

Abstract
Background
T cells control viral infection, promote vaccine durability, and in coronavirus disease 2019 (COVID-19) associate with mild disease. We investigated whether prior measles-mumps-rubella (MMR) or tetanus-diphtheria-pertussis (Tdap) vaccination elicits cross-reactive T cells that mitigate COVID-19.
Methods
Antigen-presenting cells (APC) loaded ex vivo with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), MMR, or Tdap antigens and autologous T cells from COVID-19-convalescent participants, uninfected individuals, and COVID-19 mRNA-vaccinated donors were co-cultured. T cell activation and phenotype were detected by interferon-γ (IFN-γ) enzyme-linked immunospot (ELISpot) assays and flow cytometry. ELISAs (enzyme-linked immunosorbant assays) and validation studies identified the APC-derived cytokine(s) driving T cell activation. TCR clonotyping and single-cell RNA sequencing (scRNA-seq) identified cross-reactive T cells and their transcriptional profile. A propensity-weighted analysis of COVID-19 patients estimated the effects of MMR and Tdap vaccination on COVID-19 outcomes.
Findings
High correlation was observed between T cell responses to SARS-CoV-2 (spike-S1 and nucleocapsid) and MMR and Tdap proteins in COVID-19-convalescent and -vaccinated individuals. The overlapping T cell population contained an effector memory T cell subset (effector memory re-expressing CD45RA on T cells [TEMRA]) implicated in protective, anti-viral immunity, and their detection required APC-derived IL-15, known to sensitize T cells to activation. Cross-reactive TCR repertoires detected in antigen-experienced T cells recognizing SARS-CoV-2, MMR, and Tdap epitopes had TEMRA features. Indices of disease severity were reduced in MMR- or Tdap-vaccinated individuals by 32%–38% and 20%–23%, respectively, among COVID-19 patients.
Conclusions
Tdap and MMR memory T cells reactivated by SARS-CoV-2 may provide protection against severe COVID-19.
Funding
This study was supported by a National Institutes of Health (R01HL065095, R01AI152522, R01NS097719) donation from Barbara and Amos Hostetter and the Chleck Foundation.
Keywords: SARS-CoV-2, heterologous immunity, vaccines, antigens, antibody titers, memory T cells, TCR repertoire, transcriptome, antigen presenting cells, risk prediction
Graphical abstract
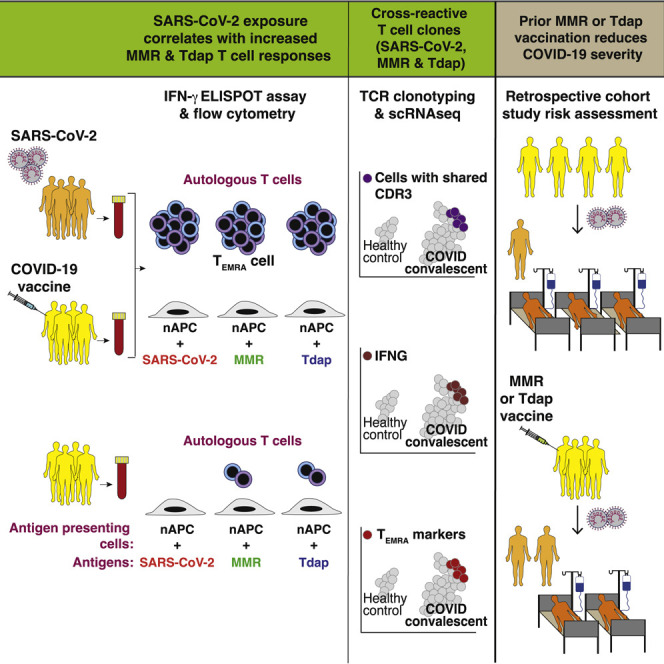
Context and significance
A diverse T cell response controls viral infection. Thus, a major goal of vaccines is the induction of strong and durable T cell memory. Reactivation of memory T cells generated against a different microbe may enhance immunity to novel pathogens. Here, the authors show that pre-existing memory T cells, specific for antigens in previously administered MMR (measles-mumps-rubella) and Tdap (tetanus-diphtheria-pertussis) vaccines, are reactivated by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) antigens following coronavirus disease 2019 (COVID-19) or COVID-19 vaccination and have the phenotype of a memory T cell subset implicated in anti-viral immunity. The analysis of a large and well-characterized COVID-19 patient cohort revealed that prior MMR or Tdap vaccination associates with reduced disease severity and death. Thus, MMR or Tdap vaccination may protect against severe COVID-19.
T cells critically control infection and effective vaccination. In COVID-19-convalescent or COVID-19-vaccinated individuals, memory T cells previously generated by MMR or Tdap vaccines are reactivated by SARS-CoV-2 antigens and have features of TEMRA, implicated in anti-viral immunity. Prior MMR or Tdap vaccination may protect against severe COVID-19.
Introduction
A diverse T cell response is essential for early control of acute viral infection and for the generation of B cells producing protective antibodies. CD4+ T helper (Th) cells induce B cells to produce high-affinity antibodies to viral protein antigens. Effector CD8+ T cells (cytotoxic T lymphocytes [CTLs]) and CD4+ T cells eradicate infected, virus-producing cells via direct killing or by secreting cytokines, such as interferon-γ (IFN-γ), which enhances inflammatory functions that support viral clearance. Antigen-presenting cells (APCs), such as classical dendritic cells (DC), play a critical role in initiating the cellular immune response by processing and presenting internalized antigen to T cells, which then become activated and proliferate. T cell expansion following productive immunity usually produces a memory T cell population that can persist for decades. Compared with their naive precursors, memory T cells are more abundant, have a lower threshold for activation, and more rapidly reactivate effector functions following antigen encounter. They are also maintained in barrier tissues to rapidly respond to reinfection. Thus, a major goal of vaccines is the induction of strong and durable T and B cell memory.1
The appearance of SARS-CoV-2 specific CD4+ and CD8+ T cells early after symptom onset2, 3, 4, 5 is associated with rapid viral clearance and mild disease,3 whereas delayed T cell responses correlated with worse clinical outcomes.6 Antigen-specific T cell responses evaluated by exposing peripheral blood mononuclear cells (PBMCs) to peptide pools7, 8, 9, 10 suggest that spike (the target of most coronavirus disease 2019 [COVID-19] vaccines), nucleocapsid, and M envelope proteins are the most relevant CD4+ and CD8+ T cell targets.7 , 8 , 10, 11, 12 In contrast, peptide-MHC tetramer staining, used to screen epitopes for T cell recognition across various HLA alleles, revealed that CD8+ T cells specific to nucleocapsid were present at a higher frequency than those specific for spike- or non-structural proteins.13 , 14 In several studies, the magnitude of SARS-CoV-2-specific IgG and IgA titers correlated with the SARS-CoV-2 T cell response.10 , 11 , 15 Interestingly, memory T cells specific for related coronaviruses, such as those that cause the common cold, that cross-react with SARS-CoV-2 antigens are present in a large percent of SARS-CoV-2 naive individuals.7 , 8 , 11 , 16 , 17 Moreover, profiling of the TCR repertoire of T cells isolated from naive or COVID-19-convalescent patients and expanded in vitro with predicted immunodominant SARS-CoV-2 peptides show clonal expansion of T cells with TCR sequences recognizing peptides from other viruses, including human cytomegalovirus (HCMV), human herpes virus-5 (HHV-5), and influenza A.18 The impact of these pre-existing, cross-reactive memory T cells on COVID-19 outcomes is largely unknown.19
Heterologous immunity is a response to a microbe mediated by memory T cells generated against the antigens of a different microbe that may provide enhanced immunity to novel pathogens.20 , 21 Here, we performed two complementary sets of analyses to seek evidence of a role for heterologous immunity in the host response to SARS-CoV-2. First, we used a sensitive, recently developed assay for antigen-specific T cell responses22 to determine whether SARS-CoV-2-specific T cells in the blood of COVID-19-convalescent patients or COVID-19 mRNA-vaccinated individuals cross-react with antigens in trivalent MMR (measles-mumps-rubella) and Tdap (tetanus-diphtheria-pertussis) vaccines, known to be highly effective in eliciting long-lasting protective T and B cell memory responses.23 Second, we interrogated a large, well-characterized cohort of COVID-19 patients to determine whether prior trivalent MMR or Tdap vaccination was associated with decreased disease severity.
Results
Correlation of T cell responses to SARS-CoV-2 and Tdap and MMR vaccine antigens in COVID-19-convalescent patients
COVID-19-convalescent patients with PCR-confirmed SARS-CoV-2 infection and uninfected controls (confirmed by absence of SARS-CoV-2 antibodies) were studied (Table 1 ). Plasma cytokine profiles were similar in both groups (Figure S1A). The strategy for assessing T cell recall responses to SARS-CoV-2 and Tdap and MMR vaccine antigens and anti-SARS-CoV-2 antibody profiles is summarized in Figure 1 A. Prior studies examining human T cell responses in vitro have primarily relied on DCs derived from monocytes24 , 25 or PBMCs26 that contain a mixture of poorly immunogenic APCs that are loaded with pre-selected viral peptide pools, which may not be representative of the physiological peptide repertoire that is generated by cellular antigen processing. Here, we exploited APCs that were generated by engaging FcγRIIIB on neutrophils with a complexed anti-FcγRIIIB antibody (referred to as AAC; see STAR Methods) and culturing them in granulocyte-macrophage colony-stimulating factor (GM-CSF), which converts a fraction of neutrophils into highly immunogenic APCs;22 hereafter, these cells are referred to as neutrophil-derived APCs (nAPCs). nAPCs expressing CD11c, HLA-DR, T cell co-stimulatory molecules, and the migration receptor CCR7 (Figure S1B) were pulsed with native SARS-CoV-2, MMR, or Tdap antigens and co-cultured with autologous T cells on IFN-γ ELISpot plates. IFN-γ is a sensitive marker of activated memory T cells and is produced in low to undetectable amounts by TCR-activated naive T cells.27 , 28 nAPCs pulsed with spike-S1 resulted in robust T cell activation only in samples from SARS-CoV-2-infected individuals (Figure 1B). No IFN-γ-secreting T cells were observed with unpulsed nAPCs, isotype antibody-treated neutrophils pulsed with antigen, or T cells incubated with antigen alone (Figure S1C). Most SARS-CoV-2-infected individuals seroconvert within 7–14 days of infection,29 , 30 and IgG antibodies persist for at least 9 months after exposure.31 SARS-CoV-2-specific IgG, using a sensitive assay,32 was present in the sera of all patients screened 14 days or more after a positive SARS-CoV-2 test (Figure S1D), whereas it was detectable in only a subset of patients (Figure 1C) whose blood samples were taken within 2–6 days of a SARS-CoV-2-positive test or 5–11 days of developing symptoms (Table 1). By contrast, T cell responses were detected at any time point following infection (Figure 1B), suggesting that effector memory T (TEM) cells reactive to SARS-CoV-2 antigens predate SARS-CoV-2 infection.
Table 1.
Characteristics of COVID-19-convalescent patients, uninfected donors, and uninfected COVID-19-vaccinated donors for cellular and molecular assays
| Donors for IFN-γ ELISpot assays and flow cytometric analysis | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SARS-CoV-2-infected |
Uninfected |
Uninfected, COVID-19-vaccinated |
|||||||||
| No. | Sex | Age (years) | Days from 1st COVID+ test to blood draw | Days from 1st symptoms to blood draw | No. | Sex | Age (years) | No. | Sex | Age (years) | Days from 2nd COVID-19 vaccine dose to blood draw |
| 1 | M | 74 | 22 | 11 | 1 | F | 32 | 1 | F | 32 | 74 |
| 2 | F | 89 | 4 | 5 | 2 | F | 27 | 2 | M | 67 | 80 |
| 3 | M | 52 | 37 | 36 | 3 | M | 38 | 3 | F | 35 | 71 |
| 4 | F | 61 | 5 | 5 | 4 | F | 35 | ||||
| 5 | F | 25 | 94 | 98 | 5 | F | 58 | ||||
| 6 | F | 25 | 6 | 11 | 6 | M | 26 | ||||
| 7 | M | 36 | 69 | n.k. | 7 | M | 67 | ||||
| 8 | F | 47 | 100 | 109 | 8 | M | 56 | ||||
| 9 | F | 68 | 2 | 5 | |||||||
| 10 | M | 72 | 5 | 9 | |||||||
| 11 | F | 52 | 124 | 133 | |||||||
| 12 | F | 37 | 94 | 96 | |||||||
| 13 | F | 51 | 120 | 120 | |||||||
| 14 | F | 64 | 131 | 139 | |||||||
| 15 | M | 32 | 204 | n.k. | |||||||
| 16 | M | 54 | 13 | 13 | |||||||
| 17 | M | 52 | 96 | n.k. | |||||||
| 18 |
F |
50 |
112 |
n.k. |
|||||||
| Donors for V(D)J clonotyping and scRNA-seq studies | |||||||||||
| SARS-CoV-2-infected | Uninfected | ||||||||||
| No. | Sex | Age (years) | Days from first COVID+test to blood draw | Days from first symptoms to blood draw | No. | Sex | Age (years) | ||||
| 1 | F | 25 | 192 | 196 | 1 | F | 32 | ||||
| 2 | M | 52 | 156 | 155 | 2 | M | 67 | ||||
| 3 | M | 32 | 204 | n.k. | 3 | F | 35 | ||||
Demographics of COVID-19-convalescent patients (SARS-CoV-2-infected as assessed by positive COVID-19 test), uninfected participants, and uninfected COVID-19-mRNA-vaccinated participants for IFN-γ ELISpot assays and flow cytometry studies, and SARS-CoV-2-infected and uninfected donors for V(D)J clonotyping and scRNA-seq studies. F, female; M, male; N/A, not applicable; n.k., not known.
Figure 1.

T cell responses to SARS-CoV-2, MMR, and Tdap antigens in SARS-CoV-2-infected and uninfected donors
(A) Experimental scheme for (B)–(G). Blood was collected from uninfected (UIN) and PCR-confirmed, SARS-CoV-2-infected (INF) donors. Plasma was analyzed for antibodies to SARS-CoV-2 antigens and cytokines. Blood was divided and treated with isotype control (Iso) or AAC for 2 h, and neutrophils were then isolated and cultured in GM-CSF for 2 days. AAC treatment generates nAPCs while isotype-treated controls remain neutrophils. PBMCs were harvested to isolate CD3+ T cells and monocytes, which were cultured in cytokines to generate dendritic cells (moDCs). Neutrophils (isotype), nAPCs (AAC), and moDCs were loaded without (−) or with (+) antigen and co-cultured with autologous T cells on IFN-γ ELISpot plates for 18 h. Loaded antigens included spike-S1 subunit (Sp1), spike-S2 subunit (Sp2), receptor binding domain (RBD), nucleocapsid (NC), measles (Measl.), mumps, rubella (Rubel.), diphtheria (Dipth), tetanus (Tetan.), or pertussis (Pertu.). Stimulation of T cells alone with PMA plus ionomycin (PMA+) served as a positive control.
(B) nAPCs from uninfected (1–4) and infected (1–9) individuals were co-cultured at a 1:5 ratio with autologous T cells and spike-S1 on IFNγ-ELISpot plates, and the number of IFNγ+ spots was counted.
(C) IgG titers in sera of uninfected and infected donors to SARS-CoV-2 antigens, Sp1, NC, spike-S1 and -S2 (spike), and RBD. Red arrows in (B) and (C) identify samples with IFN-γ+ T cells but no detectable SARS-CoV-2 specific IgG titers.
(D) nAPCs generated from 8 uninfected and 18 infected donors were loaded with vehicle (−) or indicated individual SARS-CoV-2, MMR, or Tdap antigens were analyzed for T cell responses as in (B). Representative images of wells with IFN-γ+ spots are shown (right). ∗p < 0.05; ∗∗p < 0.005 by two-tailed Mann-Whitney test with Bonferroni correction for multiple comparisons.
(E) A correlation of spike-S1 or nucleocapsid-derived IFN-γ+ spots with indicated vaccine antigens (circles) and percent of nAPCs generated (diamonds), which varies between individuals, in infected donors was conducted using Spearman’s rank correlation coefficients (r).
(F) Cytokine levels detected in the supernatants of neutrophils treated with isotype (neut) or AAC (to generate nAPCs) and cultured for 48 h and monocyte-derived dendritic cells (moDCs). ∗∗p < 0.005 using two-way analysis of variance and Bonferroni’s multiple comparison test.
(G) As in (B), ELISpot assays measuring IFN-γ secretion by T cells co-cultured with nAPCs pulsed with combined SARS-CoV-2 (spike-S1, nucleocapsid, RBD), MMR, or TDP (Tdap) antigens were evaluated in the presence of two independent functional blocking anti-IL-15 (top panel), anti-IL-1β/anti-IL-18, or anti-TNF-α (bottom panel) antibodies. Two-way analysis of variance and Dunnett’s multiple comparison test were used. ∗∗p < 0.005.
Data in (D), (F), and (G) are average ± SEM, and individual values are plotted. See also Figures S1 and S2.
To elucidate whether SARS-CoV-2 infection results in the activation and subsequent expansion of memory T cells generated by prior MMR or Tdap vaccination, we examined T cell responses to a range of SARS-CoV-2 antigens and antigens present in the MMR and Tdap vaccines in infected, primarily convalescent patients and uninfected individuals (Table 1). Robust T cell responses to spike-S1 and nucleocapsid were observed in infected individuals. The response to spike-S2 was low and variable, consistent with prior reports,16 , 17 while no response to the receptor binding domain (RBD) (within spike-S1) was detected. Responses to MMR and Tdap antigens were present in all individuals, but the frequency of T cells reactive to MMR and Tdap antigens trended higher in the infected versus the uninfected group (Figure 1D), leading us to examine whether there was a correlation between T cell recall responses to spike-S1 and nucleocapsid and individual MMR or Tdap vaccine antigens. A strong correlation was observed in SARS-CoV-2-infected individuals (Figure 1E), suggesting that memory T cells generated by prior MMR and Tdap vaccination are reactivated by SARS-CoV-2 infection. DCs derived from monocytes (moDCs) are a well-characterized source of APCs for in vitro T cell assays. In a subset of infected and naive subjects, moDCs were loaded with SARS-CoV-2 or vaccine antigens and assessed for their ability to stimulate autologous T cells. The overall T cell response to moDCs was markedly lower (Figure S2A) than for nAPCs (Figure 1D), despite comparable expression of HLA-DR and T cell co-stimulatory molecules (Figure S2B); nevertheless, a significant correlation of T cell responses to spike-S1 and nucleocapsid with MMR and pertussis (but not diphtheria and tetanus) antigens was observed (Figure S2C).
Superior T cell activation by nAPCs is related to IL-15 production
The higher frequency of T cell activation by nAPCs versus moDCs may reflect differences in expression of immunomodulatory cytokines33 that might sensitize cross-reactive T cells to non-cognate antigen. For example, IL-15 enhances memory CD8+ T cell TCR affinity and avidity,34, 35, 36 promotes IFN-γ production by CD8+ TEMRA,37 and counteracts CD4+ T cell suppression by T regulatory cells.38 We found that supernatants from cultures with nAPCs derived from infected and uninfected individuals had log-fold higher amounts of IL-15, IL-1β, and TNF-α cand lower levels of IL-18 compared to moDCs generated from a subset of the same individuals (Figure 1F). To investigate the contribution of these cytokines to T cell activation, we treated nAPCs and T cell co-cultures with neutralizing antibodies. Two independent IL-15 antibodies significantly reduced the number of IFN-γ-secreting T cells, whereas IL-1β plus IL-18 antibodies and a TNF-α blocking antibody had no effect (Figure 1G). A similar analysis of moDCs showed that blocking IL-15 led to a small reduction in T cell activation (Figure S2D). Together, these data suggest that higher levels of IL-15 secretion explain the superiority of nAPC-containing samples over moDCs in promoting T cell recall responses.
Phenotype of memory T cells reactive to SARS-CoV-2, MMR, and Tdap
To characterize the CD4+ and CD8+ T cell lineages activated by SARS-CoV-2, MMR, and Tdap antigens in infected individuals, we used flow cytometry to (1) assess cell surface markers that define naive (T naive), central memory (TCM), TEM, and effector memory re-expressing CD45RA (TEMRA) on CD4+ and CD8+ T cells (gating strategy, Figures S3A and S3B); and (2) measure markers of activation, homing, function, and proliferation in T cells with intracellular IFN-γ.39 We found that the majority of SARS-CoV-2, Tdap, and MMR antigen-responsive IFN-γ+ CD4+ and CD8+ T cell populations were TEMRA (Figures 2 A and 2B). Antigen-activated CD4+ TEMRA cells expressed GPR56, a marker of cytotoxicity,40 and CX3CR1, a marker associated with peripheral surveillance of infected tissue41 , 42 (Figure 2A), while CD8+ TEM and TEMRA cells expressed the activation marker CD69 (Figure 2B). A small degree of CD3+ T cell proliferation (2.87% ± 0.12%) was also observed. IL-15 blockade caused a significant decrease in the percentage of IFN-γ+ CD4+ and CD8+ TEM and TEMRA cells (Figure 2C). A similar analysis with SARS-CoV-2-loaded moDCs showed that IFN-γ+ T cells were primarily CD4+ TEM cells and a small population of CD4+ and CD8+ TEMRA cells; anti–IL-15 had no effect on the observed frequency of any of these populations (Figure S3C).
Figure 2.

Phenotype of T cells responding to SARS-CoV-2, MMR, and Tdap antigens
(A–D) T cells from the ELISpot wells were harvested after 18-h co-culture with nAPCs loaded with combined SARS-CoV-2, MMR, or Tdap (TDP) antigens. The cells were treated with brefeldin A, stained for phenotypic surface and intracellular markers, and analyzed by flow cytometry. (A) Proportions of CD4+ T cells classified as naive (Tnaive, CCR7+CD45RA+), central memory (TCM, CCR7+CD45RA−), effector memory (TEM, CCR7−CD45RA−), and effector memory re-expresses CD45RA (TEMRA, CCR7−CD45RA+) and expressing IFN-γ (left) and additional cytolytic markers, CX3CR1 (middle) and GPR56 (right), in TEMRA are shown. (B) CD8+ T cells expressing IFN-γ were classified as Tnaive (CCR7+CD45RA+CD27+), TCM (CCR7+CD45RA−CD27+), TEM (CCR7−CD45RA−CD27−), and TEMRA (CCR7−CD45RA+CD27−) (left). TEM (middle) and TEMRA (right) were further evaluated for CD69. (A) and (B) were analyzed using three-way analysis of variance and Tukey’s multiple comparisons test. ∗p < 0.05, ∗∗p < 0.005. (C) Anti-IL-15 or vehicle control was included during T cell co-cultures with nAPCs pulsed with SARS-CoV-2 antigens, and subsets of CD4+ (top panel) and CD8+ (bottom panel) T cells producing IFN-γ were determined. Two-way analysis of variance and Dunnett’s multiple comparisons test were used. ∗p < 0.05, ∗∗p < 0.005. Data in (A)–(C) are average ± SEM, and individual values are plotted. (D) FLOWsom-based visualization on tSNE plots of flow cytometry datasets for CD4+ live T cells from two individual infected donors. Cells shown in gray correspond to total CD4+ T cells from down-sampled and concatenated specimens stimulated with indicated antigens with each in the presence or absence of anti-IL-15 antibody, to create a single tSNE map. Cell populations defined by FLOWsom for each antigen were then projected onto tSNE space, and a population was identified that overlapped between SARS-CoV-2 (SARS), MMR, and TDP antigens (purple). The phenotype of the overlapping population between the antigens was defined by evaluating the following markers: CD4, CD45RA, CCR7, CD27, GPR56, CX3CR1, and IFN-γ and shown as heatmaps along with a continuous scale.
See also Figure S3.
To further define nAPC-stimulated T cell populations, we visualized the profile of CD4+ T cells using viSNE, which uses t-stochastic neighbor embedding (t-SNE) to generate a two-dimensional map of cell relatedness based on marker profile similarity.43 viSNE depicted an IFN-γ+ CD4+ T cell cluster that was responsive to SARS-CoV-2, MMR, or Tdap antigens and had features of TEMRA (Figure 2D; Figure S3D). The prevalence of CD4+ TEMRA cells ranged from 5% to 13%, which is consistent with the reported variability in the frequency of TEMRA cells (<0.3%–18% of total CD4+ T cells) even in the absence of infection.44 The cluster containing CD4+ TEMRA cells was significantly reduced after anti–IL-15 treatment (Figure 2D). An overlapping region among the three sets of antigen was not detected for CD8+ T cells (data not shown), although inclusion of antibodies to additional markers could identify such overlaps. Collectively, this analysis provides evidence of a distinct population of responsive TEMRA cells that is similarly enriched in co-cultures with nAPCs presenting SARS-CoV-2, MMR, or Tdap antigens and whose activation depends on IL-15 stimulation.
TEMRA cells are prevalent in COVID-19-convalescent patients.45 Our data suggest that TEMRA in SARS-CoV-2-infected individuals include a cross-reactive, memory T cell population whose detection ex vivo relied on nAPC-derived IL-15.
Increase in IFN-γ+ T cells reactive to Tdap and MMR vaccine antigens in uninfected individuals immunized with COVID-19 mRNA vaccine
To determine whether COVID-19-vaccinated individuals also exhibit an increase in MMR and Tdap-specific T cells, we evaluated three uninfected healthy controls before and 2.5 months after receiving the second dose of the Moderna (mRNA-1273) COVID-19 vaccine encoding the SARS-CoV-2 spike protein. Vaccination produced a marked increase in the frequency of IFN-γ+-secreting T cells reactive to spike-S1 that correlated with a pronounced enhancement in the number of T cell clones reactive to MMR and Tdap antigens (Figure 3 A); similar results were obtained when antigen was presented by moDCs derived from these same individuals (Figure S4). As expected, there was no response to nucleocapsid in COVID-19-vaccinated subjects (Figure 3A). A high proportion of the IFN-γ+, spike-S1 reactive CD4+ and CD8+ T cells expressed TEMRA markers (Figures 3B and 3C), as was observed with SARS-CoV-2-infected individuals (Figures 2A and 2B). However, compared to infected individuals, SARS-CoV-2-reactive IFN-γ+ CD4+ TEM was higher, whereas the frequency of IFN-γ+ CD8+ TEMRA cells that are CD69+ was lower in COVID-19-vaccinated subjects.
Figure 3.

T cell responses to SARS-CoV-2, MMR, and Tdap antigens in uninfected individuals before and after COVID-19 mRNA vaccination
(A) Blood was analyzed from three individuals approximately 3 months before and 2.5 months after receiving the Moderna mRNA-based COVID-19 vaccine. nAPCs generated from blood neutrophils were loaded with indicated individual SARS-CoV-2, MMR, or Tdap (TDP) antigens, and IFN-γ secretion by co-cultured autologous T cells was evaluated on ELISpot plates as described in Figure 1. Images of one well with IFN-γ+ spots representative of triplicates are shown. Asterisk (∗) denotes too many IFN-γ+ spots to count.
(B and C) The phenotype of T cells was examined by flow cytometry as in Figures 2A and 2B. Data are average ± SEM, and individual values are plotted. ∗p < 0.05, ∗∗p < 0.005 using three-way analysis of variance and Tukey’s multiple-comparisons test.
TCR clonotype and transcriptome of SARS-CoV-2, Tdap, and MMR antigen-reactive T cells
T cell-dependent antigen recognition relies on the interaction of TCRs in CD8+ and CD4+ T cells with peptide-loaded class I or II HLA, respectively. TCRα and β chains contain three hypervariable loops, termed complementary determining regions (CDRs), of which CDR3 is unique for each clone and the main contributor to peptide-MHC specificity. Thus, T cells that express the same pair of CDR3 nucleotide sequences are highly likely to recognize the same antigen and to be derived from the same clonally expanded T cell. To identify and characterize cells with cross-reactive TCR clonotypes, we performed single-cell RNA sequencing (scRNA-seq) coupled with T cell receptor V(D)J sequencing. We profiled three replicate batches, each containing T cells isolated from a COVID-19-convalescent patient and stimulated with SARS-CoV-2, MMR, or Tdap antigen-loaded nAPCs, and T cells from a healthy, uninfected individual stimulated with SARS-CoV-2 antigen-loaded nAPCs, which served as our control for non-antigen-specific responses. The characteristics of the profiled convalescent patient and healthy, uninfected control samples are detailed in Table 1. After data processing and filtering (see STAR Methods), 15,931 cells remained (range, 833–1,663 per sample). Principal-component analysis was used to reduce the dimensionality of the dataset for graph-based clustering and uniform manifold approximation and projection (UMAP) visualization. This resulted in two large groups of T cells, one from the healthy controls and the other from the COVID-19-convalescent patients (Figure 4 A), suggesting that the major source of variation in gene expression was antigen-specific T cell activation.
Figure 4.

TCR clonotyping and single-cell RNA sequencing of T cells
(A) Uniform manifold approximation and projection (UMAP) plots of single-cell gene expression data of T cells from uninfected (healthy) control and COVID-19-convalescent donors colored by the antigen to which they were exposed.
(B) UMAP plot highlighting cells with shared CDR3 sequences (left). Number of clonotypes and cells across SARS-CoV-2 (SARS), MMR, and Tdap (TDP) antigens overall and by each COVID-19-convalescent donor (in parentheses) is given.
(C) UMAP plot of single-cell gene expression data (left) and violin plots (right) showing the distribution of expression for indicated genes. Adjusted p values for differential expression were obtained from Wilcoxon rank-sum tests, as implemented in the FindMarkers function in the Seurat R package. ∗∗∗p < 0.001 compared with other groups.
(D) UMAP plot displaying the clustering scheme.
(E) Heatmap of mean expression of top 50 marker genes for cells with shared CDR3 sequences by COVID-convalescent status and antigen. The black bars on the left-hand side indicate that the gene was identified as a TEMRA marker in Patil et al.46 (column labeled “Patil”) or in Szabo et al.47 (column labeled “Szabo”). Rows and columns were grouped by overall expression pattern using hierarchical clustering. Lollipops (right) highlight cytotoxic T cell effector molecule genes.
(F) Circos plot of sequences with shared CDR3 sequences. Each sector on the innermost track corresponds to an α chain (TRA, blue) or β chain (TRB, red). The width of a sector is proportional to the number of times the α or β chain sequence occurs. An arc between an α and β chain indicates these sequences are combined in a single CDR3 sequence. The antigen is shown in the third track. The Seurat cluster is shown in the fourth track; not all clusters have cells with shared CDR3 sequences, so fewer than 30 clusters are shown. The preponderance of straight lines spanning across the plot shows that in nearly all cases, a given α chain is combined with only one β chain and vice versa.
Next, we determined whether T cell clones responsive to SARS-CoV-2 antigens in COVID-19-convalescent patients express the same clonotype CDR3 sequences as T cells responsive to MMR and Tdap antigens. Sequence analysis identified 12,613 unique clonotypes, of which 90 clonotypes shared TCRs (Table S1). These 90 clonotypes clustered in UMAP space (Figure 4B) and were expressed by 1,323 cells (8.3% of the cells profiled) (Figure 4B). Each clonotype was unique to a replicate batch. All three replicate batches had cells with shared CDR3 sequences, but such cells were most prevalent in replicate 2, which contained 84 of the 90 shared clonotypes (among 1,301 total cells) (Figure 4B). The percent of cross-reactive T cells was 0.15%, 10%, and 0.023% in subjects 1, 2, and 3, respectively. The majority (60 of 90) of cross-reactive clonotypes were observed after stimulation with each of the three antigens (SARS-CoV-2, MMR, Tdap), with the remaining 30 being observed after stimulation with SARS-CoV-2 or MMR and SARS-CoV-2 or Tdap (Figure 4B). In addition to these candidate heterologous SARS-CoV-2 CDR3 clonotypes, 10 additional clonotypes were common with MMR and Tdap stimulation (Figure 4B). To determine whether the T cells with shared CDR3 sequences are bona fide antigen-responsive T cells, we assessed the expression of mRNAs encoding IL-2RA (CD25), a canonical CD4+ and CD8+ T cell activation marker that encodes for the IL-2 receptor,47 and activation-induced markers CD134 (OX40, TNFRS4) and CD137 (TNFRS9, 4-1BB),48 previously reported to be expressed by SARS-CoV-2-reactive T cells.11 We found that IL-2RA, CD134, and CD137 expression overlapped with antigen-stimulated T cells and, interestingly, within this group was significantly higher in those clusters with shared CDR3 sequences as assessed by differential expression analysis. IFN-γ mRNA (IFNG) is well correlated with protein levels of IFN-γ, a known activation marker for a subset of TEM cells following TCR engagement 27 and IL-2 is upregulated by signals from TCR and CD28.49 Thus, we examined IFNG and IL-2 expression levels. Notably, IFNG expression overlapped with clusters sharing CDR3 sequences, while IL-2 expression did not. Also, differential expression analyses showed that the level of IFNG was markedly higher, while IL-2 is significantly lower, in clusters with shared CDR3 compared with other activated or control T cells (Figure 4C; Table S2). Together, these studies suggest that high expression of CD134, CD137, and IFN-γ and low IL-2 distinguishes activated T cells with shared CDR3 sequences from other antigen-activated T cells in the COVID-convalescent group.
We next used graph-based clustering to partition cells with the characteristics of cells with shared CDR3 sequences to examine their gene expression profile. This identified 30 cell populations (clusters). Clusters 2, 15, and 18 (Figure 4D) were markedly enriched for cells with shared CDR3 sequences (69%, 59%, and 54% of these clusters, respectively). Twelve other clusters contained a smaller (≤15%) fraction of cells with shared CDR3 sequences (Table S1). We next sought to identify a gene expression signature for cells in clusters 2, 15, and 18. These clusters contained a total of 1,810 cells, of which 1,154 (63.7%) were cells with shared CDR3 sequences (84 of the 90 identified clonotypes). Differential expression analysis of these clusters relative to all other clusters identified 386 genes that were expressed at significantly higher levels (Table S2). The top 50 genes for cells with shared CDR3 sequences (by statistical significance) are shown in a heatmap (Figure 4E). Notably, of these genes, 13 were identified as TEMRA markers by Patil et al.,46 and 6 were identified as TEMRA markers by Szabo et al.47 (Figure 4E; Table S2).
Investigation of TCRα and β chain combinations within cells with shared CDR3 sequences shows they are mostly unique pairings, with only one β chain pairing with two α chains (represented by the “curved” arcs) (Figure 4F; Table S1).
Effect of Tdap and MMR immunization on COVID-19 outcomes
There is growing epidemiological evidence that vaccinations can impact morbidity and mortality beyond their effect on the diseases they prevent.20 , 21 In COVID-19, a significant association between MMR vaccination status and lower COVID-19 severity was observed,50 and high titers of mumps antibodies were more likely to be associated with asymptomatic or less severe COVID-19.51 These studies were, however, limited by small sample size50 , 51 or survey research methodology50 and may be confounded by co-variables that influence both the likelihood of getting vaccinated with MMR or Tdap and the risk for progressing to severe COVID. To address these challenges, we performed a retrospective cohort study with overlap propensity score weighting. All patients were seen at the Cleveland Clinic Health System in Ohio or Florida and tested positive for COVID-19 between March 8, 2020, and March 31, 2021 (73,582 COVID-19-positive patients). The cohort included 11,483 patients vaccinated with MMR and 36,793 patients vaccinated with Tdap (Table S3), a skewing that is consistent with vaccination scheduling, because the single trivalent MMR is given in childhood and only became available in 1971,52 whereas the Tdap is given as a booster every 10 years. Our propensity score matching53 was effective at making the two groups comparable as evidenced by the identical scores across patient groups for both the MMR and Tdap comparisons (Table 2 ). After adjusting for 44 patient characteristics (Table 2), two primary endpoints reflecting disease severity (COVID-related hospitalization and COVID-related admission to the intensive care unit or death) were decreased in patients previously vaccinated for MMR by 38% and 32%, respectively, and in patients previously vaccinated for Tdap by 23% and 20%, respectively, at the 5% level of significance (Figure 5 A; Table 2). Differential effects of sex and age on the observed relationships between Tdap and MMR and disease outcomes were not hypothesized a priori but were explored given the emerging literature on the topic.54 , 55 At the 5% level of significance, we found that receiving the MMR or Tdap was more highly associated with a decreased rate of hospitalization in women than in men (Figure 5B). We also found that Tdap was more highly associated with a decrease in rate of hospitalization in younger (age < 50 years) versus older individuals (Figure 5C). If we conservatively apply the Bonferroni method to adjust for multiple testing, only the sex difference for MMR and hospitalization remains significant at the 5% level. The time interval from vaccination (either MMR or Tdap) to positive COVID-19 test was not significantly associated with outcome, possibly because this cohort is dominated by individuals who had MMR or Tdap vaccines within the past 20 years. Thus, this may not be the ideal dataset to test the effect of interval from vaccination to disease.
Table 2.
Overlap propensity score-weighted characteristics and disease severity markers (hospitalization, ICU admission/death) among MMR and Tdap vaccination status groups in all COVID-19-test-positive patients
| No prior MMR (62,099) | With prior MMR (11,483) | No prior Tdap (36,789) | With prior Tdap (36,793) | |
|---|---|---|---|---|
| Demographics | ||||
| Race (%) | ||||
| Asian | 1.5 | 1.5 | 1.1 | 1.1 |
| Black | 18.6 | 18.6 | 20 | 20 |
| Other | 11.5 | 11.5 | 10 | 10 |
| White | 68.4 | 68.4 | 68.9 | 68.9 |
| Ethnicity (%) | ||||
| Hispanic | 6.9 | 6.9 | 7.1 | 7.1 |
| Non-Hispanic | 88.3 | 88.3 | 88.1 | 88.1 |
| Unknown | 4.8 | 4.8 | 4.8 | 4.8 |
| Male (%) | 32.2 | 32.2 | 44.4 | 44.4 |
| Age | 32.89 | 32.89 | 48.92 | 48.92 |
| BMI | 29.45 | 29.45 | 29.8 | 29.8 |
| Risk factors | ||||
| Smoking (%) | ||||
| Current smoker | 5.8 | 5.8 | 7.3 | 7.3 |
| Former smoker | 14.9 | 14.9 | 22.4 | 22.4 |
| No | 58.9 | 58.9 | 48.4 | 48.4 |
| Unknown | 20.4 | 20.4 | 21.9 | 21.9 |
| Median income | $62,213.54 | $62,213.54 | $60,771.82 | $60,771.82 |
| Exposed to COVID-19? Yes (%) | 45.9 | 45.9 | 40.2 | 40.2 |
| Family with COVID-19? Yes (%) | 12.2 | 12.2 | 10.9 | 10.9 |
| Presenting COVID symptoms | ||||
| Cough? Yes (%) | 47.7 | 47.7 | 46.7 | 46.7 |
| Fever? Yes (%) | 32.1 | 32.1 | 30.9 | 30.9 |
| Fatigue? Yes (%) | 34.8 | 34.8 | 35.8 | 35.8 |
| Sputum production? Yes (%) | 6.9 | 6.9 | 8.8 | 8.8 |
| Flu-like symptoms? Yes (%) | 37.2 | 37.2 | 34.7 | 34.7 |
| Diarrhea? Yes (%) | 8.1 | 8.1 | 9.6 | 9.6 |
| Loss of appetite? Yes (%) | 8.4 | 8.4 | 10.4 | 10.4 |
| Vomiting? Yes (%) | 14.8 | 14.8 | 14.8 | 14.8 |
| Co-morbidities | ||||
| COPD emphysema? Yes (%) | 1.3 | 1.3 | 5.3 | 5.3 |
| Asthma? Yes (%) | 14.8 | 14.8 | 12 | 12 |
| Diabetes? Yes (%) | 6.2 | 6.2 | 14.2 | 14.2 |
| Hypertension? Yes (%) | 13.7 | 13.7 | 31.6 | 31.6 |
| Coronary artery disease? Yes (%) | 1.4 | 1.4 | 7.4 | 7.4 |
| Heart failure? Yes (%) | 1.2 | 1.2 | 5.7 | 5.7 |
| Cancer? Yes (%) | 3.6 | 3.6 | 9.1 | 9.1 |
| Transplant history? Yes (%) | 0.5 | 0.5 | 0.7 | 0.7 |
| Multiple sclerosis? Yes (%) | 0.4 | 0.4 | 0.5 | 0.5 |
| Connective tissue disease? Yes (%) | 1.6 | 1.6 | 2.2 | 2.2 |
| Inflammatory bowel disease? Yes (%) | 1.3 | 1.3 | 1.4 | 1.4 |
| Immunosuppressive disease? Yes (%) | 3.9 | 3.9 | 7.2 | 7.2 |
| Other vaccination history | ||||
| Influenza vaccine? Yes (%) | 65.4 | 65.4 | 49.8 | 49.8 |
| Pneumovax vaccine? Yes (%) | 14.7 | 14.7 | 18.1 | 18.1 |
| Laboratory values upon presentation | ||||
| Pre-testing platelets | 231.05 | 231.05 | 228.02 | 228.02 |
| Pre-testing aspartate aminotransferase | 26.11 | 26.11 | 28.03 | 28.03 |
| Pre-testing chloride | 101.02 | 101.02 | 100.89 | 100.89 |
| Pre-testing creatinine | 0.95 | 0.95 | 1.02 | 1.02 |
| Pre-testing hematocrit | 39.86 | 39.86 | 39.78 | 39.78 |
| Pre-testing potassium | 3.99 | 3.99 | 4.01 | 4.01 |
| Baseline medication use at time of COVID diagnosis | ||||
| NSAIDs? Yes (%) | 14.2 | 14.2 | 14.3 | 14.3 |
| Steroids? Yes (%) | 9.3 | 9.3 | 11.5 | 11.5 |
| Carvedilol? Yes (%) | 0.7 | 0.7 | 2.3 | 2.3 |
| ACE inhibitor? Yes (%) | 3.5 | 3.5 | 8.5 | 8.5 |
| Angiotensin receptor blocker? Yes (%) | 2.1 | 2.1 | 6.1 | 6.1 |
| Melatonin? Yes (%) | 1.1 | 1.1 | 2.7 | 2.7 |
| Hospitalization (%) | 8.5 | 5.5 | 19.0 | 15.3 |
| ICU/death (%) | 1.6 | 1.1 | 6.0 | 4.9 |
ACE, angiotensin-converting enzyme; COPD, chronic obstructive pulmonary disease; NSAID, nonsteroidal anti-inflammatory drug.
Figure 5.

Association of MMR and Tdap vaccination history with hospitalization and progression to ICU stay or death from SARS-CoV-2 infection
(A–C) Overlap propensity score-weighted analysis odds ratios with 95% confidence intervals presented for the whole cohort (A) and then stratified by gender (B) and age (C). The upper limit of the 95% confidence intervals for the adjusted odds ratios was less than 1 for both risk for hospitalization and risk for transfer to the ICU or death for patients with a history of prior vaccination for either MMR or Tdap.
See also Table S3.
Discussion
Our findings provide definitive cellular and molecular evidence that heterologous adaptive immunity exists between SARS-CoV-2 and antigens present in Tdap and MMR vaccines. We observe enhanced in vitro T cell recall responses to Tdap and MMR antigens in individuals with a history of SARS-CoV-2 infection or uninfected individuals immunized with the COVID-19 vaccine, and a strong correlation between the magnitude of the TEM cell response following T cell exposure to APCs loaded ex vivo with SARS-CoV-2 antigens and Tdap or MMR vaccine antigens. We identify identical TCR clonotypes in T cells activated by SARS-CoV-2, Tdap, or MMR antigens, thus providing clear molecular evidence for adaptive heterologous immunity. We also demonstrate that the cross-reactive T cells resemble cytotoxic TEMRA, known to contribute to anti-viral immunity. Heterologous immunity can variously alter disease outcomes by providing enhanced immunity or by exacerbating immunopathology or lessening viral control.20 Our propensity-weighted analysis of a large COVID-19 patient cohort adjusted for multiple patient characteristics revealed that severe disease outcomes were reduced in MMR- or Tdap-vaccinated individuals.
Our ability to detect cross-reactive T cells was enabled by three features of our approach: (1) readout on IFNγ generation, which predominantly identifies activated TEM versus Tnaive cells;27 , 28 (2) use of highly immunogenic nAPCs that generate IL-15,22 which sensitizes T cells with low TCR binding affinity for antigens;34, 35, 36, 37, 38 , 56 and (3) an epitope unbiased approach in which the relevant peptide epitopes were generated by physiological antigen processing rather than exposure to a limited set of viral peptide epitopes,7, 8, 9, 10 which may not represent the specificities of cross-reactive clones generated during a natural SARS-CoV-2 infection. To date, T cell functional responses to vaccines have been difficult to evaluate because of the lack of sensitive in vitro assays. Our approach, described herein, may help bridge this gap.
Heterologous immunity with examples of protection against widely divergent pathogens has been documented in mice and humans.21 , 57 , 58 Innate heterologous immunity can result from antigen-nonspecific functional epigenetic rewiring of monocyte/macrophage precursors in response to one pathogen that induces immunity to a second unrelated infection.57 In humans, this has been best studied in the context of Bacillus Calmette-Guérin (BCG) vaccine for tuberculosis, which may afford some protection against SARS-CoV-2.59, 60, 61 Adaptive heterologous immunity is mediated by memory T cells and antibodies and has been documented for infections by or immunization against bacteria and viruses in experimental mouse models and in humans.20 , 62 Yet, to our knowledge, definitive identification of the relevant epitopes or the cross-reacting lymphocyte clones in humans has not been previously achieved. Our data provide direct molecular evidence of overlapping TCRs in T cell clones that respond to SARS-CoV-2 proteins and Tdap and MMR vaccine antigens. The high frequency of overlap in TCR CDR3 sequences across viral (MMR) and bacterial (Tdap) antigens in SARS-CoV-2-infected individuals suggests that heterologous immunity is prevalent in humans. This is consistent with the estimate that effective immunity during a human’s lifetime requires each of the unique 107–108 TCRs to recognize up to 106 theoretical peptides,63, 64, 65 the finding that TCR polyspecificity is a general characteristic of T cell recognition,66 and the presence of large numbers of memory CD4+ T cells that recognize viral peptides in unexposed adults.62 A study of human T cell cross-reactivity between unrelated HLA-A2-restricted influenza A virus (IAV)- and Epstein-Barr virus-encoded epitopes suggests that cross-reactive epitopes bind with lower affinity and may theoretically lead to the broadening of the TCR repertoire.67 Our studies using highly immunogenic nAPCs that release IL-15, which can lower the threshold for TCR activation, likely facilitated the in vitro detection of expanded low-affinity, cross-reactive MMR and Tdap memory T cells in SARS-CoV-2-infected individuals.
In mice, a prior infection with various unrelated pathogens, lymphocytic choriomeningitis virus (LCMV), Pichinde virus (PICV), murine cytomegalovirus (MCMV), IAV, or BCG resulted in a 1–2 log reduction in organ viral titers in vaccinia virus (VACV)-infected mice at 3–4 days after infection.68, 69, 70 However, protective heterologous immunity was observed in only a subset of mice, even though they were genetically identical and depended on the repertoire of CDR3 sequences that bind a given epitope in the cross-reactive CD8+ T cell memory pool of each animal.21 , 71 Interestingly, protective immunity was seen with as little as 0.6% cross-reactive CD8+ T cells72 , 73 and was reliant on IFN-γ generated by CD8+ and CD4+ memory T cells.70 , 74 Thus, both individual private CDR3 specificity and the number of cross-reactive IFN-γ+ memory T cells can determine the outcome of heterologous infection between viruses. These studies align with our findings in human samples; we show that the cross-reactive T cell pool is comprised of IFN-γ-producing CD8+ and CD4+ T cells and observe low (0.023%) to high (10%) frequency of cross-reactive T cells with shared TCR-CDR3 clonotypes in the three individuals evaluated. In mice, heterologous immunity is not necessarily reciprocal. For example, mice previously infected with VACV were not protected from LCMV, MCMV, or PICV;70 this was linked to an increase in the number of cross-reactive memory CD8+ T cells in LCMV- versus VACV-immunized mice.72 Moreover, a T cell clone recognizing a cross-reactive epitope in one virus does not necessarily react with that epitope in the heterologous virus.21 Thus, prior COVID-19 vaccination will not necessarily provide immunity to measles, mumps, and rubella or diphtheria, tetanus, and pertussis toxins.
The presence of T cell cross-reactivity in COVID-19 is strengthened by reported comparative in silico analyses of SARS-CoV-2 proteins and vaccine peptides. A sequence in tetanus toxin protein significantly matched a region of SARS-CoV-2 spike predicted to interact with an MHC class I receptor encoded by the corresponding HLA, while a segment of the measles virus hemagglutinin significantly overlapped with a segment of the SARS-CoV-2 ORF7b protein with predicted T cell antigenicity.75 A comprehensive study by Reche76 indicates that combination diphtheria-tetanus-pertussis vaccines are significant sources of T cell cross-reactivity to the SARS-CoV-2 orferome. Overall sequence similarities between SARS-CoV-2 and MMR vaccine antigens have also been reported,77, 78, 79 albeit CD4+ or CD8+ T cell epitopes were not specifically examined in these studies.
We found that the phenotype and transcriptional profile of the cross-reactive T cell clusters primarily comprise IFN-γ+ T cells that are IL-2− and have features of TEMRA, a cytotoxic TEM cell subset unique to humans that are implicated in protective anti-viral immunity.41 , 42 , 80 , 81 For example, the frequency of CD4+ TEMRA in dengue virus correlates with vaccine-elicited protection,40 , 80 , 82 and CD8+ TEMRA may control viral load during early HIV infection.81 In influenza, pre-existing IFN-γ+, IL-2− CD8+ TEMRA T cells cross-reactive with different viral subtypes correlate with less severe illness in individuals lacking pre-existing humoral immunity,83 and a large percent of these TEMRA express CCR5 upon viral exposure, which is critical for recruitment of CD8+ T cells to the lung during respiratory viral infection.84 Supporting evidence that the lung-localized TEMRA may elicit viral clearance comes from a study in tissue harvested from organ donors, which shows that CD8+ T cell TEMRA represent 20%–30% of CD8+ T cells in the lung, a frequency that is comparable with that in spleen and blood.85
Prior epidemiological studies suggest that human childhood MMR vaccine may reduce the incidence of other infections,86 albeit study limitations have been noted.87 MMR was also reported to lower COVID-19 severity;50 , 51 , 88 , 89 these small observational studies generated interesting correlations but failed to fully adjust for confounders, limiting the ability to draw meaningful conclusions. Here, we used a large, deeply phenotyped COVID-19 cohort of close to 74,000 patients, with >300 data points per patient, and adjusted for 44 patient characteristics that include demographics, social determinants of health, symptoms upon initial presentation with COVID-19, co-morbidities, additional vaccination history (influenza and pneumococcal vaccination), laboratory values, and home medication use prior to COVID-19. Our propensity-weighted methodology allows us to isolate differences attributed to MMR and Tdap vaccination and revealed that severe disease outcomes (hospitalization, transfer to intensive care unit, or death) were reduced in MMR- or Tdap-vaccinated individuals by 32%–38% and 20%–23%, respectively. A recent study using the UK Biobank supports our finding that prior Tdap vaccination associates with reduced COVID-19 severity,90 but only covaried for age, sex, respiratory disease diagnosis, and socioeconomic status, while our propensity scoring methodology adjusted for significantly more confounding variables. Our exploratory analyses suggest that the women were protected more than men by prior MMR and Tdap vaccination, and younger patients were protected more than older, which may reflect the role of gender on vaccination outcomes and the general decline in response to vaccines that occurs with aging.54 , 55 , 91
Although there is evidence that SARS-CoV-2-specific memory T cells are generated during natural infection and following COVID-19 vaccination, not enough time has lapsed since the start of the pandemic to assess the longevity (half-life) of these memory T cells.7 , 92 Effective T cell memory is essential for productive anti-viral immunity, particularly to pathogens that evolve to evade recognition by neutralizing antibodies.1 In COVID-19, additional SARS-CoV-2 variants in the spike protein may emerge, such as the highly transmissible delta variant.93 This, in turn, could trigger additional waves of resurgence94 and potentially inter-species transmission. In our studies, the robust correlation of MMR and Tdap reactive memory T cells to spike-S1 and the more invariant nucleocapsid protein that contains the highest density of SARS-CoV-2 epitopes recognized by CD8+ T cells13 predicts that nucleocapsid-mediated reactivation of MMR and Tdap memory cells could provide immunity to SARS-CoV-2 spike variants. The markedly enhanced T cell responses to MMR and Tdap antigens not only in infected but also vaccinated individuals suggests that reactivated MMR and Tdap memory T cells may provide protective heterologous T cell immunity following COVID-19 vaccination. The observed prevalence of heterologous immunity in our studies may have implications for vaccine development against future novel pathogens, because the effectiveness of vaccines may correlate with their ability to harness pre-existing memory T cells generated by prior infections or vaccines. Interestingly, in mice, T cells specific for cross-reactive epitopes in the memory pool are maintained, while non-cross-reactive epitopes are selectively lost, which could shape the response to future viral infections.95 We posit that intentional MMR or Tdap vaccine-induced heterologous immunity to SARS-CoV-2 could enhance the effectiveness and durability of COVID-19 vaccines by generating an expanded population of SARS-CoV-2-specific memory T cells that respond vigorously to the vaccines and, in countries where COVID-19 vaccines are not yet available, provide protection from severe disease. This possibility could be explored in future clinical trials.
In conclusion, our studies provide evidence of broad cross-reactivity between T cells responsive to SARS-CoV-2, MMR, and Tdap antigens in humans. The breadth of cross-reactive CDR3 sequences to these three distinct pathogens suggests that adaptive heterologous immunity is prevalent in humans. The correlation of MMR and Tdap vaccination in COVID-19 patients with a decrease in disease severity may reflect the observed cross-reactivity of MMR and Tdap vaccine antigens not only with spike-S1 protein but also with the relatively invariant nucleocapsid protein. A signature of cytotoxic TEMRA cells in responding cross-reactive cells and the reported prevalence of these cells in recovered COVID-19 patients45 suggest that this T cell subset may promote robust, heterologous anti-viral immunity in COVID-19. Finally, our studies predict that MMR or Tdap vaccination together with approved COVID-19 vaccines may afford greater and more durable protection, particularly against emerging spike variants, than the vaccines alone.
Limitations of the study
Our ex vivo correlation analysis of T cell activation on nAPCs loaded with viral antigens from MMR, Tdap, and SARS-CoV-2 indicates that responses to these antigens are interdependent, and thus infers that heterologous immunity exists between MMR, Tdap, and SARS-CoV-2 antigens. However, definitive evidence of cross-reactivity requires the stimulation of T cells with APCs loaded with viral antigen A, isolation of the activated clones, and restimulation with APCs loaded with viral antigen B. We were unable to conduct this well-validated conventional method because, to date, we have been unable to preserve nAPCs over the weeks required to examine the response of T cells to the first and then second antigen challenge. Nonetheless, our correlative analysis along with the TCR clonotyping that showed that CDR3 sequences, known to be unique for each T cell clone and the main contributor of peptide-MHC specificity, are present in T cells activated by three independent sets of antigens (SARS-CoV-2, MMR, and Tdap) provides strong evidence that cross-reactive T cells are present. Another limitation of our in vitro approach is the use of highly immunogenic nAPCs, which may overestimate the extent of cross-reactivity that occurs in individuals if antigen presentation in vivo is not primarily driven by similarly potent APCs. Inter-individual variability in this parameter is also likely. Although our risk assessment was completed with a large, well-characterized cohort of COVID-19 patients, a limitation is that the history of MMR and/or Tdap vaccination may be underreported, which could lead to some degree of misclassification. Our study also cannot definitively determine whether remote vaccination with MMR or Tdap associates with disease outcomes because the patient cohort was dominated by individuals with MMR or Tdap vaccinations within the past 20 years. Finally, we cannot distinguish the contribution of adaptive versus innate heterologous immunity in the estimated effects of MMR and Tdap vaccination on disease outcomes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD10, clone AHN1.1 | Ancell Technologies | Cat#164-040 |
| CD11c, clone 3.9 | Biolegend | Cat#301619; RRID: AB_439792 |
| HLA-DR, clone L243 | Biolegend | Cat#307639; RRID: AB_11219187 |
| CD40, clone 5C3 | Biolegend | Cat#334321; RRID: AB_10643414 |
| CD86, clone IT2.2 | Biolegend | Cat#305419; RRID: AB_1575070 |
| CCR7, clone G043H7 | Biolegend | Cat#353214; RRID: AB_10917387 |
| CD4, clone SK3 | Biolegend | Cat#344646; RRID: AB_2734348 |
| CD8, clone SK1 | Biolegend | Cat#344708; RRID: AB_1967149 |
| CD45RA, clone HI100 | Invitrogen | Cat#11-0458-42; RRID: AB_11219672 |
| CX3CR1, clone 2A9-1 | Invitrogen | Cat#341612; RRID: AB_10900816 |
| GPR56, clone CG4 | Biolegend | Cat#358206; RRID: AB_2562090 |
| IFN-γ, clone 4S.B3 | Invitrogen | Cat#12-7319-42; RRID: AB_1311247 |
| CD66b, clone Tet2 | MACS Miltenyi biotech | Cat#130-117-811; RRID: AB_2733834 |
| CD3, clone UCHT1 | Biolegend | Cat#300456; RRID: AB_2564150 |
| CD27, clone M-T271 | BD Biosciences | Cat#560609; RRID: AB_1727456 |
| CD69, clone FN50 | Biolegend | Cat#310938; RRID: AB_2562307 |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 S1 protein, Fc-Tag | Acro Biosystems | Cat#S1N-C5255 |
| SARS-CoV-2 S protein RBD, Fc-Tag | Acro Biosystems | Cat#SPD-C5255 |
| SARS-CoV-2 S2 protein, His-Tag | Acro Biosystems | Cat#S2N-C52H |
| SARS-CoV-2 Nucleocapsid protein, His-Tag | Acro Biosystems | Cat#NUN-C5227 |
| Rubella Grade III Antigen Protein | Aviva Systems Biology | Cat#OPMA04522 |
| Inactivated (gamma radiated) Native Measles Virus | Bio-Rad | Cat#PIP013 |
| Inactivated (gamma radiated) Native Mumps Virus | Bio-Rad | Cat#PIP014 |
| Pertussis Toxin (heat inactivated before use) | GIBCO Invitrogen | Cat#PHZ1174 |
| Tetanus Toxoid | EMD Millipore | Cat#582231 |
| Diphtheria Toxin | SIGMA | Cat#D0564 |
| Deposited data | ||
| Processed sequencing data | Gene Expression Omnibus (GEO) database | GEO: GSE181046 |
Resource availability
Lead contact
Further information and requests for resources and reagents will be fulfilled by the lead contact, Tanya N. Mayadas (tmayadas@rics.bwh.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Participants
Blood samples for in vitro studies were obtained from consented healthy, self-reporting SARS-CoV-2 uninfected volunteers under a Mass General Brigham Institutional Review Board (IRB)-approved protocol (1999P001694). COVID-19 patients signed informed consent to participate in a Mass General Brigham IRB-approved COVID-19 observational sample collection protocol (2020P000849).
The retrospective cohort study risk assessment used the Cleveland Clinic COVID-19 Enterprise Registry, which was created on March 17, 2020 as a resource for COVID-19 research across the health system. More than 300 data points are extracted from the electronic health record through a combination of manual pulls and validated natural language processing algorithms on all patients tested for COVID-19 in our facilities in Ohio and Florida (18 regional hospitals and 220 outpatient locations).96 , 97 A waiver of informed consent (oral or written) from study participants in the COVID-19 registry was granted by the Cleveland Clinic Health System institutional review board. For this study, we included all COVID positive patients diagnosed between March 2020 and March 31, 2021. Infection with SARS-CoV-2 was confirmed by laboratory testing using the Centers for Disease Control and Prevention reverse transcription–polymerase chain reaction SARS-CoV-2 assay.
Method details
Serum immunoglobulin Simoa Assays
SARS-CoV-2 serological Simoa assays for IgG against four viral antigen spike 1 subunit (S1), spike (stabilized ectodomain of spike with mutated furin cleavage site), Nucleocapsid, and RBD were prepared and preformed as previously described.32 Briefly, plasma samples were diluted 4000-fold in Homebrew Detector/Sample Diluent (Quanterix Corp.). Four antigen-conjugated capture beads were mixed and diluted in Bead Diluent, with a total of 500,000 beads per reaction (125,000 of each bead type). Biotinylated anti-human IgG antibodies (Bethyl Laboratories A80-148B) were diluted in Homebrew Detector/Sample Diluent to final concentrations of 7.73ng/mL. Streptavidin-β-galactosidase (SβG) was diluted to 30 pM in SβG Diluent (Quanterix). The serology assay was performed on an HD-X Analyzer (Quanterix) in an automated three-step assay. Average Enzyme per Bead (AEB) values were calculated by the HD-X Analyzer software. All samples were measured in duplicates.
Plasma cytokine assays
Plasma cytokines were measured in plasma samples using the CorPlex Cytokine Panel (Quanterix Corp), which included sample diluent buffer. Plasma samples were diluted 4-fold in sample diluent buffer and assays were performed following the CorPlex manufacturer protocols. Each CorPlex cytokine panel kit was analyzed by the SP-X Imaging and Analysis System (Quanterix Corp.). All samples were measured in duplicates.
Cell isolation, treatment, and culture
Blood and serum collection
Peripheral blood was drawn into tubes containing trisodium citrate, citric acid and dextrose (Vacutainer ACD Solution A, BD). Serum was obtained by drawing blood into BD Vacutainer Venous Blood Collection Tubes SST, followed by centrifugation at 2500xg for 30 min and removal of the resulting supernatant.
Generation of complexed anti-FcγRIIIB (AAC)
Anti-FcγRIIIB (3G8) (Biolegend) was conjugated to FITC-Ovalbumin (#O23020, Thermofisher) by Biolegend as a custom order and referred to as antibody-antigen conjugate (AAC). Importantly, Ovalbumin in the AAC served as a model antigen in mouse models but is irrelevant for our human studies.
Human blood treatments to generate neutrophil derived APC (nAPC)
10mls human blood was supplemented with GM-CSF (10 ng/ml) for 30 min at 37C followed by addition of 30μg AAC or FITC-IgG isotype control for 2 hr at 37C. Blood was then incubated with Hetasep (STEMCELL Technologies) according to manufacturer protocols to deplete red blood cells and enrich leukocytes. Neutrophils were isolated from the leukocyte-rich plasma layer using a Easysep Neutrophil enrichment kit (STEMCELL Technologies) and placed in RPMI media, which was supplemented with 10% autologous serum, penicillin/streptomycin (50 U/ml penicillin and 50 mg/ml streptomycin) and 20ng/ml GM-CSF. AAC-treated samples were additionally incubated with 10 μg/mL AAC. After 48 hours, cells were harvested using Accutase and evaluated by flow cytometry for surface markers of APCs.
Monocyte isolation and culture to generate monocyte-derived DCs (moDC)
Peripheral blood mononuclear cells were isolated using Lymphoprep (Stemcell technologies, Canada). Monocytes were positively selected by anti-CD14-coated magnetic beads (Miltenyi biotec, Germany) to > 98% purity. Monocytes were cultured in complete medium supplemented with GM-CSF (50ng/ml) and IL-4 (10ng/ml). Cells were harvested after 7 days and evaluated for surface markers CD11c, HLA-DR and CD14.
T cell isolation
PBMCs were isolated from peripheral blood using Lymphoprep (Stemcell technologies, Vancouver, Canada) density gradient medium, aliquoted in 1ml cryopreservation tubes at a concentration of 5-10 million cells/ml and frozen. The tubes were thawed after 2 days to isolate CD3+ T cells for co-culture studies. For isolation of CD3+ T cells, negative selection was performed using EasySep Human T cell isolation kit (#17951, Stemcell technologies, Vancouver, Canada). The CD3+ T cells were labeled with 1μM Cell Trace Violet dye (#C34557, ThermoFisher Sientific) according to manufacturer’s instructions just prior to setting up their co-cultures.
Co-culturing nAPC/moDC and T cells
nAPCs derived from neutrophils and moDCs were harvested and co-cultured with Cell trace Violet labeled CD3+ T cells isolated from PBMCs at a ratio of 1:5 (nAPC:T cells) on a IFNγ ELISpot plate and incubated for 18h. Additionally, the co-cultures were incubated with vehicle alone (PBS) or the following antigens individually or in combinations as indicated in the Result section: SARS-COV-2 antigens 5μg/ml spike-S1 subunit (S1-fc), 8.85μg/ml spike S2 subunit (S2-his), 7.8μg/ml spike S1 receptor binding domain (RBD-fc), 2.35μg/ml Nucleocapsid protein (NC his); inactivated Measles, Mumps, Rubella viral preparations at 5μg of total protein/ml; 5μg/ml of heat inactivated Pertussis toxin, Diphtheria toxin, Tetanus toxoid. T cells alone were incubated with a cocktail of PMA and ionomycin (Biolegend, Cat #423301) as a positive control.
Antibody blocking treatments
Neutralizing antibodies were used as follows. Anti-IL15(1) (Invitrogen, #16-0157-82) at 5μg/ml, anti-IL15(2) (R&D systems, #MAB247) at 5μg/ml, anti-IL1β (R&D systems, #MAB201) 1.6μg/ml and anti-IL18 (MBL, #D044-3) at 1.8μg/ml. Antibodies remained during the entire period of co-culture.
Interferon Gamma (IFNγ) ELISpot assay
ELISpot kits to measure the secretion of human IFN-γ (R&D Systems, #EL285) were used according to manufacturer’s protocols. Fresh CD3+ T cells (Cell trace violet labeled) isolated from PBMCs and nAPCs were plated in triplicates at a ratio of 1:5 (nAPC:CD3+ T cells) per well and incubated with indicated antigens and/or blocking antibodies for 18h. Samples were processed according to manufacturer’s protocol and results were quantitated using an ELISpot reader (CTL ImmunoSpot® S6 Fluorescent Analyzer). Results are a mean of triplicate wells and reported as number of spots per million T cells.
Cell cytokine detection and analysis
Forty-eight hour cultures of isotype or AAC (also containing unconverted neutrophils because unlike mouse,22 human nAPCs are non-adherent) treated neutrophils and 7 day generated monocyte-derived DCs (moDC) were cultured for an additional 72h and supernatants were collected and analyzed for cytokine and chemokine levels using human cytokine 42-plex discovery assay (Eve Technologies, Calgary, AB).
Flow cytometry
Flow cytometry was performed on a FACSCanto II. FCS (flow cytometry standard format) 3.0 data file was used to export data that was analyzed using FlowJo (Mac version 10.7). Compensation controls were created for each fluorochrome. BD multicolor compensation beads and cells were used to set up compensation for the individual fluorochromes. For all experiments, cells were stained with the Fixable Viability Dye eFluor 780 (ThermoFisher) to gate out dead cells. Forward and side scatter gates were used to discriminate doublets and debris (FSC-A, FSC-H, SSC-A x SSC-H). Matched isotypes were used as controls and negative gating was based on FMO (fluorescence minus one) strategy. Only viable cells were included for the studies.
For surface staining, single cell suspensions in FACS buffer (PBS supplemented with 2% FCS and 2mM EDTA) were incubated with human TruStain FcX for 10 min at 4C. Samples were incubated with the indicated fluorochrome-conjugated antibodies for 30 min at 4C, washed with PBS and fixed with 1% paraformaldehyde.
To evaluate surface markers on nAPCs, antibodies to the following were used: CD15, CD66b, CD11c, HLA-DR, CD40, CD86 and CCR7. Within the viable single cell population, CD11c+ and HLA-DR+ events were further gated for CD66b and CD15 expression. The CD11c+, HLA-DR+, CD15+ and CD66b+ population was further analyzed for CD40, CD86 and CCR7 markers.
To evaluate cell surface markers on T cells, antibodies to the following markers were used: CD3, CD4, CD8, CD45RA, CCR7, CD27, GPR56, CX3CR1, IFN-γ. Subsets of T cells were classified based on CD45RA, CCR7 and CD27. Live, singlet CD3+ cells were assessed for proliferation by monitoring CFSE stained T cells for Cell trace violet signal dilution. For detection of intracellular IFN-γ, T cells were treated with Brefeldin A (3ug/ml) for 5h, stained with surface markers, fixed for 30min, permeabilized with BD Perm/Wash (BD Biosciences) and stained with anti-IFN-γ. Cells were washed with permeabilization buffer and analyzed by flow cytometry.
Flow cytometric data analysis
Sample files were exported as FCS 3.0 files from FACSDiva and imported into flowJo v.10.7.1 software for subsequent analysis. The following plug-ins were used: Downsample (1.1), t-distributed stochastic neighbor embedding (tSNE) and FlowSOM (2.6). To visualize the high-dimensional data in two dimensions, the t-SNE algorithm was applied on data. Cells were selected for each sample at random, downsampled and merged into a single expression matrix prior to tSNE analysis. tSNE was performed unsupervised from a maximum of 5,000 randomly selected cells from each sample, with a perplexity set to 80, using the implementation of tSNE plugin in flowJo. Events were identified by gating on live, singlet intact CD3+CD4+ or CD8+ T cells and were included in generating the tSNE plots. The Barnes-Hut implementation of t-SNE with 1,000 iterations, a perplexity parameter of 30, and a trade-off θ of 0.5, was used for applying the dimensionality reduction algorithm. The output was in the form of 2 columns corresponding to t-SNE dimension 1 and dimension 2. t-SNE maps were generated by plotting each event by its t-SNE dimensions in a dot-plot.
Intensities for markers of interest were overlaid on the dot-plot to show the expression of those markers on different cell islands and facilitate assignment of cell subsets to these islands using FlowSOM plugin. Samples were examined by running tSNE with the following markers: CD4, CD45RA, CCR7, CD27, GPR56, CX3CR1, IFN-γ. Phenotypic characteristics of the cell island are shown as heatmaps.
V(D)J clonotyping and scRNA-seq
Sample preparation
nAPCs were co-cultured with CD3+ T cells isolated from PBMCs at a ratio of 1:5 (nAPC: T cells) and incubated for 18h. The co-cultures were incubated with SARS-COV-2 antigens (5μg/ml S1-fc, 7.8μg/ml RBD-fc, 2.35μg/ml NC his) or Measles, Mumps and Rubella viral preparations (5μg of total protein/ml) or 5μg/ml of Diphtheria toxin, Tetanus toxoid and Pertussis toxin. After 18hrs, the cells were harvested using Accutase, aliquoted in 1ml cryopreservation tubes at a concentration of 1.2 million cells/ml and frozen at −80C. Samples were sent to MedGenome, Inc. Foster City, CA, for library preparation and sequencing.
Library preparation and sequencing
Cryopreserved cells were thawed and washed twice with Dulbecco’s phosphate-buffered saline (Fisher Scientific cat# SH3026402) plus 0.04% bovine serum albumin (Fisher Scientific cat# AM2618) then stained with hashtag antibodies (BioLegend, Cat#s 394661, 394663, 394665, or 394666) according to the manufacturer’s protocol. Cells were counted and checked for viability > 80%. Stained cells were brought to ∼1000 cells/ul, pooled equally in sets of four samples, and loaded onto the 10x Chromium instrument (10x Genomics) to generate single-cell gel beads in emulsion. For 5′ multiomics including gene expression, TCR-sequencing, and cell-hashing seq, we used the 10x Single Cell V(D) Kit with Feature Barcoding (10x Genomics, Cat# 1000006) along with the Human V(D) Enrichment Kit (10x Genomics, Cat# 10000005) following the manufacturer’s protocol. Libraries were checked for concentration using Qubit dsDNA HS assay kit (Fisher Scientific, Cat# Q32854) and size distribution using TapeStation (Agilent). Libraries were then sequenced on NovaSeq (Illumina) to a sequencing depth of 50,000 reads per cell for gene expression libraries and 10,000 reads per cell for TCR and hashtag libraries.
Data analysis
scRNA-seq and V(D)J data preprocessing. 10X Genomics Cell Ranger Software Suite (v.4.0.0)98 was used to process raw fastq files to expression tables and assemble V(D)J clonotypes, performing all the necessary barcode processing, mapping to the Human reference genome (GRCh38-2020-A, GENCODE v32/Ensembl 98) and unique molecular identifier (UMI) expression counting; each batch contained an estimated > 7K cells. The gene-cell matrix of all cells was analyzed in R v4.0.3 with Seurat v3.9.9.99 Individual antibody sample hashtags were used to distinguish cells from the four samples within batch (Normal, Convalescent Covid, MMR and Tdap), requiring at least 100 antibody reads present per cell and tag specificity of > 80% to a single sample. V(D)J Clonotypes were linked to the scRNA expression by common cell barcode. The following cell filtering criteria were then applied: gene number greater than 1,000, unique UMI count between 1,000 and 50,000 and mitochondrial gene content < 10%. After filtering, a total of 15,931 cells were left across all 12 samples for the subsequent analysis, of which 14,362 have a V(D)J clonotype defined. All samples were processed together and the matrix was normalized using ‘LogNormalize’ method with default parameters. Then, the top 2,000 variable genes were identified using the ‘vst’ method from Seurat’s FindVariableFeatures function which is used for scaling, dimensionality reduction and clustering. The variables batch, S.Score and G2M.Score from the CellCycleScoring function, percent.mito, and nFeature_RNA were regressed out using ScaleData and PCA was performed. Finally, UMAP and graph-based clustering was performed using the top 50 principal components for visualizing the cells. A cluster resolution of 1.75 with 23 clusters was chosen for downstream analysis.
Identification of candidate heterologous T cells
The dataset contained a total of 12,613 unique clonotypes (alpha and beta chain amino acid sequences) in 15,931 cells. Clonotypes which 1) occurred in a minimum of 3 cells (118 clonotypes); 2) were present in a SARS-CoV-2 sample and at least one of MMR or Tdap sample (92 clonotypes); and 3) and was absent from the healthy control sample (2 clonotypes) were considered. The filter resulted in 90 clonotypes in 1,323 cells highly enriched in a few clusters (clusters 2 – 69% of cells, 10 – 15% of cells, 15 – 59% of cells, 18 – 54% of cells). Batch 2 contained the most heterologous clonotypes with 84, with Batch 1 having 5 clonotypes and Batch 3 just 1.
Differential gene expression analysis
Wilcoxon rank-sum tests as implemented in Seurat v. 4.0.0 (FindMarkers function) were used to perform differential gene expression (DEG) analysis. For enriched clusters (2, 15,19) containing cells with candidate heterologous T cells (as outlined above), DEGs were generated relative to all of the other clusters. A gene was considered significant with adjusted p < 0.05. Only genes upregulated relative to all other cells were considered as markers for the purposes of this analysis.
The heatmap plot was created using the R package ComplexHeatmap, version 2.6.2. The Circos plot was generated using the R package circlize, version 0.4.12.
Quantification and statistical analysis
Statistical analyses for cell-based assays were performed using Graphpad prism 8 (LaJolla, CA), and JMP10 software (SAS Institute, Inc, USA). All the data included in the studies are expressed as mean ± SEM. ∗p < 0.05 and ∗∗p < 0.005 was considered significant.
All descriptive statistics were reported as counts (percentages) or median (interquartile ranges [IQRs]). For comparison of demographic variables and comorbidities among cohorts, Wilcoxon signed-rank tests were used for numeric variables, while χ2 or Fisher exact tests were used for categorical variables.
Overlap propensity score weighting was performed to address potential confounding in comparing non-MMR/Tdap-vaccinated and MMR/Tdap-vaccinated patients given their baseline differences. The overlap propensity score weighting method was chosen given its benefits of preservation of numbers of individuals in each group and of achieving higher levels of precision in the resulting estimates. This methodology is preferred when the propensity score distributions among the groups are dissimilar and when the propensity scores are clustered near the extremes (i.e., close to zero or one). A propensity score for being MMR/Tdap vaccinated was estimated from a multivariable logistic regression model. For the outcomes of hospital and intensive care unit (ICU) admission or death of COVID-19 test-positive patients, the propensity score logistic regression model included covariates that were found to be associated with the outcome in our previous work.
The overlap propensity score weighting method was then applied where each patient’s statistical weight is the probability of that patient being assigned to the opposite group. Overlap propensity score weighted logistic regression models were used to investigate associations between vaccine status and the probability of hospital admission for COVID-19, and ICU admission or death for COVID-19 illness. The results are thus reported as weighted proportions, odds ratios, and 95% confidence intervals.
To address the effect of the time interval between date of vaccine and date of COVID test to the outcomes, we used the time interval as a covariate into multivariable logistic regression models, adjusting for the same covariates as with the overlap propensity scoring models. The time interval is modeled with restricted cubic splines because of suspected nonlinear effects.
Statistical analyses were performed using R 4.0. P values were 2-sided, with a significance threshold of 0.05.
Acknowledgments
We would like to thank Brigham and Women’s Hospital and MassCPR for their support in collecting samples from SARS-CoV-2-infected patients; Colin Heyson for statistical advice; Pascal Yazbeck and Chi-An Cheng for technical assistance; and Jon Aster for scientific discussion and advice. The work was supported by National Institutes of Health grants R01HL065095 (to T.N.M.), R01AI152522 (to T.N.M.), and R01NS097719 (to L.J.); a generous donation from Barbara and Amos Hostetter (to D.R.W.); and the Chleck Foundation (to D.R.W.).
Author contributions
V.M. and X.C. performed and V.M. designed and analyzed all cell-based studies and completed all the statistical analyses for cell-based studies; T.G. performed studies on plasma samples and related statistics; D.R.W. and T.G. designed the studies on plasma samples; M.L.S. and B.D.-J. completed the scRNA sequencing and TCR clonotyping analysis and related statistics; M.D. and L.R.B. collected and managed clinical blood samples from COVID-19-positive participants; X.J. participated in data analysis; M.W.K. and L.J. participated in study design, data interpretation, and data analysis and related statistics; A.H.L. provided valuable advice; T.N.M. conceptualized the research and designed and supervised the cell-based experiments; T.N.M., V.M., X.C., A.H.L., M.L.S., M.W.K., and L.J. wrote the manuscript. All authors edited the manuscript.
Declaration of interests
M.L.S., B.D.-J., M.D., L.R.B., A.H.L., X.J., M.W.K., and L.J. declare that they have no competing interests to disclose. D.R.W. has a financial interest in Quanterix Corporation, a company that develops an ultra-sensitive digital immunoassay platform. He is an inventor of the Simoa technology, founder of the company, and serves on its Board of Directors. The anti-SARS-CoV-2 Simoa assays in this publication have been licensed by Brigham and Women’s Hospital to Quanterix Corporation. T.G. receives royalty payments from Brigham and Women’s Hospital for the antibody assay technology. T.N.M. has a financial interest in neuAPC Therapeutics, a company that will develop AAC for the generation of nAPCs and serves as one of its scientific advisors. V.M., X.C., and T.N.M. have a provisional patent on the generation of immunogenic nAPCs and methods of use thereof.
Published: August 14, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.medj.2021.08.004.
Supplemental information
List of the 90 shared CDR3 sequences and cells count presence within sample and cluster.
List of 386 marker genes identified comparing clusters 2, 15, and 18 against all other clusters. Columns include gene symbol, raw p value, average log fold change, percent presence within clusters of interest, percent presence outside clusters of interest, adjusted p value and presence of genes associated with TEMRA cell type identified in Patil et al.49 and Szabo et al.46
Data and code availability
Sequence data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) database and are publicly available as of the date of publication. An accession number is listed in the Key resource table.
Original code has not been deposited due to patient privacy concerns.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
The patient datasets used in the current study are available upon request under appropriate data use agreements with the specific parties interested in academic collaboration. For further information, please contact Drs. Lara Jehi (JEHIL@ccf.org) or Michael Kattan (kattanm@ccf.org).
References
- 1.Jarjour N.N., Masopust D., Jameson S.C. T Cell Memory: Understanding COVID-19. Immunity. 2021;54:14–18. doi: 10.1016/j.immuni.2020.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J., et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell. 2020;183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan A.T., Linster M., Tan C.W., Le Bert N., Chia W.N., Kunasegaran K., Zhuang Y., Tham C.Y.L., Chia A., Smith G.J.D., et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021;34:108728. doi: 10.1016/j.celrep.2021.108728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiskopf D., Schmitz K.S., Raadsen M.P., Grifoni A., Okba N.M.A., Endeman H., van den Akker J.P.C., Molenkamp R., Koopmans M.P.G., van Gorp E.C.M., et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020;5:eabd2071. doi: 10.1126/sciimmunol.abd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulien I., Kemming J., Oberhardt V., Wild K., Seidel L.M., Killmer S., Sagar, Daul F., Salvat Lago M., Decker A., et al. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat. Med. 2021;27:78–85. doi: 10.1038/s41591-020-01143-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhou R., To K.K., Wong Y.C., Liu L., Zhou B., Li X., Huang H., Mo Y., Luk T.Y., Lau T.T., et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity. 2020;53:864–877.e5. doi: 10.1016/j.immuni.2020.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Bert N., Tan A.T., Kunasegaran K., Tham C.Y.L., Hafezi M., Chia A., Chng M.H.Y., Lin M., Tan N., Linster M., et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584:457–462. doi: 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- 8.Nelde A., Bilich T., Heitmann J.S., Maringer Y., Salih H.R., Roerden M., Lübke M., Bauer J., Rieth J., Wacker M., et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat. Immunol. 2021;22:74–85. doi: 10.1038/s41590-020-00808-x. [DOI] [PubMed] [Google Scholar]
- 9.Oja A.E., Saris A., Ghandour C.A., Kragten N.A.M., Hogema B.M., Nossent E.J., Heunks L.M.A., Cuvalay S., Slot E., Linty F., et al. Divergent SARS-CoV-2-specific T- and B-cell responses in severe but not mild COVID-19 patients. Eur. J. Immunol. 2020;50:1998–2012. doi: 10.1002/eji.202048908. [DOI] [PubMed] [Google Scholar]
- 10.Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Oxford Immunology Network Covid-19 Response T cell Consortium. ISARIC4C Investigators Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S., et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekine T., Perez-Potti A., Rivera-Ballesteros O., Strålin K., Gorin J.B., Olsson A., Llewellyn-Lacey S., Kamal H., Bogdanovic G., Muschiol S., et al. Karolinska COVID-19 Study Group Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell. 2020;183:158–168.e14. doi: 10.1016/j.cell.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferretti A.P., Kula T., Wang Y., Nguyen D.M.V., Weinheimer A., Dunlap G.S., Xu Q., Nabilsi N., Perullo C.R., Cristofaro A.W., et al. Unbiased Screens Show CD8+ T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 that Largely Reside outside the Spike Protein. Immunity. 2020;53:1095–1107.e3. doi: 10.1016/j.immuni.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kared H., Redd A.D., Bloch E.M., Bonny T.S., Sumatoh H., Kairi F., et al. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J. Clin. Invest. 2021;131(5):e145476. doi: 10.1172/JCI145476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ni L., Ye F., Cheng M.L., Feng Y., Deng Y.Q., Zhao H., Wei P., Ge J., Gou M., Li X., et al. Detection of SARS-CoV-2-Specific Humoral and Cellular Immunity in COVID-19 Convalescent Individuals. Immunity. 2020;52:971–977.e3. doi: 10.1016/j.immuni.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braun J., Loyal L., Frentsch M., Wendisch D., Georg P., Kurth F., Hippenstiel S., Dingeldey M., Kruse B., Fauchere F., et al. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature. 2020;587:270–274. doi: 10.1038/s41586-020-2598-9. [DOI] [PubMed] [Google Scholar]
- 17.Mateus J., Grifoni A., Tarke A., Sidney J., Ramirez S.I., Dan J.M., Burger Z.C., Rawlings S.A., Smith D.M., Phillips E., et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science. 2020;370:89–94. doi: 10.1126/science.abd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reed S.G. Cross-viral protection against SARS-CoV-2? Nat. Rev. Immunol. 2021;21:3. doi: 10.1038/s41577-020-00483-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyerholz D.K., Perlman S. Does common cold coronavirus infection protect against severe SARS-CoV-2 disease? J. Clin. Invest. 2021;131:e144807. doi: 10.1172/JCI144807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balz K., Trassl L., Härtel V., Nelson P.P., Skevaki C. Virus-Induced T Cell-Mediated Heterologous Immunity and Vaccine Development. Front. Immunol. 2020;11:513. doi: 10.3389/fimmu.2020.00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gil A., Kenney L.L., Mishra R., Watkin L.B., Aslan N., Selin L.K. Vaccination and heterologous immunity: educating the immune system. Trans. R. Soc. Trop. Med. Hyg. 2015;109:62–69. doi: 10.1093/trstmh/tru198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mysore V., Cullere X., Mears J., Rosetti F., Okubo K., Liew P.X., Zhang F., Madera-Salcedo I., Rosenbauer F., Stone R.M., et al. FcgR engagement reprograms neutrophils into antigen cross-presenting cells that elicit acquired anti-tumor immunity. Nat. Commun. 2021;12:4791. doi: 10.1038/s41467-021-24591-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amanna I.J., Slifka M.K. Successful Vaccines. Curr. Top. Microbiol. Immunol. 2020;428:1–30. doi: 10.1007/82_2018_102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sung S.J. Monocyte-Derived Dendritic Cells as Antigen-Presenting Cells in T-Cell Proliferation and Cytokine Production. Methods Mol. Biol. 2019;2020:131–141. doi: 10.1007/978-1-4939-9591-2_9. [DOI] [PubMed] [Google Scholar]
- 25.Tang-Huau T.L., Segura E. Human in vivo-differentiated monocyte-derived dendritic cells. Semin. Cell Dev. Biol. 2019;86:44–49. doi: 10.1016/j.semcdb.2018.02.018. [DOI] [PubMed] [Google Scholar]
- 26.Harding C.V., Canaday D., Ramachandra L. Choosing and preparing antigen-presenting cells. Curr. Protoc. Immunol. 2010;Chapter 16:Unit 16.1. doi: 10.1002/0471142735.im1601s88. [DOI] [PubMed] [Google Scholar]
- 27.Nicolet B.P., Guislain A., Wolkers M.C. Combined Single-Cell Measurement of Cytokine mRNA and Protein Identifies T Cells with Persistent Effector Function. J. Immunol. 2017;198:962–970. doi: 10.4049/jimmunol.1601531. [DOI] [PubMed] [Google Scholar]
- 28.Cano-Gamez E., Soskic B., Roumeliotis T.I., So E., Smyth D.J., Baldrighi M., Willé D., Nakic N., Esparza-Gordillo J., Larminie C.G.C., et al. Single-cell transcriptomics identifies an effectorness gradient shaping the response of CD4+ T cells to cytokines. Nat. Commun. 2020;11:1801. doi: 10.1038/s41467-020-15543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thevarajan I., Nguyen T.H.O., Koutsakos M., Druce J., Caly L., van de Sandt C.E., Jia X., Nicholson S., Catton M., Cowie B., et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat. Med. 2020;26:453–455. doi: 10.1038/s41591-020-0819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He Z., Ren L., Yang J., Guo L., Feng L., Ma C., Wang X., Leng Z., Tong X., Zhou W., et al. Seroprevalence and humoral immune durability of anti-SARS-CoV-2 antibodies in Wuhan, China: a longitudinal, population-level, cross-sectional study. Lancet. 2021;397:1075–1084. doi: 10.1016/S0140-6736(21)00238-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Norman M., Gilboa T., Ogata A.F., Maley A.M., Cohen L., Busch E.L., Lazarovits R., Mao C.P., Cai Y., Zhang J., et al. Ultrasensitive high-resolution profiling of early seroconversion in patients with COVID-19. Nat. Biomed. Eng. 2020;4:1180–1187. doi: 10.1038/s41551-020-00611-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curtsinger J.M., Mescher M.F. Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol. 2010;22:333–340. doi: 10.1016/j.coi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deshpande P., Cavanagh M.M., Le Saux S., Singh K., Weyand C.M., Goronzy J.J. IL-7- and IL-15-mediated TCR sensitization enables T cell responses to self-antigens. J. Immunol. 2013;190:1416–1423. doi: 10.4049/jimmunol.1201620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh S., Perera L.P., Burke D.S., Waldmann T.A., Berzofsky J.A. IL-15/IL-15Ralpha-mediated avidity maturation of memory CD8+ T cells. Proc. Natl. Acad. Sci. USA. 2004;101:15154–15159. doi: 10.1073/pnas.0406649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X., Yu J., Cheng X., Zhao B., Manyam G.C., Zhang L., Schluns K., Li P., Wang J., Sun S.C. The deubiquitinase Otub1 controls the activation of CD8+ T cells and NK cells by regulating IL-15-mediated priming. Nat. Immunol. 2019;20:879–889. doi: 10.1038/s41590-019-0405-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Setoguchi R. IL-15 boosts the function and migration of human terminally differentiated CD8+ T cells by inducing a unique gene signature. Int. Immunol. 2016;28:293–305. doi: 10.1093/intimm/dxw004. [DOI] [PubMed] [Google Scholar]
- 38.Van Belle T.L., Dooms H., Boonefaes T., Wei X.Q., Leclercq G., Grooten J. IL-15 augments TCR-induced CD4+ T cell expansion in vitro by inhibiting the suppressive function of CD25 High CD4+ T cells. PLoS ONE. 2012;7:e45299. doi: 10.1371/journal.pone.0045299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hagen J., Zimmerman R., Goetz C., Bonnevier J., Houchins J.P., Reagan K., Kalyuzhny A.E. Comparative Multi-Donor Study of IFNγ Secretion and Expression by Human PBMCs Using ELISPOT Side-by-Side with ELISA and Flow Cytometry Assays. Cells. 2015;4:84–95. doi: 10.3390/cells4010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tian Y., Babor M., Lane J., Schulten V., Patil V.S., Seumois G., Rosales S.L., Fu Z., Picarda G., Burel J., et al. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re-expressing CD45RA. Nat. Commun. 2017;8:1473. doi: 10.1038/s41467-017-01728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian Y., Sette A., Weiskopf D. Cytotoxic CD4 T Cells: Differentiation, Function, and Application to Dengue Virus Infection. Front. Immunol. 2016;7:531. doi: 10.3389/fimmu.2016.00531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiskopf D., Bangs D.J., Sidney J., Kolla R.V., De Silva A.D., de Silva A.M., Crotty S., Peters B., Sette A. Dengue virus infection elicits highly polarized CX3CR1+ cytotoxic CD4+ T cells associated with protective immunity. Proc. Natl. Acad. Sci. USA. 2015;112:E4256–E4263. doi: 10.1073/pnas.1505956112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amir E.D., Davis K.L., Tadmor M.D., Simonds E.F., Levine J.H., Bendall S.C., Shenfeld D.K., Krishnaswamy S., Nolan G.P., Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burel J.G., Qian Y., Lindestam Arlehamn C., Weiskopf D., Zapardiel-Gonzalo J., Taplitz R., Gilman R.H., Saito M., de Silva A.D., Vijayanand P., et al. An Integrated Workflow To Assess Technical and Biological Variability of Cell Population Frequencies in Human Peripheral Blood by Flow Cytometry. J. Immunol. 2017;198:1748–1758. doi: 10.4049/jimmunol.1601750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dan J.M., Mateus J., Kato Y., Hastie K.M., Yu E.D., Faliti C.E., Grifoni A., Ramirez S.I., Haupt S., Frazier A., et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. 2021;371:eabf4063. doi: 10.1126/science.abf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patil V.S., Madrigal A., Schmiedel B.J., Clarke J., O’Rourke P., de Silva A.D., Harris E., Peters B., Seumois G., Weiskopf D., et al. Precursors of human CD4+ cytotoxic T lymphocytes identified by single-cell transcriptome analysis. Sci. Immunol. 2018;3:eaan8664. doi: 10.1126/sciimmunol.aan8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szabo P.A., Levitin H.M., Miron M., Snyder M.E., Senda T., Yuan J., Cheng Y.L., Bush E.C., Dogra P., Thapa P., et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 2019;10:4706. doi: 10.1038/s41467-019-12464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bowyer G., Rampling T., Powlson J., Morter R., Wright D., Hill A.V.S., Ewer K.J. Activation-induced Markers Detect Vaccine-Specific CD4+ T Cell Responses Not Measured by Assays Conventionally Used in Clinical Trials. Vaccines (Basel) 2018;6:50. doi: 10.3390/vaccines6030050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fraser J.D., Irving B.A., Crabtree G.R., Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. 1991;251:313–316. doi: 10.1126/science.1846244. [DOI] [PubMed] [Google Scholar]
- 50.López-Martin I., Andrés Esteban E., García-Martínez F.J. Relationship between MMR vaccination and severity of Covid-19 infection. Survey among primary care physicians. Med. Clin. (Barc.) 2021;156:140–141. doi: 10.1016/j.medcle.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gold J.E., Baumgartl W.H., Okyay R.A., Licht W.E., Fidel P.L., Jr., Noverr M.C., Tilley L.P., Hurley D.J., Rada B., Ashford J.W. Analysis of Measles-Mumps-Rubella (MMR) Titers of Recovered COVID-19 Patients. MBio. 2020;11:e02628-20. doi: 10.1128/mBio.02628-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lievano F., Galea S.A., Thornton M., Wiedmann R.T., Manoff S.B., Tran T.N., Amin M.A., Seminack M.M., Vagie K.A., Dana A., Plotkin S.A. Measles, mumps, and rubella virus vaccine (M-M-R™II): a review of 32 years of clinical and postmarketing experience. Vaccine. 2012;30:6918–6926. doi: 10.1016/j.vaccine.2012.08.057. [DOI] [PubMed] [Google Scholar]
- 53.Thomas L.E., Li F., Pencina M.J. Overlap Weighting: A Propensity Score Method That Mimics Attributes of a Randomized Clinical Trial. JAMA. 2020;323:2417–2418. doi: 10.1001/jama.2020.7819. [DOI] [PubMed] [Google Scholar]
- 54.Aaby P., Benn C.S., Flanagan K.L., Klein S.L., Kollmann T.R., Lynn D.J., Shann F. The non-specific and sex-differential effects of vaccines. Nat. Rev. Immunol. 2020;20:464–470. doi: 10.1038/s41577-020-0338-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gustafson C.E., Kim C., Weyand C.M., Goronzy J.J. Influence of immune aging on vaccine responses. J. Allergy Clin. Immunol. 2020;145:1309–1321. doi: 10.1016/j.jaci.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nolz J.C., Richer M.J. Control of memory CD8+ T cell longevity and effector functions by IL-15. Mol. Immunol. 2020;117:180–188. doi: 10.1016/j.molimm.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mantovani A., Netea M.G. Trained Innate Immunity, Epigenetics, and Covid-19. N. Engl. J. Med. 2020;383:1078–1080. doi: 10.1056/NEJMcibr2011679. [DOI] [PubMed] [Google Scholar]
- 58.Agrawal B. Heterologous Immunity: Role in Natural and Vaccine-Induced Resistance to Infections. Front. Immunol. 2019;10:2631. doi: 10.3389/fimmu.2019.02631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Neill L.A.J., Netea M.G. BCG-induced trained immunity: can it offer protection against COVID-19? Nat. Rev. Immunol. 2020;20:335–337. doi: 10.1038/s41577-020-0337-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Covián C., Fernández-Fierro A., Retamal-Díaz A., Díaz F.E., Vasquez A.E., Lay M.K., Riedel C.A., González P.A., Bueno S.M., Kalergis A.M. BCG-Induced Cross-Protection and Development of Trained Immunity: Implication for Vaccine Design. Front. Immunol. 2019;10:2806. doi: 10.3389/fimmu.2019.02806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Netea M.G., Giamarellos-Bourboulis E.J., Domínguez-Andrés J., Curtis N., van Crevel R., van de Veerdonk F.L., Bonten M. Trained Immunity: a Tool for Reducing Susceptibility to and the Severity of SARS-CoV-2 Infection. Cell. 2020;181:969–977. doi: 10.1016/j.cell.2020.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Su L.F., Kidd B.A., Han A., Kotzin J.J., Davis M.M. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity. 2013;38:373–383. doi: 10.1016/j.immuni.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arstila T.P., Casrouge A., Baron V., Even J., Kanellopoulos J., Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–961. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- 64.Wooldridge L., Ekeruche-Makinde J., van den Berg H.A., Skowera A., Miles J.J., Tan M.P., Dolton G., Clement M., Llewellyn-Lacey S., Price D.A., et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem. 2012;287:1168–1177. doi: 10.1074/jbc.M111.289488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coles C.H., Mulvaney R.M., Malla S., Walker A., Smith K.J., Lloyd A., Lowe K.L., McCully M.L., Martinez Hague R., Aleksic M., et al. TCRs with Distinct Specificity Profiles Use Different Binding Modes to Engage an Identical Peptide-HLA Complex. J. Immunol. 2020;204:1943–1953. doi: 10.4049/jimmunol.1900915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wucherpfennig K.W. T cell receptor crossreactivity as a general property of T cell recognition. Mol. Immunol. 2004;40:1009–1017. doi: 10.1016/j.molimm.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 67.Clute S.C., Naumov Y.N., Watkin L.B., Aslan N., Sullivan J.L., Thorley-Lawson D.A., Luzuriaga K., Welsh R.M., Puzone R., Celada F., Selin L.K. Broad cross-reactive TCR repertoires recognizing dissimilar Epstein-Barr and influenza A virus epitopes. J. Immunol. 2010;185:6753–6764. doi: 10.4049/jimmunol.1000812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen H.D., Fraire A.E., Joris I., Welsh R.M., Selin L.K. Specific history of heterologous virus infections determines anti-viral immunity and immunopathology in the lung. Am. J. Pathol. 2003;163:1341–1355. doi: 10.1016/S0002-9440(10)63493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mathurin K.S., Martens G.W., Kornfeld H., Welsh R.M. CD4 T-cell-mediated heterologous immunity between mycobacteria and poxviruses. J. Virol. 2009;83:3528–3539. doi: 10.1128/JVI.02393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Selin L.K., Varga S.M., Wong I.C., Welsh R.M. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J. Exp. Med. 1998;188:1705–1715. doi: 10.1084/jem.188.9.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim S.K., Cornberg M., Wang X.Z., Chen H.D., Selin L.K., Welsh R.M. Private specificities of CD8 T cell responses control patterns of heterologous immunity. J. Exp. Med. 2005;201:523–533. doi: 10.1084/jem.20041337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Che J.W., Selin L.K., Welsh R.M. Evaluation of non-reciprocal heterologous immunity between unrelated viruses. Virology. 2015;482:89–97. doi: 10.1016/j.virol.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ehl S., Klenerman P., Aichele P., Hengartner H., Zinkernagel R.M. A functional and kinetic comparison of antiviral effector and memory cytotoxic T lymphocyte populations in vivo and in vitro. Eur. J. Immunol. 1997;27:3404–3413. doi: 10.1002/eji.1830271240. [DOI] [PubMed] [Google Scholar]
- 74.Chen H.D., Fraire A.E., Joris I., Brehm M.A., Welsh R.M., Selin L.K. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat. Immunol. 2001;2:1067–1076. doi: 10.1038/ni727. [DOI] [PubMed] [Google Scholar]
- 75.Haddad-Boubaker S., Othman H., Touati R., Ayouni K., Lakhal M., Ben Mustapha I., Ghedira K., Kharrat M., Triki H. In silico comparative study of SARS-CoV-2 proteins and antigenic proteins in BCG, OPV, MMR and other vaccines: evidence of a possible putative protective effect. BMC Bioinformatics. 2021;22:163. doi: 10.1186/s12859-021-04045-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reche P.A. Potential Cross-Reactive Immunity to SARS-CoV-2 From Common Human Pathogens and Vaccines. Front. Immunol. 2020;11:586984. doi: 10.3389/fimmu.2020.586984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sidiq K.R., Sabir D.K., Ali S.M., Kodzius R. Does Early Childhood Vaccination Protect Against COVID-19? Front. Mol. Biosci. 2020;7:120. doi: 10.3389/fmolb.2020.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marakasova E., Baranova A. MMR Vaccine and COVID-19: Measles Protein Homology May Contribute to Cross-Reactivity or to Complement Activation Protection. MBio. 2021;12:e03447-20. doi: 10.1128/mBio.03447-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Young A., Neumann B., Mendez R.F., Reyahi A., Joannides A., Modis Y., Franklin R.J.M. Homologous protein domains in SARS-CoV-2 and measles, mumps and rubella viruses: Preliminary evidence that MMR vaccine might provide protection against COVID-19. medRxiv. 2020 doi: 10.1101/2020.04.10.20053207. [DOI] [Google Scholar]
- 80.Graham N., Eisenhauer P., Diehl S.A., Pierce K.K., Whitehead S.S., Durbin A.P., Kirkpatrick B.D., Sette A., Weiskopf D., Boyson J.E., Botten J.W. Rapid Induction and Maintenance of Virus-Specific CD8+ TEMRA and CD4+ TEM Cells Following Protective Vaccination Against Dengue Virus Challenge in Humans. Front. Immunol. 2020;11:479. doi: 10.3389/fimmu.2020.00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Northfield J.W., Loo C.P., Barbour J.D., Spotts G., Hecht F.M., Klenerman P., Nixon D.F., Michaëlsson J. Human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T(EMRA) cells in early infection are linked to control of HIV-1 viremia and predict the subsequent viral load set point. J. Virol. 2007;81:5759–5765. doi: 10.1128/JVI.00045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tian Y., Babor M., Lane J., Seumois G., Liang S., Goonawardhana N.D.S., De Silva A.D., Phillips E.J., Mallal S.A., da Silva Antunes R., et al. Dengue-specific CD8+ T cell subsets display specialized transcriptomic and TCR profiles. J. Clin. Invest. 2019;129:1727–1741. doi: 10.1172/JCI123726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sridhar S., Begom S., Bermingham A., Hoschler K., Adamson W., Carman W., Bean T., Barclay W., Deeks J.J., Lalvani A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013;19:1305–1312. doi: 10.1038/nm.3350. [DOI] [PubMed] [Google Scholar]
- 84.Kohlmeier J.E., Miller S.C., Smith J., Lu B., Gerard C., Cookenham T., Roberts A.D., Woodland D.L. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity. 2008;29:101–113. doi: 10.1016/j.immuni.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sathaliyawala T., Kubota M., Yudanin N., Turner D., Camp P., Thome J.J., Bickham K.L., Lerner H., Goldstein M., Sykes M., et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38:187–197. doi: 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Benn C.S., Netea M.G., Selin L.K., Aaby P. A small jab - a big effect: nonspecific immunomodulation by vaccines. Trends Immunol. 2013;34:431–439. doi: 10.1016/j.it.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 87.Kandasamy R., Voysey M., McQuaid F., de Nie K., Ryan R., Orr O., Uhlig U., Sande C., O’Connor D., Pollard A.J. Non-specific immunological effects of selected routine childhood immunisations: systematic review. BMJ. 2016;355:i5225. doi: 10.1136/bmj.i5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Beric-Stojsic B., Kalabalik-Hoganson J., Rizzolo D., Roy S. Childhood Immunization and COVID-19: An Early Narrative Review. Front. Public Health. 2020;8:587007. doi: 10.3389/fpubh.2020.587007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pawlowski C., Puranik A., Bandi H., Venkatakrishnan A.J., Agarwal V., Kennedy R., O’Horo J.C., Gores G.J., Williams A.W., Halamka J., et al. Exploratory analysis of immunization records highlights decreased SARS-CoV-2 rates in individuals with recent non-COVID-19 vaccinations. Sci. Rep. 2021;11:4741. doi: 10.1038/s41598-021-83641-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Monereo-Sanchez J., Luykx J.J., Pinzon-Espinosa J., Richard G., Motazedi E., Westlye L.T., Andreassen O.A., van der Meer D. Vaccination history for diphtheria and tetanus is associated with less severe COVID-19. medRxiv. 2021 doi: 10.1101/2021.06.09.21257809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jehi L., Ji X., Milinovich A., Erzurum S., Merlino A., Gordon S., Young J.B., Kattan M.W. Development and validation of a model for individualized prediction of hospitalization risk in 4,536 patients with COVID-19. PLoS ONE. 2020;15:e0237419. doi: 10.1371/journal.pone.0237419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Long Q.X., Jia Y.J., Wang X., Deng H.J., Cao X.X., Yuan J., Fang L., Cheng X.R., Luo C., He A.R., et al. Immune memory in convalescent patients with asymptomatic or mild COVID-19. Cell Discov. 2021;7:18. doi: 10.1038/s41421-021-00250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lazarevic I., Pravica V., Miljanovic D., Cupic M. Immune Evasion of SARS-CoV-2 Emerging Variants: What Have We Learnt So Far? Viruses. 2021;13:1192. doi: 10.3390/v13071192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sabino E.C., Buss L.F., Carvalho M.P.S., Prete C.A., Jr., Crispim M.A.E., Fraiji N.A., Pereira R.H.M., Parag K.V., da Silva Peixoto P., Kraemer M.U.G., et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet. 2021;397:452–455. doi: 10.1016/S0140-6736(21)00183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brehm M.A., Pinto A.K., Daniels K.A., Schneck J.P., Welsh R.M., Selin L.K. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat. Immunol. 2002;3:627–634. doi: 10.1038/ni806. [DOI] [PubMed] [Google Scholar]
- 96.Jehi L., Ji X., Milinovich A., Erzurum S., Rubin B.P., Gordon S., Young J.B., Kattan M.W. Individualizing Risk Prediction for Positive Coronavirus Disease 2019 Testing: Results From 11,672 Patients. Chest. 2020;158:1364–1375. doi: 10.1016/j.chest.2020.05.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mehta N., Kalra A., Nowacki A.S., Anjewierden S., Han Z., Bhat P., Carmona-Rubio A.E., Jacob M., Procop G.W., Harrington S., et al. Association of Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Testing Positive for Coronavirus Disease 2019 (COVID-19) JAMA Cardiol. 2020;5:1020–1026. doi: 10.1001/jamacardio.2020.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zheng G.X., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of the 90 shared CDR3 sequences and cells count presence within sample and cluster.
List of 386 marker genes identified comparing clusters 2, 15, and 18 against all other clusters. Columns include gene symbol, raw p value, average log fold change, percent presence within clusters of interest, percent presence outside clusters of interest, adjusted p value and presence of genes associated with TEMRA cell type identified in Patil et al.49 and Szabo et al.46
Data Availability Statement
Sequence data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) database and are publicly available as of the date of publication. An accession number is listed in the Key resource table.
Original code has not been deposited due to patient privacy concerns.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
The patient datasets used in the current study are available upon request under appropriate data use agreements with the specific parties interested in academic collaboration. For further information, please contact Drs. Lara Jehi (JEHIL@ccf.org) or Michael Kattan (kattanm@ccf.org).