

Abstract
Aberrant neuronal cell cycle re-entry (CCR) is a phenomenon that precedes and may mechanistically lead to a majority of the neuronal loss observed in Alzheimer’s disease (AD). Recent developments concerning the regulation of aberrant neuronal CCR in AD suggest that there are potential intracellular signaling “hotspots” in AD, cancer, and brain insulin resistance, the latter of which is characteristically associated with AD. Critically, these common signaling nodes across different human diseases may represent currently untapped therapeutic opportunities for AD. Specifically, repurposing of existing US Food and Drug Administration-approved pharmacological agents, including experimental therapeutics that target the cell cycle in cancer, may be an innovative avenue for future AD-directed drug discovery and development. In this review we discuss overlapping aspects of AD, cancer, and brain insulin resistance from the perspective of neuronal CCR, and consider strategies to exploit them for prevention or therapeutic intervention of AD.
Keywords: Alzheimer’s disease, amyloid, cell cycle re-entry, tau
Alzheimer’s disease (AD) is the sixth leading cause of death in the US, yet it may actually represent the third overall cause of death in the elderly [1]. AD causes irreparable neurodegeneration that destroys memory and cognition. Although extracellular amyloid plaques, intraneuronal neurofibrillary tangles, and significant synaptic and neuronal loss in the brain define AD [2], the pathogenesis of AD is complex and its dysfunctional metabolic pathways continue to be mapped [3, 4]. Two such seminal pathways are those for aberrant neuronal cell cycle re-entry (CCR) [5] and reduced insulin signaling [6], which together form the focus of this review.
NEURONAL DEATH IN AD
In the brain regions affected by AD, neuronal death is a massive, irreversible pathological feature. Indeed, up to 65% of hippocampal CA1 neurons and 90% of the neurons in select neocortical areas degenerate and die in typical AD cases [7, 8]. There is substantial evidence that most of the neuron death in AD is prompted by aberrant cell cycle re-entry (CCR), which is typified by the re-expression of signaling effectors commonly associated with cell cycle progression and cell proliferation [9, 10]. While neurons generally are considered post-mitotic, and permanently arrested in the G0 phase of the cell cycle (Fig. 1), in vulnerable brain regions, roughly 10% of the neurons exhibit biochemical and histological evidence of CCR beginning in presymptomatic AD stages. However, the neurons never complete cytokinesis and their gradual disappearance from the brain parallels a commensurate net loss of neurons in the same regions [10–12].
Fig. 1.
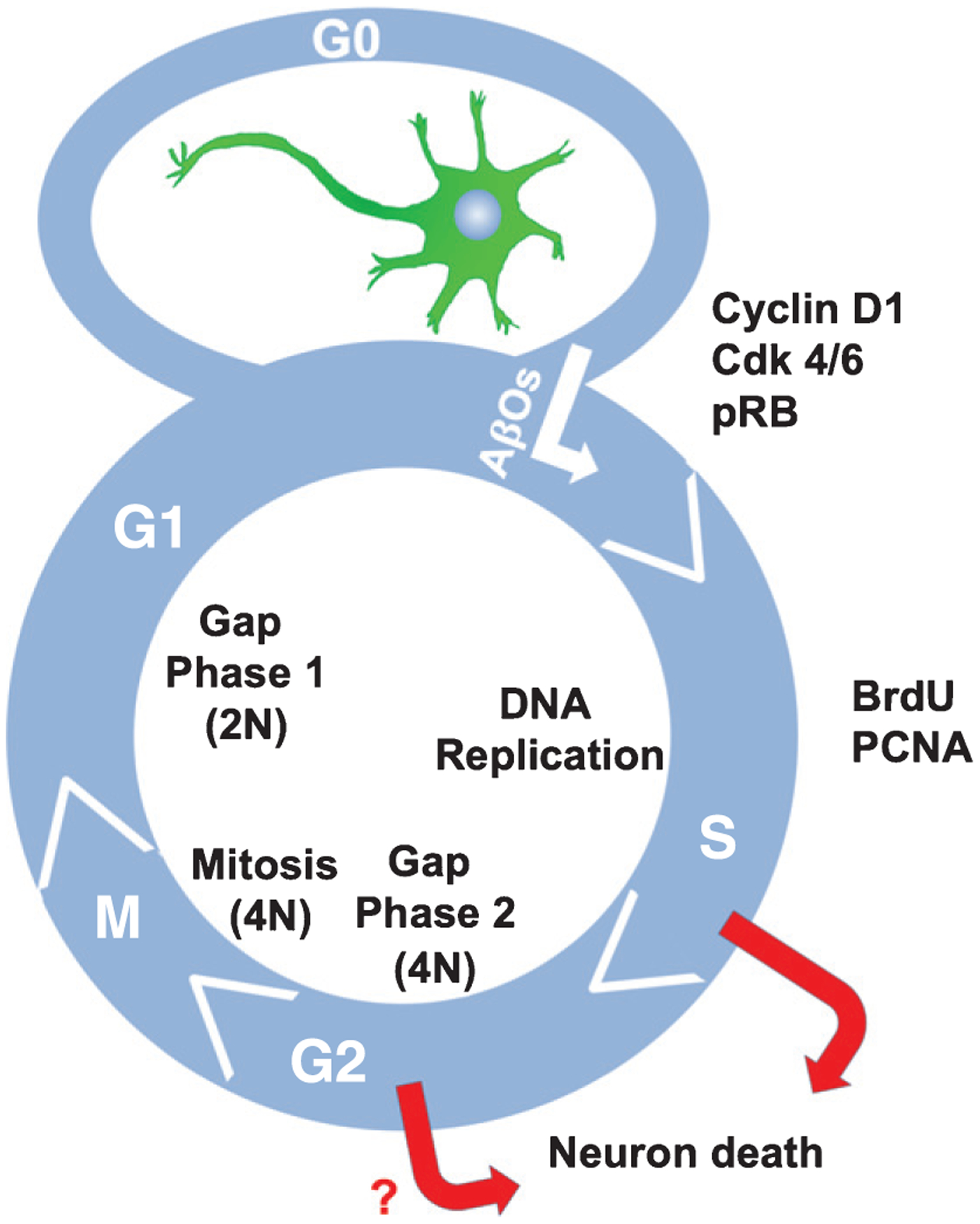
Cell cycle re-entry in AD neurons. AβO exposure induces neurons to exit G0 and proceed through the cell cycle at least as far as S phase. Markers of AβO-mediated CCR include Cyclin D1, Cdk4/6, phosphorylated retinoblastoma (pRB), BrdU incorporation, and PCNA expression. Neurons never complete cytokinesis, but instead eventually die. N refers to the haploid DNA content in the cell.
The precise signaling mechanisms propelling this neuronal loss are not well defined, although our deeper understanding of the molecular pathogenesis of an array of human diseases, including AD, indicates that many seemingly unrelated pathologies share common aberrant pathways. For example, IκB kinase (IKK) and c-Jun N-Terminal kinase (JNK) have been linked to cancer, diabetes, and inflammation [13–15]; serine/threonine glycogen synthase kinase 3 (GSK3) has been implicated in bipolar disease, AD, type II diabetes, cardiac hypertrophy, and cancer [16–18]; and both the PI3 kinase-mTOR and Erk1/2 pathways have been linked to cancer, type II diabetes, and AD [19–23]. Likewise, insulin resistance and reduced glucose uptake are pathological phenomena that link diabetes with AD [24]. Thus, signaling convergence points among seemingly unrelated diseases may provide opportunities for accelerated approaches to prevent and treat AD.
NEURONAL CCR AND AD
Proliferating eukaryotic cells have readily detectable mitotic (M) and DNA synthesis (S) phases separated by two gap (G1 and G2) phases. In contrast, terminally differentiated neurons have permanently exited the cell cycle and thus reside as mitotically quiescent cells in a phase called G0 (Fig. 1). Classically, it was thought that exposure of neurons to various stresses can cause their re-entry back into G1, although recent results surprisingly indicate that some neural stem cells in Drosophila exit the cell cycle from G2 in response to nutritional cues using the insulin signaling pathway [25].
In the brains of individuals with AD, re-expression of several cell cycle or proliferation-associated proteins is indicative of neuronal exit from G0. Examples of such cell cycle proteins include cyclins B, D, and E [26–28], cyclin-dependent kinases 1 and 4 (Cdk1 and Cdk4) [29, 30], PCNA [26, 31], Ki67 [27, 31], and Cdk inhibitors, such as p16 and p21 [32, 33]. Cell cycle-related protein overexpression has been observed in the brains of individuals with mild cognitive impairment, which is a clinical predecessor of AD, and in brain regions that experience substantial neuronal loss in AD [12]. DNA replication was also observed using fluorescence in situ hybridization of four different loci on three chromosomes. In this study, DNA replication was detected in a significant fraction of neurons in the “at risk” regions of the brains of individuals with AD, but not in non-demented aged-matched controls [34]. Furthermore, in AD model mouse brains, abnormal increases in cell cycle markers and DNA replication have been detected in regions corresponding to sites where there is neuronal loss in humans with AD [35, 36]. These findings imply that aberrant neuronal CCR precedes neuronal loss in AD. Notably, there is growing evidence that several other neurodegenerative conditions, including stroke, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease, exhibit extensive neuronal CCR [37–40], suggesting that CCR is a common pathological feature of neurodegeneration.
AMYLOID-β OLIGOMERS AND TAU AS MEDIATORS OF NEURONAL CCR IN AD
Amyloid-β (Aβ) peptides and tau protein, which are the main components of the amyloid plaques and neurofibrillary tangles that respectively accumulate in AD brain, are considered to be central to AD pathogenesis [2]. Although poorly soluble amyloid plaques are hallmarks of AD, soluble Aβ oligomers (AβOs) correlate much better with cognitive impairment in AD and are early drivers of AD pathogenesis [41]. Aβ peptides are generated from amyloid-β protein precursor (AβPP) by sequential cleavage mediated by β- and γ-secretases. The Aβ peptides are variable in length, but those that end with a C-terminal valine (Aβ40) or alanine (Aβ42), and are typically 40 or 42 amino acids long, respectively, are among the principal Aβ species that increase over time in AD brain [2, 42]. Aβ peptides and aberrant neuronal CCR were first detected in the brains of an AD mouse model (R1.40) expressing Swedish mutant human APP [43]. However, direct genetic evidence that amyloidogenic cleavage of AβPP and subsequent Aβ peptide generation is required for CCR induction was provided by crossing R1.40 and Bace1 knockout mice. The resulting β-secretase-deficient, mutant APP-positive mice did not exhibit CCR [43]. Moreover, CCR was detected in R1.40 mouse brains long before deposition of the poorly soluble Aβ fibrils that populate plaques, suggesting that the trigger for neuronal CCR is soluble AβOs. Consistent with that notion is the observation that AβOs, but not monomeric or fibrillar Aβ, potently induce CCR in primary neurons in vitro [43]. All of these findings support the proposition that soluble AβOs are the inducer of aberrant neuronal CCR within the context of AD.
Several membrane receptors have been proposed to bind AβOs (reviewed in [44, 45]); however, only NMDA receptor (NMDAR) has been shown to be involved in AβO-induced CCR. In cultured mouse neurons, NMDAR antagonists or knock down of NMDAR subunit NR1 prevents AβO induced CCR [46]. Moreover, treatment of Tg2576 AD mice model with an NMDAR antagonist, memantine, prevented neuronal CCR in the mice brains [46]. Possible contribution of other receptors on AβO-induced neuronal CCR and subsequent cell death remains to be an intriguing question.
Significantly, almost all of the known adverse effects of AβOs depend on the presence of tau, which is the other protein most conspicuously associated with AD [3]. We found that AβO-induced neuronal CCR requires site-specific tau phosphorylation at Y18, S262, S409, and S416 (relative to the largest human CNS tau isoform), and that phosphorylation at those sites is dependent on Fyn, mTORC1, cAMP-regulated kinase A (PKA), and calcium-calmodulin-dependent kinase II (CaMKII), respectively [46–48] (Fig. 2). Additional tau phosphorylation sites and tau kinases that are required for neuronal CCR may also exist, but as yet have not been identified. While our findings firmly place tau in the AβO-mediated neuronal CCR pathway in AD (Fig. 2), the mechanism of this tau dependence is incompletely understood.
Fig. 2.
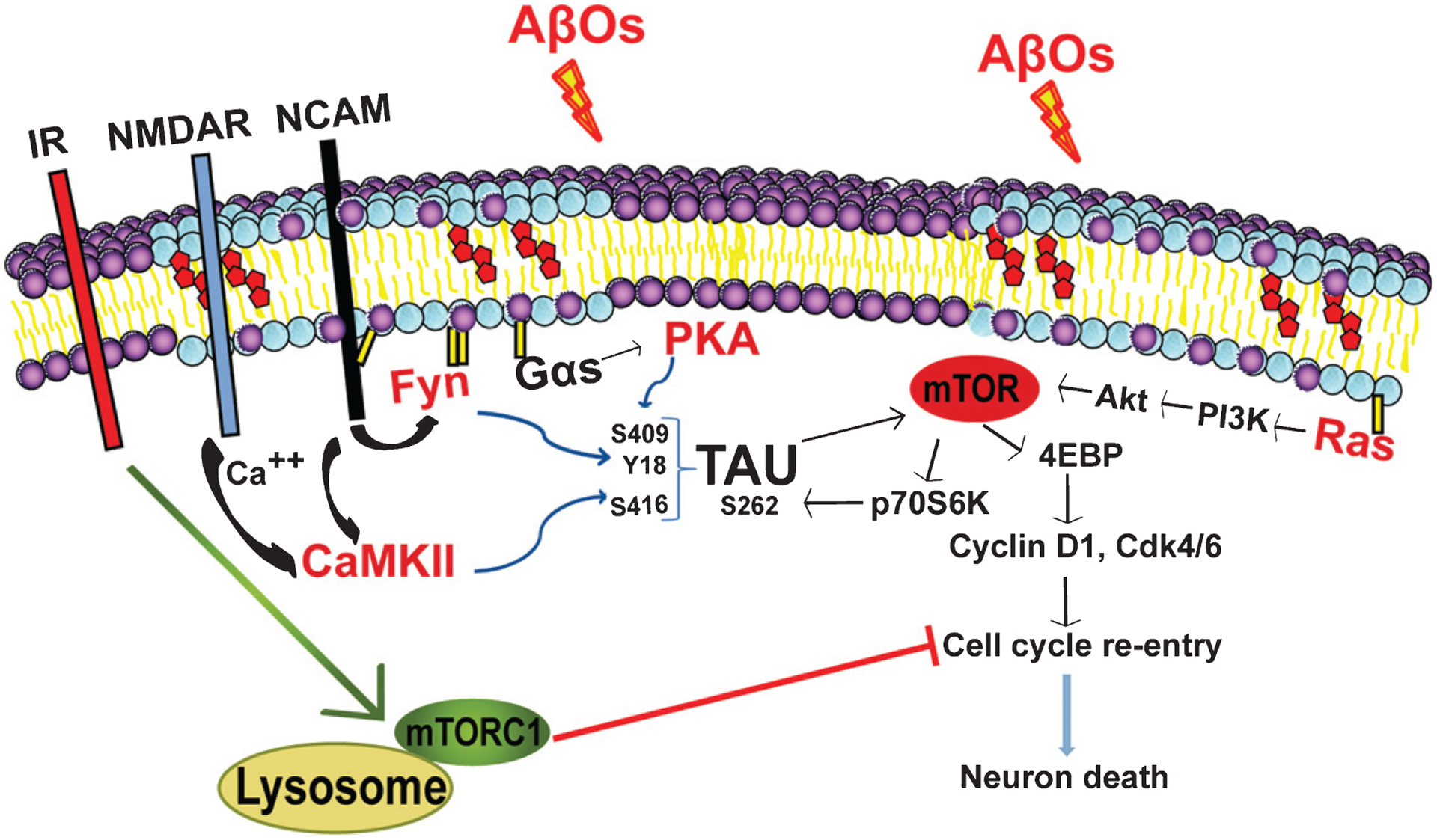
AβOs trigger several parallel events that cumulatively induce cell cycle re-entry (CCR) in AD. While inhibiting insulin signaling, AβOs induce mTORC1 activation at the plasma membrane, which triggers neuronal CCR. AβO-induced tau phosphorylation at multiple sites is required for neuronal CCR. IR (insulin receptor), NCAM (neuronal cell adhesion molecule); all other abbreviations are defined in the main text.
PARALLELS WITH CANCER BIOLOGY
The CCR observed in AD prompts comparison with the major proliferative pathways that are hyperactive in cancers [20, 23, 49], although the fate of cancer cells is significantly different than that of neurons. Whereas cancer cells entering the cell cycle evade cell death and continue to proliferate [50], in AD, neurons degenerate and die in a manner reminiscent of oncogene-induced cell death [34]. Studies have shown that aberrant activation of oncogenes, such as Ras, initially induce CCR in cancer, but eventually the DNA damage response becomes activated and cells are arrested in the cell cycle prior to mitosis, and eventually die via apoptotic or senescence pathways [51]. Similarly, ectopic expression of Ras and/or Myc in neurons triggers CCR, but in both cases neurons eventually degenerate without dividing, which resembles what is seen in the brains of individuals with AD [52, 53]. Significantly, Ras is overexpressed in the brains of AD patients [54, 55] and Ras activity has been shown to be required for AβO-induced CCR and subsequent death of differentiated neuroblastoma cells [49]. These data align with the intriguing hypothesis that neurons in AD patients undergo events similar to oncogene-induced cell cycle arrest, which eventually leads to neuron death, and suggest that Ras is a critical upstream regulator of AβO-induced neuronal CCR.
PI3K-AKT-mTOR
The PI3K-AKT-mTOR signaling pathway regulates the cell cycle and directly mediates cellular quiescence, proliferation, metabolism, motility, growth, and survival [56]. It also is one of the most frequently dysregulated pathways in human cancers [56]. Bhaskar et al. showed that the PI3K-Akt-mTOR pathway is also required for AβO-induced neuronal CCR and subsequent neurodegeneration. They demonstrated that AβO treatment activates Akt and mTOR, and that pharmacological inhibition of PI3K with wortmannin, of Akt with Akt inhibitor VIII, or of mTOR with rapamycin prevents AβO-induced CCR [20].
ERK1/2-MAPK
The Erk1/2-MAPK circuit is hyperactive in almost one third of human cancers [57]. Overexpression and subcellular translocation of MAPKK and MAPK were detected in the brains of AD patients at the early stages, suggesting that MAPK pathway is active as an early feature of AD [58]. Intriguingly, the Erk1/2 pathway has also been shown to be involved in AβO-induced neurotoxicity. Chong et al. [23] showed that the Erk1/2 pathway mediates AβO-induced neuronal death. Specifically, exposure to low micromolar concentrations of AβOs induced cell death in rat organotypic hippocampal brain slices concomitantly with detection of phospho-activated ERK1/2. Moreover, pharmacological inhibition of the ERK1/2 signaling pathway with U0126 attenuated AβO-mediated neuronal toxicity, further supporting a role for ERK1/2 in AβO-mediated effects on neurons. Significantly, Ras proteins are upstream of both the ERK1/2 and PI3K-Akt-mTOR pathways [59]. Both genetic and chemical inhibition of Ras (i.e., salirasib) prevents neuronal CCR in vitro [49] suggesting that Ras is a nodal mediator of AβO-induced CCR.
Cell cycle and proliferation-associated proteins
Other cancer-associated proteins that may be involved in AβO-induced CCR and subsequent cell death include the cyclins, B, D and E; the protein kinases, Cdk 1, 2, 4, and 5 [26–28, 60]; and the dual specificity phosphatases, CDC25A, B and C, which dephosphorylate and active Cdks [60, 61]. Cdk4 and the closely related Cdk6 are key kinases that trigger CCR, whereas Cdk2 is responsible for S phase entry and progression and Cdk1 regulates cell entry into mitosis. The importance of some of these proteins has been documented in pharmacological and shRNA-based studies targeting Cdk4/6 or Cdk2, in which inhibition prevents AβO-induced CCR and neurotoxicity in human induced pluripotent stem (iPS) cell-derived neurons and primary mouse cortical neurons [23, 62, 63].
NEURONAL CCR AND BRAIN INSULIN RESISTANCE
The Rotterdam Study provided compelling epidemiological evidence for a correlation between diabetes and AD, but could not establish whether the two distinctive pathological conditions are mechanistically linked [64, 65]. While type II diabetes is a strong risk factor for AD, brain insulin resistance (BIR) is common among AD patients even in the absence of systemic diabetes, prompting the idea that AD is a brain-specific, or type 3 form of diabetes [6, 24]. Recent evidence strongly suggests that BIR, as defined by the failure of brain cells to respond to insulin, leads to compromised synaptic, metabolic, and immune response functions, but the mechanisms by which BIR leads to synaptic dysfunction and neuron death have been difficult to unravel [66]. Moreover, whether BIR reflects neurons being unable to respond properly to insulin due to deficiencies in insulin receptor expression or the failure of systemic insulin to infiltrate the brain is still under discussion [67]. Nonetheless, postmortem studies of AD patient brains have revealed dramatically reduced expression of receptors for insulin and insulin-like growth factor-1 (IGF1) in the hippocampus and hypothalamus [6], and BIR also impacts the brain independently of those receptors. For example, BIR alters membrane trafficking of the AMPA glutamate receptor subunit, GluA1, in the hypothalamus, thereby hindering synaptic plasticity in that brain region, and impairs hippocampal-dependent memory as well. [68]. In addition, a recent study links BIR and AD through the use of the diabetes drug, liraglutide. Specifically, liraglutide, blocks AD phenotypes in mouse and non-human primate model systems [69].
Despite persistent mysteries about the diabetes-AD connection, recently reported evidence indicates that AβOs are responsible for two relevant phenomena: rapid downregulation of insulin receptors from the neuronal cell surface [70] and AβO-mediated activation of a signaling network that stimulates neuronal CCR [48]. The latter mechanism involves AβO-induced activation of mTORC1 at the plasma membrane, but not at lysosomes, whereas insulin preferentially activates lysosomal mTORC1 [48]. We have shown that AβO-induced CCR is functionally connected to insulin resistance as the process is blocked by rescuing lysosomal mTORC1 activity through genetic manipulations that bypass normal insulin signaling or by modulating insulin availability in primary neuron cultures [48]. The mechanism by which AβO-mediated dysregulation of mTORC1 leads to neuronal CCR remains to be fully understood, but interestingly, insulin and mTORC1 have also been shown to regulate mitochondrial biogenesis and metabolism [71, 72], and mitochondrial dysfunction has been linked to BIR [73].
In this line, we have recently shown that activation of lysosomal mTORC1 by insulin regulates mitochondrial activity not only in mouse and human neurons in culture, but also in the live mouse brain [73]. Strikingly, we also found that activation of lysosomal mTORC1 regulates mitochondrial DNA (mtDNA) synthesis. AβOs, which activate mTORC1 at the plasma membrane, but not at lysosomes, blocks this nutrient-induced mitochondrial activity (NiMA) pathway by a mechanism dependent on tau, and deregulates mtDNA replication [73]. Collectively, these results suggest that lysosomal mTORC1 couples nutrient availability to mtDNA replication and mitochondrial activity, thus functionally connecting these two organelles. As alterations in neuronal mtDNA maintenance account for brain energy metabolism deficiencies in AD [74, 75], the NiMA pathway might represent a mechanistic link connecting metabolic alterations, such as Type II diabetes (insulin resistance), to mtDNA maintenance and dysfunction in AD.
Thus, BIR combined with the AβO-mediated activation of mTORC1 at the plasma membrane converge on mitochondria affecting their activity [76]. As insulin and mTORC1 have been shown to regulate mitochondrial biogenesis and metabolism [71, 72], and mitochondrial dysfunction has also been linked to BIR [73, 76], we speculate that BIR combined with the AβO-mediated activation of mTORC1 at the plasma membrane alters neuronal energy metabolism by a mechanism that directly disrupts mitochondrial functions and thereby contributes indirectly to neuronal CCR. In fact, nucleotide biosynthesis necessary for DNA replication and CCR requires the coordinate action of mitochondria and mTOR [77, 78]. While the role of nucleotide biosynthetic pathways has not been comprehensively addressed in the context of either AD or BIR, these observations open the possibility that signaling pathways acting on lysosomal mTORC1 (i.e., insulin) directly regulate mitochondrial metabolic pathways involved in controlling cell cycle events. In this scenario, AβO-mediated activation of mTORC1 at the PM in neurons might disrupt proper nutrient signaling and mitochondrial metabolism. This process may also be relevant under other metabolic conditions. TNFα, a known proinflammatory cytokine, blocks insulin signaling through activation of JNK [79] and AβOs trigger the release of TNFα from microglia present in primary neuron cultures [80, 81]. In neurons, activation of TNFα receptors stimulate JNK-mediated phosphorylation of insulin receptor kinase substrate-1 (IRS-1), which inhibits IRS-1 activity and downregulates insulin receptor mediated activation of PI3K and Akt [80].
PHARMACOLOGICAL TARGETING OF NEURONAL CCR IN AD
The failure rate of clinical trials for AD during the past decade has been remarkable, particularly when one considers the preclinical evidence that formed the foundation for these trials [82]. A regrettable consequence has been the abandonment of efforts by several major pharmaceutical companies to develop new treatments for AD. It has been argued, for example, that broad knowledge gaps and poor clinical trial designs are responsible for the failures of γ-secretase inhibitors [83], and that cell-based and animal models of AD do not accurately recapitulate key features of bona fide AD in humans [84, 85]. Regardless of why so many potential AD drugs have failed, the fact remains that the last such drug to gain Food and Drug Administration (FDA) approval is memantine, in 2003, and the world still awaits regulatory approval of any drug that actually prevents or slows symptom progression. It is notable that nearly all experimental drug development for AD has been focused on reducing the amyloid burden in brain, though some recent efforts have targeted tau. With these considerations in mind, alternative drug discovery and development strategies, including those directed at AβO-induced neuronal CCR, seem reasonable and timely.
In the past decade and a half, there has been a growing interest in formally investigating the repurposing of drugs that have already been approved by regulatory agencies, such as the FDA or the European Medicines Agency (EMA), for possible use against neurodegenerative diseases, such as AD [86–88]. Likewise, rescuing of experimental therapeutics that have failed their primary disease indication provides an additional pool of compounds for evaluation. One obvious benefit of the repurposing approach is the pre-existing body of knowledge about the safety and pharmacokinetics of approved compounds. One large class of drugs that might be repositioned for use against AD would be those currently used for solid and hematological malignancies [89, 90]. Other drugs that might be repurposed for AD prevention or therapy include those developed to treat diabetes.
Cancer drugs and CCR in AD
Most anti-cancer drugs have the capacity to block cell proliferation and, thus, would likely inhibit neuronal CCR. FDA-approved cytotoxic cancer chemotherapeutics with good (e.g., carmustine) or poor (e.g., paclitaxel) brain penetrance have been shown to have some potential positive effects, at least in existing preclinical AD models [89].
As has been already mentioned, newer, more targeted, anti-cancer drugs that inhibit PI3K, Akt, and mTOR have also been shown to prevent AβO-induced neuronal CCR (Fig. 3A) [20]. The investigational PI3K/mTOR inhibitor dactolisib has been observed to protect mice against Aβ42-induced neurotoxicity and memory loss [91]. Similarly, pharmacological and genetic inhibition of Ras prevented AβO-induced CCR and subsequent cell death in vitro [49]. Finally, pharmacological and genetic inhibitors of Cdk2, Cdk4, and Cdk6 reduced AβO-induced CCR and neurotoxicity in human iPS-derived neurons and primary mouse cortical neurons [62, 63].
Fig. 3.

Chemical structures of FDA-approved drugs that potentially can be re-purposed to target neuronal cell cycle re-entry in AD. A) Inhibitors of mTOR. B) Inhibitor of Src family kinases. C) Inhibitor of Ras. D) Inhibitors of Cdk4/6. E) Inhibitor of NMDA receptor. F) Tanimoto analysis of the two-dimensional similarity of the chemical structures of potential regulators of CCR. 1) Memantine. 2) Saracatinib. 3) Abemaciclib. 4) Palbociclib. 5) Ribociclib. 6) Salirasib. 7) Rapamycin. 8) Temsirolimus. 9) Everolimus. Tanimoto analysis was performed with PubChem software (https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?p=clustering).
More than 35 tyrosine kinase inhibitors have been approved by the FDA or EMA for cancer and several have the potential to inhibit neuronal CCR. The investigational tyrosine kinase inhibitor saracatinib (Fig. 3B) is known to be a potent inhibitor of the tyrosine kinase Fyn, which is expressed in the brain, phosphorylates tau, and mediates Aβ toxicity [92], including neuronal CCR [49]. In an initial repurposing Phase 1b clinical trial, saracatinib was found to be generally safe and well tolerated [92]. The National Center for Advancing Translational Sciences just completed a multicenter Phase IIa clinical trial with 159 participants called CONNECT (ClinicalTrials.gov Identifier: NCT02167256) to evaluate the clinical potential of saracatinib for AD. There is considerable interest in the results of this clinical trial. While CCR was not an evaluated endpoint, mechanistically it is reasonable to speculate that saracatinib can block neuronal CCR. Similarly, some of the tyrosine kinase inhibitors also have been investigated for use in diabetes [93] and some well-established agents used for diabetes, such as metformin, are being examined for their potential use in cancer [94].
Rapamycin, which inspired approval by the FDA and EMA of the anti-cancer mTOR inhibitors, temsirolimus and everolimus (Fig. 3A), have been shown to reduce Aβ levels and cognitive loss in mouse AD models [95]. The authors linked the delay or inhibition of AD-like symptoms to an increase in autophagy but they did not exclude the possibility that inhibition of CCR was also involved.
The Ras inhibitor, salirasib (Fig. 3C), has been shown to inhibit AβO-induced CCR in vitro [49]. Salirasib passes through the blood-brain barrier and is neuroprotective in mouse closed head injury models [96, 97]. Furthermore, targeting Ras has shown to diminish excitotoxic stimulus-triggered brain damage. Salirasib has shown low efficacy against lung cancer in Phase II trial [98], but it has potential to suppress AD pathogenesis by inhibiting AβO-induced CCR.
There have been reports of neuroprotective effects in Aβ-mediated in vitro models of Cdk4/6 inhibitors, including palbociclib, which is approved by the FDA and EMA [62, 63]. Considering the increasing numbers of next generation Cdk4/6 inhibitors that are emerging for use in cancer, including abemaciclib and ribociclib (Fig. 3D), and the apparent complexity of their actions, there may be an opportunity to exploit these agents for use in the treatment of AD [99].
Diabetes drugs and CCR in AD
Several diabetes drugs, including insulin itself, have been repurposed to treat BIR in AD [100, 101]. Our recent findings connect the insulin signaling to AβO-induced CCR as an important component of AD pathology. AβO signaling highjacks mTORC1 to the PM to trigger CCR, while the insulin pathway is diminished due to BIR [48]. Our findings suggest that targeting BIR with diabetes drugs, such as GLP-1 agonists, metformin, or insulin [100, 101], may inhibit CCR, which precedes most of the neuronal death in AD.
Other drugs targeting CCR
We recently showed that NMDA receptor antagonist, memantine, prevents AβO-induced CCR both in vitro and in vivo [46]. Memantine (Fig.3E) is an FDA-approved drug for modest symptom relief in moderate to severe AD, but our findings suggest that memantine can be a disease modifying drug to forestall AD progression if administered beginning at preysmptomatic stages of the disease. A chemoinformatics analysis of the chemical structures of these inhibitors of CCR reveals considerable diversity in their chemical fingerprint as measure by Tanimoto score (Fig. 3F). These results suggest multiple other chemotypes in chemical space could be available for exploitation as possible agents for use against AD and we believe this is worthy of further investigation.
CONCLUSIONS
The emerging findings about intracellular signaling of AβO-mediated neuronal CCR reveals convergence points among AD, cancer and BIR. This signaling pathway commonality that connects different diseases raises the possibility that pharmacological tools developed to combat cancer or diabetes can be re-purposed for AD prevention or therapy by targeting AβO-mediated signaling, including that which leads to neuronal CCR.
ACKNOWLEDGMENTS
The authors greatly appreciate the generous financial support from the Owens Family Foundation (GSB, and JSL), NIH (grant RF1 AG051085 to GSB and P30 CA044579 to JSL), the Cure Alzheimer’s Fund (GSB, ERS, and JSL), the Alzheimer’s Association (grants 4079 and ZEN-16-363266 to GSB), Webb and Tate Wilson (GSB), The Virginia Chapter of the Lady’s Auxiliary of the Fraternal Order of Eagles (GSB), the University of Virginia President’s Fund for Excellence (GSB), the Department of Defense (grant BC170507 to JSL), the Fiske Drug Discovery Fund (JSL and ERS), and the Alzheimer’s and Related Diseases Research Award Fund (grant 17-5 to AN).
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0874r1).
REFERENCES
- [1].James BD, Leurgans SE, Hebert LE, Scherr PA, Yaffe K, Bennett DA (2014) Contribution of Alzheimer disease to mortality in the United States. Neurology 82, 1045–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- [3].Khan SS, Bloom GS (2016) Tau: The center of a signaling nexus in Alzheimer’s disease. Front Neurosci 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bloom GS, Lazo JS, Norambuena A (2018) Reduced brain insulin signaling: A seminal process in Alzheimer’s disease pathogenesis. Neuropharmacology 136(Pt B), 192–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bonda DJ, Lee HP, Kudo W, Zhu X, Smith MA, Lee HG (2010) Pathological implications of cell cycle re-entry in Alzheimer disease. Expert Rev Mol Med 12, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J Alzheimers Dis 7, 63–80. [DOI] [PubMed] [Google Scholar]
- [7].West MJ, Coleman PD, Flood DG, Troncoso JC (1994) Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 344, 769–772. [DOI] [PubMed] [Google Scholar]
- [8].Bussiere T, Giannakopoulos P, Bouras C, Perl DP, Morrison JH, Hof PR (2003) Progressive degeneration of nonphosphorylated neurofilament protein-enriched pyramidal neurons predicts cognitive impairment in Alzheimer’s disease: Stereologic analysis of prefrontal cortex area 9. J Comp Neurol 463, 281–302. [DOI] [PubMed] [Google Scholar]
- [9].Herrup K, Yang Y (2007) Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat Rev Neurosci 8, 368–378. [DOI] [PubMed] [Google Scholar]
- [10].Arendt T, Bruckner MK, Mosch B, Losche A (2010) Selective cell death of hyperploid neurons in Alzheimer’s disease. Am J Pathol 177, 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Delobel P, Lavenir I, Ghetti B, Holzer M, Goedert M (2006) Cell-cycle markers in a transgenic mouse model of human tauopathy: Increased levels of cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1. Am J Pathol 168, 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang Y, Mufson EJ, Herrup K (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci 23, 2557–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tornatore L, Thotakura AK, Bennett J, Moretti M, Franzoso G (2012) The nuclear factor kappa B signaling pathway: Integrating metabolism with inflammation. Trends Cell Biol 22, 557–566. [DOI] [PubMed] [Google Scholar]
- [14].Moretti M, Bennett J, Tornatore L, Thotakura AK, Franzoso G (2012) Cancer: NF-kappaB regulates energy metabolism. Int J Biochem Cell Biol 44, 2238–2243. [DOI] [PubMed] [Google Scholar]
- [15].Kumar A, Singh UK, Kini SG, Garg V, Agrawal S, Tomar PK, Pathak P, Chaudhary A, Gupta P, Malik A (2015) JNK pathway signaling: A novel and smarter therapeutic targets for various biological diseases. Future Med Chem 7, 2065–2086. [DOI] [PubMed] [Google Scholar]
- [16].Cohen P, Goedert M (2004) GSK3 inhibitors: Development and therapeutic potential. Nat Rev Drug Discov 3, 479–487. [DOI] [PubMed] [Google Scholar]
- [17].Khan I, Tantray MA, Alam MS, Hamid H (2017) Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur J Med Chem 125, 464–477. [DOI] [PubMed] [Google Scholar]
- [18].Lal H, Ahmad F, Woodgett J, Force T (2015) The GSK-3 family as therapeutic target for myocardial diseases. Circ Res 116, 138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- [20].Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT (2009) The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kohno M, Tanimura S, Ozaki K (2011) Targeting the extracellular signal-regulated kinase pathway in cancer therapy. Biol Pharm Bull 34, 1781–1784. [DOI] [PubMed] [Google Scholar]
- [22].Ozaki KI, Awazu M, Tamiya M, Iwasaki Y, Harada A, Kugisaki S, Tanimura S, Kohno M (2016) Targeting the ERK signaling pathway as a potential treatment for insulin resistance and type 2 diabetes. Am J Physiol Endocrinol Metab 310, E643–E651. [DOI] [PubMed] [Google Scholar]
- [23].Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ (2006) ERK1/2 activation mediates Abeta oligomer-induced neurotoxicity via caspase-3 activation and tau cleavage in rat organotypic hippocampal slice cultures. J Biol Chem 281, 20315–20325. [DOI] [PubMed] [Google Scholar]
- [24].de la Monte SM (2014) Type 3 diabetes is sporadic Alzheimers disease: Mini-review. Eur Neuropsychopharmacol 24, 1954–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Otsuki L, Brand AH (2018) Cell cycle heterogeneity directs the timing of neural stem cell activation from quiescence. Science 360, 99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Busser J, Geldmacher DS, Herrup K (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J Neurosci 18, 2801–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Smith MZ, Nagy Z, Esiri MM (1999) Cell cycle-related protein expression in vascular dementia and Alzheimer’s disease. Neurosci Lett 271, 45–48. [DOI] [PubMed] [Google Scholar]
- [28].Hoozemans JJ, Bruckner MK, Rozemuller AJ, Veerhuis R, Eikelenboom P, Arendt T (2002) Cyclin D1 and cyclin E are co-localized with cyclo-oxygenase 2 (COX-2) in pyramidal neurons in Alzheimer disease temporal cortex. J Neuropathol Exp Neurol 61, 678–688. [DOI] [PubMed] [Google Scholar]
- [29].Pei JJ, Braak H, Gong CX, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF (2002) Up-regulation of cell division cycle (cdc) 2 kinase in neurons with early stage Alzheimer’s disease neurofibrillary degeneration. Acta Neuropathol 104, 369–376. [DOI] [PubMed] [Google Scholar]
- [30].Dranovsky A, Vincent I, Gregori L, Schwarzman A, Colflesh D, Enghild J, Strittmatter W, Davies P, Goldgaber D (2001) Cdc2 phosphorylation of nucleolin demarcates mitotic stages and Alzheimer’s disease pathology. Neurobiol Aging 22, 517–528. [DOI] [PubMed] [Google Scholar]
- [31].Nagy Z, Esiri MM, Cato AM, Smith AD (1997) Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol 94, 6–15. [DOI] [PubMed] [Google Scholar]
- [32].Arendt T, Holzer M, Gartner U (1998) Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J Neural Transm (Vienna) 105, 949–960. [DOI] [PubMed] [Google Scholar]
- [33].Arendt T, Rodel L, Gartner U, Holzer M (1996) Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. Neuroreport 7, 3047–3049. [DOI] [PubMed] [Google Scholar]
- [34].Yang Y, Geldmacher DS, Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci 21, 2661–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yang Y, Varvel NH, Lamb BT, Herrup K (2006) Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models. J Neurosci 26, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li L, Cheung T, Chen J, Herrup K (2011) A comparative study of five mouse models of Alzheimer’s disease: Cell cycle events reveal new insights into neurons at risk for death. Int J Alzheimers Dis 2011, 171464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rashidian J, Iyirhiaro GO, Park DS (2007) Cell cycle machinery and stroke. Biochim Biophys Acta 1772, 484–493. [DOI] [PubMed] [Google Scholar]
- [38].Pelegri C, Duran-Vilaregut J, del Valle J, Crespo-Biel N, Ferrer I, Pallas M, Camins A, Vilaplana J (2008) Cell cycle activation in striatal neurons from Huntington’s disease patients and rats treated with 3-nitropropionic acid. Int J Dev Neurosci 26, 665–671. [DOI] [PubMed] [Google Scholar]
- [39].Maccioni RB, Munoz JP, Barbeito L (2001) The molecular bases of Alzheimer’s disease and other neurodegenerative disorders. Arch Med Res 32, 367–381. [DOI] [PubMed] [Google Scholar]
- [40].Ranganathan S, Bowser R (2003) Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am J Pathol 162, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sakono M, Zako T (2010) Amyloid oligomers: Formation and toxicity of Abeta oligomers. FEBS J 277, 1348–1358. [DOI] [PubMed] [Google Scholar]
- [42].Murphy MP, LeVine H 3rd (2010) Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis 19, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Varvel NH, Bhaskar K, Patil AR, Pimplikar SW, Herrup K, Lamb BT (2008) Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease. J Neurosci 28, 10786–10793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kam TI, Gwon Y, Jung YK (2014) Amyloid beta receptors responsible for neurotoxicity and cellular defects in Alzheimer’s disease. Cell Mol Life Sci 71, 4803–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jarosz-Griffiths HH, Noble E, Rushworth JV, Hooper NM (2016) Amyloid-beta receptors: The good, the bad, and the prion protein. J Biol Chem 291, 3174–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kodis EJ, Choi S, Swanson E, Ferreira G, Bloom GS (2018) N-methyl-D-aspartate receptor–mediated calcium influx connects amyloid-β oligomers to ectopic neuronal cell cycle reentry in Alzheimer’s disease. Alzheimers Dement 14, 1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Seward ME, Swanson E, Norambuena A, Reimann A, Cochran JN, Li R, Roberson ED, Bloom GS (2013) Amyloid-beta signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J Cell Sci 126, 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Norambuena A, Wallrabe H, McMahon L, Silva A, Swanson E, Khan SS, Baerthlein D, Kodis E, Oddo S, Mandell JW, Bloom GS (2017) mTOR and neuronal cell cycle reentry: How impaired brain insulin signaling promotes Alzheimer’s disease. Alzheimers Dement 13, 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Koseoglu MM, Ozdilek BA, Djakbarova U, Gulusur A (2016) Targeting Ras activity prevented amyloid beta-induced aberrant neuronal cell cycle re-entry and death. Curr Alzheimer Res 13, 1267–1276. [DOI] [PubMed] [Google Scholar]
- [50].Hanahan D, Weinberg RA (2011) Hallmarks of cancer: The next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- [51].Liu XL, Ding J, Meng LH (2018) Oncogene-induced senescence: A double edged sword in cancer. Acta Pharmacol Sin 39, 1553–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].McShea A, Lee HG, Petersen RB, Casadesus G, Vincent I, Linford NJ, Funk JO, Shapiro RA, Smith MA (2007) Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim Biophys Acta 1772, 467–472. [DOI] [PubMed] [Google Scholar]
- [53].Lee HG, Casadesus G, Nunomura A, Zhu X, Castellani RJ, Richardson SL, Perry G, Felsher DW, Petersen RB, Smith MA (2009) The neuronal expression of MYC causes a neurodegenerative phenotype in a novel transgenic mouse. Am J Pathol 174, 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gartner U, Holzer M, Heumann R, Arendt T (1995) Induction of p21ras in Alzheimer pathology. Neuroreport 6, 1441–1444. [DOI] [PubMed] [Google Scholar]
- [55].Gartner U, Holzer M, Arendt T (1999) Elevated expression of p21ras is an early event in Alzheimer’s disease and precedes neurofibrillary degeneration. Neuroscience 91, 1–5. [DOI] [PubMed] [Google Scholar]
- [56].Janku F, Yap TA, Meric-Bernstam F (2018) Targeting the PI3K pathway in cancer: Are we making headway? Nat Rev Clin Oncol 15, 273–291. [DOI] [PubMed] [Google Scholar]
- [57].Mandal R, Becker S, Strebhardt K (2016) Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: An emerging threat to anticancer therapy. Oncogene 35, 2547–2561. [DOI] [PubMed] [Google Scholar]
- [58].Arendt T, Holzer M, Grossmann A, Zedlick D, Bruckner MK (1995) Increased expression and subcellular translocation of the mitogen activated protein kinase kinase and mitogen-activated protein kinase in Alzheimer’s disease. Neuroscience 68, 5–18. [DOI] [PubMed] [Google Scholar]
- [59].Karnoub AE, Weinberg RA (2008) Ras oncogenes: Split personalities. Nat Rev Mol Cell Biol 9, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chang KH, Vincent F, Shah K (2012) Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J Cell Sci 125, 5124–5137. [DOI] [PubMed] [Google Scholar]
- [61].Ding XL, Husseman J, Tomashevski A, Nochlin D, Jin LW, Vincent I (2000) The cell cycle Cdc25A tyrosine phosphatase is activated in degenerating postmitotic neurons in Alzheimer’s disease. Am J Pathol 157, 1983–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sanphui P, Pramanik SK, Chatterjee N, Moorthi P, Banerji B, Biswas SC (2013) Efficacy of cyclin dependent kinase 4 inhibitors as potent neuroprotective agents against insults relevant to Alzheimer’s disease. PLoS One 8, e78842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Xu X, Lei Y, Luo J, Wang J, Zhang S, Yang XJ, Sun M, Nuwaysir E, Fan G, Zhao J, Lei L, Zhong Z (2013) Prevention of beta-amyloid induced toxicity in human iPS cell-derived neurons by inhibition of Cyclin-dependent kinases and associated cell cycle events. Stem Cell Res 10, 213–227. [DOI] [PubMed] [Google Scholar]
- [64].Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM (1999) Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53, 1937–1942. [DOI] [PubMed] [Google Scholar]
- [65].Ikram MA, Brusselle GGO, Murad SD, van Duijn CM, Franco OH, Goedegebure A, Klaver CCW, Nijsten TEC, Peeters RP, Stricker BH, Tiemeier H, Uitterlinden AG, Vernooij MW, Hofman A (2017) The Rotterdam Study: 2018 update on objectives, design and main results. Eur J Epidemiol 32, 807–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, Craft S, Gandy S, Buettner C, Stoeckel LE, Holtzman DM, Nathan DM (2018) Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat Rev Neurol 14, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gray SM, Meijer RI, Barrett EJ (2014) Insulin regulates brain function, but how does it get there? Diabetes 63, 3992–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Spinelli M, Fusco S, Mainardi M, Scala F, Natale F, Lapenta R, Mattera A, Rinaudo M, Li Puma DD, Ripoli C, Grassi A, D’Ascenzo M, Grassi C (2017) Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat Commun 8, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Batista AF, Forny-Germano L, Clarke JR, Lyra ESNM, Brito-Moreira J, Boehnke SE, Winterborn A, Coe BC, Lablans A, Vital JF, Marques SA, Martinez AM, Gralle M, Holscher C, Klein WL, Houzel JC, Ferreira ST, Munoz DP, De Felice FG(2018) The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J Pathol 245, 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL (2008) Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J 22, 246–260. [DOI] [PubMed] [Google Scholar]
- [71].Cheng Z, Tseng Y, White MF (2010) Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab 21, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St-Pierre J, Topisirovic I, Sonenberg N (2013) mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab 18, 698–711. [DOI] [PubMed] [Google Scholar]
- [73].De Felice FG, Lourenco MV, Ferreira ST (2014) How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement 10, S26–32. [DOI] [PubMed] [Google Scholar]
- [74].Mosconi L, Brys M, Switalski R, Mistur R, Glodzik L, Pirraglia E, Tsui W, De Santi S, de Leon MJ (2007) Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci U S A 104, 19067–19072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli R, Kukull W, Morris JC, Hulette CM, Schmechel D, Rogers J, Stephan DA (2008) Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A 105, 4441–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Norambuena A, Wallrabe H, Cao R, Wang DB, Silva A, Svindrych Z, Periasamy A, Hu S, Tanzi RE, Kim DY, Bloom GS (2018) A novel lysosome-to-mitochondria signaling pathway disrupted by amyloid-β oligomers. EMBO J. doi: 10.15252/embj.2018100241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ben-Sahra I, Hoxhaj G, Ricoult SJ, Asara JM, Manning BD mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].French JB, Jones SA, Deng H, Pedley AM, Kim D, Chan CY, Hu H, Pugh RJ, Zhao H, Zhang Y, Huang TJ, Fang Y, Zhuang X, Benkovic SJ Spatial colocalization and functional link of purinosomes with mitochondria. Science 351, 733–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK (2005) Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 54, 2939–2945. [DOI] [PubMed] [Google Scholar]
- [80].Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL, Holscher C, Arnold SE, Talbot K, Klein WL, Munoz DP, Ferreira ST, De Felice FG (2012) An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J Clin Invest 122, 1339–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bhaskar K, Maphis N, Xu G, Varvel NH, Kokiko-Cochran ON, Weick JP, Staugaitis SM, Cardona A, Ransohoff RM, Herrup K, Lamb BT (2014) Microglial derived tumor necrosis factor-alpha drives Alzheimer’s disease-related neuronal cell cycle events. Neurobiol Dis 62, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Doody RS, Aisen PS, Iwatsubo T (2013) Semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369, 1661. [DOI] [PubMed] [Google Scholar]
- [83].De Strooper B (2014) Lessons from a failed gamma-secretase Alzheimer trial. Cell 159, 721–726. [DOI] [PubMed] [Google Scholar]
- [84].Toyn J (2015) What lessons can be learned from failed Alzheimer’s disease trials? Expert Rev Clin Pharmacol 8, 267–269. [DOI] [PubMed] [Google Scholar]
- [85].Mullard A (2018) BACE failures lower AD expectations, again. Nat Rev Drug Discov 17, 385. [DOI] [PubMed] [Google Scholar]
- [86].Ashburn TT, Thor KB (2004) Drug repositioning: Identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3, 673–683. [DOI] [PubMed] [Google Scholar]
- [87].Langedijk J, Mantel-Teeuwisse AK, Slijkerman DS, Schutjens MH (2015) Drug repositioning and repurposing: Terminology and definitions in literature. Drug Discov Today 20, 1027–1034. [DOI] [PubMed] [Google Scholar]
- [88].Duraes F, Pinto M, Sousa E (2018) Old drugs as new treatments for neurodegenerative diseases. Pharmaceuticals (Basel) 11, E44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Araki W (2013) Potential repurposing of oncology drugs for the treatment of Alzheimer’s disease. BMC Med 11, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Monacelli F, Cea M, Borghi R, Odetti P, Nencioni A (2017) Do cancer drugs counteract neurodegeneration? Repurposing for Alzheimer’s disease. J Alzheimers Dis 55, 1295–1306. [DOI] [PubMed] [Google Scholar]
- [91].Bellozi PM, Lima IV, Doria JG, Vieira EL, Campos AC, Candelario-Jalil E, Reis HJ, Teixeira AL, Ribeiro FM, de Oliveira AC (2016) Neuroprotective effects of the anti-cancer drug NVP-BEZ235 (dactolisib) on amyloid-beta 1–42 induced neurotoxicity and memory impairment. Sci Rep 6, 25226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Nygaard HB, Wagner AF, Bowen GS, Good SP, MacAvoy MG, Strittmatter KA, Kaufman AC, Rosenberg BJ, Sekine-Konno T, Varma P, Chen K, Koleske AJ, Reiman EM, Strittmatter SM, van Dyck CH (2015) A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimers Res Ther 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fountas A, Diamantopoulos LN, Tsatsoulis A (2015) Tyrosine kinase inhibitors and diabetes: A novel treatment paradigm? Trends Endocrinol Metab 26, 643–656. [DOI] [PubMed] [Google Scholar]
- [94].Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F (2010) Metformin in cancer therapy: A new perspective for an old antidiabetic drug? Mol Cancer Ther 9, 1092–1099. [DOI] [PubMed] [Google Scholar]
- [95].Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V (2010) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One 5, e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Shohami E, Yatsiv I, Alexandrovich A, Haklai R, Elad-Sfadia G, Grossman R, Biegon A, Kloog Y (2003) The Ras inhibitor S-trans, trans-farnesylthiosalicylic acid exerts long-lasting neuroprotection in a mouse closed head injury model. J Cereb Blood Flow Metab 23, 728–738. [DOI] [PubMed] [Google Scholar]
- [97].Marciano D, Shohami E, Kloog Y, Alexandrovitch A, Brandeis R, Goelman G (2007) Neuroprotective effects of the Ras inhibitor S-trans-trans-farnesylthiosalicylic acid, measured by diffusion-weighted imaging after traumatic brain injury in rats. J Neurotrauma 24, 1378–1386. [DOI] [PubMed] [Google Scholar]
- [98].Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA, Kris MG, Pietanza MC, Azzoli CG, Krug LM, Pao W, Ginsberg MS (2011) A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol 6, 1435–1437. [DOI] [PubMed] [Google Scholar]
- [99].Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A (2018) CDK4/6 Inhibitors: The mechanism of action may not be as simple as once thought. Cancer Cell 34, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ribaric S (2016) The rationale for insulin therapy in Alzheimer’s disease. Molecules 21, 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Yarchoan M, Arnold SE (2014) Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes 63, 2253–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]