

Abstract
Introduction:
Alzheimer’s disease (AD) symptoms reflect synaptic dysfunction and neuron death. Amyloid-β oligomers (AβOs) induce excess calcium entry into neurons via N-methyl-D-aspartate receptors (NMDARs), contributing to synaptic dysfunction. The study described here tested the hypothesis that AβO-stimulated calcium entry also drives neuronal cell cycle reentry (CCR), a prelude to neuron death in AD.
Methods:
Pharmacologic modulators of calcium entry and gene expression knockdown were used in cultured neurons and AD model mice.
Results:
In cultured neurons, AβO-stimulated CCR was blocked by NMDAR antagonists, total calcium chelation with 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), or knockdown of the NMDAR subunit, NR1. NMDAR antagonists also blocked the activation of calcium-calmodulin-dependent protein kinase II and treatment of Tg2576 AD model mice with the NMDAR antagonist, memantine, prevented CCR.
Discussion:
This study demonstrates a role for AβO-stimulated calcium influx via NMDAR and CCR in AD and suggests the use of memantine as a disease-modifying therapy for presymptomatic AD.
Keywords: Amyloid-β oligomers, Tau, Alzheimer’s disease, Neuronal cell cycle reentry, NMDA receptor, Calcium
1. Introduction
Alzheimer’s disease (AD) is a devastating neurological disorder characterized by memory loss and cognitive decline. These behavioral symptoms are caused at the cellular level by synaptic dysfunction and loss and neuron death and at the molecular level by toxic forms of amyloid-β (Aβ) and tau that work coordinately to damage synapses [1–3], reduce insulin signaling [4], impair axonal transport [5], and kill neurons [6–8]. While poorly soluble, fibrillar forms of Aβ and tau that are found in plaques and tangles, respectively, are histopathological hallmarks of AD, soluble oligomeric forms of Aβ and tau are now recognized as being far more toxic [9–11]. Naturally, the efforts to prevent and treat AD will benefit from advances in our still primitive understanding of the pathogenic signaling mechanisms that underlie the breakdown of normal neuronal homeostasis caused by toxic oligomers of Aβ and tau.
As much as 90% of neuron death in AD may follow ectopic reentry of neurons into the cell cycle [12,13]. Whereas fully differentiated, healthy neurons are permanently post-mitotic, affected neurons in AD and other neurodegenerative disorders often express molecular markers of the G1 and S-phase stages of the cell cycle [14–17]. Instead of dividing, however, these neurons apparently die after a delay of up to hundreds of days following cell cycle reentry (CCR) [18]. Previous research from our laboratory defined a CCR signaling network in AD, whereby Aβ oligomers (AβOs) induce activation of multiple protein kinases that catalyze site-specific tau phosphorylation and a positive feedback loop between phosphotau and the multisubunit protein kinase complex, mechanistic target of rapamycin complex 1, that leads to mechanistic target of rapamycin complex 1 dysregulation and drives neurons from G0 into G1 [19,20].
Besides triggering CCR and neuron death, AβOs cause excitotoxicity by stimulating excess calcium influx into neurons [21–23]. This disruption of normal calcium homeostasis affects numerous signaling pathways and can damage and destroy synapses and lead to abrupt neuron death [24]. A major contributor to this pathological process is the N-methyl-D-aspartate receptor (NMDAR), which permits toxic levels of calcium to enter neurons exposed to AβOs [3,25]. Interestingly, one of the few United States Food and Drug Administration–approved treatments for AD is memantine, which works by blocking excess calcium entry into neurons via NMDAR [26].
The study described here tested the hypothesis that neuronal CCR and excitotoxic calcium influx via NMDAR share a common mechanistic origin initiated by AβOs. Using AβO-treated primary mouse neuron cultures, we show that CCR is prevented by chelating total cellular calcium, by pharmacologically blocking AβO-mediated calcium influx through NMDAR, or by reducing expression of an NMDAR subunit protein. Moreover, we found that neuronal CCR in vivo in Tg2576 AD model mice can be blocked by treating the mice prophylactically with memantine. Taken together, these results mechanistically link AβO-induced calcium influx and neuronal CCR. Moreover, they suggest that memantine, which is used as a drug for modestly relieving symptoms in patients with a clinical AD diagnosis, but does not apparently act as a disease-modifying drug in that context, has the potential to forestall disease progression if administered during presymptomatic stages of the disease.
2. Methods
2.1. Animals and usage
All animal usage and protocols were approved by the Institutional Animal Care and Use Committee of the University of Virginia. Animals were housed in a barrier facility with ad libitum access to food and water on a 12-hour light/dark cycle.
2.2. Neuron dissections and cultures
Cortical neuron cultures derived from E17/18 wild type (WT) C57/BL6 mouse embryos prepared and maintained as described previously [20]. Cultures were maintained for 16–18 days before experimental manipulations.
2.3. Aβ oligomerization and treatment
AβOs were made from lyophilized Aβ(1–42) (AnaSpec, AS-20276-5), which first was resuspended in 1,1,1,3,3,3-hexafluoro-2-propanol (Sigma, 105228-5G) and incubated for 4 hours at room temperature to yield a solution of monomeric peptide. Twenty microliters aliquots were stored at −80°C until ready to use. To prepare oligomers, an aliquot was dried in a speedvac for 3 hours, solubilized to a final concentration of 1 mM in dimethyl sulfoxide (Sigma, D2650-5X5ML), placed in a water bath sonicator for 5 minutes, and then supplemented with Neurobasal medium (GIBCO/Life Technologies, 21103-049) to yield a final peptide concentration of 100 μM. The solution was then placed on an orbiting rocker at 4°C for 48 hours, after which it was centrifuged at 14,000 × g in a tabletop microcentrifuge for 15 minutes to pellet large aggregates. AβO solutions were diluted into neuron cultures to yield a final total Aβ concentration of ~2 μM, and the cultures were processed for immunofluorescence microscopy or Western blotting 16–18 hours later. Controls demonstrating that AβOs, but not Aβ monomers or fibrils, cause CCR were described in a previous publication from our laboratory [19].
2.4. Immunofluorescence microscopy
All steps were performed at room temperature unless indicated otherwise. Primary mouse cortical neurons grown on #1.5 thickness, 12 mm round glass coverslips were rinsed once with ice-cold phosphate buffered saline (PBS), and then were fixed in 4% paraformaldehyde in PBS for 12 minutes. Following fixation, the cells were rinsed three times for 5 minutes each with ice-cold PBS, permeabilized for 15 minutes with PBS containing 0.2% Triton X-100 (Fisher, 9002-93-1), and rinsed three times for 5 minutes each with PBS. Next, the cells were blocked with PBST (PBS supplemented with 2% bovine serum albumin (Roche, 03116956001); and 0.1% Tween 20 (Fisher, 9005-64-5) for 1 hour, after which they were labeled overnight with primary antibodies followed by secondary antibodies for 1 hour. Antibodies were diluted into 2% bovine serum albumin in PBS, and after each antibody incubation step the cells were rinsed three times for 5 minutes each with PBS. Finally, the coverslips were rinsed with ultrapure water and mounted onto slides using Fluoromount-G (Southern Biotech, 0100-01) containing 1% 1,4-diazabicyclo[2.2.2]octane (Sigma, D27802-25MG), an anti-quenching agent.
Brain tissue sections were obtained from mice that were transcardially perfused first with 10 mL of phosphate buffer (0.1 M sodium phosphate, pH 7.6), followed by 10 ml of 4% paraformaldehyde in phosphate buffer at a flow rate of 1.5 mL/minute. Next, brains were removed and placed in 4% paraformaldehyde in phosphate buffer for 24 hours, washed three times in PBS, incubated in 30% sucrose overnight, and flash-frozen using dry ice. Frozen tissue was stored at −80°C until ready for sectioning to 40 μm thickness on a Microm HM 505 E cryostat.
Sagittal brain sections were placed into wells of a 12-well tissue culture dish, rinsed briefly with PBS, then rinsed three times for 15 minutes each in PBS +0.3% Triton X-100 followed by PBS +0.3% Triton X-100 and 2% normal goat serum for 2 hours with gentle rocking. Sections were then labeled with primary and secondary antibodies as described for cultured neurons.
Cultured neurons and brain sections were imaged on either of two microscopes: (1) Nikon Eclipse Ti equipped with a Yokogawa CSU-X1 spinning disk head, 20× 0.75 NA CFI Plan Apo, 40× 1.3 NA CFI S Fluor and 60× 1.4 NA Plan Apo objectives; 405, 488, 561 and 640 nm lasers; and a Hamamatsu Flash 4.0 scientific CMOS camera; or (2) EVOS FL imaging system (Invitrogen). Micrographs were captured using either Nikon Elements or EVOS software. CCR-positive cells were counted using the ImageJ cell counter plugin for brain sections (https://imagej.nih.gov/ij/plugins/cell-counter.html) and were counted manually using the EVOS microscope for primary neuron cultures. For cultured neurons (Figs. 1–4, and Supplementary Figs. S1 and S2), four independent, biological replicates were used per experiment, and for each independent replicate, more than 300 neurons were counted.
Fig. 1.
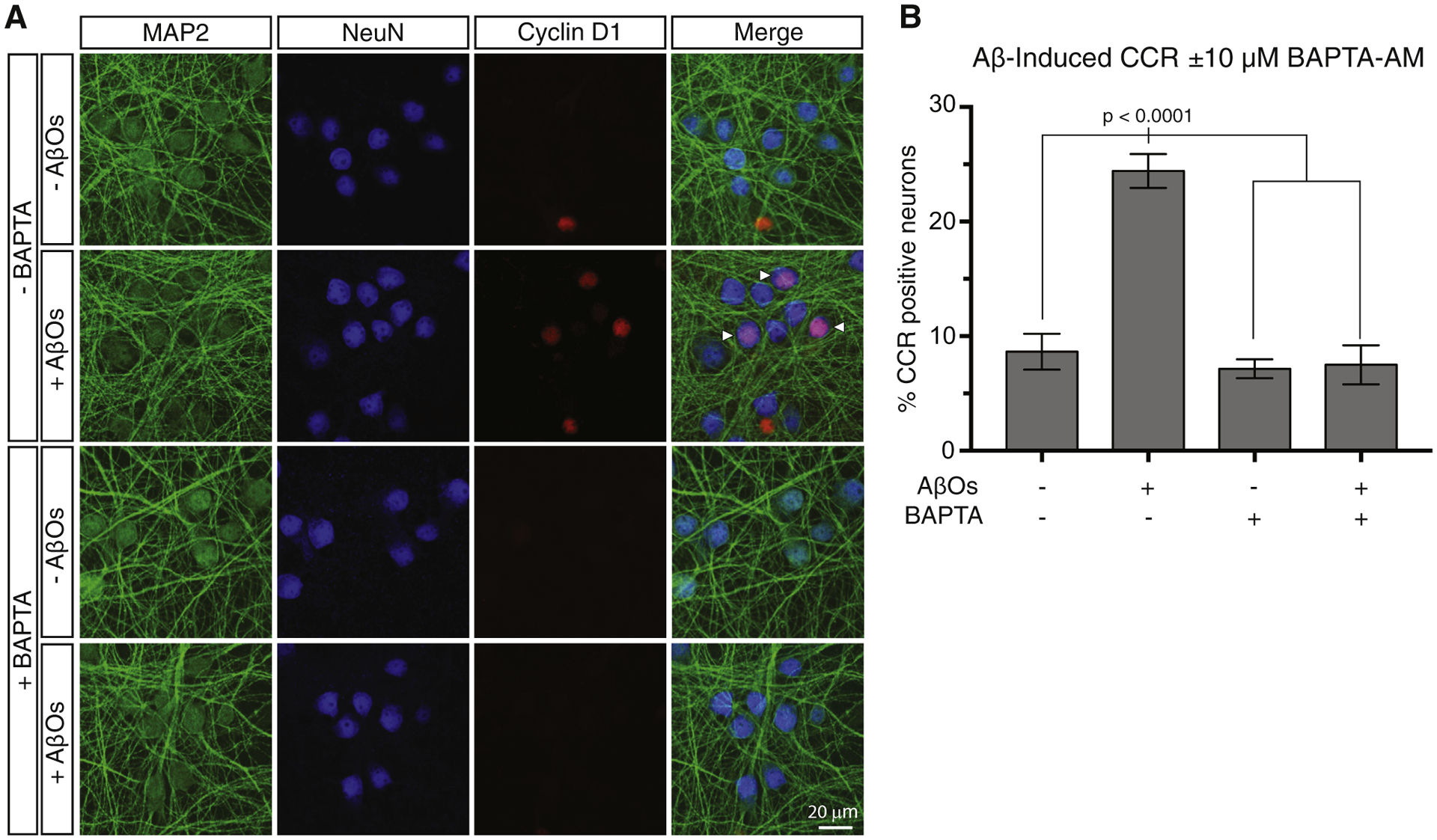
Intracellular calcium chelation by BAPTA-AM prevents AβO-induced neuronal CCR. (A) Primary cortical neurons were treated for 18 hours with AβOs with or without 10 μM BAPTA-AM to chelate intracellular calcium. CCR neurons were identified by their immunoreactivity with antibodies to both cyclin D1, which marks nuclei during G1 of the cell cycle, and NeuN, which marks nuclei in all neurons. Neuronal somatodendritic compartments were labeled with an antibody to MAP2. (B) Quantification of the immunofluorescence results and statistical analysis by one-way ANOVA. Error bars indicate S.E.M. Abbreviations: AβO, amyloid-β oligomer; ANOVA, analysis of variance; CCR, cell cycle reentry.
Fig. 4.
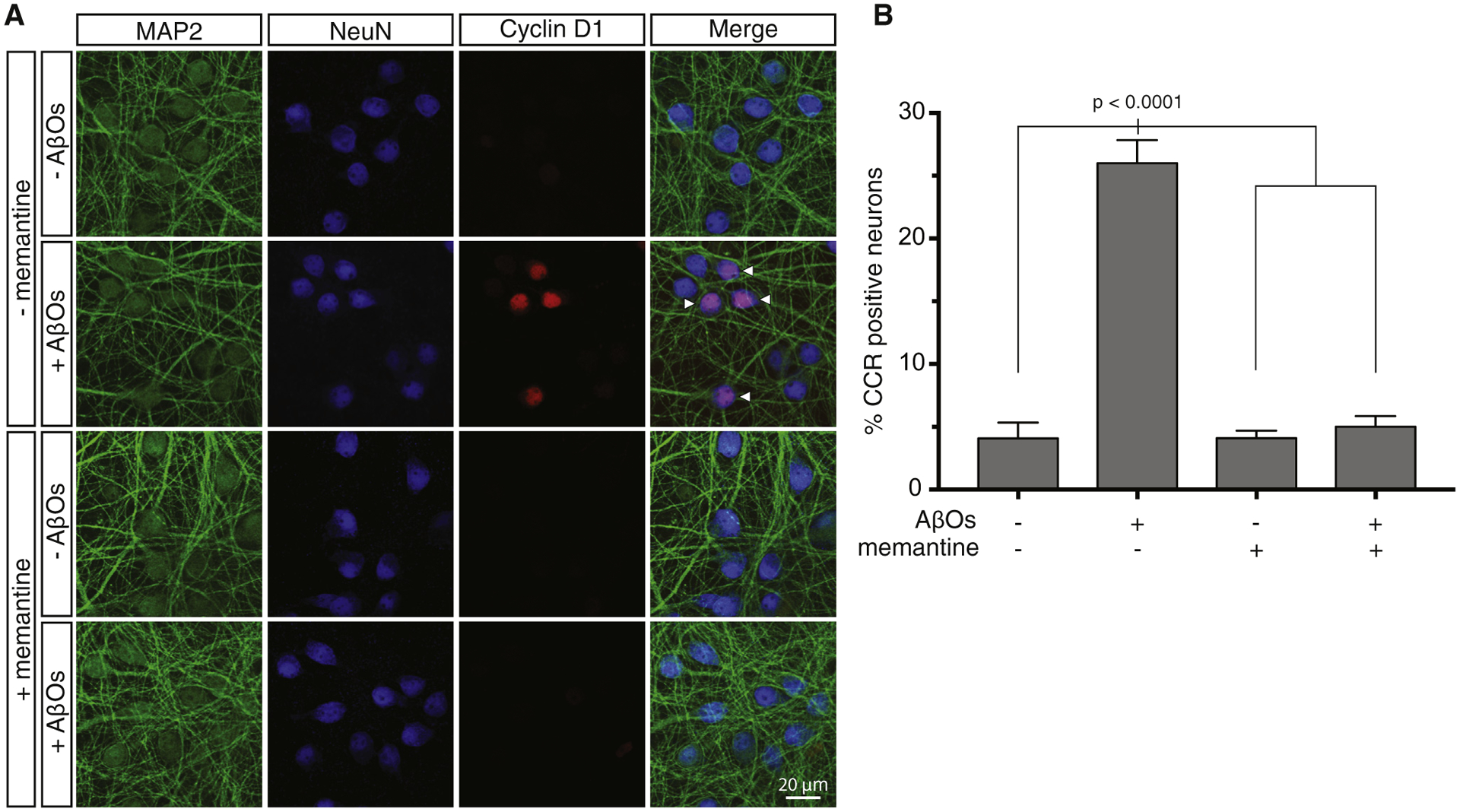
Memantine blocks AβO-induced neuronal CCR. (A) Primary cortical neurons were treated with AβOs with or without pretreatment with 10 μM memantine. Cells were then stained with antibodies to MAP2, NeuN, and cyclin D1 to identify CCR neurons. (B) Quantification of the immunofluorescence results and statistical analysis by one-way ANOVA. Error bars indicate S.E.M. Abbreviations: ANOVA, analysis of variance; AβO, amyloid-β oligomer; CCR, cell cycle reentry.
For brain sections (Fig. 5), four brains from each condition (WT with and without memantine, and Tg2576 with and without memantine) were used. To ensure thorough neuroanatomical coverage, three images for each of three cortical sections (lateral, medial, and middle) were counted per brain to quantify cyclin D1-positive neurons. ~500 neurons per section were counted, yielding ~1500 each of lateral, medial, and middle cortical neurons, or a total of ~4500 neurons per mouse. Because four brains per condition were counted in this manner, ~18,000 total neurons were counted for each condition. At a qualitative level, CCR neurons appeared to be evenly distributed throughout the cortical regions described here.
Fig. 5.
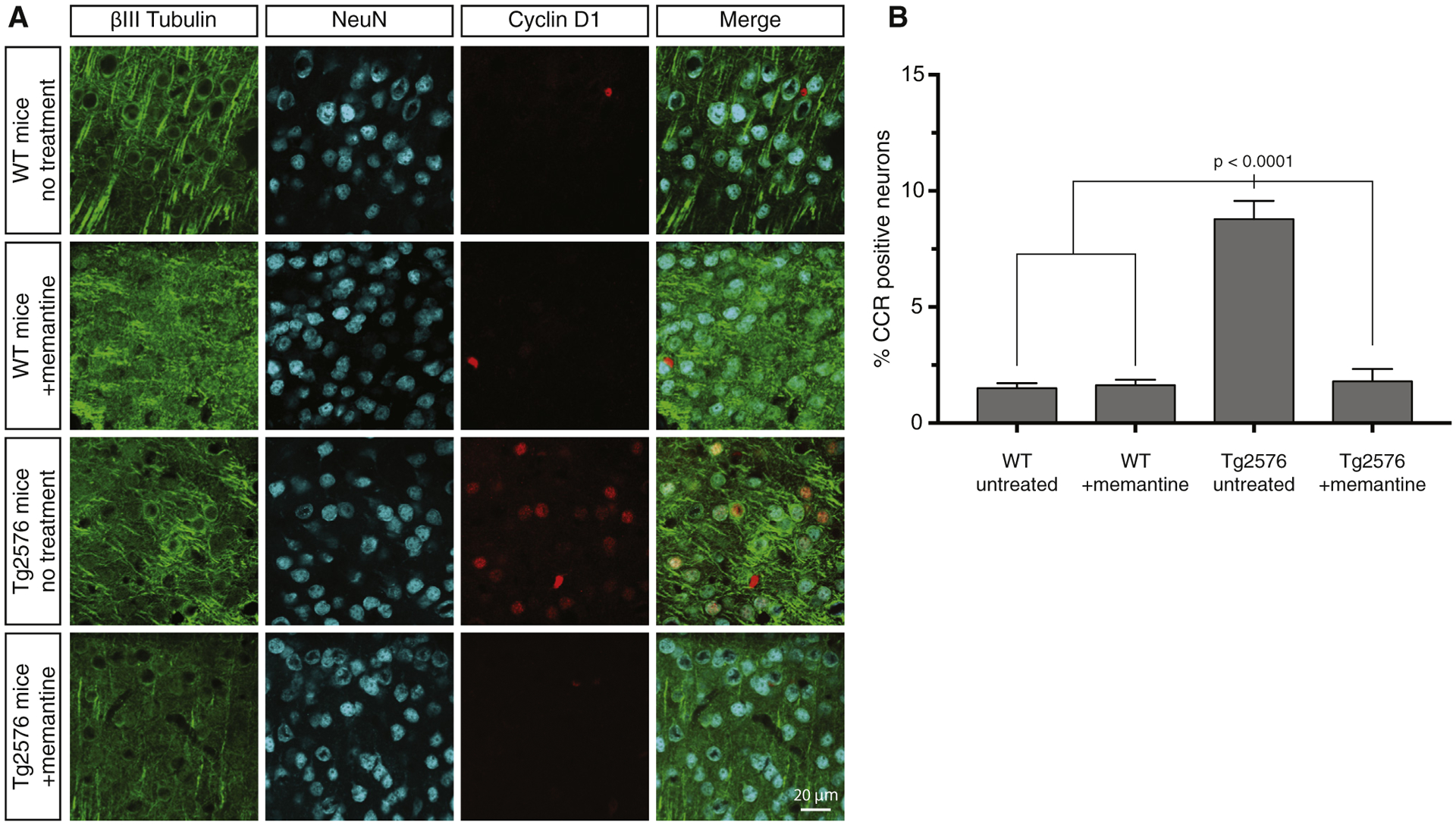
Treatment of Tg2576 AD model mice with memantine prevents CCR in vivo. (A) Tg2576 and wild type mice were provided ad libitum access to drinking water with or without memantine from the time of weaning at 3 weeks until 2 months of age. Brain sections from each condition were stained by triple immunofluorescence for the neuron-specific protein, βIII-tubulin, cyclin D1, and NeuN to identify CCR-positive neurons. (B) Quantification of the immunofluorescence results and statistical analysis by one-way ANOVA. Error bars indicate S.E.M. Abbreviations: AD, Alzheimer’s disease; ANOVA, analysis of variance; CCR, cell cycle reentry.
2.5. Western blotting
Samples were resolved on 12% SDS polyacrylamide gels, then transferred at 100 V for 1.75 hours onto 0.22 μm pore size nitrocellulose membranes using a Bio-Rad Mini-PROTEAN Tetra electrophoresis cell. Following transfer, blots were rinsed once in tris-buffered saline (TBS), blocked for 30–60 minutes in blocking buffer (LI-COR, 927-50000), sequentially incubated in primary antibodies overnight at 4°C in antibody buffer (1:1 mix of blocking buffer, and TBST: TBS +0.1% Tween-20), then LI-COR infrared-labeled secondary antibodies for 30 minutes. Blots were rinsed three times for 5 minutes each using TBST after the primary antibody or TBS after the secondary antibody. Finally, membranes were scanned using a LI-COR Odyssey imaging station.
2.6. Lentivirus production
HEK-293T cells were grown in culture to 90% confluency, then transfected with envelope, packaging, and shRNA plasmids using Lipofectamine 3000 (Invitrogen, L3000-015). Mission shRNAs for the knockdown vectors were obtained from Sigma-Aldrich (NR1 knockdown vector: TRCN0000233327). Media collected at 2–3 and 4–5 days post-transfection was pooled, and virus was pelleted in a Beckman Coulter Optima LE-80K ultracentrifuge for 2 hours at 23,000 rpm (95,389 × gmax) at 4°C in an SW28 rotor. The supernatant was then removed, and the pellets containing the virus were resuspended in 300 μL Neurobasal medium. Viruses were then aliquoted and stored at −80°C until ready for use.
2.7. Memantine treatment
Tg2576 AD model mice that overexpress human β-amyloid precursor protein (APP) with the K670N/M67IL Swedish mutation [27] and WT mice of the same background (50% SLW, 50% C57/BL6) were treated with memantine at ~30 mg/kg/day based on a previously described protocol [28] from the time they were weaned (3 weeks) until 2 months of age. The memantine concentration in water, ~1 mM, was chosen based on mouse weight and the assumption of mice consuming 0.15 mL of water per g body weight per day. Mice were given ad libitum access to food and water.
2.8. Statistics
All statistical analysis was performed with Prism 7 software. For 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), MK-801, memantine, and knockdown experiments, one-way analysis of variances with Bonferroni post-hoc tests were used for analysis. For the phosphorylated calcium-calmodulin-dependent protein kinase II (pCaMKII) time course, a two-way analysis of variance with Bonferroni post-hoc test was used to give P values for individual time points. For Tg2576 memantine experiments, one-way analysis of variance with Bonferroni post-hoc test was used. At least 4000 neurons were counted per mouse per condition.
2.9. Reagents used
Drugs: 2-Aminoethoxydiphenyl borate (Sigma; D9754-10G), BAPTA-AM (Thermo Fisher; B1205), 6-cyano-7-nitroquinoxaline-2,3-dione (Sigma; C239-100MG), dantrolene (Sigma; D9175-100MG), MK-801 (Sigma; M107-5MG), memantine (Sigma; M9292-100MG). Antibodies: chicken anti-MAP2 (Abcam; ab92434), mouse anti-NeuN (Millipore; MAB377), rabbit anti-Cyclin D1 (Abcam; ab16663), rabbit anti-NR1 (CST; D65B7), rabbit anti-pCaMKII (CST; D21E4), mouse anti-total CaMKII (BD Labs; 611292), mouse anti-βIII-tubulin; (TuJ1; courtesy of T. Spano and T. Frankfurter, University of Virginia), goat anti-mouse IgG Alexa Fluor 568 (Life Technologies; A11041), goat anti-mouse IgG Alexa Fluor 405 (Invitrogen; 35501BID), goat anti-rabbit IgG Alexa Fluor 488 (Life Technologies; A11034), goat anti-chicken IgG Alexa Fluor 647 (Life Technologies; A21235), goat anti-mouse IRDye 680LT (Licor; 925-68070), goat anti-rabbit IRDye 800 CW (Licor; 925-32,211).
3. Results
3.1. Intracellular calcium is necessary for CCR
To test the hypothesis that neuronal CCR is calcium-dependent, we first treated primary mouse cortical neurons with BAPTA-AM, a cell-permeant chelator of intracellular calcium, beginning 30 minutes before addition of AβOs to the cultures. After 16–18 hours of AβO exposure, neurons were fixed and stained by triple immunofluorescence microscopy with antibodies to the G1 marker, cyclin D1, the neuron-specific nuclear protein, NeuN, and the neuronal somatodendritic protein, MAP2. As shown in Fig. 1, AβO treatment increased the fraction of cyclin D1-positive neurons from ~8% to ~25%, but the rise in cyclin D1-positive neurons was prevented by pretreatment with BAPTA-AM. These results demonstrate that calcium is necessary for neuronal CCR.
3.2. CCR is blocked by pharmacologically inhibiting NMDAR, but not AMPA receptor or ER calcium stores
We next investigated which specific calcium sources contribute to the initiation of CCR. Given the numerous deficits induced by AβOs at the synapse, and that AβOs induce calcium influx through NMDAR, we tested whether this AβO-mediated calcium influx is required for CCR. Accordingly, primary neuron cultures were treated with MK-801, an NMDAR inhibitor that blocks calcium entry through the channel pore, beginning 30 minutes before AβOs were added. After 16–18 hours of AβO treatment, neurons were fixed and stained for NeuN, MAP2, and cyclin D1 to enable quantitation of neuronal CCR. As shown in Fig. 2A and B, pretreatment of neurons with MK-801 blocked the induction of CCR by AβOs. Otherwise, identical experiments were performed using 6-cyano-7-nitroquinoxaline-2,3-dione to block plasma membrane α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (Fig. S1), 2-aminoethoxydiphenyl borate to block endoplasmic reticulum–associated inositol 1,4,5-trisphosphate receptors and transient receptor potential channels (Fig. S2), or dantrolene to inhibit endoplasmic reticulum–associated ryanodine receptors (Fig. S2). Because none of those inhibitors blocked CCR, we conclude that NMDAR is the major and possibly exclusive source of the excess calcium that enters neurons in response to AβO exposure and initiates CCR.
Fig. 2.
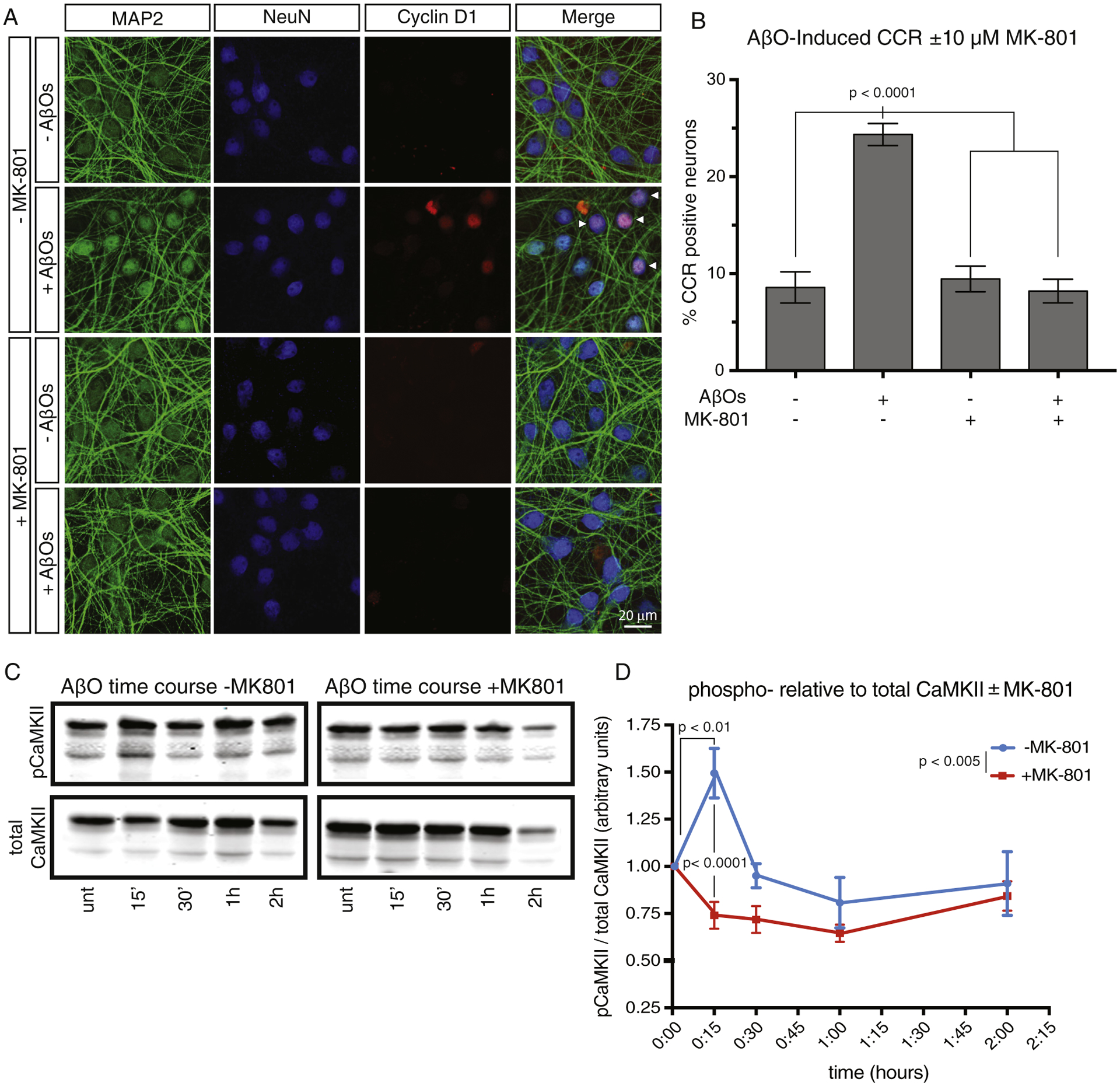
The NMDAR inhibitor, MK-801, blocks AβO-induced neuronal CCR and early activation of CaMKII. (A) Primary cortical neurons were treated overnight with AβOs with or without 10 μM MK-801. After 18 hours of exposure to AβOs, neurons were stained for MAP2, NeuN, and Cyclin D1 to mark neurons that reenter the cell cycle. (B) Quantification of the immunofluorescence results and statistical analysis by one-way ANOVA. Error bars indicate S.E.M. (C) Primary cortical neurons were treated for the indicated times with AβOs, with or without 10 μM MK-801, after which phospho-activated and total CaMKII levels were monitored by Western blotting. (D) Quantification of phosphorylated CaMKII relative to total CaMKII at each time point. Note that MK-801 prevented the transient rise in phospho-activation of CaMKII. Results are significant by two-way ANOVAwhen comparing the plus versus minus MK-801 data sets (P < .005), and by Bonferroni post hoc analysis when comparing the 0 and 15-minute time points without MK-801 (P < .01), and when comparing plus and minus MK-801 at the 15 minute time point (P < .0001). Error bars indicate S.E.M. Abbreviations: ANOVA, analysis of variance; AβO, amyloid-β oligomer; CaMKII, calciumcalmodulin-dependent protein kinase II; CCR, cell cycle reentry; NMDAR, N-methyl-D-aspartate receptor.
Previous work from our laboratory demonstrated that AβO-mediated CCR requires activation of at least four protein kinases: CaMKII, the src-family kinase, fyn, protein kinase A, and mTOR [19,20]. CaMKII is of particular interest, as it can be activated via calcium influx through NMDAR. We thus tested the hypothesis that AβO-mediated calcium influx through NMDAR activates CaMKII as a mediator of CCR. Again, we treated primary neurons with AβOs with or without a 30 minute MK-801 pretreatment and collected the cells at various time points from 0 to 2 hours after AβO stimulation for Western blotting with antibodies to phospho-activated and total CaMKII. As shown in Fig. 2C and D, we found that CaMKII was transiently activated 15 minutes after AβO addition and that activation was prevented by MK-801. The AβO-mediated activation of CaMKII necessary for CCR [19] is therefore dependent on calcium influx through NMDAR.
3.3. Knockdown of NR1 blocks CCR
To provide independent, nonpharmacological evidence for the role of NMDAR in AβO-stimulated neuronal CCR, we used antisense short hairpin ribonucleic acid (shRNA) to knock down expression of the constitutive NMDAR subunit, NR1. Ninety-six hours before AβO addition, primary neuron cultures were transduced with lentivirus containing an empty vector or an shRNA vector to NR1. Following a 16–18 hour exposure to AβOs, the cells were processed for triple immunofluorescence with antibodies to MAP2, NeuN, and cyclin D1. As shown in Fig. 3, we found that reducing the neuronal level of NR1 to 30% of normal blocked the ability of AβOs to cause CCR. These results validate the pharmacological evidence based on the use of MK-801 (Fig. 2) that NMDAR is essential for enabling AβOs to drive normally postmitotic neurons back into the cell cycle.
Fig. 3.
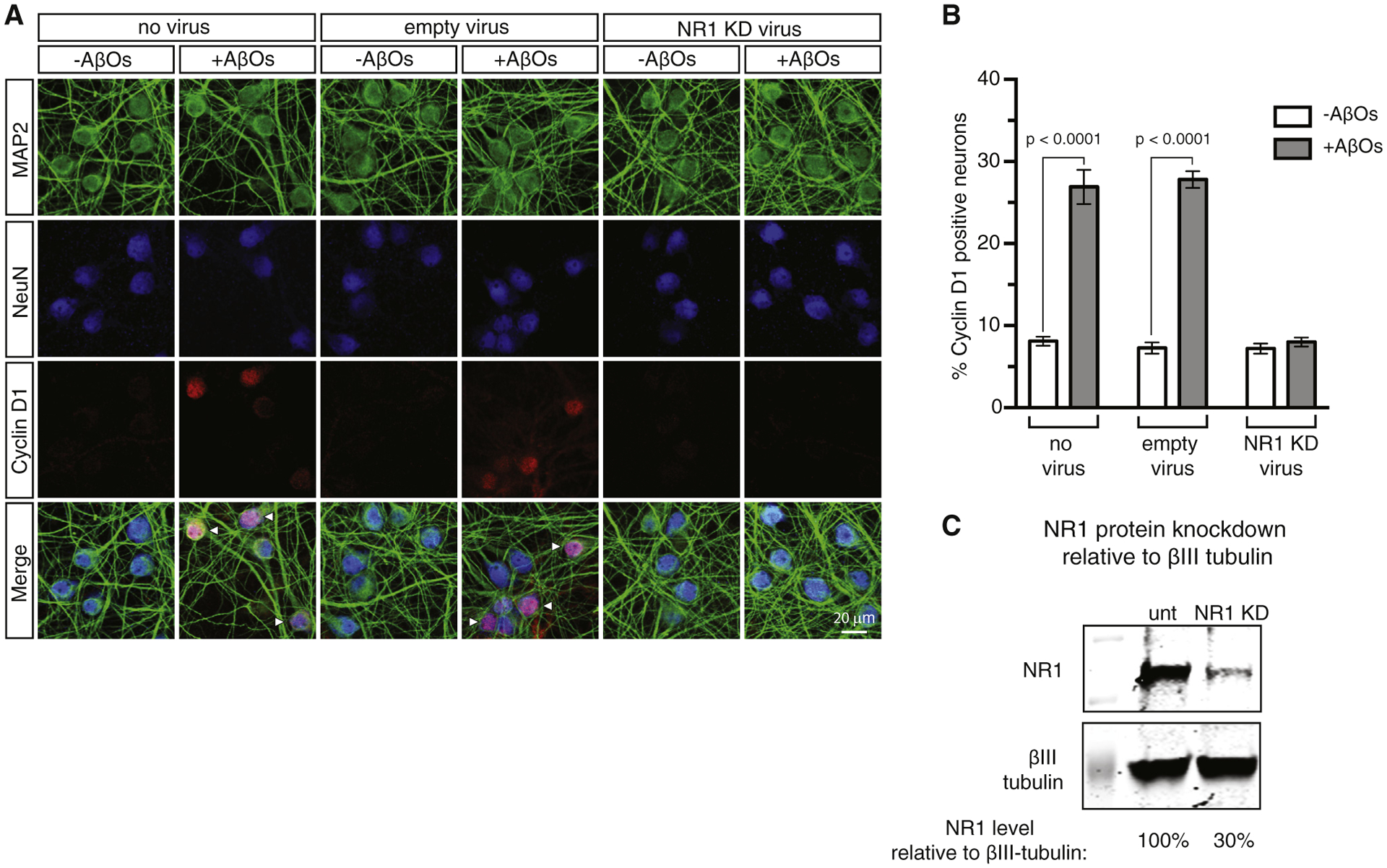
Knockdown of the NR1 subunit of NMDAR prevents AβO-induced neuronal CCR. (A) Primary cortical neurons were transduced for 96 hours prior to AβO addition with lentivirus expressing shRNA to NR1, or as a control, with lentivirus comprising an empty expression vector. After 16–18 hours of AβO exposure, the cells were stained by triple immunofluorescence for NeuN and MAP2 to mark neurons, and for cyclin D1 to assess CCR. (B) Quantification of the immunofluorescence results. Quantification of the immunofluorescence results and statistical analysis by one-way ANOVA. Error bars indicate s.e.m. (C) Quantitative western blot showing a 30% knockdown of NR1. Abbreviations: ANOVA, analysis of variance; AβO, amyloid-β oligomer; CCR, cell cycle reentry; NMDAR, N-methyl-D-aspartate receptor.
3.4. Memantine blocks CCR in cultured neurons and in vivo
Memantine is an United States Food and Drug Administration–approved drug for AD treatment and works by preventing excess calcium influx through NMDAR while still allowing normal calcium-mediated synaptic transmission through the receptor. To gain further insight into the mechanism by which excess calcium induces CCR, we tested whether specifically blocking excess, but not normal calcium, entry through NMDAR could block CCR. Similar to experiments with MK-801, we treated primary neurons with AβOs for 16–18 hours with or without a 30 minute pretreatment with memantine, followed by triple immunofluorescence labeling with antibodies to NeuN, MAP2, and cyclin D1. As shown in Fig. 4, memantine, similar to MK-801 (Fig. 2), prevented AβOs from inducing neuronal CCR. Although memantine is typically used to treat late-stage AD and is not considered to be a disease-modifying drug, these results with cultured neurons raised the possibility that memantine can interfere with CCR in vivo and might therefore be able to block neuron death in AD.
To test that possibility, WT and Tg2576 AD model mice [27], which overexpress human APP with the Swedish mutation (K670M/N671L), were provided ad libitum access to memantine-containing water from the time they were weaned (3 weeks) until 2 months of age, when abundant neuronal CCR is normally evident in the Tg2576 strain [20]. Following the 5 weeks of memantine treatment, the animals were euthanized, and brain sections were stained with antibodies to NeuN, cyclin D1, and the neuron-specific protein, βIII-tubulin. As shown in Fig. 5, WT mice with or without memantine treatment had a basal level of 1.6% neuronal CCR along cortical regions, as determined by cyclin D1 immunoreactivity. In contrast, the basal level of CCR in similar cortical regions of Tg2576 mice was 8.8%, which was reduced to WT levels by memantine. These results show that treating Tg2576 mice with memantine before symptom onset acts prophylactically to prevent CCR.
4. Discussion
The behavioral symptoms of AD are directly caused by the impaired function and loss of synapses by neurons that control memory and cognition and by the death of those neurons. Using CCR as a surrogate for eventual neuronal death in primary neuron cultures and in vivo, we demonstrate here that AβO-stimulated entry of excess calcium via NMDAR, which has been shown by others to trigger synaptotoxicity in vivo [3,29,30], also initiates neuronal CCR. We found that AβO-induced CCR could be prevented in cultured neurons by chelating total intracellular calcium with BAPTA-AM (Fig. 1), blocking calcium entry via NMDAR with MK-801 (Fig. 2), shRNA knockdown of the constitutive NMDAR subunit NR1 (Fig. 3), or using memantine for specifically blocking the entry of excess calcium into neurons via NMDAR (Fig. 4). In contrast, inhibitors of cytoplasmic calcium elevation by AMPA receptor (Fig. S1) or endoplasmic reticulum calcium stores (Fig. S2) did not block AβO-induced CCR. Most importantly in terms of clinical relevance, we also show that memantine treatment of Tg25756 AD model mice blocks neuronal CCR in vivo (Fig. 5). Together, these results indicate that AβO-stimulated excitotoxic calcium influx through NMDAR and CCR/cell death share a common mechanistic origin. Furthermore, they suggest that memantine, which is widely considered to relieve symptoms without attacking processes that lead to neuronal decline, may indeed slow or prevent AD progression if administered at early, presymptomatic disease stages.
While the pathways for both synaptotoxicity [11,31] and CCR/cell death proceed following AβO-stimulated entry of excess calcium via NMDAR, it is not yet clear if these dual causes of AD symptoms are connected serially or represent bifurcating processes with a common origin. As illustrated in Fig. 6, AβO-mediated synaptic deficits and CCR stemming from aberrant NMDAR calcium influx could be connected by a linear pathway in either direction, or alternatively, AβO-mediated calcium influx could contribute to synaptic deficits and CCR independently, eventually causing neuron death. These two schemes are not necessarily mutually exclusive, and further work will be needed to resolve this issue. Although CCR neurons are abundant in Tg2576 mice by 2 months of age [20] and the earliest reported synaptic problem in this strain is dendritic spine loss by 4.5 months of age [32], it is possible that synaptic deficits that are difficult to detect in Tg2576 mice occur before CCR or contribute to CCR at later time points.
Fig. 6.
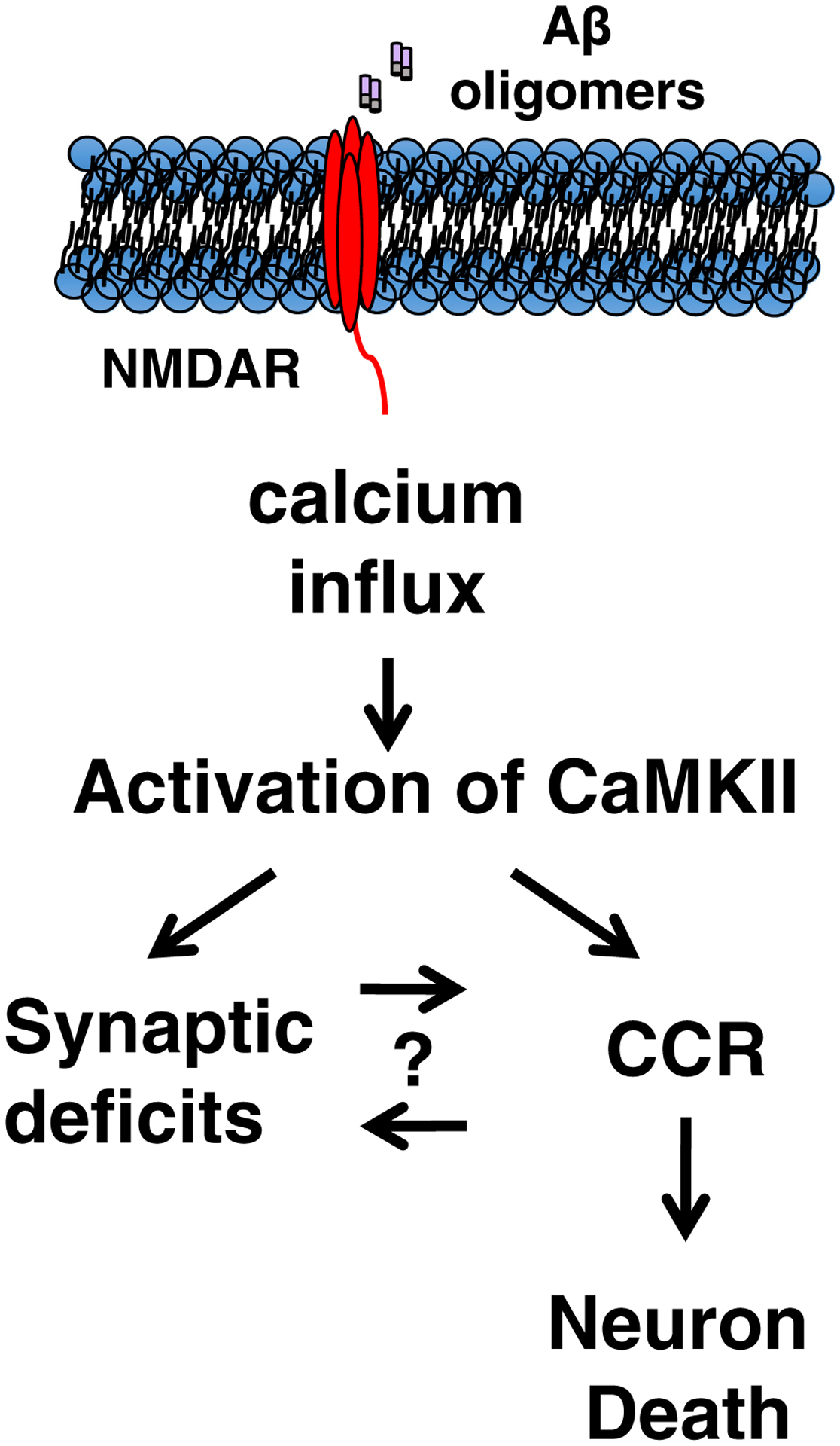
AβO-induced calcium influx via NMDAR is necessary for neuronal CCR. AβO-induced excitotoxicity contributes to many neuronal dysfunctions in AD, including synaptic deficits. Here, we show that AβO-mediated calcium influx via NMDAR is also necessary for initiating CCR. AβO-mediated synaptic dysfunction involving NMDAR could be initiating neuronal CCR directly, or alternatively, these pathways could diverge after AβO-mediated calcium influx, each contributing to neuron death independently. These two schemes are not necessarily mutually exclusive. Abbreviations: AβO, amyloid-β oligomer; AD, Alzheimer’s disease; CCR, cell cycle reentry; NMDAR, N-methyl-D-aspartate receptor.
It has been known since the 1990s that vulnerable neurons in AD frequently enter into a paradoxical pathway before dying: ectopic CCR. Differentiated neurons are permanently postmitotic, but in AD and several other neurodegenerative disorders, neuron death often appears to follow expression of various cell markers, such as cyclin D1 and duplication of large portions of the genome [18,33]. Furthermore, studies of human brain have shown a peak in polyploidy between preclinical AD and mild AD, followed by a decline in polyploidy and neuronal numbers in severe AD, presumably because CCR neurons preferentially die [17].
AβOs are the dominant species causing the excess calcium influx that contributes to excitotoxicity in AD, particularly through NMDARs [32,34]. The data shown here imply that excitotoxicity and CCR are mechanistically connected to NMDAR activity by tau. For instance, tau plays an essential role in effects downstream of excitotoxic calcium influx, as knocking out tau in an AD model mouse model protects against learning and memory deficits, seizures, and premature death [35]. In addition, it has been shown that NMDAR can be influenced by tau upstream of calcium influx, by recruiting the nonreceptor tyrosine kinase, fyn, which phosphorylates NMDAR and thereby facilitates excess calcium influx provoked by AβOs [3,29,36]. We have also shown tau to be necessary for CCR to occur by a mechanism that requires fyn-dependent tau phosphorylation [19]. Although our results imply that excess calcium entry into neurons via NMDAR is sufficient to drive CCR, they do not exclude possible contributions to cytoplasmic calcium level increases from at least one other source, mitochondria [37].
There are additional ways that calcium is likely to be connected to CCR. Calcium is one of the most versatile second messengers [38], not only for controlling functions of post-mitotic neurons but also for regulating division of proliferative cells. In fact, calcium is involved at multiple stages of the cell cycle, including conversion from the quiescent state of G0–G1 and from G1 to S phase [39,40]. These transitions are germane to neuronal CCR because neurons undergoing CCR apparently proceed into S phase before their eventual death. In other excitotoxic paradigms, such as stroke and ischemia, NMDAR overexcitation by kainic acid treatment is sufficient to induce CCR in neurons before their eventual death by excitotoxic shock [41–43]. It is possible that this already established pathway has been repurposed in the neuron as a response to stress or toxicity.
Memantine alleviates many AD symptoms, including loss of long term potentiation/long term depression, learning and memory deficits [28,44], synapse loss, and abnormal tau phosphorylation [45]. Memantine is additionally protective against more canonic excitotoxic insults beyond AD, including preventing neuron damage after ischemic shock in mice and rats [46,47]. Our results show that memantine also works prophylactically to prevent CCR when used before disease signs are evident. This raises the possibility that memantine has disease-modifying properties that have not been realized until now and that it might slow or halt disease progression if administered at a sufficiently early presymptomatic disease stage.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic review: The behavioral features of Alzheimer’s disease (AD) are caused in part by the death of neurons that mediate memory and cognition. The goal of this study was to define seminal biochemical processes that cause neuron death in AD.
Interpretation: Using cell cycle reentry (CCR) as a surrogate for neuron death, we show that neuronal exposure to amyloid-β oligomers and the resultant excess calcium entry via N-methyl-D-aspartate receptor (NMDAR) causes CCR.
Future directions: Our finding that the NMDAR antagonist, memantine, blocks CCR in vivo raises the possibility that memantine can serve as a disease-modifying therapy for AD if administered presymptomatically.
Acknowledgments
This work was supported by the following: The Owens Family Foundation (G.S.B.); the University of Virginia President’s Fund for Excellence (G.S.B.); Webb and Tate Wilson (G.S.B.); the Virginia Chapter of the Lady’s Auxiliary of the Fraternal Order of Eagles (G.S.B.); NIH/NIA grant RF1 AG051085 (G.S.B.); Alzheimer’s Association Zenith Fellowship ZEN-16-363266 (G.S.B.), and the Cure Alzheimer’s Fund (G.S.B. and John S. Lazo).
Footnotes
Conflicts of interest: The authors have declared that no conflict of interest exists.
Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.jalz.2018.05.017.
References
- [1].Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol 2012;4:a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science 2002; 298:789–91. [DOI] [PubMed] [Google Scholar]
- [3].Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010;142:387–97. [DOI] [PubMed] [Google Scholar]
- [4].la Monte de SM. Type 3 diabetes is sporadic Alzheimer׳s disease:mini-review. Eur Neuropsychopharmacol 2014;24:1954–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science 2010;330:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A 2002;99:6364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bloom GS. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 2014;71:505–8. [DOI] [PubMed] [Google Scholar]
- [8].Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 2012;485:651–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297:353–6. [DOI] [PubMed] [Google Scholar]
- [10].Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416:535–9. [DOI] [PubMed] [Google Scholar]
- [11].Lacor PN, Buniel MC, Furlow PW, Sanz Clemente A, Velasco PT, Wood M, et al. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci 2007;27:796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci 2007;8:368–78. [DOI] [PubMed] [Google Scholar]
- [13].Arendt T, Brückner MK, Mosch B, Lösche A. Selective cell death of hyperploid neurons in Alzheimer’s disease. Am J Pathol 2010; 177:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci 2003; 23:2557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Greene LA, Liu DX, Troy CM, Biswas SC. Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim Biophys Acta 2007;1772:392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Varvel NH, Bhaskar K, Patil AR, Pimplikar SW, Herrup K, Lamb BT. Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease. J Neurosci 2008;28:10786–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Arendt T Cell cycle activation and aneuploid neurons in Alzheimer’s disease. Mol Neurobiol 2012;46:125–35. [DOI] [PubMed] [Google Scholar]
- [18].Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci 2001; 21:2661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Seward ME, Swanson E, Norambuena A, Reimann A, Cochran JN, Li R, et al. Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J Cell Sci 2013;126:1278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Norambuena A, Wallrabe H, McMahon L, Silva A, Swanson E, Khan SS, et al. mTOR and neuronal cell cycle reentry: How impaired brain insulin signaling promotes Alzheimer’s disease. Alzheimer’s Dement 2017;13:152–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement 2017;13:178–82. [DOI] [PubMed] [Google Scholar]
- [22].Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 1992; 12:376–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Workgroup AACH. Calcium hypothesis of Alzheimer’s disease and brain aging:A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement 2017; 13:178–182.e17. [DOI] [PubMed] [Google Scholar]
- [24].Wang Y, Qin Z-H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010;15:1382–402. [DOI] [PubMed] [Google Scholar]
- [25].Malinow R New developments on the role of NMDA receptors in Alzheimer’s disease. Curr Opin Neurobiol 2011;22:559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat Rev Drug Discov 2006; 5:160–70. [DOI] [PubMed] [Google Scholar]
- [27].Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996;274:99–102. [DOI] [PubMed] [Google Scholar]
- [28].Minkeviciene R Memantine improves spatial learning in a transgenic mouse model of Alzheimer’s disease. J Pharmacol Exp Ther 2004; 311:677–82. [DOI] [PubMed] [Google Scholar]
- [29].Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci 2011;31:700–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harbor Perspect Med 2012; 2:a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, et al. Abeta Oligomers induce neuronal oxidative stress through an N-Methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 2007;282:11590–601. [DOI] [PubMed] [Google Scholar]
- [32].Lanz TA, Carter DB, Merchant KM. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis 2003;13:246–53. [DOI] [PubMed] [Google Scholar]
- [33].Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci 1997;17:3588–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Alberdi E Sánchez-Gómez MV, Cavaliere F, Pérez-Samartín A, Zugaza JL, Trullas R, et al. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010;47:264–72. [DOI] [PubMed] [Google Scholar]
- [35].Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 2007;316:750–4. [DOI] [PubMed] [Google Scholar]
- [36].Zempel H, Thies E, Mandelkow E, Mandelkow E-M. Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J Neurosci 2010;30:11938–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 2008; 31:454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 2000;1:11–21. [DOI] [PubMed] [Google Scholar]
- [39].Chafouleas JG, Lagace L, Bolton WE, Boyd AE. Changes in calmodulin and its mRNA accompany reentry of quiescent (G0) cells into the cell cycle. Cell 1984;36:73–81. [DOI] [PubMed] [Google Scholar]
- [40].Machaca K Ca2+ signaling, genes and the cell cycle. Cell Calcium 2010;48:243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kuan CY. Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci 2004; 24:10763–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wen Y, Yang S, Liu R, Simpkins JW. Cell-cycle regulators are involved in transient cerebral ischemia induced neuronal apoptosis in female rats. FEBS Lett 2005;579:4591–9. [DOI] [PubMed] [Google Scholar]
- [43].Marathe S, Liu S, Brai E, Kaczarowski M, Alberi L. Notch signaling in response to excitotoxicity induces neurodegeneration via erroneous cell cycle reentry. Cell Death Differ 2015;22:1775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G, et al. Memantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic mice. Am J Pathol 2010;176:870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett 2004; 566:261–9. [DOI] [PubMed] [Google Scholar]
- [46].Seif el Nasr M, Peruche B, Rossberg C, Mennel HD, Krieglstein J. Neuroprotective effect of memantine demonstrated in vivo and in vitro. Eur J Pharmacol 1990;185:19–24. [DOI] [PubMed] [Google Scholar]
- [47].López-Valdés HE, Clarkson AN, Ao Y, Charles AC, Carmichael ST, Sofroniew MV, et al. Memantine enhances recovery from stroke. Stroke 2014;45:2093–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.