

Abstract
Multicellular organisms use mitogens to regulate cell proliferation, but how fluctuating mitogenic signals are converted into proliferation-quiescence decisions is poorly understood. In this work, we combined live-cell imaging with temporally controlled perturbations to determine the time scale and mechanisms underlying this system in human cells. Contrary to the textbook model that cells sense mitogen availability only in the G1 cell cycle phase, we find that mitogenic signaling is temporally integrated throughout the entire mother cell cycle and that even a 1-hour lapse in mitogen signaling can influence cell proliferation more than 12 hours later. Protein translation rates serve as the integrator that proportionally converts mitogen history into corresponding levels of cyclin D in the G2 phase of the mother cell, which controls the proliferation-quiescence decision in daughter cells and thereby couples protein production with cell proliferation.
Cells convert extracellular mitogen availability into cell decisions by means of mitogen signaling pathways. One central branch is the mitogen-activated protein kinase (MAPK) pathway, in which growth factors bind their receptors at the plasma membrane and activate the Raf-Mek-Erk MAPK cascade. Erk activation leads to the activation of several transcription factors that promote transcription of cyclin D, activation of cyclin-dependent kinases 4/6 and 2 (CDK4/6 and CDK2), and cell cycle entry (Fig. 1A) (1). Although signal transduction from the plasma membrane to Erk takes only 3 min (2), commitment to the cell cycle occurs only once per cell cycle (Fig. 1A). Therefore, it is unclear how short–time scale MAPK signals control long–time scale cellular proliferation. Early studies based on quiescent cells released from serum starvation led to the textbook model that cells sense mitogen levels in the early G1 phase before crossing the restriction point (R point), a point marked by the buildup of CDK2 activity after which cells become mitogen independent for the remainder of the cell cycle (3–6). In cycling cells, by contrast, blocking mitogenic signals during the mother cell G2, but not during the daughter cell G1, impedes cell cycle entry (6–8). This observation led to the model of a G2-specific window in the mother cell cycle where cells sense mitogen availability (Fig. 1B) (6–8), and this model is consistent with the recent observation that the proliferation-quiescence decision is already made in late anaphase of the mother cell cycle in unperturbed cells (6, 9). However, in experiments where the MAPK pathway is continuously inhibited, two variables are changing simultaneously—the duration and the cell cycle timing of MAPK inhibition. Therefore, an alternative model to the G2-specific window is that cells sense the duration of mitogenic signaling throughout the entire mother cell cycle (Fig. 1B).
Fig. 1. MAPK activity is temporally integrated throughout the mother cell cycle to control daughter cell proliferation, whereas CDK4/6 activity is only required in early G1.
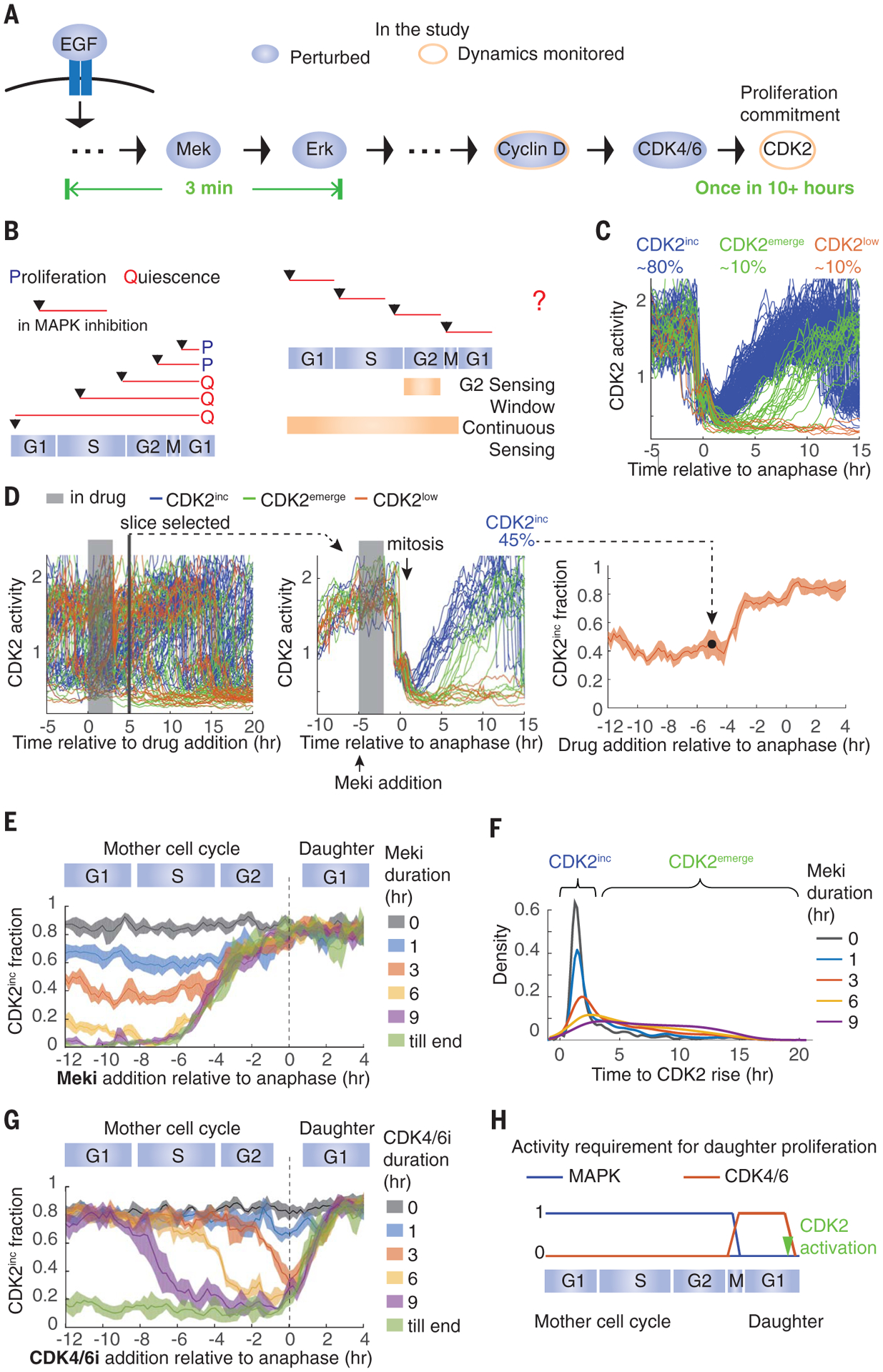
. (A) Transduction of mitogen signals to cell cycle machinery. EGF, epidermal growth factor. (B) (Left) Depiction of previous MAPK inhibition experiments. (Right) Two potential models of mitogen sensing: Cells sense mitogen availability only in the mother cell G2 or continuously throughout the mother cell cycle. (C) Typical cell CDK2 activity in optimal growth conditions. hr, hour. (D) Data processing for establishing daughter cell fate as a function of the time of drug addition, read out as CDK2 activity. (Left) CDK2 activity in asynchronously cycling cells. In this example, cells were treated with Meki for 3 hours before the drug was washed off. (Middle) Traces from cells that underwent mitosis 5 hours after drug addition were extracted and the fraction of CDK2inc daughter cells was calculated. (Right) Repeating this process for every time slice generates the plot of fraction of CDK2inc daughter cells versus the time of drug addition relative to anaphase. (E) Fraction of CDK2inc daughter cells treated with 0, 1, 3, 6, or 9 hours Meki or Meki until the end (till end) of the experiment, at various times relative to anaphase. (F) Density distribution of the time between anaphase and the rise of CDK2 activity in CDK2inc and CDK2emerge daughter cells. Cells were treated with Meki for the indicated durations starting from the G1 phase of the mother cell cycle. Areas under the curves were normalized to 1. (G) Same as in (E) except with CDK4/6i treatment. (H) Summary of the data. All data are from MCF10A cells; all CDK2inc fractions are plotted as means ± 95% confidence intervals shown as shaded bands, where nonoverlapping shading indicates a statistically significant difference as determined by t test, with P < 0.05.
To assess these two models, we used live-cell imaging of a CDK2 activity sensor (6) to follow the commitment to proliferation in conditions where the cell cycle timing and the duration of MAPK inhibition are decoupled. If mitogenic signaling is only required in G2, we should be able to inhibit the MAPK pathway for the duration of G1 and S (4 to 12 hours before mitosis), re-enable MAPK signaling at the end of the S phase, and see no effect on proliferation commitment. Alternatively, if cells continuously integrate mitogens during the entire mother cell cycle, MAPK inhibition in any phase would reduce the fraction of proliferating daughter cells (fig. S1A).
In control conditions, ~80% of MCF10A mammary epithelial cells immediately increase CDK2 activity after the completion of mitosis and are committed to the cell cycle (80% CDK2inc; Fig. 1C, blue), as has been seen previously (6, 10). The remainder enter a transient state of quiescence characterized by low CDK2 activity, where they can remain for the rest of the experiment (CDK2low; Fig. 1C, orange), or they emerge from this transient quiescence by building up CDK2 activity to reenter the cell cycle (CDK2emerge; Fig. 1C, green). To test the two models, we reversibly inhibited the MAPK pathway using a Mek inhibitor (Meki) in asynchronously cycling MCF10A cells for a fixed duration (1, 3, 6, or 9 hours) before washing out the inhibitor. We then computationally grouped cells that received the inhibition in different cell cycle phases on the basis of the time of drug addition relative to anaphase (fig. S1, B and C), and we examined the fraction of proliferating daughter cells in each group by monitoring CDK2 activity (Fig. 1D). We confirmed that Meki does not notably alter the length of the ongoing cell cycle (fig. S1, D and E) and that adding in and washing off Meki rapidly modulates Erk activity, as has been shown previously (fig. S2A) (11, 12). Consistent with previous observations (6, 8), continuous treatment of Meki (“till end”; Fig. 1E) beginning at least 6 hours before mitosis completely blocks proliferation of daughter cells (0% CDK2inc).
Notably, mother cells receiving a pulse of Meki as short as 1 hour in length at any time during their cell cycle generate fewer CDK2inc daughter cells (65% CDK2inc; Fig. 1E and fig. S2, B and C). This indicates that Mek activity is required throughout interphase in mother cells for efficient daughter cell proliferation and that cells carry the memory of a 1-hour absence in Mek activity all the way through the mother cell cycle and into the daughter cell cycle. The reduction of the CDK2inc fraction coincides with an increase of the CDK2emerge fraction, which suggests that these daughter cells need extra time to commit to the cell cycle (Fig. 1F and fig. S2, B to E). Although the fraction of CDK2inc daughter cells does not vary with the cell cycle timing of the Meki treatment, it decays with the duration of treatment (Fig. 1, E and F, and fig. S2, C and E). The same treatment duration–dependent effect on proliferation is also seen with Erk inhibition (fig. S2F), and upon withdrawal of epidermal growth factor (fig. S2G). Taken together, these results show that MAPK signaling is temporally integrated throughout the mother cell interphase to guide the proliferation of daughter cells.
We next examined where the integration of MAPK signaling occurs in the pathway. MAPK signaling promotes cell cycle entry by means of the activation of cyclin D–CDK4/6 (13–16). We therefore tested whether CDK4/6 activity is also temporally integrated throughout the entire mother cell cycle. We used palbociclib to inhibit CDK4/6 (CDK4/6i) for 1, 3, 6, or 9 hours or continuously. Contrary to Meki, CDK4/6i shows a cell cycle phase–dependent effect on proliferation—CDK4/6i covering 0 to 3 hours after anaphase maximally blocks proliferation, whereas treatments that cover other time windows show no effect on proliferation (Fig. 1G and fig. S3). This result reveals that cells only require CDK4/6 activity from anaphase until after CDK2 activation (fig. S3). Thus, the integration of MAPK signaling occurs upstream of CDK4/6 (Fig. 1H).
We next set out to determine the molecular nature of the integrator. We reasoned that the integrator should satisfy three criteria: (i) regulate proliferation; (ii) sense a 1- to 3-hour lapse of MAPK activity throughout the cell cycle; and (iii) carry memory of MAPK inhibition (fig. S4A). As first candidates, we considered CDK inhibitor proteins p21 and p27 (17) and cyclin D, the nexus between MAPK signaling and cell cycle entry (13, 14). The inhibitor protein p21 does not satisfy criterion (ii) (fig. S4B), and p27 does not satisfy criteria (i) and (iii) (fig. S4, C to E). Cyclin D satisfies criterion (i)—knockdown of all three cyclin D genes in mother cells impairs proliferation of daughter cells (20 to 30% CDK2inc; Fig. 2A and fig. S5, A and B), and overexpression of cyclin D1 rescues the Meki-induced proliferation defect (fig. S5, C and D). We and others have reported that cyclin D1 protein levels increase in the G2 phase of the cell cycle (10, 18, 19) (Fig. 2B). This G2 rise of cyclin D1 protein is attenuated by prior Mek inhibition in a duration-dependent (Fig. 2B, left) and cell cycle phase–independent manner (Fig. 2B, right), satisfying criterion (iii) that cyclin D1 protein levels in G2 carry memory of MAPK inhibition. However, cyclin D1 protein levels do not sense instantaneous Mek activity because Mek inhibition fails to rapidly alter cyclin D1 protein levels in G1 and only later blocks cyclin D1 rise in G2 (Fig. 2C). Thus, criterion (ii) is not satisfied, and cyclin D1 is not the integrator. Nonetheless, these results reveal that the integrator relays the information of MAPK activity in the mother cell G1 to cyclin D protein levels in G2 (fig. S4A).
Fig. 2. Cyclin D acts as a downstream effector of the MAPK integrator.
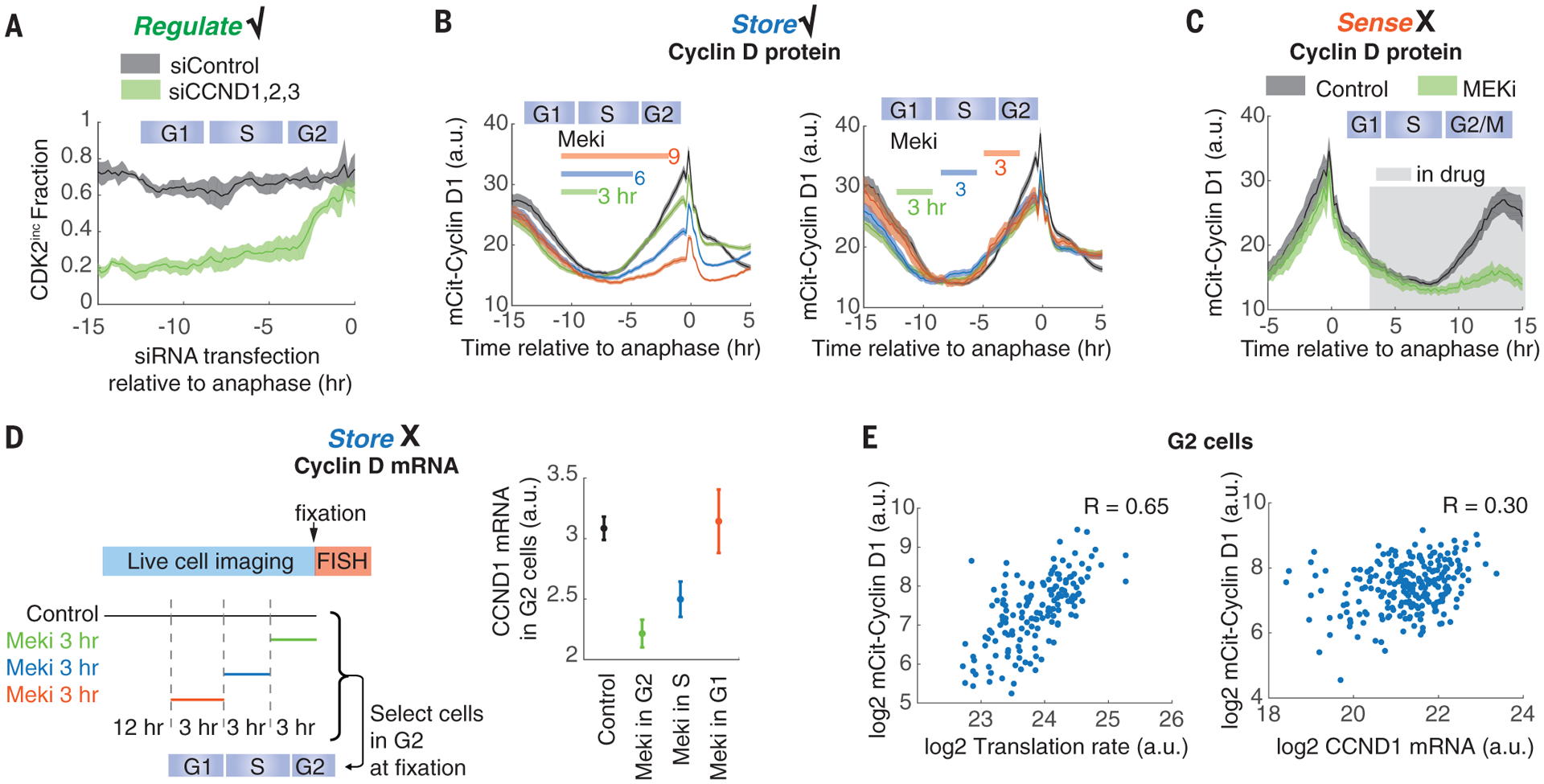
(A) Cyclin D regulates cell proliferation. The fraction of CDK2inc daughter cells is plotted against the time of small interfering RNA (siRNA) addition. siControl, nontargeting siRNA; siCCND1,2,3, siRNAs against CCND1, CCND2, and CCND3. (B) Cyclin D1 protein levels in G2 correlate negatively with the duration of the Meki treatment in the ongoing cell cycle, regardless of the cell cycle phase of the treatment. Cells expressing mCitrine-cyclin D1 from the endogenous locus were treated for 0 (black curve), 3, 6, or 9 hours (left), or for 3 hours (right) with Meki during imaging; the timing of treatment is indicated by colored bars. a.u., arbitrary units. (C) Cyclin D1 protein levels in G1 do not sense Meki treatment. The experimental setup is the same as in (B), except that Meki was left in once added. (D) Meki treatment in G1 does not reduce cyclin D1 mRNA levels in G2. Time-lapse imaging of CDK2 activity in asynchronous cells was followed by a 3-hour Meki treatment 0, 3, or 6 hours before fixation. Correspondingly, G2 cells at the time of fixation (10 to 12 hours after anaphase) received Meki treatment in the G2, S, or G1 phase, respectively. Cyclin D1 mRNA levels were measured by RNA fluorescence in situ hybridization (FISH) as the total signal in individual G2 cells. (E) Cyclin D1 protein levels strongly correlate with protein translation rate in G2 cells. Single-cell translation rates were measured with the O-propargyl-puromycin (OPP) assay, where cells were pulsed with OPP for 24 min before fixation (26) and quantified as the integrated intensity of incorporated OPP within each cell. G2 cells were identified, and cyclin D1 mRNA levels were measured as in (D). Each dot represents a single cell. R value is calculated as the Pearson correlation coefficient. mCit, mCitrine. With the exception of (E), all data are plotted as means ± 95% confidence intervals shown as shaded bands [(A), (B), and (C)] or error bars (D). All data are from MCF10A cells.
It has long been established that the MAPK pathway regulates cyclin D transcription. However, the five proposed cyclin D transcription factors—Ets1, Fos, Fra1, Jun, and Myc (20–22)—either do not satisfy the three criteria above or do not rescue the Meki-induced proliferation defect on overexpression (figs. S6 and S7). Moreover, cells receiving Meki for 3 hours in G1 alone do not show reduced cyclin D1 mRNA in G2 (Fig. 2D), which suggests that cyclin D1 mRNA levels, in contrast to cyclin D1 protein levels, do not store a cell’s MAPK history. This discrepancy points to a post-transcriptional regulation step of cyclin D in relaying MAPK history to cyclin D protein levels in G2.
Cyclin D is an unstable protein and thus is acutely sensitive to changes in translation rate (fig. S8). Its yeast homolog, Cln3, has been proposed to act as a translational sizer, the abundance of which reflects the translation rate of cells (23, 24). The level of cyclin D protein strongly correlates with the translation rate in individual G2 cells, much more so than it correlates with its mRNA levels (Fig. 2E). The building of protein mass is naturally an integration process. We therefore tested whether protein synthesis might act as the integrator that reflects the history of MAPK signaling.
Direct inhibition of translation with puromycin or cycloheximide impairs cell cycle progression and blocks mother cell mitoses and therefore cannot be used to study daughter cell proliferation (fig. S9, A and B). We circumvented this problem by indirectly reducing translation in mother cells using the mTor (mammalian target of rapamycin) inhibitor torin, which only mildly extends the length of the mother cell cycle (fig. S9, C and D). Torin treatment impairs proliferation of daughter cells in a duration-dependent, cell cycle phase–independent manner (Fig. 3A), which suggests that unperturbed translation throughout the mother cell cycle is required for maximal daughter cell proliferation [criterion (i)]. Translation rates rapidly decrease after a short Meki treatment in a cell cycle phase–independent manner, which suggests that translation rates sense Mek activity throughout the cell cycle [criterion (ii); Fig. 3, B and C]. Longer Meki treatment leads to further decrease of translation (Fig. 3, B and C), a duration-dependent effect similar to that observed on cyclin D1 levels in G2 and on the fraction of proliferative daughter cells (Fig. 3D). Cells receiving a brief Meki treatment in the G1 or S phase maintain lower translation rates in G2, which suggests that protein translation stores the history of MAPK activity [criterion (iii); Fig. 3E] and can function as an integrator of mitogen signaling.
Fig. 3. Global protein translation rate integrates MAPK activity.
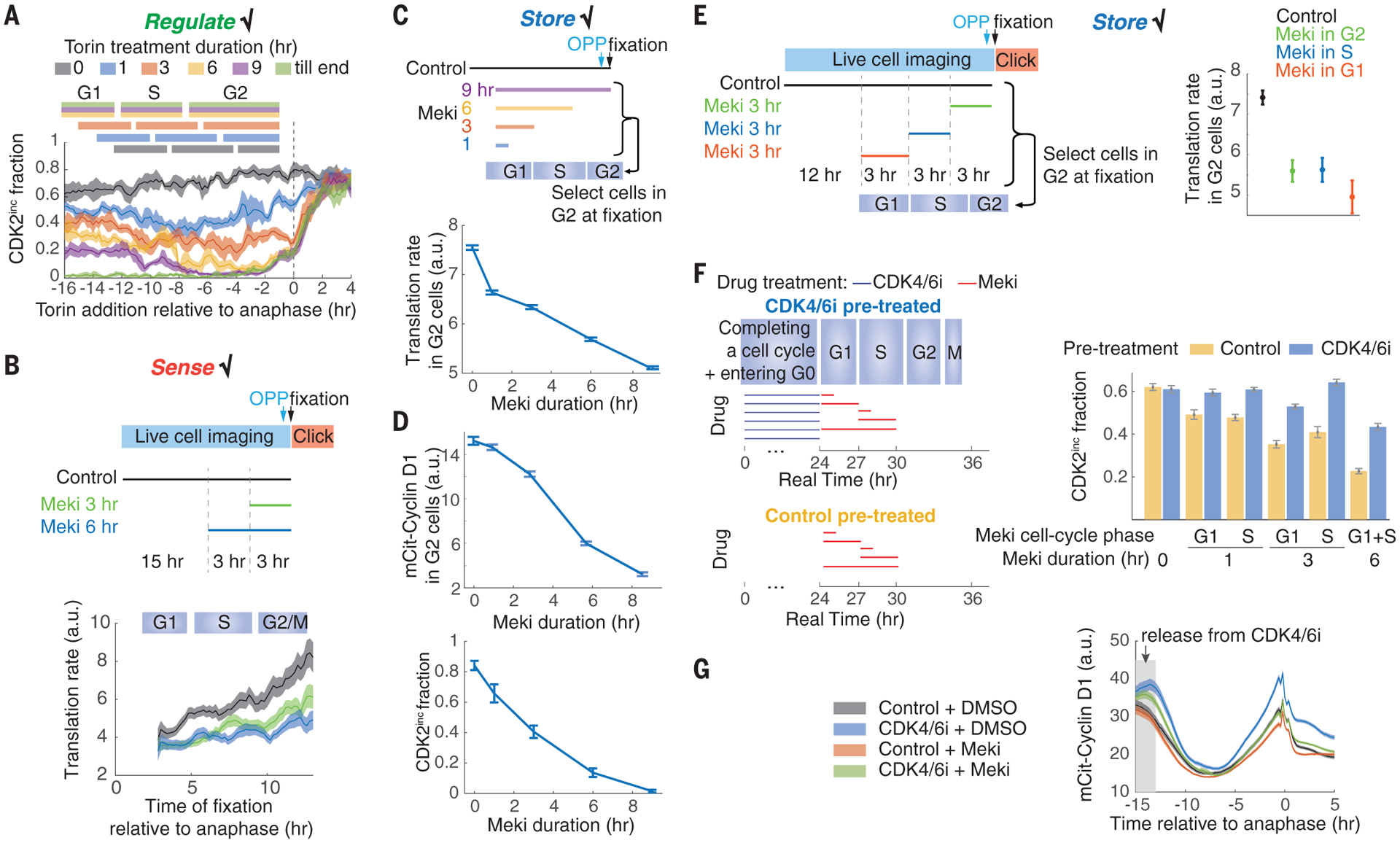
(A) Perturbation of protein translation by torin in the mother cell cycle impairs daughter cell cycle entry in a duration-dependent manner. The experimental setup is the same as in Fig. 1E except that torin is used. Cell cycle phases are derived from fig. S9, C and D. (B) Translation rate can rapidly sense MAPK activity throughout the cell cycle. Time-lapse imaging of CDK2 activity in asynchronous cells was followed by an OPP assay, as in Fig. 2E. The OPP signal was then reconstructed as a function of the time since anaphase. (C) Meki reduces the G2 phase translation rate in a duration-dependent manner. (D) Duration-dependent effect of Meki on the G2 phase cyclin D1 protein levels and on the fraction of CDK2inc daughter cells. (E) Translation rates can store past MAPK activity. The experimental setup is the same as in (B) except that cells were fixed at 0, 3, or 6 hours after the 3-hour Meki treatment. Correspondingly, G2 cells at the time of fixation (10 to 12 hours after anaphase) received Meki treatment in the G2, S, or G1 phase, respectively. (F) Enhancing translation by pre-enlarging cells using CDK4/6i can rescue the Meki-induced proliferation defect. (G) CDK4/6i pretreatment can rescue the Meki-induced reduction of cyclin D1 levels. All data are from MCF10A cells and are plotted as means ± 95% confidence intervals, shown as shaded bands [(A), (B), and (G)] or error bars [(C), (D), (E), and (F)].
To determine whether protein translation is the integrator that regulates the cyclin D rise in G2 and consequent proliferation decisions in daughter cells, we sought to test whether enhanced translation could rescue the Meki-induced proliferation defect. Inspired by the observation that CDK4/6 inhibition is the only condition out of many tested where cells accumulate protein mass in the absence of cell cycle progression (25), we pretreated cells with palbociclib for 24 hours to increase their mass (and hence ribosome number) (Fig. 3F and fig. S9E). We then released these cells into the cell cycle; treated them with Meki in the G1 or S phase for 1, 3, or 6 hours; and tracked daughter cell proliferation (Fig. 3F). Pretreating cells with CDK4/6 inhibitor before Meki treatment restores cyclin D1 levels in G2 and proliferation in daughter cells (Fig. 3, F and G). Therefore, we conclude that the history of MAPK activity is stored in the translation rate, which in turn influences the proliferation of daughter cells.
Contrary to the long-standing paradigm that cells evaluate their mitogenic environment in a window before the R point in G1, our data indicate that cells integrate mitogen signaling throughout the entire mother cell cycle to modulate the proliferation-quiescence decision in daughter cells (Fig. 4A). This proposed model can be illustrated by a simple mechanical analogy, where water constantly drips into a bucket, and it is the integral of the dripping—the weight of the bucket—that eventually flips the proliferation-quiescence switch (Fig. 4B). We propose that protein synthesis functions as the bucket to record the history of mitogenic signals throughout the mother cell cycle. Given that translation rate is strongly correlated with cell size and cell growth, cell growth itself may be the bucket (the integrator); however, it is not currently possible to measure single-cell growth at the precision required to test this idea. The MAPK history is decoded into cyclin D protein levels in the mother cell G2 to regulate the proliferation of daughter cells. This ensures that cells achieve a threshold protein synthesis rate before committing to the cell cycle. Notably, by virtue of the temporal integration, this system constitutes a form of cellular memory, in which the past experience of mitogenic signals during the entire mother cell cycle influences the fraction of proliferating daughter cells.
Fig. 4. Models.
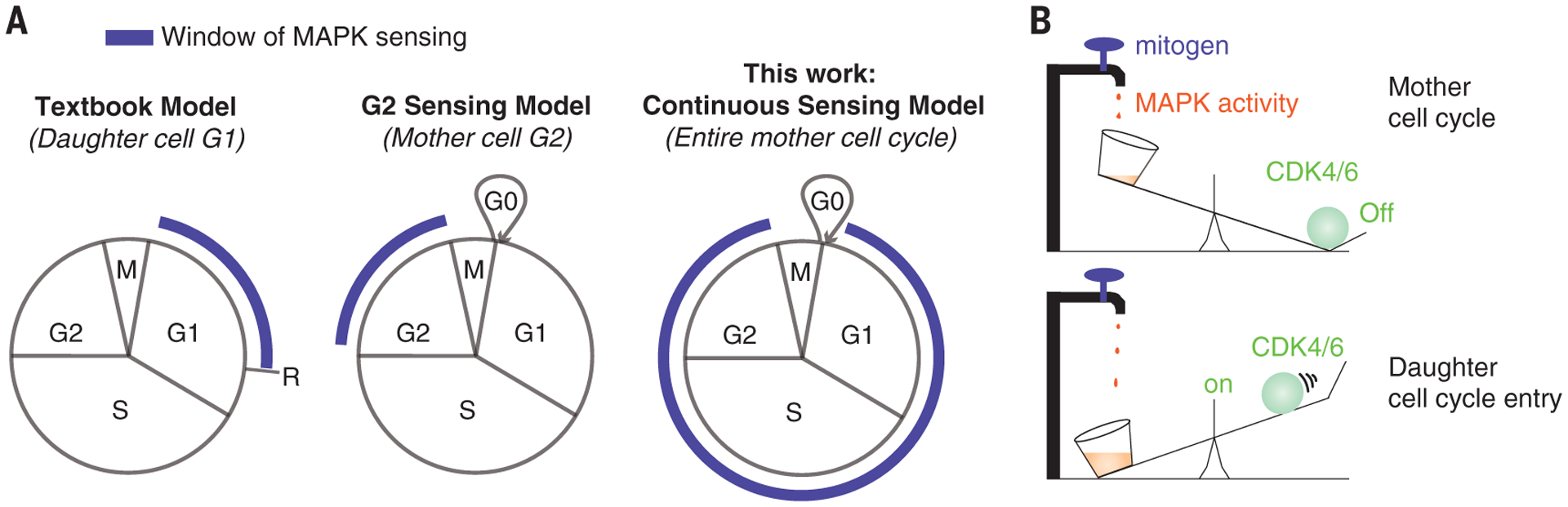
(A) An updated model of mitogen sensing. R, restriction point. (B) A mechanical metaphor of how MAPK signaling promotes cell cycle entry.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Mansfeld for RPE-hTERT Flp-In parental and RPE-hTERT cyclin D1-mVenus cell lines; J. Albeck for MCF10A Erk-sensor cell line; N. Ahn and J. Albeck for insightful discussions and comments on the manuscript; BioFrontiers Computing Core for computing support; and the Spencer laboratory for general discussion and assistance.
Funding:
This work was supported primarily by an NIH K22 Early-Career Investigator award (1K22CA188144-01) and an NIH Director’s New Innovator award (DP2-CA238330), as well as an American Cancer Society Research Scholar grant (RSG-18-008-01), a Beckman Young Investigator award, a Boettcher Webb-Waring Early-Career Investigator award, a Kimmel Scholar award (SKF16-126), a Pew-Stewart Scholar award, and a Searle Scholar award (SSP-2016-1533) to S.L.S.
Footnotes
Competing interests: The authors declare no conflict of interest.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/368/6496/1261/suppl/DC1
Materials and Methods
Figs. S1 to S9
References (27–36)
MDAR Reproducibility Checklist
Data and materials availability:
All numerical data are available in the BioStudies database under accession number S-BSST314. Materials are available by contacting S.L.S.
REFERENCES AND NOTES
- 1.Meloche S, Pouysségur J, Oncogene 26, 3227–3239 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Toettcher JE, Weiner OD, Lim WA, Cell 155, 1422–1434 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan DO, The Cell Cycle, Principles of Control (New Science Press, 2007). [Google Scholar]
- 4.Pardee AB, Proc. Natl. Acad. Sci. U.S.A 71, 1286–1290 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinberg RA, Cell 81, 323–330 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Spencer SL et al. , Cell 155, 369–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hitomi M, Stacey DW, Mol. Cell. Biol 19, 4623–4632 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang HW, Chung M, Kudo T, Meyer T, Nature 549, 404–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moser J, Miller I, Carter D, Spencer SL, Proc. Natl. Acad. Sci. U.S.A 115, E8219–E8227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gookin S et al. , PLOS Biol. 15, e2003268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albeck JG, Mills GB, Brugge JS, Mol. Cell 49, 249–261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillies TE, Pargett M, Minguet M, Davies AE, Albeck JG, Cell Syst. 5, 549–563.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu JJ et al. , Mol. Cell. Biol 15, 3654–3663 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lavoie JN, L’Allemain G, Brunet A, Müller R, Pouysségur J, J. Biol. Chem 271, 20608–20616 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Hinds PW et al. , Cell 70, 993–1006 (1992). [DOI] [PubMed] [Google Scholar]
- 16.Ezhevsky SA et al. , Proc. Natl. Acad. Sci. U.S.A 94, 10699–10704 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherr CJ, Roberts JM, Genes Dev. 9, 1149–1163 (1995). [DOI] [PubMed] [Google Scholar]
- 18.Hitomi M, Stacey DW, Curr. Biol 9, 1075–1084 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Zerjatke T et al. , Cell Rep. 19, 1953–1966 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albanese C et al. , J. Biol. Chem 270, 23589–23597 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Burch PM, Yuan Z, Loonen A, Heintz NH, Mol. Cell. Biol 24, 4696–4709 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daksis JI, Lu RY, Facchini LM, Marhin WW, Penn LJ, Oncogene 9, 3635–3645 (1994). [PubMed] [Google Scholar]
- 23.Polymenis M, Schmidt EV, Genes Dev. 11, 2522–2531 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Litsios A et al. , Nat. Cell Biol 21, 1382–1392 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Ginzberg MB et al. , eLife 7, e26957 (2018).29889021 [Google Scholar]
- 26.Liu J, Xu Y, Stoleru D, Salic A, Proc. Natl. Acad. Sci. U.S.A 109, 413–418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All numerical data are available in the BioStudies database under accession number S-BSST314. Materials are available by contacting S.L.S.