Google Scholar]
- 21.van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG & van Hinsbergh VW Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res 87, 335–340 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Blanchoin L, Boujemaa-Paterski R, Sykes C & Plastino J Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 94, 235–263 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Goddard LM & Iruela-Arispe ML Cellular and molecular regulation of vascular permeability. Thromb Haemost 109, 407–415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehta D & Malik AB Signaling mechanisms regulating endothelial permeability. Physiol Rev 86, 279–367 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Vandenbroucke E, Mehta D, Minshall R & Malik AB Regulation of endothelial junctional permeability. Ann N Y Acad Sci 1123, 134–145 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Tang M, et al. Brain microvasculature defects and Glut1 deficiency syndrome averted by early repletion of the glucose transporter-1 protein. Nat Commun 8, 14152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fessel JP, et al. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ 2, 201–213 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sasmal DK, et al. TCR-pMHC bond conformation controls TCR ligand discrimination. Cell Mol Immunol 17, 203–217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sasmal DK, Pulido LE, Kasal S & Huang J Single-molecule fluorescence resonance energy transfer in molecular biology. Nanoscale 8, 19928–19944 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraccaroli A, et al. Endothelial alpha-parvin controls integrity of developing vasculature and is required for maintenance of cell-cell junctions. Circ Res 117, 29–40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan S, et al. EndoDB: a database of endothelial cell transcriptomics data. Nucleic Acids Res 47, D736–D744 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu C, et al. Mechanosensitive PPAP2B Regulates Endothelial Responses to Atherorelevant Hemodynamic Forces. Circ Res 117, e41–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao L, Xue X, Yu P, Ni Y & Chen F Evans Blue Dye: A Revisit of Its Applications in Biomedicine. Contrast Media Mol Imaging 2018, 7628037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riedl J, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods 5, 605–607 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pertz O, Hodgson L, Klemke RL & Hahn KM Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 440, 1069–1072 (2006). [DOI] [PubMed] [Google Scholar]
- 36.De Bock K, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Wilhelm K, et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529, 216–220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karmaus PWF, et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature 565, 101–105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bantug GR, Galluzzi L, Kroemer G & Hess C The spectrum of T cell metabolism in health and disease. Nat Rev Immunol 18, 19–34 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Hensley Christopher T., et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Heerden JH, et al. Lost in transition: start-up of glycolysis yields subpopulations of nongrowing cells. Science 343, 1245114 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Huang J, et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity 39, 846–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masters C Interactions between glycolytic enzymes and components of the cytomatrix. J Cell Biol 99, 222s–225s (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y, et al. Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1alpha-dependent function. Proc Natl Acad Sci U S A 109, 17478–17483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuo A, Lee MY & Sessa WC Lipid Droplet Biogenesis and Function in the Endothelium. Circ Res 120, 1289–1297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu P, et al. FGF-dependent metabolic control of vascular development. Nature 545, 224–228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sorbara LR, et al. Thrombin-induced translocation of GLUT3 glucose transporters in human platelets. Biochem J 328 ( Pt 2), 511–516 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang RT, et al. Experimental Lung Injury Reduces Kruppel-like Factor 2 to Increase Endothelial Permeability via Regulation of RAPGEF3-Rac1 Signaling. Am J Respir Crit Care Med 195, 639–651 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang D, et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li L, et al. Artificial Virus Delivers CRISPR-Cas9 System for Genome Editing of Cells in Mice. ACS Nano 11, 95–111 (2017). [DOI] [PubMed] [Google Scholar]
Additional References
- 51.Barbee KA, Davies PF & Lal R Shear stress-induced reorganization of the surface topography of living endothelial cells imaged by atomic force microscopy. Circ Res 74, 163–171 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Carpenter AE, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Gene Expression Omnibus datasets (GEOD) were downloaded from EndoDB (https://endotheliomics.shinyapps.io/endodb/): E-GEOD-15760, E-GEOD-20741, E-MTAB-5921, E-GEOD-40999, E-GEOD-16067, E-GEOD-11870, E-GEOD-1576, E-GEOD-47067. Original data that supports our findings in this study are available from the corresponding author upon reasonable request or at doi:10.5281/zenodo.4638059.