

Abstract
There is currently no cure or effective treatment available for mucopolysaccharidosis type IIID (MPS IIID, Sanfilippo syndrome type D), a lysosomal storage disorder (LSD) caused by the deficiency of α-N-acetylglucosamine-6-sulfatase (GNS). The clinical symptoms of MPS IIID, like other subtypes of Sanfilippo syndrome, are largely localized to the central nervous system (CNS), and any treatments aiming to ameliorate or reverse the catastrophic and fatal neurologic decline caused by this disease need to be delivered across the blood-brain barrier. Here, we report a proof-of-concept enzyme replacement therapy (ERT) for MPS IIID using recombinant human α-N-acetylglucosamine-6-sulfatase (rhGNS) via intracerebroventricular (ICV) delivery in a neonatal MPS IIID mouse model. We overexpressed and purified rhGNS from CHO cells with specific activity of 3.9x104 units/mg protein, with a maximal enzymatic activity at lysosomal pH (pH 5.6), and which was stable for over one month at 4°C in artificial cerebrospinal fluid (CSF). We demonstrated that rhGNS was taken up by MPS IIID patient fibroblasts via the mannose 6-phosphate (M6P) receptor and reduced intracellular glycosaminoglycans to normal levels. Delivery of 5 micrograms of rhGNS into the lateral cerebral ventricle of neonatal MPS IIID mice resulted in normalization of the enzymatic activity in brain tissues; rhGNS was found to be enriched in lysosomes in MPS IIID treated mice relative to control. Furthermore, a single dose of rhGNS was able to reduce the accumulated heparan sulfate and β-hexosaminidase. Our results demonstrate that rhGNS delivered into CSF is a potential therapeutic option for MPS IIID worthy of further development.
Keywords: MPS IIID, enzyme replacement therapy, GNS, CHO cells, recombinant protein production, DHFR
GRAPHICAL ABSTRACT
INTRODUCTION
Mucopolysaccharidosis (MPS) III (also known as Sanfilippo syndrome) is caused by mutations in enzymes that degrade heparan sulfate, a glycosaminoglycan (GAG) that has important structural and functional roles in the brain and elsewhere. There are four subtypes of MPS III that are categorized by deficiencies of different lysosomal enzymes: MPS IIIA (OMIM: 252900; N-sulfoglucosamine sulfohydrolase, SGSH; EC 3.10.1.1); MPS IIIB (OMIM:252920; α-N-acetyl glucosaminidase, NAGLU; EC 3.2.1.50); MPS IIIC (OMIM: 252930; heparan sulfate acetyl CoA: α-glucosaminide N-acetyltransferase, HGSNAT; EC 2.3.1.78); and MPS IIID (OMIM: 252940, α-N-acetylglucosamine-6-sulfatase, GNS; EC 3.1.6.14) 1. MPS IIID (also known as Sanfilippo syndrome type D) was first characterized in 1980 2 and is one of the rarest autosomal recessive MPS disorders with an estimated prevalence of 0.02-0.10 per 100,000 live births worldwide 3. Due to deficiency of GNS, patients with MPS IIID accumulate heparan sulfate within lysosomes, triggering autophagy and cell death 4. All MPS III subtypes result in childhood neurodegeneration and premature death 5. Additionally, affected individuals can manifest severe behavioral problems, poor sleep, seizures and loss of ability to walk 1. Unlike some other MPS types, children impacted by MPS III do not manifest significant symptoms early on or develop developmental problems until later in childhood 6.
Despite more than 45 years of efforts to develop treatments for MPS III 5, 7-10, there are as yet no approved therapies. However, preclinical studies in MPS III animal models show promise that restoration of enzymatic activity and GAG catabolism can improve the phenotype.
Previous studies that administrated of a NAGLU-IGF-II fusion protein ICV to adult MPS IIIB mice showed clearance of heparan sulfate and reduction of secondary lysosomal defects and neuropathology, which lasted for at least 28 days following the last dose of 4 treatments over 2 weeks 11. A clinical trial based on this study is underway 12. Aside from enzyme replacement therapy (ERT), there are several other alternative approaches under investigation for treatment of MPSIIIB. Adeno-associated viral gene therapy led to sustained NAGLU enzymatic activity in MPS IIIB mice 13, and combined intravenous and an intracisternal injection following intravenous infusion of mannitol increased lifespan and improved behavior in mice 14, 15. Bone marrow transplant and intracranial AAV2/5-NAGLU resulted in synergistic effects on hearing and CNS lysosomal inclusions but were antagonistic for motor function and lifespan, while gene therapy alone was the most efficacious for lifespan in MPS IIIB mice 16. In 2017 the results from a phase 1/2 clinical trial with intracerebral gene therapy for MPS IIIB using a rAAV2/5 vector suggested this was well tolerated with the best results obtained in the youngest of 7 patients 17.
Injection of rhSGSH into the CSF via the cerebellomedullary cistern resulted in reduction of heparan sulfate, reduced lysosomal vesicle formation, and improved behavior in MPS IIIA mice 18. In addition, MPS IIIA mice dosed with rhSGSH via several routes of administration into the CSF (intrathecal lumbar, cisternal and ventricular) suggested ventricular and cisternal injection most effectively delivered enzyme to brain and spinal cord regions. Ventricular delivery of rhSGSH decreased heparan sulfate levels and reduces microglial activation 19. Delivery of rhSGSH to the MPS IIIA dog model via slow infusion produced significant reductions in heparan sulfate 20. In 2016, a phase 1/2 clinical study of intrathecal recombinant human heparan-N-sulfatase (rhHNS) was well tolerated and declines in a CSF heparan sulfate was observed in patients 21. In a less invasive approach, IV administration of a rhSGSH fused with the IgG domain of a monoclonal antibody against the human insulin receptor derived in a rhesus monkey has been shown to reduce sulfate incorporation into GAGs and uptake of rhSGSH into cells 22.
Gene therapy for MPS IIIA using a retroviral vector was able to transduce and correct MPS IIIA fibroblasts resulting in phenotypic correction 23. Intracerebral delivery of AAV2/5-CMV-SGSH-IRES-SUMF1 vector resulted in reduction of lysosomal storage, inflammatory regions, and improved motor and cognitive functions 24. Hydrodynamic delivery of SGSH encoding the vector pFAR4 into MPSIIIA mice led to high serum levels of the protein and reduced heparan sulfate 25. Intraparenchymal administration of AAVrh10-SGSH-IRES-SUMF1 vector into MPS IIIA mice improved heparan sulfate catabolism in regions only near the site of injection 26. Another approach such as siRNA has been used to silence 4 genes whose products are involved in GAG synthesis in MPS IIIA fibroblasts 27. Efforts to correct MPS IIIC are limited because the deficient enzyme is membrane bound. However, gene therapy with a novel AAV-TT vector corrected the neurological phenotype in MPS IIIC mice 28.
As described above, there are multiple approaches currently in development to treat lysosomal storage disorders, the most prevalent approach amongst approved treatments is ERT. The ERT approach administers the deficient enzyme, often from a recombinant source, to correct the metabolic deficiency. This therapeutic approach has had considerable success in several forms of MPS 29, 30, as well as Gaucher 31, 32, Pompe 33, Fabry diseases 34 and Batten disease 35. The success of ERT relies on the cellular uptake of the exogenous protein and transport to the lysosomal compartment, which occurs primarily via the 300 kDa, cation-independent M6P receptor. The M6P receptor is expressed throughout the CNS in addition to several other cell types 36 and has been shown to bind at the cell surface to multiple ligands, including M6P-containing lysosomal enzymes. Therefore, the presence of glycosylation rich M6P has been shown to be critical to the success of a candidate enzyme for ERT 37.
ICV is a well-tolerated and long-term approach for the administration of drug directly into the CNS 38-40. Even so, there are concerns about safety and patient compliance 41. To address these issues, several non- and minimally-invasive CNS drug delivery systems via trans-nasal or trans-vascular are under development 42. A therapeutic protein fused with a ligand or antibody that targets receptors at the blood-brain barrier (BBB) is another potential route for brain drug delivery 43-45. Target-based nanoparticles are another way to deliver therapeutic proteins cross BBB 46. Overall, there are multiple alternative drug targeting or delivery systems that can be developed to treat MPS III with ERT.
The post-translational signal for mannose 6-phosphorylation during transport through the cis-Golgi network remains to be elucidated. However, the utilization of mammalian expression systems, such as Chinese hamster ovary (CHO) cells, has been shown to produce M6P modification of recombinant lysosomal enzymes and thereby improving uptake by cells 37. GNS has been found to be glycosylated with M6P 47, enter cells efficiently, and reduce lysosomal storage in vitro 48. An early study of recombinant caprine GNS in a single MPS IIID goat found that the intravenously-administered enzyme reduced liver heparan sulfate accumulation 49. In addition, an adeno-associated viral GNS gene therapy delivered into CSF corrected pathological storage, improved lysosomal functionality in the CNS and somatic tissues, resolved neuroinflammation, restored normal behavior and extended lifespan of treated MPS IIID mice 50. Overall, these studies strongly support delivery of functional GNS to MPS IIID patients as a potential treatment. Both gene therapy delivering DNA and ERT delivering a recombinant protein are critical options for patients. Here, we developed a proof-of-concept ERT to treat MPS IIID patients.
In this study, recombinant human GNS (rhGNS) was expressed and purified from CHO cells, characterized to determine glycosylation and M6P content, and shown to be enzymatically active using two optimized in vitro assays. Delivery of rhGNS to MPS IIID patient fibroblasts demonstrated intracellular uptake and reduction of GAG storage in vitro. In MPS IIID knock-out mouse model we demonstrated in vivo normalization of brain GNS, reduction of accumulated heparan sulfate, and lysosomal localization of rhGNS throughout the brain.
EXPERIMENTAL PROCEDURE
1. Molecular cloning of rhGNS
An expression cassette containing the full-length of human GNS cDNA coding for the 552 amino acids (NM_002076.3), a short unstructured (GGGGS)4 linker, and c-myc tag (EQKLISEED) was synthesized using codon optimization in Chinese hamster ovary (CHO) cells by Genscript USA Inc. (Piscataway, NJ). The C-terminal c-myc tag allows affinity purification using anti-myc agarose. The expression cassette was further subcloned into an expression vector pCI-Neo driven by a CMV promoter (Promega Corporation, Madison, WI). We introduced a TEV protease cleavage site with a longer linker in the C-terminus of this construct using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Inc., Santa Clara, CA) with primers 1 and 2 listed in Table S1. The final cDNA sequence was confirmed by Sanger-based sequencing (Laragen, Inc., Culver City, CA). The G-418 selection rhGNS plasmid number is TCB469 (Table S2) for which a map is shown in Figure S1A.
We subcloned rhGNS into the pOptiVEC™-TOPO® vector using the pOptiVEC™-TOPO™ TA Cloning™ Kit (Gibco, Cat # 12744017) with primers 3 and 4 listed in Table S1. Sequencing Primers 5 to 11 listed in Table S1 were used, and the final cDNA sequence was confirmed by Sanger-based sequencing (Laragen, Inc., Culver City, CA). The methotrexate selection rhGNS plasmid number is TCB616 (Table S2) and a map is illustrated in Figure S3A.
2. Cell culture and CHO cell expression lines
CHO-K1 (American Type Culture Collection [ATCC], Manassas, VA) cells were cultured in Ham’s F12/DMEM, which contains glutamine (Corning Incorporated, Corning, NY) supplemented with 10% fetal bovine serum, 100 units/mL Penicillin and 100 μg/mL Streptomycin (Lonza, Walkersville, MD) at 37°C with 5% CO2. The first rhGNS expression vector pCI-Neo (Figure S1 A) was transfected into CHO-K1 cells using BioT Transfection Reagent (Bioland Scientific, Paramount, CA), and transfected cells were selected by 700 μg/ml G418 (Sigma-Aldrich, St. Louis, MO) for 14 days. Transfected cells were dissociated by trypsin and stained by propidium iodide (Thermo Scientific, Waltham, MA) for viability and sorted by FACSAria III (BD Biosciences, Franklin Lakes, NJ) with gating based on cell viability (negative for propidium iodide staining). Single cells were isolated into a 96-well plate and cultured with media containing 700 μg/mL G418 until colonies were formed. Stable CHO cell clones were maintained and expanded in culture media with 300 μg/mL G418.
Single colonies were cultured in protein expression media (HyClone serum-free PF CHO media [GE Healthcare Life Sciences, Logan, UT] supplemented with 4 mM L-glutamine, 200 μg/mL G418, and nucleosides [10 mg/L each of adenosine, cytosine, guanosine, hypoxanthine, thymidine, and uridine]). Secreted culture media were used for initial screening by GNS enzyme activity and western blotting with anti c-myc antibody (Thermo Scientific, Waltham, MA).
For methotrexate (MTX) selection, the second rhGNS expression vector pOptiVEC™ TOPO (Figure S3A) was transfected into CHO-K1 cells using BioT Transfection Reagent (Bioland Scientific, Paramount, CA). Forty-eight hours after transfection, cells were replated and treated with 100 nM of MTX. MTX-resistant clones formed after 6 weeks under MTX treatment and clones were picked up with Cloning Cylinders (Bel-Art™, Cat # F378470100).
For protein production, stable clones were inoculated into roller bottles (Thermo Scientific, Waltham, MA) and grown until confluence, at which time the medium was replaced with protein expression media. Secreted rhGNS was monitored daily by both GNS assay and western blot until full-length GNS expression reached a plateau (day 10-15) before media were harvested for rhGNS purification.
3. Purification of rhGNS-myc protein
Following harvest, conditioned media were concentrated using Vivaflow® Crossflow devices with a molecular weight cut-off of 50 kDa (Sartorius Corporation, Bohemia, NY) to 10 % of original volume. The concentrated media was desalted/buffer-exchanged to artificial CSF (150 mM NaCl, 0.8 mM MgSO4, 3 mM KCl, 1.4 mM CaCl2, 0.8 mM Na2HPO4, and 0.2 mM NaH2PO4) at 4°C. Approximately 50 mL of concentrated media was mixed with 500 μL of c-myc affinity beads (50% slurry; Medical & Biological Laboratories Co., Ltd., Nagoya, Japan) and incubated overnight at 4°C under rotation.
The slurry was then loaded onto a 25 mL chromatography column for rhGNS purification. The flow-through was collected and loaded back into the column one more time. The column was washed with artificial CSF (10 mL x 3 times). Bound rhGNS was eluted using 3 mL of 0.2 mg/mL c-myc peptide (Medical & Biological Laboratories Co., Ltd., Nagoya, Japan) and concentrated to a final concentration of approximately 1 mg/mL protein by Microcon 50,000 MWCO Centrifugal Filter (Merck Millipore, Billerica, MA).
4. Enzymatic activity assays for rhGNS
A two-step fluorometric measurement of GNS activity was performed as described previously 51 with modifications. Media or purified enzyme (2.5 μL) was incubated with 2.5 μL of 10 mM 4-methylumbelliferyl alpha-N-acetylglucosaminide-6-sulfate (4-MUGNS; Toronto Research Chemicals, Toronto, Canada) in the reaction buffer (0.2 M sodium acetate, pH 5.6; 20 mM lead acetate, 0.01% Triton X-100) at 37°C for 1 hour, and the reaction was then stopped by addition of 10 μL of phosphate-citrate buffer (0.4 M Na2HPO4 / 0.2 M citric-acid buffer, pH 4.7). To release 4-MU fluorophore from the reaction mixture, 5 μL of concentrated rhNAGLU-IGFII conditioned medium (specific activity > 100 nmol/hr/μL) or 5 μL of purified, myc-tagged rhNAGLU-IGFII was added and incubated for 2 hours at 37°C. Reactions were quenched by the addition of 50 μL of glycine carbonate buffer (pH 10.5). Fluorescence measurements were obtained using a SpectraMax iD5 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA) at excitation and emission wavelengths of 360 nm and 450 nm, respectively. One activity unit of GNS was defined as 1 nmol of 4-MU converted substrate per hour (first step) at 37 °C. Protein concentration was determined using Bradford Reagent (Bio-Rad Laboratories, Inc, Irvine, CA). Specific activity was normalized with the protein concentration and presented in units/mg protein. Commercial rhGNS (R&D Systems; Minneapolis, MN) was used as a positive control, and 4-MU standard curves were present on all readings to account for differences in read conditions between assays. In addition to the two-step fluorescence assay, we also used a colorimetric sulfatase assay that directly measures GNS activity using para-nitrocatechol sulfate as a substrate. The detailed assay optimization is described in the supplemental method.
5. Glycosylation analysis
Deglycosylation of rhGNS was performed with two different enzymes: PNGase F and Endo H (New England BioLabs, Ipswich, MA). Approximately 3.5 μg of purified rhGNS underwent glycosidase digestion with either enzyme for one hour at 37°C, according to manufacturer instructions. The digestion was then visualized via SDS-PAGE, followed by staining with Imperial™ Protein Stain or further by immunostaining with anti-c-myc antibody, as described in the supplemental method sections.
6. Composition analysis of N-glycan isolated from rhGNS sample using HPAEC-PAD analysis
HPAEC-PAD analysis was performed at GlycoAnalytics (UCSD). Purified rhGNS (100 μg) was used for N-glycan isolation using PNGase F, followed by purification of N-glycans using SPE extraction. Purified N-glycan was hydrolyzed using 2 N trifluoroacetic acid at 100°C for 4 h, the acid removed by dry nitrogen flush, and monosaccharides were finally dissolved in milli-Q water and injected on HPAEC-PAD. A Carbo-Pac PA-1 column (4 x 250 mm) was used with NaOH-NaOAc gradient as a mobile phase for separation. A standard M6P was used to quantify and assign the retention time.
7. Steady-state kinetic analysis
A Michaelis-Menten kinetics curve was generated when 3 ng/mL rhGNS was incubated for 1 hour at 37 °C at 8 different concentrations of 4-MUGNS ranging from 0.2 to 25 mM with a 4 h 4-MU release step under standard reaction conditions. All calculations of Vmax, Kcat, and Km were made using GraphPad Prism 7.0 (GraphPad, Inc., La Jolla, CA) by fitting data to Equation 1.
| Equation (1) |
In order to determine the optimal GNS activity over a range of pH, we use three buffer conditions to cover the range of pH values (pH 3.8 to 5.8 using acetate/acetic acid buffer (pKa = 4.76) and pH 6.2 to 7.6 using HEPES buffer (pKa = 7.5). rhGNS (2.5 μL of 3 ng/uL) was incubated with 10 mM 2.5 μL of 4-MUGNS (10 mM made with at different pH value in proper buffer at 37°C for 1 h. The remaining steps are described above.
8. Enzyme Uptake in MPS IIID fibroblasts
Four fibroblast cell lines used in this study were purchased from the Coriell Institute (Camden, NJ). GM05093 is homozygous for a C>T transition at nucleotide 1063 in exon 9 of the GNS gene (1063C>T) resulting in the substitution of a termination signal for arginine at codon 355 [Arg355Ter (R355X)]. The genotype of GM17495 is not available from Coriell Institute. We verified there is no GNS activity present in both GM05093 and GM17495. Intracellular rhGNS enzyme uptake was determined using fully-confluent MPS IIID human skin fibroblasts (GM17495) in DMEM supplemented with 1% L-Glutamine, 1% Penicillin Streptomycin solution, and treated with purified rhGNS at 37°C for 2.5 or 5 hours. Cells were then washed with PBS and harvested using TrypLE Select (Thermo Scientific, Waltham, MA), and cell pellets were further washed with artificial CSF buffer before lysing in GNS Lysis buffer (GNS reaction buffer supplemented with 0.1% Triton X-100 and 0.1 mM DTT) by vigorous pipetting followed by incubation on ice for 20 minutes. Samples were centrifuged at 10,000 g for 15 minutes, and supernatants were recovered for GNS activity assay as described above. Intracellular GNS enzyme activity was normalized to the protein concentration from the lysed pellet.
Uptake inhibition was performed by pre-incubating MPS IIID human skin fibroblasts (GM17495) with 10 mM M6P (Sigma-Aldrich, St. Louis, MO) for 10 minutes before applying an equal volume of medium containing 3.9x103 units/mL purified rhGNS for 5 hours at 37 °C, reaching a final concentration of 1.95x103 units/mL for rhGNS and 5 mM for M6P. Following treatment, intracellular GNS activity in treated cells was measured as described above.
9. Intracellular GAG quantification
GAG in cultured MPS IIID human skin fibroblasts (GM05093 and GM17495) and control skin fibroblasts (IMR90 and GM01392) was measured by labeling newly synthesized GAG with H235SO4 as described previously with minor modifications 52, 53. In brief, cells were grown to confluence in 6-well plates supplemented with 25 μCi/mL H235SO4 (Perkin-Elmer, Waltham, MA) in the presence of purified 2 units/mL rhGNS at 37°C and 5% CO2. After 72 hours of labeling, cells were washed and then harvested by trypsinization and centrifugation. Intracellular GAG was extracted from cell pellets by re-suspending and boiling in 85% ethanol supplemented with 1μg/mL glycogen. The extracted GAG was centrifuged and dissolved in 10% sodium hydroxide and neutralized with 2 M acetic acid. Radiolabeled GAG was measured via scintillation counting (Tri-Carb 2800 TR, Perkin-Elmer, Waltham, MA). Radioactive counts per minute were normalized to protein concentration. The estimate of Kcorrection for GAG reduction was further performed with different concentrations (0, 0.0011, 0.0054, 0.011, 0.054 and 0.27 units/mL) of rhGNS with MPS IIID human skin fibroblasts (GM17495) as described above. Data were normalized and presented as percentage of cpm/mg protein compared to untreated MPS IIID cells and were best fit to an exponential decay using GraphPad Prism 7.0. The half-maximal concentration for correction was calculated using the pharmacokinetics module (IC50) in GraphPad Prism 7.0.
10. In vivo Study using MPS IIID constitutive knockout mouse
Animal experiments were approved by the Institutional Animal Care and Use Committee at the Lundquist Institute (formally Los Angeles Biomedical Research Institute) at Harbor-UCLA Medical Center, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
A MPS IIID constitutive knockout mouse model was generated by Taconic Biosciences (Rensselaer, NY) and maintained on an inbred background (C57BL/6), which has been shown to display deficiency of GNS activity and accumulates lysosomal storage 54. Mutant male MPS IIID mice (Gns−/−) were mated with heterozygous (Gns+/−) females to obtained homozygous affected mice and heterozygous controls. Genotype was determined with MPS IIID primers 12 to 14 listed in Table S1.
Neonatal mice (both genders) were toe-clipped and genotyped at birth. Sterile-filtered (0.2 μm), purified rhGNS (5 μL; 13.4 units/μL or 40.2 units /μL) or vehicle (5 μL) was injected in the left lateral ventricle as previously described 55. The injection site was ~2.5 mm between the bregma and eyes, 2 mm away laterally from the sagittal suture, and 2.5 mm in depth.
We harvested brains from MPS IIID mutant (Gns−/−) mice treated with or without rhGNS along with age-matched carrier control (Gns+/−) mice. The lysosome fractions from mouse brains were enriched using the Lysosome Enrichment Kit for Tissue and Cultured Cells (Thermo Scientific, Waltham, MA) according to the manufacturer’s instructions. Isolated lysosomes were lysed in GNS lysis buffer for the enzyme activity assays and western blots
11. Biochemical evaluations in MPS IIID mice with rhGNS ERT
Tissues were homogenized using a bullet blender (Next Advance, Inc., Troy, NY) with GNS assay buffer containing 0.1% TritonX-100. Homogenized tissues were assayed for GNS and β-hexosaminidase activities. The catalytic activity of GNS was described above, and β-hexosaminidase (combined A and B isoforms) was determined by hydrolysis of 4-methylumbelliferyl-N-acetyl-β-glucosaminide (EMD Millipore, Burlington, MA) using 1.25 mM substrate in the incubation mixture.
12. Brain heparan sulfate detection
This experiment was performed at GlycoAnalytics Core (UCSD) as described previously 56. Mouse brains were homogenized and digested overnight with Pronase (Sigma-Aldrich) in phosphate-buffered at 37°C and centrifuged at 14,000 rpm for 20 min. The supernatant was passed through a DEAE column and the bound GAG was eluted with 2 M NaCl. The GAG was then desalted on PD10-size exclusion column and purified GAG was lyophilized. Dried GAG disaccharides were dissolved in aniline and reacted with freshly prepared sodium cyanoborohydride solution in dimethyl sulfoxide: acetic acid mixture (7:3, v/v). Reactions were carried out at 65°C for 1 h followed by 37°C for 16 h. The samples were then dried using speed vacuum at room temperature and used for further analysis by glycan reductive isotope labeling (GRIL) LTQ-MS. Aniline-tagged disaccharides were separated on a C18 column using an ion pairing solvent mixture and analyzed by mass spectrometry in negative ion mode.
13. Statistical analysis
Experiments were performed in triplicate, and data are shown as mean ± SD. Statistical comparisons were made using one-way analysis of variance (ANOVA) for the mouse study.
Additional experimental details can be found in the supplemental method sections.
RESULTS
1. Expression and characterization of rhGNS-myc from CHO-K1 cell line using G418 selection
Different organisms exhibit bias towards use of certain codons that encode the same amino acid; such bias can significantly impact recombinant protein expression. To increase the protein level we used GenScript Condon Optimization algorithm to optimize GNS codon for expressing in CHO cells 57. Drug resistance strategies are a highly efficient and less time consuming method to generate stable recombinant protein expression cell lines 58. To determine whether we can purify rhGNS using the codon optimized sequence in CHO cells and to characterize rhGNS in a shorter time frame, pCI-Neo was used as the expression vector (Figure S1A). Fifty G418-resistant clones were isolated and 12 were positive for rhGNS secretion (Figure S1B). Clone TCB469 B2 was used for subsequent purification. A continuous increase of GNS activity in cell culture supernatant was observed over 11 days (Figure S1C). Supernatant was harvested and concentrated at day 15, and analyzed by SDS-PAGE (Figure S1D). After affinity purification using myc-agarose (Table S3), a western blot assay was performed using both anti-GNS and anti-myc antibody (Figure S2A) to identify rhGNS-myc. Commercial rhGNS proteins are a mixture of two proteins, Val37-Leu552 and Thr44-Leu552, both with a C-terminal 10-His tag purified from CHO cells. Due to difference in amino acid sequence and the affinity tag, R&D GNS is slightly higher molecular weight on the western blot (Figure S2A). To confirm identify of rhGNS and detect impurity, trypsin digested purified rhGNS was analyzed by LC/MS/MS and successfully mapped to human GNS (Table S4). The SDS-PAGE showed a band with the expected molecular weight (80 kDa) as detected in western blot (Figure 1A). The purity of rhGNS was estimated to be 49% based on the quantification of band area on the gel using Image Lab (Table S5); two major contaminant proteins are Histone H4 and Isoform 3 of T-complex protein 1 subunit eta (CCT7) (Table S4).
Figure 1.

Identification and glycosylation of rhGNS. A) SDS-PAGE of purified rhGNS. B) Western blots of purified rhGNS treated by glycosidase PNGase F and Endo H using anti-myc antibody (a: c-myc). The extra bands in lane 1 are the glycosylated forms of rhGNS; the extra bands in lanes 2 and 3 are incomplete de-glycosylated forms of rhGNS. C) Composition of N-glycans isolated from purified rhGNS using HPAEC-PAD analysis. Fuc: fucose; GlcNH2: glucosamine; Gal: galactose; Glc: glucose; Man: mannose; Man-6-P: M6P.
The protein secreted from CHO cells is approximately 80 kDa as determined by SDS-PAGE potentially from N-glycosylation at one or more of the 13 potential N-glycosylation sites (Figure 1A) 59. To understand the glycosylation pattern of rhGNS, the purified enzyme was treated with endoglycosidases PNGase F or Endo H to remove attached carbohydrate molecules. PNGase F hydrolyzes almost all types of N-linked oligosaccharides, whereas Endo H cleaves mannose rich N-linked oligosaccharides within the chitobiose core 60. The rhGNS was sensitive to digestion with both PNGase F and Endo H, producing a molecular weight shift from 80 kDa to 58 kDa, which suggests that the protein was N-glycosylated with high-mannose residues (Figure 1B). Additionally, high performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD) showed the secreted rhGNS was mannose-6 phosphorylated at approximately 0.87% (W/W) (Figure 1C) 61. A native polyacrylamide gel electrophoresis was performed to resolve rhGNS in a non-denaturing environment; the molecular weight was estimated to be ~160-200 kDa, indicating a dimeric structure (Figure S2B).
2. Biochemical characterization of purified rhGNS-myc protein
To characterize the enzymatic activity of rhGNS, we optimized a two-step GNS activity assay which used 4-MUGNS as a specific GNS substrate 51. This is a coupled assay that requires a subsequent enzyme, NAGLU to release the fluorogenic product, 4-MU, and we optimized this to a 384-well plate format. We then determined the steady state kinetic constants of rhGNS by performing a GNS activity assay with increasing amounts of 4-MUGNS and generated a Michaelis-Menten kinetics curve (Figure 2A). The calculated Km was 4.0 mM, and the estimated kcat was determined to be 1.68x104 units/mg (1 unit = 1 nmol converted substrate per h at 37°C).
Figure 2.
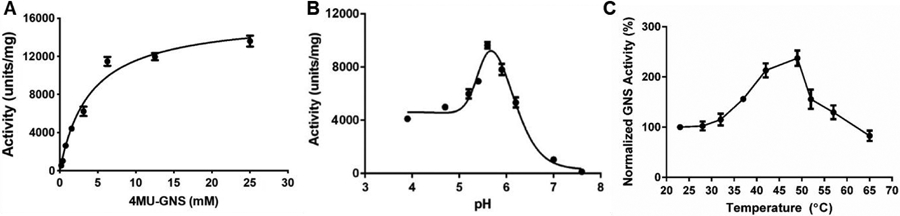
Biochemical characterization of purified rhGNS. A) rhGNS activity towards the fluorogenic substrate 4-MUGNS was measured at increasing substrate concentrations. B) GNS assay was performed from pH 3.8-5.8 (Acetate/Acetic Acid Buffer) and 6.2-7.6 (HEPES Buffer) at 37°C to assess the optimal pH range. C) Purified rhGNS activity was assessed at different temperatures to assess its thermal stability (GNS activity is normalized to 24°C). Units represent nmol of 4-MU released by rhGNS in an hour (nmol/h). The entire experiment was performed in triplicates, and data was shown as mean ± SD.
We also performed pH rate profiling to determine how pH affects enzymatic activity of rhGNS. Consistent with the acidic pH in lysosome, the purified rhGNS in this study exhibited a peak activity at pH 5.6, with GNS enzymatic activity at neutral pH 7 approximately 10-fold lower than at the pH 5.6 (Figure 2 B). To avoid the pH effect on the step 2 NAGLU enzyme and subsequently have an effect on the final 4MU release, we also performed a colorimetric sulfatase activity assay using para-nitrocatechol sulfate as the substrate and observed similar optimal pH as the GNS activity assay (Figure S2C). The subsequent GNS activity assay in this study was performed with 5 mM of 4-MUGNS, which is close to Km and at the optimal pH of 5.6. Under this condition, the specific activity of rhGNS purified from G418 selected CHO cells stably expressed rhGNS was 1.34x104 units/mg. We estimated the titer as 0.4 mg/L (Table S3).
To determine how sensitive rhGNS is to temperature, we assayed rhGNS over a range of temperatures (24 to 65°C) and found rhGNS was active at wide range of temperatures. GNS activity was 1.5-fold at 37°C compared to 24°C and was almost double at 49°C compared to room temperature. Overall, GNS exhibited good enzymatic activity at body temperature (37°C) and remained active up to 65°C (Figure 2C).
Artificial CSF is the most common buffer formulation used in FDA approved drugs for brain injection. Therefore, we have chosen to determine the stability of rhGNS in artificial CSF by using the thermal shift assay, which utilizes intrinsic tryptophan fluorescence to measure the inflection temperature (Ti) of rhGNS at 83.6°C (Figure 3A). In addition, the GNS activity assay also showed that purified rhGNS was active for one month at 4°C in artificial CSF, with greater than 80% of initial enzyme activity remaining (Figure 3B). Both results indicated rhGNS was stable in artificial CSF.
Figure 3.
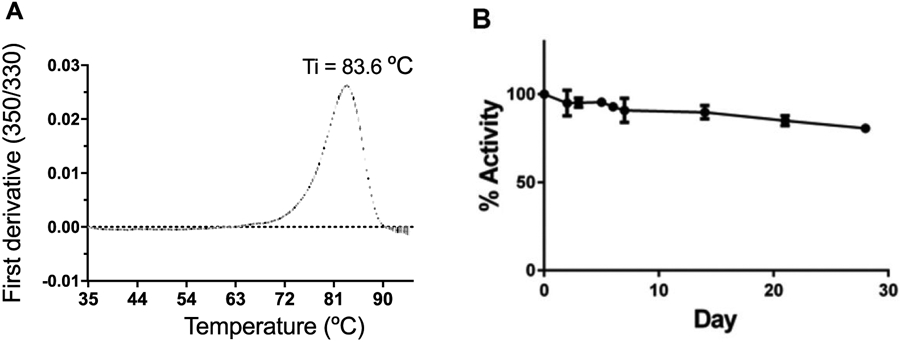
Stability of rhGNS in artificial CSF buffer. A) Thermal stability of rhGNS in artificial CSF was detected utilizing intrinsic tryptophan fluorescence. B) Purified rhGNS was stored in artificial CSF at 4°C and activity was measured over a 28-day period. Means (black circle) and standard deviation of three independent experiments are shown for each figure. Activity assay was performed in triplicates and data is shown as mean ± SD.
3. Enhanced expression of rhGNS-myc from CHO-K1 cell line utilizing dihydrofolate reductase (dhfr) gene amplification using methotrexate (MTX) selection
Large-scale recombinant protein production is crucial for biopharmaceutics development since both efficacy and safety evaluation demand large amounts of protein. In addition, industrial manufacturing processes of biopharmaceuticals are dependent on high-yielding cell-based production platforms in order to maximize production yields but also to reduce associated costs 62. The factors which affect the productivity of recombinant proteins include cell lines, expression constructs, gene optimization, selection methods and cell culture conditions 58.
Our initial results demonstrated that the codon optimized sequence encoding rhGNS was able to express in CHO cells but had low productivity. The yield was 0.16 mg with specific activity of 1.34x104 units/mg and purity of 49% when rhGNS was purified from the G418 selected CHO cells stably expressing rhGNS. To improve the productivity, rhGNS encoding sequence was cloned into pOptiVEC™-TOPO vector (TCB616, Figure S3A), which utilizes dihydrofolate reductase (dhfr) gene amplification using methotrexate (MTX) selection. The highest expression clone C2 was identified from screening 13 clones and used for subsequent purification (Figure S3B). Cell culture supernatant was harvested at day 6 when the increase of total GNS activity reached plateau (Figure S3C). Compared to TCB469 B2, the amount of rhGNS secreted from TCB616 C2 increased 100-fold (Figure 4A), and the GNS specific activity of cell culture supernatant increased 28-fold (Figure 4B). After anti-myc affinity purification, the GNS specific activity was 3.9x104 units/mg, which was 4.5-fold higher than that of cell culture supernatant (Table S6).
Figure 4.
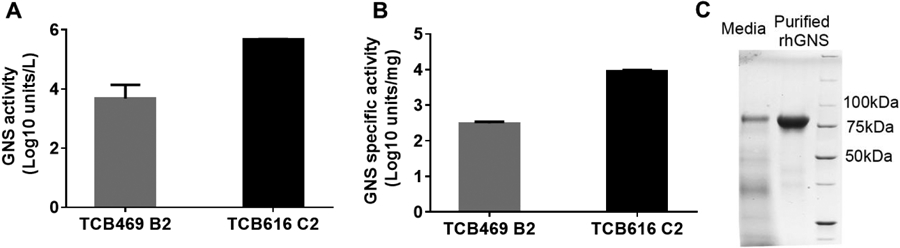
Overexpression and purification of rhGNS utilizing the dhfr gene amplification system. A) The total amount of rhGNS secreted from CHO cells. B) GNS specific activity of secreted rhGNS. C) SDS-PAGE of rhGNS in media and in the myc peptide eluted fraction. In addition to the rhGNS band, the other bands detected in the media sample were CHO proteins released to the media. All activity assays were performed in triplicate, and data are shown as mean ± SD.
The percentage of secreted rhGNS in media was 22% or 28% of total proteins estimated from specific activity measurement and Western blot quantification, respectively (Figure S3D). The titer of rhGNS secreted from TCB616 C2 was 26 mg/L, estimated from GNS specific activity, and this is a 65-fold increase over the G418 selected clone (0.4 mg/L, TCB469 B2). From the SDS-PAGE gel, we observed a prominent band at molecule weight of 80 kDa in cell culture supernatant from TCB616 C2 (Figure 4C) but not from TCB469 B2 (Figure S1D). This observation was consistent with a higher level of rhGNS produced in media. The purity of rhGNS was 95.2% quantified by SDS-PAGE (Figure 4C), 99% by size exclusion chromatography (Figure S3E), and 98% by LC/MS/MS analysis (Table S7); this purity was almost 2-fold higher than rhGNS purified from TCB469 B2 (Figure 1A). The improved purity of rhGNS purified from TCB616 C2 explained the higher specific activity as well as kcat (Figure S3F). In addition, the Km, optimal pH, temperature effect, and glycosylation pattern of rhGNS purified from TCB616 C2 were similar to rhGNS purified from TCB469 B2 (Figure S3G to J). These data also suggested that the impurity from previous purification does not affect the biochemical profile of rhGNS and only affects the specific activity and kcat due to the difference of 49% versus 95% purity. The high inflection temperature (83.7°C) indicated the high thermal stability and rigid conformation of rhGNS. This stability indicated that higher temperature might promote the conformational change of rhGNS to bind substrate or release product and therefore, the optimal temperature of its enzymatic activity was at 50°C. Overall, our results indicated that utilizing the dhfr gene amplification system dramatically increased the productivity of rhGNS with a similar biochemical profile.
4. rhGNS entered into MPS IIID fibroblasts by M6P receptors and cleared accumulated GAGs
The cellular uptake of lysosomal enzymes is thought to primarily rely on cell-surface receptors recognizing N-glycan features, including the M6P receptors (MPRs), Ashwell–Morell receptor (AMR), and mannose receptor (MR) 63. We have identified rhGNS was N-glycosylated and mannose-6 phosphorylated as described above. To determine the cellular uptake of rhGNS, MPS IIID patient fibroblasts were incubated for 2.5 or 5 hours with 3.9x103 units/mL of rhGNS, and the intracellular rhGNS activity was measured. The intracellular rhGNS activity detected in the 5-hour treatment group was almost 2-fold higher than that of the 2.5-hour group (Figure 5A), which indicated that the uptake of rhGNS by fibroblasts was time-dependent. When we treated fibroblasts with a different concentration of rhGNS for 5 hours (Figure 5B), the amount of rhGNS taken up by fibroblasts increased dose-dependently. The uptake of rhGNS into MPS IIID human fibroblasts was inhibited 72% by the addition of M6P at the final concentration of 5 mM, thus suggesting that the majority of intracellular rhGNS uptake is mediated by the M6P receptor.
Figure 5.
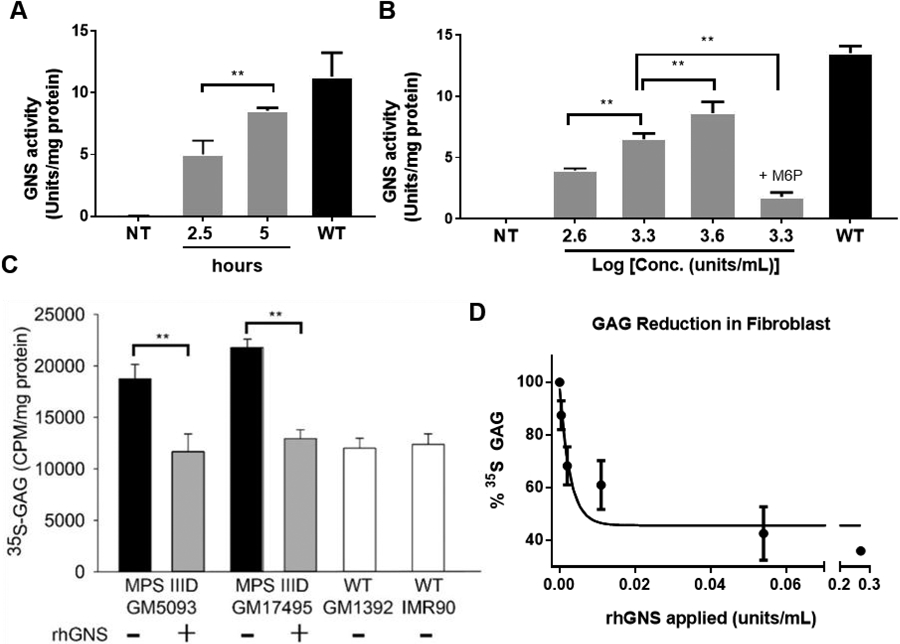
rhGNS enters and reduces GAG accumulation in human MPS IIID fibroblasts. A) Cellular uptake of rhGNS in vitro. MPS IIID human fibroblasts GM05093 were seeded in 6-well plates. Purified rhGNS was applied and incubated with cells for 2.5 h or 5 h at 37°C. B) Three amounts of purified rhGNS were applied to GM05093 cells for 5 h at 37°C with and without M6P. NT: not treated; M6P: 5 mM M6P. C) 35S-GAG assay. Two human MPS IIID patient fibroblast lines (GM05093 and GM17495) and two normal human fibroblast lines (IMR90 and GM1392) were labeled with 25 μCi/ml H235SO4 in a culture medium without serum for 72 hours at 37°C. Purified rhGNS (2 units/mL) was then added to experimental wells and intracellular counts per minute (CPM) normalized to intracellular protein concentration. D) Dose-response study of 35S-GAG incorporation in MPS IIID fibroblasts (GM17495). Radioactive counts per minute were normalized to protein concentration and expressed as a percent of untreated MPS IIID fibroblasts (“% GAG Storage”). Kcorrection was calculated using exponential decay with a bottom of 40% (equal to WT levels). Experiments were performed in triplicate, and data are shown as mean ± SD. Statistical analysis was performed using one-way ANOVA. ** p < 0.01.
We next assessed whether the rhGNS taken up by the MPS IIID fibroblasts was able to catabolize intracellular GAG using a 35S incorporation assay. Two different MPS IIID patients’ fibroblast cell lines, GM05093 and GM17495, accumulated consistently higher levels of 35S-labeled GAGs after 72-hour labelling, as compared to wild-type cell lines, IMR90 and GM01392. In the presence of 2 units/mL purified rhGNS during labelling, the total intracellular 35S-labeled GAGs in these MPS IIID lines were restored to wild-type levels (Figure 5C). We further titrated rhGNS in the labelling media and observed that a level as low as 0.01 units/mL rhGNS was sufficient to reduce GAG storage to levels similar to wild-type. The calculated concentration required for half-maximal correction was 0.005 units/mL (Figure 5D).
5. Intracerebroventricular (ICV) enzyme replacement in MPS IIID mice with rhGNS
Administering therapeutic enzyme directly into the left cerebral ventricle to bypass the blood brain barrier had been proven successful in an animal model of MPS IIIB 64. To assess its efficacy in vivo, we performed ICV injection of rhGNS into one lateral cerebral ventricle of newborn GNS knockout mice (Gns−/−) 54 (Figure 6A). A child with MPS III inherits two copies of the altered Gns gene, one from each parent. Both parents are non-affected carriers, who have one copy of the altered gene and one normal copy of WT gene. Therefore, we used carriers (Gns+/−) as positive control in our in vivo experiments. We detected a statistically significant return of brain GNS activity to 100% of the carrier levels 24 hours after single dosing with 5μL of 67 units purified rhGNS (Figure 6B). To assess the bio-distribution of rhGNS in the brain, we treated a separate cohort of MPS IIID mice as described above. After harvesting the tissue, we sliced the brain into 3 sections along the rostrocaudal axis for biochemical assays separately (Figure 6A). We were able to detect GNS activity throughout the neonatal brain in all 3 sections at 24 h post-injection (Figure 6C), which indicated that rhGNS spread throughout the brain from the injection site. To estimate the in vivo half-life of rhGNS in the brain, we measured GNS activity at 1, 2, and 3 days post-injection in MPS IIID mice, which resulted in a tissue half-life estimate of 1.1 days (Figure 6D).
Figure 6.
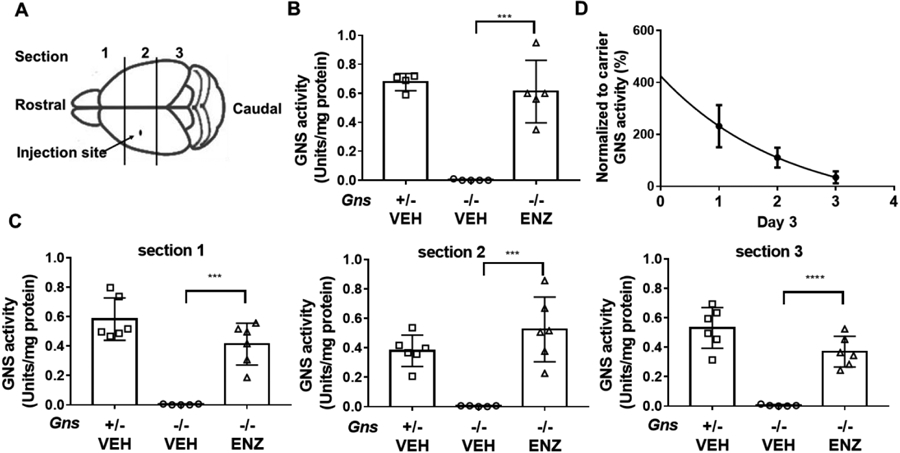
ICV injection of rhGNS in MPS IIID (Gns−/−) mice. A) Schematic of neonatal mouse brain showing the site of injection. B) GNS activity in mouse brain lysate 1 day after treatment, Gns+/− VEH (n=4), Gns−/− VEH (vehicle treated, n=5), Gns−/− ENZ (rhGNS treated, n=5). C) rhGNS distribution in 3 sections of the brain as determined by GNS activity in mouse brain lysate 1 day after treatment in 3 sections. Gns+/− VEH (n=6), Gns−/− VEH (n=5), Gns−/− ENZ (n=6.) D) Estimated half-life of rhGNS in the mouse brain. GNS activity in whole brain lysate 1, 2, and 3 days after treatment with a single dose of 201 units rhGNS delivered ICV at PND 2 of Gns−/− mice. Decay plot of GNS activity in treated mice to estimate the half-life of rhGNS, n= 6. Data was shown as mean ± SD, and individual animal means are indicated by symbols. Statistical analysis was performed using one-way ANOVA.*** p < 0.001; **** p < 0.0001.
To determine whether rhGNS reached lysosomes, we separated the lysosomal fraction from fresh brain homogenates of MPS IIID mice 1-day following ICV rhGNS, using a discontinuous density gradient centrifugation. Our western blot results showed increased relative level of lysosome marker-LAMP1 in lysosome-enriched fraction than in total lysate, which indicated the enrichment of lysosome (Figure 7A). Strong GNS bands detected in the lysosome-enriched fraction of the ERT group proved that rhGNS is targeted to the lysosomes. The relative GNS activity in the lysosome-enriched fraction was higher than levels in the crude lysate, also supporting lysosomal localization of rhGNS (Figure 7B). Interestingly, we detected stronger GNS signal by western blot but similar GNS activity in rhGNS treated mice compared to the carrier control (Gns+/−), which might due to difference of rhGNS and endogenous GNS in binding to anti-GNS antibody or their specific activity. Since the brain lysate and lysosome fraction were obtained 2 days after treatment with rhGNS, it could be the endogenous GAG in rhGNS-treated MPS IIID mice is inhibiting rhGNS activity in hydrolysing the 4-MUGNS artificial substrate.
Figure 7.
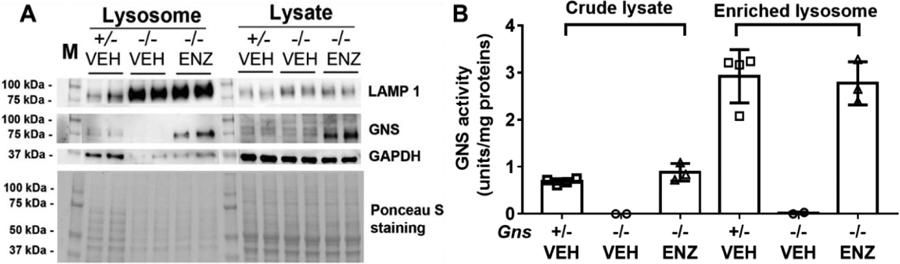
Lysosomal localization of rhGNS in the mouse brain. A) Western blot and Ponceau S staining of crude lysates and lysosome fraction of whole brain 1 day following a single dose of 201 units rhGNS administered into the lateral cerebral ventricle of 2-day-old mice. Lysosomal-associated membrane protein 1 (LAMP 1) was used as a marker for lysosome to confirm lysosomal enrichment, and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a marker for cytoplasm. B) GNS activity in crude lysates or lysosome-enriched fraction. Gns+/− VEH (n=4), Gns−/− VEH (n=2), Gns−/− ENZ (n=3). Data was shown as mean ± SD, and individual animal means are indicated by symbols.
Deficiency of GNS in the MPS IIID mouse brain led to the accumulation of heparan sulfate (Figure 8A) and was accompanied by increased activity of other lysosomal enzymes, including β-hexosaminidase (Figure 8B) as shown in other MPS III models 49, 50, 64. Seven days after the mice were treated with a single dose of ICV rhGNS, we still detected a significant decrease of heparan sulfate in the ERT group. In addition, we observed significantly lower β-hexosaminidase activity in brains of mice treated with a single dose of ICV rhGNS as compared to vehicle-treated control mice at 3 days and 7 days following injection.
Figure 8.
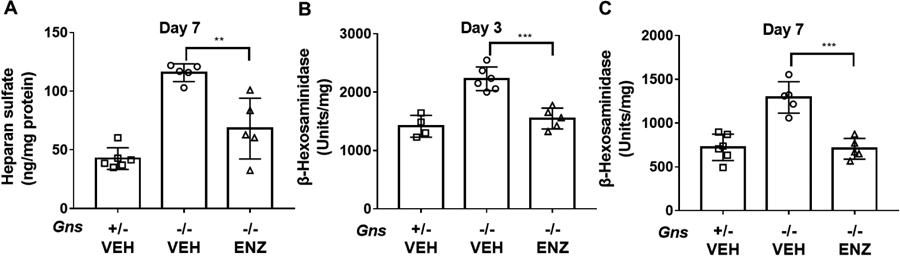
Efficacy of ICV ERT in MPS IIID mice with rhGNS. A) The amount of heparan sulfate in the mouse brain 7 days following a single dose of 67 units, Gns+/− VEH (n=6), Gnsr−/− VEH (n=5), Gnsr−/− ENZ (n=5). B, C) β-hexosaminidase activity in mouse brain lysate 3 and 7 days after treatment. Gns+/− VEH (n=4), Gns−/− VEH (n=5), Gns−/− ENZ (n=5) for day 3 and Gns+/− VEH (n=6), Gns−/− VEH (n=5), Gns−/− ENZ (n=5) for day 7. Data are shown as mean ± SD, and individual animal means are indicated by symbols. Statistical analysis was performed using one-way ANOVA. * * p < 0.05, *** p < 0.001.
DISCUSSION
There is currently no FDA or EMA approved treatment for any form of MPS III including MPS IIID. MPS IIID has perhaps the mildest phenotype and slowest progression of all the forms. While each type of MPS III is individually rare, MPS IIID is the rarest with an estimated prevalence of 0.02-0.10 per 100,000 live births worldwide 3, which would indicate at least a world-wide number of 7,500 patients. Publications referring to Sanfilippo syndrome type D usually refer to the number reported as around 15-20 65, 66. An analysis of papers has identified patients in the Netherlands 67, Turkey 66, Italy 65, Pakistan 68, Poland 69, USA 70, Australia 71, Belgium 72 and Saudi Arabia 73. We are aware of at least four MPS IIID patients in the USA, two in Australia, one in Argentina, one in Bulgaria, two in France, one in Mexico, one in the UK and one in Spain currently. Because MPS IIID is generally difficult to recognize (due to relatively mild physical features) and is known to be underdiagnosed, we estimate there are more MPS IIID patients globally that might be identified.
Of the possible therapeutic approaches to treat MPS IIID, ERT is appealing as there are examples of successfully commercialized therapeutic enzymes for MPS and other lysosomal diseases. Arguably, no therapy for any inborn error of metabolism comes close to the commercial success that ERT has achieved for lysosomal diseases 74. These include laronidase (MPS I), idursulfase (MPS II), galsulfase (MPS VI), alglucosidase alfa (Pompe disease), imiglucerase (Gaucher disease), and agalsidase beta (Fabry disease), and more recent approval for ICV ERT with cerliponase alfa for late-infantile neuronal ceroid lipofuscinosis (CLN2 Batten Disease) 75. Research on the canine model of MPS type I shows that intrathecal ERT can distribute broadly throughout the neuraxis, penetrate into deep brain structures, and achieve therapeutic concentrations there 76, 77. The success of the intrathecal approach in animal models has led to clinical trials of intrathecal ERT for MPS I, II and IIIA, metachromatic leukodystrophy 75. These experimental therapies demonstrate the proof of principle for pursuing the CSF to deliver enzyme to the human brain. ERT also avoids potential barriers to gene therapy such as the prevalence of pre-existing anti-capsid antibodies which can profoundly impact the transduction efficiency 78. Furthermore, an advantage to ERT is that it can be dosed repeatedly, allowing the possibility of adjusting dose and frequency as is traditionally the case for modulating medications. Adverse effects with other approved ERT seem to be mild 79, however there remains the potential for immune reactions as with other protein therapeutics 80.
The full-length of the engineered rhGNS fusion protein is 579 amino acids with a calculated molecular weight of 64.8 kDa and pI of 8.60, and the mature rhGNS is 543-amino acid long with the first 36 amino acid being cleaved as signal peptide 59 and the expected molecular weight is approximately ~58 kDa without glycosylation. We found properties of rhGNS favorable for an ERT in MPS IIID. The uptake of rhGNS into human fibroblasts was inhibited by addition of M6P, which indicates the uptake is mediated by M6P receptors (Figure 5B). The rhGNS protein was likely N-glycosylated with high-mannose residues (Figure 1 C), showed an optimal pH in the acidic range (pH 5.6), catabolized the natural, GAG substrate of GNS, was thermostable up to 65°C and showed stability for a month at 4°C in artificial CSF, an ideal formulation for ICV delivery.
We generated a stable CHO cell line expressing TCB616 using the dhfr gene amplification system. The titer of rhGNS secreted from this clone (TCB616 C2) reached 26 mg/L and can be potentially increased further by modifying culture conditions. These data indicated that the dhfr gene amplification system is capable of increasing the productivity of rhGNS. However, there are some limitations of our current method to produce rhGNS. 1) We used affinity purification of rhGNS with a carboxyl terminal myc tag for our proof of concept in vivo study. The product for clinical study should not contain the tag; 2) The dhfr system is more suitable to CHO cell lines deficient in dhfr gene than the WT CHO k1; 3) TCB616 C2 is an adherent cell line while the large scale production of recombinant proteins in industry usually requires suspension culture; 4) Purification using anti-myc beads is not cost-effective for large scale production of rhGNS. To address these limitations, we have established new clones using the dhfr gene amplification system in CHO/dhfr− cells to produce rhGNS without a myc tag. The new clones were also adapted to suspension culture.
MPS III types A to D cause paediatric neurodegeneration and some cause premature death 5. The earlier the intervention the better the outcome is for MPS III patients to prevent their neurodegeneration. Therefore, our initial in vivo studies were performed on a neonatal GNS KO mouse model. Our in vivo studies are the first attempt to reveal the pharmacokinetic (PK) and pharmacodynamics (PD) profile of rhGNS. To be able to detect the activity and distribution of rhGNS in the brain, we chose one day post injection as endpoint, and by separating brain into three sections we demonstrated that rhGNS distribute throughout the brain from the injection site (Figure 6A and C). The in vivo half-life of rhGNS in the brain was estimated to be 1.1 days (Figure 6D). Immunostaining is a direct and widely used method to detect co-localization. We were unable to find any anti-GNS antibody suitable to determine lysosome co-localization using immunostaining or IHC; hence, we used subcellular fractionation to identify co-localization of rhGNS in lysosome 81, 82. The increased ratio of GNS/total proteins and specific activity indicated that the isolation of lysosomes enable us enrich GNS, supporting the notion that rhGNS localizes in lysosome. To reveal the PK and PD profile of rhGNS, we used GNS activity to estimate the concentration and the reduction of β-hexosaminidase activity to reflect PD response after ICV. The estimated rhGNS brain half-life was approximately 1.1 days but the PD response last until 7 days post ICV.
Our preliminary in vivo studies with a single dose of rhGNS showed therapeutic GNS activity in all areas of the brain and localized the lysosomal compartment. Single-dose recombinant enzyme studies are limited in their estimation of the therapeutic potential, as repeated dosing will significantly reduce the GAG substrate accumulation over time, as well as “even out” biodistribution irregularities that are inevitable with the use of the slow-flowing, unpredictable CSF route for delivery. The estimated rhGNS tissue half-life of approximately 1.1 days appears short. However, the dosing interval for this lysosomal storage disease will likely depend on the rate of substrate accumulation, not enzymatic half-life. In our in vivo studies we found that the GNS substrate heparan sulfate in MPS IIID mice accumulated to 115.8 ng/mg protein and was significantly reduced 7 days post-treatment with a single dose of rhGNS to 68.1 ng/mg protein; this level is still about 150% of the unaffected carrier level, which is 42.6 ng/mg protein (Figure 8A). Our results suggested that heparan sulfate was cleared out quickly after ERT and the reaccumulation occurred slowly. Interestingly, the β-hexosaminidase activity remained at the unaffected carrier level 7 days post-treatment (Figure 8C). The accumulation of heparan sulfate likely needs to cross a threshold to cause pathological cellular response such as increased β-hexosaminidase activity; crossing this threshold occurs slowly in the absence of rhGNS activity. These results indicate that an optimal dosing regime in patients with rhGNS can improve the enzymatic activity, reduce lysosomal storage, and rescue the metabolic phenotype.
In our ongoing in vivo studies, repeated dosing with rhGNS will be performed on adult MPS IIID mice at three different doses up to 200 μg. According to the half-life (1.1 days) and PD response (last for 7 days) detected from our initial studies, ICV is performed twice per week for 2 weeks. The reduction of HS in both CSF and brain is considered as the primary PD endpoint and the β-hexosaminidase activity as the secondary PD endpoint. The long term follow up of biochemical and phenotypic analyses on MPS IIID mice will be completed in due course. In addition, we are generating human cortical neurons derived from MPS IIID patient cells to evaluate the effects of rhGNS on human neuronal cells.
For the potential clinical translation, repeat dosing with rhGNS will be performed via ICV. The dose and dosing interval will be decided according to the results of dose-responsive and toxic studies. Considering the small size of patients, the clinical trial design will be n of 1 study. The reduction of HS in CSF will be the primary PD endpoint.
CONCLUSIONS
We have developed a proof of concept ERT for MPS IIID in which rhGNS protein was purified from CHO cells. We generated high expression CHO cell lines utilizing the dhfr gene amplification system. The titer of rhGNS reached 26 mg/L and can be potentially increased further by modifying culture conditions. The specific activity of rhGNS was 3.9x104 units/mg with maximal enzymatic activity at pH 5.6, which is closer to lysosomal pH. The rhGNS purified from the CHO TCB616 C2 clone reached 95% purity. The rhGNS was stable for over one month at 4°C in artificial CSF buffer. We also demonstrated cellular uptake of rhGNS and GAG clearance in MPS IIID patient’s fibroblasts treated with rhGNS. Moreover, ICV injection of rhGNS in neonatal mice returned GNS activity to the unaffected carrier levels throughout the whole brain. Furthermore, rhGNS localized to the lysosome, and cleared the accumulated heparan sulfate; this effect lasted longer than the estimated half-life of rhGNS (1.1 days). The β-hexosaminidase activity remained at the unaffected carrier levels up to 7 days. Overall, our results demonstrate the therapeutic potential of ICV-administered rhGNS for treatment of MPS IIID.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the National Institutes of Health 1R41NS89061, R42NS089061, R44NS089061, and U44NS089061 to Phoenix Nest Inc., and R01NS088766 to P.I.D. S.-h.K. was supported by a T32 fellowship in the UCLA Medical Genetics Training Program (GM008243). The MPS IIID mouse model was developed by Taconic as a generous prize to Jonah’s Just Begun as part of the Rare Disease Science Challenge 2012, hosted by Assay Depot and the Rare Genomics Institute. The authors gratefully acknowledge the monthly meetings and encouragement of Dr. Emily Caporello, Dr. Jill Morris, and colleagues (NIH/NINDS). We appreciate Ms. Rachel Dokko and Caitlin Yumori for their initial work on this project. The authors also kindly acknowledge the support of MPSIIID families and the MPS Society. We thank the anonymous reviewers for constructive criticism.
ABBREVIATIONS
- MPS IIID
mucopolysaccharidosis type IIID
- LSD
lysosomal storage disorder
- GNS
α--N--acetylglucosamine-6-sulfatase
- rhGNS
recombinant human α-N-acetylglucosamine-6-sulfatase
- ERT
enzyme replacement therapy
- CSF
cerebrospinal fluid
- M6P
mannose 6-phosphate
- CHO cells
Chinese hamster ovary cells
- ICV
intracerebroventricular
Footnotes
DISCLOSURE
CS, DM, and SE are former Phoenix Nest Inc. employees. SS and JWare current Phoenix Nest Inc. employees.
ASSOCIATED CONTENT
Supporting Information for this article is given via a link at the end of the document.
Production rhGNS using pCI-neo vector (Figure S1); characterization of purified rhGNS (Figure S2); production and characterization of rhGNS using popvec vector (Figure S3). PCR and sequence primers (Table S1); plasmid information (Table S2); purification of rhGNS produced in CHO cells transfected with TCB469 (Table S3); LC/MS/MS analysis of trypsin digested rhGNS further confirmed the identity of rhGNS (Table S4); quantification the purity of rhGNS purified from TCB469 B2 using ImageJ (Table S5); purification of rhGNS produced in TCB616 C2 (Table S6); LC/MS/MS analysis of trypsin digested rhGNS further confirmed the identity of rhGNS purified from TCB616 C2 (Table S7); supplemental methods include assay optimization GNS assay, sulfatase assay, assay optimization sulfatase assay, SDS-PAGE and western blotting, native polyacrylamide gel electrophoresis, thermal shift assay, lysosome purification, and LC/MS/MS.
REFERENCES
- 1.Ghosh A; Shapiro E; Rust S; Delaney K; Parker S; Shaywitz AJ; Morte A; Bubb G; Cleary M; Bo T; Lavery C; Bigger BW; Jones SA, Recommendations on clinical trial design for treatment of Mucopolysaccharidosis Type III. Orphanet J Rare Dis 2017, 12 (1), 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kresse H P. E; von Figura K; Gilberg W; Fuchs W, Sanfilippo disease type D: deficiency of N-acetylglucosamine-6-sulfate sulfatase required for heparan sulfate degradation. Proceedings of the National Academy of Sciences of the United States of America 1980, 77 (11), 6822–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heron B; Mikaeloff Y; Froissart R; Caridade G; Maire I; Caillaud C; Levade T; Chabrol B; Feillet F; Ogier H; Valayannopoulos V; Michelakakis H; Zafeiriou D; Lavery L; Wraith E; Danos O; Heard JM; Tardieu M, Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A 2011, 155A (1), 58–68. [DOI] [PubMed] [Google Scholar]
- 4.Archer LD; Langford-Smith KJ; Bigger BW; Fildes JE, Mucopolysaccharide diseases: a complex interplay between neuroinflammation, microglial activation and adaptive immunity. J Inherit Metab Dis 2014, 37 (1), 1–12. [DOI] [PubMed] [Google Scholar]
- 5.Valstar MJ; Ruijter GJ; van Diggelen OP; Poorthuis BJ; Wijburg FA, Sanfilippo syndrome: a mini-review. J Inherit Metab Dis 2008, 31 (2), 240–52. [DOI] [PubMed] [Google Scholar]
- 6.Zhou J; Lin J; Leung WT; Wang L, A basic understanding of mucopolysaccharidosis: Incidence, clinical features, diagnosis, and management. Intractable Rare Dis Res 2020, 9 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Brien JS; Miller AL; Loverde AW; Veath ML, Sanfilippo disease type B: enzyme replacement and metabolic correction in cultured fibroblasts. Science 1973, 181 (4101), 753–5. [DOI] [PubMed] [Google Scholar]
- 8.Weber B; Blanch L; Clements PR; Scott HS; Hopwood JJ, Cloning and expression of the gene involved in Sanfilippo B syndrome (mucopolysaccharidosis III B). Hum Mol Genet 1996, 5 (6), 771–7. [DOI] [PubMed] [Google Scholar]
- 9.Yu WH; Zhao KW; Ryazantsev S; Rozengurt N; Neufeld EF, Short-term enzyme replacement in the murine model of Sanfilippo syndrome type B. Mol Genet Metab 2000, 71 (4), 573–80. [DOI] [PubMed] [Google Scholar]
- 10.Fedele AO, Sanfilippo syndrome: causes, consequences, and treatments. Appl Clin Genet 2015, 8, 269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aoyagi-Scharber M; Crippen-Harmon D; Lawrence R; Vincelette J; Yogalingam G; Prill H; Yip BK; Baridon B; Vitelli C; Lee A; Gorostiza O; Adintori EG; Minto WC; Van Vleet JL; Yates B; Rigney S; Christianson TM; Tiger PMN; Lo MJ; Holtzinger J; Fitzpatrick PA; LeBowitz JH; Bullens S; Crawford BE; Bunting S, Clearance of Heparan Sulfate and Attenuation of CNS Pathology by Intracerebroventricular BMN 250 in Sanfilippo Type B Mice. Mol Ther Methods Clin Dev 2017, 6, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alverez C; Arkin MR; Bulfer SL; Colombo R; Kovaliov M; LaPorte MG; Lim C; Liang M; Moore WJ; Neitz RJ; Yan Y; Yue Z; Huryn DM; Wipf P, Structure-Activity Study of Bioisosteric Trifluoromethyl and Pentafluorosulfanyl Indole Inhibitors of the AAA ATPase p97. ACS medicinal chemistry letters 2015, 6 (12), 1225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu H; Samulski RJ; McCown TJ; Picornell YJ; Fletcher D; Muenzer J, Neurological correction of lysosomal storage in a mucopolysaccharidosis IIIB mouse model by adeno-associated virus-mediated gene delivery. Mol Ther 2002, 5 (1), 42–9. [DOI] [PubMed] [Google Scholar]
- 14.Fu H; Kang L; Jennings JS; Moy SS; Perez A; Dirosario J; McCarty DM; Muenzer J, Significantly increased lifespan and improved behavioral performances by rAAV gene delivery in adult mucopolysaccharidosis IIIB mice. Gene Ther 2007, 14 (14), 1065–77. [DOI] [PubMed] [Google Scholar]
- 15.McCarty DM; DiRosario J; Gulaid K; Muenzer J; Fu H, Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice. Gene Ther 2009, 16 (11), 1340–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heldermon CD; Ohlemiller KK; Herzog ED; Vogler C; Qin E; Wozniak DF; Tan Y; Orrock JL; Sands MS, Therapeutic efficacy of bone marrow transplant, intracranial AAV-mediated gene therapy, or both in the mouse model of MPS IIIB. Mol Ther 2010, 18 (5), 873–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tardieu M; Zerah M; Gougeon ML; Ausseil J; de Bournonville S; Husson B; Zafeiriou D; Parenti G; Bourget P; Poirier B; Furlan V; Artaud C; Baugnon T; Roujeau T; Crystal RG; Meyer C; Deiva K; Heard JM, Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: an uncontrolled phase 1/2 clinical trial. Lancet Neurol 2017, 16 (9), 712–720. [DOI] [PubMed] [Google Scholar]
- 18.Hemsley KM; King B; Hopwood JJ, Injection of recombinant human sulfamidase into the CSF via the cerebellomedullary cistern in MPS IIIA mice. Mol Genet Metab 2007, 90 (3), 313–28. [DOI] [PubMed] [Google Scholar]
- 19.Beard H; Luck AJ; Hassiotis S; King B; Trim PJ; Snel MF; Hopwood JJ; Hemsley KM, Determination of the role of injection site on the efficacy of intra-CSF enzyme replacement therapy in MPS IIIA mice. Mol Genet Metab 2015, 115 (1), 33–40. [DOI] [PubMed] [Google Scholar]
- 20.King B; Marshall NR; Hassiotis S; Trim PJ; Tucker J; Hattersley K; Snel MF; Jolly RD; Hopwood JJ; Hemsley KM, Slow, continuous enzyme replacement via spinal CSF in dogs with the paediatric-onset neurodegenerative disease, MPS IIIA. J Inherit Metab Dis 2017, 40 (3), 443–453. [DOI] [PubMed] [Google Scholar]
- 21.Jones SA; Breen C; Heap F; Rust S; de Ruijter J; Tump E; Marchal JP; Pan L; Qiu Y; Chung JK; Nair N; Haslett PA; Barbier AJ; Wijburg FA, A phase 1/2 study of intrathecal heparan-N-sulfatase in patients with mucopolysaccharidosis IIIA. Mol Genet Metab 2016, 118 (3), 198–205. [DOI] [PubMed] [Google Scholar]
- 22.Boado RJ; Lu JZ; Hui EK; Pardridge WM, Insulin receptor antibody-sulfamidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in Sanfilippo type A cells. Mol Pharm 2014, 11 (8), 2928–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bielicki J; Hopwood JJ; Anson DS, Correction of Sanfilippo A skin fibroblasts by retroviral vector-mediated gene transfer. Hum Gene Ther 1996, 7 (16), 1965–70. [DOI] [PubMed] [Google Scholar]
- 24.Fraldi A; Hemsley K; Crawley A; Lombardi A; Lau A; Sutherland L; Auricchio A; Ballabio A; Hopwood JJ, Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet 2007, 16 (22), 2693–702. [DOI] [PubMed] [Google Scholar]
- 25.Quiviger M; Arfi A; Mansard D; Delacotte L; Pastor M; Scherman D; Marie C, High and prolonged sulfamidase secretion by the liver of MPS-IIIA mice following hydrodynamic tail vein delivery of antibiotic-free pFAR4 plasmid vector. Gene Ther 2014, 21 (12), 1001–7. [DOI] [PubMed] [Google Scholar]
- 26.Winner LK; Beard H; Hassiotis S; Lau AA; Luck AJ; Hopwood JJ; Hemsley KM, A Preclinical Study Evaluating AAVrh10-Based Gene Therapy for Sanfilippo Syndrome. Hum Gene Ther 2016, 27 (5), 363–75. [DOI] [PubMed] [Google Scholar]
- 27.Dziedzic D; Wegrzyn G; Jakobkiewicz-Banecka J, Impairment of glycosaminoglycan synthesis in mucopolysaccharidosis type IIIA cells by using siRNA: a potential therapeutic approach for Sanfilippo disease. Eur J Hum Genet 2010, 18 (2), 200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tordo J; O'Leary C; Antunes A; Palomar N; Aldrin-Kirk P; Basche M; Bennett A; D'Souza Z; Gleitz H; Godwin A; Holley RJ; Parker H; Liao AY; Rouse P; Youshani AS; Dridi L; Martins C; Levade T; Stacey KB; Davis DM; Dyer A; Clement N; Bjorklund T; Ali RR; Agbandje-McKenna M; Rahim AA; Pshezhetsky A; Waddington SN; Linden RM; Bigger BW; Henckaerts E, A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain 2018, 141 (7), 2014–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sifuentes M; Doroshow R; Hoft R; Mason G; Walot I; Diament M; Okazaki S; Huff K; Cox GF; Swiedler SJ; Kakkis ED, A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab 2007, 90 (2), 171–80. [DOI] [PubMed] [Google Scholar]
- 30.Fox JE; Volpe L; Bullaro J; Kakkis ED; Sly WS, First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol Genet Metab 2015, 114 (2), 203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabacam G; Kabacam G; Topcuoglu P; Kuzu I; Arat M, Enzyme replacement therapy in type 1 Gaucher disease and a review of the literature. Turk J Haematol 2010, 27 (3), 190–5. [DOI] [PubMed] [Google Scholar]
- 32.Davies EH; Erikson A; Collin-Histed T; Mengel E; Tylki-Szymanska A; Vellodi A, Outcome of type III Gaucher disease on enzyme replacement therapy: review of 55 cases. J Inherit Metab Dis 2007, 30 (6), 935–42. [DOI] [PubMed] [Google Scholar]
- 33.Toscano A; Schoser B, Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol 2013, 260 (4), 951–9. [DOI] [PubMed] [Google Scholar]
- 34.Rombach SM; Smid BE; Linthorst GE; Dijkgraaf MG; Hollak CE, Natural course of Fabry disease and the effectiveness of enzyme replacement therapy: a systematic review and meta-analysis: effectiveness of ERT in different disease stages. J Inherit Metab Dis 2014, 37 (3), 341–52. [DOI] [PubMed] [Google Scholar]
- 35.Vuillemenot BR; Kennedy D; Cooper JD; Wong AM; Sri S; Doeleman T; Katz ML; Coates JR; Johnson GC; Reed RP; Adams EL; Butt MT; Musson DG; Henshaw J; Keve S; Cahayag R; Tsuruda LS; O'Neill CA, Nonclinical evaluation of CNS-administered TPP1 enzyme replacement in canine CLN2 neuronal ceroid lipofuscinosis. Mol Genet Metab 2015, 114 (2), 281–93. [DOI] [PubMed] [Google Scholar]
- 36.Hawkes C; Kar S, Insulin-like growth factor-II/mannose-6-phosphate receptor: widespread distribution in neurons of the central nervous system including those expressing cholinergic phenotype. J Comp Neurol 2003, 458 (2), 113–27. [DOI] [PubMed] [Google Scholar]
- 37.McVie-Wylie AJ; Lee KL; Qiu H; Jin X; Do H; Gotschall R; Thurberg BL; Rogers C; Raben N; O'Callaghan M; Canfield W; Andrews L; McPherson JM; Mattaliano RJ, Biochemical and pharmacological characterization of different recombinant acid alpha-glucosidase preparations evaluated for the treatment of Pompe disease. Mol Genet Metab 2008, 94 (4), 448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen-Pfeffer JL; Gururangan S; Lester T; Lim DA; Shaywitz AJ; Westphal M; Slavc I, Intracerebroventricular Delivery as a Safe, Long-Term Route of Drug Administration. Pediatr Neurol 2017, 67, 23–35. [DOI] [PubMed] [Google Scholar]
- 39.Slavc I; Cohen-Pfeffer JL; Gururangan S; Krauser J; Lim DA; Maldaun M; Schwering C; Shaywitz AJ; Westphal M, Best practices for the use of intracerebroventricular drug delivery devices. Molecular Genetics and Metabolism 2018, 124 (3), 184–188. [DOI] [PubMed] [Google Scholar]
- 40.Atkinson AJ Jr., Intracerebroventricular drug administration. Transl Clin Pharmacol 2017, 25 (3), 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marsalkova D; Tesarova A; Limanova Z; Slavickova M; Moravcova A, [The significance of diagnostic puncture of the thyroid gland in children and adolescents]. Cesk Pediatr 1988, 43 (11), 658–60. [PubMed] [Google Scholar]
- 42.Pardridge WM, Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front Aging Neurosci 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boado RJ; Lu JZ; Hui EK; Lin H; Pardridge WM, Bi-functional IgG-lysosomal enzyme fusion proteins for brain drug delivery. Sci Rep 2019, 9 (1), 18632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pulgar VM, Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front Neurosci 2018, 12, 1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naqvi S; Panghal A; Flora SJS, Nanotechnology: A Promising Approach for Delivery of Neuroprotective Drugs. Front Neurosci 2020, 14, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Islam Y; Leach AG; Smith J; Pluchino S; Coxonl CR; Sivakumaran M; Downing J; Fatokun AA; Teixidò M; Ehtezazi T, Peptide based drug delivery systems to the brain. Nano Express 2020, 1 (1). [Google Scholar]
- 47.Sleat DE; Lobel P, Ligand binding specificities of the two mannose 6-phosphate receptors. J Biol Chem 1997, 272 (2), 731–8. [DOI] [PubMed] [Google Scholar]
- 48.Litjens T; Bielicki J; Anson DS; Friderici K; Jones MZ; Hopwood JJ, Expression, purification and characterization of recombinant caprine N-acetylglucosamine-6-sulphatase. Biochem J 1997, 327 ( Pt 1), 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Downs-Kelly E; Jones MZ; Alroy J; Cavanagh KT; King B; Lucas RE; Baker JC; Kraemer SA; Hopwood JJ, Caprine Mucopolysaccharidosis IIID: A Preliminary Trial of Enzyme Replacement Therapy. Journal of Molecular Neuroscience 2000, 15 (3), 251–262. [DOI] [PubMed] [Google Scholar]
- 50.Roca C; Motas S; Marco S; Ribera A; Sanchez V; Sanchez X; Bertolin J; Leon X; Perez J; Garcia M; Villacampa P; Ruberte J; Pujol A; Haurigot V; Bosch F, Disease correction by AAV-mediated gene therapy in a new mouse model of mucopolysaccharidosis type IIID. Hum Mol Genet 2017, 26 (8), 1535–1551. [DOI] [PubMed] [Google Scholar]
- 51.He W; Voznyi Ya V; Boer AM; Kleijer WJ; van Diggelen OP, A fluorimetric enzyme assay for the diagnosis of Sanfilippo disease type D (MPS IIID). J Inherit Metab Dis 1993, 16 (6), 935–41. [DOI] [PubMed] [Google Scholar]
- 52.Barton RWN, E. F., The Hurler corrective factor. Purification and some properties. The Journal of biological chemistry 1971, 246 (24), 7773–9. [PubMed] [Google Scholar]
- 53.Kakkis ED; Matynia A; Jonas AJ; Neufeld EF, Overexpression of the human lysosomal enzyme alpha-L-iduronidase in Chinese hamster ovary cells. Protein Expr Purif 1994, 5 (3), 225–32. [DOI] [PubMed] [Google Scholar]
- 54.Jamil M; Snella EM; Le SQ; Kan S-H; Birtcil BC; Dickson PI; Ellinwood NM; Smith JD, Initial characterization of a murine model of Sanfilippo syndrome type IIID shows similar pathology to other murine models of Sanfilippo syndrome. Molecular Genetics and Metabolism 2016, 117 (2), S61 [Google Scholar]
- 55.Clarke D; Pearse Y; Kan SH; Le SQ; Sanghez V; Cooper JD; Dickson PI; Iacovino M, Genetically Corrected iPSC-Derived Neural Stem Cell Grafts Deliver Enzyme Replacement to Affect CNS Disease in Sanfilippo B Mice. Mol Ther Methods Clin Dev 2018, 10, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lawrence R; Olson SK; Steele RE; Wang L; Warrior R; Cummings RD; Esko JD, Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J Biol Chem 2008, 283 (48), 33674–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mauro VP; Chappell SA, A critical analysis of codon optimization in human therapeutics. Trends Mol Med 2014, 20 (11), 604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hunter M; Yuan P; Vavilala D; Fox M, Optimization of Protein Expression in Mammalian Cells. Curr Protoc Protein Sci 2019, 95 (1), e77. [DOI] [PubMed] [Google Scholar]
- 59.Robertson DA; Freeman C; Morris CP; Hopwood JJ, A cDNA clone for human glucosamine-6-sulphatase reveals differences between arylsulphatases and non-arylsulphatases. Biochem J 1992, 288 ( Pt 2), 539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freeze HH; Kranz C, Endoglycosidase and glycoamidase release of N-linked glycans. Curr Protoc Mol Biol 2010, Chapter 17, Unit 17 13A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Orvisky E; Stubblefield B; Long RT; Martin BM; Sidransky E; Krasnewich D, Phosphomannomutase activity in congenital disorders of glycosylation type Ia determined by direct analysis of the interconversion of mannose-1-phosphate to mannose-6-phosphate by high-pH anion-exchange chromatography with pulsed amperometric detection. Anal Biochem 2003, 317 (1), 12–8. [DOI] [PubMed] [Google Scholar]
- 62.Fischer S; Handrick R; Otte K, The art of CHO cell engineering: A comprehensive retrospect and future perspectives. Biotechnol Adv 2015, 33 (8), 1878–96. [DOI] [PubMed] [Google Scholar]
- 63.Tian W; Ye Z; Wang S; Schulz MA; Van Coillie J; Sun L; Chen YH; Narimatsu Y; Hansen L; Kristensen C; Mandel U; Bennett EP; Jabbarzadeh-Tabrizi S; Schiffmann R; Shen JS; Vakhrushev SY; Clausen H; Yang Z, The glycosylation design space for recombinant lysosomal replacement enzymes produced in CHO cells. Nat Commun 2019, 10 (1), 1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kan SH; Aoyagi-Scharber M; Le SQ; Vincelette J; Ohmi K; Bullens S; Wendt DJ; Christianson TM; Tiger PM; Brown JR; Lawrence R; Yip BK; Holtzinger J; Bagri A; Crippen-Harmon D; Vondrak KN ; Chen Z; Hague CM; Woloszynek JC; Cheung DS; Webster KA; Adintori EG; Lo MJ; Wong W; Fitzpatrick PA; LeBowitz JH; Crawford BE; Bunting S; Dickson PI; Neufeld EF, Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc Natl Acad Sci U S A 2014, 111 (41), 14870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beesley CE; Concolino D; Filocamo M; Winchester BG; Strisciuglio P, Identification and characterisation of an 8.7 kb deletion and a novel nonsense mutation in two Italian families with Sanfilippo syndrome type D (mucopolysaccharidosis IIID). Mol Genet Metab 2007, 90 (1), 77–80. [DOI] [PubMed] [Google Scholar]
- 66.Elcioglu NH; Pawlik B; Colak B; Beck M; Wollnik B, A novel loss-of-function mutation in the GNS gene causes Sanfilippo syndrome type D. Genet Couns 2009, 20 (2), 133–9. [PubMed] [Google Scholar]
- 67.Valstar MJ; Bertoli-Avella AM; Wessels MW; Ruijter GJ; de Graaf B; Olmer R; Elfferich P; Neijs S; Kariminejad R; Suheyl Ezgu F; Tokatli A; Czartoryska B; Bosschaart AN; van den Bos-Terpstra F; Puissant H; Burger F; Omran H; Eckert D; Filocamo M; Simeonov E; Willems PJ; Wevers RA; Niermeijer MF; Halley DJ; Poorthuis BJ; van Diggelen OP, Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat 2010, 31 (5), E1348–60. [DOI] [PubMed] [Google Scholar]
- 68.Beesley CE; Burke D; Jackson M; Vellodi A; Winchester BG; Young EP, Sanfilippo syndrome type D: identification of the first mutation in the N-acetylglucosamine-6-sulphatase gene. J Med Genet 2003, 40 (3), 192–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tylki-Szymanska A; Czartoryska B; Gorska D; Piesiewicz-Grzonkowska E, Type III D mucopolysaccharidosis (Sanfilippo D): clinical course and symptoms. Acta Paediatr Jpn 1998, 40 (5), 492–4. [DOI] [PubMed] [Google Scholar]
- 70.Jones MZ; Alroy J; Rutledge JC; Taylor JW; Alvord EC Jr.; Toone J; Applegarth D; Hopwood JJ; Skutelsky E; Ianelli C; Thorley-Lawson D; Mitchell-Herpolsheimer C; Arias A; Sharp P; Evans W; Sillence D; Cavanagh KT, Human mucopolysaccharidosis IIID: clinical, biochemical, morphological and immunohistochemical characteristics. J Neuropathol Exp Neurol 1997, 56 (10), 1158–67. [PubMed] [Google Scholar]
- 71.Alroy J; Jones MZ; Rutledge JC; Taylor JW; Toone J; Applegarth D; Hopwood JJ, The ultrastructure of skin from a patient with mucopolysaccharidosis IIID. Acta Neuropathol 1997, 93 (2), 210–3. [DOI] [PubMed] [Google Scholar]
- 72.Jansen AC; Cao H; Kaplan P; Silver K; Leonard G; De Meirleir L; Lissens W; Liebaers I; Veilleux M; Andermann F; Hegele RA; Andermann E, Sanfilippo syndrome type D: natural history and identification of 3 novel mutations in the GNS Gene. Arch Neurol 2007, 64 (11), 1629–34. [DOI] [PubMed] [Google Scholar]
- 73.Ozand PT; Thompson JN; Gascon GG; Sarvepalli SB; Rahbeeni Z; Nester MJ; Brismar J, Sanfilippo type D presenting with acquired language disorder but without features of mucopolysaccharidosis. J Child Neurol 1994, 9 (4), 408–11. [DOI] [PubMed] [Google Scholar]
- 74.Talele SS; Xu K; Pariser AR; Braun MM; Farag-El-Massah S; Phillips MI; Thompson BH; Cote TR, Therapies for inborn errors of metabolism: what has the orphan drug act delivered? Pediatrics 2010, 126 (1), 101–6. [DOI] [PubMed] [Google Scholar]
- 75.Platt FM; d'Azzo A; Davidson BL; Neufeld EF; Tifft CJ, Lysosomal storage diseases. Nat Rev Dis Primers 2018, 4 (1), 27. [DOI] [PubMed] [Google Scholar]
- 76.Kakkis E; McEntee M; Vogler C; Le S; Levy B; Belichenko P; Mobley W; Dickson P; Hanson S; Passage M, Intrathecal enzyme replacement therapy reduces lysosomal storage in the brain and meninges of the canine model of MPS I. Mol Genet Metab 2004, 83 (1-2), 163–74. [DOI] [PubMed] [Google Scholar]
- 77.Dickson P; McEntee M; Vogler C; Le S; Levy B; Peinovich M; Hanson S; Passage M; Kakkis E, Intrathecal enzyme replacement therapy: successful treatment of brain disease via the cerebrospinal fluid. Mol Genet Metab 2007, 91 (1), 61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fitzpatrick Z; Leborgne C; Barbon E; Masat E; Ronzitti G; van Wittenberghe L; Vignaud A; Collaud F; Charles S; Simon Sola M; Jouen F; Boyer O; Mingozzi F, Influence of Pre-existing Anti-capsid Neutralizing and Binding Antibodies on AAV Vector Transduction. Mol Ther Methods Clin Dev 2018, 9, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baldo BA, Enzymes approved for human therapy: indications, mechanisms and adverse effects. BioDrugs 2015, 29 (1), 31–55. [DOI] [PubMed] [Google Scholar]
- 80.Dimitrov DS, Therapeutic proteins. Methods Mol Biol 2012, 899, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taguwa S; Kambara H; Fujita N; Noda T; Yoshimori T; Koike K; Moriishi K; Matsuura Y, Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J Virol 2011, 85 (24), 13185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi Y; Lin S; Staats KA; Li Y; Chang WH; Hung ST; Hendricks E; Linares GR; Wang Y; Son EY; Wen X; Kisler K; Wilkinson B; Menendez L; Sugawara T; Woolwine P; Huang M; Cowan MJ; Ge B; Koutsodendris N; Sandor KP; Komberg J; Vangoor VR; Senthilkumar K; Hennes V; Seah C; Nelson AR; Cheng TY; Lee SJ; August PR; Chen JA; Wisniewski N; Hanson-Smith V; Belgard TG; Zhang A; Coba M; Grunseich C; Ward ME; van den Berg LH; Pasterkamp RJ; Trotti D; Zlokovic BV; Ichida JK, Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 2018, 24 (3), 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.