

SUMMARY
Tumor-associated tertiary lymphoid structures (TA-TLS) are associated with enhanced patient survival and responsiveness to cancer therapies, but the mechanisms underlying their development are unknown. We show here that TA-TLS development in murine melanoma is orchestrated by cancer-associated fibroblasts (CAF) with characteristics of lymphoid tissue organizer cells that are induced by tumor necrosis factor receptor signaling. CAF organization into reticular networks is mediated by CD8 T cells, while CAF accumulation and TA-TLS expansion depend on CXCL13-mediated recruitment of B cells expressing lymphotoxin-α1β2. Some of these elements are also overrepresented in human TA-TLS. Additionally, we demonstrate that immunotherapy induces more and larger TA-TLS that are more often organized with discrete T and B cell zones, and that TA-TLS presence, number, and size are correlated with reduced tumor size and overall response to checkpoint immunotherapy. This work provides a platform for manipulating TA-TLS development as a cancer immunotherapy strategy.
Graphical Abstract
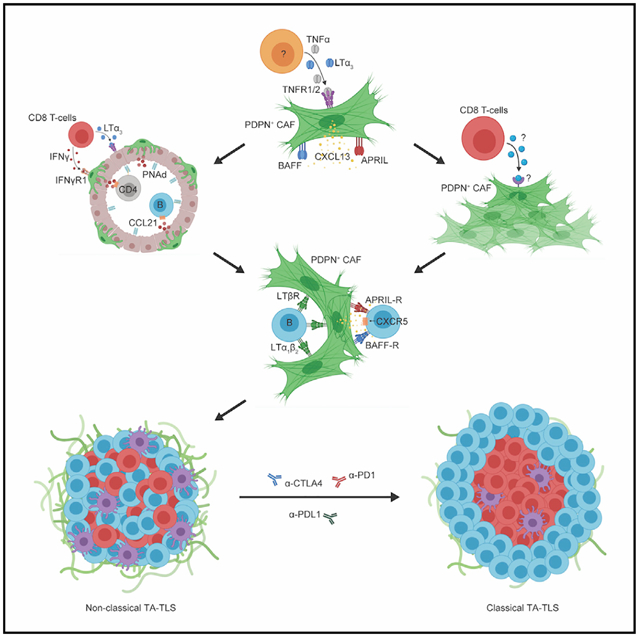
In brief
Rodriguez et al. describe the cellular and molecular mechanisms driving development of tumor-associated tertiary lymphoid structures and the importance of these structures as mediators of anti-tumor immunity and response to checkpoint immunotherapy.
INTRODUCTION
Tertiary lymphoid structures (TLS) are ectopic aggregates with morphological, cellular, and molecular similarities to secondary lymphoid organs (SLO) and commonly found with chronic infection, autoimmunity, and organ transplantation (Neyt et al., 2012; Koenig and Thaunat, 2016). TLS are found in human tumors (tumor-associated TLS [TA-TLS]) and are usually associated with higher representations of tumor-infiltrating lymphocytes (TIL), enhanced patient survival, and clinical responses to chemotherapy and immunotherapies (Engelhard et al., 2018; Sautès-Fridman et al., 2019; Rodriguez and Engelhard, 2020). It has been suggested that TA-TLS are sites for sustained antitumor immunity, and that TA-TLS augmentation could be a new strategy for cancer immunotherapy. This depends on understanding the mechanisms driving TA-TLS development.
Mechanisms governing development of SLOs are well established (reviewed in van de Pavert and Mebius, 2010; Onder and Ludewig, 2018; Mueller et al., 2018). SLO development depends on interaction between innate lymphoid type 3 (ILC3) lymphoid tissue inducer (LTi) cells expressing surface lymphotoxin-α1β2 (LTα1β2) and mesenchymal lymphoid tissue organizer (LTo) cells expressing lymphotoxin-β receptor (LTβR). LTo cells mature into CXCL13-producing follicular dendritic cells (FDC) and CCL19/21-producing fibroblastic reticular cells (FRC), which facilitate recruitment and compartmentalization of T and B cells. Although some chronic inflammation-associated TLS also depend on LTβR signaling (Furtado et al., 2007; Gatumu et al., 2009; GeurtsvanKessel et al., 2009; Gräbner et al., 2009; Motallebzadeh et al., 2012; Pikor et al., 2015), others depend on IL-5, IL-6, IL-17, IL-22, IL-23, and/or TNF-α, either in conjunction with (Lötzer et al., 2010; Rangel-Moreno et al., 2011; Pikor et al., 2015) or independent of (Lee et al., 1997; Goya et al., 2003; Khader et al., 2011; Peters et al., 2011; Furtado et al., 2014; Guedj et al., 2014; Barone et al., 2015; Bénézech et al., 2015; Cañete et al., 2015) LTβR signaling. CCL19, CCL21, CXCL12, and CXCL13 are found in TLS (Rangel-Moreno et al., 2007; Fleige et al., 2014; Barone et al., 2015; Sato et al., 2016), along with cells expressing FRC (Peduto et al., 2009; Link et al., 2011) or FDC (Drayton et al., 2003; Bombardieri et al., 2007) surface markers, and some of the inflammatory cytokines identified above can upregulate chemokine expression in cultured smooth muscle cells (Lötzer et al., 2010; Guedj et al., 2014) and fibroblasts purified from inflamed tissues containing TLSs (Khader et al., 2011; Rangel-Moreno et al., 2011; Barone et al., 2015). However, only one report has directly demonstrated that LTo-like fibroblasts could support TLS development (Nayar et al., 2019). Inflammatory TLS development can also depend on a variety of LTi cells, including dendritic cells (DC) (GeurtsvanKessel et al., 2009; Halle et al., 2009; Muniz et al., 2011), B cells (McDonald et al., 2005; Dubey et al., 2016), macrophages (Furtado et al., 2014; Guedj et al., 2014; Bénézech et al., 2015), TH17 cells (Peters et al., 2011; Rangel-Moreno et al., 2011; Pikor et al., 2015), natural killer T (NKT) cells (Bénézech et al., 2015), γδ T cells (Fleige et al., 2014), and multiple populations of IL-22-secreting adaptive and innate lymphoid cells (Barone et al., 2015). The heterogeneous drivers of TLS development likely depend on microenvironmental context. However, the cellular and molecular drivers of TA-TLS are undefined.
Vaccination can induce TA-TLS development in pancreatic tumors (Lutz et al., 2014) and cervical neoplastic lesions (Maldonado et al., 2014). TA-TLS development in murine tumors has been induced by transgenic overexpression of LTβR ligands (Schrama et al., 2001; Kim et al., 2004; Yu et al., 2004, 2007), injection of recombinant LIGHT (Johansson-Percival et al., 2017), and intratumoral administration of CCL21 (Kirk et al., 2001; Turnquist et al., 2007). However, there are few reports of spontaneous TA-TLS development in murine tumors (Finkin et al., 2015; Joshi et al., 2015; Peske et al., 2015; Rodriguez et al., 2018). We previously demonstrated that naive T cells infiltrated murine tumors, underwent in situ activation, and augmented tumor control (Thompson et al., 2010; Peske et al., 2015). Naive T cell entry depended on tumor endothelial cells expressing peripheral node addressin (PNAd) and CCL21, which in turn depended on effector CD8 T cells and natural killer (NK) cells secreting LTα3 and IFN-γ (Peske et al., 2015). We subsequently showed that tumors growing in the peritoneal cavity (intraperitoneally [I.P.]), but not those growing subcutaneously (S.C.), had TA-TLS adjacent to PNAd+ vasculature (Peske et al., 2015; Rodriguez et al., 2018). Here, we identify the cellular and molecular mechanisms driving TA-TLS development. Our results highlight the pivotal role of cancer-associated fibroblasts (CAF) as surrogate LTo cells and highlight multiple cell types, including intratumoral CD8 T cells and LTα1β2+ B cells, that synergistically act as LTi cells. They also demonstrate that TA-TLS development and organization are augmented by checkpoint blockade immunotherapy and are associated with reduced tumor size.
RESULTS
TA-TLS in I.P. tumors are associated with altered distributions of activated T cells and naïve B cells
By seven-color immunofluorescence (IF), we observed dense multicellular structures adjacent to PNAd+ vessels in I.P. B16-OVA tumors (Figure 1A; Figures S1A–S1C). However, these structures never formed in S.C. tumors. TA-TLS were dominated by a dense aggregate of B cells. In most cases, CD4 T cells were scattered throughout the B cell aggregate, while CD8 T cells and DCs were found predominantly on the periphery and also were not well organized (Figures S1D–S1G). These structures resemble “non-classical” TA-TLS seen in murine and human hepatocellular carcinoma (Finkin et al., 2015). I.P. tumors also contained less frequent “classical” TA-TLS with distinct T and B cell compartments (Figure 1A). TA-TLS were also present in parental B16-F1 I.P. tumors, although they were smaller and fewer (Figures S1H and S1I). TA-TLS were also present in B16-OVA tumors grown in lung and in I.P., but not S.C., MC38 and LLC tumors (Figures S1J–S1L). Thus, TA-TLS quantity and quality are associated with antigen strength, but their presence is due to tumor microenvironmental factors that depend on anatomical location.
Figure 1. TA-TLSs in I.P. tumors are associated with enhanced representations of distinctly differentiated T cells and naive B cells.
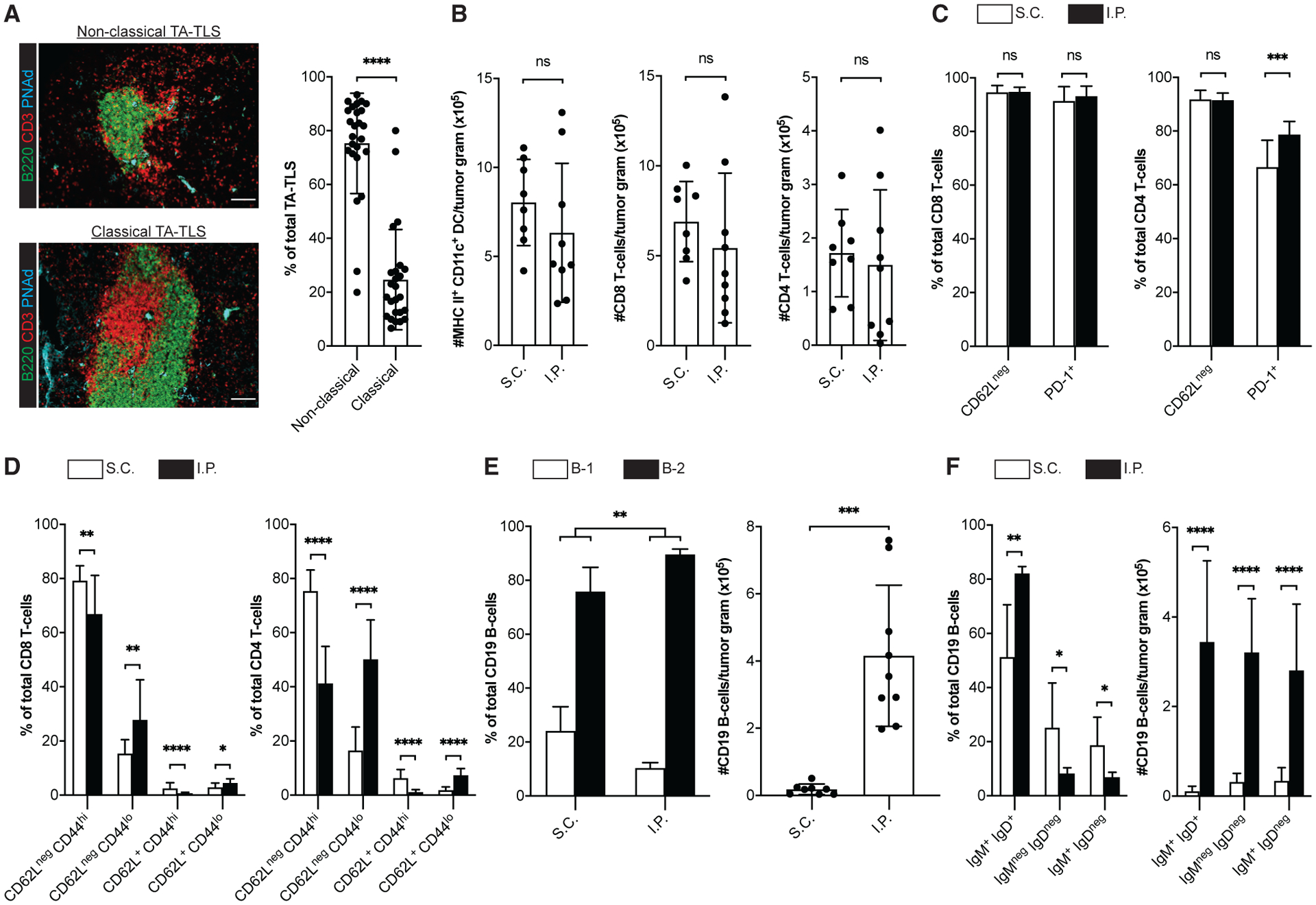
(A–F) Day 14 S.C. or I.P. B16-OVA tumors from C57BL/6 (WT) mice were prepared for IF (A) or flow cytometry (B–F) as described in STAR Methods. (A) Representative images and summary data for TA-TLS organizational types in I.P. tumors. Scale bar: 100 μm. Data are from five experiments; n = 25 tumors. (B–F) CD45+ single-cell suspensions were stained with the indicated markers to define subpopulations (DC = CD3neg CD19neg CD11c+ MHC II+; T cells = CD19neg CD3+ CD8+ or CD4+; B cells = CD3neg CD19+ CD5+ or CD5neg) and activation state and were quantitated by flow cytometry.
Data are from two to five experiments; n = 8–18 tumors per group. Results are mean ± standard deviation (SD), analyzed by unpaired Welch’s t test. nsp > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
I.P. B16 tumors grow as single pigmented masses, not ascites, juxtaposed to stomach, cecum, spleen, pancreas, and omentum (Figure S1M). They are connected by a blood vessel to the spleen and superficially associated with the omentum, but not other surrounding organs, and easily removed intact with forceps (Figure S1M). Human melanoma frequently metastasizes to visceral organs (Kawashima et al., 1991; Trout et al., 2013). TA-TLS frequently develop in gut melanoma metastases but are infrequent in primary cutaneous tumors (Cipponi et al., 2012). Thus, I.P. B16 tumors are relevant models to study TA-TLS development.
To assess the impact of TA-TLSs on intratumoral immune cell composition, we compared CD45+ cells from S.C. and I.P. tumors by flow cytometry. When normalized for tumor size, the numbers of DCs and CD8 and CD4 T cells were not significantly different (Figure 1B). In both tumor types, 92%–95% of CD8 and CD4 T cells were CD62Lneg, and thus antigen experienced (Figure 1C). Supporting this, 91%–94% of CD8 T cells and 65%–80% of CD4 T cells were PD-1+. Most CD62Lneg CD8 and CD4 T cells were also CD44hi, again in keeping with an antigen-experienced phenotype (Figure 1D). However, I.P. tumors contained a larger fraction of CD62Lneg CD44lo T cells, particularly in the CD4 compartment. These cells in both tumor types were largely PD-1+, although this was more variable on S.C. CD4+ T cells (Figure S2). Thus, despite being CD44lo, they seem activated. Both tumors also contained small populations of CD62L+ CD44hi presumptive central memory and CD62L+ CD44lo naive cells (Figure 1D), and their expression of PD-1, LAG-3, and TIM-3 was consistent with this classification (Figure S2). However, I.P. tumors contained more naive and fewer central memory CD8 T cells than S.C. tumors, and this was more pronounced in the CD4 compartment (Figure 1D). These results suggest that TA-TLS in I.P. tumors enhance representation of naive and distinctly differentiated T cells, particularly CD4 T cells.
B cells from both tumor types were mainly B-2, and this was significantly enriched in I.P. relative to S.C. tumors (Figure 1E). This contrasts with the dominance of B-1 cells in the peritoneal cavity (Hayakawa et al., 1985) and omental fat-associated lymphoid clusters (FALC) (Cruz-Migoni and Caamaño, 2016) and suggests that the origin of B cells in TA-TLS and/or the factors driving their accumulation are distinct from those of these two peritoneal sites. Consistent with the large aggregates observed by IF, I.P. tumors contained ~20× more B cells than S.C. tumors. In both tumor types, the largest population was IgM+ IgD+ and presumptively naive (Figure 1F), and this population was significantly elevated in I.P. tumors. When normalized for size, I.P. tumors contained ~30× more naive, ~10× more presumptive class-switched (IgMneg IgDneg), and 8× more memory (IgM+ IgDneg) B cells (Figure 1F). Thus, TA-TLS presence in I.P. tumors is associated with more robust B cell immunity and enhanced naive B cell representation.
A CAF population acts as LTo cells to orchestrate TA-TLS formation
When analyzed by flow cytometry, CD45+-depleted tumor suspensions contained many cells that were CD31neg and podoplanin (PDPN) positive. PDPN is a conventional CAF marker (Pula et al., 2013), but B16 tumor cells express it at a low level (Figure S3A). However, only PDPNhi cells expressed canonical CAF markers, particularly CD140α, Thy1, and fibroblast-activating protein (FAP) (Figure S3B), and these were analyzed as CAF in subsequent experiments. Thy1, FAP, and intracellular collagen type I (Col Type I) were all elevated on CAF from S.C. compared with I.P. tumors (Figure 2A and S3B), while α-smooth muscle actin (αSMA) expression was comparable (Figure S3C). I.P. tumors contained substantially more CAF than S.C. tumors, and a larger percentage expressed the proliferation marker Ki67+ (Figure 2B). Interestingly, the absolute number of FAP+ CAF was the same between S.C. and I.P. tumors (Figure 2B). Thus, the higher number of CAF in I.P. tumors is due to an expanded number of FAPneg CAF.
Figure 2. A population of CAF from I.P. tumors exhibit lymphoid tissue organizer cell characteristics.
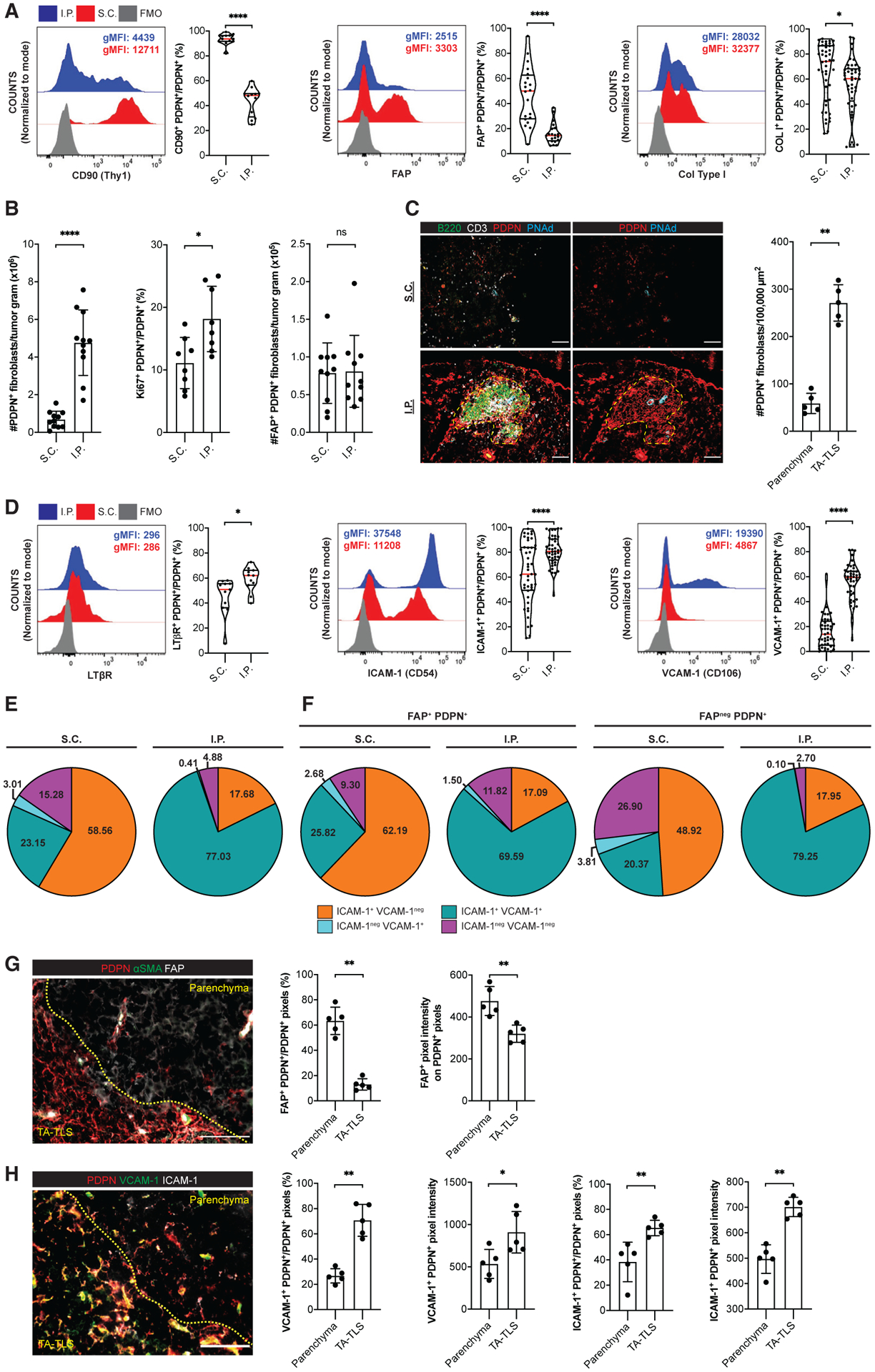
(A–E, G, and H) Day 14 S.C. or I.P. B16-OVA tumors from WT mice were prepared for flow cytometry (A, B, D, and E) or IF (C, G, and H) as described in STAR Methods. (A and D) Left, representative histograms and geometric mean fluorescence intensities (gMFIs) of indicated markers on PDPNhi CD31neg CD45neg Ter119neg CAF. gMFIs calculated on cells gated above the fluorescence minus one (FMO) control. Right, percentage of CAF expressing indicated markers. Data are from two to five experiments; n = 11–45 tumors per group.
(B, E, and F) CAF subpopulations were quantitated by flow cytometry. Data are from two to three experiments; n = 8–11 tumors per group.
(C, G, and H) Left, representative images of tumors stained with indicated markers. Yellow dashes delimit TA-TLS area. Scale bar: 100 μm. Right, summary data of PDPN+ CAF densities (C) and marker expression in parenchymal and TA-TLS regions (G and H) from one experiment; n = 5 tumors per group. Pixel intensities were calculated using a PDPN mask.
Results are mean ± SD analyzed by unpaired Welch’s t test. nsp > 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
CAF and tumor cells expressing high and low levels of PDPN, respectively, were also observed in tumor sections by IF. We established exposure times to visualize only PDPN bright CAF (Figure S3D). In S.C. tumors, CAF were scattered throughout the parenchyma (Figure 2C). In contrast, CAF in I.P. tumors formed reticular networks that were co-extensive with B and T cell aggregates of TA-TLS. By quantitative image analysis, CAF density in TA-TLS was substantially higher than in surrounding tumor parenchyma (Figure 2C), suggesting that their overall higher number in I.P. tumors was due to TA-TLS formation.
VCAM-1, ICAM-1, LTβR, and tumor necrosis factor receptors (TNFRs) I and II are canonical LTo cell markers (Mebius, 2003; Ruddle and Akirav, 2009; van de Pavert and Mebius, 2010). A larger fraction of I.P. CAF expressed TNFR I (Figure S3E) and LTβR (Figure 2D). Also, a significantly larger fraction of I.P. CAF expressed ICAM-1 and VCAM-1, and their surface expression levels were much higher (Figure 2D). The majority of I.P. CAF co-expressed ICAM-1 and VCAM-1, while those from S.C. tumors mainly expressed only ICAM-1 (Figure 2E). Although FDC-like cells are often found in TA-TLS, I.P. CAF did not express the FDC marker CD35 (Figure S3F). These results were consistent with the possibility that S.C. and I.P. tumors are dominated by mutually exclusive populations of CAF with FAP+ pro-tumor (Liu et al., 2019) and FAPneg LTo phenotypes. However, even FAP+ CAF from I.P. tumors were much more likely to co-express VCAM-1 and ICAM-1 than FAP+ or FAPneg CAF from S.C. tumors (Figure 2F). To provide more direct evidence for the association of these CAF populations with TA-TLS, we performed quantitative image analysis. Less than 15% of PDPN+ pixels in TA-TLS co-stained with FAP, while over 60% of those in the tumor parenchyma were FAP+, and their FAP expression was higher (Figure 2G). Conversely, 65%–70% of PDPN+ pixels in TA-TLS stained for VCAM-1 and ICAM-1, compared with only 25%–38% in the parenchyma, and their expression level was also higher (Figure 2H). Thus, despite the presence of both FAP+ and FAPneg cells expressing ICAM-1 and VCAM-1 in I.P. tumors, the latter population is enriched in TA-TLSs, consistent with a role in TA-TLS development.
To directly test a role for CAF in orchestrating TA-TLS formation, we flow-sorted these cells from resected S.C. and I.P. tumors and injected them with B16-OVA cells into a S.C. site. The resulting tumors that contained CAF from S.C. tumors, or mouse embryonic fibroblasts, did not develop TA-TLS, lymphoid aggregates, or PDPN+ reticular networks (Figures 3A and 3B). Conversely, S.C. tumors that contained CAF from I.P. tumors contained PDPN+ reticular networks that were co-extensive with aggregates of B220+ B cells and CD3+ T cells and adjacent to PNAd+ vasculature (Figure 3A and 3B). Although the number of these structures and their size were significantly less than TA-TLS in I.P. tumors (Figure 3B), their composition, organization, and localization established them as TA-TLS. We also flow-sorted CAF from I.P. tumors grown in mice that ubiquitously express mCherry. When injected with B16-OVA cells, they persisted in the resulting s.c. tumors and were mainly found in TA-TLS (Figure 3C). Based on their association with TA-TLS (Figure 2G), we hypothesized that only FAPneg CAF would promote TA-TLS formation. Indeed, when co-injected with B16-OVA cells, purified FAPneg CAF promoted formation of small TA-TLS, analogous in size and number to those induced by bulk I.P. CAF (Figure 3D). FAP+ CAF induced less frequent and even smaller TA-TLS-like structures that were associated with PNAd but contained small numbers of poorly organized PDPN+ cells. These results establish that FAPneg CAF from I.P. tumors act as surrogate LTo cells that promote TA-TLS formation. I.P. FAP+ CAF appear to have a minimal LTo capability that is nonetheless greater than that of S.C. CAF, suggesting that this property is induced by the I.P. tumor microenvironment.
Figure 3. CAF act as surrogate lymphoid tissue organizer cells that orchestrate TA-TLS formation.
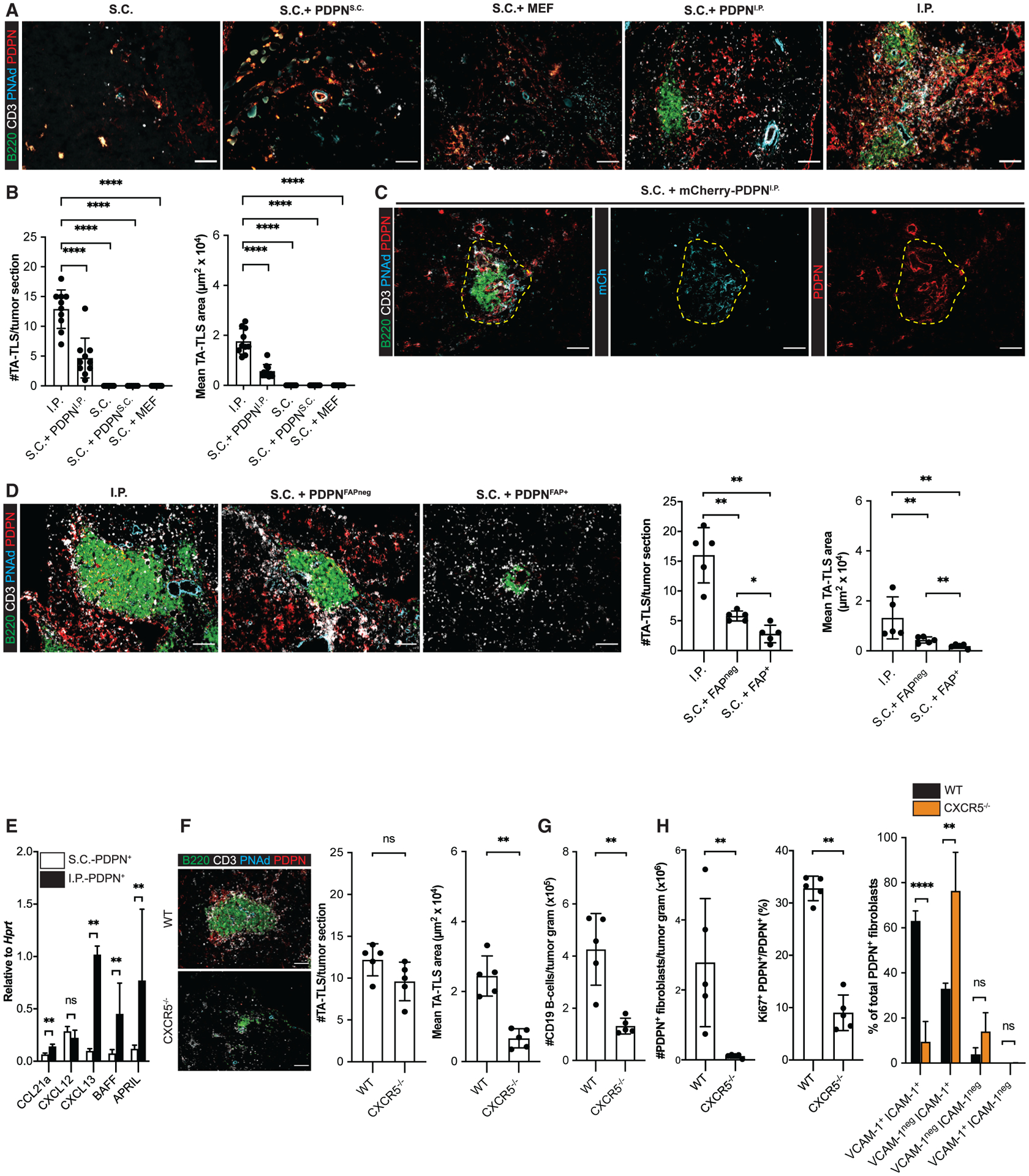
(A–D) S.C. tumors were induced by co-injection of B16-OVA cells together with the indicated populations of fibroblasts. Day 14 S.C. or I.P. tumors were prepared for IF as described in STAR Methods. (A, C, and D, left) Representative images. Yellow dashed region (C) represents TA-TLS area. Scale bar: 100 μm. (B and D, right) Summary data from one to two experiments; n = 5 or 10 tumors per group.
(E) CAF from day 14 S.C. and I.P. tumors were purified, and expression of the indicated RNA transcripts was determined as described in STAR Methods. Data from two experiments presented as 2−ΔCT relative to Hprt; n = 6 tumors per group.
(F–H) Day 14 I.P. tumors from WT or CXCR5−/− mice were prepared for IF (F) or flow cytometry (G and H) as described in STAR Methods. (F) Left: representative images. Scale bar: 100 μm. Right: summary data from one experiment; n = 5 tumors per group. (G and H) Cell populations were quantitated as outlined in Figures 1 and 2. Data represent one experiment; n = 5 tumors per group.
Results are mean ± SD, analyzed using Kruskal-Wallis H test with Dunn’s post-test (B and D) or unpaired Welch’s t test (E–H). nsp > 0.05, *p < 0.05, **p < 0.01, ****p < 0.0001.
We previously showed that both S.C. and I.P. CAF are a major source of CCL21 that recruits naive T cells (Peske et al., 2015). By quantitative real-time PCR, CAF from both tumor types expressed comparable levels of another homeostatic chemokine, CXCL12. However, I.P. CAF expressed 5- to 10-fold higher levels of the B cell organizer chemokine CXCL13 and B cell survival factors BAFF and APRIL (Figure 3E). To provide direct evidence for the importance of CXCL13 in driving TA-TLS formation, we implanted I.P. tumors into mice lacking CXCR5, the cognate receptor for CXCL13. Although the numbers of TA-TLS in these tumors were comparable with those in wild-type (WT) tumors, they were significantly smaller (Figure 3F). This was associated with a reduced number of intratumoral B cells and CAF (Figures 3G and 3H). Significantly fewer of these CAF were proliferating, and the fraction co-expressing ICAM-1 and VCAM-1 was also reduced (Figure 3H). Thus, an intact CXCR5-CXCL13 chemotactic axis supports the proliferation of CAF with LTo characteristics and normal TA-TLS development.
Intratumoral CD8 T cells and B cells act coordinately as LTi cells driving TA-TLS development
We next investigated the roles of adaptive immune cells in TA-TLS development. I.P. tumors grown in Rag1−/− mice lack PNAd+ vasculature (Peske et al., 2015). These tumors also contained a smaller number of CAF than I.P. tumors from WT mice, which was similar to that of S.C. tumors (Figure 4A). These CAF were sometimes found in a low number of small, loosely organized clusters with CD45+ cells (Figure 4A; Figure S3G), which were not observed in WT S.C. tumors but did not form PDPN+ reticular networks. Per-cell expression of CCL21 and CXCL12 in CAF from these tumors was reduced by 11- and 6-fold, respectively, but that of CXCL13, BAFF, and APRIL was unchanged (Figure 4B). Per-cell expression of CXCL13, BAFF, and APRIL in CAF from I.P. tumors grown in Rag2−/− IL2Rγ−/− mice, which lack mature NK and ILC cells, was also comparable with that of CAF from WT mouse tumors (Figure S3H). Thus, CAF expression of molecules known to drive B cell accumulation and survival is controlled by a non-adaptive, non-NK, non-ILC element in the I.P. tumor microenvironment, but their accumulation and organization into reticular networks is controlled by adaptive immune cells.
Figure 4. CD8 T cells and B cells act coordinately as lymphoid tissue inducer cells driving TA-TLS development.
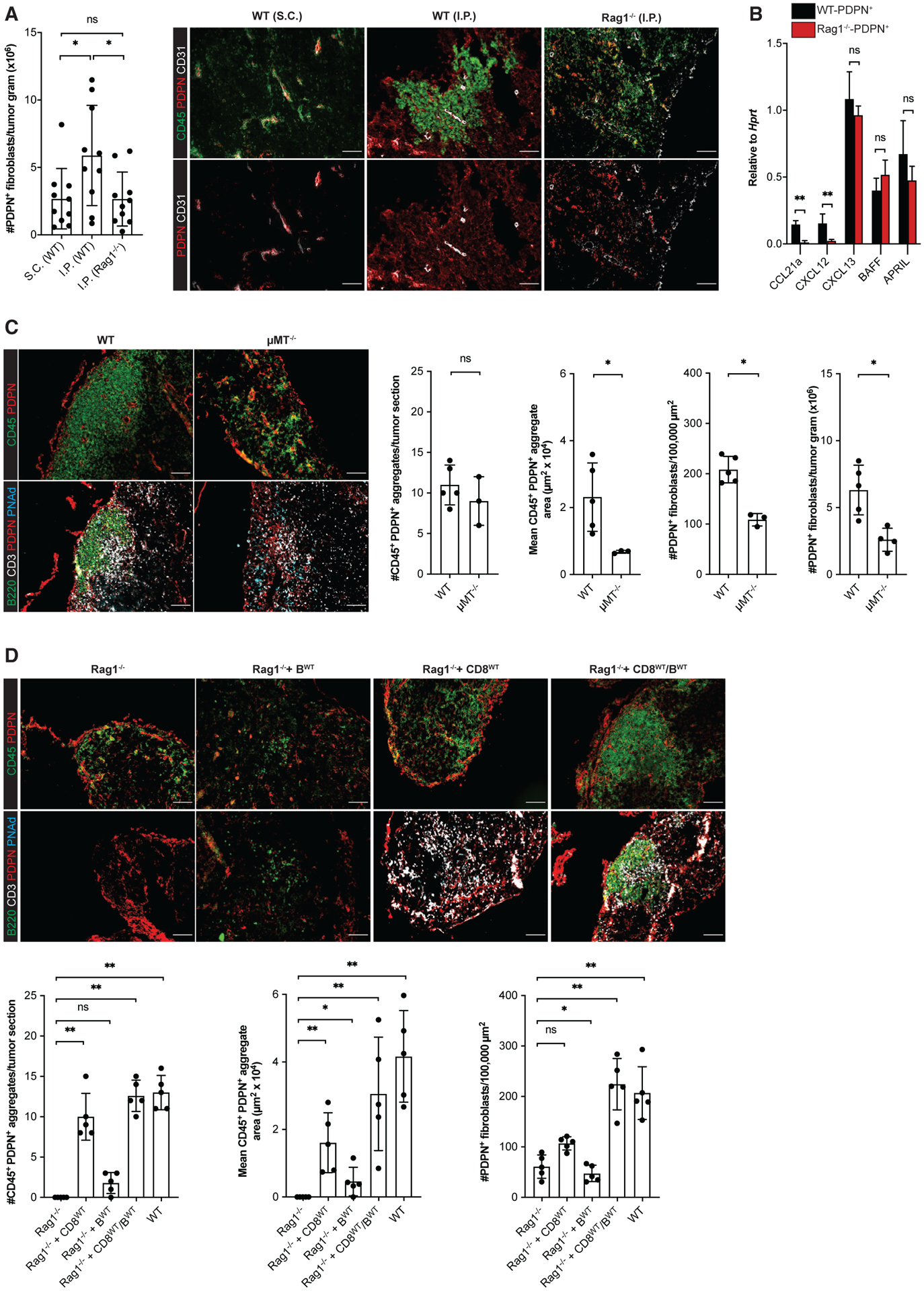
(A, C, and D) Day 14 I.P. B16-OVA tumors from WT mice, μMT−/− mice, Rag1−/− mice, and Rag1−/− mice repleted with CD8 T cells and/or B cells 3 days prior to tumor implantation were prepared for and analyzed by flow cytometry or IF as described in STAR Methods. Scale bars: 100 μm. Data are from one to two experiments; n = 3–10 tumors per group.
(B) CAF from day 14 i.p. tumors from WT or Rag1−/− mice were purified, and expression of the indicated RNA transcripts was determined as described in STAR Methods. Data from two experiments presented as 2−ΔCT relative to Hprt; n = 6 tumors per group.
Results are mean ± SD analyzed using Kruskal-Wallis H test with Dunn’s post-test (A and D) or unpaired Welch’s t test (B and C). nsp > 0.05, *p < 0.05, **p < 0.01, ****p < 0.0001.
To identify the adaptive immune cells driving CAF accumulation and organization, we utilized I.P. tumors grown in B cell-deficient μMT−/− mice and in Rag1−/− mice that had been repleted with bulk resting lymphocyte subpopulations prior to tumor implantation. Because these mice lack some TA-TLS elements, we evaluated tumors for PDPN+ reticular networks co-extensive with tightly organized aggregates of CD45+ cells. Tumors from μMT−/− mice contained such organized CD45+/PDPN+ aggregates in numbers comparable with TA-TLS in WT mouse tumors, but they were significantly smaller (Figure 4C). Consistent with this, tumors from μMT−/− mice had a lower density of CAF by IF and significantly fewer CAF by flow cytometry (Figure 4C). However, tumors from μMT−/− and WT mice contained similar number of T cells and the small loosely organized CD45+/PDPN+ clusters (Figure S3I). I.P. tumors from Rag1−/− mice repleted with CD8 T cells contained organized CD45+/PDPN+ aggregates similar in size and number to those in μMT−/− mouse tumors (Figure 4D). Tumors from Rag1−/− mice repleted with B cells contained an insignificantly different number of aggregates relative to tumors from unrepleted Rag1−/− mice, although there was a slight increase in aggregate area. Importantly, tumors from Rag1−/− mice repleted with both CD8 T cells and B cells resulted in structures comparable in number, size, and cellular composition and organization to TA-TLS in WT mouse tumors. The area per section of the structures in tumors from these different recipients was reflected in the density of CAF (Figure 4D). Tumors from all of these mice contained a similar number of small, loosely organized CD45+/PDPN+ clusters (Figure S3G). Thus, T cells and B cells act as complementary LTi cells mediating TA-TLS development: while T cells initiate reticular network formation, B cells drive expansion.
Intratumoral B cells drive TA-TLS expansion through LTβR signaling
LTβR signaling mediates the development of SLOs and some TLS. We found that I.P. tumors expressed significantly higher transcript levels of LTβR ligands LTα, LTβ, and LIGHT than S.C. tumors (Figure S4A). To test their relevance, tumor-bearing mice were treated with an LTβR-Ig fusion protein, which binds LTα1β2 and LIGHT and antagonizes LTβR signaling. Consistent with other work (Browning et al., 2005), LTβR-Ig treatment significantly reduced CXCL13 expression in tumor-reactive lymph nodes (Figure S4B). TA-TLSs in I.P. tumors from LTβR-Ig treated mice were comparable in number with WT mouse tumors, but significantly smaller, and these tumors contained fewer CAF (Figures 5A and 5B). Consistent with this, I.P. tumors from LTβR-Ig-treated mice contained fewer CD8 T cells and B cells, and the fractions of naive CD8 T cells, naive and central memory CD4 T cells, and naive B cells were significantly reduced (Figure 5C; Figures S4C–S4E). However, per-cell expression of CXCL13, BAFF, and APRIL in CAF from these tumors was unaffected (Figure 5D). Thus, LTβR signaling promotes expansion of TA-TLS-associated PDPN+ reticular networks, but not initial TA-TLS formation or CAF expression of LTo molecular characteristics.
Figure 5. Induction and robust development of PDPN+ reticular networks in TA-TLS are regulated by TNFR and LTbR signaling, respectively.
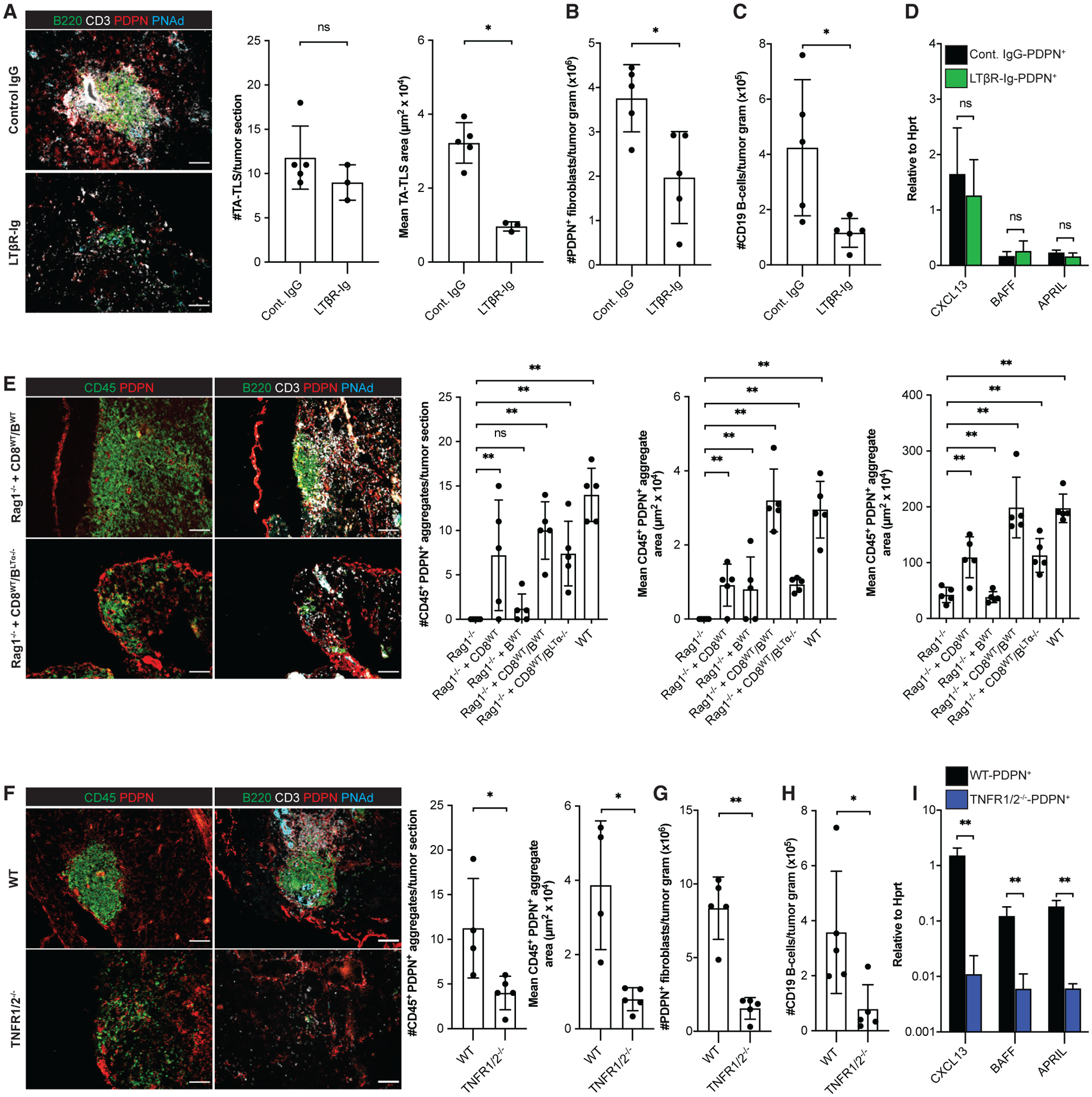
Day 14 I.P. B16-OVA tumors from WT mice, WT mice treated with LTβR-Ig fusion protein, TNFR1/2−/− mice, Rag1−/− mice, and Rag1−/− mice repleted with CD8 T cells and/or B cells 3 days prior to tumor implantation were analyzed by IF or flow cytometry as described in STAR Methods. (A, E, and F) Scale bars: 100 μm. Data are from one experiment; n = 3–5 tumors per group.
(B, C, G, and H) Data are from one experiment; n = 5 tumors per group.
(D and I) CAF from day 14 i.p. tumors from WT mice, WT mice treated with LTβR-Ig, or TNFR1/2−/− mice were purified and expression of the indicated RNA transcripts determined as described in STAR Methods. Data are from two experiments presented as 2−ΔCT relative to Hprt; n = 6 tumors per group.
Results are mean ± SD analyzed using unpaired Welch’s t test (A–D and F–I) or Kruskal-Wallis H test with Dunn’s post-test (E). nsp > 0.05, *p < 0.05, **p < 0.01.
Because CAF accumulation and reticular network expansion were impaired in the absence of either B cells or LTβR signaling, we tested whether it was mediated by LTα1β2+ B cells. Tumors in Rag1−/− mice repleted with WT CD8 T cells and LTα−/− B cells developed organized CD45+/PDPN+ aggregates comparable in size and number to those in tumors from Rag1−/− mice repleted with CD8 T cells alone and in tumors from LTβR-Ig treated mice (Figure 5E). Also, the density of CAF in these tumor sections was comparable with I.P. tumors from Rag1−/− mice repleted with CD8 T cells alone (Figure 5E). Tumors from all of these mice contained a similar number of small loosely organized CD45+/PDPN+ clusters (Figure S4F). Thus, CAF accumulation and TA-TLS expansion is mediated by intratumoral LTα1β2+ B cells.
TNFR signaling promotes PDPN+ reticular network formation and expression of LTo molecules
Next, we determined whether TNFR signaling mediates TA-TLS development. In keeping with the models described above, we observed organized CD45+/PDPN+ aggregates in I.P. tumors grown in TNFR1/2−/− mice that were smaller than TA-TLS in WT mouse tumors (Figure 5F). However, they were fewer, and the number of small loosely organized CD45+/PDPN+ clusters was also reduced (Figure S4G). As with TA-TLS in LTβR-Ig-treated mice, TNFR1/2−/− mouse tumors contained fewer CAF (Figure 5G), and the fractions of naive CD4 and CD8 T cells were reduced (Figures S4H and S4I). These tumors also contained fewer B cells (Figure 5H), although the distribution of naive, class-switched and memory B cells was unaltered (Figure S4J). Most importantly, per-cell expression of CXCL13, BAFF, and APRIL in CAF was substantially reduced (Figure 5I). These results demonstrate that TNFR signaling promotes PDPN+ reticular network development and LTo molecular characteristics of CAF.
Relationship of TA-TLS-associated i.p. CAF to peritoneal fibroblasts
The proximity of I.P. tumors to peritoneal organs suggested that CAF with LTo characteristics might derive from them. PDPNhi cells from spleen, omentum, lung, pancreas, and skin of WT mice showed varying levels of CXCL13, BAFF, and APRIL expression (Figure S5). Similar to I.P. CAF, CXCL13 expression in PDPNhi cells from the omentum was significantly reduced in TNFR1/2−/− mice (Figure S5A) but was unaltered in cells from other organs (Figure S5A). However, in contrast with WT i.p. CAF, BAFF and APRIL expression was not reduced in PDPNhi cells from any organ in TNFR1/2−/− mice (Figures S5B and S5C). These results do not identify any peritoneal organ as the direct source of I.P. CAF. To the extent that they do originate from a peritoneal organ, they indicate that elements within the I.P. tumor microenvironment alter their TNFR-dependent expression of BAFF, APRIL, and in some cases, CXCL13.
Human TA-TLS contain elevated densities of LT+ B cells co-extensive with a reticular network of LTo-like CAF
To establish translational relevance of mechanisms driving murine TA-TLS development, we evaluated three human melanomas by quantitative image analysis using seven-color IF. We identified TA-TLS as aggregates of PNAd+ cells, CD20+ B cells, CD8+ T cells, and gp36+ CAF (Figure 6A; Figure S6A). TA-TLS in all samples were enriched for APRIL+, VCAM-1+, and LTα+ cells relative to the surrounding parenchyma, and TA-TLS in two samples showed higher densities of CXCL13+ cells (Figure 6B). TA-TLS in all samples were enriched for B cells expressing LTα, and these cells represented 60%–80% of the LTα+ population inside the TA-TLS, as opposed to 0%–15% of those in the parenchyma (Figure 6C). TA-TLS in all samples also contained higher densities of CAF expressing APRIL+ and VCAM-1+ than the tumor parenchyma. The fraction of APRIL+ CAF was significantly higher in the TA-TLS of all samples, and the fraction of VCAM-1+ CAF was significantly higher in TA-TLS of two-thirds of samples (Figure 6C). However, only one sample showed higher densities of CXCL13+ CAF in TA-TLS, and there was no enrichment of CXCL13+ CAF in TA-TLS versus parenchyma (Figure 6C). The densities of B cells in TA-TLS strongly correlated with the densities of CAF overall and the densities of VCAM-1+ or APRIL+ CAF, but not those expressing CXCL13 (Figure 6D). However, the densities of B cells in TA-TLS also strongly correlated with the fractions of CAF in TA-TLS expressing VCAM-1 and APRIL, as well as CXCL13. Finally, the densities of LTα+ B cells in TA-TLS correlated with the densities of CAF expressing VCAM-1 and APRIL, but not CXCL13. These results are consistent with the idea that human melanoma-associated TA-TLS are driven by similar cellular and molecular mechanisms as those driving murine TA-TLS development.
Figure 6. Human TA-TLS are associated with LT+ B cells and a co-extensive reticular network of CAF with lymphoid tissue organizer characteristics.
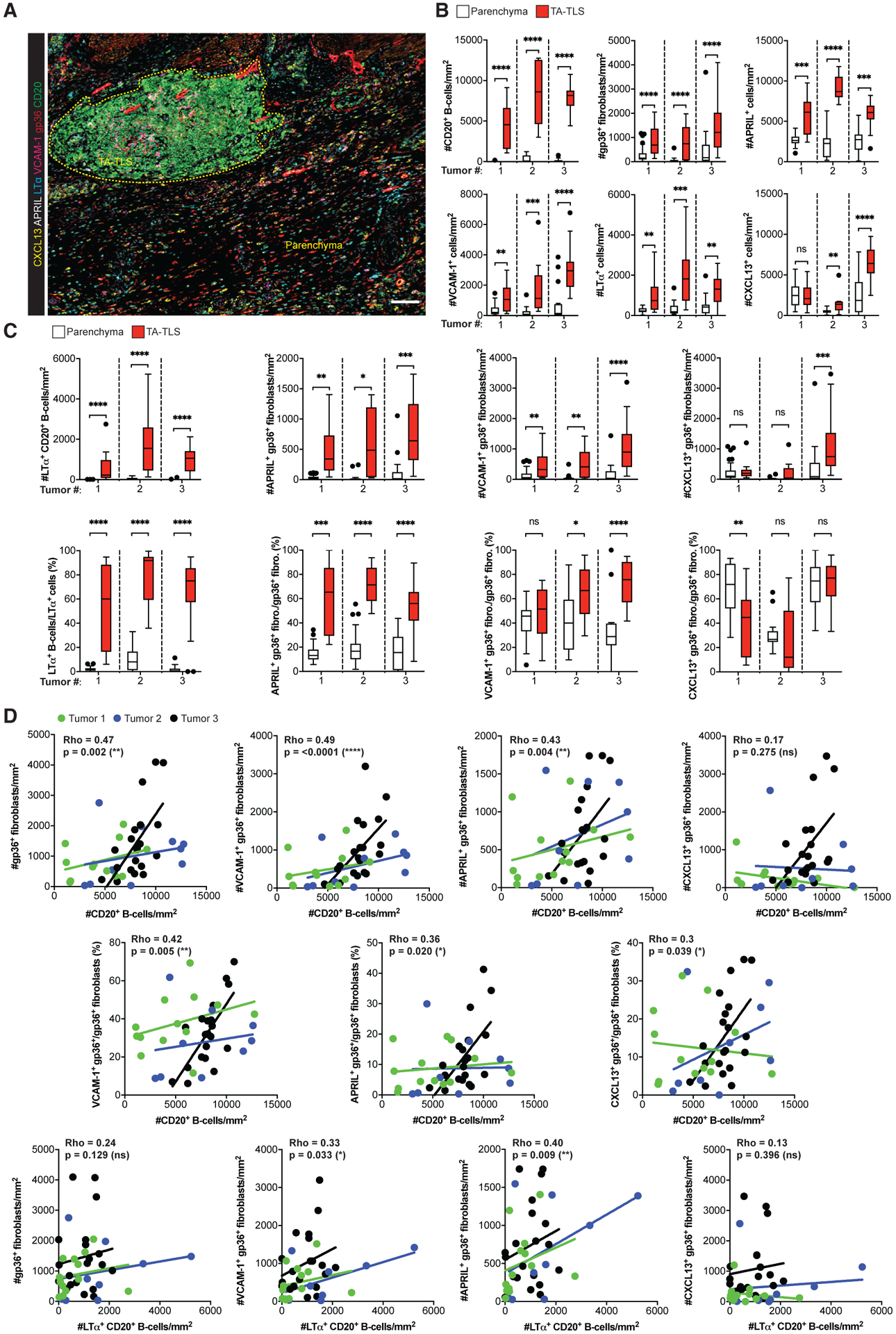
Human melanoma biopsies containing TA-TLS were collected, prepared, stained, and analyzed as described in STAR Methods.
(A) Representative image of a TA-TLS-containing melanoma sample. Dashed yellow line represents TA-TLS area. Scale bar: 100 μm.
(B and C) Densities and percentages of single- (B) and dual-stained (C) cell populations in parenchyma and TA-TLS regions of tumors from three patients (n = 12–39 parenchyma and TA-TLS regions of interest). Data were analyzed by Wilcoxon rank-sum test, and boxplots were generated according to Tukey’s method.
(D) Correlations between density of B cell populations and density or fraction of fibroblast populations. Dots represent individual TA-TLS. Each line represents the correlation for an individual tumor. Rho represents the multilevel correlation coefficient for the three tumors. Spearman’s linear mixed-models multilevel correlation analysis was determined for all tumors together to account for the nested data structure. nsp > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
TA-TLS are associated with reduced tumor outgrowth and response to checkpoint immunotherapy
Increased number and size of TA-TLS strongly correlated with diminished i.p. tumor size in WT mice (Figure S7A), and deficiencies in TA-TLS development as a result of implantation into CXCR5−/−, μMT−/−, LTβR-Ig-treated, and TNFR1/2−/− mice were all associated with tumors that were significantly larger (170%–235%) than tumors in WT mice (Figure S7B). Also, a larger fraction of I.P. CAF expressed PD-L1 than their S.C. counterparts (Figure S7C). Thus, we assessed the impact of checkpoint immunotherapy on TA-TLS development and control of tumor outgrowth. We treated tumor-bearing WT mice with anti-PD-L1 monotherapy or the combination of anti-CTLA4 and anti-PD1 and analyzed tumors after 14 days of outgrowth. Both treatments increased intratumoral T cell numbers in both S.C. and I.P. tumors, but neither increased intratumoral B cell numbers nor promoted TA-TLS formation in S.C. tumors (Figures 7A and 7B). However, both treatments increased the number of intratumoral B cells in I.P. tumors. They also induced significant increases in TA-TLS number and size, suggesting that they initiate TA-TLS development and also operate on existing TA-TLS (Figures 7A–7C). Both treatments also promoted the development of TA-TLS with a “classical” (discrete T and B cell zones) organization (Figures 7B and 7C). Also, both treatments significantly reduced I.P. tumor size, but neither significantly reduced S.C. B16-OVA tumor size at this day 14 time point, despite the increased number of intratumoral T cells (Figure 7D). As with untreated mice, increased TA-TLS size and number were correlated with reduced tumor size. However, this correlation was more significant in both treatment groups (Figure 7E; Table S1). Similarly, B cell density correlated with reduced I.P. tumor size in untreated mice, and this correlation was also more significant in treated mice. In contrast, neither the density of total intratumoral T cells in S.C. or I.P. tumors nor the density of intratumoral B cells in S.C. tumors correlated with day 14 tumor size under any treatment condition (Figure 7E; Table S1). These results suggested that TA-TLS are an important determinant of anti-tumor activity in this model. To provide support for this hypothesis, we treated TNFR1/2−/− mice bearing I.P. tumors in the same way. Both treatments again increased the number of intratumoral T and B cells at day 14 but did not promote TA-TLS development (Figures 7F and 7G). In contrast with WT mouse I.P. tumors, neither treatment reduced day 14 tumor size (Figure 7F), and the correlation between intratumoral B cell density and tumor size was lost (Figure 7H). These results demonstrate that checkpoint immunotherapy targets TA-TLS, leading to increases in their size and number. The extent of these changes is strongly correlated with tumor control. The potential importance of TA-TLS is heightened by a lack of correlation between tumor size and overall intratumoral T cell density.
Figure 7. TA-TLS number, size, and organization are augmented by checkpoint immunotherapy and correlated with tumor control.
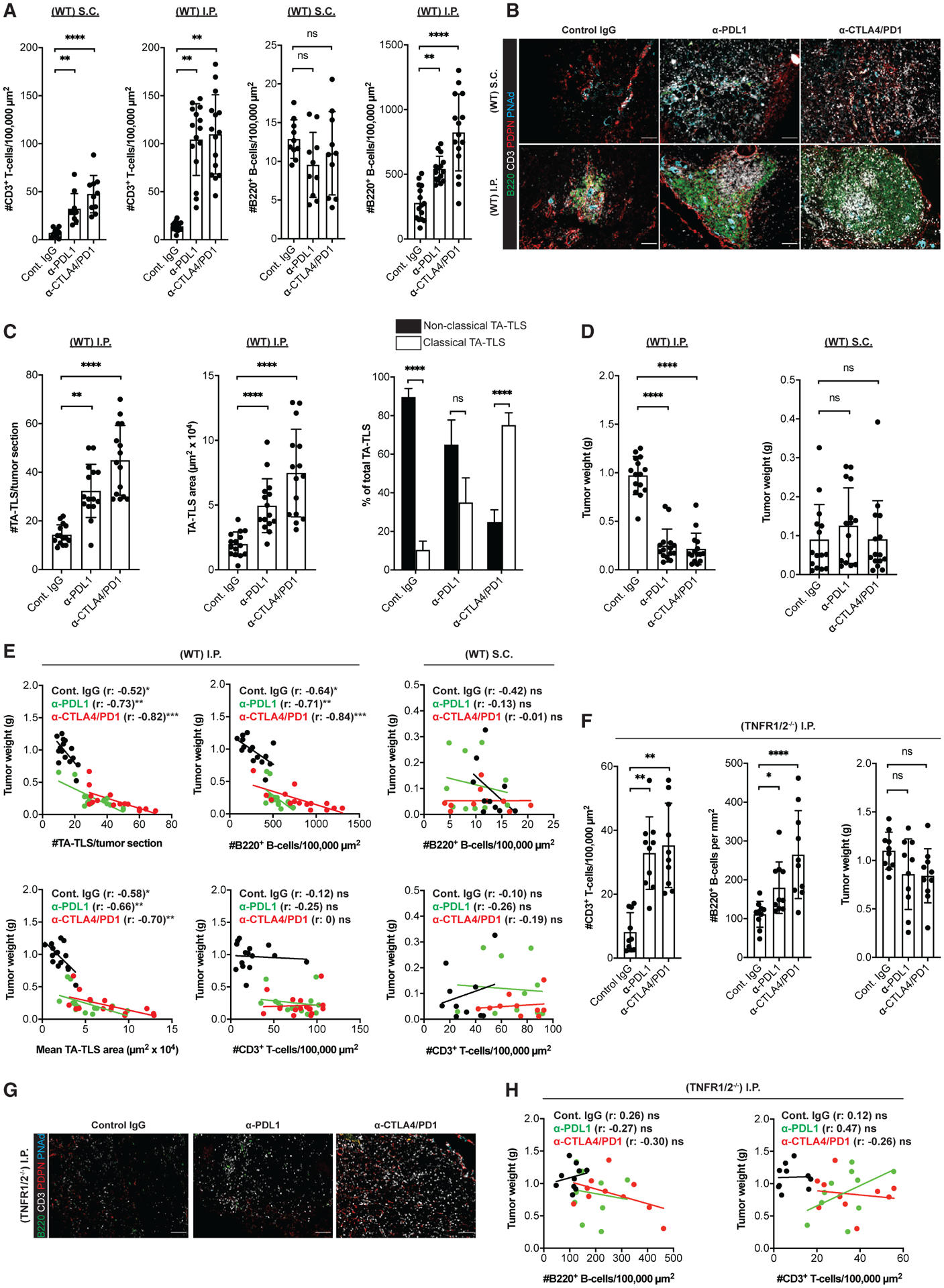
WT or TNFR1/2−/− mice were treated with control IgG, anti-PDL1, or anti-CTLA4/PD1 beginning 3 days after S.C. or I.P. tumor implantation. Tumors were harvested on day 14, weighed, and prepared for IF.
(A) Summary data for intratumoral parenchymal (non-TA-TLS) T and B cell densities in WT mouse tumors.
(B) Representative images showing typical TA-TLS size and organization in tumors from WT mice treated as indicated. Scale bar: 100 μm.
(C) Summary data of TA-TLS characteristics in tumors from WT mice. Classical TA-TLS are distinguished from non-classical by the presence of distinct T and B cell compartments.
(D) Tumor weights determined at harvest on day 14.
(E) Spearman correlation analysis of WT mouse tumor weights with TA-TLS number, size, or densities of intratumoral parenchymal T and B cells. Each dot represents an individual tumor.
(F) Left and middle: summary data for intratumoral parenchymal (non-TA-TLS) T and B cell densities in TNFR1/2−/− mouse tumors. Right: tumor weights were determined at harvest on day 14.
(G) Representative intratumoral images of i.p. tumors from TNFR1/2−/− mice treated as indicated. Scale bars: 100 μm.
(H) Spearman correlation analysis of TNFR1/2−/− mouse tumor weights with densities of T and B cells. Each dot represents an individual tumor.
(A–E) Data are from two to three experiments; n = 10–15 tumors per group. (F–H) Data are from two experiments; n = 10 tumors per group. Results shown as mean ± SD analyzed using Kruskal-Wallis H test with Dunn’s post-test (A, C, D, and F) or Spearman’s multilevel correlation analysis (E and H). nsp > 0.05, *p < 0.05, **p < 0.01.
DISCUSSION
This report identifies cellular and molecular mechanisms driving TA-TLS development in murine melanoma. A discrete population of CAF acquire LTo molecular characteristics in response to TNFR signals from a cell that is neither an adaptive nor innate immune lymphoid cell, and this population responds to intratumoral CD8 T cells and B cells that act coordinately as LTi cells. Although CD8 T cells promote initial aggregation and reticular network formation, B cells are recruited by CXCL13 expressing CAF, subsequently driving CAF proliferation and TA-TLS expansion through LTβR signaling. Some of these cellular and molecular elements are overrepresented in human TA-TLS and associated with one another. Lastly, TA-TLS number, size, and organization increase in response to checkpoint immunotherapy, and these aspects of TA-TLS are strongly correlated with reduced tumor size. This work provides a platform for manipulating TA-TLS formation as a cancer immunotherapy strategy.
Our findings identify a previously undescribed role of a population of FAPneg CAF as TA-TLS organizers. CAF that express elevated levels of FAP inversely correlate with patient survival, and their depletion results in diminished murine tumor outgrowth (Kraman et al., 2010). The numbers of FAP+ CAF in both S.C. and I.P. tumors were comparable, and these cells were rarely found in TA-TLS. In contrast, FAPneg CAF were elevated in I.P. tumors and the dominant population in TA-TLS reticular networks. FAPneg CAF promoted TA-TLS development in non-TA-TLS-containing S.C. tumors. The number and size of these structures was lower than in I.P. tumors, and this may reflect constraints imposed by the S.C. tumor microenvironment. Both FAPneg and FAP+ CAF in i.p. tumors co-expressed ICAM-1 and VCAM-1 that were significantly higher than those expressed by FAP+ CAF in S.C. tumors. In keeping with this, FAP+ CAF from I.P. tumors have a minimal LTo capability that is nonetheless greater than that of CAF from S.C tumors. Although PDPN+ cells expressing elevated levels of ICAM-1 and VCAM-1 have been documented in TLSs associated with chronically inflamed tissues or murine pancreatic carcinoma (Peduto et al., 2009; Link et al., 2011), only a single report has demonstrated that such fibroblasts could support TLS formation in a murine model of Sjögren’s syndrome, and these cells were FAP+ (Nayar et al., 2019). It is possible that FAP+ CAF from I.P. tumors express lower levels of CXCL13, BAFF, and APRIL than their FAPneg counterparts, presumably as a consequence of TNFR signaling. These FAP+ CAF may also be less proliferative than their FAPneg counterparts. However, neither of these possibilities explains the observed differential accumulation of FAPneg and FAP+ CAF in TA-TLS and parenchyma, respectively. Our findings point to the importance of continuing work to characterize CAF subpopulations in different human tumors.
CAF can originate by trans-differentiation from tumor or endothelial cells, but typically from tissue resident PDPN+ mesothelial cells (LeBleu and Kalluri, 2018; Liu et al., 2019). CAF in human and murine peritoneal carcinomas arise from PDPN+ mesothelial cells lining the peritoneal cavity (Rynne-Vidal et al., 2015). The omentum contains a substantial number of PDPN+ CXCL13+ mesothelial cells (Jackson-Jones et al., 2020), and we identified PDPN+ CXCL13+ cells in the spleen, lung, and pancreas. Any or all of these cells could contribute to CAF in I.P. tumors, but none showed the TNFR-dependent regulation of BAFF and APRIL of I.P. tumor CAF. PDPN+ cells from spleen, lung, and pancreas also did not show TNFR-dependent regulation of CXCL13. This suggests either another source of I.P. tumor CAF or changes in TNFR signaling in these populations under the influence of the tumor microenvironment. Regardless, the peritoneal cavity is a site for the growth or metastasis of many kinds of tumors, including melanoma (Kawashima et al., 1991; Trout et al., 2013). We also observed TA-TLS in tumors growing in lung, and PDPN+ CXCL13+ cells have been identified in several adipose tissues (Bénézech et al., 2015). Consequently, we believe that this I.P. tumor model reflects a physiologically relevant TA-TLS biology that should be explored further.
Our results also provided insight into the mechanisms used by the CAF population to promote TA-TLS development. CAF from I.P. tumors expressed substantially higher levels of CXCL13, BAFF, and APRIL than those from S.C. tumors, and an intact CXCR5-CXCL13 axis promoted B cell accumulation in tumors and increased proliferation of CAF and TA-TLS expansion. Although B cell follicles are disrupted in CXCR5−/− mice, the overall number of B cells in SLO is unaffected (Förster et al., 1996), making it likely that our result is due to a role for CXCL13 in promoting B cell accumulation. It is possible that CXCL13 enhances B cell proximity to CAF that express BAFF and APRIL, promoting B cell organization and/or survival in tumors. Elevated expression of CXCL13 is a common feature of TLS (Rangel-Moreno et al., 2007; Fleige et al., 2014; Barone et al., 2015; Sato et al., 2016), and fibroblasts purified from inflamed tissues containing TLS can express this molecule (Khader et al., 2011; Rangel-Moreno et al., 2011; Barone et al., 2015). The CXCR5-CXCL13 axis controls B cell accumulation in FALCs (Bénézech et al., 2015) and B cell organization, but not number, in TLS associated with a murine model of type 1 diabetes (Henry and Kendall, 2010). However, neither report identified the source of CXCL13. Although the precise mechanism by which CXCL13 promotes TLS development may vary based on anatomic location and/or immune stimulus, our results point to its central importance in TA-TLS formation in melanoma.
Our results also provide insight into the molecular and cellular control of CXCL13, BAFF, and APRIL expression in CAF. Expression of these molecules depended on signaling via TNFR, and not via LTβR, which controls them in SLO (Browning et al., 2005). Upregulation of CXCL13 in cultured smooth muscle cells from TLS-associated inflamed tissue was mediated by either LTβR or TNFR signaling alone, although the combination had a greater effect (Lötzer et al., 2010; Guedj et al., 2014). These differences point to the importance of determining the cellular sources of these LTo-associated molecules, as well as the molecules that control their expression in additional TLS models and human diseases. Interestingly, we found that the expression of CXCL13, BAFF, and APRIL was not dependent on adaptive immune cells, NK cells, or ILCs. We previously showed that CCL21 expression depended on TNFR ligands expressed by effector CD8 T cells (Peske et al., 2015). Thus, although TNFR signaling induces expression of all of these molecules, they depend on different cell types expressing TNFR ligands. The cells responsible for TNFR-induced upregulation in CAF may be present and active only transiently during the course of tumor outgrowth, prior to the entry of CD8 effectors capable of upregulating CCL21 and PNAd. Alternatively, because there are two TNFR isoforms that vary in their signaling pathways (MacEwan, 2002; Wajant and Siegmund, 2019) and their engagement with TNF-α and LTα3 (Medvedev et al., 1996), it is possible that these two cell types signal via distinct receptor-ligand pairs.
We found that effector CD8 T cells and LTα1β2+ B cells act as complementary LTi cells that coordinately induce TA-TLS development. While effector CD8 T cells promoted the organization of CAF into the reticular networks, along with upregulation of PNAd and CCL21 (Peske et al., 2015), LTα1β2+ B cells drove CAF accumulation and TA-TLS expansion. It has been shown that macrophages and NK cells can act as complementary LTi, although their exact roles were not defined (Bénézech et al., 2015). However, a role for effector CD8 T cells in promoting TLS development has not been previously reported. LTβR signaling promotes TLS development in several infection/inflammation models, but LTα1β2+ B cells have been shown to act as LTi only in development of isolated lymphoid follicles in small intestine, and this is independent of T cells (McDonald et al., 2005; Dubey et al., 2016). The presence of B cells in human tumors, particularly in association with TA-TLS, has a positive prognostic significance (Ladányi et al., 2011; Mahmoud et al., 2012; Cipponi et al., 2012; Germain et al., 2014), and it has been suggested that this is due to B cell activation and antibody production, or enhanced antigen-presenting cell function. Our results suggest that B cells might also promote tumor immunity by acting as LTi cells to drive TA-TLS development.
The cellular and molecular elements driving murine TA-TLS development were also evident in TA-TLS of human melanoma. We identified LTα+ B cells and APRIL+ and VCAM-1+ CAF as dominant components in human melanoma TA-TLS, and these phenotypes are suggestive of surrogate LTi and LTo cells, respectively. Supporting this, the densities of LTα+ B cells in TA-TLSs strongly correlated with the densities and fractions of CAF in TA-TLS expressing VCAM-1 and APRIL. How differences in cellular densities within TA-TLS influence their organization and functionality is not yet clear. Nonetheless, these data are consistent with our model in which LTα+ B cells and LTo CAF promote one another’s accumulation. The densities of B cells in TA-TLS strongly correlated with the fraction of CXCL13+ CAF, but not CXCL13+ CAF density. Also, CXCL13+ CAF were elevated in TA-TLS relative to parenchyma in only one of three melanomas, although the TA-TLS in two out of three melanomas contained elevated densities of CXCL13+ cells overall. This indicates the presence of alternative CXCL13-producing cells that are concentrated in the TA-TLS. CXCL13-expressing TFH cells and CD8 T cells have been identified in human breast tumors and non-small cell lung cancer, respectively (Gu-Trantien et al., 2017; Thommen et al., 2018), and it is likely that such cells are also found in melanoma TA-TLS. Thus, although CXCL13 is likely to be an important contributor to human TA-TLS development, its cellular source may evolve over time.
Recent publications reported that TA-TLS presence prior to treatment was associated with a favorable response to immunotherapy in patients with cancer (Griss et al., 2019; Cabrita et al., 2020; Petitprez et al., 2020; Helmink et al., 2020). Although two of these papers associated TA-TLS “maturity” with measurements of activated B cells, none correlated TA-TLS maturity and clinical responses. One paper demonstrated that checkpoint immunotherapy was associated with an increase in intratumoral B cells, but no evident change in the number of TA-TLS. We identified a strong correlation between tumor control in response to checkpoint immunotherapy and TA-TLS development. In several genetic models in which TA-TLS development was compromised, average I.P. tumor size was significantly greater. The consistency of this observation coupled with the diversity of immune defects targeted suggests an association of tumor control with TA-TLS development. The number and size of TA-TLS in WT mice were strongly correlated with tumor size, suggesting that TA-TLS played a role in limiting tumor outgrowth. Most importantly, checkpoint immunotherapy of I.P., but not S.C., tumor-bearing mice promoted tumor control, an increase in intratumoral B cell number, the development of more and larger TA-TLS, and the formation of TA-TLS with discrete T and B cell compartments. Intratumoral B cell number and TA-TLS size and number in checkpoint immunotherapy-treated mice were even more significantly correlated with a reduction in tumor size than in control mice. Although checkpoint immunotherapy also increased the overall number of intratumoral T cells, there was no correlation between these numbers and size of either I.P. or S.C. tumors. I.P. tumor control was lost in both untreated and treated TNFR1/2−/− mice, which lack TA-TLS, despite an increase in parenchymal T cell accumulation similar to that of WT mice. Multivariate studies in non-small cell lung cancer (Goc et al., 2014) and colorectal cancer (Di Caro et al., 2014) have also established a beneficial prognostic value of TA-TLS independent of TIL density. Although it is possible that checkpoint blockade-induced tumor control in TNFR1/2−/− mice is compromised for other reasons, these mice develop normal SLO and have a normal immune cell distribution and number (Peschon et al., 1998). Additionally, these mice generate adequate effector T cells (Peske et al., 2015), and total T cell infiltration into tumors in WT and TNFR1/2−/− mice was similar. We do not assert that S.C. B16-OVA tumors lacking TLS are entirely unresponsive to checkpoint immunotherapy, as there is evidence of this in other studies, albeit with considerable variation (Binder et al., 2013; Curran et al., 2010; Perez-Ruiz et al., 2019; Stark et al., 2017; Reilley et al., 2019; De Henau et al., 2016; Heidegger et al., 2019; Miller et al., 2019). Their response may be delayed relative to that of I.P. tumors that contain TA-TLS, or it may be quantitatively smaller. Regardless, in our model on day 14, it is simply not evident, either as an effect on tumor size or in the association between tumor size and the number of infiltrating B and T cells. We reach a similar conclusion regarding I.P. tumors grown in TNFR1/2−/− mice. Collectively, our work suggests that TA-TLS are major targets of checkpoint immunotherapy, and their presence and organization may be important determinants of the antitumor immune response engendered by this treatment. This conclusion supports and extends conclusions reached in correlative studies in patients.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information, and request for resources and reagents should be directed to the lead contact, Dr. Victor H. Engelhard (vhe@virginia.edu)
Materials availability
This study did not generate any unique reagents or mouse lines.
Data and code availability
This paper does not report original datasets.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
Mice used in this study and their respective vendor are detailed in Key resources table. All mice were bred and maintained on a C57BL/6 background, and kept in specific pathogen-free conditions. All experiments were carried out on female mice that were ~8–12 weeks of age. All protocols and experiments were approved by the University of Virginia Institutional Animal Care and Use Committee.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| eFluor 450 anti-mouse CD4 | ThermoFisher Scientific | Catalog #: 48-0042-82; Clone:RM4-5; RRID:AB_1272194 |
| Brilliant Violet 605 anti-mouse PD1 | Biolegend | Catalog #: 135220; Clone: 29F.1A12; RRID:AB_2562616 |
| Brilliant Violet 650 anti-mouse CD62L | Biolegend | Catalog #: 104453; Clone: MEL-14; RRID:AB_2800559 |
| Brilliant Violet 785 anti-mouse CD45 | Biolegend | Catalog #: 103149; Clone: 30-F11; RRID:AB_2564590 |
| FITC anti-mouse TIM-3 | ThermoFisher Scientific | Catalog #: 11-5870-82; Clone: RMT3-23; RRID:AB_2688129 |
| PerCP-eFluor 710 anti-mouse CD8 alpha | ThermoFisher Scientific | Catalog #: 46-0081-82; Clone: 53-6.7; RRID:AB_1834433 |
| PE anti-mouse LAG-3 | Biolegend | Catalog #: 125208; Clone: C9B7W; RRID:AB_2133343 |
| PE/Dazzle 594 anti-mouse CD44 | Biolegend | Catalog #: 103056; Clone: IM7; RRID:AB_2564044 |
| PE-Cy7 anti-mouse Ki67 | ThermoFisher Scientific | Catalog #: 25-5698-82; Clone: SolA15; RRID:AB_11220070 |
| Alexa Fluor 647 anti-mouse CD3 | Biolegend | Catalog #: 100209; Clone: 17A2; RRID:AB_389323 |
| APC-Cy7 anti-mouse CD19 | Biolegend | Catalog #: 115530; Clone: 6D5; RRID:AB_830707 |
| eFlour 450 anti-mouse IgM | ThermoFisher Scientific | Catalog #: 48-5890-82; Clone: eB121-15F9; RRID:AB_10671539 |
| PE/Dazzle 594 anti-mouse IgD | Biolegend | Catalog #: 405742; Clone: 11-26c.2a; RRID:AB_2571985 |
| PerCP-Cy5.5 anti-mouse CD11c | Biolegend | Catalog #: 117328; Clone: N418; RRID:AB_2129641 |
| PE anti-mouse I-A/I-E (MHC II) | Biolegend | Catalog #: 107608; Clone: M5/114.15.2; RRID:AB_313323 |
| Pacific Blue anti-mouse ICAM-1 (CD54) | Biolegend | Catalog #: 116116; Clone: YN1/1.7.4; RRID:AB_2121771 |
| Brilliant Violet 605 anti-mouse VCAM-1 (CD106) | BD Bioscience | Catalog #: 745193; Clone: 429; RRID:AB_2742789 |
| Alexa Fluor 488 polyclonal Collagen Type I | SoutherBiotech | Catalog #: 1310-30; Clone: Poly; RRID:AB_2794728 |
| PerCP-eFlour 710 anti-mouse CD31 | ThermoFisher Scientific | Catalog #: 46-0311-82; Clone: 390; RRID:AB_1834429 |
| Unconjugated anti-mouse alpha smooth muscle actin | ThermoFisher Scientific | Catalog #: 14-9760-82; Clone: 1A4; RRID:AB_2572996 |
| PE anti-mouse IgG2a | Biolegend | Catalog #: 407108; Clone: RMG2a-62; RRID:AB_10549456 |
| PE/Dazzle 594 anti-mouse CD140 alpha | Biolegend | Catalog #: 135922; Clone: APA5; RRID:AB_2814035 |
| PE-Cy7 anti-mouse podoplanin (gp38) | Biolegend | Catalog #: 127412; Clone: 8.1.1; RRID:AB_10613648 |
| eFlour 660 anti-mouse Ki67 | ThermoFisher Scientific | Catalog #: 50-5698-82; Clone: SolA15; RRID:AB_2574235 |
| Unconjugated anti-mouse FAP | Millipore Sigma | Catalog #: MABC1145; Clone: 73.3: RRID:AB_2892562 |
| APC-Cy7 anti-mouse IgG1 | Biolegend | Catalog #: 406620; Clone: RMG1-1; RRID:AB_2571947 |
| PE/Dazzle 594 anti-mouse PD-L1 | Biolegend | Catalog #: 124324; Clone: 10F.9G2; RRID:AB_2565639 |
| APC anti-mouse CD120 alpha (TNF R Type I/p55) | Biolegend | Catalog #: 113006; Clone: 55R-286; RRID:AB_2208779 |
| Brilliant Violet 421 anti-mouse CD120 beta (TNF R Type II/p75) | BD Bioscience | Catalog #: 564088; Clone: TR75-89; RRID:AB_2738585 |
| PE/Dazzle 594 anti-mouse CD90.2 (Thy-1.2) | Biolegend | Catalog #: 140329; Clone: 53-2.1; RRID:AB_2650965 |
| APC anti-mouse lymphotoxin beta receptor (LTβR) | Biolegend | Catalog #: 134408; Clone: 5G11; RRID:AB_2728151 |
| DyLight 550 Anti-Mouse/Human CD45R (B220) | Leinco Technologies (Discontinued) | Catalog #: C1944; Clone: RA3-6B2; RRID:AB_2829026 |
| Unconjugated anti-mouse Podoplanin (gp38) | Biolegend | Catalog #: 127402; Clone: 8.1.1; RRID:AB_1089187 |
| Biotin anti-mouse/human PNAd | Biolegend | Catalog #: 120804; Clone: MECA-79; RRID:AB_493557 |
| Alexa Fluor 488 anti-mouse CD45 | Biolegend | Catalog #: 103122; Clone: 30-F11; RRID:AB_493531 |
| Biotin anti-mouse ICAM-1 (CD54) | Biolegend | Catalog #: 116103; Clone: YN1/1.7.4; RRID:AB_313694 |
| Alexa Fluor 647 anti-mouse VCAM-1 (CD106) | Biolegend | Catalog #: 105712; Clone: 429; RRID:AB_493429 |
| Alexa Fluor 488 AffiniPure Goat Anti-Syrian Hamster IgG (H+L) | Jackson Labs | Catalog #: 107-545-142; Clone: Poly; RRID:AB_2337478 |
| Rhodamine (TRITC) AffiniPure Goat Anti-Syrian Hamster IgG (H+L) | Jackson Labs | Catalog #: 107-025-142; Clone: Poly; RRID:AB_2337452 |
| Rhodamine (TRITC) AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Labs | Catalog #: 715-025-150; Clone: Poly; RRID:AB_2340766 |
| Alexa Fluor 350 Streptavidin | ThermoFisher Scientific | Catalog #: S11249; Clone: N/A |
| InVivoPlus anti-mouse PD-L1 (B7-H1) | BioXcell | Catalog #: BP0101; Clone: 10F.9G2; RRID:AB_10949073 |
| InVivoPlus anti-mouse PD-1 (CD279) | BioXcell | Catalog #: BP0273; Clone: 29F.1A12; RRID:AB_2687796 |
| InVivoPlus anti-mouse CTLA-4 (CD152) | BioXcell | Catalog #: BP0032; Clone: UC10-4F10-11; RRID:AB_1107598 |
| Biotin anti-mouse Podoplanin | Biolegend | Catalog #: 127404; Clone: 8.1.1; RRID:AB_1134222 |
| Biotin anti-mouse CD31 | Biolegend | Catalog #: 102404; Clone: 390; RRID:AB_312899 |
| CD45 MicroBeads, mouse | Miltenyi Biotec | Catalog #: 130-097-153 |
| Anti-Biotin MicroBeads | Miltenyi Biotec | Catalog #: 130-097-046 |
| InVivomAb anti-mouse CD16/CD32 | BioXcell | Catalog #: BE0307; Clone: 2.4G2; RRID:AB_2736987 |
| Unconjugated anti-mouse/human Lymphotoxin alpha (LTA) | Abcam | Catalog #: ab100844; Clone: AT15A3; RRID:AB_10672573 |
| Unconjugated anti-human VCAM-1 | Nordic Biosite | Catalog #: LS-C391116-20; Clone: 1.4C3; RRID:AB_2892563 |
| Unconjugated anti-human CXCL13 | Novus Biological | Catalog #: NBP2-16041; Clone: Poly; RRID:AB_2892564 |
| Unconjugated anti-human/Rat/Mouse APRIL | Abcam | Catalog #: ab200836; Clone: EPR14588; RRID:AB_2892565 |
| Unconjugated anti-human gp36 | Abcam | Catalog #: ab236529; Clone: EPR22182; RRID:AB_2889051 |
| Unconjugated anti-human CD20 | Agilent | Catalog #: M075501-2; Clone: L26; RRID:AB_2892566 |
| Unconjugated anti-mouse CD4 | Abcam | Catalog #: ab183685; Clone: EPR19514; RRID:AB_2686917 |
| Unconjugated anti-mouse CD8 | Abcam | Catalog #: ab237723; Clone: CAL38; RRID:AB_2864723 |
| Unconjugated anti-mouse CD19 | Cell Signaling Technology | Catalog #: 90176S; Clone: D4V4B; RRID:AB_2800152 |
| Unconjugated anti-human/rat/mouse CD34 | Abcam | Catalog #: ab81289; Clone: EP373Y; RRID:AB_1640331 |
| Unconjugated anti-mouse CD11c | Abcam | Catalog #: ab219799; Clone: EPR21826; RRID:AB_2864725 |
| Biological samples | ||
| Melanoma tissue samples from patients | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant LTβR-Ig fusion protein | Wu etal. (1999) J. Exp. Med. 190, 629 | N/A |
| DNase I | Sigma | Catalog #: 11284932001 |
| Collagenase/Dispase | Sigma | Catalog #: 11097113001 |
| Collagenase P | Sigma | Catalog #: 11213865001 |
| Red Blood Cell Lysing Buffer Hybri-Max | Sigma | Catalog #: R7757-100ML |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | ThermoFisher Scientific | Catalog #: L34966 |
| 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI) | Sigma | Catalog #: 10236276001 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma | Catalog #: EDS-1KG |
| Sodium azide | Sigma | Catalog #: S2002-500G |
| Methanol | Sigma | Catalog #: 34860-1L-R |
| Formalin solution, neutral buffered, 10% | Sigma | Catalog #: HT501128-4L |
| ProLong Gold Antifade Mountant with DAPI | ThermoFisher Scientific | Catalog #: P36931 |
| ProLong Gold Antifade Mountant | ThermoFisher Scientific | Catalog #: P36930 |
| Critical commercial assays | ||
| Fixation/Permeabilization Solution Kit | BD Bioscience | Catalog #: 554714 |
| Transcription Factor Buffer Set | BD Bioscience | Catalog #: 562574 |
| RNAprotect Cell Reagent | QIAGEN | Catalog #: 76526 |
| RPMI 1640 Medium (Mod.) 1X with L-Glutamine | Fisher Scientific | Catalog #: MT-10043CV |
| DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate | Fisher Scientific | Catalog #: MT10013CV |
| IMDM, GlutaMAX | ThermoFisher Scientific | Catalog #: 31980030 |
| L-Glutamine (200 mM) | ThermoFisher Scientific | Catalog #: 25030081 |
| HEPES (1 M) | ThermoFisher Scientific | Catalog #: 15630080 |
| CD8a T Cell Isolation Kit, mouse | Miltenyi Biotec | Catalog #: 130-104-075 |
| B Cell Isolation Kit, mouse | Miltenyi Biotec | Catalog #: 130-090-862 |
| Biotin Tyramide signal amplification | PerkinElmer | Catalog #: NEL700001KT |
| Avidin/Biotin Blocking Kit | Vector Laboratories | Catalog #: SP-2001 |
| Opal 7-Color IHC Kits | Akoya Biosciences | Catalog #: NEL811001KT |
| AR9 Antigen Retrieval Buffer | Akoya Biosciences | Catalog #: AR9001KT |
| RNeasy Micro Kit | QIAGEN | Catalog #: 74004 |
| RNeasy Mini Kit | QIAGEN | Catalog #: 74104 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher Scientific | Catalog #: 4368814 |
| TaqMan Fast Advanced Master Mix | ThermoFisher Scientific | Catalog #: 4444557 |
| Experimental models: Cell lines | ||
| B16-OVA | Hargadon et al. (2006) J. Immunol. 177, 6081 | N/A |
| MC38-OVA | Hargadon et al. (2006) J. Immunol. 177, 6081 | N/A |
| LLC-OVA | Gift of E. Podack (Univ of Miami) | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6 | Charles River/NCI | Catalog #: 027 |
| Rag1−/− | Jackson Labs | Catalog #: 002216 |
| μMT−/− | Jackson Labs | Catalog #: 002288 |
| LTa−/− | Jackson Labs | Catalog #: 002258 |
| TNFR1/2−/− | Jackson Labs | Catalog #: 003243 |
| Human ubiquitin C Promoter (UBC)-mCherry | Jackson Labs | Catalog #: 017614 |
| CXCR5−/− | Jackson Labs | Catalog #: 006659 |
| Rag2−/−Il2rg−/− | Taconic Labs | Catalog #: 4111-F |
| Oligonucleotides | ||
| CCL21a | ThermoFisher Scientific | Assay ID: Mm03646971_gH |
| CXCL12 | ThermoFisher Scientific | Assay ID: Mm00445552_m1 |
| CXCL13 | ThermoFisher Scientific | Assay ID: Mm00444533_m1 |
| TNFSF13 (APRIL) | ThermoFisher Scientific | Assay ID: Mm03809849_s1 |
| TNFSF13b (BAFF) | ThermoFisher Scientific | Assay ID: Mm01218923_g1 |
| Software and algorithms | ||
| FlowJo v10.7 software | BD Bioscience | https://www.flowjo.com/ |
| ImageJ software | NIH | https://imagej.nih.gov/ij/ |
| Imaris image analysis software | Bitplane | https://imaris.oxinst.com/ |
| Photoshop 2021 | Adobe | https://www.adobe.com/ |
| Vectra 3 Automated Quantitative Pathology Imaging System | Akoya Biosciences | https://www.akoyabio.com/ |
| Phenochart Whole Slide Viewer | Akoya Biosciences | https://www.akoyabio.com/ |
| InForm Tissue Analysis Software | Akoya Biosciences | https://www.akoyabio.com/ |
| HALO Image Analysis Platform Software | IndicaLabs | https://indicalab.com/halo/ |
| QuantStudio 6 Flex Real-Time PCR system | Applied Biosystem | https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-instruments/quantstudio-systems/models/quantstudio-6-7-flex.html |
| R version 4 | R Studios | https://www.r-project.org/ |
| Prism version 7.0 | GraphPad | https://www.graphpad.com/ |
| SAS software suite version 9.4 | SAS | https://www.sas.com/en_us/home.html |
Tumor models
B16-F1 mouse melanoma and MC38 murine colon adenocarcinoma cells were obtained from American Type Culture Collection (ATCC). The generation of B16-OVA and MC38-OVA has previously been described (Hargadon et al., 2006). B16-OVA tumor cells were cultured in RPMI-1640 (Corning) supplemented to a final concentration of 10% (v/v) fetal bovine serum (FBS), 2 mM L-glutamine (ThermoFisher Scientific) and 15 mM HEPES (ThermoFisher Scientific). MC38-OVA were cultured in DMEM (Corning) supplemented to a final concentration of 10% (v/v) FBS, 2 mM L-glutamine (ThermoFisher Scientific) and 15 mM HEPES (ThermoFisher Scientific). Lewis lung carcinoma transfected to express ovalbumin (LLC-OVA) was a gift of Dr. E. Podack (University of Miami). These cells were cultured in IMDM (ThermoFisher Scientific) supplemented to a final concentration of 10% (v/v) fetal bovine serum, 2 mM L-glutamine (ThermoFisher Scientific) and 15 mM HEPES (ThermoFisher Scientific). All tumor cells were cultured and maintained at 37°C and 5% carbon dioxide. For tumor studies, approximately 4 × 105 tumor cells were I.P. or S.C. (loose neck scruff) injected into recipient mice. Both tumor types were allowed to establish for 14 days prior to harvesting.
METHOD DETAILS
Treatment of tumor-bearing mice
For LTβR blocking experiments, LTβR-Ig fusion protein (Wu et al., 1999) (100 μg per injection, gift from Y.X.F) was injected I.P. into C57BL/6 mice one day prior and one day after tumor implantation, and then every four days until tumor harvest. For checkpoint immunotherapy experiments, either monotherapy α-PDL1 (250 μg per injection, 10F.9G2, BioXcell) or dual therapy α-PD1 (250 μg per injection, RMP1–14, BioXcell) and α-CTLA4 (250 μg per injection, 9D9, BioXcell) was injected I.P. into C57BL/6 mice three days after tumor implantation and then every three days until tumor harvest.
Adoptive transfer of T cells and B cells
Lymph nodes and/or spleen were resected from either C57BL/6 or LTα−/− mice. Single-cell suspensions of resected lymphoid organs were prepared via Dounce homogenization. Bulk CD8 T cells and CD19 B cells were purified from pooled lymph nodes and/or spleen suspensions by magnetic bead isolation kits (Miltenyi Biotec) on an AutoMACS Pro Separator (Miltenyi Biotec) according to manufactures instructions. CD8 T cells (5 × 106), CD19 B cells (5 × 106), or the combination were injected intravenously into Rag1−/− recipient mice 3 days prior to tumor implantation.
Digestion of resected tissues
Resected lymph nodes, spleen, omentum, lung, pancreas, skin, and tumors were minced and digested with a solution containing 0.1 mg/ml DNase I (Sigma), 0.8 mg/ml Collagenase Dispase (Sigma), and 0.2 mg/ml Collagenase P (Sigma) for 30 minutes at 37°C. Every 5 minutes, tissue suspensions were pipetted up-and-down several times. Digested tissues were depleted of red blood cells by Red Blood Cell Lysing Buffer Hybri-Max (Sigma) according to manufactures instructions.
Enrichment of cells from digested tissues
Hematopoietic cells from digested tumor suspensions were enriched by CD45 magnetic beads (Miltenyi Biotec) on an AutoMACS Pro Separator (Miltenyi Biotec) according to manufactures instructions. In some cases, hematopoietic depleted tissue suspensions were stained with α-gp38 (0.5 μg/mL) biotinylated antibody for 15 minutes at 4°C. Then, CAF and tissue resident fibroblasts were enriched from stained hematopoietic depleted suspensions by α-biotin magnetic beads (Miltenyi Biotec) on an AutoMACS Pro Separator (Miltenyi Biotec) according to manufactures instructions.
Flow cytometry and cell sorting
Antibodies used for flow cytometry analysis are detailed in Key resources table. Cell surface staining was done in PBS containing 2% FBS, 2 mM EDTA (Sigma), and 2 mM NaN3 (Sigma) for 30 minutes at 4°C. For the detection of intracellular proteins, Cytofix/Cytoperm Fixation/Permeabilization kit (BD Bioscience) or Transcription Faactor Buffer Set (BD Bioscience) was used according to the manufacturer’s instructions. Live/Dead Aqua (Invitrogen) or 4,6-diamidino-2-phenylindole (Sigma) were used to exclude dead cells from analysis. In all experiments, CAF and/or tissue derived fibroblasts were defined as live, singlet, Ter119neg, CD45neg, CD31neg, PDPNhi cells. B16 tumor cells express ten-fold lower levels of PDPN. Thus, only PDPNhi cells were analyzed as CAF in all experiments. Samples were run on a FACSCanto II (BD) or Attune NxT (ThermoFisher/Invitrogen) and analyzed using FlowJo Software (BD Bioscience). For quantitative real-time PCR experiments, pre-enriched CAF and/or organ derived fibroblasts were sorted to high purity on an Influx Cell Sorter (BD) directly into RNAProtect (QIAGEN) or PBS.
S.C. tumor model with exogenous fibroblasts
B16-OVA cells were S.C. or I.P. injected into female C57BL/6 or UBC-mCherry recipient mice and allowed to establish for 14 days. Preparation of single cell suspensions from resected tumor masses is described above. Live, singlet, Ter119−, CD45−, CD31−, PDPNhi CAF from pre-enriched suspensions of bulk PDPN+ CAF were sorted to high purity on Influx Cell Sorter (BD) directly into DMEM (Corning) supplemented to a final concentration of 10% (v/v) FBS, 2 mM L-glutamine (ThermoFisher Scientific) and 15 mM HEPES (ThermoFisher Scientific). In some cases, FAP+ PDPNhi and FAPneg PDPNhi CAF were purified from a pre-enriched suspension of bulk PDPN+ CAFs were sorted to high purity on Influx Cell Sorter (BD) directly into DMEM (Corning) supplemented to a final concentration of 10% (v/v) FBS, 2 mM L-glutamine (ThermoFisher Scientific) and 15 mM HEPES (ThermoFisher Scientific). In all cases, B16-OVA cells (4 × 105) and purified CAF (5 × 104) or mouse embryonic fibroblasts (5 × 104) were S.C. injected into female C57BL/6 mice. Tumor were allowed to establish for 14 days prior to harvesting.
Immunofluorescence microscopy
Antibodies used for immunofluorescence microscopy (IF) analysis are detailed in Key resources table. Preparation of tumor tissue for frozen sectioning, immunofluorescence staining procedures, and Tyramide signal amplification (PerkinElmer) for PNAd detections has previously been described (Rodriguez et al., 2018). For image acquisition, 10–15 low magnification images per tumor section were used for TA-TLS, CAF and intratumoral T-/B cell quantification. For the detection of intratumoral CAF, exposure times and thresholds were set to visualize bright PDPN+ cells located in perivascular and TA-TLS areas, while eliminating the dim tumor cell signal. For CAF and LTo marker expression on intratumoral fibroblasts, the average of five high magnification images per parenchyma and TA-TLS area of a tumor section were used for quantification. All images were captured on an AxioImager with Apotome (Zeiss) and images were analyzed either by ImageJ Software (NIH) or Imaris image analysis software (Bitplane). For image presentation, brightness and contrast were linearly adjusted and color-merged images were generated using Photoshop CS6 Software (Adobe).
Definition of TA-TLS by IF
In I.P. tumors grown in C57BL/6, CXCR5−/−, LTβR-Ig and checkpoint treated mice, and in S.C. tumors containing PDPN+ CAF or mouse embryonic fibroblasts, intratumoral TA-TLS were identified as tight aggregates of 50 or more B cells in juxtaposition to PNAd+ vasculature and associated with a reticular network of elongated PDPN+ CAF that are not tightly apposed to tumor endothelium (Rodriguez et al., 2018). T cells and DC were either intermingled with B cell aggregates (non-classical structure) or organized as a separate zone (classical structure). Using an AxioImager with Apotome (Zeiss) and ImageJ Software (NIH) or Imaris image analysis software (Bitplane), B cell and PDPN staining pattern were used to draw a boundary of the TA-TLS, and quantitate its surface area.
In I.P. tumors grown in μMT−/−, TNFR1/2−/−, or in I.P. tumors grown in Rag1−/− mice repleted with bulk CD8 T cells and/or B cells, we identified tight clusters of CD45+ –hematopoietic cells associated with a network of elongated PDPN+ CAF that were not tightly apposed to tumor endothelium as elements of TA-TLS development. Using an AxioImager with Apotome (Zeiss) and ImageJ Software (NIH), clusters of CD45+ and PDPN+ staining pattern is used to draw a boundary and quantitate its surface area.
Multispectral Imaging
Antibodies used for multispectral imaging analysis are detailed in Key resources table. Staining of murine and human melanoma for multispectral imaging has previously been described (Mauldin et al., 2020). In brief, 4-μm thick sections were cut from formalin fixed paraffin embedded murine tumor specimens or human melanoma biopsies, and murine spleen and human lymph node biopsies was used as a positive control. Antigen retrieval was performed using AR9 buffer (PerkinElmer) according to the manufacturer instructions. OPAL Multiplex IHC Staining (PerkinElmer) was performed according to the manufacturer instructions. Slides were mounted using prolong diamond antifade (Life Technologies) and scanned at low magnification using the Vectra 3.0 system and software (PerkinElmer). Regions of interest were identified in Phenochart software, and high-powered magnification images were acquired with the Vectra 3.0 system (Akoya Biosciences). These images were spectrally unmixed using single stain positive controls and analyzed using the InForm software (PerkinElmer). Human specimen images were quantified using HALO software (Indica Labs, Albuquerque, NM) and murine specimens were quantified by eye.
Quantitative real-time PCR
Probes used for quantitative real-time PCR analysis are detailed in Key resources table. RNA was purified from whole tumor or sorted cells using RNAEasy kits (QIAGEN) according to manufactures instructions. High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and purified RNA was used to generate cDNA. Amplification was performed using TaqMan Fast Advanced Master Mix (Applied Biosystems) and QuantStudio 6 Flex Real-Time PCR system (Applied Biosystems) with the following program: 50°C for 2 minutes; 95°C for 2 minutes; 40 cycles of 95°C for 1 s, 60°C for 20 s. Data is presented as 2−ΔCt relative to Hprt.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of each experiment in this work are reported in the main and supplementary figure legends. P values are shown in each figure, and defined in the associated figure legend. Normality of data distribution was determined by D’Agostino-Pearson omnibus normality test and variance between groups was assessed by the F-test. P values for the comparison between two or more independent groups were calculated by Welch’s t test and Kruskal-Wallis h-test with Dunn’s post-test, respectively. For human boxplot data, the Wilcoxon rank-sum test was used for non-parametric comparison of two independent groups, and P values were corrected by controlling the False Discovery Rate (FDR) according to the method of Benjamini and Hochberg. For human correlation analysis, a linear mixed-models approach available in the R package Correlations (Makowski et al., 2020) was used in order to account for the nested data structure (multiple data points taken from an independent tumor). For murine checkpoint immunotherapy correlation analysis, a Spearman correlation analysis with 95% confidence intervals was used for the non-parametric comparison of two independent groups. In most cases, error bars shown in graphical data represents mean ± standard deviation (s.d.) for normally distributed data or median ± IQR for non-normal data. For human boxplots, data were generated using Tukey’s method. p < 0.05 was considered statistically significant. All graphs and statistics were calculated using Graph Pad Prism version 7.0, R version 4, and the SAS software suite version 9.4.
Supplementary Material
Highlights.
Cancer-associated fibroblasts orchestrate tertiary lymphoid structures in tumors
CD8 T cells and B cells drive tertiary lymphoid structure development via fibroblasts
Tertiary lymphoid structure development is mediated by TNF and lymphotoxin receptors
Checkpoint blockade alters tertiary lymphoid structures together with tumor control
ACKNOWLEDGMENTS
We thank the Engelhard and Dudley labs for discussion and suggestions. This work was supported by USPHS Grants CA78400 and CA181794 (to V.H.E.) and the University of Virginia Cancer Center Schiff Foundation (to C.L.S. and I.S.M.). The University of Virginia Histology and Flow Cytometry Cores are supported by USPHS Grant P30 CA44579. A.B.R. and A.N.W. were supported by USPHS Training Grant AI007496, J.D.P. by USPHS Training Grant GM007267, and K.M.L. by USPHS Training Grant CA163177. J.D.P. was the recipient of a Farrow Fellowship, and A.B.R. was the recipient of a Wagner Fellowship, both from the University of Virginia.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109422.
REFERENCES
- Barone F, Nayar S, Campos J, Cloake T, Withers DR, Toellner K-M, Zhang Y, Fouser L, Fisher B, Bowman S, et al. (2015). IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc. Natl. Acad. Sci. USA 112, 11024–11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénézech C, Luu N-T, Walker JA, Kruglov AA, Loo Y, Nakamura K, Zhang Y, Nayar S, Jones LH, Flores-Langarica A, et al. (2015). Inflammation-induced formation of fat-associated lymphoid clusters. Nat. Immunol 16, 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DC, Engels B, Arina A, Yu P, Slauch JM, Fu Y-X, Karrison T, Burnette B, Idel C, Zhao M, et al. (2013). Antigen-specific bacterial vaccine combined with anti-PD-L1 rescues dysfunctional endogenous T cells to reject long-established cancer. Cancer Immunol. Res 1, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombardieri M, Barone F, Humby F, Kelly S, McGurk M, Morgan P, Challacombe S, De Vita S, Valesini G, Spencer J, and Pitzalis C (2007). Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in Sjögren’s syndrome. J. Immunol 179, 4929–4938. [DOI] [PubMed] [Google Scholar]
- Browning JL, Allaire N, Ngam-Ek A, Notidis E, Hunt J, Perrin S, and Fava RA (2005). Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity 23, 539–550. [DOI] [PubMed] [Google Scholar]
- Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. (2020). Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565. [DOI] [PubMed] [Google Scholar]
- Cañete JD, Celis R, Yeremenko N, Sanmartí R, van Duivenvoorde L, Ramírez J, Blijdorp I, García-Herrero CM, Pablos JL, and Baeten DL (2015). Ectopic lymphoid neogenesis is strongly associated with activation of the IL-23 pathway in rheumatoid synovitis. Arthritis Res. Ther 17, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipponi A, Mercier M, Seremet T, Baurain J-F, Théate I, van den Oord J, Stas M, Boon T, Coulie PG, and van Baren N (2012). Neogenesis of lymphoid structures and antibody responses occur in human melanoma metastases. Cancer Res. 72, 3997–4007. [DOI] [PubMed] [Google Scholar]
- Cruz-Migoni S, and Caamaño J (2016). Fat-associated lymphoid clusters in inflammation and immunity. Front. Immunol 7, 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, and Allison JP (2010). PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 107, 4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, et al. (2016). Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 539, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A, Laghi L, Allavena P, Mantovani A, and Marchesi F (2014). Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res 20, 2147–2158. [DOI] [PubMed] [Google Scholar]
- Drayton DL, Ying X, Lee J, Lesslauer W, and Ruddle NH (2003). Ectopic LT α β directs lymphoid organ neogenesis with concomitant expression of peripheral node addressin and a HEV-restricted sulfotransferase. J. Exp. Med 197, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey LK, Lebon L, Mosconi I, Yang C-Y, Scandella E, Ludewig B, Luther SA, and Harris NL (2016). Lymphotoxin-Dependent B Cell-FRC Crosstalk Promotes De Novo Follicle Formation and Antibody Production following Intestinal Helminth Infection. Cell Rep. 15, 1527–1541. [DOI] [PubMed] [Google Scholar]
- Engelhard VH, Rodriguez AB, Mauldin IS, Woods AN, Peske JD, and Slingluff CL Jr. (2018). Immune Cell Infiltration and Tertiary Lymphoid Structures as Determinants of Antitumor Immunity. J. Immunol 200, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, Browning JL, Goossens N, Nakagawa S, Gunasekaran G, et al. (2015). Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol 16, 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleige H, Ravens S, Moschovakis GL, Bölter J, Willenzon S, Sutter G, Häussler S, Kalinke U, Prinz I, and Förster R (2014). IL-17-induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs. J. Exp. Med 211, 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster R, Mattis AE, Kremmer E, Wolf E, Brem G, and Lipp M (1996). A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 87, 1037–1047. [DOI] [PubMed] [Google Scholar]
- Furtado GC, Marinkovic T, Martin AP, Garin A, Hoch B, Hubner W, Chen BK, Genden E, Skobe M, and Lira SA (2007). Lymphotoxin beta receptor signaling is required for inflammatory lymphangiogenesis in the thyroid. Proc. Natl. Acad. Sci. USA 104, 5026–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furtado GC, Pacer ME, Bongers G, Bénézech C, He Z, Chen L, Berin MC, Kollias G, Caamaño JH, and Lira SA (2014). TNFα-dependent development of lymphoid tissue in the absence of RORγt+ lymphoid tissue inducer cells. Mucosal Immunol. 7, 602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatumu MK, Skarstein K, Papandile A, Browning JL, Fava RA, and Bolstad AI (2009). Blockade of lymphotoxin-beta receptor signaling reduces aspects of Sjögren’s syndrome in salivary glands of non-obese diabetic mice. Arthritis Res. Ther 11, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, Lepelley A, Becht E, Katsahian S, Bizouard G, et al. (2014). Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am. J. Respir. Crit. Care Med 189, 832–844. [DOI] [PubMed] [Google Scholar]
- GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D, Osterhaus AD, Hendriks R, Rimmelzwaan GF, and Lambrecht BN (2009). Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J. Exp. Med 206, 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goc J, Germain C, Vo-Bourgais TKD, Lupo A, Klein C, Knockaert S, de Chaisemartin L, Ouakrim H, Becht E, Alifano M, et al. (2014). Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. 74, 705–715. [DOI] [PubMed] [Google Scholar]
- Goya S, Matsuoka H, Mori M, Morishita H, Kida H, Kobashi Y, Kato T, Taguchi Y, Osaki T, Tachibana I, et al. (2003). Sustained interleukin-6 signalling leads to the development of lymphoid organ-like structures in the lung. J. Pathol 200, 82–87. [DOI] [PubMed] [Google Scholar]
- Gräbner R, Lötzer K, Döpping S, Hildner M, Radke D, Beer M, Spanbroek R, Lippert B, Reardon CA, Getz GS, et al. (2009). Lymphotoxin β receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J. Exp. Med 206, 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier-Pfisters-hammer K, Maurer-Granofszky M, Roka F, Penz T, Bock C, et al. (2019). B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun 10, 4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu-Trantien C, Migliori E, Buisseret L, de Wind A, Brohée S, Garaud S, Noël G, Dang Chi VL, Lodewyckx J-N, Naveaux C, et al. (2017). CXCL13-producing TFH cells link immune suppression and adaptive memory in human breast cancer. JCI Insight 2, e91487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedj K, Khallou-Laschet J, Clement M, Morvan M, Gaston A-T, Fornasa G, Dai J, Gervais-Taurel M, Eberl G, Michel J-B, et al. (2014). M1 macrophages act as LTβR-independent lymphoid tissue inducer cells during atherosclerosis-related lymphoid neogenesis. Cardiovasc. Res 101, 434–443. [DOI] [PubMed] [Google Scholar]
- Halle S, Dujardin HC, Bakocevic N, Fleige H, Danzer H, Willenzon S, Suezer Y, Hämmerling G, Garbi N, Sutter G, et al. (2009). Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J. Exp. Med 206, 2593–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargadon KM, Brinkman CC, Sheasley-O’neill SL, Nichols LA, Bullock TNJ, and Engelhard VH (2006). Incomplete differentiation of antigen-specific CD8 T cells in tumor-draining lymph nodes. J. Immunol 177, 6081–6090. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Hardy RR, Herzenberg LA, and Herzenberg LA (1985). Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J. Exp. Med 161, 1554–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidegger S, Wintges A, Stritzke F, Bek S, Steiger K, Koenig P-A, Göttert S, Engleitner T, Öllinger R, Nedelko T, et al. (2019). RIG-I activation is critical for responsiveness to checkpoint blockade. Sci. Immunol 4, eaau8943. [DOI] [PubMed] [Google Scholar]
- Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. (2020). B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RA, and Kendall PL (2010). CXCL13 blockade disrupts B lymphocyte organization in tertiary lymphoid structures without altering B cell receptor bias or preventing diabetes in nonobese diabetic mice. J. Immunol 185, 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Jones LH, Smith P, Portman JR, Magalhaes MS, Mylonas KJ, Vermeren MM, Nixon M, Henderson BEP, Dobie R, Vermeren S, et al. (2020). Stromal Cells Covering Omental Fat-Associated Lymphoid Clusters Trigger Formation of Neutrophil Aggregates to Capture Peritoneal Contaminants. Immunity 52, 700–715.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson-Percival A, He B, Li Z-J, Kjellén A, Russell K, Li J, Larma I, and Ganss R (2017). De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat. Immunol 18, 1207–1217. [DOI] [PubMed] [Google Scholar]
- Joshi NS, Akama-Garren EH, Lu Y, Lee D-Y, Chang GP, Li A, Du-Page M, Tammela T, Kerper NR, Farago AF, et al. (2015). Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity 43, 579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima A, Fishman EK, Kuhlman JE, and Schuchter LM (1991). CT of malignant melanoma: patterns of small bowel and mesenteric involvement. J. Comput. Assist. Tomogr 15, 570–574. [DOI] [PubMed] [Google Scholar]
- Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Junecko BAF, Fountain JJ, Martino C, Pearl JE, Tighe M, Lin YY, et al. (2011). IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J. Immunol 187, 5402–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-J, Kammertoens T, Janke M, Schmetzer O, Qin Z, Berek C, and Blankenstein T (2004). Establishment of early lymphoid organ infrastructure in transplanted tumors mediated by local production of lymphotoxin α and in the combined absence of functional B and T cells. J. Immunol 172, 4037–4047. [DOI] [PubMed] [Google Scholar]
- Kirk CJ, Hartigan-O’Connor D, and Mulé JJ (2001). The dynamics of the T-cell antitumor response: chemokine-secreting dendritic cells can prime tumor-reactive T cells extranodally. Cancer Res. 61, 8794–8802. [PubMed] [Google Scholar]
- Koenig A, and Thaunat O (2016). Lymphoid Neogenesis and Tertiary Lymphoid Organs in Transplanted Organs. Front. Immunol 7, 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, and Fearon DT (2010). Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330, 827–830. [DOI] [PubMed] [Google Scholar]
- Ladányi A, Kiss J, Mohos A, Somlai B, Liszkay G, Gilde K, Fejös Z, Gaudi I, Dobos J, and Tímár J (2011). Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol. Immunother 60, 1729–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, and Kalluri R (2018). A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis. Model. Mech 11, dmm029447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, McGarry MP, Farmer SC, Denzler KL, Larson KA, Carrigan PE, Brenneise IE, Horton MA, Haczku A, Gelfand EW, et al. (1997). Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J. Exp. Med 185, 2143–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link A, Hardie DL, Favre S, Britschgi MR, Adams DH, Sixt M, Cyster JG, Buckley CD, and Luther SA (2011). Association of T-zone reticular networks and conduits with ectopic lymphoid tissues in mice and humans. Am. J. Pathol 178, 1662–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, and Yin R (2019). Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J. Hematol. Oncol 12, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lötzer K, Döpping S, Connert S, Gräbner R, Spanbroek R, Lemser B, Beer M, Hildner M, Hehlgans T, van der Wall M, et al. (2010). Mouse aorta smooth muscle cells differentiate into lymphoid tissue organizer-like cells on combined tumor necrosis factor receptor-1/lymphotoxin β-receptor NF-kappaB signaling. Arterioscler. Thromb. Vasc. Biol 30, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, Solt S, Dorman A, Wamwea A, Yager A, et al. (2014). Immunotherapy converts non-immunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res 2, 616–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEwan DJ (2002). TNF receptor subtype signalling: differences and cellular consequences. Cell. Signal 14, 477–492. [DOI] [PubMed] [Google Scholar]
- Mahmoud SMA, Lee AHS, Paish EC, Macmillan RD, Ellis IO, and Green AR (2012). The prognostic significance of B lymphocytes in invasive carcinoma of the breast. Breast Cancer Res. Treat 132, 545–553. [DOI] [PubMed] [Google Scholar]
- Makowski D, Ben-Shachar MS, Patil I, and Lüdecke D (2020). Methods and Algorithms for Correlation Analysis in R. J. Open Source Softw 5, 2306. [Google Scholar]
- Maldonado L, Teague JE, Morrow MP, Jotova I, Wu TC, Wang C, Desmarais C, Boyer JD, Tycko B, Robins HS, et al. (2014). Intramuscular Therapeutic Vaccination Targeting HPV16 Induces T Cell Responses That Localize in Mucosal Lesions. Sci. Transl. Med 6, 221ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauldin IS, Sheybani ND, Young SJ, Price RJ, and Slingluff CL (2020). Deconvolution of the immunological contexture of mouse tumors with multiplexed immunohistochemistry. In Methods in Enzymology, Galluzzi L and Rudqvist N-P, eds. (Academic Press; ), pp. 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald KG, McDonough JS, and Newberry RD (2005). Adaptive immune responses are dispensable for isolated lymphoid follicle formation: antigen-naive, lymphotoxin-sufficient B lymphocytes drive the formation of mature isolated lymphoid follicles. J. Immunol 174, 5720–5728. [DOI] [PubMed] [Google Scholar]
- Mebius RE (2003). Organogenesis of lymphoid tissues. Nat. Rev. Immunol 3, 292–303. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Espevik T, Ranges G, and Sundan A (1996). Distinct roles of the two tumor necrosis factor (TNF) receptors in modulating TNF and lymphotoxin alpha effects. J. Biol. Chem 271, 9778–9784. [DOI] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motallebzadeh R, Rehakova S, Conlon TM, Win TS, Callaghan CJ, Goddard M, Bolton EM, Ruddle NH, Bradley JA, and Pettigrew GJ (2012). Blocking lymphotoxin signaling abrogates the development of ectopic lymphoid tissue within cardiac allografts and inhibits effector antibody responses. FASEB J. 26, 51–62. [DOI] [PubMed] [Google Scholar]
- Mueller CG, Nayar S, Campos J, and Barone F (2018). Molecular and Cellular Requirements for the Assembly of Tertiary Lymphoid Structures. Adv. Exp. Med. Biol 1060, 55–72. [DOI] [PubMed] [Google Scholar]
- Muniz LR, Pacer ME, Lira SA, and Furtado GC (2011). A critical role for dendritic cells in the formation of lymphatic vessels within tertiary lymphoid structures. J. Immunol 187, 828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayar S, Campos J, Smith CG, Iannizzotto V, Gardner DH, Mourcin F, Roulois D, Turner J, Sylvestre M, Asam S, et al. (2019). Immunofibroblasts are pivotal drivers of tertiary lymphoid structure formation and local pathology. Proc. Natl. Acad. Sci. USA 116, 13490–13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, and Lambrecht BN (2012). Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. 33, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder L, and Ludewig B (2018). A Fresh View on Lymph Node Organogenesis. Trends Immunol. 39, 775–787. [DOI] [PubMed] [Google Scholar]
- Peduto L, Dulauroy S, Lochner M, Späth GF, Morales MA, Cumano A, and Eberl G (2009). Inflammation recapitulates the ontogeny of lymphoid stromal cells. J. Immunol 182, 5789–5799. [DOI] [PubMed] [Google Scholar]
- Perez-Ruiz E, Minute L, Otano I, Alvarez M, Ochoa MC, Belsue V, de Andrea C, Rodriguez-Ruiz ME, Perez-Gracia JL, Marquez-Rodas I, et al. (2019). Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 569, 428–432. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, and Mohler KM (1998). TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol 160, 943–952. [PubMed] [Google Scholar]
- Peske JD, Thompson ED, Gemta L, Baylis RA, Fu Y-X, and Engelhard VH (2015). Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat. Commun 6, 7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, Wucherpfennig K, Turley S, Carroll MC, Sobel RA, et al. (2011). Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity 35, 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitprez F, de Reyniès A, Keung EZ, Chen TW-W, Sun C-M, Calderaro J, Jeng Y-M, Hsiao L-P, Lacroix L, Bougoüin A, et al. (2020). B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560. [DOI] [PubMed] [Google Scholar]
- Pikor NB, Astarita JL, Summers-Deluca L, Galicia G, Qu J, Ward LA, Armstrong S, Dominguez CX, Malhotra D, Heiden B, et al. (2015). Integration of Th17- and lymphotoxin-derived signals initiates meningeal-resident stromal cell remodeling to propagate neuroinflammation. Immunity 43, 1160–1173. [DOI] [PubMed] [Google Scholar]
- Pula B, Witkiewicz W, Dziegiel P, and Podhorska-Okolow M (2013). Significance of podoplanin expression in cancer-associated fibroblasts: a comprehensive review. Int. J. Oncol 42, 1849–1857. [DOI] [PubMed] [Google Scholar]
- Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Kusser K, and Randall TD (2007). Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc. Natl. Acad. Sci. USA 104, 10577–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, Kolls JK, Khader SA, and Randall TD (2011). The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat. Immunol 12, 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilley MJ, Morrow B, Ager CR, Liu A, Hong DS, and Curran MA (2019). TLR9 activation cooperates with T cell checkpoint blockade to regress poorly immunogenic melanoma. J. Immunother. Cancer 7, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AB, and Engelhard VH (2020). Insights into tumor-associated tertiary lymphoid structures: novel targets for antitumor immunity and cancer immunotherapy. Cancer Immunol. Res 8, 1338–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AB, Peske JD, and Engelhard VH (2018). Identification and characterization of tertiary lymphoid structures in murine melanoma. Methods Mol. Biol 1845, 241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddle NH, and Akirav EM (2009). Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J. Immunol 183, 2205–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rynne-Vidal A, Jiménez-Heffernan JA, Fernández-Chacón C, López-Cabrera M, and Sandoval P (2015). The Mesothelial Origin of Carcinoma Associated-Fibroblasts in Peritoneal Metastasis. Cancers (Basel) 7, 1994–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Mii A, Hamazaki Y, Fujita H, Nakata H, Masuda K, Nishiyama S, Shibuya S, Haga H, Ogawa O, et al. (2016). Heterogeneous fibroblasts underlie age-dependent tertiary lymphoid tissues in the kidney. JCI Insight 1, e87680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautès-Fridman C, Petitprez F, Calderaro J, and Fridman WH (2019). Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325. [DOI] [PubMed] [Google Scholar]
- Schrama D, thor Straten P, Fischer WH, McLellan AD, Bröcker EB, Reisfeld RA, and Becker JC (2001). Targeting of lymphotoxin-α to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity 14, 111–121. [DOI] [PubMed] [Google Scholar]
- Stark FC, Weeratna RD, Deschatelets L, Gurnani K, Dudani R, McCluskie MJ, and Krishnan L (2017). An Archaeosome-Adjuvanted Vaccine and Checkpoint Inhibitor Therapy Combination Significantly Enhances Protection from Murine Melanoma. Vaccines (Basel) 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, et al. (2018). A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med 24, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson ED, Enriquez HL, Fu Y-X, and Engelhard VH (2010). Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J. Exp. Med 207, 1791–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trout AT, Rabinowitz RS, Platt JF, and Elsayes KM (2013). Melanoma metastases in the abdomen and pelvis: Frequency and patterns of spread. World J. Radiol 5, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist HR, Lin X, Ashour AE, Hollingsworth MA, Singh RK, Talmadge JE, and Solheim JC (2007). CCL21 induces extensive intratumoral immune cell infiltration and specific anti-tumor cellular immunity. Int. J. Oncol 30, 631–639. [PubMed] [Google Scholar]
- van de Pavert SA, and Mebius RE (2010). New insights into the development of lymphoid tissues. Nat. Rev. Immunol 10, 664–674. [DOI] [PubMed] [Google Scholar]
- Wajant H, and Siegmund D (2019). TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol 7, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Wang Y, Wang J, Hedgeman EO, Browning JL, and Fu YX (1999). The requirement of membrane lymphotoxin for the presence of dendritic cells in lymphoid tissues. J. Exp. Med 190, 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, Schietinger A, Philip M, Schreiber H, and Fu YX (2004). Priming of naive T cells inside tumors leads to eradication of established tumors. Nat. Immunol 5, 141–149. [DOI] [PubMed] [Google Scholar]
- Yu P, Lee Y, Wang Y, Liu X, Auh S, Gajewski TF, Schreiber H, You Z, Kaynor C, Wang X, and Fu YX (2007). Targeting the primary tumor to generate CTL for the effective eradication of spontaneous metastases. J. Immunol 179, 1960–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report original datasets.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request