

Abstract
Background
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in children is generally milder than in adults, but a proportion of cases result in hyperinflammatory conditions often including myocarditis.
Methods
To better understand these cases, we applied a multiparametric approach to the study of blood cells of 56 children hospitalized with suspicion of SARS-CoV-2 infection. Plasma cytokine and chemokine levels and blood cellular composition were measured, alongside gene expression at the bulk and single-cell levels.
Findings
The most severe forms of multisystem inflammatory syndrome in children (MIS-C) related to SARS-CoV-2 that resulted in myocarditis were characterized by elevated levels of pro-angiogenesis cytokines and several chemokines. Single-cell transcriptomics analyses identified a unique monocyte/dendritic cell gene signature that correlated with the occurrence of severe myocarditis characterized by sustained nuclear factor κB (NF-κB) activity and tumor necrosis factor alpha (TNF-α) signaling and associated with decreased gene expression of NF-κB inhibitors. We also found a weak response to type I and type II interferons, hyperinflammation, and response to oxidative stress related to increased HIF-1α and Vascular endothelial growth factor (VEGF) signaling.
Conclusions
These results provide potential for a better understanding of disease pathophysiology.
Funding
Agence National de la Recherche (Institut Hospitalo-Universitaire Imagine, grant ANR-10-IAHU-01; Recherche Hospitalo-Universitaire, grant ANR-18-RHUS-0010; Laboratoire d’Excellence ‘‘Milieu Intérieur,” grant ANR-10-LABX-69-01; ANR-flash Covid19 “AIROCovid” and “CoVarImm”), Institut National de la Santé et de la Recherche Médicale (INSERM), and the “URGENCE COVID-19” fundraising campaign of Institut Pasteur.
Keywords: COVID-19, SARS-CoV-2, scRNA-seq, MIS-C, PIMS-TS, Kawasaki Disease, myocarditis, TNF-α and NF-κB signaling, lack of responses to type I and type II IFN secretion
Graphical abstract
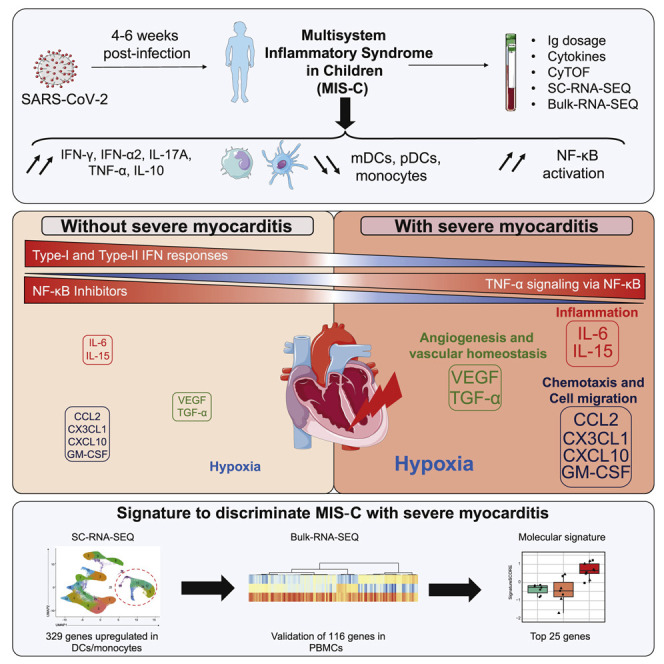
Context and significance
Children with SARS-CoV-2 infection were initially thought to have only mild COVID-19 symptoms. However, several weeks into the first wave of SARS-CoV-2 infections, there was a surge of a postacute pathology called multisystem inflammatory syndrome in children (MIS-C). The authors recruited a cohort of children with suspicion of SARS-CoV-2 infection and uncovered hyperinflammation, hypoxic conditions, exacerbation of TNF-α signaling via NF-κB, and absence of responses to type I and type II IFN secretion in the most severe forms of MIS-C with severe myocarditis. This work led the authors to identify in monocytes and validate in peripheral blood mononuclear cells a molecular signature of 25 genes that allows discrimination of the most severe forms of MIS-C with myocarditis.
Multiparametric analysis identifies, in monocytes and dendritic cells, a molecular signature of the most severe forms of multisystem inflammatory syndrome in children caused by SARS-CoV-2 infection. Severe myocarditis is characterized by an excess of TNF-α signaling via NF-κB, hypoxic conditions, and hyperinflammation in the absence of type I and type II interferon responses.
Introduction
In adults, critical forms of coronavirus disease 2019 (COVID-19) are typically characterized by severe pneumonia and acute respiratory distress syndrome.1 In children, symptomatic COVID-19 occurs much less frequently and is milder than in adults for reasons that remain poorly understood.2, 3, 4, 5, 6 However, in regions with high incidence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, some children have presented a postacute hyperinflammatory illness.7 In these cases, diagnostic evidence of recent SARS-CoV-2 infection has been consistently reported.8, 9, 10, 11 This condition was named multisystem inflammatory syndrome in children (MIS-C) or, alternatively, PIMS-TS (pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2).12 MIS-C cases can present with symptoms similar to Kawasaki disease (KD), a hyperinflammatory illness characterized by clinical features such as a strawberry-like tongue and red and dry lips, bulbar conjunctival injection, diffuse rash, swollen extremities, and cervical lymphadenopathy.13 KD complications can develop as myocarditis or shock syndrome in a minority of cases.14 KD is thought to be triggered by viral or bacterial pathogens, but the precise pathophysiological mechanisms remain elusive.15 Compared with classic KD, MIS-C occurs in individuals who are older; often have gastrointestinal symptoms, myocarditis, and shock syndrome; and exhibit higher levels of inflammatory markers.7 , 8 , 10 , 11
The inflammatory features of MIS-C in part overlap with those of KD and acute SARS-CoV-2 infection in children as well as severe COVID-19 in adults.7 , 16, 17, 18 Very high levels of C-reactive protein (CRP), procalcitonin (PCT), and interleukin-6 (IL-6), might reflect a strong immunological response to a pathogenic SARS-CoV-2 superantigen.19
To further decipher SARS-CoV-2-related conditions in children, we performed a detailed multiparametric study combining sensitive cytokine measurements, deep immune cell phenotyping, and transcriptomics analyses at the single-cell level on peripheral blood mononuclear cells (PBMCs). We first compared data from children with SARS-CoV-2 acute infection and postacute hyperinflammation and then analyzed pathways and molecular signatures characteristic of the most severe form of MIS-C with severe myocarditis.
Results
Clinical description of the cohort
The study cohort consisted of 56 children hospitalized during the first peak of the SARS-CoV-2 pandemic (from April 6 to May 30, 2020) and 34 healthy controls (n = 26 children and n = 8 adults recruited before the COVID-19 pandemic) (Figure 1 ; Table S1). Among the 13 children with acute respiratory infection suspected to have SARS-CoV-2 (Table S1; Figure S1), 9 had a confirmed SARS-CoV-2 infection (RT-PCR on nasopharyngeal aspiration or swab) (Acute-Inf (CoV-2+) group). Six of these 9 individuals had pneumonia, and one had an uncomplicated febrile seizure. Antibiotic therapy was given to 3 of 9 individuals. Only one individuals with a history of recent bone marrow transplantation for sickle cell disease required intensive care support and received tocilizumab. The 4 other individuals (Acute-Inf (CoV-2−) group) had pneumonia associated with a positive RT-PCR test for Mycoplasma pneumoniae or rhinovirus/enterovirus and negative RT-PCR for SARS-CoV-2.
Figure 1.

Timeline and experimental designs
(A) Timeline depicting when the different groups of children were enrolled.
(B) Description of the different types of analyses performed on whole blood samples, peripheral blood mononuclear cells (PBMCs), and plasma. CyTOF: mass cytometry (cytometry by time of flight). scRNA-seq: single-cell RNA sequencing; Simoa: single-molecule array, digital ELISA; Luminex: cytokine bead array assays; Ig dosage: quantification of SARS-CoV-2-specific immunoglobulins; Control (CTL): healthy donors, green; Acute-Inf (CoV-2−): individuals with acute respiratory infection but no evidence of SARS-CoV-2 infection, gray; Acute-Inf (CoV-2+): individuals with acute respiratory infection and evidence of SARS-CoV-2 infection, blue; MIS-C (CoV-2+): individuals with postacute multi-inflammatory syndrome and evidence of SARS-CoV-2 infection, orange; MIS-C_MYO (CoV-2+): individuals with postacute hyperinflammatory syndrome, severe myocarditis, and evidence of SARS-CoV-2 infection, red; KD (CoV-2−): individuals with postacute hyperinflammatory syndrome, no evidence of SARS-CoV-2 infection, but criteria for Kawasaki disease (KD), pink. Illustrations were obtained from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Unported License (https://smart.servier.com/).
Forty-three children displayed features of postacute hyperinflammatory illness (Figure S1; Table S1). The SARS-CoV-2 infection status of all samples was confirmed by specific antibody determination (immunoglobulin G [IgG] and IgA) in the plasma, using ELISA and flow cytometry-based techniques (Figure S2A). Most (n = 30) had confirmed SARS-CoV-2 infection (with 14 also positive for concomitant nasopharyngeal RT-PCR testing) and were therefore considered cases of MIS-C (MIS-C (CoV-2+) group); all 30 cases of MIS-C presented clinical features of KD, and 14 of them fulfilled clinical criteria for a complete form of KD according to the American Heart Association.13 Of note, 21 of 30 cases had severe myocarditis (i.e., with elevated high-sensitivity cardiac troponin I and/or regional wall motion abnormalities on echocardiography and clinical signs of circulatory failure requiring intensive care support; MIS-C_MYO (CoV-2+)). Thirteen tested negative for SARS-CoV-2 and fulfilled clinical criteria for complete (n = 6) or incomplete (n = 7) KD, and were therefore considered to have KD-like illness (KD (CoV-2−) group) (Figure S1; Table S1). Clinical and biological characteristics at time of disease activity and before treatment or within 24 h of treatment onset are presented in Table S1. Most children (41 of 43) with postacute hyperinflammation received intravenous immunoglobulin (IVIG) injections, many of them shortly after admission and, therefore, before inclusion and sampling (n = 7 of 9 in MIS-C (CoV-2+), n = 20 of 21 in MIS-C_MYO (CoV-2+), n = 11 of 13 in KD (CoV-2−)). Adjunction of corticosteroids was decided by the clinician in 16 of the 43 cases, mostly because of initial severity, high risk of IVIG unresponsiveness,20 or IVIG unresponsiveness. They were administered before sampling for some of them (n = 1 in MIS-C (CoV-2+), n = 7 in MIS-C_MYO (CoV-2+), n = 4 in KD CoV-2−)). All individuals responded favorably to IVIG alone or in combination with glucocorticosteroids. Individuals with MIS-C had low lymphocyte counts, and those with severe myocarditis had abnormally increased neutrophil counts compared with other groups, along with high levels of CRP, PCT, serum alanine transaminase (ALT) and ferritin (Table S1). Multiparametric analyses were performed at a median fever persistence of 9–10 days (Figures 1A and 1B) and focused on children with acute infection and postacute hyperinflammation related to confirmed SARS-CoV-2 infection.
Elevated inflammatory cytokine levels in pediatric acute infection and postacute hyperinflammatory conditions
We investigated plasma cytokine and chemokine levels in all individuals by multiplexed or ultrasensitive ELISAs. Hierarchical clustering analysis and stratification by group revealed overall elevated levels of immune and inflammatory markers, with 40 of 46 measured proteins elevated significantly (q < 0.05) compared with healthy controls (Figures 2 A and S2B; global heatmap). Thirteen cytokines were found to be elevated in all groups of affected individuals compared with healthy controls (Figure S2C). High IL-8 and CXCL1 (Figure 2C) were more specific to children with acute infection. Cytokine levels did not differ significantly in children with acute infection with or without evidence of SARS-CoV-2 infection (Figures S2B). Interferon (IFN)-α2, IFN-γ, IL-17A, TNF-α, and IL-10 were higher in children with postacute hyperinflammation (MIS-C (CoV-2+) and MIS-C_MYO (CoV-2+)) than in pediatric healthy donors (CTL) and individuals with acute infection (Acute-Inf (CoV-2+)) and were also elevated in the KD (CoV-2−) group (Figures 2A, 2B, and S2B).
Figure 2.

Analyses of cytokine/chemokine plasma levels
(A) Heatmap of all cytokines/chemokines measured in the different clinical groups: CTL, green; Acute-Inf (CoV-2+), blue; MIS-C (CoV-2+), orange; MIS-C_MYO (CoV-2+), red. On the x axis, blood donors are organized by groups and immune-modulatory treatment (untreated, blue; treated, yellow), and on the y axis, cytokines/chemokines are displayed following hierarchical clustering. Cytokines/chemokines were expressed as pg/mL and log transformed, with blue to orange colors representing lower to higher expression, respectively.
(B) Dot plots of cytokines/chemokines elevated in postacute hyperinflammatory groups (MIS-C (CoV-2+) and MIS-C_MYO (CoV-2+)) compared with Acute-Inf (CoV-2+) and healthy blood donors (CTL).
(C) Dot plots of cytokines/chemokines elevated in Acute-Inf (CoV-2+) compared with postacute hyperinflammatory groups (MIS-C (CoV-2+) and MIS-C_MYO (CoV-2+)) and healthy blood donors (CTL).
(B and C) p values are calculated by Kruskal-Wallis test for multiple comparisons, followed by a post hoc Dunn’s test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
See also Figure S2.
High inflammatory cytokine levels were detected in acute infection and postacute inflammatory cases. Even though most individuals with postacute hyperinflammation received IVIG and/or corticosteroids before sampling, the strongest inflammatory profile was observed in MIS-C. Because IVIG and corticosteroids are known immune modulators, differences between acute infection and postacute hyperinflammation are unlikely to be due to the treatments.21 , 22 IVIG treatment could account for the increase in anti-inflammatory mediators such as IL-10, IL-1RA, and transforming growth factor β1 (TGF-β1), as reported previously.23, 24, 25, 26 We could not assess the specific effects of IVIG treatment because almost all postacute cases were treated with IVIG before sampling, but the inflammatory profile was much reduced in intensity in individuals with MIS-C under combined glucocorticosteroid and IVIG treatment compared with IVIG alone (Figure 2A).
Low monocyte and dendritic cell frequencies in individuals with postacute hyperinflammatory illness
To better decipher the blood immune cell composition of each group, PBMCs were analyzed by cytometry by time of flight (CyTOF) mass spectrometry and by single-cell analyses at the transcriptomics level (single-cell RNA sequencing [scRNA-seq]) (Figure 1). Regarding CyTOF and single-cell analyses, all individuals with MIS-C were treated with IVIG, and only one individual in the group with severe myocarditis also received glucocorticosteroids before sampling. Clustering analyses of the data obtained from CyTOF and scRNA-seq revealed consistent results, with most of the alterations observed in clusters composed of monocytes or dendritic cells (DCs) (Figures 3 A, 3B, and S3A–S3D). The most drastic changes were a decrease in conventional DCs (cDCs) and plasmacytoid DCs (pDCs) in individuals with a postacute hyperinflammatory illness (MIS-C_MYO (CoV-2+), MIS-C (CoV-2+), and KD (CoV-2−)). As reported previously,18 we also observed a trend toward a decrease in monocyte clusters in children with postacute hyperinflammatory illness that was found independent of SARS-CoV-2 status (Figures 3A, 3B and S3A–S3D). It has been reported that, in some cases, IVIG treatment could impair DC function and decrease monocyte and DC proportions.23 , 25 , 27, 28, 29, 30, 31 In addition, some heterogeneity was observed in the proportions of non-classical monocytes in Acute-Inf (CoV-2+) cases and additional heterogeneity in the proportions of classical and intermediate monocytes in individuals with severe myocarditis (MIS-C_MYO (CoV-2+)) (Figures 3A and 3B), but there was no correlation with clinical data, including treatments, age (Table S1), and cytokine/chemokine measurements (Figures 2 and S2). Additional modifications were detected in individuals with acute SARS-CoV-2 infection (Acute-Inf (CoV-2+) cases), consisting of a decrease in mucosal-associated invariant T cells (MAIT) and an excess of naive and central memory CD4+ T cells (Figures 3A, 3B, S3C, and S3D). Because the median age is very low in this group (0.2 years; Table S1) compared with other groups, we cannot exclude an age effect.
Figure 3.

CyTOF and scRNA-seq characterization of PBMCs distribution
(A) Top panel: UMAP of 1,150,000 single cells from PBMCs of 7 CTL, 1 Acute-Inf (CoV-2−), 4 Acute-Inf (CoV-2+), 2 MIS-C (CoV-2+), 6 MIS-C_MYO (CoV-2+), and 3 KD (CoV-2−) donors following analyses by CyTOF and displayed as 23 clusters identified using the individual expression of 29 proteins, as described in Figure S3A. Bottom panel: boxplots of clusters with differences observed between SARS-CoV-2+ groups and CTL (Acute-Inf (CoV-2+), MIS-C (CoV-2+), and MIS-C_MYO (CoV-2+)).
(B) Top panel: Uniform Manifold Approximation and Projection (UMAP) of 152,201 single cells following extraction from PBMCs (9 CTL, 1 Acute-Inf (CoV-2−), 4 Acute-Inf (CoV-2+), 2 MIS-C (CoV-2+), 6 MIS-C_MYO (CoV-2+), and 3 KD (CoV-2−)) and processed by scRNA-seq. A resolution of 0.8 allows us to segregate cells into 26 clusters identified based on the expression of several markers and gene signatures, as shown in Figure S4B. Bottom panel: boxplots of clusters with significant differences between SARS-CoV-2+ groups and CTL (Acute-Inf (CoV-2+), MIS-C (CoV-2+), and MIS-C_MYO (CoV-2+)). See also Figure S3.
Groups are represented by the following colors: CTL, green; Acute-Inf (CoV-2+), blue; MIS-C (CoV-2+), orange; MIS-C_MYO (CoV-2+)). In the boxplots, each dot represents a sample. Boxes range from the 25th to the 75th percentiles. The upper and lower whiskers extend from the box to the largest and smallest values, respectively. Any sample with a value at most ×1.5 the interquartile range of the hinge is considered an outlier and plotted individually. p values are calculated by Kruskal-Wallis test for multiple comparisons, followed by a post hoc Dunn’s test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Overexpression of inflammatory pathways, NF-κB signaling, and metabolic changes related to hypoxia in acute infection and postacute hyperinflammatory conditions
To gain further insight into the mechanisms behind acute infection and postacute hyperinflammation driven by SARS-CoV-2 in children, we assessed pathways modulated in each group by looking at the differentially expressed genes obtained from the scRNA-seq dataset. In individuals with acute infection (Acute-Inf (CoV-2+) and Acute-Inf (CoV-2−)), the numbers of differentially expressed genes were distributed homogeneously among monocytes/DCs and T and B cells (Figures 4 A and S4A). Pathway enrichment analyses revealed a decrease in oxidative phosphorylation coupled with an increase in HMGB1 signaling, HIF-1α signaling, and hypoxia signaling (Figure 4B). Production of nitric oxide was observed in both groups of acute infection, independent of SARS-CoV-2 infection, compared with healthy controls (Figure S4B). These observations suggest a metabolic switch potentially driven by hypoxic conditions. Nuclear factor κB (NF-κB) signaling, VEGF signaling, and inflammatory pathways (type I and type II IFNs, IL-1, IL-6, and IL-17 signaling pathways) were also found to be overrepresented in both groups (Figure S4B).
Figure 4.

Genes and pathways differentially regulated in acute infection and postacute hyperinflammation following SARS-CoV-2 infection
(A) Bar charts of the number of up- and downregulated genes in Acute-Inf (CoV-2+) (left panel) and All MIS-C (MIS-C (CoV-2+) and MIS-C_MYO (CoV-2+)) (right panel) compared to CTL in PBMCs, monocyte/cDC/pDC, and T and B cell clusters obtained following scRNA-seq experiments as displayed in Figure 3B. PBMCs represent all clusters. Monocytes/cDCs/pDCs represent clusters 5, 11, 12, 17, 20, 21, and 24. T cells represent clusters 0, 1, 2, 4, 6, 7, 10, 13, 14, 15, 16, 18, and 23. B cells represent clusters 3, 8, 9, 19, and 22. The top value on the light-colored bars represents the upregulated genes, and the bottom value (dark) represents the downregulated genes. Median age for each group: CTL, 15 years; MIS-C (CoV-2+), 3.7 years; MIS-C_MYO (CoV-2+), 8.4 years.
(B) Heatmap of the canonical pathways, enriched in the differentially expressed genes (DEGs) from the comparisons performed in (A) in PBMCs, monocytes/cDCs/pDCs, and T and B cells, obtained by using Ingenuity Pathways Analysis (IPA). Left panel: part 1. Right panel: part 2. Symbols are used in front to represent pathways of the same functional groups. Pathways with an absolute Z score ≤ 2 or adjusted p > 0.05 under all conditions were filtered out. Z score > 2 means that a function is increased significantly (orange) whereas Z score < −2 indicates a significantly decreased function (blue). Gray dots indicate non-significant pathways (p > 0.05).
(C) Heatmap of activation of the NF-κB signaling pathway, as predicted by IPA, in Acute-Inf (CoV-2+) and in All MIS-C (MIS-C (CoV-2+) and MIS-C_MYO (CoV-2+)) compared with controls. The color scale represents the Z score of the prediction. The higher the score, the more activated the NF-κB signaling pathway. NS, non-significant comparison.
(D) Dot plot of the expression in monocytes/cDCs/pDCs of the negative regulators of the NF-κB complex. The color scale shows scaled average expression in all monocytes/cDCs/pDCs, with red and blue being the highest and lowest expression, respectively. Sizes of dots show the percentage of cells that express the gene.
Interestingly, alterations in the same pathways were also identified in all cases of children with SARS-CoV-2-related postacute illnesses (All MIS-C (CoV-2+): MIS-C_MYO (CoV-2+) and MIS-C (CoV-2+)). However, in these cases, alterations were mostly restricted to monocytes and DCs (Figures 4A and 4B). Comparisons of genes differentially expressed between children with postacute hyperinflammatory illness with or without evidence of SARS-CoV-2 infection (All MIS-C (CoV-2+) versus KD (CoV-2−)) did not reveal significant differences except for type I and type II IFN signaling (Figures S4C and S4D).
The NF-κB signaling pathway was identified to be activated in monocytes and DCs of all individuals with acute infection and postacute hyperinflammatory illness, independent of SARS-CoV-2 infection (Figure 4C). Although monocytes and DCs of individuals with acute infection (Acute-Inf (CoV-2+)) highly expressed genes of the NF-κB complex (REL, RELA, RELB, NFKB1, and NFKB2) (Figure 4D), monocytes and DCs from all individuals with MIS-C (MIS-C_MYO (CoV-2+) and MIS-C (CoV-2+)) exhibited a strong decrease in expression of NF-κB inhibitors, such as NFKBIA, NFKBID, NFKBIE, and NFKBIZ (Figure 4D).
Pathways dysregulated in acute infection or postacute hyperinflammatory illness reflected an inflammatory status based on NF-κB signaling combined with changes in metabolism driven by a hypoxic environment. In acute respiratory disease, changes in gene expression reflected involvement of all PBMCs, whereas in postacute hyperinflammatory illnesses, monocytes/DCs were the most affected populations. These results further support involvement of monocyte/DC populations in MIS-C.
Exacerbated TNF-α and NF-κB signaling in MIS-C with severe myocarditis
To identify differences between individuals with and without severe myocarditis, we compared cytokines/chemokines and gene expression profiles in individuals treated with IVIG only before sampling. Slightly higher expression of TRAIL, IL-7, IL-2, IL-13, IFN-γ, IFN-α2, IL-17A, and Granzyme B was found in MIS-C without myocarditis (MIS-C (CoV-2+)) (Figures 5 A and S5A). In contrast, 17 cytokines and chemokines were higher in MIS-C with severe myocarditis (MIS-C_MYO (CoV-2+)) (Figures 5A, 5B, and S5B). 9 of them are known to be associated with TNF-α and NF-κB signaling (Figure 5B). They are involved in propagation of inflammation (IL-6), angiogenesis and vascular homeostasis (VEGF and TGF cytokines) and activation, and chemotaxis and migration of myeloid cells (CCL2, CX3CL1, CXCL10, CCL20, and CCL3).32 , 33 Increased levels of CCL19 (cell migration and chemotaxis) and IL-1 antagonist (IL-1RA) were also observed, as well as increased soluble PD-L1 (Figure S5B). Other noticeably elevated cytokines were CSF2 and CSF3, known to be involved in myeloid cell differentiation and migration (Figure S5B).
Figure 5.

Cytokine/chemokine and gene expression analyses reveal exacerbation of TNF-α and NF-κB signaling pathways in MIS-C_MYO (CoV-2+) compared with MIS-C (CoV-2+)
(A) Heatmap of the cytokines/chemokines showing differences between MIS-C (CoV-2+) (orange) and MIS-C_MYO (CoV-2+) (red) in individuals not treated with corticosteroids before sampling. On the x axis, blood donors are organized by group and treatment received before sampling. On the y axis, cytokines/chemokines are displayed following hierarchical clustering. Cytokines/chemokines were expressed as pg/mL and log transformed, with blue to orange colors representing lower to higher expression, respectively.
(B) Dot plots of cytokines/chemokines elevated in MIS-C_MYO (CoV-2+) compared with MIS-C (CoV-2+) and related to TNF-α and NF-κB signaling. p values are calculated by Kruskal-Wallis test for multiple comparisons, followed by a post hoc Dunn’s test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(C) Dot plot of the expression in monocytes/cDCs/pDCs of the 49 genes from TNF-α signaling via the NF-κB pathway (pathway enrichment analysis by MSigDB Hallmark 2020 obtained from the upregulated genes of in the MIS-C_MYO (CoV-2+) group compared with MIS-C (CoV-2+); Figure S6B).
(D) Dot plot of the expression in monocytes/cDCs/pDCs of the negative regulators of the NF-κB complex in the MIS-C groups.
Regarding differentially expressed genes, most changes were observed in monocytes/DCs, which led us to focus on these populations for the following analyses (Figures S5C and S5D). Strong overexpression of genes belonging to TNF-α and NF-κB signaling was found in monocytes/DCs of MIS-C with severe myocarditis (MIS-C_MYO (CoV-2+)) (Figure 5C). Strikingly, the decrease in expression of NF-κB inhibitors observed in all individuals with MIS-C (Figure 4D) appeared to be specific to monocytes and DCs of individuals with MIS-C with severe myocarditis (MIS-C_MYO (CoV-2+)) (Figure 5D). Among the other pathways upregulated in MIS-C_MYO (CoV-2+), we identified inflammatory responses, hypoxia, and response to oxidative stress (HIF1A, HMOX1, HMBG1, etc.) (Figures S6A and S6B). TGF-β signaling and VEGF signaling were also enriched in monocytes and DCs of individuals with myocarditis and to a lesser magnitude in B cells (Figures S6A and S6B).
NF-κB activation, decreased expression of NF-κB inhibitors, TNF-α signaling, and a hypoxic response to oxidative stress and VEGF signaling characterize the monocytes and DCs of children with MIS-C and severe myocarditis.
A lack of response to type I and type II IFN in MIS-C with severe myocarditis
Pathway enrichment performed with Ingenuity Pathway Analysis (IPA) and EnrichR34 , 35 highlighted the modulation of type I and type II IFN signaling pathways, with upregulation of several IFN-stimulated genes (ISGs) (JAK2, STAT1, STAT2, IFITM1, IFITM2, IFI35, IFIT1, IFIT3, MX1, IRF1) in monocytes/DCs and T and B cells of individuals with MIS-C without myocarditis only (Figures 6 A–6D, S6A, and S6C). However, both groups of individuals with MIS-C showed elevated plasma IFN-α2 and IFN-γ proteins (Figures 2A, 2B, and 6A). Gene expression downregulation in monocytes/DCs of individuals with MIS-C with severe myocarditis (including most of the major histocompatibility complex [MHC] class II genes) suggests a decrease in antigen processing and presentation pathways (Figure S6 C) along with downregulation of genes linked with oxidative phosphorylation, nitric oxide production, and inducible Nitric Oxide Synthase (iNOS) signaling, which could be related to establishment of hypoxic conditions and response to oxidative stress (Figure S6A). Because all individuals with MIS-C analyzed in single-cell experiments had received IVIG prior to sampling, changes observed at the level of gene expression are unlikely to be biased because of this treatment.
Figure 6.

Differences in IFN responses between MIS-C (CoV-2+) and MIS-C_MYO (CoV-2+)
(A) IFN-α and IFN-γ protein levels measured by Simoa (top two panels) and ISGs (type I and type II ISG) expression measured by scRNA-seq and displayed as signatureSCORE on all PBMCs (bottom two panels).
(B and C) Violin plots of type I (B) and type II (C) IFN signaling signatures analyzed in monocytes/cDCs/pDCs and B and T cells.
(D) Dot plot of the expression in monocytes/cDCs/pDCs cells of 60 genes from the IFN signaling pathway enrichment analysis by MSigDB Hallmark 2020 obtained from the downregulated genes in the MIS-C_MYO (CoV-2+) group compared with MIS-C (CoV-2+) (Figure S6E). See also Figure S6.
(A and D) Color scales show scaled average expression in all cells, with red and blue being the highest and lowest expression, respectively. Sizes of dots show the percentage of cells that express the gene.
Identification of a molecular signature specific to MIS-C with severe myocarditis
To identify the potential clinical relevance of our study, we searched for a molecular signature that correlated with the appearance of severe myocarditis among the monocytes/DCs of children with SARS-CoV-2-related MIS-C. By using several scRNA-seq comparison strategies (Figure 7 A), we identified 329 genes upregulated in monocytes/DCs of the MIS-C group with myocarditis (n = 6) compared with all other groups (Figure 7A). To validate this molecular signature, RNA from PBMCs was sequenced from individuals enrolled in our study but not analyzed by scRNA-seq. A scoring system was generated, based on normalized expression represented by a Z score, coupled with hierarchical clustering, to identify genes that were overexpressed in children with myocarditis (MIS-C_MYO (CoV-2+) group) compared with the other groups (Figure S7A). Within the 329 genes identified by scRNA-seq in monocytes and DCs of individuals with severe myocarditis, expression of 116 genes was upregulated in PBMCs from a group of 9 individuals belonging to the MIS-C_MYO (CoV-2+) group with myocarditis and not analyzed by scRNA-seq (Figures 7B). From these genes, a signature score (SignatureSCORE) was determined for each sample processed by bulk RNA SEQ (Figure 7C). We then further developed a RankingSCORE (Figures S7A and S7B) to identify the top genes that contributed the most to the monocyte and DC myocarditis signature. This led to identification of a set of 25 genes that clearly segregate individuals with severe myocarditis from other MIS-C (Figure 7D). Most of these 25 genes belong to functional pathways that have been identified previously (Figures 5, S5, 6, and S6), such as inflammation, oxidative stress, TNF-α and/or NF-κB signaling, and, in some cases, already known markers of myocarditis or MIS-C and/or COVID-19, such as genes coding for S100 proteins (Figures S7C–S7E). S100 proteins and calcium-binding cytosolic proteins are known to serve as danger signals to regulate cell migration, homeostasis, and inflammation and have been reported recently as new biomarkers for the most severe forms of COVID-19 in adults with acute severe respiratory syndrome.36
Figure 7.

Molecular signature and proposed mechanism associated with severe myocarditis in children with MIS-C
(A) Schematic of the 3 strategies used to extract 329 markers of the MIS-C_MYO (CoV-2+) group from the monocyte/cDC/pDC clusters using the single-cell dataset. Strategy 1: direct comparison of the monocytes/cDCs/pDCs of the MIS-C_MYO (CoV-2+) group with all other samples. Strategy 2: direct comparison of the monocytes/cDCs/pDCs of MIS-C_MYO (CoV-2+) with other samples with postacute hyperinflammation (MIS-C (CoV-2+) and KD (CoV-2−)). Strategy 3: selection of genes upregulated only in the monocytes/cDCs/pDCs of MIS-C_MYO (CoV-2+) compared with CTL. Below each strategy is the corresponding dot plot obtained from scRNA-seq with the number of upregulated genes. The average expression is represented by the centered scaled expression of each gene. On the left, names of each group with their corresponding color are shown.
(B) Heatmap of expression of the 116 of 329 genes with a higher expression in MIS-C_MYO (CoV-2+) than in other groups in the bulk dataset generated from PBMCs (7 CTL, 7 MIS-C (CoV-2+), 9 MIS-C_MYO (CoV-2+), and 9 KD (CoV-2−) donors). The color scale indicates the scaled GeneSCORE (mean Z score of the gene in all samples of a group), with red and blue representing the highest and lowest expression, respectively. Hierarchical clustering of the genes was computed with a Pearson’s correlation as a distance.
(C) Boxplot of the expression of the 116 genes validated in (C), calculated as a SignatureSCORE, which represents the mean Z score in each sample of the 116 genes selected in (B) in the bulk RNA-seq dataset (Figure S7A).
(D) Boxplot of the SignatureSCORE of the top 25 genes, as ranked in Figure S7B, in the bulk RNA-seq dataset.
(E) Graphical representation based on cytokines,cellular, and transcriptomics analyses (above the black dotted line), combined with known literature (below the black dotted line), illustrating a putative model explaining the occurrence of myocarditis among children in the MIS-C (CoV-2) group. Black represents genes and functions modulated in the MIS-C (CoV-2+) and MIS-C_MYO (CoV-2+) groups compared with CTL, whereas red highlights genes and pathways differentially modulated in the MIS-C (CoV-2+) and MIS-C_MYO (CoV-2+) groups, respectively. See also Figure S7 and Data S3.
(C and D) Each mark represents a sample. Dots represent untreated samples, triangles represent IVIG-treated samples, and squares represent IVIG- and steroid-treated individuals. Boxes range from the 25th to the 75th percentiles. The upper and lower whiskers extend from the box to the largest and smallest values, respectively. Any sample with a value at most ×1.5 the inter-quartile range of the hinge is considered an outlier and plotted individually.
Discussion
Multi-parametric analysis of PBMCs from children with acute respiratory infection and postacute hyperinflammation, collected during the COVID-19 pandemic, allowed us to detect an inflammatory profile associated with a loss of circulating monocytes and DCs, as well as upregulation of genes and pathways involving NF-κB signaling, oxidative stress with establishment of hypoxic conditions, and VEGF signaling. These pathways were upregulated in acute and postacute groups of individuals, independent of SARS-CoV-2 infection. However, significant features of MIS-C with severe myocarditis were detected specifically in monocytes and DCs, including increased TNF-α and NF-κB signaling, decreased expression of NF-κB inhibitors, transcriptional responses corresponding to hypoxic conditions, and low type I and type II IFN responses, despite elevated cytokines detected in the plasma.
Acute cases were characterized by detection of inflammatory markers in the plasma with a particularly strong elevation of IL-8 and CXCL1, two chemokines known to mediate neutrophil migration to the lungs37, 38, 39 and a modest elevation of IFN-α2 levels. These findings suggest that, in some children, a suboptimal anti-viral type I IFN response, along with a hyperinflammatory response (IL-6 levels and exacerbation of the NF-κB pathway), could account for SARS-CoV-2 disease with pneumonia compared with the very usual benign or even asymptomatic clinical course of SARS-CoV-2 infection in children. This has been observed previously in severe respiratory syncytial virus infections.40
In postacute individuals treated with IVIG alone, elevated levels of plasma IFN-γ, IFN-α2, IL-10, IL-17, and, to a lesser extent, TNF-α, were found, as described previously for other cohorts.16, 17, 18 , 41 , 42 These findings are typical of an ongoing anti-viral immune response not directly related to SARS-CoV-2 infection. In addition, elevated chemokines, such as CCL2, CCL3, and CCL4, may recruit monocytes and DCs to tissues, possibly accounting for their reduced numbers observed in the blood of these individuals. Additional mechanisms, such as apoptosis or other cell death pathways, may also be involved, and we cannot exclude an effect of the IVIG treatment, as reported previously in some studies.23 , 27 , 28 , 30 , 31 Compared with children with acute infection, most of the individuals with postacute hyperinflammation received IVIG treatment before sampling, some combined with glucocorticosteroids. The immunosuppressive effects of glucocorticoids are identified at the cytokine/chemokine level. Nevertheless, IVIG treatment cannot account for the observed increase in inflammatory cytokines. However, IVIG treatment could explain the increase in anti-inflammatory mediators, such as IL-10, IL-1RA, and TGF-β1, observed in MIS-C.22, 23, 24, 25, 26 , 29
Cellular phenotypes that distinguish MIS-C from classic KD have been reported previously.17 , 41 , 42 Consoglio et al.17 described several key differences, such as elevated IL-17, IL-6, and CXCL10, that were only observed in KD, associated with decreased naive CD4+ T cells and increased central memory and effector memory CD4+ T cells in MIS-C. In the present study, high levels of IL-17, IL-6, and CXCL10 were found in MIS-C and in our KD (CoV-2−) groups. Differences observed in previous reports17 may be due to the clinical homogeneity of our MIS-C group because all had KD criteria. Centers for Disease Control and Prevention (CDC) and World Health Organization (WHO) case definitions show a much broader spectrum of disease, and Consoglio et al.17, like some other clinical studies,8 may have included individuals with multisystem involvement along with laboratory evidence of inflammation of MIS-C but without KD. Another explanation may be the time of blood sampling relative to admission to hospital and medical treatments. Third, we cannot exclude that our KD (CoV-2−) individuals were different from individuals with “classic” KD enrolled before the start of the COVID-19 pandemic. Our data still support the hypothesis that individuals with MIS-C with KD features exhibit a molecular phenotype close to the one seen in those with KD, suggesting overlapping pathogenesis mechanisms,18 but the effect of treatments received prior to sampling cannot be excluded.
Strikingly, we did find noticeable differences when comparing MIS-C with MIS-C cases associated with severe myocarditis and circulatory failure that required intensive care. Expression of several cytokines/chemokines was increased further in these cases—most of them related to the NF-κB−TNF-α signaling axis. Elevated VEGF and TGF-α and TGF-β are potential drivers of angiogenesis and vascular homeostasis, whereas elevated chemokines (CCL2, CCL3, CCL20, CX3CL1, and CXCL10) could mediate increased cell migration toward inflamed tissues. Molecular analysis confirmed upregulation of genes belonging to the TNF-α and NF-κB signaling pathways that were found specifically in monocytes and DCs of individuals with MIS-C with severe myocarditis. Lower expression of NF-κB complex inhibitors, including TNFAIP3 (A20), TNFAIP2, NFKBIA, and NFKBIZ, was detected, suggesting a possible mechanism for NF-κB sustained activation, which could then potentially lead to exacerbated TNF-α signaling. These results point to a potential role of monocytes and DCs in the pathogenesis of MIS-C with severe myocarditis, which might not be driven directly by SARS-CoV-2 infection but, rather, the consequence of a defect in a regulatory process limiting a pathological immune response, as already observed for other pathogens.43 It would be interesting to investigate the presence of genetic variants among MIS-C with severe myocarditis in genes such as TNFAIP3, as discussed previously.43 The apparent hypoxic conditions detected in children with myocarditis could also account for the exacerbation of NF-κB signaling. HIF-1α, a sensor of oxidative stress, is well known for being able to induce a switch from oxidative phosphorylation to glycolysis to limit generation of reactive oxygen species (ROS). It can also activate NF-κB signaling.44 , 45 Additional environmental factors and/or genetic predispositions could also be involved. Another striking feature was the low expression of genes involved in type I and type II IFN responses, specifically in monocytes and DCs of children with myocarditis, although IFN-γ and IFN-α2 proteins were elevated in the plasma of all individuals with MIS-C. Although an absence of type II IFN responses could account for reduced Human Leucocyte Antigen-DR (HLA-DR) cell surface expression by monocytes/DCs, the reduced response to type I IFN in the most severe forms of MIS-C (with myocarditis and circulatory failure) is in part reminiscent of the impaired type I IFN activity observed in the most severe forms of COVID-19 in adults.46, 47, 48 The search for auto-antibodies against IFN-α2 were negative (data not shown), but a presence of autoantibodies to ISGs cannot be excluded.49 Because all individuals with MIS-C analyzed by scRNA-seq had received IVIG prior to sampling, the effect of the treatment is unlikely to explain the differences observed between MIS-C with or without severe myocarditis. Furthermore, IVIG has been described to downregulate rather than upregulate TNF-α and NF-κB signaling.24, 25, 26 , 29 Abrogation of type I IFN responses following IVIG treatment has been described,50 although it is unlikely to explain here the differences between both groups of MIS-C.
Overall, our findings depict a model, supported by previous publications,51, 52, 53 where myocarditis is associated with an attenuated negative feedback loop of TNF-α-driven NF-κB activation together with an excess of proangiogenic cytokines and chemokines that could attract activated myeloid and T cells to the myocardium tissue (Figure 7E). Locally, it could lead to production of inflammatory cytokines known to promote differentiation of cardiac fibroblasts into cardiac myofibroblasts (TNF-α, TGF-β, IL1β, IL-13, IL-4, and VEGF). Cardiac myofibroblasts, as reported previously, may secrete chemokines, leading to further activation and recruitment of myeloid cells, creating a feedforward loop of locally sustained inflammation and myocarditis.51 , 54, 55, 56, 57
Using scRNA-seq data, we defined a gene signature specific for SARS-CoV-2-related postacute hyperinflammatory illness with severe myocarditis that was validated by a global transcriptomics analysis on PBMCs from individuals not analyzed by scRNA-seq. The genes defining this signature were consistently enriched in genes associated with inflammation, TNF-α and NF-κB signaling, oxidative stress, and myocarditis (Figure S7C). Interestingly, among these genes, the S100 proteins and the calprotectin complex (S100A8/S100A9) in particular have been reported previously and proposed as biomarkers for the most severe adult form of COVID-19 with acute respiratory syndrome (Figure S7D).36 Moreover, recent deep immune profiling of adult and pediatric individuals with SARS-CoV-2 highlighted similarities between MIS-C and moderate to severe adult COVID-19 profiles.58 Despite different clinical symptoms and disease temporality between adults infected with SARS-CoV-2 and children with MIS-C, our study underscored striking similarities at the cellular and molecular levels. First, as observed in adults, increased leucocyte counts, activated neutrophils combined with lymphopenia, and decreased myeloid cells are characteristic of the most severe forms of MIS-C with myocarditis.17 , 41 The hypercytokinemia described in adults is also found in children with MIS-C (elevated TNF-α, IL-6, IL-10, granulocyte-macrophage colony-stimulating factor (GM-CSF), monocyte chemoattractant protein 1 [MCP1]/CCL2, and macrophage inflammatory protein [MIP-1α]/CCL3).36 , 59 , 60 At the gene expression level, as reported in adults, the most severe disease forms in children are associated with TNF-α, NF-κB signaling, genes associated with hypoxia and/or oxidative stress (HIF1A and HMGB1), and reduced type I IFN responses.
Interestingly, cardiac involvement in adults hospitalized with COVID-19 occurs frequently, with echocardiographic aspects similar to pediatric MIS-C with myocarditis.61 , 62 In some cases, they are associated with myocardial injury with dysfunction and elevated troponin levels, more often associated with poor outcomes.63, 64, 65, 66 In autopsy studies, cardiac infection was common in individuals who died from COVID-19, although cells infected by SARS-CoV-2 were rare. Cardiac infection was often associated with myocardial inflammatory cell infiltration by macrophages and lymphocytes, alongside myocarditis, in rare cases.67
Limitations of the study
Our study has several limitations, including the relatively low number of cases in each group and lack of a comparison with asymptomatic or mildly symptomatic non-hospitalized children positive for SARS-CoV-2 and a longitudinal study of children with “classic” KD enrolled before the COVID-19 pandemic. Our KD group was not homogeneous because it included complete and incomplete KD; although we used the American Heart Association (AHA) algorithm and excluded other alternative diagnoses, we cannot exclude that incomplete KD cases were over-diagnosed and, therefore, misclassified. However, comparison of their biological and cytokine data with those of complete KD cases did not reveal any significant differences (data not shown), and we thought it important to consider all KD cases with which clinicians are confronted in their daily practice. Also, because of the severity of the illness in MIS-C, which requires immediate treatment, blood samples were almost exclusively collected after immunomodulatory treatment with IVIG and, in some cases, following addition of glucocorticoids. Potential effects of treatments were discussed throughout the manuscript. Differences in median age between groups exist and were taken into consideration during analyses. All of our cellular data were generated from frozen peripheral mononuclear cells, which does not allow direct assessment of neutrophils and cannot exclude any bias in cell proportions and immune analyses, although all samples were processed using the same methods. A parallel analysis of polymorphonuclear leukocytes will be required. Endothelial and myocardiac cells are at least targets of the disease but may also contribute to the pathophysiology, as described above. Also, additional data supporting gene expression findings will be necessary in future studies. Nevertheless, our study provides a further in-depth molecular analysis of MIS-C with severe myocarditis. These severe forms were found to be associated with excessive activation of the TNF-α and NF-κB signaling axis and poor response to type I and type II IFNs in monocytes and DCs, secretion of cytokines promoting angiogenesis, and chemotaxis and potential migration of activated myeloid cells and neutrophils in the myocardiac tissue. This may help to identify potential new clinical biomarkers and open new therapeutic strategies, including drugs targeting the TNF-α or NF-κB pathways.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Human CD45 (HI30) - 89Y - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD196 /CCR6 (G034E3) - 141Pr - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD123 (6H6) - 143Nd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD19 (HIB19) - 144Nd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD4 (RPA-T4) - 145Nd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD8a (RPA-T8) - 146Nd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD11c (Bu15) - 147Sm - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD16 (3G8) - 148Nd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD45RO (UCHL1) - 149Sm - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD45RA (HI100) - 150Nd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD161 (HP-3G10) - 151Eu - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD194/CCR4 (L291H4) - 152Sm - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD25 (BC96) - 153Eu - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD27 (O323) - 154Sm - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD57 (HCD57) - 155Gd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD183/CXCR3 (G025H7) - 156Gd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD185/CXCR5 (J252D4) - 158Gd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD28 (CD28.2) - 160Gd - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD38 (HB-7) - 161Dy - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD56/NCAM (NCAM16.2) - 163Dy - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human TCRγδ (B1) - 164Dy - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD294 (BM16) - 166Er - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD197/CCR7 (G043H7) - 167Er - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD14 (63D3) - 168Er - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD3 (UCHT1) - 170Er - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD20 (2H7) - 171Yb - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD66b (G10F5) - 172Yb - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human HLA-DR (LN3) - 173Yb - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human IgD (IA6-2) - 174Yb - 25 tests | Fluidigm | Cat#201325 |
| Anti-Human CD127 (A019D5) - 176Yb - 25 tests | Fluidigm | Cat#201325 |
| Cell-ID Intercalator-103Rh - 103Rh - 25 tests | Fluidigm | Cat#201325 |
| Biological samples | ||
| Human blood samples | Adult and Pediatric patients | This paper, Table S1 |
| Chemicals, peptides, and recombinant proteins | ||
| Ficoll | Eurobio Scientific | Cat#CMSMSL01-01 |
| Fetal Bovine Serum | GIBCO, Thermo Fisher Scientific | Cat#10270106 |
| DMSO | Sigma Aldrich | Cat#D2650 |
| Sodium Heparin Salt | Sigma Aldrich | Cat# H3149-10KU |
| TruStain FcX | Biolegend | Cat#422302 |
| Cal-LyseTM Lysing Solution (with formaldehyde and EGTA) | Thermo Fisher Scientific | Cat# GAS-010S100 (100 ml) |
| PierceTM 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher Scientific | Cat# 28906 (10 × 1 mL) |
| RNeasy Mini Kit (50) | QIAGEN | Cat# 74104 |
| EQ Beads | Fluidigm | Cat# 201078 |
| Universal Plus mRNA-Seq kit | Nugen | Cat# 0530-32 |
| Critical commercial assays | ||
| Luminex multi-analyte assay | Biotechne, R&D systems | Cat# LKTM014 |
| Homebrew assay | Quanterix | Cat# 101076 |
| Triplex assay | Quanterix | Cat# 101160 |
| Maxpar Direct Immune Profiling Assay | Fluidigm | Cat# 201325 |
| Chromium Single Cell 3′ Library & Gel Bead Kit v3 | 10X Genomics | PN-1000075 |
| RNase-Free DNase Set | QIAGEN | Cat# 79254 |
| Deposited data | ||
| Single-Cell RNA-sequencing data | This paper | GEO: GSE167029 |
| Bulk RNA-sequencing data | This paper | GEO: GSE167028 |
| Software and algorithms | ||
| Ingenuity Pathway Analysis v57662101 | QIAGEN | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Seurat v3.1 | Stuart et al.68 | https://satijalab.org/seurat/ |
| Flowjo v10.7 | BD bioscience | https://www.flowjo.com/solutions/flowjo/downloads |
| Flowjo pluggin: Downsample v3.3 | BD bioscience | https://www.flowjo.com/exchange/#/plugin/profile?id=25 |
| Flowjo pluggin: UMAP v3.1 | BD bioscience; McInnes et al.69 | https://www.flowjo.com/exchange/#/plugin/profile?id=6 |
| Flowjo pluggin: FlowSom v2.9 | BD bioscience; Van Gassen et al.70 | https://www.flowjo.com/exchange/#/plugin/profile?id=7 |
| Enrich R | Chen et al.34; Kuleshov et al.35 | https://maayanlab.cloud/Enrichr/ |
| CyTOF software version 6.7.1014 | Fluidigm | https://www.fluidigm.com/software |
| CellRanger V3.1 | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Hisat2 | Kim et al.71 | https://daehwankimlab.github.io/hisat2/ |
| DESeq2 (version 1.28.1) | Love et al.72 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Qlucore OMICS explore | Qlucore | https://www.qlucore.com/omics-explorer |
| GraphPAD Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact and corresponding author, Mickaël Ménager (mickael.menager@institutimagine.org)
Material availability
This study did not generate new unique reagents.
Experimental model and subject details
Patients and cohorts
This prospective multicenter cohort study included children (age ≤ 18 years at the time of admission) suspected of infection with SARS-CoV-2 between April 6, 2020 and May 30, 2020. Clinical aspects of 22 of the included patients were previously reported.10 , 11 Children admitted with fever in general pediatric wards or pediatric intensive care units of Tertiary French hospitals involved in the research program, suspected of SARS-CoV-2 related illness and who underwent routine nasopharyngeal swabs for SARS-CoV-2 RT-PCR (R-GENE, Argene, Biomerieux, Marcy l’Etoile) or SARS-CoV-2 IgG serology testing (Architect SARS-CoV-2 chemiluminescent microparticle immunoassay; Abbott Core Laboratory, IL, USA), were eligible. The study was approved by the Ethics Committee (Comité de Protection des Personnes Ouest IV, n° DC-2017-2987). All parents provided written informed consent.
Case definition for pediatric COVID-19 acute infection was presence of fever, fatigue, neurological abnormalities, gastro-intestinal or respiratory signs, associated with a concomitant nasopharyngeal swab positive for SARS-CoV-2 RT-PCR, and absence of MIS-C criteria.73 Case definition for postacute hyperinflammatory illness (Figure 1) was presence of fever, laboratory evidence of inflammation and clinically severe illness with multisystem involvement, during the SARS-CoV-2 epidemic period.7 This may include children with features of KD; criteria of the American Heart Association was used to define for complete (Fever > 4 days and ≥ 4 principal criteria) or incomplete KD (Fever > 4 days and 2 or 3 principal criteria, and without characteristics suggestive of another diagnosis).13 Among cases with postacute hyperinflammatory illness, children with a positive SARS-CoV-2 testing (RT-PCR or serology) were considered to have MIS-C according to CDC and WHO criteria to define MIS-C.74 Patients with postacute hyperinflammatory illness, negative SARS-CoV-2 testing (RT-PCR or serology), and criteria for KD, were considered as patients with KD-like illness. Patients with MIS-C with clinical signs of circulatory failure requiring intensive care, with elevated high-sensitivity cardiac troponin I levels (> 26 ng/mL) and/or decreased cardiac function (diastolic or systolic ventricular dysfunction at echocardiography), were considered to have MIS-C with severe myocarditis.75 , 76
For each included patient, we collected demographic data, symptoms, results of SARS-CoV-2 testing and other laboratory tests, echocardiograms, and treatments. All patient data are available in Table S1. Introduction of specific treatments for MISC and KD cases was decided by the pediatrician in charge of the patient; it generally consisted of intravenous polyvalent immunoglobulins [IVIG] alone (2g/kg in one or two infusions), or IVIG associated with methylprednisolone (2-10 mg/kg/day for at least 3 days) as first or second-line therapy. Patients with negative initial serology testing were retested after an interval of at least 3 weeks (Architect SARS-CoV-2 chemiluminescent microparticle immunoassay; Abbott Core Laboratory).
Healthy controls were recruited before the COVID-19 pandemic (before November 2019).
Samples
For each patient and healthy donor, peripheral blood samples were collected on EDTA and lithium heparin tubes. After a centrifugation of the EDTA tube at 2300rpm for 10 minutes, plasma was taken and stored at −80°C before cytokine quantification. PBMCs were isolated from the lithium heparin samples, frozen as described below and stored at −80°C and were used for both bulk and single-cell RNaseq, as well as cell phenotyping by CyTOF. The number of samples included in each dataset is summarized in the metadata table (Data S1) and the workflow is summarized in Figure 1B.
Method details
Isolation of PBMCs
Peripheral blood samples were collected on lithium heparin. PBMCs were isolated by density gradient centrifugation (2,200 rpm without break for 30 minutes) using Ficoll (Eurobio Scientific, Les Ulis, France). After centrifugation, cells were washed with Phosphate-buffered saline (PBS) (Thermo Fisher scientific, Illkirch, France). The pellet was resuspended in PBS and cells were centrifuged at 1,900 rpm for 5 minutes. Finally, the PBMCs pellet was frozen in a medium containing 90% of Fetal Bovine Serum (FBS) (GIBCO, Thermo Fisher scientific, Illkirch, France) and 10% of dimethyl sulfoxide (DMSO) (Sigma Aldrich, St. Quentin Fallavier, France).
Cytokine measurements
Prior to protein analysis plasma samples were treated in a BSL3 laboratory for viral decontamination using a protocol previously described for SARS-CoV,77 which we validated for SARS-CoV-2. Briefly, samples were treated with TRITON X100 (TX100) 1% (v/v) for 2h at Room Temperature. IFN-α2, IFN-γ, IL-17A, (triplex) and IFN-β (single plex) protein plasma concentrations were quantified by Simoa assays developed with Quanterix Homebrew kits as previously described.78 The limit of detection of these assays were 0.6 pg/mL for IFN-β, 2 fg/mL for IFN-α2, 0.05 pg/ml for IFN-γ and 3 pg/mL for IL-17A including the dilution factor. IL-6, TNF-α, and IL-10 were measured with a commercial triplex assay (Quanterix). Additional plasma cytokines and chemokines (44 analytes) were measured with a commercial Luminex multi-analyte assay (Biotechne, R&D systems).
Serology assays
SARS-CoV-2 specific antibodies were quantified using assays previously described.79 Briefly, a standard ELISA assay using as target antigens the extracellular domain of the S protein in the form of a trimer (ELISA tri-S) and the S-Flow assay, which is based on the recognition of SARS-CoV-2 S protein expressed on the surface of 293T cells (293T-S), were used to quantify SARS-CoV-2 specific IgG and IgA subtypes in plasma. Assay characteristics including sensitivity and specificity were previously described.79
Cell Phenotyping
To perform high-dimensional immune profiling of PBMCs, we used the Maxpar® Direct Immune Profiling System (Fluidigm, Inc France) with a 30-marker antibody panel, for CyTOF (Cytometry by Time Of Flight). Briefly, 3x106 PBMCs resuspended in 300 μl of MaxPar Cell Staining Buffer were incubated for 20 minutes at room temperature after addition of 3 μL of 10 KU/mL heparin solution and 5 μl of Human TruStain FcX (Biolegend Europ, Netherland). Then 270 μL of the samples were directly added to the dry antibody cocktail for 30 minutes. 3 mL of MaxPar Water was added to each tube for an additional 10-min incubation. Three washes were performed on all the samples using MaxPar Cell Staining Buffer and they were fixed using 1.6% paraformaldehyde (Sigma-Aldrich, France). After one wash with MaxPar Cell Staining Buffer, cells were incubated one hour in Fix and Perm Buffer with 1:1,000 of Iridium intercalator (pentamethylcyclopentadienyl-Ir (III)-dipyridophenazine, Fluidigm, Inc France). Cells were washed and resuspended at a concentration of 1 million cells per mL in Maxpar Cell Acquisition Solution, a high-ionic-strength solution, and mixed with 10% of EQ Beads immediately before acquisition.
Acquisition of the events was made on the Helios mass cytometer and CyTOF software version 6.7.1014 (Fluidigm, Inc Canada) at the “Plateforme de Cytométrie de la Pitié-Salpetriere (CyPS).” An average of 500,000 events were acquired per sample. Dual count calibration, noise reduction, cell length threshold between 10 and 150 pushes, and a lower convolution threshold equal to 10 were applied during acquisition. Mass cytometry standard files produced by the HELIOS were normalized using the CyTOF Software v. 6.7.1014. For data cleaning, 4 parameters (center, offset, residual and width) are used to resolve ion fusion events (doublets) from single events from the Gaussian distribution generated by each event.80 After data cleaning, the program produces new FCS files consisting of only intact live singlet cells. These data were analyzed in FlowJo v10.7.1 using 3 plugins (DownSampleV3, UMAP and FlowSOM) with R v4.0.2. To increase efficiency of the analysis, samples were downsampled to 50,000 cells, using the DownSample V3 plugin. All samples were concatenated and analyzed in an unsupervised manner. Anti-CD127 antibody had to be excluded due to poor staining. Clustering was performed using FlowSOM.70 The number of clusters was set to forty-five in order to overestimate the populations and detect smaller subpopulations. Grid size of the self-organizing map was set to 20x20. Resulting clusters were annotated as cell populations following the kit manufacturer’s instruction. When several clusters were identified as the same cell types, they were concatenated into a single cell population. For visualization purposes, UMAP was computed with the UMAP pluggin69 with the following parameters: metric (Euclidean), nearest neighbors (15), minimum distance (0.5) and number of components (2).
Single-cell transcriptomic (scRNA-SEQ)
scRNA-SEQ analyses were performed on frozen PBMCs isolated from heparin blood samples. PBMCs were thawed according to 10X Genomics protocol. The scRNA-SEQ libraries were generated using Chromium Single Cell 3′ Library & Gel Bead Kit v.3 (10x Genomics) according to the manufacturer’s protocol. Briefly, cells were counted, diluted at 1,000 cells/μL in PBS+0.04% and 20,000 cells were loaded in the 10x Chromium Controller to generate single-cell gel-beads in emulsion. After reverse transcription, gel-beads in emulsion were disrupted. Barcoded complementary DNA was isolated and amplified by PCR. Following fragmentation, end repair and A-tailing, sample indexes were added during index PCR. The purified libraries were sequenced on a Novaseq 6000 (Illumina) with 28 cycles of read 1, 8 cycles of i7 index and 91 cycles of read 2.
Sequencing reads were demultiplexed and aligned to the human reference genome (GRCh38, release 98, built from Ensembl sources), using the CellRanger Pipeline v3.1. Unfiltered RNA UMI counts were loaded into Seurat v3.168 for quality control, data integration and downstream analyses. Apoptotic cells and empty sequencing capsules were excluded by filtering out cells with fewer than 500 features or a mitochondrial content higher than 20%. Data from each sample were log-normalized and scaled, before batch correction using Seurat’s FindIntegratedAnchors. For computational efficiency, anchors for integration were determined using all control samples as reference and patient samples were projected onto the integrated controls space. On this integrated dataset, we computed the principal component analysis on the 2000 most variable genes. UMAP was carried out using the 20 most significant principal components (PCs), and community detection was performed using the graph-based modularity-optimization Louvain algorithm from Seurat’s FindClusters function with a 0.8 resolution. Cell types labels were assigned to resulting clusters based on a manually curated list of marker genes as well as previously defined signatures of the well-known PBMCs subtypes.81 Despite filtering for high quality cells, five clusters out of the twenty-six stood out as poor quality clusters and were removed from further analysis, namely: one erythroid-cell contamination; one low UMI cluster from a single control; two clusters of proliferating cells originating from a patient with EBV co-infection and one megakaryocytes cluster. In total 152,201 cells were kept for further analysis.
After extraction and reclustering of high-quality cells, differential expression was performed separately on all PBMCs, monocytes/DCs, T cells or B cells. Differential expression testing was conducted using the FindMarkers function of Seurat on the RNA assay with default parameters. Genes with log(FC) > 0.25 and adjusted p values 0.05 were selected as significant. Differential analysis results and links to the pathways analysis in EnrichR34 , 35 can be found in Data S2. Transcriptomic signatures for Type I and Type II interferon signaling were performed using Seurat’s AddModuleScore function, based on interferon-stimulated gene lists extracted from Rosenberg et al.82 and Reactome database respectively. Violin plots were performed using Seurat’s VlnPlot function.
Bulk RNA-sequencing (Bulk-RNA-SEQ)
Bulk-RNA-SEQ analyses were performed on frozen PBMCs extracted from heparin samples. RNA was extracted from PBMCs following the instructions of RNeasyR Mini kit (QIAGEN, Courtaboeuf, France). To note, the optional step with the DNase was performed. RNA integrity and concentration were assessed by capillary electrophoresis using Fragment Analyzer (Agilent Technologies). RNaseq libraries were prepared starting from 100 ng of total RNA using the Universal Plus mRNA-Seq kit (Nugen) as recommended by the manufacturer. The oriented cDNA produced from the poly-A+ fraction was sequenced on a NovaSeq6000 from Illumina (Paired-End reads 100 bases + 100 bases). A total of ~50 million of passing-filters paired-end reads was produced per library.
Paired-end RNA-seq reads were aligned to the human Ensembl genome GRCh38.91 using Hisat2 (v2.0.4)71 and counted using featureCounts from the Subread R package. The raw count matrix was analyzed using DESeq2 (version 1.28.1).72 No pre-filtering was applied to the data. Differential expression analysis was performed using the “DESeq” function with default parameters. For visualization and clustering, the data was normalized using the `variant stabilizing transformation` method implemented in the “vst” function. Plots were generated using ggplot2 (version 3.3.2), and pheatmap (version 1.0.12).
During exploratory analyses, it was noted that the clustering was mainly driven by the sex of the patients. To remove this effect, it was included in the regression formula for DESeq (~sex + groups), and then removed following vst transformation, using “removeBatchEffect” from the “limma” package (version 3.44.3).
Gene signature analysis
To identify genes that could be used as markers of severe myocarditis in the SC-RNA-SEQ dataset, three initial strategies were used, all based on differential expression and selection of the upregulated genes. First, we performed the differential expression between MIS-C_MYO (CoV-2+) samples and all other samples. Second, differential analysis was computed between MIS-C_MYO (CoV-2+) and other samples with postacute hyperinflammatory illness. In the last strategy, we selected genes that were upregulated between the MIS-C_MYO (CoV-2+) and the CTL, but not upregulated in any other group compared to the CTL (Figure 7A). These three strategies allowed us to identify 329 unique genes.
To further explore whether these genes could be considered as markers of severe myocarditis, we analyzed their expression profile in our bulk RNA-SEQ dataset. This dataset excluded samples from patients of the MIS-C_MYO (CoV-2+) that were included in the SC-RNA-SEQ cohort. Vst-transformed counts were log2-normalized and converted to Z score using the scale function in R (v 4.0.2). A GeneSCORE was computed for each group as the mean Z score of the samples of a group. Heatmaps representing this GeneSCOREgroup were performed using pheatmap. Hierarchical clustering of the 329 previously identified genes was performed using the complete method on the distance measured using Pearson’s correlation, as implemented by pheatmap. The hierarchical clustering was divided into 15 main clusters, 4 of which had the expected pattern of expression: Clusters that had a higher expression in MIS-C_MYO (CoV-2+) than any other group were selected, resulting in 116 genes. A signature score for each sample was performed on these genes, corresponding to the mean expression (Z score) of these N genes in each sample (SignatureSCORE).
These genes were subsequently ranked based on the following equation:
where the SCOREs represent the mean expression (Z score) in each disease groups, and the SignatureScore was computed on the top 25 genes. All gene lists and scores can be found in Data S3.
Quantification and statistical analysis
Cytokine heatmaps were made with Qlucore OMICS explore (version 3.5(26)) and dot plots with GraphPad Prism (version 8). Differentially secreted cytokines were included in the heatmaps based on a 1.5-Fold Change (FC) comparison between groups as indicated. Dot plot differences between each group were identified by Kruskal-Wallis tests followed by post hoc multiple comparison Dunn’s test.
Statistical tests for cellular composition analysis in both the CyTOF and SC-RNA-SEQ datasets were performed in R v3.6.1. Kruskal-Wallis test followed by post hoc multiple comparison Dunn’s test was applied to assess differences in cell population proportions (∗: p ≤ 0.05; ∗∗: p ≤ 0.01; ∗∗∗: p ≤ 0.001).
Differential expression testing in the SC-RNA-SEQ dataset was conducted using the FindMarkers function in Seurat, with default Wilcoxon testing. p values were controlled using Bonferroni correction. Genes with an absolute log(fold-change) ≥ 0.25 and an adjusted p value ≤ 0.05 were selected as differentially expressed. Pathways analysis was performed using both the Ingenuity pathway analysis v57662101 software (IPA (QIAGEN Inc.) and EnrichR.34 , 35 Heatmaps were extracted from the comparison module in IPA. Pathways with an absolute Z score lower than 2 or a Bonferroni-Hochberg corrected p values higher than 0.05 were filtered out. Reactome 2016 and Molecular Signature DataBase Hallmark 2020 (MSigDB Hallmark 2020) pathway enrichment analysis was performed using EnrichR. The TRRUST transcription factors 201921 used for the transcription factors enrichment analysis was performed using Enrich R. All differential analysis performed and the links to the EnrichR results are indicated in Data S2.
Consortia
The members of the Pediatric-Biocovid Study Group are François Angoulvant, Camille Aupiais, Fanny Bajolle, Romain Basmaci, Paul Bastard, Matthieu Bendavid, Solène Blache, Stéphane Blanche, Christine Bodemer, Martin Chalumeau, Lucienne Chatenou, Anne Chauviré-Drouard, Fleur Cohen-Aubart, Agathe Debray, Albert Faye, Simon Fillatreau, Jacques Fourgeaud, Pierre Frange, Marion Grimaud, Lucile Houyel, Diala Khraiche, Hanane Kouider, Alain Lefevre-Utile, Pierre-Louis Leger, Morgane Le Gouez, Michael Levy, Manon Marchais, Soraya Matczak, Alexis Mathian, Bénédicte Neven, Perrine Parize, Olivier Pellé, Yael Pinhas, Marie Pouletty, Pierre Quartier dit Maire, Sylvain Renolleau, Anne-Sophie Romain, Laure de Saint-Blanquat, Isabelle Sermet, and Melissa Taylor.
Acknowledgments
The study was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the “URGENCE COVID-19” fundraising campaign of Institut Pasteur, the Atip-Avenir and Emergence Ville de Paris programs and Fond de Dotation Janssen Horizon, government grants managed by the Agence National de la Recherche as part of the “Investment for the Future” program (Institut Hospitalo-Universitaire Imagine grant ANR-10-IAHU-01, Recherche Hospitalo-Universitaire grant ANR-18-RHUS-0010, and Laboratoire d’Excellence ‘‘Milieu Intérieur” grant ANR-10-LABX-69-01), the Centre de Référence Déficits Immunitaires Héréditaires (CEREDIH), the Agence National de la Recherche (ANR-flash Covid19 “AIROCovid” to F.R.L. and “CoVarImm” to D.D. and J.D.S.), and the FAST Foundation (French Friends of Sheba Tel Hashomer Hospital). The LabTech Single-Cell@Imagine is supported by the Paris Region and the “Investissements d’avenir” program through 2019 ATF funding – Sésame Filières PIA (grant 3877871). C.d.C. is the recipient of a CIFRE PhD (Sanofi). L.B. was a recipient of an Imagine Institute PhD international program supported by the Fondation Bettencourt Schueller. L.B. was also supported by EUR G.E.N.E. (reference ANR-17-EURE-0013) and is part of the Université de Paris IdEx #ANR-18-IDEX-0001 funded by the French government through its “Investments for the Future” program. S.M. was a recipient of an INSERM and Institut Imagine post-doctoral program supported by the Fondation pour la Recherche Médicale (FRM SPF20170938825). N.S. was a recipient of the Pasteur-Roux-Cantarini Fellowship. V.G.-P. obtained an Imagine international PhD fellowship program supported by the Fondation Bettencourt Schueller. B.P.P. is the recipient of an ANRS post-doctoral fellowship. We thank the Imagine genomics, bioinformatics, and single-cell core facilities; the Institut Pasteur Cytometry and Biomarkers UTechS platform; and the Pitié-Salpêtrière Cytometry platform CyPS.
Author contributions
C.d.C., M. Luka, S.M., A.M., N.S., F.C., V.G.-P., and L.B. generated and analyzed data. M. Batignes, A.B., B.P.P., G.A., and T.F. analyzed data. M. Batignes, L.G., P.G., J.P.D.S., H.M., O.S., C.B.-F., and J.-L.C. generated data. C.d.C., M. Luka, and A.F. designed figures and wrote the manuscript. D.D., F.R.-L., J.T., and M.M.M. conceived the study, analyzed data, wrote the manuscript, supervised the study, and had unrestricted access to the data.
Declaration of interests
D.D., F.R.-L., J.T., and M.M.M. are listed as inventors on a patent application related to this technology (European Patent Application no. EP21305197, entitled “Methods of predicting multisystem inflammatory syndrome [MIS-C] with severe myocarditis in subjects suffering from a SARS-CoV-2 infection”).
Published: August 14, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.medj.2021.08.002.
Supplemental information
This spreadsheet includes the differential expression tables for all comparisons presented in the paper, performed on all PBMCs and on major cell types (monocytes/cDCs/pDCs, T cells, or B cells). Links to EnrichR analysis presented in Figure S6 are included.
The list of genes extracted from SC-RNA-seq data coming from the 3 different strategies used are listed, alongside the different scores generated from Bulk-RNA-seq data.
Data and code availability
Single-cell and bulk RNA-seq data have been deposited in GEO and are publicly available. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Wiersinga W.J., Rhodes A., Cheng A.C., Peacock S.J., Prescott H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA. 2020;324:782–793. doi: 10.1001/jama.2020.12839. [DOI] [PubMed] [Google Scholar]
- 2.Brodin P. Why is COVID-19 so mild in children? Acta Paediatr. 2020;109:1082–1083. doi: 10.1111/apa.15271. [DOI] [PubMed] [Google Scholar]
- 3.Castagnoli R., Votto M., Licari A., Brambilla I., Bruno R., Perlini S., Rovida F., Baldanti F., Marseglia G.L. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection in Children and Adolescents: A Systematic Review. JAMA Pediatr. 2020;174:882–889. doi: 10.1001/jamapediatrics.2020.1467. [DOI] [PubMed] [Google Scholar]
- 4.Gudbjartsson D.F., Helgason A., Jonsson H., Magnusson O.T., Melsted P., Norddahl G.L., Saemundsdottir J., Sigurdsson A., Sulem P., Agustsdottir A.B., et al. Spread of SARS-CoV-2 in the Icelandic Population. N. Engl. J. Med. 2020;382:2302–2315. doi: 10.1056/NEJMoa2006100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy C., Basmaci R., Bensaid P., Bru C.B., Coinde E., Dessioux E., Fournial C., Gashignard J., Haas H., Hentgen V., et al. Changes in RT-PCR-positive SARS-CoV-2 rates in adults and children according to the epidemic stages. medRxiv. 2020 doi: 10.1101/2020.05.18.20098863. [DOI] [PubMed] [Google Scholar]
- 6.Tagarro A., Epalza C., Santos M., Sanz-Santaeufemia F.J., Otheo E., Moraleda C., Calvo C. Screening and Severity of Coronavirus Disease 2019 (COVID-19) in Children in Madrid, Spain. JAMA Pediatr. 2020 doi: 10.1001/jamapediatrics.2020.1346. Published online April 8, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datta S.D., Talwar A., Lee J.T. A Proposed Framework and Timeline of the Spectrum of Disease Due to SARS-CoV-2 Infection: Illness Beyond Acute Infection and Public Health Implications. JAMA. 2020;324:2251–2252. doi: 10.1001/jama.2020.22717. [DOI] [PubMed] [Google Scholar]
- 8.Abrams J.Y., Godfred-Cato S.E., Oster M.E., Chow E.J., Koumans E.H., Bryant B., Leung J.W., Belay E.D. Multisystem Inflammatory Syndrome in Children Associated with Severe Acute Respiratory Syndrome Coronavirus 2: A Systematic Review. J. Pediatr. 2020;226:45–54.e1. doi: 10.1016/j.jpeds.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones V.G., Mills M., Suarez D., Hogan C.A., Yeh D., Segal J.B., Nguyen E.L., Barsh G.R., Maskatia S., Mathew R. COVID-19 and Kawasaki Disease: Novel Virus and Novel Case. Hosp. Pediatr. 2020;10:537–540. doi: 10.1542/hpeds.2020-0123. [DOI] [PubMed] [Google Scholar]
- 10.Toubiana J., Poirault C., Corsia A., Bajolle F., Fourgeaud J., Angoulvant F., Debray A., Basmaci R., Salvador E., Biscardi S., et al. Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: prospective observational study. BMJ. 2020;369:m2094. doi: 10.1136/bmj.m2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toubiana J., Cohen J.F., Brice J., Poirault C., Bajolle F., Curtis W., Moulin F., Matczak S., Leruez M., Casanova J.-L., et al. Distinctive Features of Kawasaki Disease Following SARS-CoV-2 Infection: a Controlled Study in Paris, France. J. Clin. Immunol. 2021;41:526–535. doi: 10.1007/s10875-020-00941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whittaker E., Bamford A., Kenny J., Kaforou M., Jones C.E., Shah P., Ramnarayan P., Fraisse A., Miller O., Davies P., et al. PIMS-TS Study Group and EUCLIDS and PERFORM Consortia Clinical Characteristics of 58 Children With a Pediatric Inflammatory Multisystem Syndrome Temporally Associated With SARS-CoV-2. JAMA. 2020;324:259–269. doi: 10.1001/jama.2020.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCrindle B.W., Rowley A.H., Newburger J.W., Burns J.C., Bolger A.F., Gewitz M., Baker A.L., Jackson M.A., Takahashi M., Shah P.B., et al. American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Cardiovascular Surgery and Anesthesia; and Council on Epidemiology and Prevention Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation. 2017;135:e927–e999. doi: 10.1161/CIR.0000000000000484. [DOI] [PubMed] [Google Scholar]
- 14.Kanegaye J.T., Wilder M.S., Molkara D., Frazer J.R., Pancheri J., Tremoulet A.H., Watson V.E., Best B.M., Burns J.C. Recognition of a Kawasaki disease shock syndrome. Pediatrics. 2009;123:e783–e789. doi: 10.1542/peds.2008-1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turnier J.L., Anderson M.S., Heizer H.R., Jone P.-N., Glodé M.P., Dominguez S.R. Concurrent Respiratory Viruses and Kawasaki Disease. Pediatrics. 2015;136:e609–e614. doi: 10.1542/peds.2015-0950. [DOI] [PubMed] [Google Scholar]
- 16.Carter M.J., Fish M., Jennings A., Doores K.J., Wellman P., Seow J., Acors S., Graham C., Timms E., Kenny J., et al. Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS-CoV-2 infection. Nat. Med. 2020;26:1701–1707. doi: 10.1038/s41591-020-1054-6. [DOI] [PubMed] [Google Scholar]
- 17.Consiglio C.R., Cotugno N., Sardh F., Pou C., Amodio D., Rodriguez L., Tan Z., Zicari S., Ruggiero A., Pascucci G.R., et al. CACTUS Study Team The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell. 2020;183:968–981.e7. doi: 10.1016/j.cell.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gruber C.N., Patel R.S., Trachtman R., Lepow L., Amanat F., Krammer F., Wilson K.M., Onel K., Geanon D., Tuballes K., et al. Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C) Cell. 2020;183:982–995.e14. doi: 10.1016/j.cell.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng M.H., Zhang S., Porritt R.A., Noval Rivas M., Paschold L., Willscher E., Binder M., Arditi M., Bahar I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc. Natl. Acad. Sci. USA. 2020;117:25254–25262. doi: 10.1073/pnas.2010722117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi T., Inoue Y., Takeuchi K., Okada Y., Tamura K., Tomomasa T., Kobayashi T., Morikawa A. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. 2006;113:2606–2612. doi: 10.1161/CIRCULATIONAHA.105.592865. [DOI] [PubMed] [Google Scholar]
- 21.Han H., Cho J.-W., Lee S., Yun A., Kim H., Bae D., Yang S., Kim C.Y., Lee M., Kim E., et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46(D1):D380–D386. doi: 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Séïté J.-F., Hillion S., Harbonnier T., Pers J.-O. Review: intravenous immunoglobulin and B cells: when the product regulates the producer. Arthritis Rheumatol. 2015;67:595–603. doi: 10.1002/art.38910. [DOI] [PubMed] [Google Scholar]
- 23.Bayry J., Lacroix-Desmazes S., Carbonneil C., Misra N., Donkova V., Pashov A., Chevailler A., Mouthon L., Weill B., Bruneval P., et al. Inhibition of maturation and function of dendritic cells by intravenous immunoglobulin. Blood. 2003;101:758–765. doi: 10.1182/blood-2002-05-1447. [DOI] [PubMed] [Google Scholar]
- 24.Das M., Karnam A., Stephen-Victor E., Gilardin L., Bhatt B., Kumar Sharma V., Rambabu N., Patil V., Lecerf M., Käsermann F., et al. Intravenous immunoglobulin mediates anti-inflammatory effects in peripheral blood mononuclear cells by inducing autophagy. Cell Death Dis. 2020;11:50. doi: 10.1038/s41419-020-2249-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galeotti C., Kaveri S.V., Bayry J. IVIG-mediated effector functions in autoimmune and inflammatory diseases. Int. Immunol. 2017;29:491–498. doi: 10.1093/intimm/dxx039. [DOI] [PubMed] [Google Scholar]
- 26.Kozicky L.K., Menzies S.C., Zhao Z.Y., Vira T., Harnden K., Safari K., Del Bel K.L., Turvey S.E., Sly L.M. IVIg and LPS Co-stimulation Induces IL-10 Production by Human Monocytes, Which Is Compromised by an FcγRIIA Disease-Associated Gene Variant. Front. Immunol. 2018;9:2676. doi: 10.3389/fimmu.2018.02676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cicha A., Fischer M.B., Wesinger A., Haas S., Bauer W.M., Wolf H.M., Sauerwein K.M.T., Reininger B., Petzelbauer P., Pehamberger H., Handisurya A. Effect of intravenous immunoglobulin administration on erythrocyte and leucocyte parameters. J. Eur. Acad. Dermatol. Venereol. 2018;32:1004–1010. doi: 10.1111/jdv.14671. [DOI] [PubMed] [Google Scholar]
- 28.Dyer W.B., Tan J.C.G., Day T., Kiers L., Kiernan M.C., Yiannikas C., Reddel S., Ng K., Mondy P., Dennington P.M., et al. Immunomodulation of inflammatory leukocyte markers during intravenous immunoglobulin treatment associated with clinical efficacy in chronic inflammatory demyelinating polyradiculoneuropathy. Brain Behav. 2016;6:e00516. doi: 10.1002/brb3.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwab I., Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat. Rev. Immunol. 2013;13:176–189. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- 30.Tha-In T., Metselaar H.J., Tilanus H.W., Boor P.P.C., Mancham S., Kuipers E.J., de Man R.A., Kwekkeboom J. Superior immunomodulatory effects of intravenous immunoglobulins on human T-cells and dendritic cells: comparison to calcineurin inhibitors. Transplantation. 2006;81:1725–1734. doi: 10.1097/01.tp.0000226073.20185.b1. [DOI] [PubMed] [Google Scholar]
- 31.Tha-In T., Metselaar H.J., Tilanus H.W., Groothuismink Z.M.A., Kuipers E.J., de Man R.A., Kwekkeboom J. Intravenous immunoglobulins suppress T-cell priming by modulating the bidirectional interaction between dendritic cells and natural killer cells. Blood. 2007;110:3253–3262. doi: 10.1182/blood-2007-03-077057. [DOI] [PubMed] [Google Scholar]
- 32.Holbrook J., Lara-Reyna S., Jarosz-Griffiths H., McDermott M. Tumour necrosis factor signalling in health and disease. F1000Res. 2019;8 doi: 10.12688/f1000research.17023.1. F1000 Faculty Rev-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varfolomeev E.E., Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell. 2004;116:491–497. doi: 10.1016/s0092-8674(04)00166-7. [DOI] [PubMed] [Google Scholar]
- 34.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90-7. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silvin A., Chapuis N., Dunsmore G., Goubet A.-G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N., et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kunkel S.L., Standiford T., Kasahara K., Strieter R.M. Interleukin-8 (IL-8): the major neutrophil chemotactic factor in the lung. Exp. Lung Res. 1991;17:17–23. doi: 10.3109/01902149109063278. [DOI] [PubMed] [Google Scholar]
- 38.Pease J.E., Sabroe I. The role of interleukin-8 and its receptors in inflammatory lung disease: implications for therapy. Am. J. Respir. Med. 2002;1:19–25. doi: 10.1007/BF03257159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sawant K.V., Xu R., Cox R., Hawkins H., Sbrana E., Kolli D., Garofalo R.P., Rajarathnam K. Chemokine CXCL1-Mediated Neutrophil Trafficking in the Lung: Role of CXCR2 Activation. J. Innate Immun. 2015;7:647–658. doi: 10.1159/000430914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hijano D.R., Vu L.D., Kauvar L.M., Tripp R.A., Polack F.P., Cormier S.A. Role of Type I Interferon (IFN) in the Respiratory Syncytial Virus (RSV) Immune Response and Disease Severity. Front. Immunol. 2019;10:566. doi: 10.3389/fimmu.2019.00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brodsky N.N., Ramaswamy A., Lucas C.L. The Mystery of MIS-C Post-SARS-CoV-2 Infection. Trends Microbiol. 2020;28:956–958. doi: 10.1016/j.tim.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Esteve-Sole A., Anton J., Pino-Ramírez R.M., Sanchez-Manubens J., Fumadó V., Fortuny C., Rios-Barnes M., Sanchez-de-Toledo J., Girona-Alarcón M., Mosquera J.M., et al. Similarities and differences between the immunopathogenesis of COVID-19-related pediatric inflammatory multisystem syndrome and Kawasaki disease. J. Clin. Invest. 2021;131:e144554. doi: 10.1172/JCI144554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodnow C.C. COVID-19, varying genetic resistance to viral disease and immune tolerance checkpoints. Immunol. Cell Biol. 2021;99:177–191. doi: 10.1111/imcb.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Ignazio L., Rocha S. Hypoxia Induced NF-κB. Cells. 2016;5:10. doi: 10.3390/cells5010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Ignazio L., Bandarra D., Rocha S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016;283:413–424. doi: 10.1111/febs.13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.-H., Zhang Y., Dorgham K., Philippot Q., Rosain J., Béziat V., et al. HGID Lab. NIAID-USUHS Immune Response to COVID Group. COVID Clinicians. COVID-STORM Clinicians. Imagine COVID Group. French COVID Cohort Study Group. Milieu Intérieur Consortium. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J., Ogishi M., Sabli I.K.D., Hodeib S., Korol C., et al. COVID-STORM Clinicians. COVID Clinicians. Imagine COVID Group. French COVID Cohort Study Group. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort. NIAID-USUHS/TAGC COVID Immunity Group Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Combes A.J., Courau T., Kuhn N.F., Hu K.H., Ray A., Chen W.S., Chew N.W., Cleary S.J., Kushnoor D., Reeder G.C., et al. Global absence and targeting of protective immune states in severe COVID-19. Nature. 2021;591:124–130. doi: 10.1038/s41586-021-03234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sehgal K., Guo X., Koduru S., Shah A., Lin A., Yan X., Dhodapkar K.M. Plasmacytoid dendritic cells, interferon signaling, and FcγR contribute to pathogenesis and therapeutic response in childhood immune thrombocytopenia. Sci. Transl. Med. 2013;5:193ra89. doi: 10.1126/scitranslmed.3006277. [DOI] [PubMed] [Google Scholar]
- 51.Amoah B.P., Yang H., Zhang P., Su Z., Xu H. Immunopathogenesis of Myocarditis: The Interplay Between Cardiac Fibroblast Cells, Dendritic Cells, Macrophages and CD4+ T Cells. Scand. J. Immunol. 2015;82:1–9. doi: 10.1111/sji.12298. [DOI] [PubMed] [Google Scholar]
- 52.Calabrese F., Carturan E., Chimenti C., Pieroni M., Agostini C., Angelini A., Crosato M., Valente M., Boffa G.M., Frustaci A., Thiene G. Overexpression of tumor necrosis factor (TNF)α and TNFalpha receptor I in human viral myocarditis: clinicopathologic correlations. Mod. Pathol. 2004;17:1108–1118. doi: 10.1038/modpathol.3800158. [DOI] [PubMed] [Google Scholar]
- 53.Mann D.L. Tumor necrosis factor and viral myocarditis: the fine line between innate and inappropriate immune responses in the heart. Circulation. 2001;103:626–629. doi: 10.1161/01.cir.103.5.626. [DOI] [PubMed] [Google Scholar]
- 54.Angelo L.S., Kurzrock R. Vascular endothelial growth factor and its relationship to inflammatory mediators. Clin. Cancer Res. 2007;13:2825–2830. doi: 10.1158/1078-0432.CCR-06-2416. [DOI] [PubMed] [Google Scholar]
- 55.Delprat V., Tellier C., Demazy C., Raes M., Feron O., Michiels C. Cycling hypoxia promotes a pro-inflammatory phenotype in macrophages via JNK/p65 signaling pathway. Sci. Rep. 2020;10:882. doi: 10.1038/s41598-020-57677-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hua X., Hu G., Hu Q., Chang Y., Hu Y., Gao L., Chen X., Yang P.-C., Zhang Y., Li M., Song J. Single-Cell RNA Sequencing to Dissect the Immunological Network of Autoimmune Myocarditis. Circulation. 2020;142:384–400. doi: 10.1161/CIRCULATIONAHA.119.043545. [DOI] [PubMed] [Google Scholar]
- 57.Maloney J.P., Gao L. Proinflammatory Cytokines Increase Vascular Endothelial Growth Factor Expression in Alveolar Epithelial Cells. Mediators Inflamm. 2015;2015:387842. doi: 10.1155/2015/387842. https://www.hindawi.com/journals/mi/2015/387842/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vella L.A., Giles J.R., Baxter A.E., Oldridge D.A., Diorio C., Kuri-Cervantes L., Alanio C., Pampena M.B., Wu J.E., Chen Z., et al. UPenn COVID Processing Unit Deep immune profiling of MIS-C demonstrates marked but transient immune activation compared to adult and pediatric COVID-19. Sci. Immunol. 2021;6:eabf7570. doi: 10.1126/sciimmunol.abf7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y., Xie C., Ma K., Shang K., Wang W., et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020;71:762–768. doi: 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Y., Fu B., Zheng X., Wang D., Zhao C., Qi Y., Sun R., Tian Z., Xu X., Wei H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020;7:998–1002. doi: 10.1093/nsr/nwaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Belhadjer Z., Méot M., Bajolle F., Khraiche D., Legendre A., Abakka S., Auriau J., Grimaud M., Oualha M., Beghetti M., et al. Acute Heart Failure in Multisystem Inflammatory Syndrome in Children in the Context of Global SARS-CoV-2 Pandemic. Circulation. 2020;142:429–436. doi: 10.1161/CIRCULATIONAHA.120.048360. [DOI] [PubMed] [Google Scholar]
- 62.Szekely Y., Lichter Y., Taieb P., Banai A., Hochstadt A., Merdler I., Gal Oz A., Rothschild E., Baruch G., Peri Y., et al. Spectrum of Cardiac Manifestations in COVID-19: A Systematic Echocardiographic Study. Circulation. 2020;142:342–353. doi: 10.1161/CIRCULATIONAHA.120.047971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo T., Fan Y., Chen M., Wu X., Zhang L., He T., Wang H., Wan J., Wang X., Lu Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19) JAMA Cardiol. 2020;5:811–818. doi: 10.1001/jamacardio.2020.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lala A., Johnson K.W., Januzzi J.L., Russak A.J., Paranjpe I., Richter F., Zhao S., Somani S., Van Vleck T., Vaid A., et al. Mount Sinai COVID Informatics Center Prevalence and Impact of Myocardial Injury in Patients Hospitalized With COVID-19 Infection. J. Am. Coll. Cardiol. 2020;76:533–546. doi: 10.1016/j.jacc.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lindner D., Fitzek A., Bräuninger H., Aleshcheva G., Edler C., Meissner K., Scherschel K., Kirchhof P., Escher F., Schultheiss H.-P., et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020;5:1281–1285. doi: 10.1001/jamacardio.2020.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Metkus T.S., Sokoll L.J., Barth A.S., Czarny M.J., Hays A.G., Lowenstein C.J., Michos E.D., Nolley E.P., Post W.S., Resar J.R., et al. Myocardial Injury in Severe COVID-19 Compared With Non-COVID-19 Acute Respiratory Distress Syndrome. Circulation. 2021;143:553–565. doi: 10.1161/CIRCULATIONAHA.120.050543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bearse M., Hung Y.P., Krauson A.J., Bonanno L., Boyraz B., Harris C.K., Helland T.L., Hilburn C.F., Hutchison B., Jobbagy S., et al. Factors associated with myocardial SARS-CoV-2 infection, myocarditis, and cardiac inflammation in patients with COVID-19. Mod. Pathol. 2021;34:1345–1357. doi: 10.1038/s41379-021-00790-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McInnes L., Healy J., Melville J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv. 2020 https://arxiv.org/abs/1802.03426 arXiv:1802.03426. [Google Scholar]
- 70.Van Gassen S., Callebaut B., Van Helden M.J., Lambrecht B.N., Demeester P., Dhaene T., Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87:636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 71.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zimmermann P., Curtis N. Coronavirus Infections in Children Including COVID-19: An Overview of the Epidemiology, Clinical Features, Diagnosis, Treatment and Prevention Options in Children. Pediatr. Infect. Dis. J. 2020;39:355–368. doi: 10.1097/INF.0000000000002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.CDC Multisystem Inflammatory Syndrome in Children (MIS-C) Associated with Coronavirus Disease 2019 (COVID-19) 2020. https://emergency.cdc.gov/han/2020/han00432.asp
- 75.Brissaud O., Botte A., Cambonie G., Dauger S., de Saint Blanquat L., Durand P., Gournay V., Guillet E., Laux D., Leclerc F., et al. Experts’ recommendations for the management of cardiogenic shock in children. Ann. Intensive Care. 2016;6:14. doi: 10.1186/s13613-016-0111-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Canter C.E., Simpson K.E. Diagnosis and treatment of myocarditis in children in the current era. Circulation. 2014;129:115–128. doi: 10.1161/CIRCULATIONAHA.113.001372. [DOI] [PubMed] [Google Scholar]
- 77.Darnell M.E.R., Taylor D.R. Evaluation of inactivation methods for severe acute respiratory syndrome coronavirus in noncellular blood products. Transfusion. 2006;46:1770–1777. doi: 10.1111/j.1537-2995.2006.00976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rodero M.P., Decalf J., Bondet V., Hunt D., Rice G.I., Werneke S., McGlasson S.L., Alyanakian M.-A., Bader-Meunier B., Barnerias C., et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J. Exp. Med. 2017;214:1547–1555. doi: 10.1084/jem.20161451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grzelak L., Temmam S., Planchais C., Demeret C., Tondeur L., Huon C., Guivel-Benhassine F., Staropoli I., Chazal M., Dufloo J., et al. A comparison of four serological assays for detecting anti-SARS-CoV-2 antibodies in human serum samples from different populations. Sci. Transl. Med. 2020;12:eabc3103. doi: 10.1126/scitranslmed.abc3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bagwell C.B., Inokuma M., Hunsberger B., Herbert D., Bray C., Hill B., Stelzer G., Li S., Kollipara A., Ornatsky O., Baranov V. Automated Data Cleanup for Mass Cytometry. Cytometry A. 2020;97:184–198. doi: 10.1002/cyto.a.23926. [DOI] [PubMed] [Google Scholar]
- 81.Monaco G., Lee B., Xu W., Mustafah S., Hwang Y.Y., Carré C., Burdin N., Visan L., Ceccarelli M., Poidinger M., et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019;26:1627–1640.e7. doi: 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rosenberg B.R., Freije C.A., Imanaka N., Chen S.T., Eitson J.L., Caron R., Uhl S.A., Zeremski M., Talal A., Jacobson I.M., et al. Genetic Variation at IFNL4 Influences Extrahepatic Interferon-Stimulated Gene Expression in Chronic HCV Patients. J. Infect. Dis. 2018;217:650–655. doi: 10.1093/infdis/jix593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This spreadsheet includes the differential expression tables for all comparisons presented in the paper, performed on all PBMCs and on major cell types (monocytes/cDCs/pDCs, T cells, or B cells). Links to EnrichR analysis presented in Figure S6 are included.
The list of genes extracted from SC-RNA-seq data coming from the 3 different strategies used are listed, alongside the different scores generated from Bulk-RNA-seq data.
Data Availability Statement
Single-cell and bulk RNA-seq data have been deposited in GEO and are publicly available. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.