

Abstract
The Western pattern diet is rich not only in fat and calories but also in phosphate. The negative effects of excessive fat and calorie intake on health are widely known, but the potential harms of excessive phosphate intake are poorly recognized. Here, we show the mechanism by which dietary phosphate damages the kidney. When phosphate intake was excessive relative to the number of functioning nephrons, circulating levels of FGF23, a hormone that increases the excretion of phosphate per nephron, were increased to maintain phosphate homeostasis. FGF23 suppressed phosphate reabsorption in renal tubules and thus raised the phosphate concentration in the tubule fluid. Once it exceeded a threshold, microscopic particles containing calcium phosphate crystals appeared in the tubule lumen, which damaged tubule cells through binding to the TLR4 expressed on them. Persistent tubule damage induced interstitial fibrosis, reduced the number of nephrons, and further boosted FGF23 to trigger a deterioration spiral leading to progressive nephron loss. In humans, the progression of chronic kidney disease (CKD) ensued when serum FGF23 levels exceeded 53 pg/mL. The present study identified calcium phosphate particles in the renal tubular fluid as an effective therapeutic target to decelerate nephron loss during the course of aging and CKD progression.
Keywords: Nephrology
Keywords: Chronic kidney disease

Introduction
Chronic kidney disease (CKD) is defined as any abnormality, regardless of cause, of kidney structure and/or function lasting 3 months or longer (1). CKD progression can be viewed as a process of a decrease in the number of functioning nephrons that occurs during the natural course of aging and that is accelerated by renal diseases or systemic disorders causing renal complications, most notably diabetes and hypertension. Accordingly, CKD is prevalent in the aging population. Combined, elderly individuals with a low estimated glomerular filtration rate (eGFR) and patients with CKD whose eGFR is less than 60 mL/min/1.73 m2 account for more than 10% of the total population in Japan (2, 3). Once CKD progresses to an advanced stage, renal replacement therapy (dialysis or kidney transplantation) becomes necessary and places a burden on health care systems worldwide (4). Since hyperphosphatemia was identified as a major risk for cardiovascular events and poor prognosis (5, 6), restriction of dietary phosphate intake and inhibition of intestinal phosphate absorption by administration of phosphate binders have been applied for patients with CKD who have hyperphosphatemia. However, these therapies aimed at lowering phosphate levels in the blood are not used to prevent the progression of early-stage CKD to end-stage renal disease (ESRD), because hyperphosphatemia is a terminal symptom observed only in patients with ESRD (7). No remedy is currently available to prevent CKD progression other than identifying and controlling the disorders that have caused the kidney damage in individual patients. Hence, it is of critical importance to identify a therapeutic target that would be universally applicable to the aged and to patients with early-stage CKD to prevent nephron loss.
The dietary phosphate load has been known to induce renal tubular damage and interstitial fibrosis in rodents (8), which recapitulate, at least in part, pathologies universally observed in the elderly and patients with CKD (9, 10). Notably, it is not only the amount of phosphate intake but also the number of nephrons that determine the severity of kidney damage. Specifically, rats or mice whose nephron numbers had been reduced by partial nephrectomy developed more severe kidney damage than did sham-operated animals when placed on the same high-phosphate diet (8, 11). Elderly individuals and patients with CKD were reported to be at high risk for developing acute kidney injury and the subsequent CKD induced by phosphate gavage for colon cleansing as a pretreatment for colonoscopy (12, 13). These observations are consistent with the notion that low nephron numbers constitute a risk for the kidney damage induced by phosphate intake, although the mechanism remains unclear.
In this study, we elucidate the molecular mechanism by which an increase in phosphate excretion per nephron induces renal tubular damage and identify multiple points of therapeutic interventions. In addition, we provide clinical evidence suggesting that these interventions may benefit approximately 25% of adults over the age of 45 and the majority of patients with CKD.
Results
Calcium phosphate particles, but not phosphate, damage renal tubular cells.
We previously characterized mice placed on a diet containing different amounts of inorganic phosphate and determined the time course and the dose responsiveness of renal tubular damage and interstitial inflammation/fibrosis induced by the dietary phosphate load (11). In response to an increase in dietary phosphate intake, we found that circulating levels of FGF23 were increased. FGF23 is a hormone secreted from the bone and acts on the kidney to increase urinary phosphate excretion (14). FGF23 is indispensable for maintaining phosphate homeostasis, as evidenced by the fact that mice and humans defective in FGF23 or its obligate coreceptor Klotho develop hyperphosphatemia and ectopic calcification (15–18). Because FGF23 induces phosphaturia through suppression of phosphate reabsorption in renal proximal tubules (19), FGF23 should increase the phosphate concentration in the proximal tubule fluid. Indeed, the phosphate concentration in the tubule fluid collected by micropuncture was reported to increase along the course of the proximal tubule in rats infused with phosphate (20). Therefore, we hypothesized that proximal tubule cells might be damaged when exposed to high levels of extracellular phosphate from the apical side. To test this hypothesis, we cultured renal proximal tubule cells (HK-2) and increased the phosphate concentration in the medium. We observed a dose-dependent decrease in cell viability (Figure 1A). During this experiment, we noticed that the medium became slightly cloudy as the phosphate concentration was increased. Electron microscopic observation of the high-phosphate medium identified numerous electron-dense particles (Figure 1B). These particles contained calcium phosphate crystals, because they bound to bisphosphonate (21) and disappeared following treatment with acid or a calcium chelator (Figure 1C). To test the possibility that the phosphate-induced cell damage might be attributable to these calcium phosphate particles, and not to phosphate, we centrifuged the high-phosphate medium at 16,000g for 2 hours to precipitate and remove these calcium phosphate particles (Figure 1C), cultured HK-2 cells with the supernatant, and confirmed that the cell viability was not reduced (Figure 1D). These results indicated that the calcium phosphate particles were responsible for the cell death induced by high extracellular phosphate. Furthermore, we confirmed that addition of alendronate, which inhibits the amorphous-to-crystalline phase transition of calcium phosphate, suppressed the phosphate-induced cell death in a dose-dependent manner (Figure 1A). Conversely, addition of synthesized calcium phosphate particles to the regular medium reduced cell viability in a dose-dependent manner (Figure 1E). These observations indicate that formation of calcium phosphate crystals is necessary and sufficient for high extracellular phosphate to damage proximal tubule cells.
Figure 1. Calcium phosphate particles induce renal tubular cell damage.

(A) Human HK-2 renal proximal tubule cells were cultured in control medium (DMEM containing 0.1% FBS, 3 mM calcium, and 1 mM phosphate) or in high-phosphate media (DMEM containing 0.1% FBS, 3 mM calcium, and 3, 5, or 7 mM phosphate). Cell viability was quantified 24 hours later by MTT assay and is expressed as the percentage of viable cells in the control medium. The same experiment was repeated in the presence of alendronate at the indicated concentrations. Data represent the mean ± SD. n = 4 for each culture condition. **P < 0.0001 versus the control in each culture condition, by 1-way ANOVA with Tukey’s multiple-comparison test. (B) Transmission electron microscopic observation of the medium containing 7 mM phosphate. Scale bar: 500 nm. (C) Particle size distribution of calcium phosphate particles. DMEM containing 3 mM calcium and 7 mM phosphate was incubated at room temperature for 24 hours and then subjected to nanoparticle tracking analysis before (black, CaPi particles) or after centrifugation at 16,000g for 30 minutes (CFG) or treatment with HCl (at 100 mM for 30 minutes) or EDTA (at 50 mM for 30 minutes). (D) The relative viability of HK-2 cells cultured in the control medium and in the supernatant (sup) of the high-phosphate medium (7 mM phosphate) after centrifugation at 16,000g for 2 hours was determined by MTT assay. Data represent the mean ± SD. n = 9 for each culture condition. No significant difference was observed (NS) by Student’s t test. (E) Relative viability of HK-2 cells cultured in the control medium inoculated with the indicated doses of synthesized calcium phosphate particles. Data represent the mean ± SD. n = 8 for each culture condition. *P < 0.05 and **P < 0.0001 versus control, by 1-way ANOVA with Dunnett’s multiple-comparison test.
Calcium phosphate particles appear in the proximal tubule lumen in vivo.
We hypothesized that mice that experience tubulointerstitial damage during the dietary phosphate load should have calcium phosphate crystals in the tubule lumen. To test this hypothesis, we performed ex vivo imaging of the kidney using a fluorescent bisphosphonate (OsteoSense) that binds to crystalline calcium phosphate (22). We injected OsteoSense i.v. into mice fed a high-phosphate diet and detected numerous OsteoSense signals at the cortico-medullary junction. Such signals were absent in mice fed regular diet (Figure 2, A, B, and H). To demonstrate that these calcium phosphate crystals were in the tubule lumen, we repeated ex vivo imaging using FITC–Lotus Tetragonolobus Lectin (LTL) instead of FITC-dextran to label the apical membrane of the proximal tubules and confirmed the OsteoSense signals within the tubule lumen (Figure 2C). Furthermore, we performed scanning electron microscopy (SEM) with energy-dispersive x-ray spectroscopy (EDS) and confirmed that particles consisting of calcium and phosphate were attached to the brush borders (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI145693DS1). The localization of calcium phosphate particles was consistent with the fact that interstitial inflammation and fibrosis induced by the dietary phosphate load started primarily from the cortico-medullary junction (11). We next asked whether the pathology induced by the dietary phosphate load would be alleviated by inhibiting the formation of calcium phosphate crystals with bisphosphonate. Injection of alendronate s.c. erased calcium phosphate particles from the cortico-medullary junction (Figure 2, D and H). We also asked whether lowering the urine pH would have the same effect, because calcium phosphate can be dissolved with acid. As expected, urine acidification by giving an ammonium chloride solution as the drinking water reduced the amount of calcium phosphate particles (Figure 2, E and H). Accordingly, both bisphosphonate treatment (Supplemental Figure 2) and urine acidification (Supplemental Figure 3) alleviated inflammation and fibrosis in the kidneys. We unexpectedly found that the mice treated with ammonium chloride had significantly lower FGF23 and higher phosphate levels in the blood than did the mice given tap water (Supplemental Figure 3), suggesting that the ammonium treatment might have suppressed FGF23 secretion and/or production and increased serum phosphate levels, although the mechanism is not clear. Thus, ammonium treatment may have suppressed the formation of calcium-phosphate crystals in the tubule fluid and alleviated tubule damage not only through urine acidification but also through a reduction of the phosphate load per nephron. Conversely, urine alkalization by giving 0.15 M sodium bicarbonate solution as the drinking water to mice fed the same high-phosphate diet exacerbated the kidney damage (Supplemental Figure 4), possibly by facilitating the formation of calcium phosphate crystals in the tubule fluid.
Figure 2. Calcium phosphate particles in the tubule lumen detected by ex vivo imaging.

Calcium phosphate particles, the interstitial space, and the cell nuclei are depicted in red (OsteoSense), green (FITC-dextran), and blue (Hoechst), respectively. Mice were placed either on a regular diet containing 0.35% inorganic phosphate (NP) (A) or a high-phosphate diet containing 2.0% inorganic phosphate (HP) (B) for 8–9 days. (C) To show that calcium phosphate particles were present in the tubule lumen, FITC-dextran was replaced with FITC-LTL that labeled the apical membrane of proximal tubules. (D and E) Mice fed the HP diet were treated with alendronate (10 mg/kg, s.c. injection every other day, Ale, D) or were given 0.14 M NH4Cl solution as drinking water (NH4Cl, E). Calcium phosphate particles were absent in mice lacking the Tlr4 gene (Tlr4lps-del) fed an NP diet (F) or an HP diet (G). Scale bars: 1000 μm (A, B, and D–G) and 50 μm (C). (H) The number of calcium phosphate particles for A–G is indicated as the mean ± SD. n = 6–8 for each group. *P < 0.01 and **P < 0.0001, by 1-way ANOVA with Tukey’s multiple-comparison test.
Calcium phosphate particles bind to TLR4.
We speculated that cytotoxic signals of calcium phosphate particles might be mediated by a cell-surface receptor. The putative receptor for calcium phosphate particles should meet at least 2 criteria. First, it should bind to calcium phosphate crystals and thus probably belongs to pattern-recognition receptors. Second, it must be expressed in the cortico-medullary junction in the kidneys. We identified TLR4 as a candidate that fulfilled these 2 criteria. TLR4 is known as a pattern-recognition receptor and is expressed mainly in renal tubules in the cortico-medullary junction, where the distal portion of proximal tubules (the S3 segment) is distributed (23). To determine whether calcium phosphate particles might physically interact with TLR4 in vitro, we performed surface plasmon resonance (SPR) analysis by running the extracellular domain of the TLR4 protein over synthesized calcium phosphate particles immobilized on a sensor chip. We observed direct interaction between calcium phosphate particles and the TLR4 ectodomain with a dissociation constant of 9.63 nM (Supplemental Figure 5A) in the absence of myeloid differentiation factor 2, a cofactor necessary for TLR4 to bind to LPS or fetuin-A (24). It is noteworthy that high-phosphate diet feeding induced accumulation of TLR4 to the apical membrane of tubule cells (Supplemental Figure 5, B–D).
We confirmed that mice lacking the Tlr4 gene were resistant to the tubule damage, inflammation, and fibrosis induced by a high-phosphate diet (Supplemental Figure 6). We also confirmed that deletion of the Tlr4 gene specifically in renal tubules alleviated the kidney damage. When placed on a high-phosphate diet, mice defective in renal tubule TLR4 (i.e., mice homozygous for the Tlr4-floxed allele carrying the Ksp-Cre transgene) had lower expression levels of markers for renal tubular damage (kidney injury molecule 1 [Kim1], neutrophil gelatinase-associated lipocalin [Ngal], osteopontin [OPN]), inflammation (monocyte chemotactic protein 1 [MCP1], IL-1β, IL-6), and fibrosis (TGF-β1, matrix metalloprotease 2 [MMP2], vimentin, α-smooth muscle actin [α-SMA], collagen 1α1) than did control mice (i.e., mice carrying the Ksp-Cre transgene alone) (Figure 3, A and B), despite the fact that the urinary phosphate excretion was not different between these mice (Figure 3C). Accordingly, the collagen area fraction in the mice defective in renal tubular TLR4 was smaller than that in the control mice and similar to that in the control mice fed a regular diet (Figure 3, E–G). Taken together, expression of TLR4 in renal tubules is necessary for the dietary phosphate load to induce kidney damage in vivo.
Figure 3. TLR4 expressed in renal tubules is required for a high-phosphate diet to induce kidney damage.

Mice lacking the Tlr4 gene in renal tubular cells (Tlr4-Cre) and control Cre mice (mice carrying the Cre transgene alone) at 4 weeks of age were placed on a high-phosphate diet containing 2.0% inorganic phosphate (HP) for 4 weeks. Relative renal mRNA levels of the markers for tubule damage and inflammation (A) and fibrosis (B) were determined by quantitative RT-PCR. (C) No difference was detected in the amount of urinary phosphate excretion between Tlr4-Cre mice (n = 13) and Cre mice (n = 9). (D–F) Picrosirius red staining of the kidney sections detected patchy red areas of interstitial fibrosis in the cortex and the cortico-medullary junction (CMJ) in the control mice (HP, Cre) but not in the mice lacking the Tlr4 gene in renal tubular cells (HP, Tlr4-Cre). Scale bars: 100 μm. (G) The fibrotic area was quantified in the cortex (upper panel) and the CMJ (lower panel) in the individual mice (n = 4 mice per group). Data are indicated as the mean ± SD. **P < 0.01 versus Cre mice, by Mann-Whitney U test (A and B); **P < 0.01, by 1-way ANOVA with Tukey’s multiple-comparison test (G).
Endocytosis of calcium phosphate particles disturbs endosomal trafficking.
To explore the cellular mechanism of tubule damage induced by calcium phosphate particles, we profiled gene expression in cultured proximal tubule cells (HK-2) before and after exposure to calcium phosphate particles. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis identified enrichment of pathways relevant to inflammation (TLR, TGF-β, and IL-17 signaling) and cell adhesion/junction (Rap1 signaling and gap junction) within 6 hours, followed by endocytosis, apoptosis, and necrosis pathways, among others, within 24 hours (Supplemental Figure 7). At the protein level, we observed an increase in phosphorylation and nuclear accumulation of phosphorylated p38 (p-p38) and p–NF-κB (Figure 4, A–D). We also observed increased expression and secretion of OPN, a cytokine that not only regulates biomineralization and inflammation (25) but also serves as a marker for renal tubular damage (ref. 26 and Figure 4E). These observations suggested that calcium phosphate particles might induce these inflammatory responses through binding and activation of TLR4. However, a cell-permeable inhibitor of TLR4 signaling, TAK242, attenuated the secretion of OPN but not the phosphorylation of NF-κB or p38 (Supplemental Figure 8), indicating that the inflammatory responses were induced through both TLR4-dependent and -independent signaling pathways. Prolonged exposure (6–48 hours) additionally induced endocytosis and accumulation of calcium phosphate particles in late endosomes, lysosomes, and autophagosomes around the nuclei (Figure 5, A–F, and Supplemental Figure 9). The perinuclear clustering of lysosomes, which was reminiscent of that observed in cells under starvation (27), was associated with a decrease in early and recycling endosomes (Supplemental Figure 10), a decrease in cell viability (Figure 5G), and an increase in tubule cell damage (Figure 5H). In summary, calcium phosphate particles trigger inflammatory responses in proximal tubule cells and thereafter induce endocytosis, disturbed endosomal trafficking, and cellular damage and death.
Figure 4. Calcium phosphate particles activate the p38/NF-κB pathway and induce osteopontin secretion.

(A and B) HK-2 cells were incubated with synthesized calcium phosphate particles (10 μg phosphorus/mL) for the indicated durations and subjected to immunocytochemical analysis using antibodies against p-p38 (A) or p–NF-κB (B). The confocal microscopic images were analyzed using Nikon NIS-Elements software to determine the intensity of the fluorescence signals from the nucleus (red) and cytoplasm (blue). Data are indicated as the mean ± SD. N = the number of cells analyzed for each time point. *P > 0.25 (effect size) and **P > 0.3 (effect size); #P > 0.35 (effect size) versus time 0 (without stimulation with calcium phosphate particles), by Brunner-Munzel test. (C and D) Representative confocal images of immunocytochemistry for p-p38 (C) and p-NF-κB (D) are shown. Plasma membranes are outlined in orange and nuclear membranes in blue. Scale bars: 50 μm. (E) The osteopontin concentration was measured by ELISA in conditioned medium of HK-2 cells incubated with or without calcium phosphate particles (10 μg phosphorus/mL) for the indicated durations. n = 3 for each column. #P < 0.0001 versus vehicle, by 2-way ANOVA with Šidák’s multiple-comparison test.
Figure 5. Calcium phosphate particles disturb endosomal trafficking.

HK-2 cells were incubated with calcium phosphate particles (10 μg phosphorus/mL) labeled with 5(6)-RhR-dRIS for the indicated durations and then subjected to immunocytochemical analysis using antibodies against the endosomal markers Rab7 (A), Lamp2 (B), CD63 (C), and LC3 (D). Data are indicated as the mean ± SD. N = the number of cells analyzed for each time point. (A–D) *P > 0.5 (effect size) versus time 0 (before stimulation with calcium phosphate particles) by Brunner-Munzel test. Scale bar: 25 μm. (E) Confocal microscopic images were analyzed to determine the intensity of fluorescent signals in area 1 (perinuclear region, magenta) and area 2 (submembrane region, green). (F) Overlap between signals from calcium phosphate particles and signals from endosomal markers is shown as the ratio of the overlapping area to the total area of calcium phosphate particles. Data are indicated as violin plots with the median (red lines) and quartiles (dotted lines). N = the number of cells analyzed. (G) The viability of HK-2 cells was determined by CCK-8 assay at the indicated time points after addition of calcium phosphate particles (10 μg phosphorus/mL) to the medium in the presence of the TLR4 signaling inhibitor TAK242 (final concentration of 2 μM, blue) or vehicle (DMSO, red). (H) As in G, except that the relative cellular damage was determined by lactate dehydrogenase assay and is expressed as percentages of the maximum cell damage induced by 0.2% Triton X-100. Data are indicated as the mean ± SD. n = 4 for each time point. No difference was observed between the TAK242 group and the vehicle group, by 2-way ANOVA with Šidák’s multiple-comparison test. *P < 0.0001 versus time 0.5 hours, by 2-way ANOVA with Dunnett’s multiple-comparison test.
Notably, the cell damage and death induced by calcium phosphate particles in vitro was independent of TLR4 activation, because it was not attenuated by TAK242 (Figure 5, G and H). Therefore, we conclude that the TLR4-dependent renal tubular damage in vivo was caused not by activation of TLR4 signaling by calcium phosphate particles, but rather by tethering of the calcium phosphate particles on the surface of proximal tubule cells for 6 hours or longer against the tubular fluid flow, which facilitated the endocytosis to induce disturbed endosomal trafficking and tubule cell damage. Consistent with this conclusion, calcium phosphate particles were barely detectable in the tubule lumen in mice lacking the Tlr4 gene that were fed a high-phosphate diet (Figure 2, F–H). Another finding consistent with this conclusion includes the fact that mice lacking Myd88, one of the major intracellular adaptor proteins that mediate TLR4 signal transduction, developed renal fibrosis when placed on a high-phosphate diet, unlike mice lacking the Tlr4 gene (Supplemental Figure 11). Of note, calcium phosphate particles were present in the cortico-medullary junction. This can be explained by the fact that TLR4 was expressed predominantly in the tubules in the cortico-medullary junction (Supplemental Figure 5) and that the phosphate concentration in the proximal tubule fluid in the S3 segments in the cortico-medullary junction can be higher than that in the S1 segments in the cortex upon phosphate loading (20).
FGF23 correlates with renal tubular damage.
Given that the formation of calcium phosphate particles in renal tubular fluid is required for tubule damage, a threshold should exist in the phosphate concentration of the proximal tubule fluid (PTFp), above which calcium phosphate precipitation and tubule damage occur. Because it is technically challenging to obtain tubule fluid from the cortico-medullary junction in live mice, we estimated the PTFp from the concentration of phosphate and creatinine in the blood and urine based on the following assumptions. First, phosphate concentration in the glomerular filtrate is equal to that in the blood. Second, proximal tubules reabsorb 70% of the filtrated water by default (28), which causes a 3.33-fold increase in the concentration of the solutes. Third, phosphate reabsorption takes place almost exclusively in proximal tubules (29). Last, fractional excretion of phosphate (FEp), which is defined as the ratio of phosphate clearance to creatinine clearance (CCr), indicates the fraction of phosphate that was not reabsorbed at the proximal tubules. Thus, the product of the serum phosphate concentration and the FEp multiplied by 3.33 should reflect the PTFp (Supplemental Figure 12A), which we defined as the estimated PTFp (ePTFp) (Equation 1):
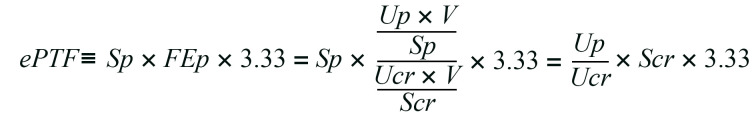
Equation 1
In Equation 1, V, Sp, Up, Scr, and Ucr represent the 24-hour urine volume and the concentrations of serum phosphate, urine phosphate, serum creatinine, and urine creatinine, respectively. We confirmed that the ePTFp served as an approximation of the actual PTFp determined by direct measurement of the phosphate concentration in the proximal tubule fluid collected by micropuncture in living rats (Supplemental Figure 12B and ref. 20). It should be noted that the serum phosphate concentration was cancelled out in Equation 1 and thus had no contribution to the ePTFp.
We changed the PTFp as an independent variable by placing mice with or without uninephrectomy on a diet containing either 0.35%, 1.0%, 1.5%, or 2.0% inorganic phosphate for 12 weeks and quantified serum FGF23 levels and relative mRNA levels of markers for renal tubular damage, inflammation, and fibrosis as dependent variables (Figure 6A). In double-logarithmic plots, the relation between ePTFp and these variables was fitted with 2-segmented linear regression, with the slope of the first segment being 0 (Figure 6, B–I). Specifically, serum FGF23 levels began to elevate when ePTFp was increased beyond 5.18 mg/dL (Figure 6B). Concurrently with the FGF23 increase, expression of tubule damage markers (Ngal and OPN) started to increase (Figure 6, B–D), which was followed by an increase in the expression of inflammatory markers (MCP1 and TNF-α). An increase in fibrosis marker expression and a decline in renal function (decrease in CCr) became evident when the ePTFp reached approximately 10 mg/mL.
Figure 6. Correlation of ePTFp with serum FGF23 levels and kidney damage in mice.

(A) Schematic of the experimental design. Urine, blood, and kidney samples were obtained from uninephrectomized mice or sham-operated mice placed on a diet containing 0.35%, 1.0%, 1.5%, or 2.0% inorganic phosphate for 12 weeks. The correlation between the ePTFp and the following parameters is shown in double-logarithmic plots: serum FGF23 levels (B); relative renal mRNA levels of Ngal (C), osteopontin (D), MCP1 (E), TNF-α (F), vimentin (G), and collagen 1α1 (H); and CCr (I). The ePTFp values of the inflection points and coefficients of determination (R2) of the segmental liner regression lines are indicated. (J) Schematic representation of the mechanism by which high FGF23 levels cause progressive nephron loss. The points of therapeutic intervention are indicated in blue.
Taken together, we propose that the mechanism of renal tubular damage is associated with increased FGF23 expression (Figure 6J). An increase in phosphate intake and/or a decrease in the number of nephrons must be accompanied by an increase in phosphate excretion per nephron to maintain phosphate homeostasis. This demand is met by increasing FGF23, a hormone that increases the excretion of phosphate per nephron. However, the increase in FGF23 raises the PTFp and increases the risk for the formation of calcium phosphate particles in the tubule fluid, which induce tubule damage through binding to TLR4 expressed on the tubule cells. If the tubule damage kills the nephron, FGF23 must be further increased to compensate for the decrease in nephron numbers unless phosphate intake is reduced, which would trigger a deterioration spiral that leads to progressive nephron loss.
Phosphate load reduces nephron numbers.
To test whether the kidney damage induced by an increase in phosphate excretion per nephron indeed causes nephron loss, we developed a method for estimating the nephron number. The same experiment as in Figure 6A was repeated, except that the duration of the phosphate load was shortened to 2 weeks instead of 12 weeks. Because mice have approximately 10,000 nephrons per kidney (30), we calculated the phosphate excretion per nephron for individual mice by dividing the amount of 24-hour urinary phosphate excretion by 10,000 in the uninephrectomized mice or by 20,000 in the sham-operated mice. In double-logarithmic plots, serum FGF23 levels started increasing when the phosphate excretion per nephron exceeded 0.80 μg/d (Figure 7A). Based on the regression line, the nephron number in mice with serum FGF23 levels higher than 290 pg/mL (= y-intercept in Figure 7A) can be calculated by the following equation:
Figure 7. A prolonged increase in phosphate excretion per nephron reduces nephron numbers in mice.

(A) Urine and blood were collected from the 8 groups of mice (uninephrectomized mice or sham-operated mice placed on either a 0.35%, 1.0%, 1.5%, or 2.0% phosphate diet for 2 weeks). The relation between phosphate excretion per nephron and serum FGF23 levels is shown in a log-log plot. The nephron number (B), serum FGF23 levels (C), phosphate excretion per nephron (D), and CCr (E) at the 2 week (white columns) and 12 weeks (cyan columns) are shown for each group. The nephron number of the mice with serum FGF23 levels lower than 290 pg/mL was assumed as 20,000 and 10,000 for the sham-operated mice and the uninephrectomized mice, respectively (shaded gray). The other data are indicated as the mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.0001, by 2-way ANOVA with Šidák’s multiple-comparison test.
Equation 2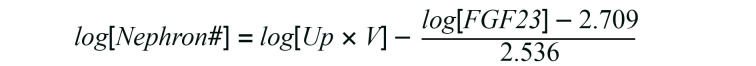
Using Equation 2, we compared the nephron number in mice receiving the short-term (2 weeks) phosphate load with that in mice receiving the long-term (12 weeks) phosphate load. During the additional 10 weeks of phosphate load, we observed a significant decrease in the nephron number (Figure 7B) and a reciprocal increase in serum FGF23 levels (Figure 7C) in the sham-operated mice and the uninephrectomized mice placed on a 1.5% or 2.0% phosphate diet. The phosphate excretion per nephron for mice in these groups was 1.1 μg/d or more on average at the 2-week point (Figure 7D). Hence, we conclude that nephron loss occurs when the phosphate excretion per nephron exceeds 1.1 μg/d. However, a higher level of phosphate excretion per nephron was required to impair renal function within the 12-week period, because a decrease in the average CCr was observed only in the uninephrectomized mice fed a 2.0% phosphate diet (Figure 7E) whose phosphate excretion per nephron exceeded 3.4 μg/d on average (Figure 7D).
FGF23 as a clinical parameter for progressive CKD.
Last, we asked whether the relation between ePTFp and FGF23 observed in mice (Figure 6B) would also be observed in humans. Non-dialysis CKD patients at various stages were recruited to measure ePTFp and serum FGF23 levels (31). In this cross-sectional study, the relation between ePTFp and FGF23 fitted with the 2-segmental linear regression observed in mice. Serum FGF23 levels started increasing when the ePTFp exceeded 2.32 mg/dL (Figure 8A). These findings suggested that Patients with FGF23 more than 53 pg/mL have already developed tubulointerstitial damage, and if left untreated, the damage will grow worse.
Figure 8. Serum FGF23 levels predict CKD progression.

(A) Relation between ePTFp and serum FGF23 levels in 148 outpatients with varying renal function is shown in a log-log plot. The ePTFp and FGF23 values of the inflection points and the coefficient of determination (R2) of the segmental liner regression line are indicated. (B) Cumulative incidence by renal event and FGF23 levels in patients who participated in the EMPATHY study. Kaplan-Meier curves of the cumulative incidence rates of renal events for 2 groups stratified by FGF23 levels of 53 pg/mL during the follow-up period. Patients with FGF23 levels above 53 pg/mL had a higher risk of renal events independently of their serum creatinine levels.
To verify this possibility, we performed a prospective study to explore the relation between serum FGF23 levels and incident kidney events using blood samples from the EMPATHY study (Standard Versus Intensive Statin Therapy for Hypercholesterolemic Patients with Diabetic Retinopathy), in which patients with diabetic retinopathy and hyperlipidemia, but without advanced CKD (eGFR < 30 mL/min/1.73 m2), were enrolled (32). The median concentration of FGF23 was 54.7 pg/mL, with an IQR from 44.0 pg/mL to 69.0 pg/mL. A total of 5039 patients were stratified into 2 groups on the basis of the cutoff point of 53 pg/mL of the serum FGF23 level. During the 5-year follow-up, the renal events defined as initiation of chronic dialysis or an increase in serum creatinine levels by at least 2-fold (and >1.5 mg/dL) occurred in 100 patients: 0.6% in the group with lower FGF23 levels (14 of 2336 patients) and 3.2% in the group with higher FGF23 levels (86 of 2703 patients). The higher FGF23 levels at baseline were associated with the increase in renal events (Figure 8B). Cox regression analysis indicated that the group with high FGF23 levels had a significantly higher risk for renal events than did the group with low FGF23 levels (HR 5.18, 95% CI, 2.94–9.11; P < 0.001, by log-rank test). This relationship remained significant after adjusting for age, sex, BMI, and, importantly, serum creatinine levels (HR 2.84, 95% CI, 1.57–5.13, P = 0.001).
Discussion
Kawasaki et al. performed a retrospective cohort study of 191 non-dialysis CKD patients and demonstrated that the higher ratio of urinary phosphate excretion to CCr was associated with the higher risk for renal events (defined as progression to ESRD and/or a 50% reduction in the eGFR) within 3 years (33). The ratio of urinary phosphate excretion to CCr was actually equivalent to the ePTFp, because:
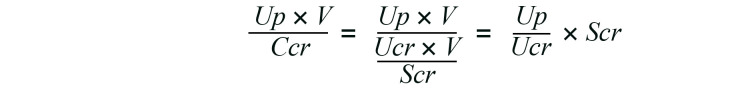
Equation 3
Equations 1 and 3 indicate that the ratio of urinary phosphate excretion to CCr can be converted to ePTFp by proportional calculation. We calculated the ePTFp of the patients in their study and found that the median ePTFp was 3.93 mg/dL, with an IQR from 2.56 to 6.81 mg/dL. Therefore, the patients in quartiles 2–4 had an ePTFp higher than the threshold for the FGF23 increase determined in our study (2.32 mg/dL) and were thus expected to have serum FGF23 levels higher than 53 pg/mL (Figure 8A), although Kawasaki et al. did not measure FGF23 levels. Their conclusion was that the patients in quartiles 2–4 exhibited a graded increase in the risk for renal events when compared with the patients in quartile 1. Another prospective study showed that an increase in Up/Ucr was independently associated with a decrease in the eGFR in patients with stage 2 or 3 CKD (34). These findings are consistent with our conclusion that an increase in the phosphate concentration in the proximal tubule fluid beyond the threshold accelerates nephron loss.
Daily phosphate intake in industrialized countries is approximately 900–1400 mg/d, roughly 60%–70% of which is absorbed from the digestive tract (35). It should be noted that this estimation does not include phosphate in food additives, which amounts to more than 30% of the phosphate in the food itself (35). Thus, the average adult on a standard diet consumes 1200–1800 mg phosphate in total and excretes approximately 1000 mg into the urine each day. The question is whether urinary phosphate excretion of this amount contributes to kidney damage in the elderly and patients with CKD. Although there are considerable individual variations, healthy young adults have 600,000–1,400,000 nephrons per kidney (36). Assuming that a healthy adult with 2 million nephrons excretes 1000 mg phosphate per day, the phosphate excretion per nephron is estimated to be 0.5 μg/d. During the natural course of aging in humans, the nephron number is decreased by approximately 50% in their seventies (36). Unless phosphate intake is reduced with age, the phosphate excretion per nephron reaches 1.0 μg/d, which is close to the level that reduces nephron numbers in mice (1.1 μg/d). Thus, in humans, dietary phosphate consumed on a daily basis is a potential driving force of progressive nephron loss.
Therefore, individuals with decreased nephron numbers, such as elderly people and patients with early-stage CKD whose serum FGF23 levels are higher than 53 pg/mL, are expected to benefit from phosphate restriction aimed at reducing urinary phosphate excretion to lower serum FGF23 levels below 53 pg/mL. Because this notion is based on the correlation between FGF23 and renal events (Figure 8), the causality remains to be determined by randomized, controlled clinical trials. Specifically, it can be expected that restriction of dietary intake and intestinal absorption of phosphate by avoiding phosphate-rich ingredients and food additives and by administration of phosphate binders will decelerate nephron loss. However, the current paradigm of phosphate restriction aims to lower phosphate levels in the blood of patients with ESRD with hyperphosphatemia to reduce cardiovascular events and mortality. We propose another paradigm of phosphate restriction that would apply not only to ESRD patients with hyperphosphatemia but also to normophosphatemic CKD patients with hyper-FGF23-emia (FGF23 > 53 pg/mL) to prevent tubulointerstitial damage and nephron loss. Such patients represent approximately one-quarter of adults over the age of 45 years and two-thirds of patients with stage 3 CKD (37, 38). Besides phosphate restriction, agents such as bisphosphonates that inhibit the formation of calcium phosphate crystals may be useful for the treatment of this patient population (Figure 2, Figure 6J, and Supplemental Figure 2).
Calcium phosphate particles in the tubule fluid are likely not only composed of calcium phosphate crystals but also of serum protein fetuin-A, because fetuin-A is present in the tubule fluid (39) and has a high capacity to adsorb calcium phosphate crystals (40). Thus far, fetuin-A molecules laden with solid-phase calcium phosphate have been reported in the blood and designated as calciprotein particles (41, 42). However, calciprotein particles in the blood are too large to be filtrated through glomeruli and unlikely to be the source of calcium phosphate particles in the tubule fluid. Therefore, we speculate that calcium phosphate particles in the tubule fluid are generated in situ in the tubule lumen independently of circulating calciprotein particles. Indeed, calciprotein particles in the blood are increased with serum phosphate levels (31), whereas the formation of particles in the tubular fluid is probably dependent on the ePTFp, which is independent of serum phosphate levels, as shown in Equation 1.
Regarding the mechanism by which the bone senses the necessity for increasing the per-nephron excretion of phosphate and induces FGF23 secretion and production, we speculate that an increase in the phosphate load excreted per nephron may enhance and/or prolong postprandial increases in blood levels of phosphate and calciprotein particles, which induces FGF23 expression and secretion in osteoblasts/osteocytes (43). This hypothesis remains to be confirmed.
We showed that bicarbonate treatment exacerbated the kidney damage induced by dietary phosphate loading in mice (Supplemental Figure 4). On the other hand, bicarbonate has been widely prescribed for patients with CKD who have metabolic acidosis and has been shown to decelerate eGFR decline and reduce urinary albumin excretion (44). Although the mechanism of kidney damage induced by metabolic acidosis remains elusive, it has been postulated that adaptive responses that increase acid excretion, including activation of ammoniagenesis and the renin-angiotensin-aldosterone system, may eventually induce inflammation and tubulointerstitial damage (45). We speculate that a net effect of bicarbonate on renal therapy outcomes in patients with CKD may be determined by a balance between the advantage obtained by correcting metabolic acidosis and the disadvantage caused by the urine alkalization that facilitates the formation of calcium phosphate crystals in the tubule fluid. Indeed, some recent randomized, controlled studies failed to show the benefits of bicarbonate treatment (46–48), raising the possibility that the disadvantage could dominate the advantage in some patients with CKD who have a high phosphate excretion load per nephron or high serum FGF23 levels.
Equation 2 was derived solely from the regression line between FGF23 levels and phosphate excretion per nephron (Figure 7A) and thus does not take into account the concentration of ions other than phosphate, such as citrate and magnesium, which potentially affect the formation of calcium phosphate crystals. Nonetheless, this equation may be useful for monitoring changes in the nephron number by simple blood and urine tests. It should be noted that the nephron number calculated using Equation 2 does not represent the actual nephron number, but a “virtual” nephron number, assuming that all nephrons had the same single-nephron GFR (snGFR). Actually, all nephrons may not have an equivalent snGFR. The hyperfiltrating nephrons with a high snGFR should have a greater phosphate load and thus more severe tubule damage than nephrons with a lower snGFR. Indeed, the fibrotic area in the kidneys of mice fed a high-phosphate diet had a nonuniform, segmented distribution along the radial tracts of the nephrons (Figure 3D).
Because the number of nephrons is highly variable from individual to individual and from species to species, the phosphate load necessary for inducing calcium phosphate precipitation in the tubule fluid also varies from individual to individual and species to species. As discussed above, the amount of phosphate excretion per nephron in young adults and older adults is approximately 0.5 μg/d and 1.0 g/d, respectively. By contrast, phosphate excretion per nephron in mice fed a regular diet was approximately 0.03 g/d (Figure 7A), which is more than one order of magnitude less than that in humans. Hence, dietary phosphate overload is necessary in mice to trigger the formation of calcium phosphate crystals in the tubule fluid. Considering our body size, humans have fewer nephrons than do mice (approximately 30 vs. 700 per gram BW) and are thus prone to phosphate-induced kidney damage.
Methods
Cell culture.
The human renal proximal tubule cell line HK-2 was obtained from the American Type Culture Collection (ATCC, CRL-2190) and cultured in DMEM (Nacalai Tesque) supplemented with 5% FBS (Biological Industry), 50 g/mL penicillin, and 50 μg/mL streptomycin (Gibco, Thermo Fisher Scientific). Calcium and phosphate concentrations were increased to the desired final concentrations by adding 1 M CaCl2 and 1 M phosphate buffer to the medium. HK-2 cell viability was quantified using the MTT assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay, Promega) or the CCK-8 assay (CK12, Dojindo Molecular Technologies) according to the manufacturer’s instructions. HK-2 cell damage was quantified using a lactate dehydrogenase assay (CytoTox96 Non-Radioactive Cytotoxicity Assay, Promega). For inhibition of TLR4 signaling, 2 mM TAK242 (Merck Millipore, 614316) stock solution in DMSO was added to cells at each time point (final concentration: 2 μM).
Synthesis of calcium phosphate particles.
Calcium phosphate particles were synthesized in DMEM containing 0.1%~10% FBS by inoculating 1 M CaCl2 and 1 M phosphate buffer (mixture of 1 M NaH2PO4 and 1 M Na2HPO4, pH 7.4) to increase calcium and phosphate concentrations to 3 mM and 3~7 mM, respectively. The mixture was incubated at 37°C for 16~24 hours with gentle rocking and then centrifuged at 16,000g for 2 hours. After removal of the supernatant, the precipitated calcium phosphate particles were suspended with regular DMEM (1.8 mM calcium, 0.9 mM phosphate) containing 5% FBS. The concentration of calcium phosphate particles is indicated as the phosphorus content (μg phosphorus/mL) determined using the ICP-MS Nexion 2000 (PerkinElmer) as previously described (43). For fluorescence labeling of calcium phosphate particles, 5(6)-RhR-dRIS (final concentration: 25 nM) (BioVinc) was added and incubated for 10~60 minutes at room temperature.
Measurement of calcium phosphate particle size.
The particle size distribution of calcium phosphate particles in the medium was determined by nanoparticle tracking analysis (NTA) using the NanoSight NS300 (Malvern Panalytical). A bisphosphonate (alendronate) conjugated with FITC was synthesized by amine coupling between FITC-carboxylic acid and the primary amine in alendronate. The FITC-alendronate was purified by HPLC and then added to DMEM containing 5% FBS, 3 mM calcium, and 7 mM phosphate, at a final concentration of 1 μM. After incubation at 25°C for 24 hours, the mixture was subjected to NTA using a 488 nm laser and a 500 nm long-pass filter to visualize calcium phosphate particles labeled with FITC-alendronate.
Animals.
WT C57BL/6 mice, mice lacking the Tlr4 gene (B6.B10ScN-Tlr4lps-del/JthJ), Tlr4-floxed mice (B6.Cg-Tlr4tm1.Karp/J), Myd88-deficient mice [B6.129P2(SJL)-Myd88tm1.1Defr/J], and Ksp-Cre mice [B6.Cg-Tg(Cdh-Cre)91Igr/J] were obtained from The Jackson Laboratory. Mice were subjected to either right uninephrectomy or sham operation (laparotomy alone) at 8 weeks of age. After a 4-week recovery period, the mice were placed on a diet containing 0.35%, 1.0%, 1.5%, or 2.0% inorganic phosphate for up to 12 weeks and then transferred individually to metabolic cages. After urine collection and measurement of food and water consumption for 3 days, the mice were sacrificed to harvest their blood and kidneys. A part of the kidney was fixed in buffered formalin for histological examination. The rest of the kidney was snap-frozen in liquid nitrogen for RNA extraction. For the bisphosphonate treatment, 4-week-old WT C57BL6 male mice were placed on a diet containing 2.0% inorganic phosphate and injected s.c. with alendronate (10 mg/kg), etidronate (100 mg/kg), or vehicle (saline) every other day for 4 weeks. For urine acidification, 4-week-old WT C57BL6 male mice were placed on a diet containing 2.0% inorganic phosphate and given 0.14 M NH4Cl solution or tap water as drinking water. For urine alkalization, 8-week-old WT C57BL/6 male mice were placed on a diet containing 2.0% inorganic phosphate and given 0.15 M NaHCO3 solution.
Ex vivo imaging of the kidney.
Mice lacking the Tlr4 gene and WT C57BL/6J mice at 8 weeks of age were fed a chlorophyll-free, normal phosphate diet (0.35% inorganic phosphate) for 7 days and subsequently switched to a chlorophyll-free, high-phosphate diet (2.0%) for 8–9 days for ex vivo imaging of the kidney. Some of the mice were treated with alendronate (10 mg/kg, every other day) by s.c. injection or with ammonium chloride (0.14 M, for the last 10 days) in the drinking water. After anesthetization by i.p. injection of urethane (1.5 g/kg), the mice were injected with OsteoSense 680EX (PerkinElmer, 50 nmol/kg), FITC-dextran (MW 150kD, 80 mg/kg), and Hoechst 33342 (20 mg/kg) into the right jugular vein to visualize calcium phosphate crystals, the interstitial space, and cell nuclei, respectively. To visualize the apical membrane of proximal tubules, FITC-labeled LTL was used instead of FITC-dextran. Thirty minutes after the injection, the kidneys were excised and cut in half coronally. The cut surface of the kidney was attached to a glass-bottomed dish (MatTek) without washing or fixation, and the dish was placed onto a Piezo drive stage (Tokai Hit, Nikon) of an inverted microscope (Eclipse Ti, Nikon). The tissue was excited at wavelengths of 405, 488, and 561 nm, and XY or XYZ images were captured using an A1R system (Nikon) in the galvano scanning mode. Nikon 2× (NA 0.10, dry), 10× (NA 0.45, dry), and 40× (NA 1.15, water immersion) objective lenses were used. The images were converted into 8-bit RGB TIFF files using NIS Elements software (Nikon), and the number of calcium phosphate particles was determined by counting the number of OsteoSense-positive particles using the “Analyze Particle” algorithm in ImageJ (NIH). OsteoSense-positive particles in the ureter were excluded.
Electron microscopic studies.
After euthanization, kidney tissues from mice fed a high-phosphate diet for 10 days were collected, cut into small pieces in Krebs-Ringer HEPES buffer, and fixed with high-pressure freezing using the EM ICE solution (Leica). The tissues were freeze-substituted in acetone containing 2% OsO4 (Nissin EM) using EM AFM2 (Leica). In the freeze substitution, the samples were kept at –80°C for 48 hours, –35°C for 6 hours, 0°C for 1 hour, and room temperature for 2 hours, and the temperature was raised at the speed of 8.2°C/h and 10°C/h when the temperature was raised to –35°C and 0°C, respectively. The substitution medium was replaced by pure acetone after the incubation at room temperature, and the samples were infiltrated with and embedded in epoxy resin. After curing, the samples in the resin were sectioned at 500 nm thickness. Some of the serial sections were stained with toluidine blue and imaged with a light microscope (AX80, Olympus). Others of the serial sections were mounted on pieces of conductive carbon nanotube tape (49), and the sections on the tape were imaged with a scanning electron microscope (Scios2, Thermo Fisher Scientific) equipped with a retractable annular backscattered electron detector and an EDS detector (Ultim Max 170, Oxford Instruments). The secondary electron and backscatter electron images were acquired at accelerating voltage 5 kV and probe current 0.4 nA, and EDS images were acquired at accelerating voltage 10 kV and probe current 0.8 nA.
Histological and immunohistochemical analysis.
The mouse kidneys fixed with 10% buffered formalin were processed to make regular paraffin sections. The kidney sections were stained with Picrosirius red to evaluate fibrosis. Fibrotic lesions were quantified as described previously (11). In addition, the kidney sections were subjected to immunohistochemical analysis using an antibody against TLR4 (Santa Cruz Biotechnology) to determine its localization.
Quantitative RT-PCR.
Quantitative RT-PCR was performed as previously described (43). The nucleotide sequences of the primers were as follows: α-SMA, 5′-CTGACAGAGGCACCACTGAA-3′ and 5′-CATCTCCAGAGTCCAGCACA-3′; collagen 1α1, 5′-GAGCGGAGAGTACTGGATCG-3′ and 5′-GTTCGGGCTGATGTACCAGT-3′; IL-6, 5′-CTGCAAGAGACTTCCATCCAGTT-3′ and 5′-AAGTAGGGAAGGCCGTGGTT-3′; MCP1, 5′-GGCTCAGCCAGATGCAGTTAAC-3′ and 5′-GCCTACTCATTGGGATCATCTTG-3′; MMP2, 5′-ACCCTGGGAGAAGGACAAGT-3′and 5′-ATCACTGCGACCAGTGTCTG-3′; vimentin, 5′-CTGCACGATGAAGAGATCCA-3′ and 5′-AGCCACGCTTTCATACTGCT-3′; TGF-β1, 5′-TTGCTTCAGCTCCACAGAGA-3′ and 5′-TGGTTGTAGAGGGCAAGGAC-3′; OPN, 5′-TCCAAAGAGAGCCAGGAGAG-3′ and 5′-GGCTTTGGAACTTGCTTGAC-3′; Kim1, 5′-CTGGAATGGCACTGTGACATCC-3′ and 5′-GCAGATGCCAACATAGAAGCCC-3′; Nlrp3, 5′-GAAATATGCACAGGTATCCT-3′ and 5′-GTAATTTTGAAGTATTGCTTGTTT-3′; TNF-α, 5′-CGAGACCTCTGGGAAAAAGCT-3′ and 5′-CGAGACCTCTGGGAAAAAGCT-3′; cyclophilin, 5′-TGGAGAGCACCAAGACAGACA-3′ and 5′-TGCCGGAGTCGACAATGAT-3′.
Microarray experiment.
Microarray analysis was performed using the SurePrint G3 Human Gene Expression 8 × 60K v3 microarray (Macrogen). Cy3-labeled cRNA was prepared from 1~5 μg total RNA using the Quick Amp Labeling Kit (Agilent Technologies). The labeled cRNA (1.65 μg) was hybridized with the Agilent expression microarray according to the protocols provided by the manufacturer. Three time points (3 hours, 6 hours, and 24 hours) after stimulation with or without synthesized calcium-phosphate particles (10 μg phosphorus/mL) were tested in 4 replications for each time point. Arrays were scanned using the G4900DA SG12494263 (Agilent Technologies). Data processing and analysis were performed using Feature Extraction software, version 11.0.1.1 (Agilent Technologies). An enrichment test based on the KEGG Pathway (https://www.kegg.jp/kegg/pathway.html) was conducted with a significant gene list. Raw P values were calculated by modified Fisher’s exact and Bonferroni’s tests. The data presented here have been deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE135977; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135977).
SPR.
Physical interaction between calcium phosphate particles and TLR4 was determined by SPR using the ProteOn XPR36 system (Bio-Rad). Alendronate was immobilized on a sensor chip (ProteOn GLC Sensor Chip, Bio-Rad) by amine coupling. Synthesized calcium-phosphate particles were applied on the chip in the running buffer (20 mM HEPES, pH 7.4, 135 mM NaCl, 1 mM Na2HPO4, 2 mM CaCl2, 0.005% Tween-20) and immobilized on the chip. After washing with the running buffer, the extracellular domain of human TLR4 (R&D Systems, Bio-Techne) was applied. Kinetics analysis was performed using the Langmuir model.
Indirect immunofluorescence.
HK-2 cells were seeded in each well of 96-well plates (655090, Greiner Bio-One). Twenty-four hours after seeding, the cells were treated with or without synthesized calcium-phosphate particles (10 μg phosphorus/mL) for the indicated durations. The cells were fixed with 4% paraformaldehyde in 10 mM PBS for 20 minutes at room temperature. The fixed cells were permeabilized with 0.2% Triton X-100 in PBS for 3 minutes for staining with primary antibodies against APPL1 (3858, Cell Signaling Technology [CST]), Rab5 (3547, CST), Rab7 (9367, CST), Rab11 (5589, CST), EEA1 (3288, CST), Lamp2 (H4B4, Hybridoma Bank), GM130 (610823, BD Transduction), p–p38-MAPK (4511, CST), and p–NF-κB (ab86299, Abcam). For staining with LC3 primary antibody (M152-3, MBL International), the cells were permeabilized with 50 μg/mL digitonin in PBS for 5 minutes at room temperature. For staining with the CD63 primary antibody (Abcam, ab37149), the cells were permeabilized with 0.02% saponin for 1 minute at room temperature. After washing with PBS, the cells were soaked in blocking solution (PBS containing 1% BSA) for 30 minutes followed by incubation with primary antibodies in blocking solution overnight at 4°C. The cells were then washed with PBS and incubated with an Alexa Fluor 488 or Alexa Fluor 647 secondary antibody (Thermo Fisher Scientific) in blocking solution for 1 hour at room temperature. For staining with the OPN primary antibody (ab8448, Abcam), the cells were permeabilized and blocked with PBS containing 1% BSA, 10% normal goat serum, 0.3 M glycine, and 0.05% Tween-20 for 1 hour at room temperature. Nuclei, cell membranes, and cytoskeletons for image analysis were stained with Hoechst 33342 (Thermo Fisher Scientific), CellMask Deep Red (Thermo Fisher Scientific), and β-tubulin (T8328, MilliporeSigma), respectively. After washing with PBS, confocal images were acquired using a confocal laser-scanning microscope (Nikon A1).
Image processing.
Confocal images were acquired at a resolution of 1024 × 1024 pixels and a Z-step resolution of 0.65 μm with a Nikon CFI Plan Apo λ 40× objective (NA = 0.95). The images of endosome marker proteins and fluorescently labeled calcium-phosphate particles were acquired using a galvano scanner (average of 4 images). The images of tubulin, Hoechst, CellMask, and signaling proteins were acquired without the averaging step. Images of 96-well plates were acquired in the JOBS mode, which can run fully automated procedure macros. The images of each well were obtained for 10 different areas.
Image analysis.
All confocal images were analyzed using NIS-Elements AR 4.5 microscope imaging software (Nikon). Maximum intensity projection was performed before the image analysis. Hoechst signals were used to measure the area of the nucleus. For detection of the cell shape, the signals of the tubulin antibody or CellMask were used. To determine subcellular localization of endosomes, 2 areas in the cell were extracted (Figure 5E). Area 1 was defined as the perinuclear area within 10 pixels of the nucleus boundary; area 2 was defined as the submembrane area within 12 pixels of the plasma membrane. To calculate the mean intensity of each signal, the sum intensity in area 1 or area 2 was divided by the sum area of area 1 or area 2, respectively.
ELISA.
OPN levels in the conditioned medium were measured using the Quantikine ELISA Kit (DOST00, R&D Systems, Bio-Techne). HK-2 cells were seeded into 6-well plates and stimulated with synthesized calcium-phosphate particles (10 g phosphorus/mL) or vehicle (DMEM containing 5% FBS, 3 mM CaCl2, and 1 mM phosphate buffer). After incubation for the indicated durations, the supernatant from each well was collected, centrifuged at 13,500g for 10 minutes at 25°C, and subjected to the ELISA. Serum FGF23 levels in mice and humans were measured using the intact FGF23 ELISA (Kinos) according to the manufacturers’ protocols. The FGF23 assay is reported to have a lower detection limit of less than 3 pg/mL, with an interassay coefficient of variation of 2.1% at 42.4 pg/mL and 3.8 % at 19.5 pg/mL. The intra-assay coefficient of variation is 2.8 % at 28.7 pg/mL and 2.0% at 33.6 pg/mL. The upper reference limit is defined as 53 pg/mL, corresponding to the cutoff value determined by the cross-sectional study.
Blood and urine tests.
Serum creatinine, urine creatinine, and urine phosphate in mice were quantified using Fuji Dri-Chem NX500V slides and the analyzer. Serum creatinine, urine creatinine, urine L-FABP, and urine β2-microglobulin levels in humans were measured as a part of standard patient care at the Jichi Medical University laboratory (Tochigi, Japan).
Clinical studies.
The cross-sectional study was performed using urine and serum samples obtained from 198 pre-dialysis CKD patients recruited among outpatients at the Nephrology Department of the Jichi Medical University Hospital between July 2014 and August 2017 (31). All study participants were Japanese: 69 were women and 129 men. Fourteen of the patients had stage 1 CKD (eGFR ≥ 90 mL/min/1.73 m2); 37 had stage 2 CKD (90 > eGFR ≥ 60); 84 had stage 3 CKD (60 > eGFR ≥ 30); 34 had stage 4 CKD (30 > eGFR ≥ 15); and 29 had stage 5 CKD (eGFR < 15). The prospective study was performed using serum samples from the EMPATHY study, which was registered with the University Hospital Medical Information clinical trials registry (UMIN000003486). In the EMPATHY study, a total of 5107 patients with hyperlipidemia and diabetic retinopathy were randomized to either intensive or standard lipid-lowering therapy. The baseline characteristics of the participants were described previously (32). Serum samples from 5039 patients were available for the measurement of FGF23 at baseline. The outcome was the renal event defined as the initiation of chronic dialysis or an increase in serum creatinine levels by at least 2-fold (and >1.5 mg/dL).
Statistics.
For statistical analysis of immunofluorescence images, the R lawstat package software was used. First, a normality test and an F test were performed on all imaging data. As all data did not show normal distribution or equal variance, a nonparametric Brunner-Munzel test was performed to obtain a P value and effect size according to a large sample size. For statistical analysis of the prospective clinical study, the cumulative endpoint was derived by the Kaplan-Meier method, and the groups were compared with a log-rank test. Cox proportional hazards regression was performed to identify associations with the endpoint. In multivariable analysis, potential confounders were adjusted by selecting for age, sex, BMI, and serum creatinine level. Statistical analyses were performed using R software, version 3.6.0. P values of less than 0.05 were considered statistically significant. Statistical analysis of the other data was performed using GraphPad Prism, version 8.4.3 (GraphPad Software). Student’s t test was 2 sided.
Study approval.
All animal experiments were approved by the IACUC of Jichi Medical University. All clinical studies were conducted in accordance with Declaration of Helsinki principles. The clinical study protocols were approved by the ethics committee of Jichi Medical University. Written informed consent was obtained from all study participants.
Author contributions
K. Shiizaki, YM, M. Miura, YI, HH, and HK conducted animal and tissue culture experiments. AT, RK, and M. Masutani conducted tissue culture experiments and image analysis. K. Seo, TK, and HH conducted ex vivo imaging. BB, NO, and TH performed electron microscopic studies. K. Shiizaki, YI, KN, K. Kario, K. Kotani, TY, DN, IK, and HI performed clinical studies. HK, M. Murata, and MK planned the experiments. AT, K. Seo, and MK wrote the manuscript. All authors reviewed, edited, and approved the manuscript. The order of the co–first authors, K. Shiizaki and AT, was determined on the basis of their relative contributions to the in vivo and in vitro experiments. The corresponding author judged that the in vivo and in vitro experiments were equally important to support the main conclusion of this manuscript.
Supplementary Material
Acknowledgments
The authors thank Yukinari Ohsaka, Yuko Shimizu, and Taeko Yamauchi (Division of Anti-aging Medicine, Jichi Medical University), Tom Kouki and Megumi Yatabe (Department of Anatomy, Jichi Medical University), and Yuka Hara (National Institute for Materials Science) for their technical assistance; Kyoko Nakamura (Division of Anti-aging Medicine, Jichi Medical University) for administrative assistance; and Satoshi Nishimura (Division of Anti-aging Medicine, Jichi Medical University) for help with ex vivo imaging. We also thank Kishiko Osaka and Naomi Okamoto (University of Tokyo) for assistance with experiments; Orson Moe, Ming-Chang Hu, Johanne Pastor, and Charles Pak (University of Texas Southwestern Medical Center) for helpful discussions. This work was supported in part by funding from the NIH (R01AG19712 and R01 DK091392, to MK); Japan Agency for Medical Research and Development-Core Research for Evolutional Science and Technology (JP19gm0610012, to MK); the Japan Society for the Promotion of Science KAKENHI (JP16K08941, to YI, JP15K09269, to K Shiizaki, and JP20K21506, to NO); a Grant-in-Aid for Scientific Research on Innovative Areas (17H05870, to M. Murata); the Project for Cancer Research and Therapeutic Evolution (19cm0106110h0004, to M. Murata); the JST-Mirai Program (JPMJMI19G5, to M. Murata); and Bayer Japan (to MK).
Version 1. 06/29/2021
In-Press Preview
Version 2. 08/16/2021
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(16):e145693.https://doi.org/10.1172/JCI145693.
See the related Commentary at null.
Contributor Information
Kazuhiro Shiizaki, Email: shiizaki@yurina-mp.com.
Asako Tsubouchi, Email: asakotsubo@gmail.com.
Yutaka Miura, Email: miura.y@jichi.ac.jp.
Kinya Seo, Email: seokin54@gmail.com.
Takahiro Kuchimaru, Email: kuchimaru@jichi.ac.jp.
Hirosaka Hayashi, Email: hirosakah@jichi.ac.jp.
Yoshitaka Iwazu, Email: iwazu@jichi.ac.jp.
Marina Miura, Email: wildcat.strawberry@gmail.com.
Batpurev Battulga, Email: d2031@jichi.ac.jp.
Nobuhiko Ohno, Email: nohno@jichi.ac.jp.
Toru Hara, Email: HARA.Toru@nims.go.jp.
Rina Kunishige, Email: r-kunishige@g.ecc.u-tokyo.ac.jp.
Mamiko Masutani, Email: Mamiko.Masutani@nikon.com.
Keita Negishi, Email: k-negishi@jichi.ac.jp.
Kazuomi Kario, Email: kkario@jichi.ac.jp.
Kazuhiko Kotani, Email: kazukotani@jichi.ac.jp.
Toshiyuki Yamada, Email: yamadanji@jichi.ac.jp.
Daisuke Nagata, Email: nagatad@jichi.ac.jp.
Issei Komuro, Email: komuro_tky2000@yahoo.co.jp.
Hiroshi Itoh, Email: hiito@keio.jp.
Hiroshi Kurosu, Email: kurosu23@jichi.ac.jp.
Masayuki Murata, Email: mmurata@bio.c.u-tokyo.ac.jp.
Makoto Kuro-o, Email: mkuroo@jichi.ac.jp.
References
- 1.Webster AC, et al. Chronic kidney disease. Lancet. 2017;389(10075):1238–1252. doi: 10.1016/S0140-6736(16)32064-5. [DOI] [PubMed] [Google Scholar]
- 2.Hill NR, et al. Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS One. 2016;11(7):e0158765. doi: 10.1371/journal.pone.0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levey AS, et al. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2005;67(6):2089–2100. doi: 10.1111/j.1523-1755.2005.00365.x. [DOI] [PubMed] [Google Scholar]
- 4.Levin A, et al. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet. 2017;390(10105):1888–1917. doi: 10.1016/S0140-6736(17)30788-2. [DOI] [PubMed] [Google Scholar]
- 5.Ganesh SK, et al. Association of elevated serum PO(4), Ca x PO(4) product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J Am Soc Nephrol. 2001;12(10):2131–2138. doi: 10.1681/ASN.V12102131. [DOI] [PubMed] [Google Scholar]
- 6.Qunibi WY, et al. Cardiovascular calcification in patients with end-stage renal disease: a century-old phenomenon. Kidney Int Suppl. 2002;(82):73–80. doi: 10.1046/j.1523-1755.62.s82.15.. [DOI] [PubMed] [Google Scholar]
- 7.Levin A, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71(1):31–38. doi: 10.1038/sj.ki.5002009. [DOI] [PubMed] [Google Scholar]
- 8.Haut LL, et al. Renal toxicity of phosphate in rats. Kidney Int. 1980;17(6):722–731. doi: 10.1038/ki.1980.85. [DOI] [PubMed] [Google Scholar]
- 9.Hommos MS, et al. Structural and functional changes in human kidneys with healthy aging. J Am Soc Nephrol. 2017;28(10):2838–2844. doi: 10.1681/ASN.2017040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Sullivan ED, et al. Renal aging: causes and consequences. J Am Soc Nephrol. 2017;28(2):407–420. doi: 10.1681/ASN.2015121308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirano Y, et al. Interleukin-36α as a potential biomarker for renal tubular damage induced by dietary phosphate load. FEBS Open Bio. 2020;10(5):894–903. doi: 10.1002/2211-5463.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ori Y, et al. Acute phosphate nephropathy-an emerging threat. Am J Med Sci. 2008;336(4):309–314. doi: 10.1097/MAJ.0b013e318167410c. [DOI] [PubMed] [Google Scholar]
- 13.Lien YH. Is bowel preparation before colonoscopy a risky business for the kidney? Nat Clin Pract Nephrol. 2008;4(11):606–614. doi: 10.1038/ncpneph0939. [DOI] [PubMed] [Google Scholar]
- 14.Kuro-o M. The Klotho proteins in health and disease. Nat Rev Nephrol. 2019;15(1):27–44. doi: 10.1038/s41581-018-0078-3. [DOI] [PubMed] [Google Scholar]
- 15.Shimada T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichikawa S, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117(9):2692–2701. doi: 10.1172/JCI29134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuro-o M, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 18.Benet-Pages A, et al. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14(3):385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 19.Shimada T, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19(3):429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 20.Bank N, et al. A micropuncture study of renal phosphate transport in rats with chronic renal failure and secondary hyperparathyroidism. J Clin Invest. 1978;61(4):884–894. doi: 10.1172/JCI109014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebetino FH, et al. The relationship between the chemistry and biological activity of the bisphosphonates. Bone. 2011;49(1):20–33. doi: 10.1016/j.bone.2011.03.774. [DOI] [PubMed] [Google Scholar]
- 22.Zaheer A, et al. In vivo near-infrared fluorescence imaging of osteoblastic activity. Nat Biotechnol. 2001;19(12):1148–1154. doi: 10.1038/nbt1201-1148. [DOI] [PubMed] [Google Scholar]
- 23.El-Achkar TM, et al. Tamm-Horsfall protein protects the kidney from ischemic injury by decreasing inflammation and altering TLR4 expression. Am J Physiol Renal Physiol. 2008;295(2):F534–F544. doi: 10.1152/ajprenal.00083.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pal D, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18(8):1279–1285. doi: 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- 25.Giachelli CM, Steitz S. Osteopontin: a versatile regulator of inflammation and biomineralization. Matrix Biol. 2000;19(7):615–622. doi: 10.1016/S0945-053X(00)00108-6. [DOI] [PubMed] [Google Scholar]
- 26.Giachelli CM, et al. Osteopontin expression in angiotensin II-induced tubulointerstitial nephritis. Kidney Int. 1994;45(2):515–524. doi: 10.1038/ki.1994.67. [DOI] [PubMed] [Google Scholar]
- 27.Korolchuk VI, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13(4):453–460. doi: 10.1038/ncb2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baum M, Quigley R. Proximal tubule water transport-lessons from aquaporin knockout mice. Am J Physiol Renal Physiol. 2005;289(6):F1193–F1194. doi: 10.1152/ajprenal.00283.2005. [DOI] [PubMed] [Google Scholar]
- 29.Blaine J, et al. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol. 2015;10(7):1257–1272. doi: 10.2215/CJN.09750913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cullen-McEwen LA, et al. Nephron number, renal function, and arterial pressure in aged GDNF heterozygous mice. Hypertension. 2003;41(2):335–340. doi: 10.1161/01.HYP.0000050961.70182.56. [DOI] [PubMed] [Google Scholar]
- 31.Miura Y, et al. Identification and quantification of plasma calciprotein particles with distinct physical properties in patients with chronic kidney disease. Sci Rep. 2018;8(1):1256. doi: 10.1038/s41598-018-19677-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ueshima K, et al. Rationale and design of the standard versus intensive statin therapy for hypercholesterolemic patients with diabetic retinopathy (EMPATHY) study: a randomized controlled trial. J Atheroscler Thromb. 2016;23(8):976–990. doi: 10.5551/jat.33563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawasaki T, et al. Urinary phosphorus excretion per creatinine clearance as a prognostic marker for progression of chronic kidney disease: a retrospective cohort study. BMC Nephrol. 2015;16:116. doi: 10.1186/s12882-015-0118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santamaria R, et al. Increased phosphaturia accelerates the decline in renal function: a search for mechanisms. Sci Rep. 2018;8(1):13701. doi: 10.1038/s41598-018-32065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mathewson AM, et al. Dietary phosphate assessment in dialysis patients. J Ren Nutr. 2010;20(6):351–358. doi: 10.1053/j.jrn.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 36.Denic A, et al. The substantial loss of nephrons in healthy human kidneys with aging. J Am Soc Nephrol. 2017;28(1):313–320. doi: 10.1681/ASN.2016020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devaraj S, et al. Evaluation of a method for fibroblast growth factor-23: a novel biomarker of adverse outcomes in patients with renal disease. Metab Syndr Relat Disord. 2010;8(6):477–482. doi: 10.1089/met.2010.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Isakova T, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370–1378. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsui I, et al. Retention of fetuin-A in renal tubular lumen protects the kidney from nephrocalcinosis in rats. Am J Physiol Renal Physiol. 2013;304(6):F751–F760. doi: 10.1152/ajprenal.00329.2012. [DOI] [PubMed] [Google Scholar]
- 40.Heiss A, et al. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J Biol Chem. 2003;278(15):13333–13341. doi: 10.1074/jbc.M210868200. [DOI] [PubMed] [Google Scholar]
- 41.Hamano T, et al. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol. 2010;21(11):1998–2007. doi: 10.1681/ASN.2009090944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith ER, et al. Phosphorylated fetuin-A-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol Dial Transplant. 2012;27(5):1957–1966. doi: 10.1093/ndt/gfr609. [DOI] [PubMed] [Google Scholar]
- 43.Akiyama K, et al. Calciprotein particles regulate fibroblast growth factor-23 expression in osteoblasts. Kidney Int. 2019;97(4):702–712. doi: 10.1016/j.kint.2019.10.019. [DOI] [PubMed] [Google Scholar]
- 44.Navaneethan SD, et al. Effects of treatment of metabolic acidosis in CKD: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2019;14(7):1011–1020. doi: 10.2215/CJN.13091118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wesson DE, et al. Mechanisms of metabolic acidosis-induced kidney injury in chronic kidney disease. J Am Soc Nephrol. 2020;31(3):469–482. doi: 10.1681/ASN.2019070677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witham MD, et al. Clinical and cost-effectiveness of oral sodium bicarbonate therapy for older patients with chronic kidney disease and low-grade acidosis (BiCARB): a pragmatic randomised, double-blind, placebo-controlled trial. BMC Med. 2020;18(1):91. doi: 10.1186/s12916-020-01542-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melamed ML, et al. Effects of sodium bicarbonate in CKD stages 3 and 4: a randomized, placebo-controlled, multicenter clinical trial. Am J Kidney Dis. 2020;75(2):225–234. doi: 10.1053/j.ajkd.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raphael KL, et al. A randomized trial comparing the safety, adherence, and pharmacodynamics profiles of two doses of sodium bicarbonate in CKD: the BASE pilot trial. J Am Soc Nephrol. 2020;31(1):161–174. doi: 10.1681/ASN.2019030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kubota Y, et al. A carbon nanotube tape for serial-section electron microscopy of brain ultrastructure. Nat Commun. 2018;9(1):437. doi: 10.1038/s41467-017-02768-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.