

Abstract
Here we describe an updated TAR cloning protocol for the selective and efficient isolation of any genomic fragment or gene of interest up to 280 kb in size from genomic DNA. The method exploits the special recombination machinery of the yeast Saccharomyces cerevisiae. TAR cloning is based on the high level of in vivo recombination that occurs between a specific genomic DNA fragment of interest and targeting sequences (hooks) in a TAR vector that are homologous to the 5′ and 3′ ends of the targeted region. Upon co‐transformation into yeast, this results in the isolation of the chromosomal region of interest as a circular YAC molecule, which then propagates and segregates in yeast cells and can be selected for. In the updated TAR cloning protocol described here, the fraction of region‐positive clones typically obtained is increased from 1% up to 35% by pre‐treatment of the genomic DNA with specifically designed CRISPR/Cas9 endonucleases that create double‐strand breaks (DSBs) bracketing the target genomic DNA sequence, thereby making the ends of the chromosomal region of interest highly recombinogenic. In addition, a new TAR vector was constructed that contains YAC and BAC cassettes, permitting direct transfer of a TAR‐cloned DNA from yeast to bacterial cells. Once the TAR vector with the hooks is constructed and genomic DNA is prepared, the entire procedure takes 3 weeks to complete. The updated TAR protocol does not require significant yeast experience or extensively time‐consuming yeast work because screening only about a dozen yeast transformants is typically enough to find a clone with the region of interest. TAR cloning of chromosomal fragments, individual genes, or gene families can be used for functional, structural, and population studies, for comparative genomics, and for long‐range haplotyping, and has potential for gene therapy. Published 2021. This article is a U.S. Government work and is in the public domain in the USA. Current Protocols published by Wiley Periodicals LLC.
Basic Protocol 1: Preparation of CRISPR/Cas9‐treated genomic DNA for TAR cloning
Basic Protocol 2: Isolation of a gene or genomic locus by TAR cloning
Basic Protocol 3: Transfer of TAR/YAC/BAC isolates from yeast to E. coli
Keywords: CRISPR/Cas9, TAR cloning, transformation‐associated recombination
INTRODUCTION
The transformation‐associated recombination (TAR) cloning method allows entire genes and chromosomal regions of up to 280 kb in size to be selectively, accurately, and efficiently isolated from total genomic DNA. It is based on in vivo recombination in the yeast S. cerevisiae (Larionov et al., 1996; Larionov, Kouprina, Solomon, Barrett, & Resnick, 1997; Kouprina, & Larionov, 2006; Kouprina, & Larionov, 2016). The isolation of full‐size genes containing coding regions (exons) as well as non‐coding regions (introns) and regulatory elements that control gene expression can facilitate gene function studies in clones that can reproduce “physiological” gene expression.
TAR cloning exploits the high level of recombination between homologous DNA sequences when transformed into yeast (Larionov et al., 1996). A specific genomic region/gene can be isolated from total genomic DNA in a TAR vector containing two unique sequences (hooks) homologous to the 5′‐ and 3′‐ends of the targeted region (see Fig. 1). The hooks can be as small as 60 bp (Noskov et al., 2001). In the original TAR protocol, the TAR vector contains a YAC cassette for proper segregation, propagation, and selection of the cloned material in yeast. Because the TAR vector does not contain a yeast origin of replication (ARS element), the vector itself produces no background clones. The propagation of TAR‐isolated regions, instead, absolutely depends on the presence of ARS‐like sequences (WWWWTTTAYRTTTWGTT) in the cloned genomic DNA fragments. Such sequences are common in mammalian and other complex genomes, with roughly one ARS‐like sequence per 20‐30 kb (Stinchcomb, Thomas, Kelly, Selker, & Davis, 1980; Noskov et al., 2002). Figure 1 illustrates the original version of TAR cloning: recombination between the vector hooks and homologous sequences in the co‐transformed genomic DNA results in selective isolation of the genomic region of interest as a circular YAC molecule. Using the original TAR cloning method, the yield of positive yeast clones for the targeted gene/region typically ranges between 0.5% and 1%. To allow users to convert a TAR/YAC isolate into a BAC form, a set of retrofitting vectors were constructed (Kouprina, Noskov, Koriabine, Leem, & Larionov, 2004a). Retrofitting the TAR/YAC into a BAC form allows the sequence of interest to be transferred to bacterial cells, to simplify DNA isolation for further structural and functional studies. This process, however, is time‐consuming.
Figure 1.
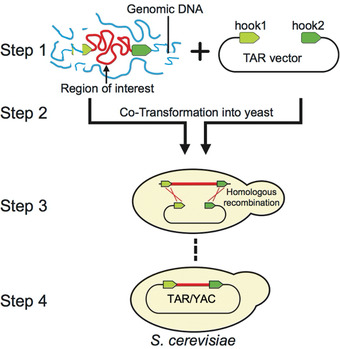
Diagram of the original TAR cloning protocol (Larionov et al., 1997; Kouprina, & Larionov, 2008). In step 1, genomic DNA and TAR vector are prepared. In step 2, genomic DNA containing a region of interest (in red) is co‐transformed with the linearized TAR vector into yeast spheroplasts. The TAR vector contains a YAC (yeast artificial chromosome) cassette for proper selection, segregation, and propagation in S. cerevisiae and two targeting sequences (hook 1 and hook 2 in green) homologous to the 3′ and 5′ ends of the target region. In steps 3 and 4, and after co‐transformation, recombination between the hooks and the target sequences leads to the rescue of a region of interest as a circular YAC molecule.
TAR cloning has become a widely useful procedure. The ability to selectively and accurately isolate genomic regions of interest and full‐size genes without constructing representative BAC genomic libraries of whole genomes as a source facilitates structural and functional analysis of complex genomes, including those of humans, non‐human primates, and mice (summarized in Kouprina, & Larionov, 2016; Kouprina, & Larionov, 2019). TAR cloning is also a powerful tool for comparative genomics and evolutionary studies. For instance, gene homologs from nonhuman primates have been TAR‐isolated using available human sequence information as the source of target sequences for hooks (summarized in Kouprina, & Larionov, 2016).
Despite its many uses, the original TAR cloning method has some limitations. The most time‐consuming step is the identification of region‐positive clones, requiring the analysis of hundreds of yeast colonies to find one of interest. This limitation is especially bothersome when the available amount of genomic DNA is limited. We had observed that DSBs specifically introduced close to the ends of the targeted genomic fragment made homologous recombination between TAR vector hooks and targeted genomic sequences more efficient (Leem et al., 2003), and we decided to explore this feature to address the aforementioned limitation.
In the updated TAR protocol described below, we use a programmable endonuclease, CRISPR/Cas9 (Mali, Esvelt, & Church, 2013; Hsu, Lander, & Zhang, 2014; Wijshake, Baker, & van de Sluis, 2014; Kim & Kim, 2014; Karvelis, Gasiunas, & Siksnys, 2013) to create double‐strand breaks at user‐defined sequences. Pre‐treatment of genomic DNA with CRISPR/Cas9 endonucleases increases the subsequent yield of gene‐ or region‐positive clones 20‐ to 35‐fold, and can thereby reach an efficiency of 20%‐35% (Lee, Larionov, & Kouprina, 2015). In addition, a newly designed TAR vector contains both YAC and BAC cassettes, allowing direct transfer of the TAR‐cloned region from yeast to bacterial cells without the time‐consuming step of retrofitting a YAC molecule into a YAC/BAC form. Finally, because of the increased efficiency, extensive experience with yeast is no longer required, because screening only about a dozen yeast transformants is typically enough to identify a colony containing a desired chromosomal fragment.
The updated TAR cloning method described in this article includes three protocols. Basic Protocol 1 describes the preparation of CRISPR/Cas9‐treated genomic DNA for TAR cloning. Basic Protocol 2 describes isolation of a gene or genomic locus by recombination in the yeast S. cerevisiae. Basic Protocol 3 provides a method to transfer the TAR isolates from yeast to E. coli for further BAC DNA isolation and sequencing. An overview of the entire protocol is shown in Figure 2.
Figure 2.
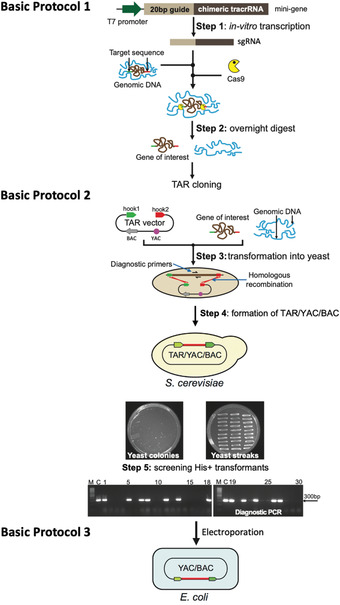
Diagram of the updated TAR‐CRISPR/Cas9 method for selective isolation of a gene/locus of interest from complex genomes. Basic Protocol 1, Step 1: The mini‐gene is in vitro transcribed to make sgRNA. Prepared genomic DNA is then cleaved in vitro at positions close to the 5′ and 3′ ends of a gene/locus by specifically designed Cas9‐sgRNA complexes. Step 2: A digestion mix containing sgRNAs, Cas9, and genomic DNA containing a gene of interest is prepared and left to digest overnight. Basic Protocol 2, Step 3: Prepared yeast spheroplasts are transformed with CRISPR/Cas9‐treated genomic DNA along with the linearized TAR vector containing BAC and YAC cassettes and the hooks corresponding to the 5′ and 3′ ends of a gene of interest (hook 1 in green and hook 2 in red). Step 4: Homologous recombination between the targeting sequences in the vector and the targeted genomic DNA fragment ends leads to the establishment of a circular TAR/YAC/BAC molecule. A representative His‐ plate with yeast His+ transformants after 5‐7 days of growth obtained by TAR cloning of the human NBS1 gene from total genomic DNA is shown, together with a plate with 30 streaked randomly chosen His+ transformants after 1 day of growth. Step 5: Analysis of the streaked His+ transformants by PCR for the presence of the gene of interest using a pair of diagnostic primers. An example of the screening of transformants harboring the human NSB1 gene is shown. M‐GeneRuler 1 kb DNA Ladder (Thermo Fisher Scientific, cat. no. SM0311); C‐control PCR with human genomic DNA. Basic Protocol 3: YAC/BAC DNAs containing a gene/region of interest are prepared in agarose plugs and then transferred from yeast to E. coli cells by electroporation for further BAC DNA isolation, sequencing, and analysis.
Overall, these protocols allow the investigator to selectively and efficiently isolate any chromosomal segment or gene for structural and functional genomic studies, among other applications.
Strategic Planning
TAR cloning experiments require genomic DNA, which may be prepared by a solution‐based DNA extraction method using, for instance, the Gentra Puregene Blood Kit (Qiagen, cat. no. 158467) or following the procedure outlined in Basic Protocol 1. However, DNA isolated using these methods does not allow TAR cloning of fragments larger than 150 kb. If the user wants to isolate larger‐size fragments, genomic DNA should be prepared in agarose blocks as previously described (Kouprina et al., 2004a).
For treating the genomic DNA in vitro with Cas9‐sgRNA complexes to induce DSBs near the targeted genomic sequences, the user will need to design guide sequences. The CRISPR design tool (https://zlab.bio/guide‐design‐resources) can be used to identify CRISPR guide sequences. This tool, however, may yield CRISPR guide sequences of different qualities. Only high‐quality CRISPR guide sequences should be considered for use. A Blat search (http://genome.ucsc.edu/cgi‐bin/hgBlat) is used to validate that the CRISPR guide sequences do not have other homologies within the region of interest. The CRISPR guide sequence must follow the ‘5‐GG’ rule (Imburgio, Rong, Ma, & McAllister, 2000; Ran et al., 2013). More specifically, it should start with a ‘G’ (guanine), followed by a purine (G/A), with ‘G’ being significantly preferred, and finally followed by an optional third ‘G’. Further, CRISPR‐Cas9 specificity may be improved by truncating the guide to a sequence as short as 17 bp (Fu, Sander, Reyon, Cascio, & Joung, 2014).
At least two CRISPR guides (sgRNAs) should be selected for each TAR hook position (i.e., two at the 5′ end of the target chromosomal region and two at the 3′ end). CRISPR guide sequences should be located outside of the 5′ and 3′ ends of the target region, but within no more than 1 kb sequence from each end. The cleavage efficiency of each Cas9‐sgRNA complex should be checked before TAR cloning experiments. Those that have off‐target sites within the region of interest should be discarded. Preference should be given to CRISPR guides located closest to the 5′ and 3′ends of the target region (Lee et al., 2015).
As an example, we show four CRISPR guide sequences that were designed for the TAR cloning of the human NBS1 gene. Two were designed at the 5′ end of the target region (Cas9‐sgRNA‐A and Cas9‐sgRNA‐B) and two at the 3′ end (Cas9‐sgRNA‐C and Cas9‐sgRNA‐D) (Fig. 3). The efficiency of the different Cas9‐sgRNA complexes was assessed beforehand, and based on the results (Fig. 4), the Cas9‐sgRNA‐B and Cas9‐sgRNA‐C pair was selected for DNA cleavage. Cas9‐sgRNA‐B and Cas9‐sgRNA‐C recognize and allow cutting the genome 47 bp upstream of the 5′ NBS1 targeting hook and 7 bp downstream of the 3′ NBS1 targeting hook, respectively (Fig. 4B; Lee et al., 2015).
Figure 3.
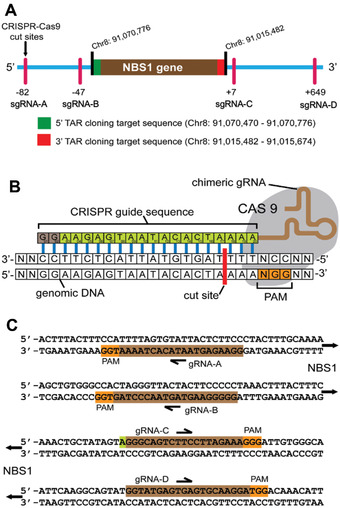
Locations of CRISPR/Cas9 cut sites relative to the position of the human NBS1 gene. (A) The NBS1 gene is located at Chr8: positions 91,015,482‐91,070,776 [Mar 2006 (NSB136/hg18)]. The 3′ and 5′ ends of the NBS1 (targeted genomic sequences) homologous to the TAR cloning vector hooks are marked in red (hook 1) and in green (hook 2), respectively. The cut sites of Cas9‐sgRNA‐A and Cas9‐sgRNA‐B complexes upstream of hook 1, and of Cas9‐sgRNA‐C and Cas9‐sgRNA‐D complexes downstream of hook 2, with their positions toward the hooks, are indicated. (B) Schematic representation of Cas9‐sgRNA binding to DNA. The CRISPR guide sequence is cloned into the 5′ end of the sgRNA molecule. The guide sequence is any 20‐bp DNA sequence upstream of an NGG, the Protospacer Adjacent Motif (PAM). The Cas9 cut site is 3 bp upstream of PAM. (C) Guide sequences used for the human NBS1 gene cloning are illustrated. This figure was adapted from Lee et al. (2015).
Figure 4.
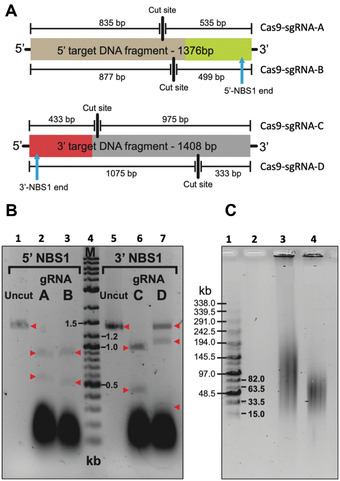
Nuclease activity of different Cas9‐sgRNA complexes. (A) Cleavage positions of Cas9‐sgRNA complexes within their respective target DNA fragments and the expected fragment sizes generated after digest are shown. (B) 200 ng of PCR‐amplified target DNA were digested for 3 hr using the Cas9‐sgRNA complexes shown in Figure 3, and the products then run on an agarose gel. Cas9‐sgRNA‐B and Cas9‐sgRNA‐C complexes cut their target sequence and were chosen for further experiments. Cas9‐sgRNA‐A and Cas9‐sgRNA‐D complexes failed to completely digest their target sequence. Red arrows indicate the fragments, uncut and after digestion. Lane 1 is uncut 1376‐bp 5′ target DNA fragment. Lane 2 is Cas9‐sgRNA‐A cut 5′ target DNA; expected fragments are 835 and 535 bp. Lane 3 is Cas9‐sgRNA‐B cut 5′ target DNA; expected fragments are 877 and 499 bp. Lane 4 is a marker, which is 8 µl of DNA ladder (New England, BioLabs, cat. no. N3200L). Lane 5 is uncut 1408‐bp 3′ target DNA fragment. Lane 6 is Cas9‐sgRNA‐C cut 3′ target DNA; expected fragments are 975 and 433 bp. Lane 7 is Cas9‐sgRNA‐D cut 3′ target DNA; expected fragments are 1075 and 333 bp. (C) Basic Protocol 1 results in untreated human genomic DNA from suspension cells with a size that ranges between 50 and 250 kb. To check the size of the untreated human genomic DNA and Cas9‐treated DNA, CHEF electrophoresis should be run. As an example, uncut and cut commercial human genomic DNA is shown. Lane 1 is a marker that corresponds to the MidRange PFG Marker I (New England BioLabs, cat. no. N0342S). Lane 3 is 1 µg of uncut commercial human genomic DNA (Promega, cat. no. G304A) used for CRISPR‐TAR cloning of the human NBS1 gene. Lane 4 is 1 µg of human genomic DNA digested overnight with 1 µl of Cas9 enzyme, 1 µl of gRNA‐B, and 1 µl of gRNA‐C in a total volume of 40 µl. This figure is adapted from Lee et al. (2015).
The pJYB TAR vector (Kim et al., 2018) contains a YAC (yeast artificial chromosome) cassette (a CEN6 sequence and a HIS3 yeast selectable marker) and a pUC polylinker with the AmpR marker and ColE1 origin of replication that allows propagation in bacterial cells as a multicopy vector, which simplifies its isolation (Fig. 5). The pJYB vector also contains a BAC (bacterial artificial chromosome) cassette. A BAC cassette includes the selectable marker CMR and F′ origin of replication for propagation of BAC molecules with a size of up to 250 kb in bacterial cells. The 5′ and 3′ hooks should be inserted as SalI/AscI and NotI/XbaI targeting sequences, correspondingly. Before TAR cloning experiments, pJYB vector is linearized by AscI/NotI to make the hooks highly recombinogenic. Upon linearization, the pUC polylinker is removed.
Figure 5.
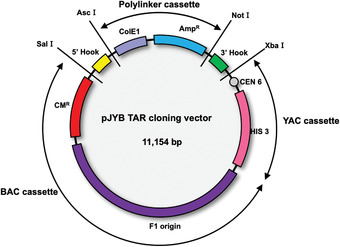
Diagram of the TAR cloning vector pJYB. The vector contains BAC and YAC (bacterial and yeast artificial chromosomes, respectively), and polylinker cassettes. A YAC cassette contains a yeast centromere (CEN6) and a yeast selectable marker, HIS3. A BAC cassette contains the chloramphenicol acetyltransferase (CMR ) gene and the F′‐ origin of replication. A polylinker cassette contains the ampicillin resistance gene (AmpR ) gene and ColE1 origin of replication that makes the vector multicopy. Targeting sequences (hooks) that have homology to 3′ and 5′ ends of the target genomic sequence are inserted into a TAR vector. Before TAR cloning experiments, the vector is linearized by endonuclease digestion to expose the hooks. The 5′ hook (in yellow) and 3′ hook (in green) are cloned in the TAR vector as SalI/AscI and NotI/XbaI fragments, respectively. As a result of linearization, the polylinker cassette is eliminated and the targeting sequences are exposed. This figure is adapted from Kim et al. (2018).
Hooks should be unique sequences. The uniqueness of the hooks chosen from the human genome can be easily checked by blasting against the genome (http://genome.ucsc.edu/cgi‐bin/hgBlat). Though the minimal size of the targeting sequences is 60 bp, longer hooks can also be used. For more details, see Critical Parameters.
The basic TAR vector pJYB is available upon request from the Developmental Therapeutics Branch, National Cancer Institute (NIH).
Basic Protocol 1. PREPARATION OF CRISPR/Cas9‐TREATED GENOMIC DNA FOR TAR CLONING
Here, the designed guide RNAs (see Strategic Planning) will be used to create an oligonucleotide‐based sgRNA DNA template (i.e., a minigene). The minigene will then be subjected to PCR, and the user will perform T7‐mediated in vitro transcription to obtain the purified sgRNAs. These sgRNAs can then be used with the Cas9 enzyme for specific genomic DNA cleavage. We also describe a protocol for the preparation of genomic DNA in solution from suspension cells.
At the end of this protocol, the user will have genomic DNA that is ready to use for TAR cloning (Basic Protocol 2).
Materials
5× HF buffer (New England Biolabs, cat. no. M0530L)
10 mM deoxynucleotide (dNTP) solution mix (New England Biolabs, cat. no. N0447L)
Phusion High‐Fidelity DNA Polymerase (New England Biolabs, cat. no. M0530L)
-
100 µM Basic Target Gene Specific Forward primer T7crTARGET (custom order; store at −20°C):
5′‐GAAATTAATACGACTCACTATAGN19‐20GTTTTAGAGCTAGAAATAGC‐3′
The N19‐20 refers to the 19 to 20 nucleotides of the CRISPR/Cas9 targeting DNA sequence. The 3′ end of the CRISPR/CAS9 targeting DNA sequence (N19‐20) must have a proto‐spacer motif (PAM) sequence (5′‐NGG‐3′) downstream, but the user should not include the PAM sequence in the Target Gene Specific Primers (Fig. 3B and 3C).
-
100 µM common reverse primer sgRNAT7common (custom order; store at −20°C):
5′‐AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC‐3′
QIAquick PCR Purification Kit (Qiagen, cat. no. 28104)
HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs, cat. no. E2050S)
Nuclease‐free water (Quality Biological, cat. no. 351‐029‐721)
DNase I (New England Biolabs, cat. no. M0303S) and DNase I buffer
Murine RNase Inhibitor (New England Biolabs, cat. no. M0314S)
E.Z.N.A. MicroElute RNA clean‐up kit (Omega Bio‐Tek, cat no. R6247‐01)
2‐mercaptoethanol (ME) (Sigma‐Aldrich, cat. no. M6250‐100ML)
100% ethanol and ice‐cold 70% ethanol (The Warner Graham Company, cat. no. 64‐17‐5)
Gentra Puregene Blood kit (Qiagen, cat. no. 158467)
Cells or tissues of interest for extraction of genomic DNA
Isopropanol
Phosphate‐buffered saline (PBS), pH 7.4 (Thermo Fisher Scientific, cat. no. 10010023)
DEPC‐treated water (Quality Biological, cat. no. 351‐068‐721)
20 µM Cas9 Nuclease, S. pyogenes (New England Biolabs, cat. no. M0386M)
10× Reaction Buffer 3.1 (New England Biolabs)
PCR tubes
PCR thermocycler
1.7‐ml microcentrifuge tubes (Thomas Scientific, cat. no. 1159M35)
Refrigerated microcentrifuge
Nanodrop spectrophotometer
15‐ml centrifuge tube (Corning Falcon, cat. no. 352196)
50‐ml centrifuge tube (Corning Falcon, cat. no. 352070)
10‐ml disposable pipettes (Corning Falcon, cat. no. 356551)
Refrigerated centrifuge.
Thermo Scientific™ Compact Microbiological Incubator (The Lab Depot, cat. no. 50125882)
Synthesis of the sgRNA DNA template by PCR
-
1
Order custom DNA oligos (T7crTARGET and sgRNAT7common) for sgRNA DNA template synthesis.
Example for the human NBS1 gene (see Fig. 3):- NBS1 Cas9‐sgRNA‐B (5′‐GAAATTAATACGACTCACTATAG GGGGGAAGTAGTAACCCTAG GTTTTAGAGC TAGAAATAGC‐3′)
- NBS1 Cas9‐sgRNA‐C (5′‐GAAATTAATACGACTCACTATAG GGGGCAGTCTTCCTTAGAAA GTTTTAGAGCTAGAAATAGC‐3′)
For TAR cloning, the user needs two unique Target Gene Specific Forward primers for the desired full‐size gene, for both the 5′ and 3′ regions (e.g., NBS1 Cas9‐gRNA‐B and NBS1 Cas9‐gRNA‐C).
CRISPR guide sequences should be located outside of 5′ and 3′ ends of the target region, but within not more than 1 kb sequence from each end. See Strategic Planning.
-
2Prepare the following mix in a PCR tube for each sgRNA template:
Reagent Amount 5× Phusion HF Buffer 20 µl dNTP mix (10 mM) 2.5 µl Phusion DNA Polymerase 1 µl Custom T7crTARGET forward primer (100 µM) 0.5 µl sgRNAT7common reverse primer (100 µM) 0.5 µl H2O (Total 100 µl) 74.5 µl PCR product size will be 98 or 99 bp, depending on the sgRNA target sequence (N19/20).
-
3Run each reaction in a thermal cycler using the following PCR conditions:
1 cycle: 98°C 30 s Initial denaturation 35 cycles: 98°C 10 s Denaturation 58°C 30 s Annealing 72°C 15 s Extension 1 cycle 72°C 5‐10 min Final extension 1 cycle 4°C ∞ Hold -
4
Purify each custom sgRNA DNA template PCR product using the Qiagen QIAquick PCR Purification Kit. To do this, first, transfer the 100‐µl PCR product to a clean 1.7‐ml microcentrifuge tube.
-
5
Add 500 µl of Buffer PB from the kit, and then mix by pipetting.
-
6
Place a QIAquick spin column in one of the provided 2‐ml collection tubes (from Qiagen QIAquick PCR Purification Kit).
-
7
To bind DNA, apply the PCR product mixture to the QIAquick column and centrifuge 60 s at 10,000 rpm at room temperature in a microcentrifuge.
-
8
Discard the flowthrough. Place the QIAquick column back into the same tube.
-
9
To wash, add 750 µl of Buffer PE to the QIAquick column and centrifuge for 60 s at 10,000 rpm at room temperature in a microcentrifuge.
-
10
Discard the flowthrough and place the QIAquick column back into the same tube.
-
11
Microcentrifuge the column for 1 min at 13,000 rpm at room temperature.
-
12
Place QIAquick column in a clean 1.7‐ml microcentrifuge tube.
-
13
To elute DNA, add 30 µl of Buffer EB (10 mM Tris·HCl, pH 8.5; provided with QIAquick kit) or water (pH 7.0‐8.5) to the center of the QIAquick membrane and incubate for 2 min at room temperature.
-
14
Microcentrifuge the column for 1 min at 13,000 rpm at room temperature.
-
15
Check DNA concentration by spectrophotometer or Nanodrop.
Final DNA concentration should be around 200 ng/µl.
In vitro transcription
-
16Assemble each reaction using items from the HiScribe T7 Quick High Yield RNA Synthesis Kit at room temperature in the following order in a clean 1.7‐ml microcentrifuge tube:
Reagent Amount Nuclease‐free water x µl (Total reaction volume 30 µl) ATP (100 mM) 1.5 µl UTP (100 mM) 1.5 µl GTP (100 mM) 1.5 µl CTP (100 mM) 1.5 µl 10× T7 reaction Buffer 3 µl T7 RNA Polymerase Mix 2 µl Purified DNA template x µl (∼ 600 ng) -
17
Mix thoroughly and microcentrifuge briefly to bring solution to bottom of tube.
-
18
Incubate at 37°C overnight.
-
19
The next day, add 60 µl of nuclease‐free water, 10 µl of 10× DNase I buffer, 1 µl of Murine RNase Inhibitor, and 2 µl of DNase I to each 1.7‐ml microcentrifuge tube.
-
20
Mix thoroughly by tapping, then incubate for 30 min at 37°C.
sgRNA purification
-
21
Add 350 µl of QLV Lysis Buffer from the Microelute RNA clean‐up kit to each sample and mix well.
2‐Mercaptoethanol should be added to QLV Lysis Buffer before use. Add 20 μl of 2‐mercaptoethanol per 1 ml of QLV Lysis Buffer.
-
22
Add 300 µl of 100% ethanol. Mix well by pipetting.
-
23
Incubate for 5 min at room temperature.
If the transcription reaction works well, a white opaque precipitate will be seen.
-
24
Place a HiBind RNA Mini column (from the E.Z.N.A. Microelute RNA cleanup kit) in one of the provided 2‐ml collection tubes.
-
25
To bind sgRNA, transfer the mix to the HiBind RNA Mini column and microcentrifuge for 30 s at 10,000 rpm at room temperature.
-
26
Discard the flowthrough. Place the HiBind RNA Mini column back into the same tube.
-
27
To wash, add 500 µl of RNA Wash Buffer 2 (from the E.Z.N.A. Microelute RNA cleanup kit).
-
28
Microcentrifuge for 1 min at 12,000 rpm at room temperature.
-
29
Discard the flowthrough. Place the HiBind RNA Mini column back into the same tube.
-
30
Add 500 µl of RNA Wash Buffer 2.
-
31
Microcentrifuge for 2 min at 14,000 rpm at room temperature.
-
32
Discard the flowthrough. Place the HiBind RNA Mini column back into the same tube.
-
33
Microcentrifuge for 5 min at maximum speed to completely dry the HiBind RNA Mini column.
It is important to dry the HiBind RNA Mini column matrix before elution. Residual ethanol may interfere with downstream applications.
-
34
Place the dry HiBind RNA Mini column in a clean 1.7‐ml microcentrifuge tube.
-
35
To elute sgRNA, add 30 µl of nuclease‐free water directly to the center of the HiBind RNA Mini column membrane and incubate for 30 min to 1 hr.
-
36
Micro centrifuge the column for 1 min at 13,000 rpm at room temperature.
-
37
Check sgRNA concentration by spectrophotometer or Nanodrop.
Final concentration should be around 2 µg/µl.
The sgRNA transcripts can now be used for treating genomic DNA with CRISPR/Cas9 endonucleases, or stored at −80°C
Genomic DNA preparation
Use the Gentra Puregene Blood kit.
-
38
Transfer ∼2 × 107 suspended cells into a 15‐ml centrifuge tube with 10 ml of culture medium.
-
39
Centrifuge 5 min at 600 × g, room temperature.
-
40
Carefully discard the supernatant and resuspend with 5 ml of PBS.
-
41
Centrifuge 5 min at 600 × g, room temperature.
-
42
Carefully discard the supernatant using a 10‐ml disposable pipette, leaving approximately 200 µl of residual PBS.
-
43
Resuspend the cells by tapping, add 3 ml of Cell Lysis Solution (from Gentra Puregene Blood kit), and mix thoroughly by tapping.
-
44
Add 15 µl of RNase A solution (from Gentra Puregene Blood kit) to the 15‐ml centrifuge tube, mix by inverting 20 times, and incubate for 30 min at 37°C.
-
45
Cool down the sample on ice for 5 min.
-
46
Add 1 ml of Protein Precipitation Solution (from Gentra Puregene Blood kit) and mix thoroughly by inverting.
-
47
Centrifuge 20 min at 9600 × g, 4°C.
-
48
Transfer the supernatant to a 50‐ml centrifuge tube using a 10‐ml disposable pipette.
-
49
Add 3 ml of isopropanol and mix by inverting 20 times.
-
50
Before transferring the white thread‐like DNA, prepare a 1.7‐ml microcentrifuge tube with 1 ml of 70% ethanol.
-
51
Transfer the white thread‐like strands of DNA from step 50 using a 200‐µl pipette tip into the 1.7‐ml microcentrifuge tube with 1 ml of 70% ethanol.
The white thread‐like strands of DNA are very sticky, so be very careful when transferring the DNA.
-
52
Invert the tube 10 times and carefully remove the 70% ethanol.
DNA will precipitate to the bottom of the tube. Remove the ethanol carefully using a micropipette.
-
53
Add 1 ml of 70% ethanol and invert 10 times. Then, remove as much 70% ethanol possible without touching the white thread‐like strands of DNA.
-
54
Allow the genomic DNA to air dry for 5 min.
-
55
Add 400 µl of DEPC‐treated water and mix a few times by tapping.
Do not use a pipette to mix.
-
56
Incubate at 60°C for 1 hr and mix a few times by tapping.
Do not use pipette to mix.
-
57
Store at 4°C or proceed to the following section.
Typically, this protocol yields 50 to 150 µg of genomic DNA, and the size of Cas9‐untreated genomic DNA ranges between 50 and 250 kb.
Genomic DNA treatment with CRISPR/Cas9 endonuclease
-
58Assemble the reaction at room temperature in the following order in a clean 1.7‐ml microcentrifuge tube:
Reagent Amount Nuclease‐free water x µl Purified custom 5′sgRNA (e.g.NBS1 sgRNA‐B) (step 37) x µl (16 µg) Purified custom 3′sgRNA (e.g., NBS1 sgRNA‐C) (step 37) x µl (16 µg) 10× Reaction Buffer 3.1 20 µl Cas9 Nuclease (20 µM) 4 µl Purified genomic DNA (step 57) x µl (5 µg) Total reaction volume 100 µl -
59
Mix thoroughly and briefly centrifuge to bring solution to bottom of tube.
-
60
Incubate at 37°C for 3 to 4 hr.
-
61
Incubate for 10 min at 65°C.
-
62
Store at 4°C.
The cleaved DNA can now be used for TAR cloning. The DNA can be kept at 4°C for 1 month.
Basic Protocol 2. ISOLATION OF A GENE OR GENOMIC LOCUS BY TAR CLONING
Here, the user will perform TAR cloning, which involves isolation of a genomic region of interest via the TAR vector using homologous recombination in yeast. First, the user will prepare highly competent yeast spheroplasts, and then transform these with genomic DNA that has been pre‐treated with CRISPR/Cas9 (Basic Protocol 1), along with a TAR vector linearized between the hooks homologous to the 5′ and 3′ ends of the region of interest. TAR cloning will produce a library of yeast colonies. The user will then screen the yeast colonies by PCR with diagnostic primers specific to the region of interest. It is expected that this updated TAR protocol will result in 20%‐35% region‐positive transformants.
Materials
Saccharomyces cerevisiae strain VL6‐48N (MAT alpha, his3− Δ 200, trp1−Δ1, ura3− Δ 1, lys2, ade2‐101, met14).
This strain is available under request from the Developmental Therapeutics Branch, National Cancer Institute (NIH).
YPD agar plates (Sigma, cat no. Y1003)
YPD medium (Sigma, cat. no. Y1375)
1 M sorbitol (G‐BIOSCIENCES, cat. no. R038)
SPE solution (see recipe)
10 mg/ml Zymolyase solution (see recipe)
14.3 M 2‐mercaptoethanol (2‐ME; Sigma, cat. no. M6250)
10% SDS solution (BioRad, cat. no. 1610416)
STC solution (see recipe)
Genomic DNA pre‐treated by CRISPR/Cas9 (Basic Protocol 1, step 62)
-
pYJB TAR cloning vector
Before use, the TAR vector should be “activated” by cutting between the targeting hooks with an appropriate restriction enzyme(s). Concentration of the TAR vector should be 0.5‐1 μg/μl. The linearized TAR vector can be kept at −20°C up to 2‐3 months. Vector DNA may be isolated by DNA Maxi kit (Qiagen). The basic TAR vector pYJB (without hooks) is available upon request from the Developmental Therapeutics Branch, National Cancer Institute (NIH).
PEG 8000 solution (see recipe)
SOS solution (see recipe)
TOP SORB‐agar‐His plates (see recipe)
SORB‐His plates (Teknova, cat. no. C3004), equilibrated to 50°C
SD‐His plates (Teknova, cat. no. C3020)
5 M potassium acetate (KAc) solution (Santa Cruz Biotechnology, cat. no. 127‐08‐2)
100% isopropanol, room temperature
-
Diagnostic PCR primers to screen yeast transformants for gene/region positive clones
The primers may be chosen from any unique internal sequences to the region of interest (i.e., exon regions).
Agarose (Sigma, cat. no. A9539)
GeneRuler 1 kb Plus DNA ladder (Thermo Fisher Scientific, cat. no. SM1331)
Qiagen Plasmid kit (Qiagen, cat. no. 10023)
30°C incubator and shaker
250‐ml Erlenmeyer flasks (e.g., Cole‐Parmer)
Spectrophotometer with visible‐light source
15‐ and 50‐ml conical screw‐cap polypropylene centrifuge tubes (Sigma, cat. no. CLS430791 and CLS430290)
Refrigerated centrifuge and microcentrifuge (e.g., Eppendorf)
Vortex
1.7‐ml microcentrifuge tubes (e.g., Dot Scientific, cat. no. RN1700‐GMT)
2.0‐ml microcentrifuge tubes (Sigma, cat. no. BR780546)
Sterile toothpicks
Sterile disposable pipette tips
Heat blocks0.2‐ml PCR strip tubes (Dot Scientific, cat. no. 415‐8PCR)
PCR thermocycler
UV gel imaging system
Preparation of competent yeast spheroplasts
-
1
Streak the host yeast strain VL6‐48N on a YPD plate to isolate single colonies. Grow at 30°C.
-
2
One day before the TAR cloning experiment, place 50‐ml aliquots of YPD medium in three separate 250‐ml Erlenmeyer flasks. Inoculate each flask with different‐size single colonies from the freshly streaked VL6‐48N and grow the cultures overnight (for 14‐16 hr) at 30°C with vigorous shaking (200‐300 rpm), to assure good aeration.
The user inoculates three flasks to ensure that at least one of them will be ready to use the next morning
-
3
The following morning, measure the optical density of the cultures every 20‐30 min. Choose the flask with an OD660 of 2.0‐4.0.
To measure, prepare a 1/10 dilution of the culture in water; the density should be between 0.2 and 0.4.
The culture with an OD660 of 2.0‐4.0 is ready for the preparation of highly competent spheroplasts. This optical density corresponds to approximately 2 × 107 cells per ml.
-
4
Transfer the yeast culture from this flask into a 50‐ml conical tube and pellet the cells by centrifugation for 5 min at 1000 × g, 5°C. Remove and discard the supernatant.
-
5
Resuspend the cell pellet in 30 ml of sterile water by vortexing and centrifuge 5 min at 3000 × g, 5°C. Remove and discard the supernatant.
-
6
Resuspend the cell pellet in 20 ml of 1 M sorbitol by vortexing and centrifuge 5 min at 3000 × g, 5°C. Remove and discard the supernatant.
Yeast cells in 1 M sorbitol may be kept overnight.
-
7
Resuspend the cell pellet in 20 ml of SPE solution. Add 20 μl of 10 mg/ml Zymolyase solution and 40 μl of 14.3 M 2‐ME, and mix well. Incubate at 30°C for ∼ 20 min with slow shaking (∼100 rpm).
-
8
Check the level of spheroplasting by comparing the optical densities of the cell suspension in 1 M sorbitol versus 2% SDS. To do this, take two 200‐μl aliquots of the Zymolyase‐treated cell suspension and dilute 10‐fold in 1 M sorbitol and 2% SDS, respectively. The spheroplasts are determined to be ready when the difference between the sorbitol and SDS OD660 readings is 3‐ to 5‐fold (e.g., OD = 0.844 vs. OD = 0.187).
This step indicates if the spheroplasts are ready for transformation. SDS lyses the spheroplasts but not intact cells.
Note that the optical density reading for the sorbitol samples should be no less than 1.5. If this value is lower, the spheroplasts have been overexposed to Zymolyase and cannot be used. Both underexposure and overexposure to Zymolyase greatly affect transformation efficiency. From this point on, extreme care must be taken to avoid lysing the delicate spheroplasts: very slow, gentle resuspensions are necessary.
-
9
Centrifuge the spheroplasts 10 min at 570 × g, 5°C. Decant the supernatant, add 50 ml of 1.0 M sorbitol, and then rock very gently to resuspend the pellet. Pellet the spheroplasts again by centrifugation for 10 min at 300‐600 × g, 5°C.
-
10
Repeat the wash with 50 ml of 1 M sorbitol one more time and gently resuspend the final pellet in 2.0 ml of STC solution.
The spheroplasts are ready for transformation and are stable at room temperature for at least 1 hr.
Transformation of spheroplasts by genomic DNA along with a TAR vector linearized between the hooks
-
11
Take 200 μl of the spheroplast suspension (step 10) and mix gently with 1‐2 μg of genomic DNA pre‐treated by CRISPR/Cas9 (Basic Protocol 1, step 62) and 1.0 μg of the linearized TAR vector in a 2.0‐ml tube. Incubate for 10 min at room temperature.
Since each transformation will require 200 μl of the spheroplast suspension (from step 10), each suspension is enough for 10 transformations.
-
12
Add 800 μl of PEG 8000 to each mix, gently mix by inverting, and incubate for 10 min at room temperature.
-
13
Pellet the spheroplasts by centrifugation for 5 min at 300‐500 × g, 5°C. Remove the supernatant and gently resuspend the spheroplasts in each tube with 800 μl of SOS solution.
-
14
Incubate the spheroplasts for 40 min at 30°C without shaking.
-
15
Transfer the spheroplasts from each tube into a 15‐ml conical tube containing 7.0 ml of melted TOP SORB‐agar‐His (equilibrated at 50°C), gently mix, and then quickly pour the agar mix onto a SORB‐His plate.
-
16
Keep the plates at 30°C for 5‐7 days until transformants become visible.
Identification of individual gene‐positive clones
Typically, 20‐35 out of 100 primary His+ transformant colonies obtained contain the sequence of interest. To identify sequence‐positive colonies, primary transformants are examined for the presence of the region of interest by PCR using a pair of primers specific for some of its internal sequences (Fig. 2).
-
17
Streak ∼50 primary transformants (from step 16) using toothpicks onto new SD‐His plates (see Fig. 2).
-
18
Incubate the plates with transformants at 30°C overnight.
-
19
Touch the streak of each His+ transformant with a sterile disposable pipette tip and then rinse the tip thoroughly in 100 μl of a mixture containing 80 μl of water, 20 μl of Zymolyase solution, and 1 μl of 2‐ME.
-
20
Incubate the resulting suspension for 1 hr at 30°C.
-
21
Add 10 μl of 2% SDS. Incubate for 15 min at 70°C.
-
22
Add 10 μl of 5 M KAc and let the tubes sit on ice for 15 min.
-
23
Spin 2 min at 20,800 × g, room temperature.
-
24
Transfer the supernatant to a new 1.7‐ml tube and add an equal volume of isopropanol. Microcentrifuge at maximum speed for 5 min to bring down the precipitate.
-
25
Dissolve the pellets in 30 μl water.
-
26
Use 1 μl of the DNA solution in a 50‐μl PCR reaction (see Current Protocols article: Kuslich, Chui, & Yamashiro, 2018) with appropriate diagnostic primers and separate the PCR products by agarose gel electrophoresis (see Current Protocols article: Armstrong & Schulz, 2008) to identify gene‐positive clones (Fig. 2).
-
27
Store the positive clones streaked on the plates at 4°C.
The gene‐positive yeast transformants may be kept at 4°C for at least 3 weeks.
Basic Protocol 3. TRANSFER OF TAR/YAC/BAC ISOLATES FROM YEAST TO E. coli
Here, the user will transfer the TAR/YAC/BAC containing the DNA region of interest, obtained in Basic Protocol 2, from yeast to E. coli. If the size of the TAR‐cloned DNA is less than 50 kb, DNA isolated from the region‐positive yeast clones (Basic Protocol 2, step 25) may be directly transformed into E. coli following standard procedures. If the size of the region of interest is larger than 50 kb; however, the user will need to prepare DNA in agarose plugs before transforming E. coli by electroporation, and this approach is described below.
Materials
Sequence‐positive yeast clones (Basic Protocol 2)
SD‐His plates (Teknova, cat. no. C3020)
YPD medium (Sigma, cat. no. Y1375)
EDTA mix (see recipe)
10 mg/ml Zymolyase solution (see recipe)
1% LMP agarose (see recipe)
LET solution (see recipe)
NDS cell lysis buffer (see recipe)
1000 U/ml β‐agarase (BioLabs, cat. no. M0392S); keep at −20°C
DH10B E. coli competent cells (Thermo Fisher Scientific, cat. no. EC0113)
SOC solution (Thermo Fisher Scientific, cat. no. 15544034)
LB agar with 20 mg/ml chloramphenicol (LB‐Cm) plates (Teknova, cat. no. 100216‐570)
BAC DNA Isolation kit (Sigma, cat. no. NA0100)
10 mg/ml sonicated salmon sperm DNA (Thermo Fisher Scientific, cat. no. AM9680), denaturated by boiling for 10 min every time before the experiment
Agarose (Sigma, cat. no. A9539)
30°C incubator and shaker
Refrigerated centrifuge and microcentrifuge (e.g., Eppendorf)
Vortex
1.5‐ml tubes (Dot Scientific. cat. no. RN2000‐GMT)
2‐ml tubes
Heat blocks at 42°C, 45°C, 50°C, 55°C, 68°C
Ultra micro pipette tips (Eppendorf, cat. no. AXYT400RS)
6‐cc luer‐lock syringe (PRACTICON, cat. no.70103232)
Gene Pulser and cuvettes (BioRad, cat. no. 1652662)
Toothpicks
GeneRuler 1 kb plus DNA ladder (Thermo Fisher Scientific, cat. no. SM1331)
0.2‐ml PCR strip tubes (Dot Scientific, cat. no. 415‐8PCR)
Refrigerated microcentrifuge
37°C incubator
PCR thermocycler
UV gel imaging system
Preparation of YAC/BAC DNAs in agarose plugs for electroporation
-
1
Streak sequence‐positive His+ transformants (Basic Protocol 2) on SD‐His plates, grow overnight, and inoculate five colonies separately in 5 ml of YPD medium in 50‐ml conical tubes. Grow overnight at 30°C with vigorous shaking (200‐300 rpm).
-
2
Pellet the cells by centrifugation. Remove and discard the supernatant.
-
3
Resuspend the cells from each colony in 100 μl of EDTA solution. Mix well by vortexing.
-
4
Transfer the mix into 1.5‐ml tubes. Add 100 μl of Zymolyase solution, vortex the cells for 4 s, and incubate the suspension for 30 min at 37°C.
-
5
Melt 1% LMP agarose (∼10 ml in the bottle), then take 1 ml and transfer it into a 2‐ml tube.
-
6
Keep the tube with the melted agarose in a 50°C water bath to cool.
-
7
Transfer the 2‐ml tube from step 5 and the resuspended cells (from step 4) to a 42°C heat block. Equilibrate for 15 min.
-
8
Add an equal volume of the melted agarose to the cell suspension (i.e., take 100 μl of the melted agarose per 100 μl of the cell suspension for each sequence‐positive colony) and mix well by vortexing. Keep the cell/agarose suspension at 42°C.
It is important that the final concentration of agarose be equal to 0.5%. With a higher concentration of agarose, it is impossible to completely melt the plugs for electroporation.
-
9
Take 50‐μl aliquots of the cell/agarose suspension and gently place each into a separate ultra micro pipette tip. Keep the tips horizontal for 10 min at 5°C until the agarose is completely solidified.
-
10
Transfer the agarose plugs (for each sequence‐positive colony) into 2‐ml tubes. To do this, take up LET solution in a 6‐cc syringe without a needle, place the tip of the ultra micro pipette tip into the syringe luer, and gently apply pressure. The plug should slide out into the 2‐ml tube. Incubate the agarose plugs for 1 hr at 37°C.
Four 50‐μl plugs can be made for each sequence‐positive colony.
-
11
Remove LET and add enough NDS cell lysis buffer to cover the plugs. Incubate the plugs for 1 hr at 55°C.
-
12
Remove the NDS solution carefully and wash the plugs five times with 2 ml of EDTA mix (just add and remove).
To wash the plugs, add 1‐2 ml of EDTA mix into each 2‐ml tube with the plugs, mix gently, keep the tubes for 20 min at room temperature, and then remove the EDTA mix and add 1‐2 ml of fresh EDTA mix again. Repeat this step five times. Dialyzed plugs may be stored at 4°C in EDTA mix for several months.
Electroporation the YAC/BACs into E. coli cells
-
13
The day before electroporation, take one plug for each sequence‐positive colony and place into a 2‐ml tube with water. Incubate the plugs in water overnight at room temperature.
-
14
Melt the plugs by placing the tube with the plugs at 68°C for 15 min, then cool them to 42°C for 10 min. Treat each plug with 1.5 U of agarase for 1 hr at 42°C. Chill the treated plugs on ice for 10 min.
-
15
Dilute the treated plug mix two‐fold with sterile water.
-
16
Use 1 μl of the mixture to electroporate 20 μl of the E. coli DH10B competent cells using a BioRad Gene Pulser with the settings 2.5 kV, 200 Ω, and 25 μF.
-
17
After electroporation, add 1 ml of SOC to the cuvette, mix well by pipetting up and down, and then transfer the mix into a microcentrifuge tube.
-
18
Incubate the cells for 1 hr at 37°C without agitation.
-
19
Spread 30, 100, and 300 μl of the cell suspension onto three separate LB‐Cm plates.
-
20
Incubate the plates at 37°C overnight.
-
21
Streak 5‐10 CmR colonies onto a LB‐Cm plate.
-
22
Isolate BAC DNAs from three colonies using the BAC DNA Isolation kit, according to the manufacturer's instructions.
For BACs with a size of ∼100 kb, the yield of chloramphenicol‐resistant transformants varies from 30 to 200 colonies per plate. The yield is lower for larger‐sized BACs. With BACs larger than 200 kb, the integrity of the material in E. coli should be checked. To check the size of the BACs in bacterial cells, the BAC DNAs are linearized by a unique restriction endonuclease and then checked using contour‐clamped homogeneous electric field (CHEF) electrophoresis. See Critical Parameters.
REAGENTS AND SOLUTIONS
EDTA mix
0.05 M EDTA (Sigma, cat. no. 03690)
0.01 M Tris·HCl, pH 7.5
Store buffer up to 6 months at room temperature
LET solution
0.5 M EDTA (Sigma, cat. no. 03690)
0.01 M Tris·HCl, pH 7.5
Store buffer up to 6 months at 4°C
LMP (low‐gelling/melting temperature) agarose
1% (w/v) LMP agarose (Thermo Fisher Scientific, cat. no. 16520050) prepared in 0.125 M EDTA, pH 7.5. Store agarose gel up to 6 months at room temperature.
NDS cell lysis buffer
Mix the following solutions:
20 ml 5% (w/v) N‐lauroylsarcosine, pH 9.5 (Sigma, cat. no. L9150)
1 ml 1.0 M Tris·HCl, pH 7.5
79 ml 0.5 M EDTA, pH 7.5 (Sigma, cat. no. 03690)
Store solution with the above components at room temperature
Immediately before use, add proteinase K to 2 mg/ml, final concentration
Store buffer up to 6 months at −20°C
Final concentrations:
0.39 M EDTA (Sigma, cat. no. 03690)
0.01 M Tris⋅HCl, pH 7.5
1% (w/v) N‐lauroylsarcosine
2 mg/ml proteinase K (Thermo Fisher Scientific, cat. no. AM2542).
PEG 8000 solution
To prepare, add 100 g of polyethylene glycol (PEG 8000; Sigma, cat. no. 25322‐68‐3) to 400 ml water. Add 5 ml of 1 M Tris·HCl, pH 7.5 and mix with magnetic stirrer until the PEG 8000 is completely dissolved (the mixture may be warmed to ∼40°C for faster dissolution). When PEG is dissolved, cool the solution to room temperature and add 5 ml of 1 M CaCl2 (a precipitate may appear, but that is normal). Filter‐sterilize using Rapid‐FlowTM Sterile Single Use Vacuum Filter Unit (Thermo Fisher Scientific, cat. no. 09‐741‐01) and store at room temperature up to 2‐3 weeks.
Final concentrations:
20% (w/v) polyethylene glycol 8000
10 mM CaCl2
10 mM Tris·HCl, pH 7.5
Store up to 2 weeks at room temperature.
SOS solution
1 M sorbitol (G‐BIOSCIENCES, cat. no. R038)
6.5 mM CaCl2
0.25% (w/v) Yeast Extract (Sigma, cat. no. Y0375)
0.5% (w/v) Bacto Peptone (Sigma, cat. no. 68971)
SPE solution
1 M sorbitol (G‐BIOSCIENCES, cat. no. R038)
10 mM disodium EDTA (Sigma, cat. no. 03690)
0.01 M sodium phosphate, pH 7.5
Filter‐sterilize using Single Use Vacuum Filter Unit (Thermo Fisher Scientific, cat. no. 09‐741‐01) and store indefinitely at room temperature.
STC solution
1 M sorbitol (G‐BIOSCIENCES, cat. no. R038)
10 mM CaCl2
10 mM Tris·HCl, pH 7.5
Filter‐sterilize and store indefinitely at room temperature
TOP agar‐His
Prepare the following:
1 M sorbitol (G‐BIOSCIENCES, cat. no. R038)
2% (w/v) d‐glucose (Fisher Scientific, cat. no., AAA1682836)
0.17% (w/v) Yeast Nitrogen Base (Difco, cat. no. 90004‐148)
0.5% (NH4)2SO4
3% (w/v) Bacto agar (Fisher Scientific, cat. no. DF0140‐15‐4)
Containing the following supplements:
0.006% adenine sulfate
0.006% uracil
0.005% l‐arginine HCl
0.008% l‐aspartic acid
0.01% l‐glutamic acid
0.005% l‐isoleucine
0.01% l‐leucine
0.012% l‐lysine HCl
0.002% l‐methionine
0.005% l‐phenylalanine
0.0375% l‐serine
0.01% l‐threonine
0.005% l‐tryptophan
0.005% l‐tyrosine
0.015% l‐valine
Store indefinitely at 4°C
Zymolyase solution, 10 mg/ml
Prepare 10 mg/ml of Zymolyase 20T (MP Biomedicals, cat. no. 08320921) in 20% (v/v) glycerol. Keep as frozen aliquots at −20°C.
COMMENTARY
Background Information
For decades, the source for full‐size genes for different studies has been clones from genomic BAC and YAC libraries. However, the isolation of a gene of interest using BAC and YAC libraries is time consuming because it relies on the isolation of fragmented chromosomal DNA followed by the identification of relevant clones. The frequency of desired gene‐positive clones in such libraries is less than 0.0003% (Asakawa et al., 1997). Even if a yeast or bacterial artificial chromosome contains an entire gene, its isolation is an arduous process involving the trimming of the insert by in vivo recombination in yeast. The process is more complicated if a gene is large and available as a set of fragments that must be pieced together in a contig to recover the entire gene. This requires multiple additional cloning steps to re‐assemble a full‐size gene clone. The entire procedure is even more cumbersome if the gene must be isolated from the genome of a particular individual (for example, from a patient). In such cases, a new library must be constructed specifically for that purpose. In sum, prior to the development of transformation‐associated recombination (TAR) cloning, selective isolation of a single‐copy genes from complex genomes was laborious and inconvenient, and often required the piecing together of cloned fragments of the gene into an intact version.
TAR cloning is based on the well‐established property of free DNA ends as efficient substrates for homologous recombination in yeast (Orr‐Weaver, Szostak, & Rothstein, 1981; Larionov et al., 1994). When the TAR vector containing the hooks homologous to the 5′ and 3′ ends of the targeted genomic region is constructed and genomic DNA is pre‐treated by CRISPR/Cas9 endonucleases and specifically designed gRNAs, a desired genomic fragment of up to 280 kb can be isolated in yeast from multiple DNA samples within 3‐5 weeks, with a yield of gene/fragment‐positive clones of as high as 35% (using the updated TAR cloning protocol described here) (Lee et al., 2015). An important auxiliary feature of the method is that the TAR‐cloned material may be directly moved from yeast cells to bacterial cells, making DNA isolation and further analysis easier.
For many animal and plant genomes, only limited sequence information is available. In such instances, if the sequence at one end of the gene is known, a “radial TAR cloning” approach may be used (Kouprina et al., 1998; Cancilla et al., 1998; Kim et al., 2000). A TAR vector with one unique hook and a common repeat sequence as a second hook (i.e., Alu for the primate genome or B1 for the mouse genome) can yield overlapping regions of different size, with the cloned material ending at any of several repeat sequences matching the hook. There is the risk that the presence of multiple targets in genomic DNA may lead to artifacts such as chimeric clones recovered by radial TAR cloning. However, such aberrant clones can be easily identified by sequencing of the ends of the TAR‐cloned fragment, and can then be excluded from further consideration.
At present, TAR cloning has become a routine and useful procedure. It has been used to isolate full‐size genes, gene clusters, and chromosomal regions from human, nonhuman primate, and mouse genomes for functional and structural analyses (Larionov et al., 1997; Kouprina et al., 1998; Annab et al., 2000; Nihei et al., 2002; Leem et al., 2002; Kouprina et al., 2004b; Kouprina, & Larionov, 2006; Kim et al., 2011; Kononenko et al., 2014; Kouprina, & Larionov, 2016; Kim et al., 2018; Kim et al., 2021). Because the targeting hooks of the TAR vector recombine at equal frequencies in both pairs of homologous chromosomes, TAR cloning allows one to separate alleles of a gene and infer haplotypes that may contribute to disease (Kim, Leem, Sunwoo, & Kouprina, 2003); even long‐range haplotypes can be analyzed (Kouprina et al., 2007; Kouprina et al., 2012). TAR cloning has also been applied to isolate gene homologs for evolutionary studies (Pavlicek et al., 2004; Kouprina et al., 2004c; Kouprina et al., 2004b; Koupriva et al., 2005). The TAR‐isolated full‐size gene homologs can help provide information on the evolution of coding as well as noncoding regions.
TAR cloning has also been used to close gaps in the human genome (Kouprina et al., 2003; Leem et al., 2004; Grimwood et al., 2004). The most recent example was the selective isolation of the rDNA repeats from chromosomes 21 and 22 (Kim et al., 2018; Kim et al., 2021). The TAR cloning strategy can also be used to isolate many human genes that reside within very similar segmental duplications (SDs). This resolves the difficulty encountered when mutational analysis of such regions is attempted by routine PCR methods. TAR cloning is similarly applicable to all gene families that map multiple members to SDs (Kouprina, & Larionov, 2016).
A modified version of TAR cloning (Noskov et al., 2003; Kouprina, Noskov, & Larionov, 2020) was adapted to clone and rapidly assemble genes, biosynthetic gene clusters (BGCs), and even entire genomes from simple organisms, including bacteria and viruses, from collections of soil‐derived eDNA (environmental DNA) clones (Gaida et al., 2011; Ross, Gulland, Dorrestein, & Moore, 2015; Cano‐Prieto et al., 2015; Yamanaka et al., 2014; Bonet, Teufel, Crüsemann, Ziemert, & Moore, 2015; Li et al., 2015a; Tang et al., 2015; Li et al., 2015b; Becker et al., 2004; Young et al., 2008; Feng, Kallifidas, & Brady, 2011; Gibson et al., 2008; Gibson et al., 2010; Lartigue et al., 2009; Karas, Tagwerker, Yonemoto, Hutchison, & Smith, 2012; Karas et al., 2013). The same version of TAR cloning provides a powerful tool to engineer synthetic viruses with novel properties as an aid to the design of a new generation of vaccines and vaccine vectors (Shang et al., 2017; Oldfield et al., 2017; Vashee et al., 2017). A recent example is its use to recover variants of SARS‐CoV‐2 (Thi Nhu Thao et al., 2020).
In all, TAR cloning is a powerful and versatile approach, and its use for isolating chromosomal fragments, individual genes, or gene families can be employed for functional, structural, and population studies and for comparative genomics and long‐range haplotyping, and has potential for gene therapy.
Critical Parameters
An important parameter of TAR cloning is the selection of the specific hook(s) for a TAR vector. Hooks should be unique sequences; their uniqueness may be checked by comparison to the genome. Hooks should also be free of yeast origin of replication (ARS)−like sequences; the absence of ARS may be checked by transformation of the TAR cloning vector with inserted hooks into the VL6‐48N strain deficient in HIS3, via LiAc yeast transformation (see Current Protocols article: Treco, Reynolds, & Lundblad, 1998). If the hooks contain no ARS‐like sequences, no or very few His+ transformants should appear.
Another important parameter of TAR cloning is the correct orientation of the hooks. Incorrect orientation of the hooks in the TAR vector can lead to failure to clone the desired gene. The hooks should be cloned in the vector in the same orientation in which they occur in the genome.
Efficiency of yeast transformation, which relies on the proper preparation of competent spheroplasts, is also important. The required density of the culture to make competent spheroplasts may vary depending on (i) the growth medium; (ii) growth conditions; and (iii) the type of spectrophotometer used to assess optical density. The optimum optical density should be determined empirically. At OD660, it may vary between 2.0 and 5.0.
Another variable of importance is the Zymolyase treatment time, which varies with the enzyme lot. New stocks of enzyme, even with the same vendor batch number, should be re‐checked empirically. Notably, the activity of the enzyme may decline even during storage at 4°C. If activity is too low, new stocks of the enzyme should be prepared and optimized once.
For TAR cloning experiments, the vector DNA should not be contaminated by chromosomal DNA. The quality of the vector DNA should be checked by electrophoresis. Similarly, the completeness of the vector linearization between the targeting hooks should be checked by endonuclease digestion followed by electrophoresis. This step is critical because nonlinearized vector molecules are inactive in homologous targeting of cognate chromosomal DNA.
The quality of genomic DNA is also critical for TAR cloning. Therefore, before treatment with CRISPR/Cas9, genomic DNA should be checked by contour‐clamped homogeneous electric field (CHEF) electrophoresis to ensure that the DNA is larger than the size of the region to be cloned (see Fig. 4B as an example).
The cleavage efficiency of each Cas9‐sgRNA pair is yet another critical parameter. It may be easily evaluated by digesting the appropriate target DNA fragment (Fig. 4A and 4B). The target DNA fragments should be designed such that Cas9‐sgRNA cleavage of each fragment will yield two distinctly sized DNA fragments clearly evident upon agarose gel electrophoresis. Of course, any Cas9‐sgRNA that does not cut the target DNA should be discarded.
Usually, TAR isolates with sizes of up to 250 kb can be efficiently and faithfully transferred from yeast cells to become BACs in E. coli by electroporation. However, some BACs may be truncated during electroporation. We highly recommend isolation of BAC DNA from several E. coli clones, linearization by endonuclease digestion, separation of the DNA by CHEF, and visualization of the separated DNA by ethidium bromide staining to select the ones with the proper size. As an example, Figure 6 shows physical characterization of the human NBS1 gene, 65 kb in size, in yeast and bacterial cells (Lee et al., 2015).
Figure 6.
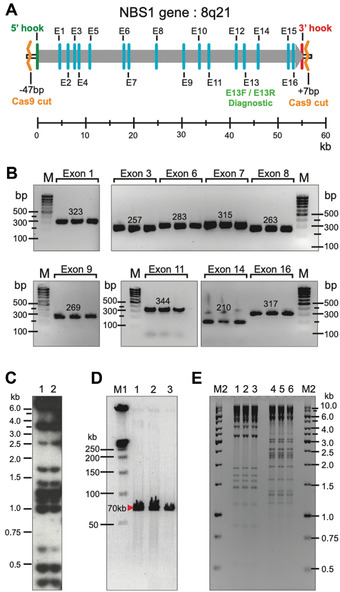
Physical characterization of YAC and BAC clones containing the human NBS1 gene isolated by CRISPR/Cas9‐mediated TAR cloning from total genomic DNA. (A) The NBS1 gene contains 16 exons and maps at 8q21. E13F/E13R are diagnostic primers for exon 13 used to screen yeast transformants to find gene‐positive clones. Positions of Cas9 sites relative to 5′ and 3′ hooks are indicated. (B) The presence of different NBS1 coding regions in three independent His+ TAR/YAC/BAC clones was examined by PCR. M corresponds to GeneRuler DNA Ladder Low Range (Sigma, cat. no. D0428). (C) Alu profiles of two YACs containing the NBS1 gene in yeast. (D) Characterization of three BAC clones containing the NBS1 gene. BAC DNAs were isolated, linearized, separated by CHEF, and visualized by staining with EtBr. Predicted sizes of the NBS1‐containing fragments are observed. M1 correspond to CHEF DNA Size Standard Ladder (BioRad, cat no. 1703635) (E) Restriction digestion profiles of BAC DNA isolated from three NBS1‐positive E. coli colonies. BAC DNA was digested by HindIII (lanes 1–3) and EcoRI (lanes 4–6). Identical profiles were observed for each restriction endonuclease. M2 corresponds to GenRuler 1 kb DNA Ladder (Thermo Fisher Scientific, cat. no. SM0311). This figure was adapted from Lee et al. (2015).
Approximately 5% of human DNA fragments cloned in TAR vectors exhibit abnormally low transformation efficiency during electroporation into E. coli, either because of toxicity or instability in bacterial cells (Kouprina et al., 2003). If E. coli clones are not obtained after electroporation, the TAR isolates can, instead, be analyzed in yeast. Using our protocol (Noskov, Chuang, Leem, Larionov, & Kouprina, 2011), microgram quantities of pure circular YAC DNA molecules up to 300 kb in size can be isolated directly from yeast cells. Such purified YAC DNA is suitable for restriction enzyme digestion and even DNA sequencing.
Troubleshooting
See Table 1 for a list of common problems with the protocols, their causes, and potential solutions.
Table 1.
Troubleshooting Guide for CRISPR/Cas9‐mediated TAR Cloning
| Problem | Possible cause | Solution |
|---|---|---|
| No His+ transformants | Medium on the plates is wrong |
Check the medium on the plates Check growth of the VL6‐48N strain on medium supplemented with histidine |
| Poor transformation efficiency | Amount of the TAR vector is less than 1 μg | Check concentration of the vector and quality of vector DNA |
| Amount of genomic DNA is less than 2 μg | Check concentration of genomic DNA | |
| PEG solution is more than 3 months old | Prepare fresh PEG 8000 solution | |
| Spheroplasts are not competent for transformation | Make spheroplasts at different OD660 | |
| Yield of region‐positive clones is low | Cleavage efficiency of sgRNA‐Cas9 is poor | Design a new sgRNA‐Cas9 pair and evaluate its efficiency |
| No region‐positive clones | Orientation of the hooks in the TAR vector is wrong | Orientation of the hooks should correspond to that illustrated in Figure 1 |
| Hooks are not unique | Choose other hooks. Uniqueness of hook sequences can be checked by blasting against genome sequence. | |
| Genomic DNA is degraded | Check genomic DNA by CHEF. Prepare genomic DNA again. | |
| Size of the target region is bigger than size of genomic DNA | Check genomic DNA by CHEF. Prepare genomic DNA again. | |
| TAR vector is not completely linearized | Check vector linearization by gel electrophoresis | |
| Transformants contain empty vector background | Hooks contains yeast ARS‐like sequences. ARS‐like sequences can be identified based on the presence of a 17‐bp ARS core consensus, WWWWTTTAYRTTTWGTT, where W = A or T; Y = T or C; R = A or G. Alternatively, carry out empty TAR vector transformation to identify. | |
| The target genomic region does not contain ARS‐like sequence | Size of the targeted region should be larger than 20 kb. Average distribution of sequence ARS‐like sequences in mammalian genomes is one per 20‐40 kb. | |
| Diagnostic PCR primers do not work | Check the primers with genomic DNA or choose other primers |
Understanding Results
The updated TAR protocol described here yields 30 to 200 His+ transformants per plate starting with 1 to 1.5 µg of the TAR vector, 1 to 2 µg of genomic DNA pre‐treated by CRISPR/Cas9, and 1 × 108 spheroplasts. Approximately 20%‐35% of these transformants typically contain the gene/region of interest. As indicated above, however, the variation in yield is normally determined by several factors: (i) uniqueness of the hooks selected for gene isolation; (ii) yeast growth conditions; (iii) level of spheroplasting; (iv) purity and size of transfected DNA; and (v) the efficiency of DNA digestion by Cas9‐sgRNAs.
Time Considerations
Construction of a TAR vector with two unique hooks usually takes 1‐2 weeks. Once the TAR vector is constructed and genomic DNA is pretreated by CRISPR/Cas9, the full procedure described here, with the analysis of the cloned inserts, can be completed in 3 weeks. More specifically, preparation of genomic DNA for TAR cloning in solution and treatment by CRISPR/Cas9 (Basic Protocol 1) requires less than 5 days once the cells are grown and the CRISPR guide sequences have been designed and checked. Transformation of spheroplasts by treated genomic DNA and the TAR vector linearized between hooks (Basic Protocol 2), and obtaining yeast transformants, can be accomplished in 5‐7 days. An additional 2 days are required to grow the individual transformants on new SD‐His plates and identify gene‐positive clones by PCR. Gene‐positive YAC/BAC transfer from yeast cells to E. coli followed by isolation of YAC/BAC DNA from bacterial cells (Basic Protocol 3) and analysis of the cloned material for size, restriction profile, and content of diagnostic primers by PCR can be accomplished within 1 week.
Author Contributions
Natasha Kouprina: conceptualization, funding acquisition, methodology, writing original draft, writing‐review and editing; Jung‐Hyun Kim: methodology, writing original draft; Vladimir Larionov: conceptualization, funding acquisition, methodology, writing original draft, writing‐review and editing.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, U.S.A. (N.K., J.H.K. and V.L.; ZIA BC010413)
Kouprina, N. , Kim, J. , & Larionov, V. (2021). Highly selective, CRISPR/Cas9‐mediated isolation of genes and genomic loci from complex genomes by TAR cloning in yeast. Current Protocols, 1, e207. doi: 10.1002/cpz1.207
Contributor Information
Natalay Kouprina, Email: kouprinn@mail.nih.gov.
Jung‐Hyun Kim, Email: kimjung@mail.nih.gov.
Vladimir Larionov, Email: larionov@mail.nih.gov.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Literature Cited
- Annab, L. , Kouprina, N. , Solomon, G. , Cable, L. , Hill, D. , Barrett, J. C. , … Afshari, C. (2000). Isolation of a functional copy of the human BRCA1 gene by transformation‐associated recombination in yeast. Gene, 250(102), 201–208. doi: 10.1016/S0378-1119(00)00180-3. [DOI] [PubMed] [Google Scholar]
- Armstrong, J. A. , & Schulz, J. R. (2008). Agarose gel electrophoresis. Current Protocols Essential Laboratory Techniques, 00, 7.2.1−7.2.20. doi: 10.1002/9780470089941.et0702s00. [DOI] [Google Scholar]
- Asakawa, S. , Abe, I. , Kudoh, Y. , Kishi, N. , Wang, Y. , Kubota, R. , … Shimizu, N. (1997). Human BAC library: Construction and rapid screening. Gene, 191(1), 69–79. doi: 10.1016/S0378-1119(97)00044-9. [DOI] [PubMed] [Google Scholar]
- Becker, M. , Aitcheson, N. , Byles, E. , Wickstead, B. , Louis, E. , & Rudenko, G. (2004). Isolation of the repertoire of VSG expression site containing telomeres of Trypanosoma brucei 427 using transformation‐associated recombination in yeast. Genome Research, 14(11), 2319–2329. doi: 10.1101/gr.2955304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonet, B. , Teufel, R. , Crüsemann, M. , Ziemert, N. , & Moore, B. S. (2015). Direct capture and heterologous expression of Salinispora natural product genes for the biosynthesis of enterocin. Journal of Natural Products, 78(3), 539–542. doi: 10.1021/np500664q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancilla, M. , Tainton, K. , Barry, A. , Larionov, V. , Kouprina, N. , Resnick, M. , … Choo, A. (1998). Direct cloning of human 10q25 neocentromere DNA transformation‐associated recombination (TAR) in yeast. Genomics, 47(3), 399–404. doi: 10.1006/geno.1997.5129. [DOI] [PubMed] [Google Scholar]
- Cano‐Prieto, C. , García‐Salcedo, R. , Sánchez‐Hidalgo, M. , Braña, A. F. , Fiedler, H. P. , Méndez, C. , … Olano, C. (2015). Genome mining of Streptomyces sp. Tü 6176: Characterization of the nataxazole biosynthesis pathway. Chembiochem, 16(10), 1461–1473. doi: 10.1002/cbic.201500153. [DOI] [PubMed] [Google Scholar]
- Feng, Z. , Kallifidas, D. , & Brady, S. F. (2011). Functional analysis of environmental DNA‐derived type II polyketide synthases reveals structurally diverse secondary metabolites. Proceedings of the National Academy of Sciences of the United States of America, 108(31), 12629–12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. , Sander, J. D. , Reyon, D. , Cascio, V. M. , & Joung, J. K. (2014). Improving CRISPR‐Cas nuclease specificity using truncated guide RNAs. Nature Biotechnology, 32(3), 279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaida, A. , Becker, M. M. , Schmid, C. D. , Bühlmann, T. , Louis, E. J. , & Beck, H. P. (2011). Cloning of the repertoire of individual Plasmodium falciparum var genes using transformation associated recombination (TAR). PLoS One, 6(3), e17782. doi: 10.1371/journal.pone.0017782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, D. G. , Benders, G. A. , Andrews‐Pfannkoch, C. , Denisova, E. A. , Baden‐Tillson, H. , Zaveri, J. , … Smith, H. O. (2008). Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science, 319(5867), 1215–1220. doi: 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- Gibson, D. G. , Glass, J. I. , Lartigue, C. , Noskov, V. N. , Chuang, R. Y. , Algire, M. A. , … Venter, J. C. (2010). Creation of a bacterial cell controlled by a chemically synthesized genome. Science, 329(5987), 52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- Grimwood, J. , Gordon, L. A. , Olsen, A. , Terry, A. , Schmutz, J. , Lamerdin, J. , … Lucas, S. M. (2004). The DNA sequence and biology of human chromosome 19. Nature, 428(6982), 529–535. doi: 10.1038/nature02399. [DOI] [PubMed] [Google Scholar]
- Hsu, P. D. , Lander, E. S. , & Zhang, F. (2014). Development and applications of CRISPR‐Cas9 for genome engineering. Cell, 157(6), 1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imburgio, D. , Rong, M. , Ma, K. , & McAllister, W. T. (2000). Studies of promoter recognition and start site selection by T7 RNA polymerase using a comprehensive collection of promoter variants. Biochemistry, 39(34), 10419–10430. doi: 10.1021/bi000365w. [DOI] [PubMed] [Google Scholar]
- Karas, B. J. , Tagwerker, C. , Yonemoto, I. T. , Hutchison, C. A. 3rd , & Smith, H. O. (2012). Cloning the Acholeplasma laidlawii PG‐8A genome in Saccharomyces cerevisiae as a yeast centromeric plasmid. ACS Synthetic Biology, 1(1), 22–28. doi: 10.1021/sb200013j. [DOI] [PubMed] [Google Scholar]
- Karas, B. J. , Molparia, B. , Jablanovic, J. , Hermann, W. J. , Lin, Y. C. , Dupont, C. L. , … Weyman, P. D. (2013). Assembly of eukaryotic algal chromosomes in yeast. Journal of Biological Engineering, 7(1), 30. doi: 10.1186/1754-1611-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karvelis, T. , Gasiunas, G. , & Siksnys, V. (2013). Programmable DNA cleavage in vitro by Cas9. Biochemical Society Transaction, 41, 1401–1406. doi: 10.1042/BST20130164. [DOI] [PubMed] [Google Scholar]
- Kim, H. , & Kim, J. S. (2014). A guide to genome engineering with programmable nucleases. Nature Reviews Genetics, 15(5), 321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- Kim, J. , Noskov, V. , Lu, X. , Bergman, A. , Ren, X. , … Stubbs, L. (2000). Discovery of a novel, paternally expressed ubiquitin‐specific processing protease gene through comparative analysis of an imprinted region of mouse chromosome 7 and human chromosome 19q13.4. Genome Research, 10(8), 1138–1147. doi: 10.1101/gr.10.8.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H. , Leem, S. H. , Sunwoo, Y. , & Kouprina, N. (2003). Separation of long‐range human TERT gene haplotypes by transformation‐associated recombination cloning in yeast. Oncogene, 22(16), 2443–2456. doi: 10.1038/sj.onc.1206316. [DOI] [PubMed] [Google Scholar]
- Kim, J. H. , Kononenko, A. , Erliandri, I. , Kim, T. A. , Nakano, M. , Iida, Y. , … Kouprina, N. (2011). Human Artificial Chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proceedings of the National Academy of Sciences, U.S.A., 108, 20048–20053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H. , Dilthey, A. , Ramaiah, N. , Lee, H. S. , Koren, S. , Dudekula, D. , … Larionov, V. (2018). Heterogeneity of human ribosomes inferred from rDNA and rRNA sequencing. Nucleic Acids Research, 46(13), 6712–6725. doi: 10.1093/nar/gky442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H. , Noskov, V. N. , Ogurtsov, A. Y. , Petrov, N. , Liskovykh, M. , Nagaraja, R. , … Larionov, V. (2021). The genomic structure of a human chromosome 22 nucleolar organizer region. Scientific Reports, 11(1), 2997. doi: 10.1038/s41598-021-82565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko, A. V. , Bansal, R. , Lee, N. C. , Grimes, B. R. , Masumoto, H. , Earnshaw, W. C. , … Kouprina, N. (2014). A portable BRCA1‐HAC (human artificial chromosome) module for analysis of BRCA1 tumor suppressor function. Nucleic Acids Research, 42(21), e164. doi: 10.1093/nar/gku870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Annab, L. , Graves, J. , Afshari, C. , Barrett, J. C. , Resnick, M. A. , & Larionov, V. (1998). Functional copies of a human gene can be directly isolated by TAR cloning with a small 3’ end target sequence. Proceedings of the National Academy of Sciences of the United States of America, 95(8), 4469–4474. doi: 10.1073/pnas.95.8.4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Leem, S. H. , Solomon, G. , Ly, A. , Koriabine, M. , Otstot, J. , … Larionov, V. (2003). Segments missing from the draft human genome sequence can be isolated by TAR cloning in yeast. EMBO Reports, 4(3), 257–262. doi: 10.1038/sj.embor.embor766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Noskov, V. N. , Koriabine, M. , Leem, S. H. , & Larionov, V. (2004a). Exploring transformation‐associated recombination cloning for selective isolation of genomic regions. Methods in Molecular Biology, 255, 69–89. [DOI] [PubMed] [Google Scholar]
- Kouprina, N. , Mullokandov, M. , Rogozin, I. B. , Collins, N. K. , Solomon, G. , Otstot, J. , … Larionov, V. (2004b). The SPANX gene family of cancer/testis‐specific antigens: Rapid evolution and amplification in African great apes and hominids. Proceedings of the National Academy of Sciences of the United States of America, 101(9), 3077–3082. doi: 10.1073/pnas.0308532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Pavlicek, A. , Mochida, G. H. , Solomon, G. , Gersch, W. , Yoon, Y. H. , … Larionov, V. (2004c). Accelerated evolution of the ASPM gene controlling brain size begins prior to human brain expansion. PLoS Biology, 2(5), 653–663. doi: 10.1371/journal.pbio.0020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Pavlicek, A. , Noskov, V. N. , Solomon, G. , Otstot, J. , Isaacs, W. , … Larionov, V. (2005). Genome Research, 15(11), 1477–1486. doi: 10.1101/gr.4212705. [DOI] [PMC free article] [PubMed]
- Kouprina, N. , & Larionov, V. (2006). TAR cloning: Insights into gene function, long‐range haplotypes and genome structure and evolution. Nature Reviews Genetics, 7(10), 805–812. doi: 10.1038/nrg1943. [DOI] [PubMed] [Google Scholar]
- Kouprina, N. , & Larionov, V. (2016). Transformation‐associated recombination (TAR) cloning for genomics studies and synthetic biology. Chromosoma, 125(4), 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Noskov, V. N. , Solomon, G. , Otstot, J. , Isaacs, W. , Xu, J. , … Larionov, V. (2007). Mutational analysis of SPANX genes in families with X‐linked prostate cancer. Prostate, 67(8), 820–828. doi: 10.1002/pros.20561. [DOI] [PubMed] [Google Scholar]
- Kouprina, N. , & Larionov, V. (2008). Selective isolation of genomic loci from complex genomes by transformation‐associated recombination cloning in the yeast Saccharomyces cerevisiae . Nature Protocols, 3(3), 371–377. doi: 10.1038/nprot.2008.5. [DOI] [PubMed] [Google Scholar]
- Kouprina, N. , Lee, N. C. , Pavlicek, A. , Samoshkin, A. , Kim, J. H. , Lee, H. S. , … Larionov, V. (2012). Exclusion of the 750‐kb genetically unstable region at Xq27 as a candidate locus for prostate malignancy in HPCX1‐linked families. Genes Chromosomes & Cancer, 51(10), 933–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Liskovykh, M. , Lee, N. C. O. , Noskov, V. N. , Waterfall, J. J. , Walke, R. L. , … Larionov, V. (2018). Analysis of the 9p21.3 sequence associated with coronary artery disease revealed a tendency to duplication in the CAD patient. Oncotarget, 9(20), 15275–15291. doi: 10.18632/oncotarget.24567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , & Larionov, V. (2019). TAR cloning: Perspectives for functional genomics, biomedicine and biotechnology. Molecular Therapy—Methods & Clinical Development, 14, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Noskov, V. N. , & Larionov, V. (2020). Selective isolation of large microbial genomic segments using transformation‐associated recombination (TAR) in yeast. Nature Protocols, 15(3), 734–749. doi: 10.1038/s41596-019-0280-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuslich, C. D. , Chui, B. , & Yamshiro, C. T. (2018). Overview of PCR. Current Protocols Essential Laboratory Techniques. 27, 1–34. doi: 10.1002/cpet.27. [DOI] [Google Scholar]
- Larionov, V. , Kouprina, N. , Eldarov, M. , Perkins, E. , Porter, G. , & Resnick, M. A. (1994). Transformation‐associated recombination between diverged and homologous DNA repeats is induced by strand breaks. Yeast, 10(1), 93–104. doi: 10.1002/yea.320100109. [DOI] [PubMed] [Google Scholar]
- Larionov, V. , Kouprina, N. , Graves, J. , Chen, X. N. , Korenberg, J. R. , & Resnick, M. A. (1996). Specific cloning of human DNA as yeast artificial chromosomes by transformation‐associated recombination. Proceedings of the National Academy of Sciences of the United States of America, 93(1), 491–496. doi: 10.1073/pnas.93.1.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larionov, V. , Kouprina, N. , Solomon, G. , Barrett, J. C. , & Resnick, M. A. (1997). Direct isolation of human BRCA2 gene by transformation‐associated recombination in yeast. Proceedings of the National Academy of Sciences of the United States of America, 94(14), 7384–7387. doi: 10.1073/pnas.94.14.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartigue, C. , Vashee, S. , Algire, M. A. , Chuang, R. Y. , Benders, G. A. , Ma, L. , … Glass, J. I. (2009). Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science, 325(5948), 1693–1696. doi: 10.1126/science.1173759. [DOI] [PubMed] [Google Scholar]
- Lee, N. C. , Larionov, V. , & Kouprina, N. (2015). Highly efficient CRISPR/Cas9‐mediated TAR cloning of genes and chromosomal loci from complex genomes in yeast. Nucleic Acids Research, 43(8), e55. doi: 10.1093/nar/gkv112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem, S. H. , Londono‐Vallejo, J. A. , Kim, J. H. , Bui, H. , Tubacher, E. , Solomon, G. , … Larionov, V. (2002). The human telomerase gene: Complete genomic sequence and analysis of tandem repeats polymorphisms in intronic regions. Oncogene, 21(5), 769–777. doi: 10.1038/sj.onc.1205122. [DOI] [PubMed] [Google Scholar]
- Leem, S. H. , Kouprina, N. , Grimwood, J. , Kim, J. H. , Mullokandov, M. , Yoon, Y. H. , … Larionov, V. (2004). Closing the gaps on human chromosome 19 revealed genes with a high density of repetitive tandemly arrayed elements. Genome Research, 14(2), 239–246. doi: 10.1101/gr.1929904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem, S. H. , Noskov, V. N. , Park, J. E. , Kim, S. I. , Larionov, V. , & Kouprina, N. (2003). Optimum conditions for selective isolation of genes from complex genomes by transformation‐associated recombination cloning, Nucleic Acids Research, 31(6), e29. doi: 10.1093/nar/gng029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Li, Z. , Yamanaka, K. , Xu, Y. , Zhang, W. , Vlamakis, H. , … Qian, P. Y. (2015a). Bacillus subtilis. Scientific Reports, 5, 9383. doi: 10.1038/srep09383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. R. , Li, Y. , Lai, J. Y. , Tang, J. , Wang, B. , Lu, L. , … Qian, P. Y. (2015b). Critical intermediates reveal new biosynthetic events in the enigmatic colibactin pathway. Chembiochem, 16(12), 1715–1719. doi: 10.1002/cbic.201500239. [DOI] [PubMed] [Google Scholar]
- Mali, P. , Esvelt, K. M. , & Church, G. M. (2013). Cas9 as a versatile tool for engineering biology. Nature Methods, 10(10), 957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihei, N. , Kouprina, N. , Larionov, V. , Oshima, J. , Martin, G. M. , Ichikawa, T. , & Barrett, J. C. (2002). Functional evidence for a metastasis suppressor gene for rat prostate cancer within a 60 kb region on human chromosome 8p21‐p12. Cancer Research, 62(2), 367–370. [PubMed] [Google Scholar]
- Noskov, V. , Koriabine, M. , Solomon, G. , Randolph, M. , Barrett, J. C. , Leem, S. H. , … Larionov, V. (2001). Defining the minimal length of sequence homology required for selective gene isolation by TAR cloning. Nucleic Acids Research, 29(6), E62. doi: 10.1093/nar/29.6.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov, V. , Kouprina, N. , Leem, S.‐H. , Koriabine, M. , Barrett, J. C. , & Larionov, V. (2002). A novel genetic system allowing to directly select gene‐positive clones during the gene capture by transformation‐associated recombination in yeast. Nucleic Acids Research, 3, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov, V. N. , Kouprina, N. , Leem, S. H. , Ouspenski, I. , Barrett, J. C. , & Larionov, V. (2003). A general cloning system to selectively isolate any eukaryotic or prokaryotic genomic region in yeast. BMC Genomics, 4(1), 16. doi: 10.1186/1471-2164-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov, V. V. , Chuang, R. Y. , Leem, S. H. , Larionov, V. , & Kouprina, N. (2011). Isolation of circular yeast artificial chromosomes for synthetic biology and functional genomics studies. Nature Protocols, 6(1), 89–96. doi: 10.1038/nprot.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield, L. M. , Grzesik, P. , Voorhies, A. A. , Alperovich, N. , MacMath, D. , Najera, C. D. , … Vashee, S. (2017). Genome‐wide engineering of an infectious clone of herpes simplex virus type 1 using synthetic genomics assembly methods. Proceedings of the National Academy of Sciences of the United States of America, 114(42), E8885–E8894. doi: 10.1073/pnas.1700534114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr‐Weaver, T. L. , Szostak, J. W. , & Rothstein, R. J. (1981). Yeast transformation: A model system for the study of recombination. Proceedings of the National Academy of Sciences of the United States of America, 78(10), 6354–6358. doi: 10.1073/pnas.78.10.6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlicek, A. , Noskov, N. , Kouprina, N. , Barrett, J. C. , Jurka, J. , & Larionov, V. (2004). Human Molecular Genetics, 13(32), 1–15. [DOI] [PubMed]
- Ran, F. A. , Hsu, P. D. , Wright, J. , Agarwala, V. , Scott, D. A. , & Zhang, F. (2013). Genome engineering using the CRISPR‐Cas9 system. Nature Protocols, 8(11), 2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, A. C. , Gulland, L. E. , Dorrestein, P. C. , & Moore, B. S. (2015). Targeted capture and heterologous expression of the Pseudoalteromonas alterochromide gene cluster in Escherichia coli represents a promising natural product exploratory platform. ACS Synthetic Biology, 4(4), 414–420. doi: 10.1021/sb500280q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, Y. , Shang, Y. , Wang, M. , Xiao, G. , Wang, X. , Hou, D. , … Hu, Z. (2017). Construction and rescue of a functional synthetic baculovirus. ACS Synthetic Biology, 6(7), 1393–1402. doi: 10.1021/acssynbio.7b00028. [DOI] [PubMed] [Google Scholar]
- Stinchcomb, D. T. , Thomas, M. , Kelly, J. , Selker, E. , & Davis, R. W. (1980). Eukaryotic DNA segments capable of autonomous replication in yeast. Proceedings of the National Academy of Sciences of the United States of America, 77(8), 4559–4563. doi: 10.1073/pnas.77.8.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X. , Li, J. , Millán‐Aguiñaga, N. , Zhang, J. J. , O'Neill, E. C. , Ugalde, J. A. , … Moore, B. S. (2015). Identification of thiotetronic acid antibiotic biosynthetic pathways by target‐directed genome mining. ACS Chemical Biology, 10(12), 2841–2849. doi: 10.1021/acschembio.5b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagwerker, C. , Dupont, C. L. , Karas, B. J. , Ma, L. , Chuang, R. Y. , Benders, G. A. , … Hutchison, C. A. 3rd (2012). Sequence analysis of a complete 1.66 Mb Prochlorococcus marinus MED4 genome cloned in yeast. Nucleic Acids Research, 40(20), 10375–10383. doi: 10.1093/nar/gks823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi Nhu Thao, T. , Labroussaa, F. , Ebert, N. , V'kovski, P. , Stalder, H. , Portmann, J. , … Thiel, V. (2020). Rapid reconstruction of SARS‐CoV‐2 using a synthetic genomics platform. Nature, 582(7813), 561–565. doi: 10.1038/s41586-020-2294-9. [DOI] [PubMed] [Google Scholar]
- Treco, D. A. , Reynolds, A. , & Lundblad, V. (1998). Growth and manipulation of yeast. Current Protocols in Protein Science, 14, A.4L.1−A.4L.6. doi: 10.1002/0471140864.psa04ls14. [DOI] [PubMed] [Google Scholar]
- Vashee, S. , Stockwell, T. B. , Alperovich, N. , Denisova, E. A. , Gibson, D. G. , Cady, K. C. , … Früh, K. (2017). Cloning, assembly, and modification of the primary human cytomegalovirus isolate toledo by yeast‐based transformation‐associated recombination. mSphere, 2(5), e00331–17. doi: 10.1128/mSphereDirect.00331-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijshake, T. , Baker, D. J. , & van de Sluis, B. (2014). Endonucleases: New tools to edit the mouse genome. Biochimica et Biophysica Acta, 1842(10), 1942–1950. doi: 10.1016/j.bbadis.2014.04.020. [DOI] [PubMed] [Google Scholar]
- Yamanaka, K. , Reynolds, K. A. , Kersten, R. D. , Ryan, K. S. , Gonzalez, D. J. , Nizet, V. , … Moore, B. S. (2014). Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proceedings of the National Academy of Sciences of the United States of America, 111(5), 1957–1962. doi: 10.1073/pnas.1319584111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, R. , Taylor, J. E. , Kurioka, A. , Becker, M. , Louis, E. J. , & Rudenko, G. (2008). Isolation and analysis of the genetic diversity of repertoires of VSG expression site containing telomeres from Trypanosoma brucei gambiense, T. b. brucei and T. equiperdum . BMC Genomics, 9, 385. doi: 10.1186/1471-2164-9-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.