

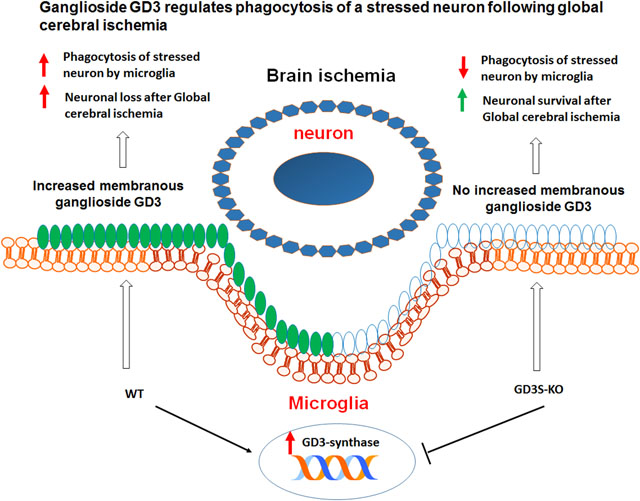
Abstract
Gangliosides, the major sialic-acid containing glycosphingolipids in the mammalian brain, play important roles in brain development and neural functions. Here, we show that the b-series ganglioside GD3 and its biosynthetic enzyme, GD3-synthase (GD3S), were up-regulated predominantly in the microglia of mouse hippocampus from 2 to 7 days following global cerebral ischemia (GCI). Interestingly, GD3S knockout (GD3S-KO) mice exhibited decreased hippocampal neuronal loss following GCI, as compared to wild-type (WT) mice. While comparable levels of astrogliosis and microglial proliferation were observed between WT and GD3S-KO mice, the phagocytic capacity of the GD3S-KO microglia was significantly compromised after GCI. At 2 and 4 days following GCI, the GD3S-KO microglia demonstrated decreased amoebic morphology, reduced neuronal material engulfment, and lower expression of the phagolysosome marker CD68, as compared to the WT microglia. Finally, by using a microglia-primary neuron co-culture model, we demonstrated that the GD3S-KO microglia isolated from mouse brains at 2 days after GCI are less neurotoxic to co-cultured hippocampal neurons than the WT-GCI microglia. Moreover, the percentage of microglia with engulfed neuronal elements in the co-cultured wells was also significantly decreased in the GD3S-KO mice after GCI. Interestingly, the impaired phagocytic capacity of GD3S-KO microglia could be partially restored by pre-treatment with exogenous ganglioside GD3. Altogether, this study provides functional evidence that ganglioside GD3 regulates phagocytosis by microglia in an ischemic stroke model. Our data also suggest that the GD3-linked microglial phagocytosis may contribute to the mechanism of delayed neuronal death following ischemic brain injury.
Keywords: Ganglioside GD3, GD3-synthase, global cerebral ischemia, microglia, phagocytosis, stroke
Graphical Abstract
INTRODUCTION
Gangliosides are a class of glycosphingolipids that contain one or more sialic acid residues. The expression of gangliosides is found to be predominantly on the outer layer of the cellular plasma membrane, and is particularly abundant in the central nervous system(Yu et al. 2011). During brain development, maturation, and aging, the quantity and species of gangliosides undergo dynamic changes in the mammalian brain (Ngamukote et al. 2007). Although the more complex gangliosides, particularly GM1, GD1a, GD1b, and GT1b are known to be the major ganglioside species in the adult brains, simple gangliosides, such as GM3 and GM2, have been shown to be up-regulated in acute brain injury models, including ischemic stroke (Caughlin et al. 2015; Woods et al. 2013). Moreover, an increase in the proportion of simple gangliosides and ganglioside metabolism imbalance has also been reported in neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease and Huntington’s disease (Kracun et al. 1992; Desplats et al. 2007; Chiricozzi et al. 2019). However, the precise role of simple gangliosides, such as GD3 in brain injury has not been well studied.
Global cerebral ischemia (GCI) is a widely used experimental stroke model that simulates human ischemic stroke during cardiac arrest, shock, and asphyxia (Yonekura et al. 2004). The well-known pathology of GCI in both animals and humans is characterized by a delayed loss of neuronal cells (3–7 days after ischemia) and progressive accumulation of glial scar in the CA1 region of the hippocampus (Baron et al. 2014). Various cell death pathways have been invoked to explain the molecular mechanisms underlying the selective hippocampal neuronal cell death following GCI, including excitotoxicity, oxidative stress, and apoptosis-dependent mechanisms (Sims 1992). There is currently no effective treatment that can prevent or reverse the neuronal damage after ischemia.
Microglia, the resident innate immune cell of the CNS, undergo rapid activation in response to cerebral ischemia (Weinstein et al. 2010) and a variety of neurodegenerative conditions, including Alzheimer’s disease, Parkinson’s disease and Huntington’s disease(Savage et al. 2020) (Bartels et al. 2020). The activated microglia become highly hypertrophic, secreting inflammatory cytokines, migrating to the lesion area, and phagocytosing cell debris or damaged neurons (Weinstein et al. 2010). Microglia-mediated phagocytosis after stroke is thought to play a beneficial role in limiting the inflammatory reaction and promoting neuronal repair by clearing harmful cellular components (Neumann et al. 2009). This conclusion is based on the assumption that only the dead or dying neurons can be tagged with “eat-me” signals for the microglia to recognize and phagocytose (Ravichandran 2003). However, recent studies have revealed that in addition to damaged components and pathogens, microglia may also engulf live neurons, neural progenitor cells, neutrophils, and glioma cells in the brain (Brown & Neher 2014; Butler et al. 2021). Importantly, viable neurons subjected to sub-lethal stimuli, such as oxidative stress, ATP depletion, and increased calcium level, may reversibly expose the “eat-me” signal phosphatidylserine, which can be recognized by the activated microglia for phagocytosis (Mari et al. 2004). The process of phagocytosis of viable cells is termed as “primary phagocytosis” or “phagoptosis”, with the defining characteristics that inhibition of phagocytosis prevents cell death (Brown & Neher 2012).
The ST8SiaI gene encodes sialyltranferase II (ST-II, namely GD3-synthase/GD3S), an enzyme that catalyzes the formation of ganglioside GD3 and its downstream b-series gangliosides (Yu et al. 2011). In the current study, we analyzed the dynamic changes of the brain ganglioside GD3 and GD3S expression in a mouse model of GCI. We detected a significant increase of ganglioside GD3 and GD3S in the injured hippocampal region, with the up-regulated GD3 primarily located in reactive microglia. To investigate the role of GD3 in the regulation of glial function and neurological outcome after GCI, we used GD3S-KO mice, in which b-series gangliosides, including GD3, are absent. Intriguingly, we found that the number of surviving neurons in the hippocampal region of GD3S-KO mouse brain was significantly increased at 2-day (R2d) and 4-day (R4d) following GCI reperfusion. Consistently, in-vitro study showed that the GD3S-KO microglia isolated from mouse brains at 2 days after GCI are less neurotoxic to co-cultured hippocampal neurons than the WT-GCI microglial. Meanwhile, microglia proliferation, astrocyte activation, and microglial proliferation were not significantly affected by GD3S deletion. Further analysis revealed an impairment of phagocytic capacity in GD3S-KO reactive microglia using both in vivo and in vitro assays. The in-vitro compromised phagocytic capacity of GD3S-KO microglia can be partially rescued by pre-treating the cells with exogenous GD3. These findings suggest that the GD3-mediated microglial phagocytosis may contribute to the delayed neuronal death after ischemic brain injury.
MATERIALS AND METHODS
Animals
All animal experiments were approved by the Augusta University Institutional Animal Care and Use Committee (Protocol #2010–1035) and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The original GD3S-KO (RRID:MMRRC_000037-MU) were kindly provided by the courtesy of Dr. Richard Proia (NIDDK, NIH, Bethesda, MD). Heterozygous male and female mice were mated, and PCR screening was performed for genotyping. Wild type littermates were used as controls. Three to four-month-old male animals were used in this study. Mice were group-housed (5 mice maximum cage capacity) in environmentally controlled conditions with 12:12/h light/dark cycle, and provided with food and water ad libitum. This study is not pre-registered in institutional registration sites. The experiment details are described in a flow sheet shown in Figure 1. In total 160 adult mice were used approximately.
Figure 1. Scheme of animal use, experiments and corresponding figures.
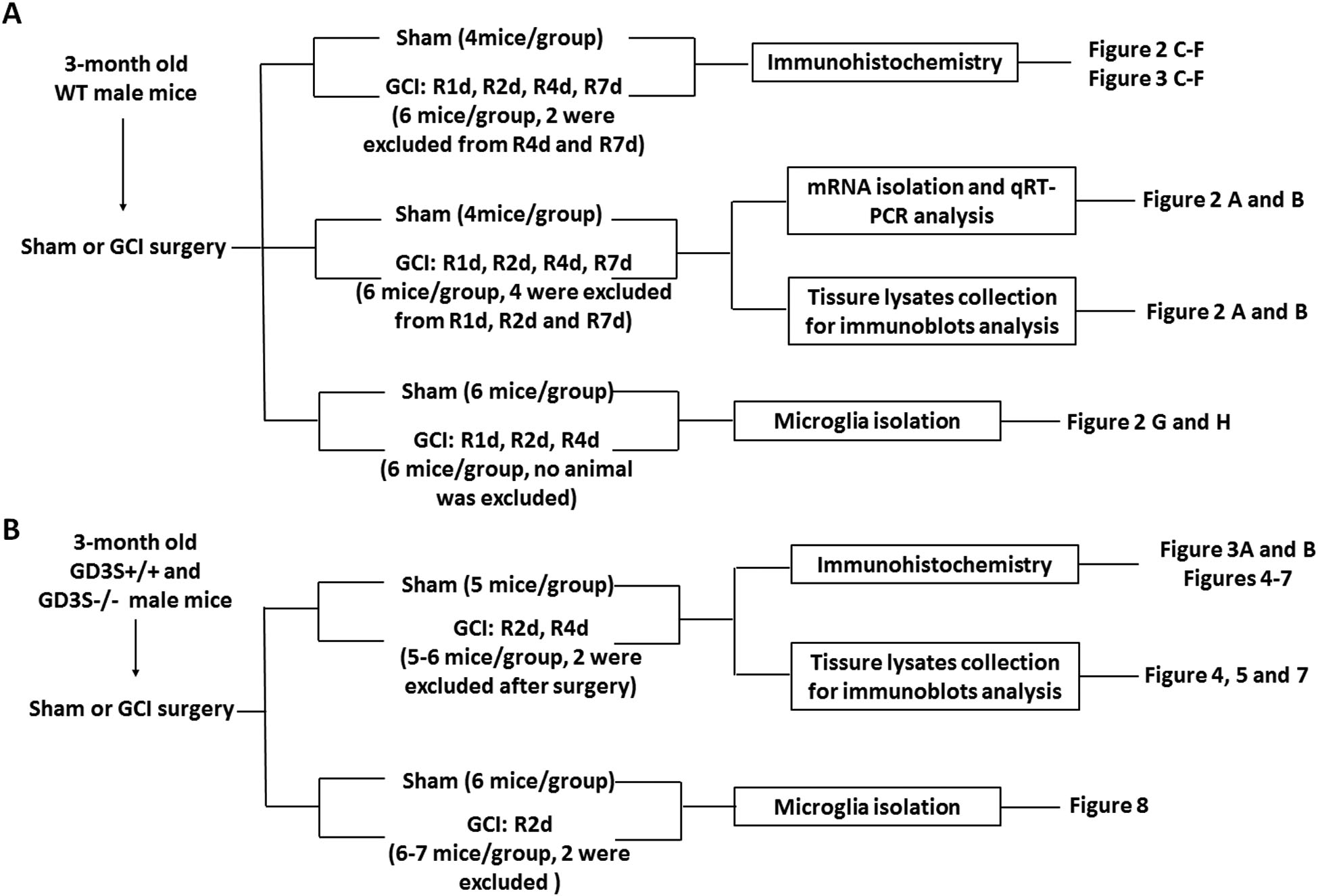
The initial number of animal assigned for sham was 4–6/group, and for GCI was 5–7/group. In the sham group, no animal was excluded. In the GCI group, about 10% of mice failed to meet the criteria of successful GCI surgery by showing dilated pupil, and loss of righting reflex for at least 1 minutes after incision closure, or experienced seizure, severe bleeding during surgery were excluded from further analysis. 2 animals in the GCI group experienced weight loss over 15% of the original body weight during the first 3 days after surgery were excluded.
Global Cerebral Ischemia
Two-vessel occlusion global cerebral ischemia was performed on WT and GD3S-KO mice as described previously (Wang et al. 2020b). Briefly, mice were anesthetized with isoflurane (4% induction, 1–2% maintenance). Via a midline incision of the neck, both common carotid arteries (CCAs) were exposed and encircled loosely with 4–0 suture to enable later occlusion with an aneurysm clip. Global ischemia was induced by bilateral occlusion of the CCAs for 30 min. After ischemia, the clips were removed, and reperfusion was confirmed by visual inspection of blood flow in CCAs. Five min after reperfusion, the incisions were closed with wound clips, and isoflurane was discontinued. The core body temperature of mice before, during and after ischemia was maintained at 37 °C by a homoeothermic blanket, with rectal temperature recorded continuously. Before being returned to their cage at room temperature, the mice were kept in a warm chamber at 33 °C for 2 h to maintain body temperature of approximately 37 °C. For the sham controls, identical procedures were performed, and CCAs were exposed without occlusion. Mice that showed dilated pupils during GCI surgery, and lost the righting reflex for at least 1 minute after incision closure were included for further experiments. In the GCI group, about 10% of the mice failed to meet the above mentioned inclusion criteria, experienced seizures, or had weight loss more than 15% of the original body weight were euthanatized with isoflurane overdoes and excluded for data collection. 2 mg of carprofen (Rimadyl, formulated in a 5 g food tablet) was given to each mouse 8 hours before surgery and after surgery for analgesia. Animals are monitored twice daily for distress and response after surgery. Simple randomization was employed to assign the mice to sham or GCI groups. The initial number of animal assigned for sham groups was 4–6, and for GCI groups was 5–7. We marked the animal on the tail with a number (e.g., from 1 to 20) in the beginning of the animal experiments. After the surgery, the animals were randomly assigned to an experimental group (R1d, R2d R4d or R7d) by the experiment conductor. No computerized randomization was performed to allocate animals in this study.
Tissue Collection
All surgery experiments were started during the morning, and sample collection were conducted at 48 hours (R2d) or 96 hour (R4d) after GCI. Mice were sacrificed by an anesthetic overdose (5% isoflurane inhalation for 10 minutes) and transcardially perfused with ice-cold saline, and brain tissues were then collected. For qRT-PCR, and Western blot analyses, the hippocampus was dissected and processed as described in subsequent sections. For immunohistochemistry, mice were transcardially perfused with 4% paraformaldehyde followed by saline perfusion before decapitation. The brains were removed, fixed overnight in 4% paraformaldehyde, and cryoprotected with 30% (w/v) sucrose in PBS for sectioning on a cryostat.
qRT-PCR
Total RNA was isolated from the cortex or hippocampus dissected from mouse brains using the SV Total RNA Isolation System (Promega, Cat. Z3100). A sample of 400 ng of RNA was used for reverse-transcription polymerase chain reaction (RT-PCR) using the SuperScript III Platinum SYBR Green One-Step qRT-PCR Kit (Invitrogen, Grand Island, NY, USA, Cat. 11736051). The reaction was performed with QuantStudio™ 7 Flex Real-Time PCR System (ThermoFisher Scientific). The threshold cycle values were first normalized to housekeeping gene encoding for GAPDH in the same sample. The relative transcript levels for each gene were then reported as “fold changes versus sham controls” by calculating the 2-ΔΔCT value. The sequences of primers used were as follows: St8sla1-forward: ATGCTAGCTCGGAAATTCCCG, St8sla1-reverse: CAGGGTCACAGCAGTCTTCC; Gapdh forward: CAACTCCCTCAAGATTGTCAGCAA, Gapdh reverse: GGCATGGACTGTGGTCATGA.
Western Blot Analysis
Tissue samples from the hippocampus of sham/GCI mouse brains or following magnetic-activated cell sorting (MACS) were collected as previously described (Wang et al. 2020b). The isolated tissue or cell samples were frozen in liquid nitrogen or kept on ice for immediate homogenization in RIPA buffer (50 mM Tris•HCl at pH 7.4, 150 mM NaCl, 1% Triton x-100, 1% sodium deoxycholate, 0.1%SDS, 1 mM EDTA) with cOmplete protease inhibitor (Roche, REF 5892970001) and PhosphoSTOP (Roche, REF 4906845001) with a tissue tearor followed by denaturation at 95°C for 10 min. Immunoblot analysis was performed as previously described (Wang & Yu 2013). Antibodies used for Western blots included GD3-synthase (sheep, 1:1000, R&D system, Cat. AF6716); GFAP (mouse, 1:2000, Millipore, Cat. MAB360); Iba1 (goat, 1:1000, Abcam, Cat. ab5076); GAPDH (mouse, 1:2000, Santa Cruz Biotechnology, Cat. sc-32233); Cleaved Caspase-3 (Asp175) (rabbit, 1:1000, Cell Signaling, Cat. 9661). Blots were visualized using a LI-COR Odyssey Imager, and the ImageJ analysis software (Version 1.49; NIH, USA) was used to determine the intensity of each band. Band densities for the indicated proteins were normalized to the corresponding loading controls.
Immunohistochemistry (IHC) and histological analysis
Coronal sections (30 μm thick, about a total of 60 sections) containing the hippocampal structure were collected between −0.94 mm to −2.80 mm from bregma. IHC was performed as previously described (Wang et al. 2017). For each immunostaining, at least 4 randomly selected sections were chosen from each animal and processed for histological analysis. Primary antibodies used are as follows: GFAP (goat, 1:2000, Abcam, Cat. ab53554); Iba1 (goat, 1:400, Abcam, Cat. ab5076); NeuN (rabbit, 1:1000, Millipore, Cat. ABN78); GFAP (mouse, 1:800, Millipore, Cat. MAB360); Cleaved Caspase-3 (Asp175) (rabbit, 1:400, Cell Signaling, Cat. 9661). Following three washes with PBS, the sections were incubated in secondary antibodies conjugated to Alexa 488 (RRID:AB_221544), Alexa 568 (RRID:AB_2535753) (1:500, Thermo Fisher Scientific). Sections were then mounted with Vectashield antifade mounting medium with DAPI (VECTOR Laboratories) and examined using confocal microscopy. Images were captured using a Zeiss 510 confocal microscope at 20x or 40x (Plan-Apochromat objective) with constant exposure for each marker in all analyzed sections. DAB stains for anti-GD3 (R24, mouse, 1:200, Abcam, Cat. ab11779) and anti-S100β (mouse, 1:400, Sigma, Cat. S2523) were conducted as previously described (Wang et al. 2020a). Cresyl violet staining and counterstains were performed as previously described (Wang et al. 2017). To build 3-D images of microglia, 30 μm of brain sections were subjected to Iba1 staining. Confocal z-stack images with a step size of 0.5 μm were obtained under a 63x objective lens of LSM510 Zeiss confocal microscopy. 3-D constructions were processed with AutoQuant X software, and cell volumes were measured with Imaris software as previously described (Wang et al. 2020b). A cell with a volume over 200 μm3 were included for analysis. Dispersion of MAP2 stained neurites in the hippocampal CA1 region was measured by the Directionality plugin of the Image J software as described previously (Lu et al. 2020). The hippocampal CA1 from at least 3 different sections of each animal were used for analysis. Blinding were performed to all histologically analysis and cell number counting. The experimenter was unware of the experimental groups analyzed by given images named by number for counting or quantification.
Microglia isolation
Microglia were isolated as previously described (Wang et al. 2017). Briefly, animals were sacrificed by isoflurane euthanasia (5% isoflurane inhalation for 10 minutes). Then, the brain hemispheres were freshly collected and dissociated into a single-cell suspension using an adult brain dissociation kit (Miltenyi Biotec, Cat. 130-107-677). MACS using CD11b MicroBeads (Miltenyi Biotec, Cat. 130-093-634) were used to positively select the CD11b expression cells, which were subject to phagocytosis assay and Western blot analysis. CD11b-negative cells were also collected and lysed for Western blot analysis.
Primary hippocampal neuron culture and co-culture with microglia
Primary hippocampal neurons culture were performed as described by Francesco T. et al. (Tomassoni-Ardori et al. 2020). Neuron-microglial co-culture and LDH neurotoxicity assay were performed as previously described with minor modifications (Wang et al. 2017). Briefly, the hippocampi from C57BL/6 mice on embryonic day 17.5–18.5 (E 17.5–18.5) were micro-dissected. Cells from therein were dissociated and seeded at a density of ~2× 105/ml in poly-D-lysine (Sigma-Aldrich, Cat.P7280) coated plates, followed by incubating at 37°C in a humidified atmosphere of 5% CO2 in air. When the neurons were in vitro for 7 days (DIV, the primary microglia isolated from sham or GCI mouse brains were plated on top of the neurons at a microglia to neuron ratio of 1:33. Immunocytochemistry was performed for morphological and phagocytosis analysis at 72 hrs after the start of the co-culture. Microglia-mediated neurotoxicity was measured with the LDH Cytotoxicity Assay Kit (Thermo Scientific. Cat. C20301) following the manufacturer’s instruction. Data were collected from two independent cultures, and each culture contained triplicated wells for each group/treatment.
Phagocytosis assay
Phagocytosis assays of isolated microglia were conducted using a phagocytosis assay kit from Abcam (Cat. ab21156) following the manufacturer’s instructions. Briefly, 1×105 cell/ml microglia were seeded on a 96-well plate and incubated overnight. The next day, cells were treated with vehicle, 0.1 μg /ml, or 1μg /ml pre-dissolved GD3 (Avanti Polar Lipids Inc, SKU# 860060P) for 2 hours. Afterwards, 10 μg of Zymosan suspension was added to each well (except the negative control) and incubated for 1 hour. After washing with cold PBS, cells were fixed and permeabilized. Then the detection reagent, reaction substrate, and stop solution were subsequently added. The signals of internalized Zymosan were read by a microplate reader at OD 405nm, and the reading was normalized to the negative control. Each group of treatment and the negative control were conducted in triplicated wells, and two independent cultures were performed for the analysis.
Statistical Analysis
GraphPad Prism 6 software was used to analyze all the data. Data are presented as mean ± SD. No test for outliers was conducted. For each statistical analysis, normality test was performed using Shapiro-Wilk test in Prism, and the data were normally distributed. A two-tailed unpaired Student t-test was performed when only comparing two groups (e.g., WT and GD3S-KO groups). A one-way ANOVA test was conducted to analyze differences within sham and the different time points following GCI. Two-way ANOVAs were used to compare the difference between WT and GD3S-KO in sham or after GCI. When the ANOVA test was found to be significant, the posthoc test was conducted to make pairwise comparisons between experimental groups. A value of p<0.05 was considered statistically significant. The sample size for each experiment is determined by a power analysis (α=0.05, β=0.02) of our pilot experiments, and also based on previous articles from our group (Wang et al. 2020b). A summary of experimental procedures is shown in Figure 1.
RESULTS
Up-regulation of GD3-synthase in mouse hippocampal region after GCI
To understand the regulation of b-series gangliosides in the mouse brain after GCI, we evaluated the mRNA level of the GD3S (also known as ST8Sia1) gene, from 1 to 7 days after GCI-reperfusion (R1d to R7d). We first measured the mRNA levels of GD3S in the hippocampal and cortical tissue samples. Quantitative real time-PCR (qRT-PCR) revealed that the mRNA level of ST8Sia1 was modestly but not significantly increased in the hippocampus at R1d as compared to the sham control, followed by a significant increase at R2d and R4d, with the highest ST8Sia1 mRNA levels observed at R2d (Fig. 2A, F(4,19)=13.40, p<0.01, one-way ANOVA followed by Dunnett’s test). In the cortex, there was no significant difference in GD3S gene expression detected between the sham control group and the GCI groups at any time points examined (Fig. 2B, F(4,19)=1.677, p=0.1969. One-way ANOVA followed by Dunnett’s test). We also conducted DAB staining of the hippocampal sections using the anti-ganglioside GD3 (R24) monoclonal antibody, and counterstained with cresyl violet. DAB staining revealed that the GD3 expression was significantly enhanced in the hippocampal region from R2d to R7d after GCI (Fig. 2C). Consistent with the qRT-PCR and IHC results, Western blot analysis revealed that hippocampal GD3S protein levels were significantly increased at R2d and R4d (Fig. 2D and 2E, F(4,19)=7.476, p<0.01. one-way ANOVA followed by Dunnett’s test). The brain hippocampal region is well known to be highly susceptible to GCI-induced neural damage, characterized by a delayed loss of neuronal cells occurring 3 to 7 days after ischemia. The up-regulation of GD3S mRNA and enhanced ganglioside GD3 immunoreactive signals in the hippocampal region indicates that the GD3 expression was specifically increased in the damage-prone brain region. The up-regulated GD3S enzyme at both mRNA and protein levels also suggests an increase of de-novo b-series gangliosides synthesis in the mouse hippocampal region following brain ischemia.
Figure 2. Up-regulation of ganglioside GD3 and GD3S expression in the hippocampus after GCI.
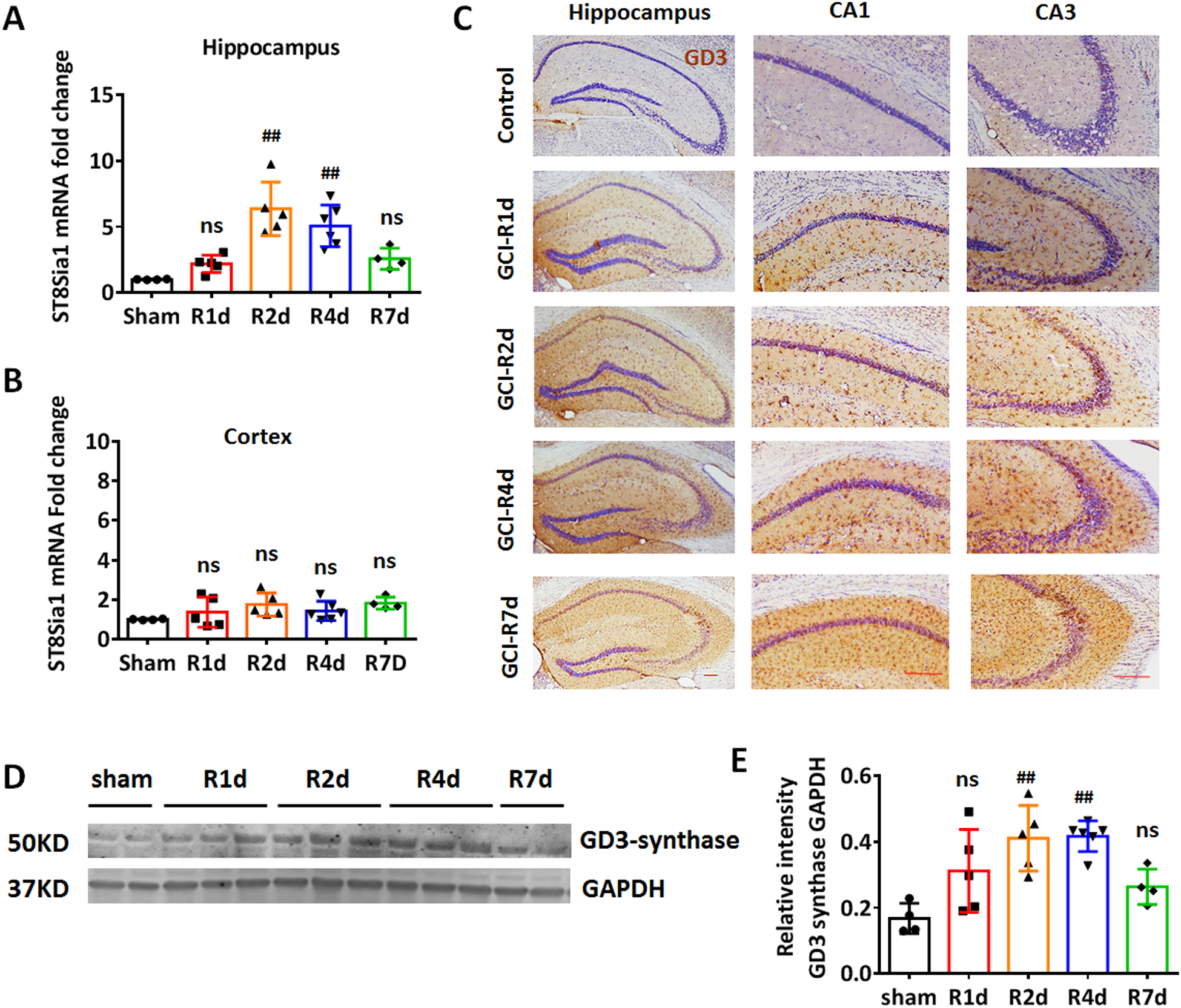
A and B. RT-PCR measurement of GD3-synthase gene mRNA levels in hippocampal (A) and cortical (B) tissue collected in sham and at 1–7 day after GCI reperfusion (R1d to R7d). C. Representative DAB immunostaining of anti-GD3 (brown) and counter stain of cresyl violet (purple) on brain sections from sham and GCI R1d to R7d. D and E. Western blot (D) and densitometry analysis (E) of GD3-synthase expression of sham and GCI R1d to R7d hippocampal tissue lysates. Sham n=4 mice/group, GCI n=6 mice/group. Scale bars: 20 μm. #p<0.05, ##p<0.01 compared to sham, one-way ANOVA followed by post-hoc. ns, not significant.
The up-regulated ganglioside GD3 and GD3S was predominantly localized in microglia after GCI
To verify the authenticity of the immunostaining signal of the anti-GD3 antibody, we stained the hippocampal section samples from WT and GD3S-KO brains using the DAB staining method. In contrast to the prominent GD3 positive signal detected in the WT-R2d hippocampus, no significant positive DAB signals were detected in the GD3S-KO hippocampus at R2d (Fig. 3A). Interestingly, cells that show a high GD3 expression exhibited a cellular morphology similar to that observed for reactive glial cells, e.g., enlarged soma volume and retracted cellular processes (Fig. 3A zoomed). Glial cells, including astrocytes and microglia, can be activated and display hypertrophic morphology in the injured brain region following ischemia. To investigate the specific cellular localization of ganglioside GD3 and its potential biological function, we performed co-immunostaining of GD3 with the astrocytic marker GLAST1 and the microglia marker CD11b. We chose these two markers because both CD11b and GLAST are located on the cell surface and can be stained without detergent treatment. The co-immunostaining results show that GD3 is predominantly localized in the CD11b expressing microglial cells, with a hypertrophic “reactive” morphology (Fig.3B, upper row). As expected, immunostaining for GD3 and CD11b in the GD3S-KO GCI hippocampus did not detect significant anti-GD3 signal or co-localization with CD11b (Fig. 3B, lower row). However, no apparent co-localization of GD3 with the astrocyte marker GLAST1 was detected, indicating that ganglioside GD3 was not significantly up-regulated in the reactive astrocytes (Fig. 3C). Co-immunostaining detected localization of GD3 in CD11b+ cells in the mouse hippocampus (including CA1, CA3 and the hilus of dentate gyrus (DG)) after GCI (Figure 3D). Quantification of the co-localization co-efficiency of GD3 with CD11b demonstrates the most significant co-localization of these two markers at R2d and R4d, and this coincides with the time point at which GD3 is most highly expressed (Fig. 3E and 3F, F(4,21)=107.9, p<0.01, one-way ANOVA followed by Dunnett’s test). To further confirm our findings, we acutely purified CD11b+ microglia and CD11b- non-microglia from the control, R1d, R2d, and R4d hemispheres, and examined the expression of GD3S by Western blot. Using these methods, we were able to obtain a purified cell population of ~95% of CD11b+ cells, which has been confirmed by flow cytometry (Wang et al. 2017). Western blot analysis using cell-specific markers also confirmed that the microglial marker Iba1 was highly expressed in the CD11b+ cell portion, and the astrocyte cell marker GFAP was detected in the CD11b- cell portion (Fig. 3G). Blots of GD3-synthase demonstrated that the expression of this enzyme was highly up-regulated in the CD11b+ microglia (Fig. 3G and 3H, F(3,8)=60.62, p<0.01, one-way ANOVA followed by Dunnett’s test), but not in CD11b- cells at R1d to R4d (Fig. 3G and 3H, F(3,8)=1.469, p=0.2943, one-way ANOVA followed by Dunnett’s test). These results confirmed that ganglioside GD3 and GD3S were predominantly up-regulated in microglial cells after GCI.
Figure 3. Ischemia induced up-regulation of ganglioside GD3 and GD3S in the hippocampus occurred predominantly in microglia.
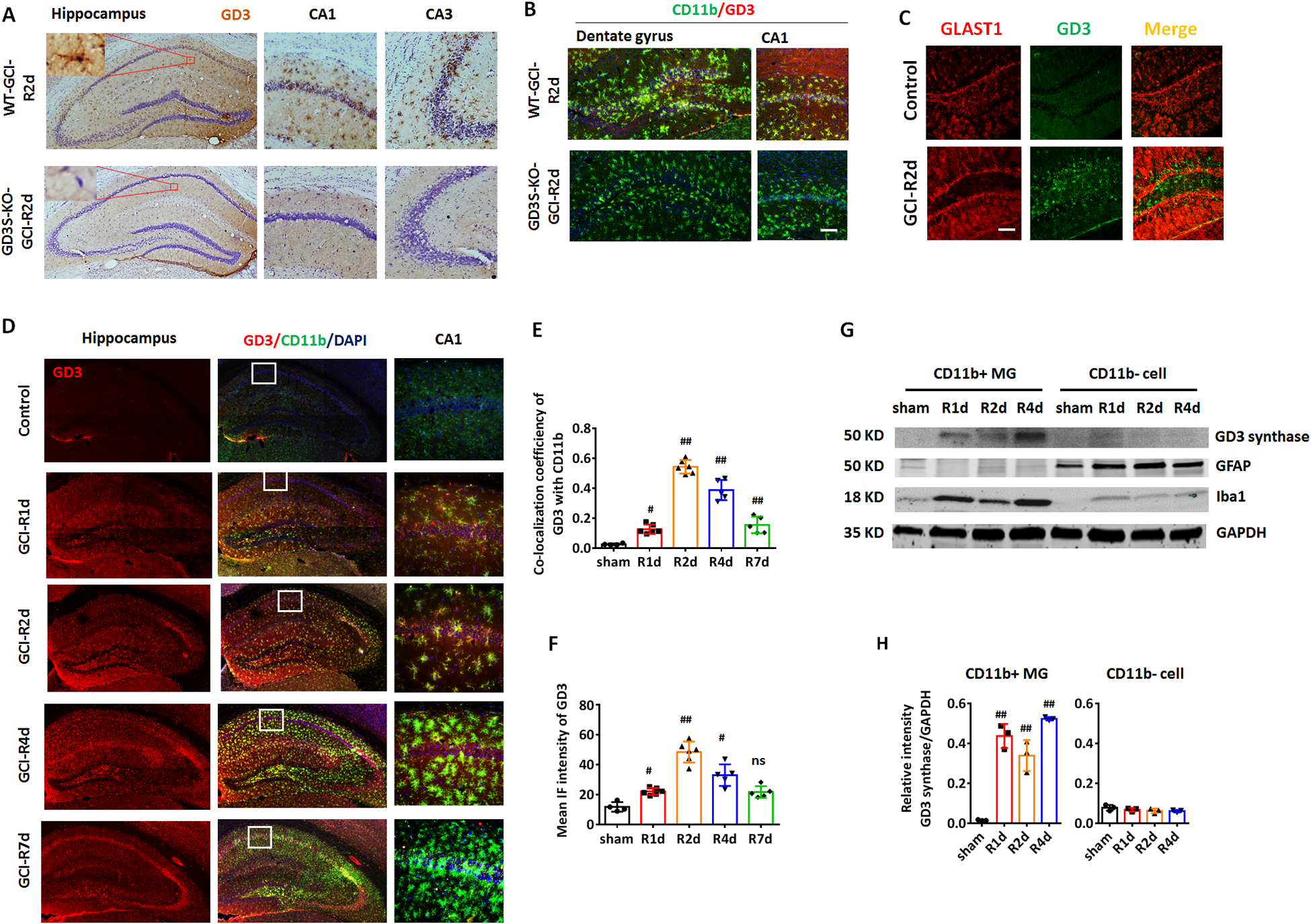
A. Representative DAB immunostaining images of anti-GD3 (brown) and counter stain of cresyl violet (purple) on brain sections from WT and GD3S-KO hippocampus at R2d. B. Representative IF image of GD3 and microglial marker CD11b on hippocampal brain sections from WT and GD3S-KO mice at R2d. C. IF staining of GD3 and astrocyte marker GLAST1 didn’t show significant co-localization. D. IF staining of GD3 and microglial marker CD11b showed localization of ganglioside GD3 in microglial cells in the injured hippocampal regions (CA1, CA3 and the hilus of dentate gyrus (DG)) after GCI. Zoomed area showed representative images of CA1. E. Quantification on the co-localization coefficiency of GD3 with CD11b showed a significant increase of the co-efficiency at 2 and 4 day after GCI (Sham n=4 mice/group, GCI n=6 mice/group). F. Quantification of the mean IF intensity of GD3 in the hippocampus (Sham n=4 mice/group, GCI n=5–6 mice/group). G and H. Western blot (G) and densitometry analysis (H) showed predominant up-regulation of GD3-synthase in lysate of CD11b+ microglia but not CD11b- cells. n=6 mouse/group, 2 mouse brains were pooled for isolating 1 sample of CD11b+/CD11b- cell lysates for Western blot. #p<0.05, ##p<0.01 compared to sham. One-way ANOVA followed by post-hoc. IF: Immunofluorescence. Scale bar: 20 μm.
Increased survival neurons in the hippocampal CA1 of GD3S-KO mouse after GCI
To investigate the role of ganglioside GD3 in the brain under ischemic brain injury, we conducted GCI models in the GD3S-KO mice and their WT littermates, and compared the neuronal damage at R2d and R4d between the two groups. As determined by cresyl violet staining of the coronal brain sections, significant neuronal damage/loss was detected in the CA1 and CA3 regions of the hippocampus in WT mice. However, the neuronal damage was less severe in the GD3S-KO-GCI hippocampus as compared to that in the WT-GCI groups (Fig. 4A). Quantification of the CA1 cellularity demonstrated that number of neurons in the GD3S-KO CA1 was significantly increased as compared to WT after GCI (Fig. 4B, F(2,24)=10.28, p<0.01, two-way ANOVA followed by Sidaks’s test), indicating that the ischemia-induced neuronal damage is significantly less in GD3S-KO hippocampus than in the WT hippocampus. To further confirm this observation by cresyl violet stain, we conducted co-immunostaining of the neuronal marker NeuN and the apoptosis marker, Cleaved-Caspase3, to measure hippocampal neuronal survival and apoptosis, respectively, at the CA1 and CA3 regions after GCI (Fig.4C and 4D). Quantification of survival neuron number based on the NeuN staining demonstrated a 60.5% and 63.5% reduction of hippocampal CA1 neuron number in the WT mice after GCI at R2d and R4d respectively, while the CA1 neuronal loss in GD3S-KO (R2d, 26.5%; R4d, 27.4%) was significantly less severe (F(2,24)=14.26, p<0.01, two-way ANOVA followed by Sidaks’s test). Similarly, GCI induced 32.1% (R2d) and 56.0% (R4d) neuronal loss in WT hippocampal CA3, while in GD3S-KO mice a 17.6% (R2d) and 31.0% (R4d) reduction in CA3 neuron was detected (F(2,24)=6.559, p<0.05, two-way ANOVA followed by Sidaks’s test). Accordingly, the immunofluorescence (IF) intensity of Cleaved-Caspase 3 was significantly decreased in the GD3S-KO hippocampal CA1 and CA3, as compared to WT at R2d and R4d after GCI (Fig. 4G and 4H, CA1: F(2,24)=19.09, p<0.01; CA3: F (2,24)=15.29, p<0.01, two-way ANOVA followed by Sidaks’s test). Consistently, Western blot analysis of the hippocampal tissue lysates detected a significant decrease of the Cleaved-Caspase 3 level in the GD3S-KO hippocampus as compared to WT at R4d after GCI (Fig. 4I and 4J, F(1,16)=36.97, p<0.01, two-way ANOVA followed by Sidaks’s test). Taken together, these results suggest that loss of GD3 and GD3S in the brain results in decreased neuronal damage and increased neuronal survival after ischemic brain injury.
Figure 4. Increased survival neurons in the hippocampus of GD3S-KO mice after GCI.
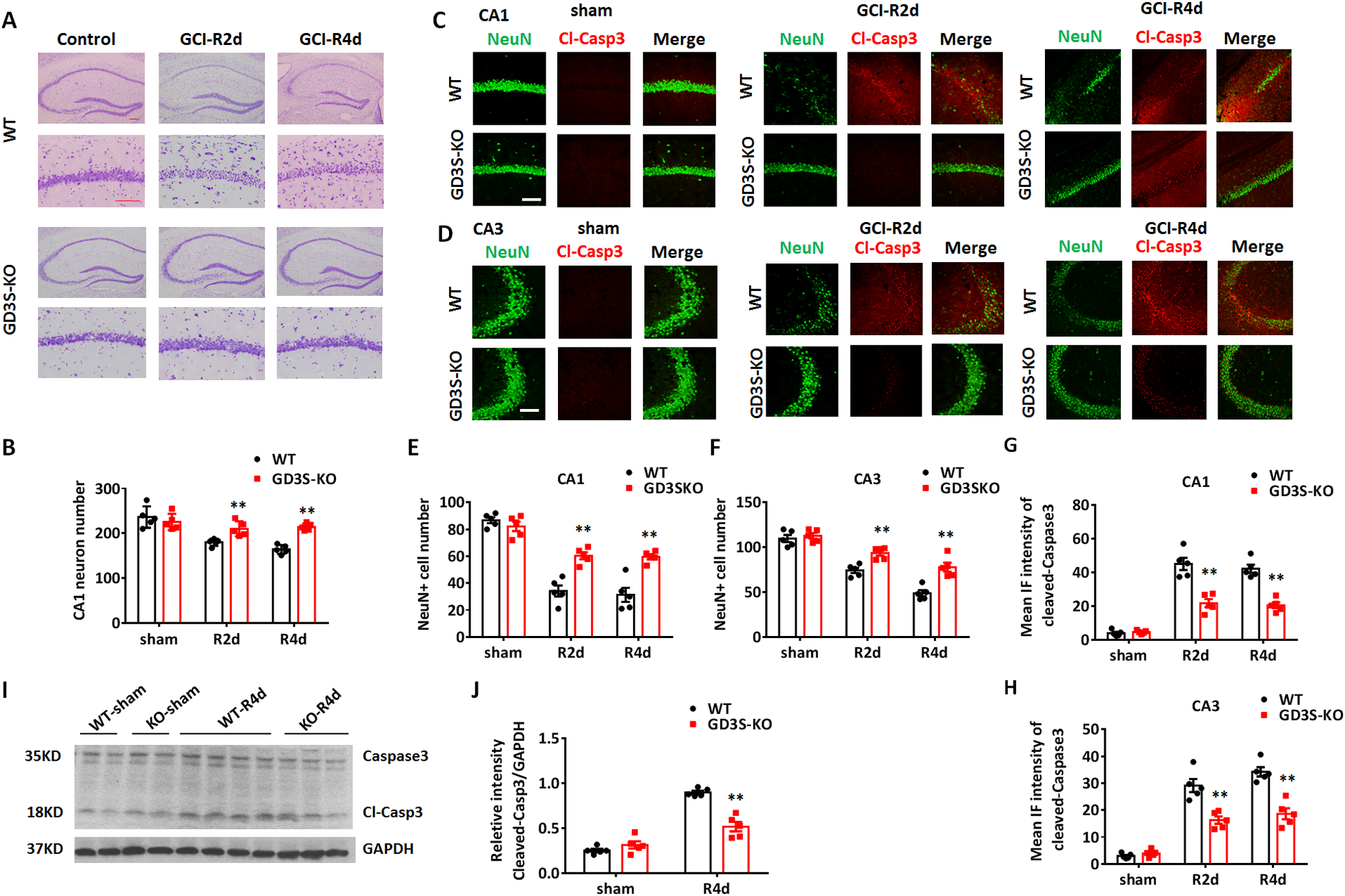
A. Representative images of hippocampus and CA1 region from WT and GD3S-KO brain sections stained with cresyl violet in sham, R2d and R4d after GCI. B. Quantification of hippocampal CA1 neuron number based on cresyl violet staining. C and D. Immunostaining of NeuN and Cleaved-Caspase 3 (Cl-Casp3) in the hippocampal CA1 (C) and CA3 (D) regions in sham, R2d and R4d. E and F. Quantification of survival neuron number in hippocampal CA1 (E) and CA3 (F) regions based on NeuN immunostaining. G and H. Quantifications of Cleaved-Caspase 3 IF intensity in hippocampal CA1 (G) and CA3 regions (H) demonstrate a significant reduction of Cleaved-Caspase 3 expression in GD3S-KO mice as compared to WT mice after GCI. I. Representative Western blot analysis of Cleaved-Caspase 3 on the hippocampal tissue lysate collected from WT and GD3S-KO mice in sham and R4d. J. Quantification of the relative expression level of Cleaved-Caspase 3 based on densitometry analysis of Western blots. n=5 mice/group. **p<0.01 GD3S-KO vs. WT. Two-way ANOVA followed by post-hoc. IF: Immunofluorescence. Scale bar: 20 μm.
Astrogliosis and microglial proliferation after GCI are not affected by GD3S deletion.
Next, we examined the glial cell response to GCI in the WT and GD3S-KO hippocampi. At R4d, significant astrogliosis was detected in the hippocampi of both the WT and GD3S-KO mouse brains, characterized by enhanced expression of the glial fibrillary acidic protein (GFAP) and hypertrophic change of the anti-GFAP stained cells (Fig. 5A). Quantification of GFAP IF intensity in the hippocampus demonstrated enhanced GFAP expression level in both WT and GD3S-KO at R4d as compared to sham controls (Fig. 5B, p<0.01, two-way ANOVA followed by Sidaks’s test). However, there is no significant difference in the GFAP IF intensity between WT and GD3S-KO in both sham and R4d (Fig. 5A and 5B, F(1,16)=2.521, p=0.1319, two-way ANOVA followed by Sidaks’s test). We also examined the astrogliosis by DAB staining with another astrocyte marker, S100β (Fig. 5C). Intensity analysis revealed no significant difference between WT and GD3S-KO in both sham and R4d (Fig.5D, F(1,16)=0.6751, p=0.4233, two-way ANOVA followed by Sidaks’s test). Western blot analysis confirmed that the GFAP expression level in hippocampal samples did not significantly differ between WT and GD3S-KO in sham animals or after GCI (Fig. 5E and 5F, F(1,16)=0.1523, p=0.7015, two-way ANOVA followed by Sidaks’s test). These results suggest that GD3S deletion does not significantly affect astrogliosis in response to GCI in the mouse brain.
Figure 5. Astrogliosis and microglial proliferation after GCI is not affected by GD3S deletion.
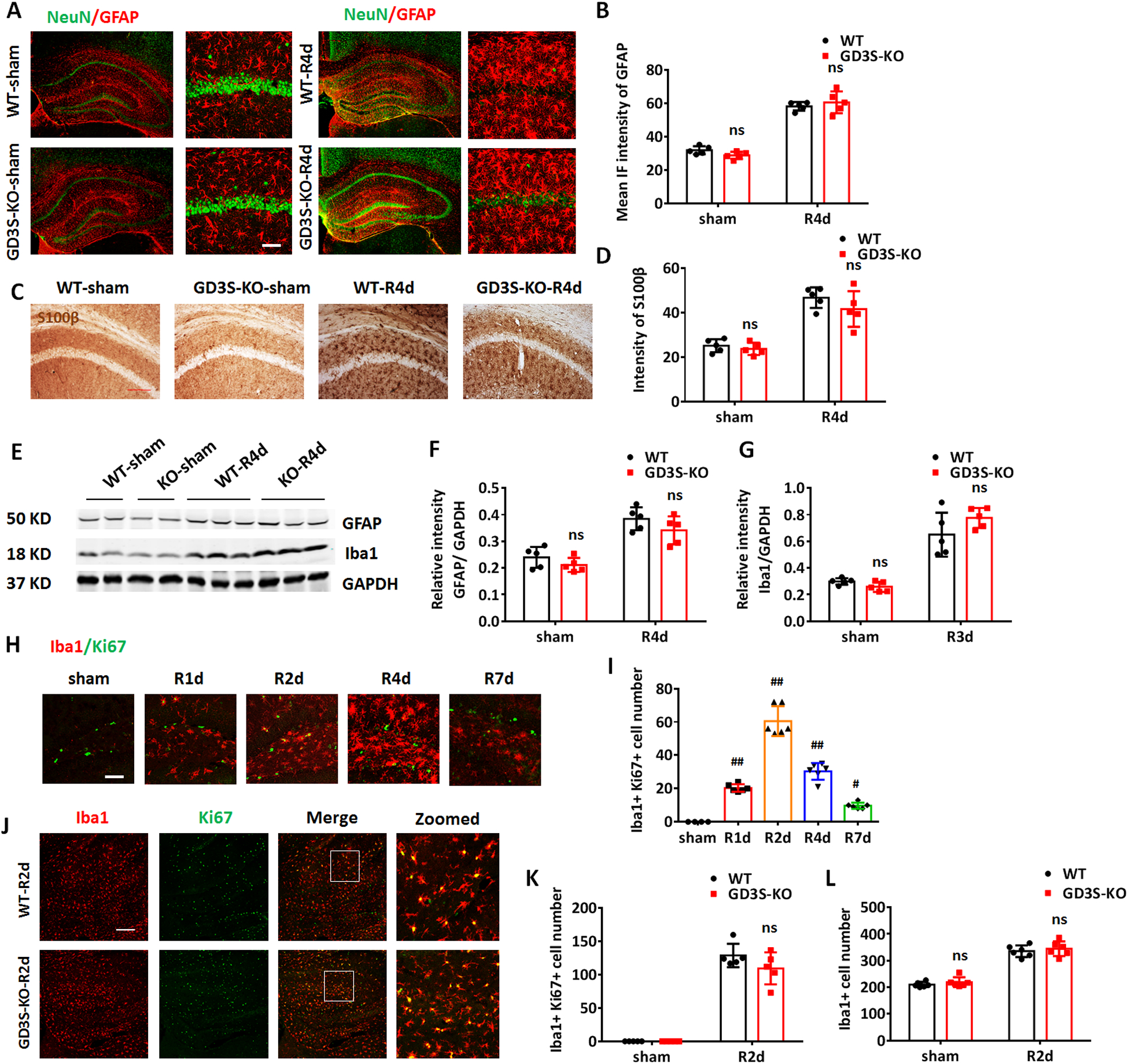
A. Representative images of co-immunostaining for NeuN (green) and GFAP (red) in the sham and GCI-R4d hippocampus and CA1. B. Quantifications of GFAP IF intensity in the hippocampus demonstrate no significant difference between WT and GD3S-KO both in sham and after GCI. C. Representative images of DAB staining for S100β in the hippocampal CA1 region. D. Quantification of the intensity of S100β demonstrates no significant difference between WT and GD3S-KO in sham and after GCI. E-G. Representative Western blots (E) and quantification of GFAP (F) and Iba1 (G) expression in the hippocampal tissue samples. H-I. Representative image of co-immunostaining for Iba1 and Ki67 (H) and quantification of Iba1+Ki67+ cell number (I) in WT hippocampal at different time points (R1d-R7d) after GCI (sham n=4 mice/group, GCI n=6 mice/group). J. Representative image of co-immunostaining for Iba1 and Ki67 in WT and GD3S-KO hippocampus at R2d. K-L. Quantification of Iba1+Ki67+ cell number (K) and Iba1+ cell number (L) demonstrates no significant difference between WT and GD3S-KO in sham and after GCI. n=5 mice/group except H and J. #p<0.05, ##p<0.01 compared with sham group, one-way ANOVA followed by post-hoc; ns. not significant, GD3S-KO vs WT. Two-way ANOVA followed by post-hoc. IF: Immunofluorescence. Scale bar: A and H, 20 μm; C and J, 50 μm.
We also examined the microglial response to GCI in the WT and GD3S-KO hippocampi. Western blot analysis demonstrated that the Iba1 expression level in the hippocampal samples did not significantly differ between WT and GD3S-KO in sham mice or after GCI (Fig. 5E and 5G, F(1,16)=3.913, p=0.0654, two-way ANOVA followed by Sidaks’s test). Proliferation by cell division is a crucial cellular response of microglia to brain ischemia. An increase in the cell population of microglia indicates an enhanced immune response within the injured brain (Zhang 2019). We therefore examined the microglial proliferation by immunostaining for the cell proliferation marker Ki67 and the microglial marker Iba1 from R1d-R7d. Following GCI, a robust microglial proliferation, as revealed by an increase of Iba1+Ki67+ cells, was detected in mouse hippocampus. The Iba1+Ki67+ cell number peaked at R2d, and decreased from R4d to R7d in the hippocampi (Fig. 5H and 5I). We next compared the microglia proliferation and the microglial cell number between WT and GD3S-KO in the hippocampus. In the sham and R2d hippocampi, the numbers of Iba1+Ki67+ cells and the Iba1+ cells do not show a statistical difference between WT and GD3S-KO (Fig. 5J–5L, Iba1+Ki67+ cell number, F(1,16)=2.111, p=0.1655; Iba1+ cell number, F(1,16)=0.005, p=0.9456; two-way ANOVA followed by Sidaks’s test). These results support the concept that GD3S deletion does not significantly affect microglia proliferation and the overall reactive response of microglia following GCI.
Phagocytic cells are decreased in the GD3S-KO hippocampus after GCI
Although the overall microglial activation is not significantly different between WT and GD3S-KO after GCI, we noticed that the activated microglia in the WT and GD3S-KO hippocampi demonstrated distinguishable morphological changes after GCI (Figure 6A). A large amount of microglia in the WT hippocampus changed from ramified into amoeboid morphology at R2d and R4d (Fig. 6A and 6C, upper row). Microglia in the GD3S-KO-GCI hippocampus were hypertrophic with thicker branches, however, most of them demonstrated bushy-like morphology rather than amoeboid shape (Fig. 6A and 6C, lower row). The amoeboid shape allows microglia more free movement and also enhances their capacity to fulfill their role as scavenger cells; therefore, the amoeboid microglia are considered phagocytic microglia in the brain (Fu et al. 2014). We next performed a 3-D reconstruction of confocal microscopy image of the Iba1 immunostaining, and quantified the percentage of amoeboid microglia in the hippocampus according to Kreutzberg’s classification of microglia morphology (Kreutzberg 1996). In the GD3S-KO hippocampus, the rate of amoeboid cells decreased by 39.4% at R2d and 28.5% at R4d as compared to WT after GCI (Fig. 6A and 6B, F(2,24)=36.79, p<0.01, two-way ANOVA followed by Sidaks’s test). This result indicates a decrease of amoeboid microglia in the GD3S-KO hippocampus after GCI.
Figure 6. Decreased phagocytic microglia in GD3S-KO hippocampus after GCI.
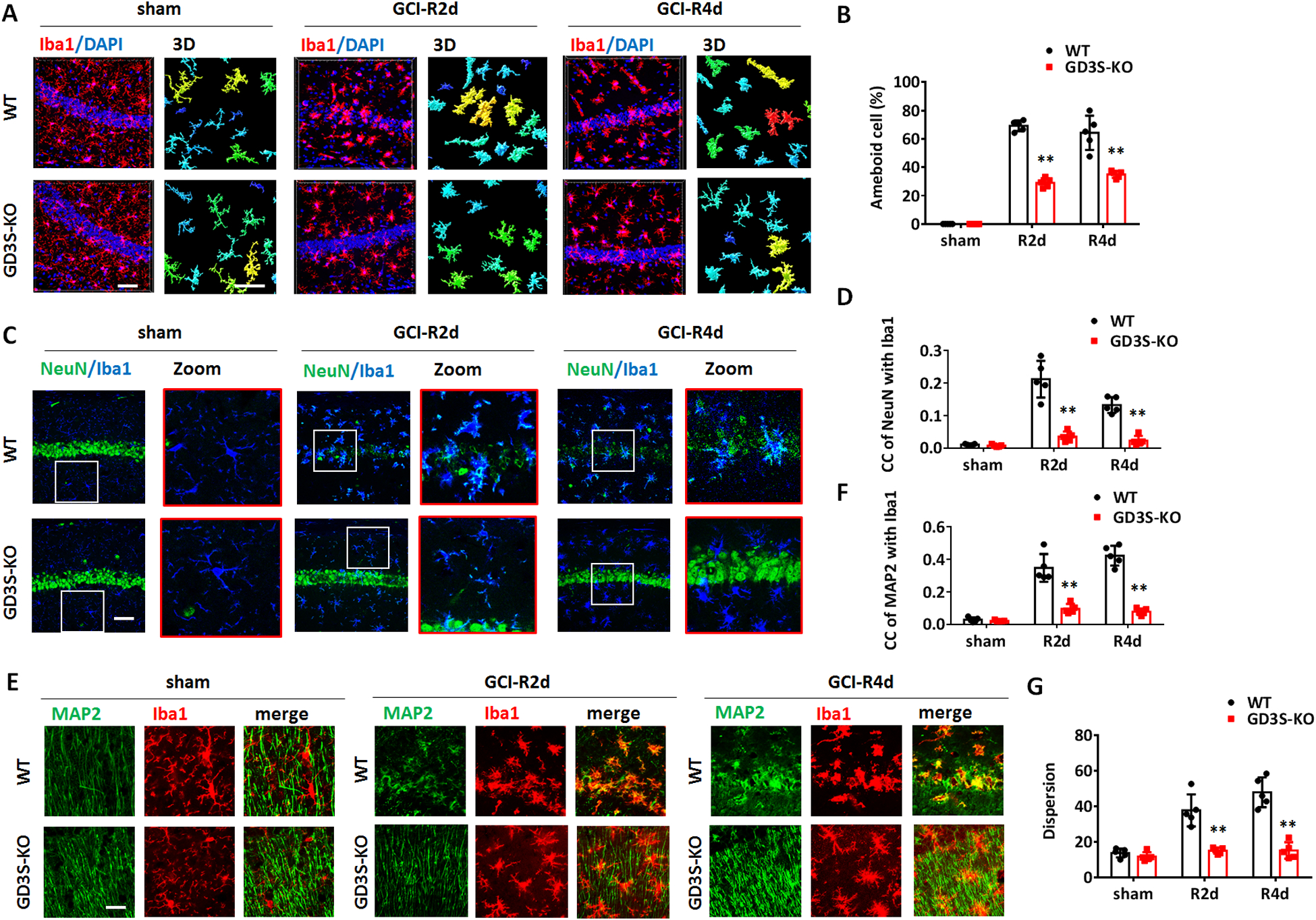
A. Representative images of immunostaining for Iba1 in hippocampal CA1 region and 3-D reconstruction of con-focal images. B. Quantification of the percentage of amoeboid cell number in the hippocampal CA1 region according to Kreutzberg’s classification of microglia morphology. C-D. Representative images of immunostaining for NeuN and Iba1 in hippocampal CA1 (C) and confocal microscopy quantification of NeuN and Iba1 co-localization co-efficiency (D). E-F. Representative images of immunostaining for MAP2 and Iba1 in the hippocampal CA1 region (E), and confocal microscopy quantification of NeuN and Iba1 co-localization co-efficiency (F). G. Dispersion of MAP2 immunostaining was analyzed and quantified on the images of the hippocampal CA1 region by Image J software. n=5 mice/group. **p<0.01 compared with sham or control group. Two-way ANOVA followed by post-hoc. CC: co-localization co-efficiency. Scale bar: 20 μm.
A major function for microglial activation and recruitment in the brain ischemic injury region is to clear cell debris and dying cells through phagocytosis. We therefore measured the in-vivo phagocytosis of neurons in WT and GD3S-KO hippocampi in sham and after GCI. Phagocytosis of neurons by microglia can be recognized by co-localization of NeuN or MAP2 with Iba1 analyzed by confocal microscopy (Neher et al. 2013). We first conducted co-immunostaining of Iba1 with the neuronal markers NeuN followed by confocal microscopy. In the sham animals, no apparent co-localization of NeuN with Iba1 was detected in both WT and GD3S-KO hippocampi (Fig. 6C and 6D). After GCI, a significant localization of NeuN in Iba1+ microglia was observed in the WT hippocampus at R2d and R4d (Fig. 6C and 6D). However, in the GD3S-KO hippocampus, co-localization of NeuN with Iba1 was not significantly detected after GCI (Fig. 6C). Z-Stack analysis of co-localization co-efficiency revealed a significant decrease of NeuN and Iba1 co-localization co-efficiency in GD3S-KO-GCI compared with WT-GCI at R2d and R4d (Fig. 6D, F(2,24)=26.99, p<0.01, two-way ANOVA followed by Sidaks’s test). We also conducted co-immunostaining for Iba1 with the neuronal cytoskeleton markers MAP2 followed by confocal Z-stack and co-localization co-efficiency assay. Consistent with the Iba1 and NeuN co-localization study, enhanced localization of MAP2 in Iba1+ microglia was detected in WT-GCI hippocampus but not in GD3S-KO-GCI hippocampus at R2d and R4d (Fig. 6E). Quantification analysis also demonstrated a significant decrease of the MAP2 and Iba1 co-localization co-efficiency in GD3S-KO after GCI (Fig. 6F, F(2,24)=36.23, p<0.01, two-way ANOVA followed by Sidaks’s test). Notice that a significant decrease of the MAP2 immunostaining dispersion was detected in GD3S-KO hippocampus at R2d and R4d after GCI, indicating increased neurite integrity compared to the WT-GCI hippocampus (Fig. 6E and 6G, F(2,24)=19.50, p<0.01, two-way ANOVA followed by Sidaks’s test). All these results indicate that the decreased phagocytosis of neurons in the GD3S-KO hippocampus is associated with increased neurite integrity and neuronal survival after GCI.
We also examined the expression of the phagolysosome marker CD68 in WT and GD3S-KO hippocampi. Quantification of the CD68+ cells demonstrated that GCI induced a robust increase of phagocytic cells at R2d and R4d in WT mice (Fig. 7A and 7B, p<0.01, two-way ANOVA followed by Sidaks’s test). Notably, the number of CD68+ cells in GD3S-KO-GCI hippocampus was significantly decreased at R2d and R4d as compared to WT-GCI (Fig. 7A and 7B, F(2,24)=43.18, p<0.01, two-way ANOVA followed by Sidaks’s test). The IF intensity of CD68 also significantly decreased in GD3S-KO-GCI as compared to WT-GCI at R2d and R4d (Fig. 7A and 7C, F(2,24)=27.07, p<0.01, two-way ANOVA followed by Sidaks’s test). Western blot analysis confirmed that the expression of CD68 in the hippocampal tissue was significantly decreased in GD3S-KO mice at R2d and R4d as compared to WT mice (R2d, Fig. 7D and 7E, F(1,16)=21.27, p<0.01; R4d, Fig. 7F and 7G, F(1,16)=58.15, p<0.01; two-way ANOVA followed by Sidaks’s test). These findings suggest that the phagocytic microglia increased by GCI in WT hippocampus, and these cells were diminished in the GD3S-KO hippocampus after GCI.
Figure 7. Decreased phagolysosome expression in GD3S-KO hippocampus after GCI.
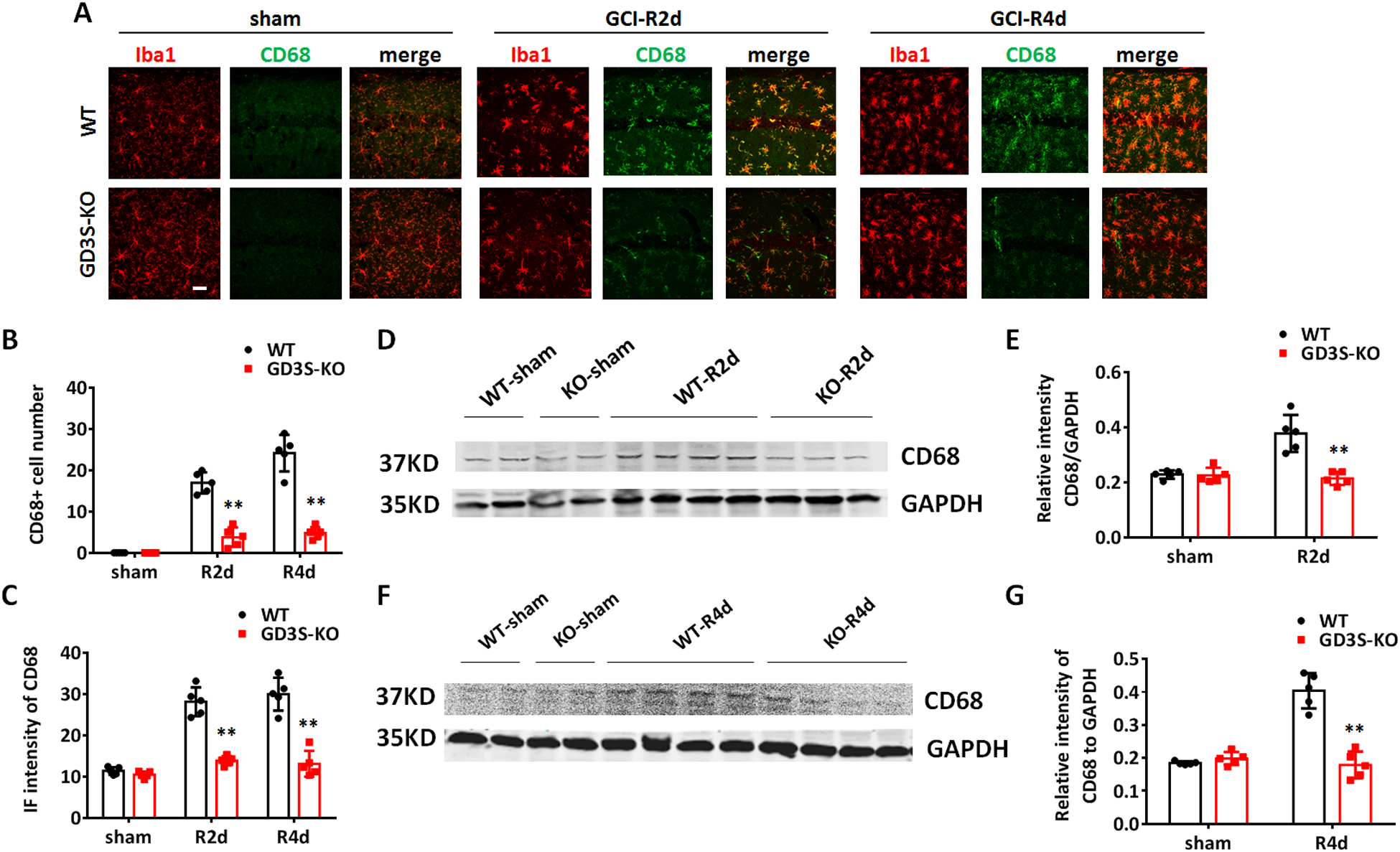
A. Representative images of immunostaining for Iba1 and phagolysosome marker CD68 in the hippocampal CA1. B-C. Quantification of CD68 IF intensity (B) and Iba1+CD68+ cell number (C) in hippocampal CA1. D-G. Western blots analysis of CD68 in hippocampal tissue lysates in sham and at R2d (D) and R4d (F) after GCI. Densitometry analysis showing relative CD68 expression level in hippocampal tissue samples after being normalized with GAPDH intensity at R2d (E) and R4d (G). n=5 mice/group. **p<0.01 GD3S-KO vs. WT group, Two-way ANOVA followed by post-hoc. IF: Immunofluorescence. Scale bar: 20 μm.
Decreased neurotoxicity and phagocytic capacity of microglia from GD3S-KO mice after GCI
To determine whether the phagocytic microglia are less neurotoxic in the absence of GD3, we examined the microglial neurotoxicity using a primary neuron and microglia co-culture model as previously described (Wang et al. 2017). We prepared primary hippocampal neurons from WT embryos (E17.5–18.5), and co-cultured the neurons (7 div) with WT or GD3S-KO microglia in a 1:33 microglia to neuron ratio for 72 hours. When these primary hippocampal neurons are co-cultured with microglia from sham animals, very low neurotoxicity (<10%) were detected from both WT and GD3S-KO microglia (Fig. 8 A and 8B). A robust increase of neurotoxicity was detected when neurons were co-cultured with microglia from WT-GCI mouse brain. However, the neurotoxicity was significantly decreased in the GD3S-KO-GCI microglia co-cultured group (Fig. 8A and 8B, 26.75% in GDS3-KO-GCI vs. 40.72% in WT-GCI, F(1,20)=23.10, p<0.01, two-way ANOVA). Meanwhile, significant localization of neuronal element (Red, stained by anti-β-Tubulin III) was detected in WT-GCI microglia (green, stained by anti-Iba1), but not in GD3S-KO-GCI microglial, indicating that the microglial phagocytic capacity was compromised in the absence of GD3. We further measured the percentage of microglia with engulfed neuronal elements in the co-culture wells. The quantification revealed a significant increase in phagocytic microglia from WT-GCI mouse brain compared with sham (Fig. 8A and 8C, p<0.01, two-way ANOVA followed by Sidaks’s test). The percentage of phagocytic microglia in the GD3S-KO-GCI microglia co-culture wells is significantly decreased than that in the WT-GCI microglia co-culture group (Fig. 8A and 8C, F(1,20)=220.6, p<0.01, two-way ANOVA followed by Sidaks’s test). The in-vitro culture results mirror our in-vivo findings, and support that microglia from GD3S-KO-GCI mice are less phagocytic and less neurotoxic than that from the WT-GCI mice, which may contribute to the increased neuronal survival in the GD3S-KO mice after GCI.
Figure 8. Evidence that ganglioside GD3 regulates the neurotoxicity and phagocytic capacity of microglia in vitro.
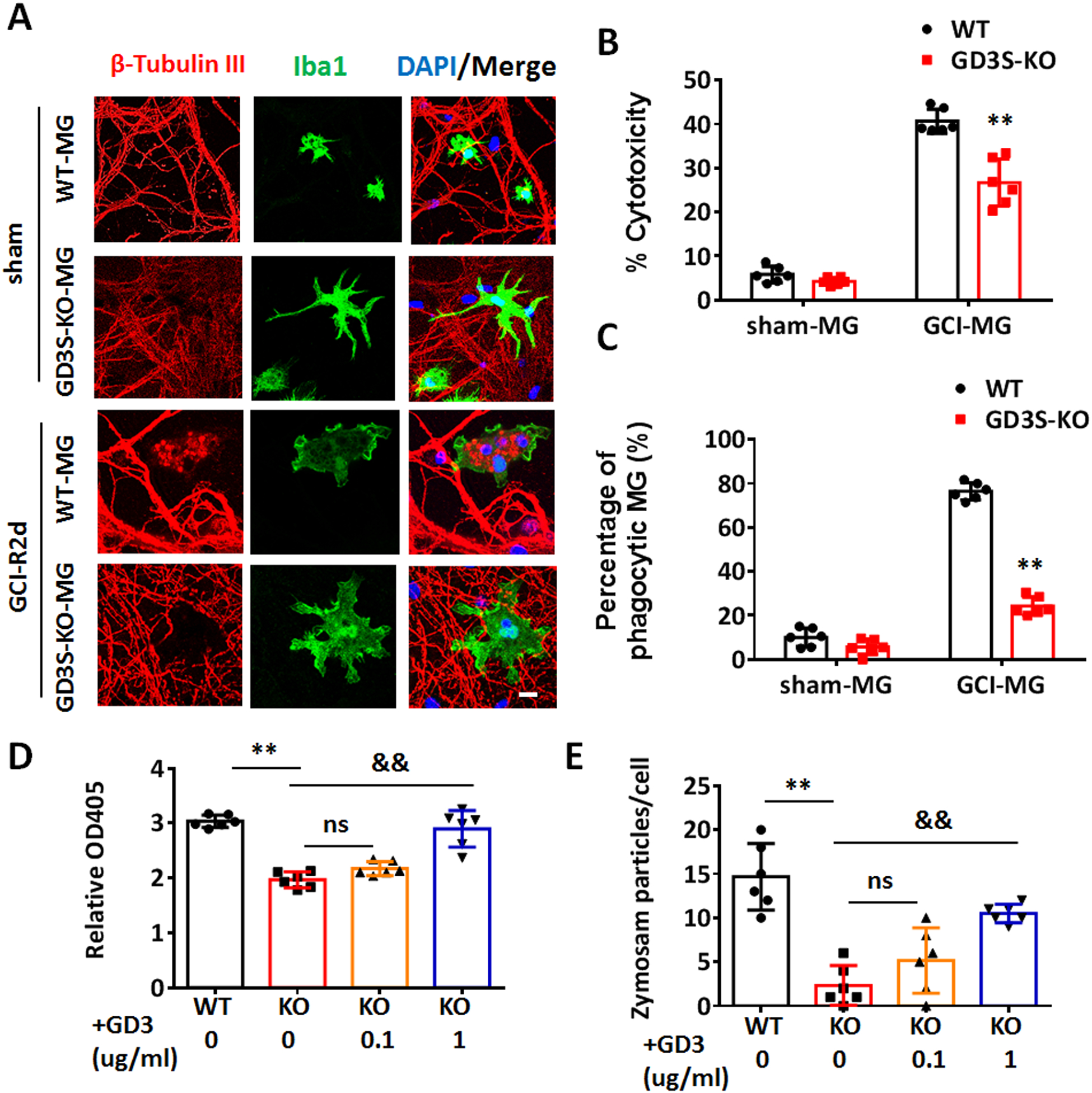
A. Representative images of primary hippocampal neuron (β-tubulin III, red) co-cultured with microglia (MG) (Iba1, green) from sham or GCI-R2d mouse brain for 72 h. B. Neurotoxicity of co-cultured microglia was measured by LDH assay compared between the control wells without microglia co-culture. C. The percentage of phagocytic MG was quantified by counting the number of MG with engulfed neuronal elements and dividing that by the total number of MG in 10 randomized fields in each well. D. Phagocytic capacity of primary microglia isolated from GCI-R2d mouse brain, with/without exogenous GD3 treatment were measured by Zymosan substrate assay at 48 hour in vitro. E. Phagocytic capacity of primary microglia isolated from GCI-R2d mouse brain, with/without exogenous GD3 treatment were measured by quantification of the average Zymosan particles number in each cell. Cells from 2 randomly captured images of each well were quantified. For the each experiments, n=6 mice/group. 2 mouse brains were pooled for isolating 1 sample of microglia for in-vitro culture. 2 independent cell cultures were performed for the neurotoxicity and phagocytic capacity assay, and data from triplicated wells were collected. **p<0.01 GD3S-KO vs. WT. &&p<0.01 vs. KO (0 ug/ml GD3). ns, not significant. Two-way ANOVA followed by post-hoc. Scale bar: 5 μm.
In the Zymosan based phagocytic capacity tests, we also detected a significant decrease in the phagocytic capacity of GD3S-KO-GCI microglia compared to the WT-GCI, as measured by both substrate colorimetric detection (Fig. 8D, F(3,20)=41.50, p<0.01, one-way ANOVA followed by Tukey’s test), and internalized Zymosan particle quantification (Fig. 8E, F(3,20)=21.22, p<0.01, one-way ANOVA followed by Tukey’s test). To test whether an increase in the GD3 level could rescue the phagocytosis deficits in GD3S-KO microglia, we pre-treated the KO-GCI microglia with 0.1 μg/ml or 1 μg/ml of exogenous GD3 for 2 hours before the phagocytosis assay. Compared to the vehicle-treated GD3-KO-GCI microglia, the phagocytic capacity of microglia pre-treated with 1 μg/ml of GD3 was significantly increased (Fig. 8D, p<0.01, one-way ANOVA followed by Tukey’s test). However, this rescue effect was not observed in the group with 0.1 μg/ml of GD3 treatment (Fig. 8D, p>0.05, one-way ANOVA followed by Tukey’s test). Consistently, pre-treatment of GD3S-KO-GCI microglia with 1 ug/ml but not 0.1 μg/ml of exogenous GD3 significantly increased the phagocytosis of Zymosan particles (Fig. 8E, KO+GD3 (1 μg/ml) vs. KO-vehicle, p<0.01; KO+GD3 (0.1 μg/ml) vs. KO-vehicle, p>0.05, one-way ANOVA followed by Tukey’s test). Altogether, these results provide unequivocal evidence that ganglioside GD3 plays a crucial role in mediating the phagocytosis activity of microglia upon reactivation.
DISCUSSION
Gangliosides are the major sialic acid-containing glycosphingolipids in the mammalian brain (Yu et al. 2004). They are predominantly localized on the outer layer of the plasma membrane of nerve cells, with various sialic-acid-bearing glycan structures extending to the extracellular space. These unique localization and structure properties enable gangliosides to function as varied cell-surface markers or receptors with specific cell-cell recognition and interaction capabilities. Gangliosides also interact laterally with adjacent surface-membrane proteins, and regulate signal transduction of target proteins in response to stimuli from their ligands (Lopez & Schnaar 2009). In the current study, we used a murine GCI model to study the role of ganglioside in ischemic brain injury. We report for the first time that ganglioside GD3 is highly up-regulated in reactive microglia within the ischemic hippocampus. Our results also revealed a novel role of GD3 in mediating microglial phagocytic capacity following GCI.
Rapid activation of local innate immune cells, primarily microglia, is a key initial event in the brain following an ischemic stroke. The switch of microglia from a “resting” homeostatic state to an activated state involves morphological transformation, proliferation, recruitment to the injury site, cytokine release, and other functional changes. Accompanying microglia activation, ischemic brains may undergo progressive neuronal damage and delayed neuronal loss at 3–7 days post-injury (Horn & Schlote 1992; Nitatori et al. 1995). The delayed neuronal loss following brain ischemia can be due to a variety of molecular mechanisms, including excitotoxicity, oxidative stress, mitochondria damage, as well as neuroinflammation, in which microglial activation plays a central role (Abe et al. 1995). Activated microglia can either exacerbate ischemic injury (e.g., by releasing proinflammatory cytokines and ROS) or help to limit and repair the damage – with the effect observed depending on different signals controlled by neuron-glial communication (Jayaraj et al. 2019; Al Mamun et al. 2020). The highly heterogeneous transcriptional and functional profiles of activated microglia make them a multifaceted cell population in the brain following an ischemic injury, allowing them to assume various roles as dictated by the local microenvironment and associated cues (Jayaraj et al. 2019).
A key function of reactive microglia is phagocytosis of irreversibly damaged cell debris, and cellular components, which may potentially be harmful to surviving neurons in the ischemic brain. In the transient whole-brain hypoxia status, several previous studies have shown that the microglia play a more predominant role in phagocytosis compared to macrophage (Schilling et al. 2005; Denes et al. 2007). The phagocytosis of tissue debris is generally considered to be a beneficial aspect of microglial reactivation, which can help to limit the damage, and facilitate the reconstruction of the neuronal network. It has also been proposed that phagocytosis may contribute to neuronal loss during ischemia by engulfing stressed-but-viable neurons, which may express an “eat me” signal (Neher et al. 2013; Butler et al. 2021). In support of this contention, mice deficient for the phagocytic proteins, Mer receptor tyrosine kinase (MerTK) and Milk fat globule EGF-like factor 8 (MFG-E8), showed strongly reduced neuronal loss and significantly improved functional outcome after cerebral ischemia. In the GD3S-KO mice, we detected a strikingly decreased neuronal loss at R2d and R4d following GCI as compared to their WT littermates. Further analysis revealed that microglial proliferation and overall glial cell reactivation are not significantly affected by GD3S deletion; however, the phagocytic capacity of GD3S-KO microglia was robustly decreased as measured by in-vivo and in-vitro assays. These results are similar to the finding in the MerTK and MFG-E8 knockout mice, indicating that up-regulation of GD3 in microglia following GCI may play a role in facilitating the phagocytosis of stressed neuron by microglia, thereby resulting in enhanced neuronal loss.
The glycosphingolipid composition of the mammalian brain can undergo dynamic changes under conditions of neurological disease or injury. Gangliosides have also been proposed to be an important messenger of the adaptive response to stress, such as apoptosis (d’Azzo et al. 2006). Early studies have reported that ramified, as well as amoeboid microglia in adult rat retina and cerebellum, express the ganglioside GD3 (Wolswijk 1995). Increased GD3 was found in plaques of multiple sclerosis (Yu et al. 1974), as well as brain tissue from patients with neurodegenerative disorders (Ando et al. 1984), such as Creutzfeldt-Jakob disease and subacute sclerosing panencephalitis. Findings in these studies suggest a role for ganglioside GD3 in brain response to injury; however, the cellular localization and specific function of the ganglioside have been controversial (Goldman & Reynolds 1996). In the plaques of multiple sclerosis and glial scars, a few studies observed R24 (the anti-GD3 antibody) binding to astrocytes (Kawai et al. 1994; Goldman & Reynolds 1996). However, in our murine GCI model, no apparent co-localization of R24 was detected to be co-localized with the astrocyte marker GLAST-1 from R1d to R7d.
GD3 is a major sialic-acid containing glycosphingolipids of the developing CNS, and is highly expressed in immature neuroectodermal cells; however, this expression dramatically diminishes when the newborn cells become mature (Goldman et al. 1984). Previous work from our group demonstrated an important role of ganglioside GD3 in maintaining adult neurogenesis in the mouse brain (Wang & Yu 2013; Wang et al. 2014). In the previous study, we detected a significant decrease of granular cell layer neurons in the DG of GD3S-KO mice at 6-month old but not in younger age (Wang et al. 2014). In the 3-month old mice, we measured the CA1 and CA3 cellularity at basal level (without GCI), and detected no significant changes in the CA1 and CA3 cellularity between WT and GD3S-KO mice (Fig. 4, sham groups). Therefore, the neuronal survival analysis between WT and GD3S-KO mice after GCI should not be confounded by the regulatory effects of GD3 on neurogenesis. It is worth noting that the embryonic origin of microglia is not from the neuroectoderm, where other neuroglia are commonly derived from. Fate-mapping analysis has revealed that adult murine microglia are derived from primitive macrophages, which originate in the yolk sac and migrate to the brain at embryonic day 9.5 (Ginhoux et al. 2010). Cellular localization of GD3 in the microglia/macrophage after brain injury (Reynolds & Wilkin 1993; Simon et al. 2002) indicated that the expression of GD3 is not restricted to the neuroectodermal lineage. In other studies, ganglioside GD3 has been proposed to induce cell apoptosis through mitochondrial damage, although many of these studies are performed in vitro (Malisan & Testi 2002; Rippo et al. 2000). However, GD3 has also been suggested to regulate mitochondria dynamics during adult neurogenesis by binding to a mitochondria fusion protein (Tang et al. 2020). Therefore, the role of ganglioside GD3 in regulating cellular functions depends on specific physiological and pathological conditions. In the post-ischemic mouse brain, we cannot rule out the possibility that GD3 or other gangliosides may directly act on neurons and regulate the neurological outcome in response to brain ischemia. It should be noted that the zymosan used in the in-vitro phagocytosis analysis is not a disease related substance. Therefore, the regulatory effects of GD3 in phagocytic cells are not specific to live-neurons. To more conclusively confirm the role of ganglioside GD3 in microglia phagocytosis of live neurons, further studies are needed. Future studies could include the use using of an inducible microglia specific GD3S-KO mice, and labeled neurons, as well as development and utilization of an inducible microglia-specific GD3S-KO mouse model.
In conclusion, our study detected predominant up-regulation of GD3-synthase and ganglioside GD3 in microglia following GCI. In a murine GCI model, we found that GD3S deletion resulted in a decreased hippocampal neuronal loss after ischemia-reperfusion. We also found that the phagocytic capacity of the GD3S-KO microglia was compromised in vivo and in vitro. These novel findings suggest that ganglioside GD3 regulates the functional phagocytosis of microglia in the ischemic brain, which may link to the delayed neuronal cell death in the hippocampus following ischemic brain injury.
Supplementary Material
Acknowledgements:
This research was supported by Career Developmental Award 19CDA34490003 (to JW) from American Heart Association and National Institute of Neurological Disorders and Stroke, Grant/Award Number: RO1 NS100839.
Abbreviations:
- GCI
global cerebral ischemia
- GFAP
glial fibrillary acidic protein
- CNS
central nervous system
- IHC
immunohistochemistry
- IF
immunofluorescence
- ip
intraperitoneal injection
- WT
wild type
- GD3S-KO
GD3-synthase knockout
- RRID
Research Resource Identifier
- CCAs
common carotid arteries
- MACS
magnetic activated cell sorting
Footnotes
Conflict of Interest:
The authors declare that there is no conflict of interest.
References
- Abe K, Aoki M, Kawagoe J, Yoshida T, Hattori A, Kogure K and Itoyama Y (1995) Ischemic delayed neuronal death. A mitochondrial hypothesis. Stroke 26, 1478–1489. [DOI] [PubMed] [Google Scholar]
- Al Mamun A, Chauhan A, Qi S et al. (2020) Microglial IRF5-IRF4 regulatory axis regulates neuroinflammation after cerebral ischemia and impacts stroke outcomes. Proceedings of the National Academy of Sciences of the United States of America 117, 1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando S, Toyoda Y, Nagai Y and Ikuta F (1984) Alterations in brain gangliosides and other lipids of patients with Creutzfeldt-Jakob disease and subacute sclerosing panencephalitis (SSPE). Jpn J Exp Med 54, 229–234. [PubMed] [Google Scholar]
- Baron JC, Yamauchi H, Fujioka M and Endres M (2014) Selective neuronal loss in ischemic stroke and cerebrovascular disease. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 34, 2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T, De Schepper S and Hong S (2020) Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 370, 66–69. [DOI] [PubMed] [Google Scholar]
- Brown GC and Neher JJ (2012) Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’. Trends in biochemical sciences 37, 325–332. [DOI] [PubMed] [Google Scholar]
- Brown GC and Neher JJ (2014) Microglial phagocytosis of live neurons. Nature reviews. Neuroscience 15, 209–216. [DOI] [PubMed] [Google Scholar]
- Butler CA, Popescu A, Kitchener E, Allendorf DH, Puigdellivol M and Brown GC (2021) Microglial phagocytosis of neurons in neurodegeneration, and its regulation. Journal of neurochemistry. [DOI] [PubMed] [Google Scholar]
- Caughlin S, Hepburn JD, Park DH, Jurcic K, Yeung KK, Cechetto DF and Whitehead SN (2015) Increased Expression of Simple Ganglioside Species GM2 and GM3 Detected by MALDI Imaging Mass Spectrometry in a Combined Rat Model of Abeta Toxicity and Stroke. PloS one 10, e0130364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiricozzi E, Mauri L, Lunghi G et al. (2019) Parkinson’s disease recovery by GM1 oligosaccharide treatment in the B4galnt1(+/−) mouse model. Sci Rep 9, 19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Azzo A, Tessitore A and Sano R (2006) Gangliosides as apoptotic signals in ER stress response. Cell Death Differ 13, 404–414. [DOI] [PubMed] [Google Scholar]
- Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA and Allan SM (2007) Proliferating resident microglia after focal cerebral ischaemia in mice. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 27, 1941–1953. [DOI] [PubMed] [Google Scholar]
- Desplats PA, Denny CA, Kass KE, Gilmartin T, Head SR, Sutcliffe JG, Seyfried TN and Thomas EA (2007) Glycolipid and ganglioside metabolism imbalances in Huntington’s disease. Neurobiology of disease 27, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R, Shen Q, Xu P, Luo JJ and Tang Y (2014) Phagocytosis of microglia in the central nervous system diseases. Molecular neurobiology 49, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M et al. (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JE, Hirano M, Yu RK and Seyfried TN (1984) GD3 ganglioside is a glycolipid characteristic of immature neuroectodermal cells. J Neuroimmunol 7, 179–192. [DOI] [PubMed] [Google Scholar]
- Goldman JE and Reynolds R (1996) A reappraisal of ganglioside GD3 expression in the CNS. Glia 16, 291–295. [DOI] [PubMed] [Google Scholar]
- Horn M and Schlote W (1992) Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta neuropathologica 85, 79–87. [DOI] [PubMed] [Google Scholar]
- Jayaraj RL, Azimullah S, Beiram R, Jalal FY and Rosenberg GA (2019) Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation 16, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai K, Kuroda S, Watarai S, Takahashi H and Ikuta F (1994) Occurrence of GD3 ganglioside in reactive astrocytes--an immunocytochemical study in the rat brain. Neurosci Lett 174, 225–227. [DOI] [PubMed] [Google Scholar]
- Kracun I, Kalanj S, Talan-Hranilovic J and Cosovic C (1992) Cortical distribution of gangliosides in Alzheimer’s disease. Neurochemistry international 20, 433–438. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19, 312–318. [DOI] [PubMed] [Google Scholar]
- Lopez PH and Schnaar RL (2009) Gangliosides in cell recognition and membrane protein regulation. Current opinion in structural biology 19, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Sareddy GR, Wang J, Zhang Q, Tang FL, Pratap UP, Tekmal RR, Vadlamudi RK and Brann DW (2020) Neuron-Derived Estrogen Is Critical for Astrocyte Activation and Neuroprotection of the Ischemic Brain. The Journal of neuroscience : the official journal of the Society for Neuroscience 40, 7355–7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malisan F and Testi R (2002) GD3 ganglioside and apoptosis. Biochimica et biophysica acta 1585, 179–187. [DOI] [PubMed] [Google Scholar]
- Mari C, Karabiyikoglu M, Goris ML, Tait JF, Yenari MA and Blankenberg FG (2004) Detection of focal hypoxic-ischemic injury and neuronal stress in a rodent model of unilateral MCA occlusion/reperfusion using radiolabeled annexin V. European journal of nuclear medicine and molecular imaging 31, 733–739. [DOI] [PubMed] [Google Scholar]
- Neher JJ, Emmrich JV, Fricker M, Mander PK, Thery C and Brown GC (2013) Phagocytosis executes delayed neuronal death after focal brain ischemia. Proceedings of the National Academy of Sciences of the United States of America 110, E4098–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Kotter MR and Franklin RJ (2009) Debris clearance by microglia: an essential link between degeneration and regeneration. Brain : a journal of neurology 132, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngamukote S, Yanagisawa M, Ariga T, Ando S and Yu RK (2007) Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. Journal of neurochemistry 103, 2327–2341. [DOI] [PubMed] [Google Scholar]
- Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E and Uchiyama Y (1995) Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. The Journal of neuroscience : the official journal of the Society for Neuroscience 15, 1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS (2003) “Recruitment signals” from apoptotic cells: invitation to a quiet meal. Cell 113, 817–820. [DOI] [PubMed] [Google Scholar]
- Reynolds R and Wilkin GP (1993) Cellular reaction to an acute demyelinating/remyelinating lesion of the rat brain stem: localisation of GD3 ganglioside immunoreactivity. J Neurosci Res 36, 405–422. [DOI] [PubMed] [Google Scholar]
- Rippo MR, Malisan F, Ravagnan L et al. (2000) GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 14, 2047–2054. [DOI] [PubMed] [Google Scholar]
- Savage JC, St-Pierre MK, Carrier M, El Hajj H, Novak SW, Sanchez MG, Cicchetti F and Tremblay ME (2020) Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J Neuroinflammation 17, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB and Kiefer R (2005) Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 196, 290–297. [DOI] [PubMed] [Google Scholar]
- Simon BM, Malisan F, Testi R, Nicotera P and Leist M (2002) Disialoganglioside GD3 is released by microglia and induces oligodendrocyte apoptosis. Cell Death Differ 9, 758–767. [DOI] [PubMed] [Google Scholar]
- Sims NR (1992) Energy metabolism and selective neuronal vulnerability following global cerebral ischemia. Neurochemical research 17, 923–931. [DOI] [PubMed] [Google Scholar]
- Tang FL, Wang J, Itokazu Y and Yu RK (2020) Ganglioside GD3 regulates dendritic growth in newborn neurons in adult mouse hippocampus via modulation of mitochondrial dynamics. Journal of neurochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassoni-Ardori F, Hong Z, Fulgenzi G and Tessarollo L (2020) Generation of Functional Mouse Hippocampal Neurons. Bio Protoc 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cheng A, Wakade C and Yu RK (2014) Ganglioside GD3 is required for neurogenesis and long-term maintenance of neural stem cells in the postnatal mouse brain. The Journal of neuroscience : the official journal of the Society for Neuroscience 34, 13790–13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ma MW, Dhandapani KM and Brann DW (2017) Regulatory role of NADPH oxidase 2 in the polarization dynamics and neurotoxicity of microglia/macrophages after traumatic brain injury. Free radical biology & medicine 113, 119–131. [DOI] [PubMed] [Google Scholar]
- Wang J, Sareddy GR, Lu Y et al. (2020a) Astrocyte-Derived Estrogen Regulates Reactive Astrogliosis and is Neuroprotective Following Ischemic Brain Injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Sareddy GR, Lu Y et al. (2020b) Astrocyte-Derived Estrogen Regulates Reactive Astrogliosis and is Neuroprotective following Ischemic Brain Injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 40, 9751–9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J and Yu RK (2013) Interaction of ganglioside GD3 with an EGF receptor sustains the self-renewal ability of mouse neural stem cells in vitro. Proceedings of the National Academy of Sciences of the United States of America 110, 19137–19142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JR, Koerner IP and Moller T (2010) Microglia in ischemic brain injury. Future neurology 5, 227–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolswijk G (1995) Strongly GD3+ cells in the developing and adult rat cerebellum belong to the microglial lineage rather than to the oligodendrocyte lineage. Glia 13, 13–26. [DOI] [PubMed] [Google Scholar]
- Woods AS, Colsch B, Jackson SN et al. (2013) Gangliosides and ceramides change in a mouse model of blast induced traumatic brain injury. ACS chemical neuroscience 4, 594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura I, Kawahara N, Nakatomi H, Furuya K and Kirino T (2004) A model of global cerebral ischemia in C57 BL/6 mice. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 24, 151–158. [DOI] [PubMed] [Google Scholar]
- Yu RK, Bieberich E, Xia T and Zeng G (2004) Regulation of ganglioside biosynthesis in the nervous system. Journal of lipid research 45, 783–793. [DOI] [PubMed] [Google Scholar]
- Yu RK, Ledeen RW and Eng LF (1974) Ganglioside abnormalities in multiple sclerosis. Journal of neurochemistry 23, 169–174. [DOI] [PubMed] [Google Scholar]
- Yu RK, Tsai YT, Ariga T and Yanagisawa M (2011) Structures, biosynthesis, and functions of gangliosides--an overview. Journal of oleo science 60, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S (2019) Microglial activation after ischaemic stroke. Stroke and vascular neurology 4, 71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.