

Abstract
Castration-resistant prostate cancer (CRPC) is driven by AR gene aberrations that arise during androgen receptor (AR)-targeted therapy. AR amplification and mutations have been profiled in circulating tumor cells (CTCs), but whether AR gene rearrangements can be assessed in CTCs is unknown. In this study, we leveraged CRPC cell lines with defined AR gene rearrangements to develop and validate a CTC DNA analysis approach that utilized whole genome amplification and targeted DNA-sequencing of AR and other genes important in CRPC. We tested the utility of this approach by analyzing matched CTC DNA and plasma cell-free DNA (cfDNA) from a case series of ten CRPC patients. One of ten CTC samples and two of ten cfDNA samples were positive for AR gene rearrangements. All AR gene rearrangements were discordant between matched liquid biopsy samples. One patient harbored separate AR gene rearrangements in CTC DNA and cfDNA, but concordant AR amplification and AR T878A mutation. This patient also displayed concordant loss of TP53 and PTEN, but loss of RB1 in cfDNA only. The overall frequency of discordant alterations in these genes between matched CTC DNA and cfDNA was high. This study establishes the technical feasibility of analyzing structural rearrangements, mutations, and copy number variants in AR and other CRPC genes using two different sources of DNA from a single blood sample. Paired CTC DNA and cfDNA analysis may have utility for capturing the heterogeneity of genetic alterations in CRPC patients.
Keywords: Androgen receptor, AR gene rearrangements, Castration-resistant prostate cancer, Cell-free DNA, Circulating tumor cells
Introduction
In the United States, prostate cancer (PC) is the second leading cause of male cancer deaths (Siegel et al. 2020). Growth of PC cells is dependent on the androgen receptor (AR). Therefore, first- and second-generation hormonal therapies that inhibit AR are effective for advanced PC. AR re-activation during hormonal therapy drives the emergence of castration-resistant PC (CRPC). Genomic studies of CRPC biopsies have revealed the landscape of mechanisms underlying AR re-activation in CRPC (Robinson et al. 2015; Quigley et al. 2018), which include AR amplification, AR mutations, AR gene rearrangements, or expression of truncated, constitutively active AR variants (AR-Vs) (Ho & Dehm 2017). However, metastatic biopsies are challenging and a high burden for patients. As a result, assessing CRPC genomic alterations via blood-based “liquid biopsy” is an attractive alternative.
One common liquid biopsy approach involves examination of circulating tumor cells (CTCs). High CTC counts are correlated with poor outcomes (Danila et al. 2007; De Bono et al. 2008). AR gene amplification has been detected in CTCs by in situ hybridization (Shaffer et al. 2007), and AR mutations have been assessed using PCR and targeted DNA sequencing (DNA-seq) (Jiang et al. 2010; Sperger et al. 2017). Development of AR mRNA and protein biomarkers in CTCs has also been robust, exemplified by detection of AR variant 7 (AR-V7) mRNA or protein in CTCs predicting resistance to the second-generation hormonal therapies abiraterone and enzalutamide (Antonarakis et al. 2014, 2015; Scher et al. 2016, 2018).
Another common liquid biopsy approach is detection of circulating tumor DNA (ctDNA) shed from tumors by identifying tumor-associated copy number alterations, mutations, and structural variants in plasma cell-free DNA (cfDNA). Targeted DNA-seq profiling of cfDNA and paired tissue biopsies in CRPC patients has shown that genomic alterations in ctDNA broadly reflect the underlying genomic alterations occurring in tumor tissue (Ulz et al. 2016; Wyatt et al. 2017). Further, detection of AR genomic alterations in ctDNA may have biomarker utility in CRPC based on the findings that AR amplification, mutation, or rearrangements correlate with poor outcomes of patients treated with various second-generation hormonal therapies (Conteduca et al. 2017; Annala et al. 2018; Dang et al. 2020).
AR gene rearrangements are DNA structural variants (SVs) impacting AR that promote expression of a diverse repertoire of AR-V mRNA and protein species in CRPC cells (Nyquist et al. 2013; Henzler et al. 2016; Li et al. 2020). Analysis of AR gene rearrangements may have potential to identify a broader fraction of CRPC driven by AR-Vs than analysis of AR-V7 alone. Clinical assays are available for AR-V7 mRNA or protein detection in CTCs (Armstrong et al. 2019). However, there have been no reports of DNA SV analysis in CRPC CTCs. Therefore, the goals of this study were to develop a strategy for detecting AR rearrangements in CTCs, assess other genomic alterations in AR as well as established CRPC driver genes, and compare genomic alteration profiles in a case series of matched CTC and ctDNA specimens.
Materials and Methods
Patient selection and sample collection
Patients with metastatic CRPC treated at the University of Wisconsin-Madison Carbone Cancer Center (UWCCC) donated blood samples under the University of Wisconsin Institutional Review Board (IRB) approved protocol 2014–1214; H-2009–0019. All patients provided written, informed consent. All patients had radiographic metastases with disease progression on most recent therapy. Clinical, pathological, and treatment data were collected by review of the electronic medical records.
CTC isolation, enumeration, and genomic interrogation
Mononuclear CD45-/EpCAM+ CTCs were isolated from blood for enumeration and DNA extraction using the Vertical Exclusion-based Rare Sample Analysis (VERSA) platform (Sperger et al. 2017). Alternatively, DNA was extracted from PC cell lines harboring known AR gene alterations. DNA was halved for parallel, independent whole genome amplification (WGA) and analysis by targeted DNA-seq. Alterations in AR and other CRPC driver genes were identified based on detection in both parallel WGA reactions. Full details of methods and data analysis approaches are in the Supplementary Materials and Methods.
Cell-free plasma DNA
cfDNA was isolated from double-spun plasma and analyzed by targeted DNA-seq. Full details of methods and data analysis approaches are detailed in Supplementary Materials and Methods.
Data Availability
Data and code are available for bona fide researchers who request it from the authors.
Results
A strategy for identifying AR gene rearrangements in DNA from CTCs
To model AR gene rearrangements in CTCs, we analyzed three isogenic prostate cancer cell lines with engineered AR rearrangements reflecting those discovered in CRPC tissues (Nyquist et al. 2013). R1-D567 cells harbor a deletion of AR exons 5–7 and R1-I567 cells harbor an inversion of exons 5–7. R1-AD1 cells harbor a structurally normal AR gene, and were used for generating R1-D567 and R1-I567 (Supplementary Fig. 1). DNA was isolated from 100 cells and each DNA sample was halved for parallel whole genome amplification (WGA) and targeted DNA-seq. DNA SVs were identified by SV detection algorithms DELLY, LUMPY, Manta, and SvABA (Rausch et al. 2012; Layer et al. 2014; Chen et al. 2016; Wala et al. 2018) (Fig. 1A, Supplementary Table 1).
Figure 1. Structural variant callers detect a wide range of AR gene rearrangements in whole genome amplified prostate cancer cell line DNA.
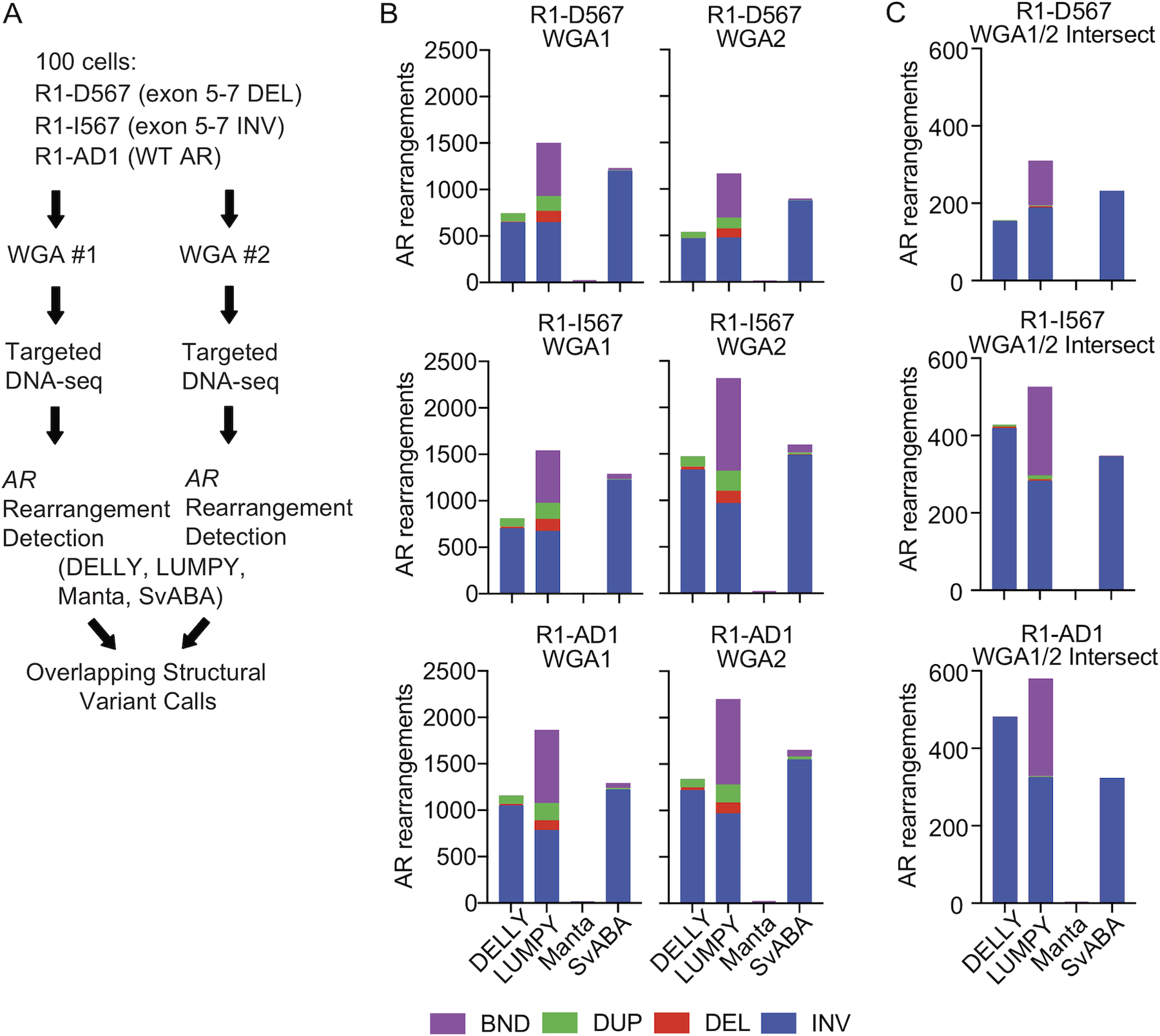
(A) AR gene rearrangement detection strategy for use with limiting numbers of cells. (B) Distribution of AR gene rearrangement calls (BND, breakend; DUP, duplication; DEL, deletion; INV, inversion) made using DNA-seq data from individual whole genome amplification (WGA) reactions with DNA from 100 R1-D567, R1-I567, and R1-AD1 cells. (C) Distribution of intersecting AR gene rearrangement calls in each WGA sample from (B).
AR gene rearrangements are DNA SVs with at least one breakpoint mapping within the AR gene. In individual WGA reactions, there was a wide range of AR gene rearrangement calls made by the four SV callers (Fig. 1B). We filtered SV calls to those that intersected between the two parallel WGA reactions, with the rationale that true SVs occurring in a DNA sample should be detected in both WGA reactions. Strikingly, despite this requirement for intersection, false positive and false negative AR gene rearrangement calls were prevalent (Fig. 1C). We noted that most false positive intersecting calls were intra-AR inversions.
WGA with phi29 DNA polymerase creates small inversions
The majority of these unexpectedly pervasive AR gene rearrangement calls were small (100–5,000 bp) with strong support from discordant read pairs and split reads (Supplementary Fig. 2A). AR gene rearrangements in this small size range were detected by all SV callers (Supplementary Fig. 2B). We hypothesized that the template switching activity of phi29 DNA polymerase used for multiple displacement amplification (MDA)-based WGA (Murthy et al. 1998; Ducani et al. 2014) was generating spurious small inversions at sufficient quantities in the individual WGA reactions to yield the observed intersecting AR gene rearrangement calls. To test this, we compared WGA with MDA to WGA with multiple annealing and looping based amplification cycles technology (MALBAC) (Zong et al. 2012). In R1-AD1 and R1-I567 cells, WGA by MALBAC resulted in fewer AR gene rearrangement calls than WGA by MDA, with no bias for inversions (Supplementary Fig. 3). However, DNA-seq coverage of AR following WGA by MALBAC was poor compared to WGA by MDA.
To remove these small inversions generated by MDA WGA, we filtered the output of each SV caller by size. These filters reduced the number of AR gene rearrangement calls in individual WGA reactions and intersecting calls between WGA reactions (Supplementary Fig. 2C–F). Comparing size-filtered WGA-intersecting inversion calls in R1-I567 and R1-AD1 cell demonstrated that a 5000 bp size filter eliminated all false positive AR inversion calls from R1-AD1 and R1-I567 cells, while retaining all inversion calls with breakend coordinates that matched the known inversion of AR exons 5–7 in R1-I567 cells (Fig. 2A–D).
Figure 2. Size filtering improves specificity of detecting an intra-AR inversion in WGA DNA.
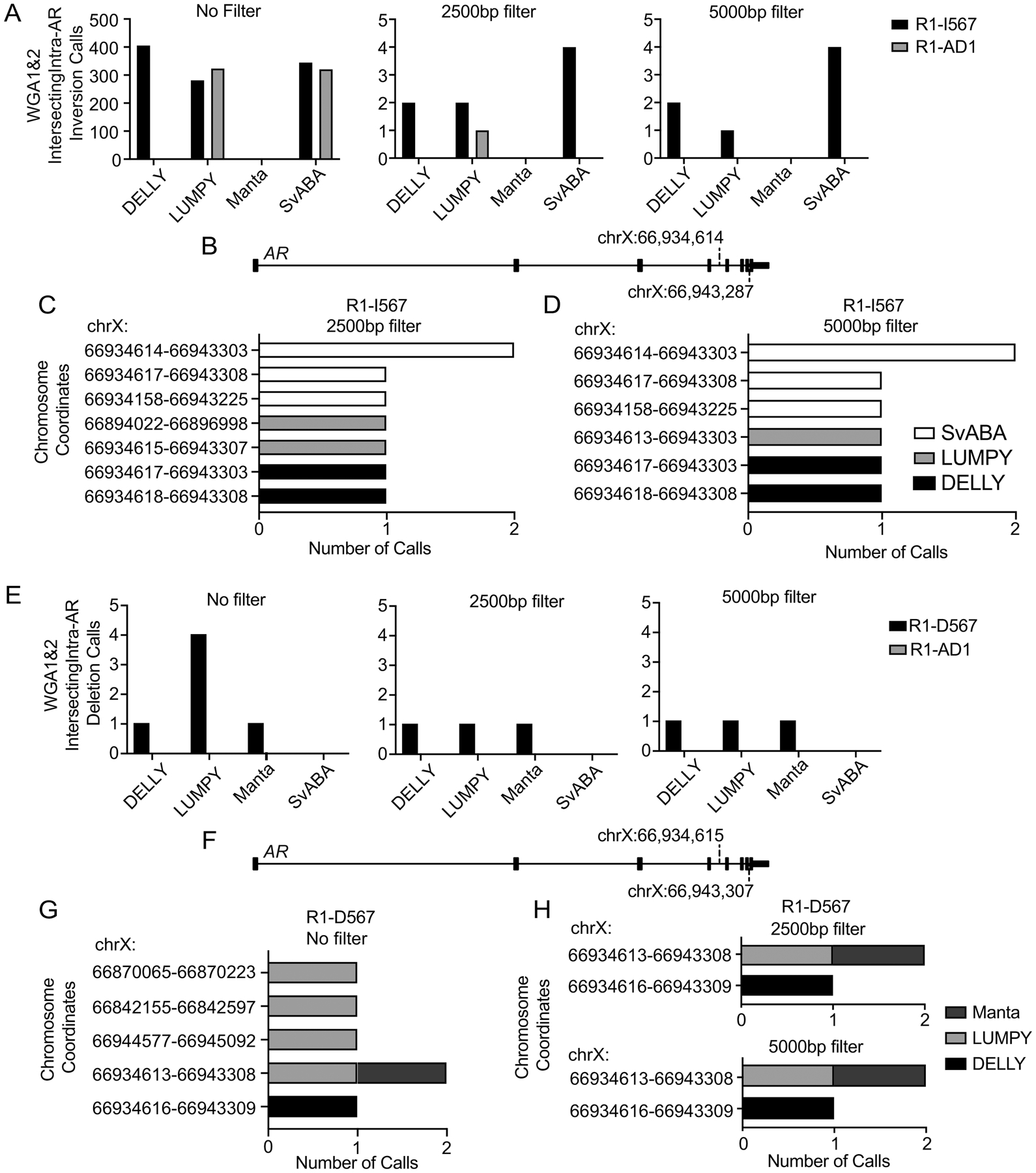
(A) Inversion calls made by DELLY, LUMPY, Manta, and SvABA in WGA DNA from R1-AD1 and R1-I567 cells after applying no size filter, a 2500bp filter, and a 5000bp. (B) AR gene schematic showing the expected inversion breakend coordinates in R1-I567 cells. (C, D) Potential inversion calls from (A) are shown based on AR gene rearrangement breakend coordinate and SV caller with a 2500bp filter (C) and a 5000bp filter (D). (E) Deletion calls made by DELLY, LUMPY, Manta, and SvABA in WGA DNA from R1-AD1 and R1-D567 cells after applying no size filter, a 2500bp filter, and a 5000bp filter. (F) AR gene schematic showing the expected deletion breakend coordinates in R1-D567 cells. (G, H) Potential deletion calls from (E) are shown based on breakend coordinate and SV caller with no filter (G), a 2500bp filter and a 5000bp filter (H).
WGA enables specific detection of intra-AR deletions and duplications
We next tested the WGA intersection strategy for detecting an intra-AR deletion (Fig. 2E). DELLY and Manta correctly identified the deletion in 100 R1-D567 cells, with no effect of restricting deletion calls by size. Conversely, LUMPY identified four intra-AR deletions with no size restrictions applied, two of which were false positives based on chromosomal breakpoint coordinates (Fig. 2E–H). The 2500 bp and 5000 bp size filters eliminated these false positive intra-AR deletions called by LUMPY and retained the true positive deletion call. Three callers correctly called zero deletions in the R1-AD1 cell line regardless of size filter. SvABA did not detect deletions in any samples. These data indicated that requiring detection by at least two of the four independent SV callers would provide sensitive and specific detection of intra-AR deletions, with no benefit of filtering deletion calls by size.
To test the WGA intersection strategy for detecting intra-AR duplications, we analyzed AR gene rearrangement calls from 100 22Rv1 cells, which harbor a 35 kb tandem duplication (Supplementary Fig. 1). The intra-AR duplication breakpoints were called by LUMPY and SvABA (Supplementary Fig. 4A–G). However, LUMPY also called false positive intra-AR duplications, which were removed by 2500bp and 5000bp size filters (Supplementary Fig. 4B, C, F, G). These data indicated that the same requirements for detecting deletions would provide sensitive and specific detection of intra-AR duplications.
To test the sensitivity of detecting AR gene rearrangements in limiting numbers of cells, we analyzed DNA isolated from 10, 100, and 1000 R1-D567, R1-I567, and R1-AD1 cells (Supplementary Fig. 5A–D). DNA-seq read coverage from parallel WGA reactions with DNA from 10 cells was inconsistent, but 100 or more cells yielded robust coverage as well as clear validation of the intra-AR deletion in R1-D567 cells. These data indicated that detection of AR gene rearrangements in CTCs would require analysis of DNA isolated from at least 100 CTCs.
Discordant AR gene rearrangements in CTCs and cfDNA
We used these optimized parameters for analysis of CTCs from a case series of 10 CRPC patients (Fig. 3A), employing the VERSA platform (Sperger et al. 2017) for CTC isolation and DNA extraction. Patients ranged in age from 63–78 years at the time of blood draw and encompassed a broad range of treatment histories (Supplementary Table 2). Patient 7 was found to harbor an AR gene rearrangement in CTC DNA, with discordant read pairs and split reads supporting a 2.6 Mb inversion with one breakpoint occurring in AR intron 1, and the other occurring within ZC4H2 (Fig. 3B, C and Supplementary Table 3). We validated the breakpoint fusion junction arising from this inversion in Patient 7 using PCR and Sanger sequencing with WGA DNA (Fig. 3D, E).
Figure 3. Discovery of 2.6 Mb AR inversion in CTC DNA from Patient 7.
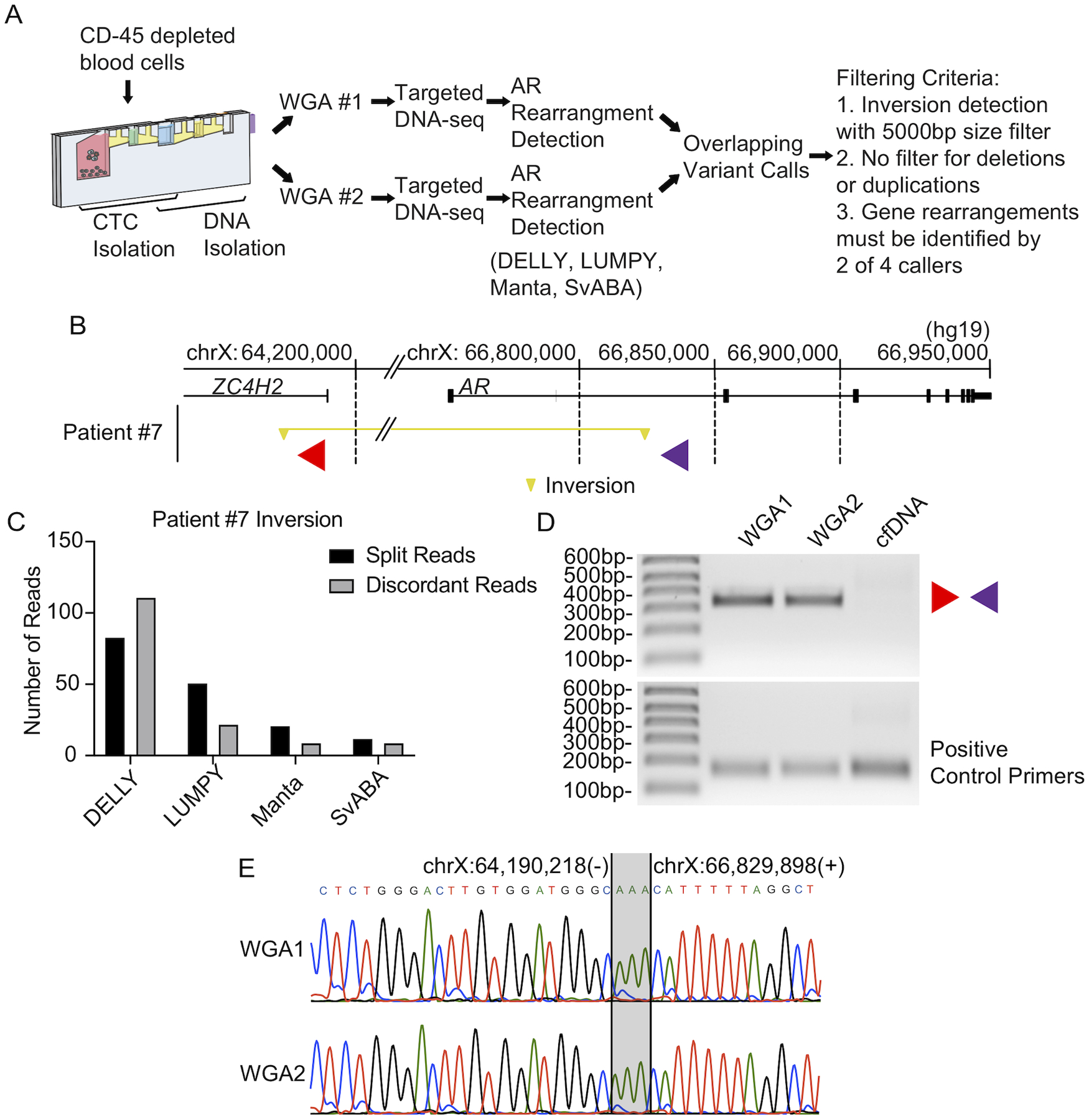
(A) AR gene rearrangement detection pipeline used for circulating tumor cells. (B) Locations of AR gene rearrangement breakpoints caused by a 2.6 Mb inversion in CTC DNA from Patient #7 are shown by yellow triangles. The inverted segment is indicated by a yellow horizontal line. Locations and direction of PCR primers used for validation are denoted with red and purple triangles. Genome coordinates are genome build hg19. (C) Split read counts, discordant read counts, and SV callers supporting the inversion called in (B). Note: LUMPY and Manta made this call as a BND. (D) PCR validating the inversion illustrated in (B) in two parallel WGA reactions of CTC DNA (WGA1 and WGA2), as well as cfDNA from Patient #7. Primers used for PCR are illustrated in (B). (E) Sanger sequencing analysis of the AR inversion PCR products from (D). Microhomology at this breakpoint is indicated with gray shading.
Prior studies have reported AR gene rearrangement breakpoints in cfDNA from CRPC patients (De Laere et al. 2017; Annala et al. 2018; Chung et al. 2019). Interestingly, cfDNA from the same Patient 7 blood sample used for CTC isolation was PCR-negative for this inversion (Fig. 3D). To explore this further, we performed targeted DNA-seq with plasma cfDNA isolated from the same 10 CRPC patient blood samples used for CTC isolation (Fig. 4A). This revealed two additional AR gene rearrangements, one occurring in Patient 2 and one occurring in Patient 7 (Fig. 4B and Supplementary Table 4). In Patient 2, the discordant read pairs and split reads supported one side of a 4.5 Mb genomic inversion, with one breakpoint in AR intron 1 (Fig. 4B, C). A 1.8 Mb duplication identified in Patient 7 had one breakpoint in AR intron 2 (Fig. 4B, D).
Figure 4. AR gene rearrangement detection in cell-free DNA from patients with metastatic CRPC.
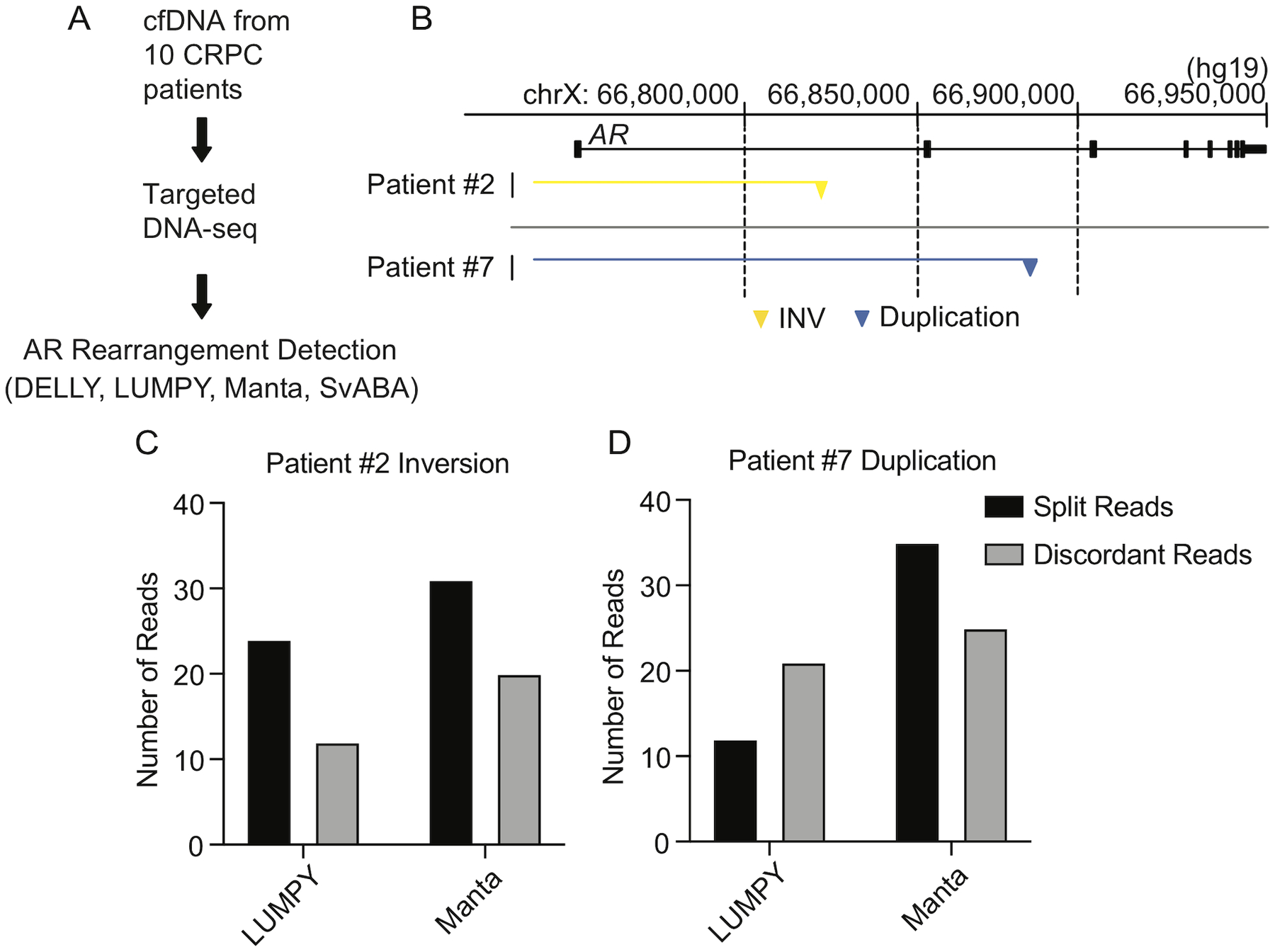
(A) AR gene rearrangement detection pipeline for use with cfDNA. (B) Locations of AR gene rearrangement breakpoints discovered in cfDNA from Patients #2 and #7. Breakend fusion junctions due to an inversion (yellow) and a duplication (blue) are shown as triangles, with the rearranged segments indicated by yellow and blue horizontal lines. Genome coordinates are genome build hg19. (C, D) Split read counts, discordant read counts, and caller combinations supporting the calls shown in (B).
Discordance in AR gene rearrangements from CTCs vs. cfDNA from the same blood samples was unexpected in this case series. We confirmed that CTC DNA and cfDNA samples were exact matches from the same patient by genotyping informative germline single nucleotide polymorphisms (SNPs) (Supplementary Fig. 6 and 7). We further assessed tumor DNA fraction with ichorCNA (Adalsteinsson et al. 2017), revealing that tumor DNA fractions were similar between CTC DNA and cfDNA samples from Patients 2 and 7 (Fig. 5A–C). Manual CTC enumeration confirmed high CTC count (Fig. 5D). DNA-seq coverage was robust across the entire AR locus for all samples (Supplementary Fig. 8, 9).
Figure 5. Genomic landscape of AR and prostate cancer-related genes in CTC DNA and cfDNA from CRPC patients.
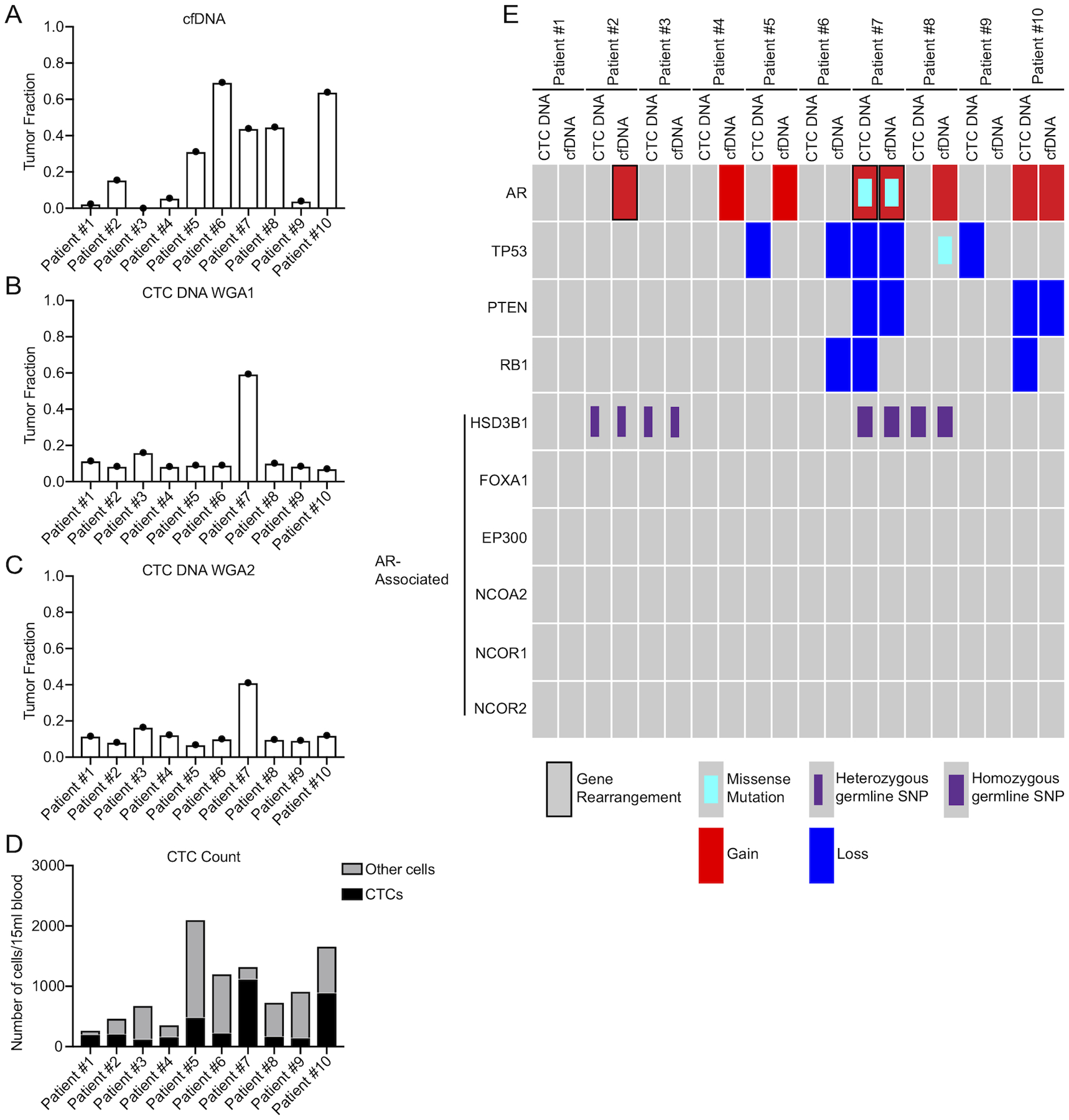
(A-C) Tumor DNA fraction estimates in cfDNA (A), CTC DNA from WGA1 (B), and CTC DNA from WGA2 (C) using ichorCNA. (D) CTC counts per 15mL of blood from 10 CRPC patients. (E) Oncoprint summary of gene alterations identified from targeted CTC DNA-seq and cfDNA-seq in the 10 CRPC patients. Alteration type is shown at the bottom.
To assess whether certain AR gene rearrangements may be evident below our imposed thresholds, we manually searched for individual reads in DNA-seq data (Fig. 3A). However, we found zero cfDNA-seq reads supporting the 2.6 Mb inversion discovered in CTCs from Patient 7 (Supplementary Fig. 10A–C) and zero CTC DNA-seq reads supporting the 4.5 Mb inversion discovered in cfDNA from Patient 2 (Supplementary Fig. 11A–C). However, we did find one DNA-seq read in each of CTC DNA WGA1 and WGA2 that supported the 1.8 Mb duplication discovered in cfDNA from Patient 7 (Supplementary Fig. 11D–F). Collectively, these data indicate that the distinct AR gene rearrangements observed in matched liquid biopsy samples are not technical artifacts and likely reflect distinct tumor clones.
CTC DNA and ctDNA provide concordant and complementary information on pathogenic DNA alterations in CRPC patients
To expand on these findings, we evaluated AR somatic mutations. AR T878A occurred in CTC DNA and ctDNA from Patient 7 (Fig. 5E and Supplementary Fig. 12, Fig. 13A, Supplementary Table 5, 6, Supplementary Data 1). We also used CNVkit (Talevich et al. 2016) to assess AR copy number, on the basis that AR gain would be detectable in cfDNA and CTC DNA samples having high AR copy number and/or a high tumor DNA fraction. To benchmark this approach for WGA DNA, we analyzed DNA-seq data from PC cell lines and correctly detected AR amplification from 100 VCaP cells, AR copy number gain from 100 LAPC4 cells, and no gain in LNCaP, 22Rv1, or R1-AD1 cells (Li et al. 2012; Hoefer et al. 2016) (Supplementary Fig. 14). In the 10 patient cohort, AR copy number gain occurred in both CTC DNA and ctDNA from Patients 7 and 10 (Supplementary Fig. 15). Interestingly, Patients 2, 4, 5, and 8 also displayed AR copy number gain, but only in ctDNA.
Next, we evaluated mutations and copy number alterations in 27 additional genes that are recurrently altered in CRPC genomes (Taylor et al. 2010; Grasso et al. 2012; Robinson et al. 2015) (Supplementary Fig. 12). We identified TP53 R158H in ctDNA from Patient 8, but there were no reads supporting this variant in corresponding CTC DNA-seq data (Fig. 5E and Supplementary Fig. 13B, Supplementary Table 5, Supplementary Data 1). TP53 copy number loss was observed in both CTC DNA and ctDNA samples from Patient 7 (Supplementary Fig. 16). However, Patients 5 and 9 displayed TP53 copy number loss in CTC DNA only, and Patient 6 displayed TP53 copy loss in ctDNA only. PTEN copy number loss was detected in both CTC DNA and ctDNA samples from Patients 7 and 10. Interestingly, RB1 copy number loss was detected in Patients 6, 7, and 10. However, in Patients 7 and 10 this evidence for RB1 copy number loss came from CTC DNA and in Patient 6 this evidence came from ctDNA, with no concordance between matched CTC DNA and ctDNA in these patients.
Discussion
AR gene rearrangements are an important mechanism of resistance to AR-targeted hormonal therapies, but are challenging to detect relative to AR amplification and mutation (Henzler et al. 2016; Li et al. 2020). In this study, we exploited CRPC cell lines with defined AR gene rearrangements to optimize a DNA-seq approach to identify AR gene rearrangements in DNA from limiting numbers of cells. We found that a common WGA method using phi29 DNA polymerase incorporated spurious small DNA inversions, which could be overcome by applying SV size-filters and requiring at least two SV callers to identify the same AR gene rearrangement in two parallel WGA reactions. To the best of our knowledge, this study represents the first optimized approach for discovery of AR gene rearrangements in CTC DNA. Importantly, this approach likely has broad utility for discovery of genomic SVs in CTC DNA.
Limitations of our genomic SV discovery approach include an inability to detect inversions smaller than 5 kb, and a requirement for DNA isolated from at least 100 cells that may be challenging to obtain for all CRPC patients (Danila et al. 2007; De Bono et al. 2008). Additionally, the small 10 patient case series that we used to test clinical performance of our approach restricts our ability to perform statistical analysis linking genomic alterations to clinical outcomes. A future study involving a larger clinical cohort will be necessary to confirm any observations noted in this case series.
The frequency of AR gene rearrangements that we found in CRPC patient CTCs (10%) was lower than the 23–39% frequency observed in previous studies of CRPC tumor tissues (Henzler et al. 2016; Li et al. 2020). This lower frequency could be due to the small case series, low tumor cell purity from CTC DNA, low CTC counts, or real differences in representation of cells with AR gene rearrangements in CTCs vs. tumor tissue. For example, Patient 7 had the highest CTC count and tumor cell purity, and was the only patient where an AR gene rearrangement was detected in CTC DNA. An expanded cohort of matched CTC/tumor specimens is required to address this discrepancy in AR gene rearrangement frequency in CTCs vs. CRPC tumor tissues.
Although AR gene rearrangements had not been previously studied in CTCs, several groups have studied AR gene rearrangements in ctDNA, which is in higher abundance and thus more suitable for SV analysis. For instance, in targeted DNA-seq studies with cfDNA, De Laere et al. observed AR gene rearrangements in 15 of 30 CRPC patients (De Laere et al. 2017) and Annala et al. detected AR gene rearrangements in 19 of 50 CRPC patients (Annala et al. 2018). Chung et al. detected AR gene rearrangements in 0.4% and 2.2% of PC and CRPC patients, respectively (Chung et al. 2019). A recent study from Annala et al. detected AR gene rearrangements in 69 of 175 cfDNA samples from CRPC patients (Annala et al. 2021). In our case series, we identified AR gene rearrangements in 2 of 10 patient cfDNA samples. An explanation for the low frequency of AR gene rearrangements detected in our case series was that we required an AR gene rearrangement to be called by at least 2 of 4 different SV callers, with support from at least 9 split reads and 9 discordant paired-end reads. De Laere et al. used one structural variant caller, and Annala et al. required only 3 split reads of support.
An important finding was that a specific AR gene rearrangement detectable in CTC DNA from Patient 7 was not detectable in ctDNA from the same blood specimen, and specific AR gene rearrangements detectable in ctDNA from Patients 2 and 7 were not detectable in CTC DNA from the same blood specimen. This is likely because AR gene rearrangements are late events in disease progression that are associated with CRPC resistance to hormonal therapies and are known to increase in abundance in serially collected cfDNA samples (Li et al. 2020; Annala et al. 2021). Thus, the ability to detect AR gene rearrangements in CTCs enhances the degree of information that can be obtained from liquid biopsy in advanced-stage disease. Our strategy for detecting genomic SVs in CTCs also opens a path for broader analysis of matched CTC DNA and cfDNA that can expand on prior studies that assessed concordance of mutations and copy number variants. For instance, a prior study using DNA-seq to evaluate somatic mutations in matched liquid biopsy specimens reported a higher degree of discordance than concordance. However, this conclusion of widespread discordance could also be attributable to one WGA reaction, which has high potential to introduce artifacts (Hodara et al. 2019). Nevertheless, our finding that a R158H TP53 mutation was detectable in ctDNA but not CTC DNA in Patient 8 from our cohort further supports a conclusion that liquid biopsy profiles from the same patient can be discordant. Moreover, a previous study that profiled chromosome-level DNA copy number in CRPC patients found that matched CTC and cfDNA samples harbored different patterns of gains and losses (Gupta et al. 2020). Our finding that gene-level copy number gains in AR and copy number losses in tumor suppressors like TP53, RB1, and PTEN are differentially detectable in CTC DNA vs. cfDNA from the same blood sample support and extend these findings.
In this study, we developed and optimized an approach for detecting AR gene rearrangements in CTC DNA. Our work demonstrates that AR gene rearrangements in CTC DNA and cfDNA can provide complementary information that captures disease heterogeneity in CRPC-stage patients. Moreover, we extended this conclusion more broadly to mutation and copy number profiles for clinically-relevant genes that are known to be altered more frequently in later disease stages. Since the goal of liquid biopsy is to capture the full spectrum of clinically-relevant genomic aberrations for informing treatment decisions, paired analysis of CTC DNA and cfDNA is likely superior to analysis of only one liquid biopsy source of tumor DNA. Future studies expanding on the trends observed in this case series are necessary for potential clinical application.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R01CA174777 and 1R01CA256157 (to S.M.D.), DOD Synergistic Idea Development Awards W81XWH-15-1-0500 (to J.M.L.) and W81XWH-15-1-0501 (to S.M.D.), and PCF Challenge Grant 17CHAL-05 (to J.M.L. and S.M.D.).
Footnotes
Declaration of Competing Interests
JML has ownership interest in Salus Discovery, LLC which has licensed the CTC technology included in this report. JML has consulted for Sanofi, Pfizer/Astellas, Immunomedics, Janssen. SMD has consulted for Janssen, Celgene/Bristol Myers Squibb, Oncternal Therapeutics and served as Principal Investigator on grants to University of Minnesota from Janssen and Pfizer/Astellas.
References
- Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, Gydush G, Reed SC, Rotem D, Rhoades J et al. 2017Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nature Communications 8. (doi: 10.1038/s41467-017-00965-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW, Sunderland K, Kollmannsberger C, Eigl BJ, Finch D et al. 2018Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discovery 8444–457. (doi: 10.1158/2159-8290.CD-17-0937) [DOI] [PubMed] [Google Scholar]
- Annala M, Taavitsainen S, Khalaf DJ, Vandekerkhove G, Beja K, Sipola J, Warner EW, Herberts C, Wong A, Fu S et al. 2021Evolution of castration-resistant prostate cancer in ctDNA during sequential androgen receptor pathway inhibition. Clinical Cancer Research clincanres.CCR-21–1625-E.2021. (doi: 10.1158/1078-0432.CCR-21-1625) [DOI] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL et al. 2014AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. The New England Journal of Medicine 3711028–1038. (doi: 10.1056/NEJMoa1315815) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, Nadal R, Paller CJ, Denmeade SR, Carducci MA et al. 2015Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA Oncology 1582–591. (doi: 10.1001/jamaoncol.2015.1341) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong AJ, Halabi S, Luo J, Nanus DM, Giannakakou P, Szmulewitz RZ, Danila DC, Healy P, Anand M, Rothwell CJ et al. 2019Prospective multicenter validation of androgen receptor splice variant 7 and hormone therapy resistance in high-risk castration-resistant prostate cancer: The PROPHECY study. Journal of Clinical Oncology 371120–1129. (doi: 10.1200/JCO.18.01731) [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, Doyle GV., Terstappen LWWM, Pienta KJ & Raghavan D 2008. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clinical Cancer Research 14 6302–6309. (doi: 10.1158/1078-0432.CCR-08-0872) [DOI] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S & Saunders CT 2016. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32 1220–1222. (doi: 10.1093/bioinformatics/btv710) [DOI] [PubMed] [Google Scholar]
- Chung JH, Dewal N, Sokol E, Mathew P, Whitehead R & Millis SZ 2019. Prospective Comprehensive Genomic Profiling of Primary and Metastatic Prostate Tumors. JCO Precision Oncology 3. (doi: 10.1200/PO.18.00283) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conteduca V, Wetterskog D, Sharabiani MTA, Grande E, Fernandez-Perez MP, Jayaram A, Salvi S, Castellano D, Romanel A, Lolli C et al. 2017Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Annals of Oncology 281–9. (doi: 10.1093/annonc/mdx155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang HX, Ph D, Chauhan PS, Ph D, Ellis H, Feng W, Harris PK, Ph D, Smith G, Qiao M et al. 2020Cell-free DNA alterations in the AR enhancer and locus predict resistance to AR-directed therapy in patients with metastatic prostate cancer. JCO Precision Oncology 4680–713. (doi: 10.1200/po.20.00047.Cell-free) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danila DC, Heller G, Gignac GA, Gonzalez-Espinoza R, Anand A, Tanaka E, Lilja H, Schwartz L, Larson S, Fleisher M et al. 2007Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clinical Cancer Research 137053–7058. (doi: 10.1158/1078-0432.CCR-07-1506) [DOI] [PubMed] [Google Scholar]
- Ducani C, Bernardinelli G & Högberg B 2014. Rolling circle replication requires single-stranded DNA binding protein to avoid termination and production of double-stranded DNA. Nucleic Acids Research 42 10596–10604. (doi: 10.1093/nar/gku737) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu Y-M, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC et al. 2012The mutational landscape of lethal castration-resistant prostate cancer. Nature 487239–243. (doi: 10.1038/nature11125) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Hovelson DH, Kemeny G, Halabi S, Foo WC, Anand M, Somarelli JA, Tomlins SA, Antonarakis ES, Luo J et al. 2020Discordant and heterogeneous clinically relevant genomic alterations in circulating tumor cells vs plasma DNA from men with metastatic castration resistant prostate cancer. Genes Chromosomes and Cancer 59225–239. (doi: 10.1002/gcc.22824) [DOI] [PubMed] [Google Scholar]
- Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, Liu G, Coleman I, Lakely B, Li R et al. 2016Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nature Communications 7. (doi: 10.1038/ncomms13668) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y & Dehm SM 2017. Androgen Receptor Rearrangement and Splicing Variants in Resistance to Endocrine Therapies in Prostate Cancer. Endocrinology 158 1533–1542. (doi: 10.1210/en.2017-00109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodara E, Morrison G, Cunha A, Zainfeld D, Xu T, Xu Y, Dempsey PW, Pagano PC, Bischoff F, Khurana A et al. 2019Multiparametric liquid biopsy analysis in metastatic prostate cancer. JCI Insight 4. (doi: 10.1172/jci.insight.125529) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefer J, Akbor M, Handle F, Ofer P, Puhr M, Parson W, Culig Z, Klocker H & Heidegger I 2016. Critical role of androgen receptor level in prostate cancer cell resistance to new generation antiandrogen enzalutamide. Oncotarget 7 59781–59794. (doi: 10.18632/oncotarget.10926) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Palma JF, Agus DB, Wang Y & Gross ME 2010. Detection of androgen receptor mutations in circulating tumor cells in castration-resistant prostate cancer. Clinical Chemistry 56 1492–1495. (doi: 10.1373/clinchem.2010.143297) [DOI] [PubMed] [Google Scholar]
- De Laere B, van Dam PJ, Whitington T, Mayrhofer M, Diaz EH, Van den Eynden G, Vandebroek J, Del-Favero J, Van Laere S, Dirix L et al. 2017Comprehensive Profiling of the Androgen Receptor in Liquid Biopsies from Castration-resistant Prostate Cancer Reveals Novel Intra-AR Structural Variation and Splice Variant Expression Patterns. European Urology 72192–200. (doi: 10.1016/j.eururo.2017.01.011) [DOI] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR & Hall IM 2014. LUMPY: A probabilistic framework for structural variant discovery. Genome Biology 15 1–19. (doi: 10.1186/gb-2014-15-6-r84) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, Hirsch B, Beckman KB, Silverstein KA & Dehm SM 2012. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene 31 4759–4767. (doi: 10.1038/onc.2011.637) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yang R, Henzler CM, Ho Y, Passow C, Auch B, Carreira S, Rodrigues DN, Bertan C, Hwang TH et al. 2020Diverse AR gene rearrangements mediate resistance to androgen receptor inhibitors in metastatic prostate cancer. Clinical Cancer Research 261965–1976. (doi: 10.1158/1078-0432.CCR-19-3023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy V, Meijer WJJ, Blanco L & Salas M 1998. DNA polymerase template switching at specific sites on the φ29 genome causes the in vivo accumulation of subgenomic φ29 DNA molecules. Molecular Microbiology 29 787–798. (doi: 10.1046/j.1365-2958.1998.00972.x) [DOI] [PubMed] [Google Scholar]
- Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KAT, Voytas DF & Dehm SM 2013. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proceedings of the National Academy of Sciences of the United States of America 110 17492–17497. (doi: 10.1073/pnas.1308587110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM et al. 2018Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 174758–769.e9. (doi: 10.1016/j.cell.2018.06.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V & Korbel JO 2012. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28 333–339. (doi: 10.1093/bioinformatics/bts378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu Y-M, Schultz N, Lonigro RJ, Mosquera J-M, Montgomery B, Taplin M-E, Pritchard CC, Attard G et al. 2015Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 1611215–1228. (doi: 10.1016/j.cell.2015.05.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Graf RP, Schreiber NA, Jayaram A, Winquist E, McLaughlin B, Lu D, Fleisher M, Orr S, Lowes L et al. 2018Assessment of the validity of nuclear-localized androgen receptor splice variant 7 in circulating tumor cells as a predictive biomarker for castration-resistant prostate cancer. JAMA Oncology 41179–1186. (doi: 10.1001/jamaoncol.2018.1621) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, Johnson A, Jendrisak A, Bambury R, Danila D, McLaughlin B, Wahl J, Greene SB, Heller G, Marrinucci D, Fleisher MDR 2016Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker with Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncology 1961124. (doi: 10.1016/j.juro.2016.07.066) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer DR, Leversha MA, Danila DC, Lin O, Gonzalez-Espinoza R, Gu B, Anand A, Smith K, Maslak P, Doyle GV et al. 2007Circulating tumor cell analysis in patients with progressive castration-resistant prostate cancer. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research 132023–2029. (doi: 10.1158/1078-0432.CCR-06-2701) [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD & Jemal A 2020. Cancer statistics, 2020. CA: A Cancer Journal for Clinicians 70 7–30. (doi: 10.3322/caac.21590) [DOI] [PubMed] [Google Scholar]
- Sperger JM, Strotman LN, Welsh A, Casavant BP, Chalmers Z, Horn S, Heninger E, Thiede SM, Tokar J, Gibbs BK et al. 2017Integrated analysis of multiple biomarkers from circulating tumor cells enabled by exclusion-based analyte isolation HHS Public Access. Clin Cancer Res 23746–756. (doi: 10.1158/1078-0432.CCR-16-1021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T & Bastian BC 2016. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Computational Biology 12 1–11. (doi: 10.1371/journal.pcbi.1004873) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B et al. 2010Integrative genomic profiling of human prostate cancer. Cancer Cell 1811–22. (doi: 10.1016/j.ccr.2010.05.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulz P, Belic J, Graf R, Auer M, Lafer I, Fischereder K, Webersinke G, Pummer K, Augustin H, Pichler M et al. 2016Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nature Communications 7. (doi: 10.1038/ncomms12008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wala JA, Bandopadhayay P, Greenwald NF, O’Rourke R, Sharpe T, Stewart C, Schumacher S, Li Y, Weischenfeldt J, Yao X et al. 2018SvABA: Genome-wide detection of structural variants and indels by local assembly. Genome Research 28581–591. (doi: 10.1101/gr.221028.117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, Foye A, Lloyd P, Nykter M, Beer TM et al. 2017Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. Journal of the National Cancer Institute 1091–9. (doi: 10.1093/jnci/djx118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR & Xie XS 2012. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338 1622–1626. (doi: 10.1126/science.1229164) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and code are available for bona fide researchers who request it from the authors.