

Summary
Allergic airway inflammation is driven by type-2 CD4+ T cell inflammatory responses. We uncover an immunoregulatory role for the nucleotide release channel, Panx1, in T cell crosstalk during airway disease. Inverse correlations between Panx1 and asthmatics, and our mouse models revealed the necessity, specificity, and sufficiency of Panx1 in T cells to restrict inflammation. Global Panx1−/− mice experienced exacerbated airway inflammation, and T cell-specific deletion phenocopied Panx1−/− mice. A transgenic designed to re-express Panx1 in T cells reversed disease severity in global Panx1−/− mice. Panx1 activation occurred in pro-inflammatory Teff and inhibitory Treg cells and mediated the extracellular nucleotide based Treg-Teff crosstalk required for suppression of Teff cell proliferation. Mechanistic studies identified a Salt inducible kinase-dependent phosphorylation of Panx1 serine 205 important for channel activation. A genetically targeted mouse expressing non-phosphorylatable Panx1S205A phenocopied the exacerbated inflammation in Panx1−/− mice. These data identify Panx1-dependent Treg:Teff cell communication in restricting airway disease.
Graphical abstract
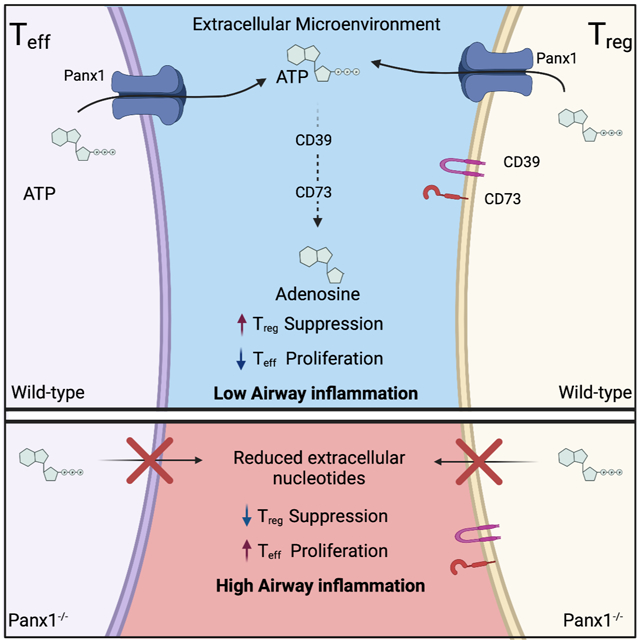
Blurb:
While extracellular nucleotides (ATP) are known to be important, the source and regulation of ATP at immune microenvironments is still unclear. Medina et. al. show that CD4+ Teff and CD4+ Treg cells use a SIK-Panx1 axis to control nucleotide release for inter-T cell communication and suppression of allergic airway inflammation.
Introduction
Allergic airway inflammation, also known as allergic asthma, affects approximately 1 in 20 individuals or about 300 million people worldwide (Lambrecht and Hammad, 2015). Many immune cells drive allergic airway inflammation including T cells, B cells, eosinophils, innate lymphoid cells, and myeloid cells, with T helper 2 (Th2) cell-dependent inflammatory response being central (Cheng and Locksley, 2014; Kaur et al., 2015; Locksley, 2010). Th2 CD4+ T cells that infiltrate the lungs produce interleukin-4 (IL-4) and IL-5 to mediate eosinophilia, IgE accumulation, mast cell degranulation, and bronchial hyperreactivity (Walker et al., 1991). While treatments such as inhaled corticosteroids can alleviate the inflammatory symptoms of disease, many patients are still refractory to such therapeutics (Umetsu and Dekruyff, 2006). Therefore, further understanding of the molecular details that influence pathogenesis is needed for developing better treatments.
Among the many cellular and acellular factors contributing to disease progression and severity, extracellular ATP (eATP) is one dynamic signaling molecule found in the bronchoalveolar lavage of asthmatic patients (Lázár et al., 2010). Both inflammatory and immunosuppressive roles for extracellular ATP during allergic airway inflammation have been described (Idzko et al., 2013). The relative instability of eATP and the conversion of ATP to adenosine (a potent immunosuppressive molecule), via CD39 and CD73, suggests that these factors are tightly regulated and likely act in a local zone. Therefore, specific microdomains of extracellular nucleotides may play pivotal roles in dictating their function under certain pathological and physiological settings. Although the actions of extracellular ATP and their metabolic breakdown products have been described, the source and specific cellular contributions for these extracellular nucleotides in different disease settings including allergic airway inflammation are unknown.
Uncontrolled tissue damage has been proposed as a main mechanism of ATP release, however, whether coordinated extracellular ATP release occurs in immune microenvironments that lack excessive cell death and what consequence this has during inflammatory settings remains unclear. One mechanism by which cells control extracellular nucleotide amounts is through heptameric plasma membrane Pannexin 1 (Panx1) channels, which are capable of ATP release (and AMP) (Chekeni et al., 2010; Michalski et al., 2020; Yamaguchi et al., 2014). Panx1-mediated nucleotide release can occur during several cellular processes including: programmed cell death, G-protein coupled receptor (GPCR)-dependent channel activation (e.g. phenylephrine stimulation of α1D receptors), and increases in intracellular calcium (Billaud et al., 2015; Chekeni et al., 2010; Medina et al., 2020). Panx1 channel opening during apoptosis or live cells are regulated in different ways. Further, while large amounts of ATP release from completely lytic cells can be pro-inflammatory, regulated ATP release via Panx1 is less than 0.1% of the cellular ATP content, and can be anti-inflammatory (Dubyak, 2019; Medina et al., 2020). Therefore, Panx1 channels may play important roles in managing extracellular ATP concentrations during allergic airway inflammation.
Here, we describe a role for Panx1 in the nucleotide-based crosstalk between Treg and Teff cells that in turn, restricts the severity of allergic airway inflammation. In the absence of Panx1, Treg cell-mediated suppression is hampered by a lack of extracellular nucleotides at the Treg -Teff cell interface, which results in unchecked Teff cell proliferation. Both T cell populations can activate Panx1 channels after stimulation, thereby allowing the release of ATP for breakdown to immunosuppressive adenosine via CD39 and CD73. Furthermore, we identify an important Panx1::Salt-inducible kinase interaction involved in Panx1 channel activation and control of airway inflammation.
Results
Panx1−/− mice exhibit exacerbated allergen-induced airway inflammation
When we examined human datasets for PANX1 expression in asthmatic patients (Raedler et al., 2015), we noted that PANX1 expression was significantly reduced in the peripheral blood mononuclear cells (PBMCs) of allergic asthmatic children, relative to healthy controls (Figure 1A). This inverse correlation was capable (beyond chance) of identifying asthmatic patients based on Panx1 expression (ROC curve – please see Methods). To directly investigate the importance of Panx1 in airway disease, we used wild-type and Panx1-deficient mice in a model of allergic airway inflammation induced by house-dust mite (HDM), a relevant pathologic allergen in developed countries (Arias-Calderón et al., 2016; Gandhi et al., 2013; Gold et al., 2015). After a priming and a challenge phase with HDM (Figure 1B), Panx1+/+ mice developed lung pathology (Figures 1C and S1A) and a typical Th2 cell-mediated inflammatory response characterized by eosinophil and CD4+ T cell recruitment into the lungs (Figures 1D and 1E). Mice with global Panx1 deletion (Panx1−/−) showed exacerbated disease severity as evidenced by worsened lung pathology (Figures 1C and S1A), increased immune cell infiltration into the lungs (Figures 1D and 1E), an increased proportion of activated (CD69+) T cells (Figure 1E) and greater numbers of IL-4 producing T cells (Figure 1F). The lower PANX1 expression in human asthmatics and exacerbated airway inflammation in Panx1−/− mice suggest that Panx1 limits the severity of airway disease.
Figure 1. Panx1 expression restricts the severity of allergic airway inflammation.

(A) Relative Panx1 expression (log2FC relative to healthy) on peripheral blood mononuclear cells (PBMC) of healthy or allergic asthmatic children (ctrl n=13, a.asthmatics n=14) (*p=0.015), revealing a decrease in Panx1 in asthmatics.
(B) Schematic representation of house dust mite (HDM)-induced allergic airway inflammation.
(C) H&E lung histology images of wild-type (n=13) and global Panx1−/−(n=8) mice during PBS or HDM challenge. Arrows highlight areas of immune cell infiltration and inflammation. Disease severity score as assessed by a pathologist blinded to the genotypes (*p=0.05).
(D) Flow plots showing the extent of eosinophil and CD4+ T cell infiltration into the airways of Panx1+/+ and Panx1−/− mice after HDM-induced airway inflammation.
(E) Absolute cellularity of eosinophils (left) (*p=0.017), total CD4+ T cells (middle) (**p=0.008), and activated CD69+ CD4+ T cells (right) (*p=0.033). Each dot represents a mouse. (PBS control– Panx1+/+ n=4, PBS control– Panx1−/− n=5, HDM– Panx1+/+ n=19, HDM– Panx1−/− n=21) and lung (PBS– Panx1+/+ n=3, PBS– Panx1−/− n=3, HDM– Panx1+/+ n=22, HDM– Panx1−/− n=20), respectively, in wild-type (Panx1+/+) and global Panx1−/−.
(F) Flow plots showing the percentage of IL4+ CD4+ T cells (left) and quantification (right) during
HDM-induced allergic airway inflammation. (Panx1+/+ n=4, Panx1−/−n=6) (*p=0.011). Unpaired Student’s t-test (A,C,E,F). Related to Figure S1.
While CD4+ T cells control the adaptive immune response during allergen challenge, innate lymphoid type 2 cells (ILC2) are a major component of the innate response (Holtzman et al., 2014; Morita et al., 2016; Ricardo-Gonzalez et al., 2020; Scanlon and McKenzie, 2012; Van Dyken et al., 2016). Both T cells and ILCs express Panx1, albeit expression is ~60% higher in CD4+ T cells (Figure S1B). To determine whether Panx1 contributed to the innate immune response and ILC2s during allergic airway inflammation, we analyzed airway inflammation on day 6, after the initial priming phase and before the complete onset of antigen-specific memory T cell responses in the lung (schematic, Figure S1C). Wild-type and Panx1−/− mice exhibited similar immune cell infiltration at day 6 (Figure S1C). Additionally, after two weeks of HDM treatment, ILC2 numbers and the percentage of IL-4 producing ILC2s were unchanged between Panx1+/+ and Panx1−/− mice (Figure S1D). Since loss of Panx1 affects T-cell numbers, but not ILC2 numbers during airway inflammation, the findings suggest a more salient role for Panx1 in the adaptive component of disease progression.
Panx1 expression in T cells is necessary for limiting allergic airway inflammation
Analysis of Panx1 expression in the Immgen database revealed CD4+ T cells as high expressors of Panx1 (Figure 2A) (Heng et al., 2008), with splenic naïve CD4 T cells expressing ~100 fold more Panx1 than mouse bone-marrow derived macrophages (Figure S2A). Furthermore, while T cells only express Panx1, macrophages had detectable expression of Panx2 and Panx3 (Figure S2A), that may functionally compensate the loss of Panx1 in some settings (Lohman and Isakson, 2014). In the whole lung, expression of Panx1 and Panx2 were detected (Figure S2B). With the highest expression of Panx1 in T cells, and the central role of CD4+ T cells in airway inflammation, we tested the relevance of Panx1 in T cells during allergic airway disease.
Figure 2. T cell Panx1 expression limits allergic airway inflammation.

(A) Gene expression data from the Immgen database assessing the relative expression of Panx1.
(B) Schematic representation of Cd4-cre mediated Panx1 deletion and efficiency of deletion via immunoblot.
(C) H&E lung histology images of Panx1fl/fCd4-cre− (n=12) and Panx1fl/fCd4-cre+ (n=14) mice during HDM challenge. Arrows highlight areas of immune cell infiltration and inflammation. Disease severity score as assessed by a pathologist blinded to genotypes is shown on the right (*p=0.029).
(D) Flow plots showing the extent of eosinophil and CD4+ T cell infiltration into the airways of Panx1fl/fCd4-cre− and Panx1fl/fCd4-cre+ mice after HDM-induced airway inflammation.
(E, F) Absolute cellularity of eosinophils (left) (**p=0.009), CD4+ T cells (middle) (*p=0.012), activated CD69+ CD4+ T cells (left) (*p=0.032), and BALF IL-4 cytokine concentration (F) (*p=0.034). (PBS–Panx1fl/fCd4-cre− n=2, PBS–Panx1fl/fCd4-cre+ n=3, HDM–Panx1fl/fCd4-cre− n=13, HDM–Panx1fl/fCd4-cre+ n=11) and lung (PBS–Panx1fl/fCd4-cre− n=3, PBS–Panx1fl/fCd4-cre+ n=4, HDM–Panx1fl/fCd4-cre− n=15, HDM–Panx1fl/fCd4-cre+ n=13), respectively.
(G)Foxp3-cre mediated Panx1 deletion and the efficiency of deletion.
(H) Absolute cellularity of eosinophils (left) and CD4+ T cells (right) (Panx1fl/fFoxp3-cre− n=17, Panx1fl/fFoxp3-cre+ n=19). Unpaired Student’s t-test (C,E,F). Related to Figure S2 and S3.
We crossed Panx1fl/fl mice to Cd4-cre mice (Panx1fl/fl Cd4-cre+) to delete Panx1 in T cells (Figure 2B) and subjected these mice to allergic airway inflammation. The severity of HDM-induced airway inflammation in Panx1fl/fl Cd4-cre+ mice phenocopied the global Panx1−/− mice, with worse lung pathology via H&E staining (Figure 2C and Figure S2C) and increased eosinophil and CD4+ T cell infiltration into the lungs relative to cre− wild-type controls (Figures 2D and 2E). Bronchoalveolar lavage fluid of Panx1fl/fl Cd4-cre+ mice displayed greater numbers of activated T cells (Figure 2E right), higher concentrations of IL-4 (Figure 2F), and a trending increase in IL-5 (Figure S2D). Increased inflammation was not due to Cre toxicity, and required elimination of both Panx1 alleles, as heterozygous Panx1fl/wt Cd4-cre+ mice showed a similar phenotype to wild-type Panx1fl/wt mice without cre (Figure S2E). We also tested the loss of Panx1 expression in the macrophage lineage by crossing Panx1fl/fl mice to Cx3cr1-cre mice. Panx1fl/f Cx3cr1-cre+ mice displayed comparable airway inflammation to cre− littermate controls (Figures S2F and S2G). These data suggest that T cell specific expression of Panx1 is required to limit disease severity during allergen-mediated immune responses.
Panx1 facilitates communication between Treg and Teff cells to achieve optimal suppression
Panx1 deletion did not affect homeostatic T cell numbers in the lung (Figures 1E and 2E, PBS control), nor did it impact thymic T cell development or basal immune cell numbers in the spleen (Figures S2H-K). We therefore asked whether the loss of Panx1 led to greater activation of CD4+ Teff cells. When naïve CD4+ Teff cells (CD4+CD25−) were treated with anti-CD3 and anti-CD28, there was no difference in activation between wild-type and Panx1−/− Teff cells, as measured by upregulation of CD69, CD44, CD25 or downregulation of CD62L (Figures S3A-D). Additionally, there was no detectable differences across genotypes when we examined downstream T cell receptor (TCR) signaling via phosphorylation of ERK (Figure S3E) or the proliferative capacity of wild-type and Panx1−/− CD4+ Teff cells (Figure S3F), suggesting that the loss of Panx1 on CD4+ Teff cells per se did not lead to enhanced Teff cell-intrinsic activation.
During allergic airway disease, the pro-inflammatory responses of effector CD4+ T cells are kept in check by regulatory CD4+ T cells (Treg), and this balance of action determines the severity of airway inflammation (Josefowicz et al., 2012). Cd4-cre mediated deletion will delete floxed genes in both Treg and Teff cells. This led us to ask whether Treg cells require Panx1 to limit CD4+ Teff cell responses during allergic airway inflammation. Although Treg cells make up ~20% of the CD4+ T cell population in the lung during airway disease, Treg cell numbers within the Panx1fl/fl Cd4-cre+ lung was unchanged (Figure S3G). Yet, loss of Panx1 in Treg cells could disrupt their suppressive function. To directly address this possibility, we generated animals with Treg specific Panx1 deletion using Foxp3-cre mice crossed to Panx1fl/fl mice (Figure 2G). However, Panx1fl/fl Foxp3-cre+ mice showed similar immune cell infiltration as control mice (Figures 2H and S3H). When we tested cultures of Treg cells mixed with Teff cells in suppression assays in vitro, both wild-type and Panx1−/− Treg cells were able to comparably suppress the proliferation of wild-type CD4+ Teff cells (Figures 3A and S4A black and yellow). This led to the conclusion that Panx1 deletion on both Teff and Treg cells (via Cd4-cre) leads to increased airway inflammation, yet the loss of Panx1 does not hamper the intrinsic activation of Teff cells or the intrinsic suppressive function of Treg cells.
Figure 3. Panx1 regulates crosstalk between Teff and Treg cells during Treg mediated suppression.

(A) Quantitative analysis of suppression across the four different cellular combinations (n=3) (*p=0.047) (top). Individual experimental line plots demonstrating the extent of suppression across Teff:Treg cell ratios (bottom).
(B) Histograms of combinatorial suppression assays with wild type and Panx1−/− Teff and Treg cells at indicated cell ratios.
(C) Concentrations of extracellular ATP, AMP, and adenosine over time during suppression assays with wild-type or Panx1−/− T cells.
(D) Schematic of conversion of ATP to adenosine by CD39 and CD73 mediated enzymatic reactions and adenosine actions on the A2AR. Conversion of adenosine to inosine with ADA treatment. ATP supplementation, adenosine supplementation, CD73 inhibition, ADA treatment, and A2AR agonist during wild-type or Panx1−/− T cell suppression assays (1:1 Teff:Treg ratio) (n=3-10) (****p<0.0001, *p<0.05).
(E) Flow plots and quantification of in vivo T cell proliferation using Edu incorporation during allergic airway inflammation in Panx1fl/fCd4-cre+ mice (n=5) and wild-type littermate controls (n=6) (*p=0.028). Data are mean ± s.e.m. Two-way ANOVA (A), One-way ANOVA (D), Unpaired Student’s t-test (E). Related to Figure S4.
Extracellular ATP released through Panx1 channels can be converted to the anti-inflammatory molecule adenosine via two ectonucleotidases, CD39 (which converts ATP and ADP to AMP) and CD73 (which converts AMP to adenosine) (Antonioli et al., 2013), both of which are highly expressed on Treg cells. Further, during antigen presentation by dendritic cells (DC) to Teff cells, Treg cells can also ‘dock’ to the same DC to simultaneously inhibit excessive Teff activation and control inflammation (Liu et al., 2015). Thus, we considered the possibility that Panx1, via local ATP release and subsequent adenosine production, could facilitate the microenvironmental communication between Teff and Treg cells to limit inflammation. To address this, we performed suppression assays in vitro with wild-type or Panx1−/− Treg cells mixed with either Panx1+/+ or Panx1−/− Teff cells. When Panx1 was deleted only on either Teff or Treg cells, suppression was minimally affected compared to controls where both Teff and Treg cells expressed Panx1 (Figures 3A and 3B and S4A). However, when Panx1 was deleted on both Teff and Treg cells (Panx1−/− Teff mixed with Panx1−/− Treg), the suppressive capacity of Treg cells was significantly diminished, evidenced by increased Teff cell proliferation (Figures 3A and 3B). This pointed to a non-cell autonomous role of Panx1 at the Teff-Treg communication interface; it also suggested the possibility that Teff cells are not ‘passive’ and can contribute to their own suppression in certain contexts by releasing ATP through Panx1.
Mechanistically, a lack of extracellular ATP in Panx1−/−Teff::Panx1−/−Treg conditions could lead to the reduced production of adenosine, and in turn, the reduced suppressive capacity of Treg cells (Dwyer et al., 2007). When we tested this possibility, we detected decreased extracellular ATP, AMP and adenosine in Panx1−/− cultures relative to wild-type settings, consistent with the role of Panx1 in ATP release (Figure 3C). To determine if the loss extracellular nucleotide release was responsible for the Panx1−/− phenotype, we supplemented ATP or adenosine to the Panx1−/− Teff ::Panx1−/− Treg assay condition and found that both ATP and adenosine could rescue Treg-mediated suppression (Figure 3D). Addition of ATP or adenosine to wild-type Teff ::Treg conditions did not further increase suppression in this setting. In contrast, inhibiting CD73 to limit the ability of Treg cells to generate adenosine from extracellular nucleotides or treating cultures with adenosine deaminase (ADA) to degrade extracellular adenosine, blunted Treg-mediated suppression in wild-type conditions, but not the Panx1−/− Teff::Panx1−/− Treg setting (Figure 3D). We did not observe any differences in CD39 or CD73 expression on Teff or Treg cells between wild-type and Panx1 deficient mice, either basally or when enhanced by HDM challenge (Figures S4B and S4C). Lastly, extracellular adenosine acts through adenosine receptors to mediate its actions. Both Teff and Treg cells express Adenosine Receptor 2a (A2AR), which can drive the immunosuppressive functions of adenosine on T cells (Figure S4D) (Deaglio et al., 2007). Consistent with this notion, treatment with CGS-21680, an A2AR agonist, was able to rescue suppression in Panx1−/− cultures (Figure 3D). These data suggested that Panx1-dependent eATP and extracellular adenosine are needed for the optimal suppressive function of Treg cells in this immune microenvironment. Importantly, both Teff and Treg cells can contribute to the extracellular accumulation of ATP at this interface, as loss of Panx1 on either cell type alone did not affect Treg-mediated suppression.
We next asked whether this in vitro phenotype translated to greater in vivo proliferation of Teff cells during airway inflammation. For this, we used the Panx1fl/fl Cd4-cre+ mice in which Panx1 is deleted in both Teff and Treg populations. We used the thymidine analog EdU to track CD4+ T cell proliferation after HDM challenge (Figure S4E). T cells harvested from the lungs of Panx1fl/fl Cd4-cre+ mice showed increased CD4+ T cell proliferation (and total CD3+ T cell proliferation), compared to wild-type littermate controls (Figures 3E and S4F). Overall, these data suggest that when Panx1 is absent from both Teff and Treg cells, a lack of extracellular nucleotides leads to inefficient control of Teff proliferation during allergen challenge.
Transgenic Panx1 re-expression in CD4 T cells is sufficient to attenuate disease in Panx1 deficient mice
ATP release during allergic airway inflammation in vivo can potentially come from other cell types (i.e. non-T cells) and such extracellular ATP could also be converted by Treg cells to adenosine to mediate the suppression of Teff cells. To directly address the sufficiency of Panx1 expression on T cells in limiting airway inflammation, we generated a Panx1 transgenic mouse line (Panx1Tg), where we engineered a ‘conditional’ human PANX1 transgene into the endogenous non-essential murine Rosa26 locus as a single copy (Soriano, 1999; Xiao et al., 2007). In this construct design, a floxed transcriptional STOP cassette upstream of PANX1 allows Cre-dependent cell type expression of the transgene; further, this bi-cistronic construct carries a downstream IRES-GFP to track transgene expression (Figures 4A and S5A). For initial mouse validation, the Panx1Tg mice were crossed to E2a-cre mice for global expression, where PANX1 protein and GFP expression were confirmed (Figures S5B and S5C). We then crossed Panx1Tg mice to Cd4-cre mice to overexpress PANX1 in T cells (Figure 4B). Functional testing of the transgene by measuring TO-PRO-3 dye uptake (a measurement of Panx1 activity) (Billaud et al., 2015; Chekeni et al., 2010) confirmed that the transgenic PANX1 can be activated in T cells (Figures S5D and S5E).
Figure 4. T cell specific rescue of Panx1 expression dampens airway inflammation.

(A) Schematic representation for the generation of Panx1Tg mice and the Cd4-cre mediated Panx1 transgene re-expression in the global Panx1−/− mice.
(B) Immuno blot validation of Panx1 over-expression in CD4+ T cells by crossing the Panx1Tg mice to Cd4-cre mice and assessing Panx1 protein from isolated splenic CD4+ T cells. Actin expression was used as a loading control. The Panx1 expression is shown in two different exposures.
(C, D) Flow plots for eosinophil and CD4+ T cell infiltration into the bronchoalveolar space. The T cells infiltrating into the lungs were GFP positive only in the Panx1−/− Panx1TgCd4-cre+ condition (C). Absolute cellularity of eosinophils (*p=0.048) (left) and CD4+ T cells (**p=0.002) (right) (D). (Panx1−/−Panx1TgCd4-cre− n=5, Panx1−/−Panx1TgCd4-cre+ n=4) in PBS and HDM-treated mice.
(E, F) Quantitative analysis of T cell suppression assays between Panx1−/−Panx1TgCd4-cre− (n=3) and Panx1−/− Panx1TgCd4-cre+ (n=3) mice across different T cell seeding ratios (*p=0.039) (E). Histograms of suppression assays. Yellow highlights the area of difference (F). Unpaired Student’s t-test (D). Two-way ANOVA, Data are mean +/− s.e.m. (E). Related to Figure S5.
Next, we crossed the Panx1TgCd4-cre mice to global Panx1−/− mice so that we could restore Panx1 expression only in T cells, in an otherwise Panx1 deficient background (Figure 4A). We induced allergic airway inflammation in Panx1−/−Panx1TgCd4-cre+ mice to ask whether we could reverse the heightened disease parameters seen in the Panx1−/− mice. Panx1−/− Panx1TgCd4-cre+ mice showed fewer eosinophils and CD4+ T cells in the BALF compared to the Panx1−/−Panx1TgCd4-cre− littermate controls (Figures 4C and 4D). Further, in ex vivo studies, Panx1−/−Panx1TgCd4-cre+ T cells restored the suppressive function of Treg cells when compared to Panx1−/−Panx1Tg T cells without cre (Figures 4E and 4F). These transgenic rescue studies coupled with Panx1 deletion in T cells (Panx1fl/flCd4-cre+ mice) establish the necessity, specificity, and sufficiency of T cell expressed Panx1 for the communication between Treg and Teff cells to limit disease severity during HDM-induced allergic airway inflammation.
A Panx1::Salt inducible kinase (SIK) axis limits airway inflammation
Panx1 can be activated (leading to ATP release) via several mechanisms including irreversible caspase cleavage during apoptosis, or a reversible phosphorylation of the channel, which can occur downstream of GPCRs (Billaud et al., 2015). We considered both of these mechanisms in leading to Panx1 activation on T cells. To address the Panx1:apoptosis link, we focused on the accumulation of apoptotic or necrotic cells in the airways during allergic airway inflammation, as defective apoptotic cell clearance can predispose individuals to greater inflammation (Bosurgi et al., 2017; Juncadella et al., 2013; Morioka et al., 2018). However, Panx1−/− mice did not display greater uncleared apoptotic cells compared to wild-type controls, per cleaved caspase-3 staining in lung tissue sections and Annexin V and 7AAD staining of bronchoalveolar lavage fluid (BALF) (Figures S6A-D). This suggested that caspase-mediated activation of Panx1 may not play a prominent role during allergic airway inflammation.
We next asked whether Panx1 activation might occur on live T cells. In prior studies of Panx1 activation on live cells, channel activation was shown to occur after GPCR-mediated signaling, such as phenylephrine (PE) stimulation of the α1D adrenergic receptor (Billaud et al., 2015). To determine whether Panx1 activation occurs on primary CD4+ T cells, we used whole cell patch clamp current recordings as a sensitive way to measure channel activation at the cellular level. While CD3 and CD28 stimulation alone did not lead to appreciable Panx1 channel activation (e.g., see low basal currents; Figure 5A), when we added phenylephrine to CD3 and CD28-stimulated cells, Panx1 currents in both Teff and Treg cells were induced (confirmed by blocking with the Panx1 inhibitor carbenoxolone) (Figure 5A). Of note, ~ 50% of T cells responded to PE stimulation, suggesting that perhaps only a subset of cells is ‘poised’ for Panx1 activation at a given time in these conditions (Figure 5A). PE stimulation alone, in the absence of CD3 and CD28, also led to detectable Panx1 currents (data not shown). While we have used phenylephrine as one example of a ligand capable of inducing Panx1 activation on CD4+ T cells, the specific or diverse GPCR ligand(s) responsible for Panx1 activation on Teff and Treg cells during airway inflammation could be different.
Figure 5. Phosphorylation of Serine 205 on Panx1 by SIK mediates channel activation.

(A) Whole cell patch-clamp recordings of Panx1 currents over time and the representative current-voltage relationship (I/V curves) from purified CD4+ T cells after phenylephrine stimulation (blue) (left). The cells were activated with CD3 and CD28 prior to PE stimulation. The specificity of the Panx1 currents were verified via the Panx1 inhibitor CBX (red). CBX-sensitive current densities of Treg and Teff cells stimulated with CD3 and CD28 ± phenylephrine (right).
(B) Schematic representation of methodologies used to investigate the mechanism of live cell Panx1 activation. After initial yeast 2-hybrid screens, co-immunoprecipitations and kinase assays were performed to validate hits. Electrophysiology studies and Panx1 mutagenesis were used to investigate SIK1 mediated activation of Panx1 at serine 205.
(C) Yeast 2-hybrid (Y2H) screen showing colony growth on plates with Panx1 and SIK1 specific plasmids.
(D) Immuno blot analysis of co-immunoprecipitations of endogenous Panx1 and SIK proteins in Jurkat T cells. Actin was used as a whole cell lysate loading (WCL) control.
(E) Whole cell patch-clamp recordings of Panx1 currents over time in HEK293T cells expressing the α1D-adrenergic receptor and either Panx1-WT (left), Panx1-S205A (middle), or Panx1-S205D (right) during phenylephrine (PE) and carbenoxolone (CBX – Panx1 inhibitor) treatment. Current density quantification of Panx1 currents across the different Panx1 mutants and treatments. Unpaired Student’s t-test (E). Related to Figure S6.
To better understand how Panx1 might get activated on live cells at the molecular level, we took several complementary approaches: a yeast two-hybrid screen, co-immunoprecipitations, kinase assays, and mutagenesis studies (Figure 5B). Unbiased identification of Panx1 intracellular interaction partners was determined using a yeast two-hybrid screen with the C-terminus of Panx1 as ‘bait’. Out of the 6 primary hits, we focused on one target, salt-inducible kinase 1 (SIK1) (Figure 5C), as GPCR-mediated Panx1 activation can be phosphorylation-dependent. SIK1 is a serine-threonine kinase belonging to the AMP-activated protein kinase family (Wang et al., 1999). SIK family kinases have been implicated in several cellular and physiological processes such as cell cycle regulation and metabolism, with only recently having their roles in immune contexts explored (Nixon et al., 2016; Sun et al., 2020). Validation of the two-hybrid screen using reciprocal co-immunoprecipitation experiments in an exogenous overexpression system confirmed the interaction between Panx1 and SIK1 (Figure S6E and S6F). Importantly, co-immunoprecipitation studies also demonstrated an endogenous Panx1-SIK1 interaction occurring in T cells (Figure 5D).
Next, to test SIK1-dependent phosphorylation of Panx1, we performed in vitro kinase assays using purified recombinant proteins. Phos-tag mobility shift gels, which separate proteins based on their phosphorylation status, showed an increased presence of high molecular weight Panx1 bands after incubation with SIK1 (Figure S6G). Thus, SIK1 may phosphorylate Panx1. To determine specific residues on Panx1 regulated by SIK, we performed computational analyses of the mouse Panx1 amino acid sequence and identified several potential phosphorylation sites, including a serine at position 205 (S205) that is conserved across species (residue S206 in human). Whole-cell patch clamp analyses of HEK293T cells (with α1D and Panx1 co-expression), provides us with an efficient way to manipulate and assess channel activation at the molecular level. Wild-type Panx1 (Panx1-WT) was activated by PE stimulation, but mutation of serine 205 to alanine (Panx1-S205A) rendered the channel unresponsive to PE (Figure 5E). We also mutated S205 to S205D, as converting a serine to aspartic acid can often mimic a phosphorylated ‘active’ state; in fact, expression of Panx1-S205D resulted in constitutively high basal currents, which could not be further increased after PE treatment (Figure 5E). Thus, S205 emerged as a key residue in Panx1 channel activation. Lastly to link the conserved Panx1 serine residue with SIK1, we transfected a constitutively active SIK1 (SIK1-CA) together with either wild type or mutant hPANX1S206A. SIK1-CA induced channel currents in cells expressing the wild type channel independent of PE and receptor stimulation, whereas it was unable to induce channel currents in cells transfected with the serine-mutated channel (Figure S6H). Overall, these data outline a model in which SIK1 mediates the phosphorylation of Panx1 at serine 205 to induce channel activation.
The identification of the Panx1::SIK1 interaction suggested that SIK1 may play a role upstream of Panx1 during airway disease. To address this, we deleted Sik1 in T cells by crossing Sik1fl/fl mice to Cd4-cre mice. However, there was no obvious difference in disease severity between Sik1fl/flCd4-cre+ mice and littermate controls (Figure 6A). Further investigation into the salt inducible kinase family showed that SIK2 and SIK3 are also expressed in both Teff and Treg cells and therefore might have compensatory functions (Figure S6I). Consistent with a possible role of the SIK family genes during disease, human asthmatic patient CD4+ T cell data showed reduced expression of all three SIK genes, with SIK2 and SIK3 significantly downregulated, relative to health controls (Figure 6B) (Tsitsiou et al., 2012). In fact, Panx1 could interact with SIK2 (but not SIK3) in co-immunoprecipitation studies in vitro (Figure S6F) and electrophysiology studies demonstrated that receptor-mediated Panx1 channel activation (currents) was efficiently inhibited by a pan-SIK family inhibitor (Figure 6C). Given that the SIK family proteins may play redundant roles in this pathway, it seemed plausible their functions may converge at the Panx1S205 residue to mediate channel activation.
Figure 6. The Panx1S205 phosphorylation axis controls the extent of airway inflammation.

(A) Absolute cellularity of eosinophils (left) and CD4+ T cells (right) in SIK1fl/flCd4-cre− (n=4), and SIK1fl/flCd4-cre+ mice (n=9).
(B) Relative SIK family gene expression (log2FC relative to healthy) on CD4+ T cells of healthy or asthmatic patients (ctrl n=8, asthmatics n=8), revealing a decrease in the expression of the SIK gene family in asthmatics.
(C) Current density quantification from whole cell patch-clamp recordings of Panx1 currents in HEK293T cells transfected with α1D and Panx1-WT before and after PE stimulation and the SIK family inhibitor HG9-91-01 (***p <0.0007). Two-way ANOVA
(D) Functional analysis of genetically targeted Panx1S205A cells by TO-PRO-3 dye uptake during apoptosis. Histograms and MFI of TO-PRO-3 dye uptake within apoptotic cells and live cells of respective mice.
(E) Flow plots showing the extent of eosinophil and CD4+ T cell infiltration into the bronchoalveolar space in Panx1+/+ and Panx1S205A mice.
(F) Absolute cellularity of eosinophils (***p=0.0002) (left) and CD4+ T cells (***p=0.0006) (right). (Panx1+/+ n=5, Panx1S205A n=4). Unpaired Student’s t-test (C,F). Related to Figure S6.
To address the role of Panx1S205 in airway inflammation, we engineered a Panx1S205A genetically targeted mouse. Using the CRISPR-Cas9 system, wherein embryos were microinjected with Panx1 specific sgRNA, Cas9 protein, and a homologous repair template encoding the S205A mutation, we generated a mouse line expressing endogenous Panx1-S205A globally (Panx1S205A). Importantly, Panx1S205A was capable of being activated in T cells during apoptosis, which involves caspase-dependent cleavage of Panx1 C-terminal tail and does not require S205 phosphorylation (Figure 6D). We then tested the Panx1S205A mice in HDM-induced allergic airway inflammation. The Panx1S205A mice showed increased inflammation compared to wild type controls, essentially phenocopying the Panx1−/− and Panx1fl/fl Cd4-cre+ mice (Figures 6E and 6F). Collectively, these data identified a Panx1-SIK signaling link, with Panx1S205 as an important residue in limiting allergic airway inflammation in vivo.
Discussion
Panx1 has been shown to play important functions during apoptotic cell clearance (Chekeni et al., 2010) and blood pressure regulation (Billaud et al., 2015). Therefore, it has been predominantly implicated in several different ischemic injuries, where extensive cell death and blood flow can regulate disease outcome (Jankowski et al., 2018; Sharma et al., 2018; Wei et al., 2015). However, in addition to Panx1 expression in the vasculature, immune cells also express the channel, yet, the role of Panx1 in inflammatory diseases, such as allergic airway inflammation have not been explored.
Our initial data mining revealed that Panx1 expression was significantly lower in human asthmatic patients, prompting us to probe the importance of Panx1 in airway inflammation via cell type-specific deletion, transgenic and Panx1 mutant mice. Using an in vivo mouse model of airway inflammation, we demonstrated the necessity, specificity, and sufficiency of Panx1 on T cells to limit disease severity. Panx1 played redundant roles on both Teff and Treg cells as deletion of the channel on either cell type alone did not affect inflammation. However, Panx1 deletion from both Teff and Treg cells led to inefficient suppression of Teff cells by Treg cells, which resulted in worse disease pathology. Mechanistically, we were able to demonstrate that Panx1 channels on both Treg and Teff cells can be activated to release ATP, which is then metabolized to immunosuppressive adenosine via CD39 and CD73 in order to inhibit proliferation of Teff cells. Our work suggests that it is possible for Treg cells to ‘salvage’ ATP released from Teff cells to limit their activation, identifying a concept in which Teff cells may indirectly contribute to their own downregulation as a fail-safe mechanism to prevent excessive proliferation in certain microenvironments. These data collectively uncover a non-cell autonomous role for Panx1 at specific T cell interfaces required for optimal Treg-mediated suppression of pro-inflammatory Teff cells and consequently control of inflammation during airway disease.
A number of elegant studies have identified different modes and molecules by which Treg cells can influence the function of Teff cells, and in turn limit inflammatory diseases such as allergic airway inflammation(Josefowicz et al., 2012). Among the many cell-autonomous actions of Treg cells, CD39 and CD73 make up an important extracellular signaling axis that breaks down pro-inflammatory ATP into the immunosuppressive adenosine (Idzko et al., 2014). However, the actual source of extracellular nucleotides for use by Treg cells has remained unknown. Additionally, the extracellular location of nucleotides dictates their effect on inflammation, as extracellular ATP that is not degraded can be pro-inflammatory. In fact, studies have shown that extracellular ATP can be detrimental during allergic airway inflammation, however, in this setting ATP was localized to the airway space, an area with very low numbers of Treg cells (Idzko et al., 2007). Therefore, the specific microenvironments and whether ATP can be broken down to the immunosuppressive adenosine are relevant. In the context of the ATP release channel Panx1, its specific function at the Treg-Teff interphase highlights its anti-inflammatory role at this microenvironment and during allergic airway inflammation.
In molecular studies aimed at understanding Panx1 communication between T cells, we determined that channel activation is likely independent of caspases, but instead relies on GPCRs. Although we use the phenylephrine and alpha 1D receptor modality to demonstrate channel activation, several GPCRs have been implicated in Panx1 function, including CCR5 and CXCR4 (Velasquez et al., 2016). Therefore, during allergic airway inflammation, several GPCRs on both Teff and Treg cells could be involved in Panx1 channel activation at this interface. Additionally, while CD3 stimulation was not required for GPCR-mediated Panx1 activation, it can lead to increased Panx1 expression on T cells, which may impact optimal Panx1 function (data not shown). GPCR-mediated activation of Panx1 has been shown to rely on kinase-driven phosphorylation of the channel, although the specific kinase(s) involved in T cells was unknown. Using a yeast-two hybrid screen we could identify the Salt-inducible kinase family as an interacting partner of Panx1, and also a positive regulator of channel activation. The SIK family kinase axis has been shown to control important aspects of Treg cell function (He et al., 2017), and therefore, Panx1 may constitute an additional mechanism of action for the immunosuppressive role of SIK on T cells during inflammation. Lastly, to further define the Panx1-SIK activation axis and its role in allergic airway inflammation, we examined the specific phosphorylation residue on Panx1 important for channel function. We uncovered that Panx1S205 phosphorylation was essential for both SIK mediated activation of the channel and control of airway disease as disruption of this phosphorylation in vivo results in exacerbated airway inflammation.
The mechanisms by which ATP and its derivatives can accumulate in the extracellular environment, especially in inflammatory settings that lack cell death, has remained an open question. The data presented here suggest that fine-tuning of extracellular ATP concentrations via Panx1 can be coordinated between T cells to properly regulate an immune response. Collectively, these data reveal that Panx1-based communication between Treg and Teff cells in microenvironments can be beneficial to limit inflammation associated tissue damage. Additionally, this work emphasizes the importance of understanding the localized actions of extracellular nucleotides.
Limitations of Study
Our model suggests that extracellular nucleotides can act at CD4+ T cell interfaces as we demonstrate in vitro. However, we were unable to measure microenvironmental extracellular ATP in vivo. Extracellular ATP is unstable, therefore, hard to measure and to our knowledge we are unaware of a tool for the accurate measurement or imaging of extracellular ATP within tissues in vivo. Furthermore, although we provide numerous mouse models to address the role of Panx1 on CD4+ T cells, our genetically targeted Panx1S205A is a global mutation. Therefore, we cannot rule out Panx1S205A effects on other cell types during allergic airway inflammation. Lastly, we identify SIK proteins as important mediators for Panx1 activation, however, our SIK1 T cell deficient mice did not have exacerbated inflammation. We describe that SIK2 and SIK3 may have compensatory functions, therefore a mouse deficient in all three SIK family kinases would provide definitive evidence of this.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kodi S. Ravichandran (ravi@virginia.edu)
Materials Availability
Plasmids and mouse lines generated in this study are available from the lead contact upon request.
Data and Code availability
This study did not generate or develop any datasets or codes.
EXPERIMENETAL MODELS AND SUBJECT DETAILS
All animal experiments were performed in animal housing facilities at the University of Virginia. Male and female 8-12-week-old mice were used in all experiments. Animal procedures were approved and performed according to the Institutional Animal Care and Use Committee (IACUC) at the University of Virginia.
METHOD DETAILS
Mice
C57BL6J mice were ordered from Jackson Laboratories. Panx1fl/fl and Panx1−/− mice have been described previously(Poon et al., 2014). SIK1fl/fl mice were a gift from the Berdeaux Lab. Panx1fl/fl and SIK1fl/fl mice were crossed to Cd4-cre mice (Taconic), Foxp3-cre mice (Jackson Laboratories), or Cx3cr1-cre mice (Jackson Laboratories) to generate deletion of Panx1 specifically in all T cells, Treg cells, or the myeloid lineage, respectively. Panx1-transgenic (Panx1Tg) mice were generated as described below. For in vivo experiments, female and male mice aged 8 weeks to 12 weeks were used. All procedures and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Virginia.
Generation of Panx1-transgenic mice and S205A mutant mouse
Flag-tagged PANX1 cDNA were cloned into previously characterized CAG-STOP-eGFP-ROSA26TV (CTV) vector containing a chicken actin promoter, a floxed Neo-STOP cassette, and an IRES-eGFP(Soriano, 1999). CTV-Panx1Tg vector (25μg) was linearized using the restriction enzyme Sgf1 (Promega) prior to transfection into C57BL/6 embryonic stem cells (JM8A3, KOMP) via electroporation (BTX ElectroSquarePorator). Electroporated ES cells were plated on mitomycin-treated MEF (Millipore) cells and incubated for 48 hours, selected using Geneticin (G418) (Gibco) at 200mg ml−1. Resistant clones were picked and expanded before being harvested for analysis and storage. Southern blotting analysis and qPCR were used to determine homologous recombination into the Rosa26 locus. Panx1Tg and eGFP expression were confirmed after transfection of Cre recombinase using an aliquot of the selected ES cells. Selected clones were injected into blastocysts and implanted into pseudopregnant females for generation of chimeric mice. The chimeric mice were then bred to C57BL/6J to determine germline transition and were subsequently backcrossed at least eight generations to C57BL/6J background. Panx1Tg mice (which do not express the Panx1 transgene until crossed to a Cre mouse line) were further bred to global Cd4-cre mice to re-express Panx1 in CD4+ cells. Panx1Tg Cd4-cre+ mice were then bred to Panx1−/− mice to specifically express Panx1 in T cells on a global Panx1−/−.
The S205A Panx1 mutant mouse was developed at the UVA GEMM (Genetically Engineered Mouse Model) Core. To successfully target Cas9 to the correct location on the Panx1 locus the highest scoring sgRNA (5’- CCA CTT CAA GTA CCC AAT CG-3’ (PAM site: TGG), closest to the region of interest, was identified using http://crispor.tefor.net. The oligos were synthesized through IDT, annealed and cloned into pX330 expression plasmid (gift from Wenhao Xu). High-fidelity PCR was completed on confirmed clones, the PCR was purified (Qiagen#28004) and an in-vitro transcription reaction was performed, using the MEGAscript™ T7 Transcription Kit (Cat# AMB13345). The products were purified with MEGAClear kit (#AM1908). To introduce a point mutation at S205 of Panx1 a DNA template was also developed to allow for homologous recombination at the sgRNA-Cas9 cut site. The DNA repair template was precisely designed, where a point mutation, S205A (TCT→GCG) also introduced a restriction site, recognized by Bsh1236I (#ER0921) for the ease of genotyping. The template also carried a silent mutation of the PAM site to cease Cas9 cutting (5’-GTGAAGAGAGGCTGAAGTAATAGCTCAAGTAGATACATGCCAACAGTATAACCACAAATGTCACCAGCCGGCAGCTAATGTATTTCATGATTAAATGACTCGCGTTCTTTTTTGTCTTCAAGTACTGCTCAACGATTGGGTACTTGAAGTGGCTTTCAGATATCTCCCACAGACTGAAAAACAAAGCAAATAAAAATAAT-3’). In-house made sgRNA, Cas9 protein (PNA Bio # CP01) and single stranded 200bp oligo template (ssDNA; IDT) were submitted to the UVA GEMM, where the microinjection was performed into an embryo of B6.SJL mouse strain, followed by transplantation into a pseudo-pregnant mother. The founder mouse was heterozygous for S205A, as confirmed by genotyping and TOPO-TA cloning, followed by sequencing. The female was bred to a wild-type C57BL/6J male (Jackson) and the offspring was bred to homozygosity.
Cloning and Plasmids
Plasmids of mouse α1D adrenergic receptor and the wild-type mouse Panx1 were described previously(Billaud et al., 2015). Plasmids expressing mutant mouse Panx1 at Ser205 were generated by performing site-directed mutagenesis with Pfu DNA polymerase (Agilent) following manufacturer’s manual. We used the following primer pairs: 5'-GCAGTACTTGAAGACAAAAAAGAACGCTAGTCATTTAATCATGAAATACATTAGC-3'/5'-GCTAATGTATTTCATGATTAAATGACTAGCGTTCTTTTTTGTCTTCAAGTACTGC-3' and 5'-GCAGTACTTGAAGACAAAAAAGAACGATAGTCATTTAATCATGAAATAC ATTAGC-3'/ 5'-GCTAATGTATTTCATGATTAAATGACTATCGTTCTTTTTTGT CTTCAAGTACTGC-3' were used to generate Ser-to-Ala (S205A) or Ser-to-Asp (S206D) mutation, respectively. To generate plasmids of wild type and constitutively-active SIK1, we first PCR amplified wild-type human SIK1 cDNA (Dharmacon, MHS6278-202807607) using a primer set of 5'-GTGCCATATGGTTATCATGTCGGAGTTCAGCGCG-3' and 5'-TCGCGGCCGCTCACTGCACCAGGACAAACGT-3', and subcloned the PCR product into an expression vector (pEBB) at Nde I and Not I sites using T4 DNA ligase (New England BioLab). The constitutively-active SIK1 (caSIK1; SIK1-T182E) was then generated by exchanging Thr182 to Glu182 using site-directed mutagenesis with Pfu polymerase (Agilent) and a primer pair of 5'-TACAAGTCAGGAGAGCCTCTGTCCGAGTGGTGTGGGAGCCC-3' and 5’-GGGCTCCCACACCACTCGGACAGAGGCTCTCCTGACTTGTA-3’. Wildtype and constitutively active SIK1 were further subcloned in pEGFP-C3 vectors at Xho I and Xba I sites using T4 DNA ligase. All constructs were verified by Sanger’s sequencing.
HDM-induced allergic airway inflammation
Mice were primed intranasally with 10μg of low endotoxin house dust mite extracts (HDM) (Indoor Biotechnologies) on days 0, 2, and 4 and challenged intranasally on days 10, 12, and 14. Mice were harvested and analyzed for allergic airway inflammation 24-36 hours after the last challenge unless otherwise noted. Bronchoalveolar lavage (BAL) was performed via delivery of 1.0 ml of PBS intratracheally through a canula. BAL fluid was centrifuged, and supernatants were frozen at −80°C for subsequent cytokine analysis via Luminex MAGPIX. Collected cells were treated with RBC lysis buffer (Sigma-Aldrich), washed, and stained for surface markers to distinguish cell populations. For immunophenotyping, harvested lungs were placed in HBSS media containing Ca2+ and Mg2+ (Gibco) and type 2 collagenase (Worthington Biochemicals Corporation). Lungs were then minced and incubated at 37°C for an hour with intermittent pipetting every 15 minutes to create a single cell suspension. Homogenates were passed through a 70μm filter, treated with RBC lysis buffer, washed and resuspended in PBS with 1% BSA for surface immunostaining.
Immunostaining and flow cytometry
Collected BAL cells, lung single cell suspension, spleen or thymus were stained for indicated immune cells using live dead and antibodies to CD45, CD11c, CD3, CD4, CD8, CD44, CD69, CD25, CD62L, Foxp3. Additional antibodies used to determine T cell and ILC2 protein expression include CD39, CD73, and IL-4. For intracellular Foxp3 and IL-4 staining, cells were fixed and permeabilized using Foxp3 Transcription Factor Staining Buffer Set (eBioscience) according to manufacturer’s protocol. For identification of ILC2s, cells were gate on Lineage− (CD3, CD4, CD11b, CD11c, CD19, Ly6G, NK1.1, TER119, TCR-β), livedead stain, CD45+, ICOS+, Thy1.2+, and ST2+. Samples were collected and analyzed on the BD Canto II or Attune Nxt flow cytometer and FlowJo 10.6.2.
Lung histology and immunohistochemical staining
For lung hematoxylin and eosin staining, mice were perfused through the heart with PBS. A canula was then inserted into the trachea and the lungs were gently inflated with 10% formalin at a constant fluid pressure of 25cm. The trachea was then tied, cut, and the lungs were removed and placed in 10% formalin for overnight fixation. After 24 hours, the lungs were placed into 70% ethanol. Paraffin embedding, sectioning, and H&E staining was performed by the Histology Core at the University of Virginia. Immunohistochemical staining for cleaved caspase 3 was then performed. Full imaging of the entire lung was performed using the Leica SCN400 at the UVA BTRF. Cleaved caspase 3 quantification was performed by counting all positively stained cells in the left lobe of the mouse lung by an investigator blinded to the genotype. H&E clinical scoring was performed by a pathologist who was blinded to experimental conditions and genotypes.
In vivo Edu T cell proliferation
For measurement of in vivo proliferation, the HDM model was modified where mice were harvested 24 hours post HDM treatment on Day10 in order to track the initial burst of proliferation. Mice received 1mg of EdU in 200μl of PBS i.p. at 12 hours post HDM challenge (12 hours prior tissue collection). Following collagenase digestion of the lung, cells were surface stained, fixed in 10% formalin, washed, and permeabilized in 0.07% saponin. Edu was detected using Click-it EdU AF647 imaging kit (Fischer), with additional azide from Click Chemistry Tools according to manufacturers’ protocol.
Quantitative RT-PCR
Total RNA was extracted from isolated tissues or cells using NucleoSpin RNA (Macherey-Nagel) and cDNA was generated using Quantitect Reverse Transcriptase kit (Qiagen) according to manufacturer’s protocols. Quantitative expression of Panx1, Panx2, Panx3, and Gapdh was performed using Taqman probes (Applied Biosystems) and the StepOnePlus RT PCR (ABI).
Immuno Blotting
Total protein extracts were prepared from specified isolated tissue or cells using RIPA lysis buffer supplemented with protease inhibitors (Calbiochem). Equal amounts of protein were loaded onto TGX-precast gels (Bio-Rad) or Phos-tag gels (Wako Pure Chemical Corporation) for in vitro kinase assay, subjected to SDS-PAGE, and transferred to PVDF membrane using the Transblot Turbo Transfer System (Bio-Rad). Antibodies to Panx1 (clone D9M1C, Cell Signaling Technologies), p-ERK, ERK, SIK1(Y-20), FLAG (M2), HA(F-7), and GFP (B-2) were used for immunoblotting at 1:1000 dilution at 4°C overnight. Actin-HRP (clone: AC15, Sigma) was used as a loading control. Western Lightning Plus ECL kit (Perkin-Elmer) and chemiluminescence was detected via ChemiDoc (Bio-Rad).
CD4 T cell Isolation
Spleens were harvested from naive 8-12-week-old mice, homogenized, and run through 70μm filters, and treated with RBC lysis buffer. Single cells suspensions were resuspended in MACS buffer and CD4 Teff and Treg cells were isolated using the CD4+ CD25+ Regulatory T cell Isolation kit (Miltenyi Biotech), according to manufacturers’ protocol. Briefly, splenocytes were incubated with an antibody cocktail to negatively select, via magnetic separation, CD4 T cells, followed by an additional positive selection to separate CD4+CD25− Teff from CD4+ CD25+ Treg cells. Isolation efficiency and purity was analyzed via flow cytometry on the Attune Nxt Flow Cytometry.
Apoptosis measurement
BALF was harvested as indicated above and cells were stained with annexin V-Pacific blue and 7AAD in the annexin V binding buffer for 15 min at room temperature. Samples were then diluted 2-fold with binding buffer, put on ice and analyzed via flow. AV+7AAD− cells were scored as apoptotic, and AV+7AAD+ cells as necrotic.
T cell Activation and Suppression Assays
Isolated Teff cells were incubated with 2.5μg ml−1 of anti-CD3 and 1.25μg ml−1 anti-CD28 to induce T cell activation in round-bottom plates. For downstream TCR analysis, T cells were incubated for the indicated times at 37°C. Cells were then spun down, flash frozen and lysed for immuno blot analysis of downstream TCR signaling proteins. Upregulation of T cell activation markers CD69, CD44, CD25, and CD62L were measured after T cells were incubated with anti-CD3 and anti-CD28 for indicated times. Cells were then stained for respective markers on ice and analyzed by flow cytometry. To measure T cell proliferation, Teff cells were stained with 5μM CFSE (Invitrogen) according to manufacturer’s protocol prior to activation, and dilution of the CFSE signal during cell division was measured via flow cytometry. For suppression assays different ratios of Teff to Treg cells were incubated for 4 days after anti-CD3 and anti-CD28 treatment and the CFSE dilution measured. Where indicated, ATP (10μM), adenosine (10μM), CD73 inhibitor AMP-CP (100μM), Adenosine Deaminase (ADA)(50U ml−1), and A2AR agonist CGS-21680 (5μM) was added to cultures on Day 0.
Extracellular nucleotide measurement
Suppression assays were performed as described above. At each timepoint cells were spun down at 300g for five minutes and 150 μl of supernatant was taken. Supernatant was spun down again at 2000xg for another five minutes and 125 μl was taken for analysis. For ATP detection CellTiter-Glo Luminescent Assay (Promega) was used. Briefly, 50 μl of supernatant was mixed with 50 μl of CellTiter-Glo reagent and luminescent signal was immediately recorded on a microplate reader (Flex Station 3). For AMP detection, AMP-Glo Assay (Promega) was used and the manufacturer’s protocol was followed. Briefly, 25 μl of supernatant was added to 25 μl of AMP-Glo reagent one and incubated for one hour to remove ATP from the sample and convert sample AMP to ADP. After one hour 50μl of AMP-Glo detection solution was added to the sample and incubated for another hour to convert the generated ADP to ATP and produce a luminescent signal. Luminescence was recorded on a plate reader (Flex Station 3). For adenosine detection, Adenosine Assay Kit (Fluoreometric) (abcam) was used according to manufacturer’s protocol (urine clarifier step was omitted). Samples (50μl) was mixed with adenosine reaction mix (with and without the adenosine detector reagent to subtract background) and incubated for fifteen minutes at room temperature. Fluorescence was measured on the Flex Station 3 (Ex/Em= 535/587).
Whole cell patch-clamp recording
Whole-cell, voltage-clamp recordings were performed at room temperature. Axopatch 200B amplifier commanded by pCLAMP10 software and Digidata 1322A digitizer were used to collect data (all from Molecular Devices). Micropipettes of 3-5 MΩ were pulled from thin-walled borosilicate glass capillaries (Harvard Apparatus) using a P-97 Flaming/Brown Micropipette Puller (Sutter Instrument) and coated with Sylgard 184 silicone elastomer (Dow Corning Corporation). Micropipettes were filled with a pipette solution containing 100 mM CsMeSO4, 30 mM TEACl, 4 mM NaCl, 1 mM MgCl2, 0.5 mM CaCl2, 10 mM HEPES, 10 mM EGTA, 3 mM ATP-Mg, and 0.3 mM GTP-Tris (pH 7.3; ~290 mOsm). A bath solution containing 140 mM NaCl, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, and 10 mM glucose (pH 7.3; ~300 mOsm) was used to obtain the whole cell recordings. Ramp voltage clamp commands were applied at 7 second intervals, with voltage ranging from −100 to 80 mV (0.26 mV/ms). Carbenoxolone (CBX; 50 μM)-sensitive current was taken as the difference in current at +80 mV before and after CBX application. Current density was calculated off-line by normalizing the whole-cell current to cell capacitance. Phenylephrine (PE; 20 μM) -induced current was determined by the difference in CBX-sensitive current at +80 mV, before and after PE application in bath.
Yeast 2-Hybrid screen
Yeast 2-hybrid screens were performed as previously described(Park et al., 2007). Briefly, HF7C yeast strain was used to screen a mouse 7-day old embryonic library with pGBT10-hPanx1 C-term (residues 298-422) as bait. Colonies that were able to grow on selective SCM plates (Trp-, Leu-, His- with 5 mM 3-amino-1,2,4-triazole) were sequenced and mSIK1 (residues 169-273) was identified as a potential binding partner of hPanx1 C-term.
In vitro kinase assay
In vitro kinase assay was performed using recombinant GST-SIK1 (1-303; Thermo Fisher Scientific) and a kinase buffer composed of 20 mM Tris-Hcl, 10 mM MgCl2, 40 mM NaCl (pH7.4), as previously reported (Yang et al., 2013). GFP-tagged hPanx1 (1-355) proteins were generous gifts from Dr. Mark Yeager group. The kinase buffer was supplemented with a cocktail of protease inhibitors (Sigma-Aldrich; P8340), 1 mM DTT, 1 mM NaF, 1 mM NaVO3, 50 mM β-glycerophosphate, and 3 mM Mg-ATP. GFP-tagged hPanx1 (1-355) proteins, with or without GST-SIK1, were mixed with the kinase buffer, followed by an incubation at 30° with constant shacking. After 30, 60, or 90 minutes, protein samples from the kinase reaction were boiled in Laemmli sample buffer (60 mM Tris-HCl, 2% SDS, 10% glycerol, 0.01% bromophenol blue, and 5% β-mercaptoethanol), and Phos-tag gels (Wako Pure Chemical Corporation) were used for electrophoresis.
Co-Immunoprecipitation Assays
For co-immunoprecipitation studies using HEK293T over expression system, cells were plated at 875x103 cells per 60mm dish. Twenty hours after plating HEK293T cells were transfected with 5ug of respective plasmids (empty vector control, hPanx1-FLAG, hSIK1-HA, hSIK2-GFP, hSIK3-GFP, tSIK1-HA, or KDSIK1-HA) using the Lipofectamine 2.0 reagent according to manufacturers’ protocol. Twenty-four hours after transfection cells were lysed in PBST (PBS 1%Tritonx100, protease inhibitors) and 500μg of protein from each sample were pulled down using FLAG or HA beads at 4° rotating overnight. The following day samples were washed three times with PBST and eluted in 60ul with either FLAG or HA peptide. Eluted samples and whole cell lysates were analyzed via immuno blot. For endogenous protein co-immunoprecipitation studies, Jurkat cells were used. Thirty million Jurkat cells were lysed in PBST and 750ug of protein were used for pulldown experiments. Cell lysates were first incubated with Protein A slurry for 1 hr at 4° to clear lysate of any non-specific binding proteins. Samples were spun down and supernatants were incubated with 1μg of Panx1 antibody (clone D9M1C, Cell Signaling Technologies) or IgG control. Samples were incubated at 4° rotating overnight and Protein A beads were added to samples the following day and incubated for another hour. Samples were spun down and washed three times. After washing 50ul of loading buffer was used to elute protein and boiled before loading for immuno blot analysis.
QUANTIFICATION AND STATICTICAL ANALYSIS
Receiver operating characteristic curve (ROC curve) was used to determine whether Panx1 expression could positively identify asthmatic disease outcome. Number of mice, definition of center and dispersion is recorded in each figure legend if applicable. Where not indicated the dispersion includes full data range. Statistical significance was determined using GraphPad Prism 7, using Student’s two-tailed t-test (paired or unpaired), one-way ANOVA, or two-way ANOVA according to test requirements. Grubbs’ Outlier Test was used to determine outliers. A p value of <.05 (indicated by *), <.01 (indicated by **), <.001 (indicated by ***), or <.0001 (indicated by ****) were considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45 (30-F11) – APC-eF780 | eBioscience | Cat# 47-0451-82; RRID: AB_1548781 |
| CD11c (N418) – APC | eBioscience | Cat# 17-0114-81; RRID: AB_469345 |
| Siglec-F (E50-2440) – PE | BD | Cat# 552126; RRID: AB_394341 |
| CD3 (145-2C11) – FITC | eBioscience | Cat# 11-0031-82; RRID: AB_464882 |
| CD4 (RM4-5) – eF450 | eBioscience | Cat# 48-0042-82; RRID: AB_1272195 |
| CD8 (53-6.7) – PeCy7 | eBioscience | Cat# 25-0081-82; RRID: AB_469584 |
| CD44 (IM7) – APC | eBioscience | Cat# 17-0041-83; RRID: AB_469321 |
| CD25 (PC61.5) – PE | eBioscience | Cat# 12-0251-82; RRID: AB_465607 |
| CD69 (H1.2F3) – PE | eBioscience | Cat# 12-0691-81; RRID: AB_465731 |
| CD62L (MEL-14) – APC | eBioscience | Cat# 17-0621-81; RRID: AB_469409 |
| Foxp3 (FJK-16s) – APC | eBioscience | Cat# 17-5773-80; RRID: AB_469456 |
| CD39 (Duha59) – AF647 | Biolegend | Cat# 143807; RRID: AB_3563977 |
| CD73 (TY/11.8) – PE | eBioscience | Cat# 12-0731-81; RRID: AB_763516 |
| IL-4 (11B11) – APC | eBioscience | Cat# 17-7041-81; RRID: AB_469493 |
| CD3 (145-2C11) – PerCP-Cy5.5 | eBioscience | Cat# 45-0031-82; RRID: AB_1107000 |
| CD4 (RM4-5) – PerCP-Cy5.5 | eBioscience | Cat# 45-0042-82; RRID: AB_1107001 |
| CD11b (M1/70) – PerCP-Cy5.5 | eBioscience | Cat# 45-0112-82; RRID: AB_953558 |
| CD11c (N418) – PerCP-Cy5.5 | eBioscience | Cat# 45-0114-82; RRID: AB_925727 |
| CD19 (1D3) – PerCP-Cy5.5 | eBioscience | Cat# 45-0193-80; RRID: AB_906215 |
| Ly6G/C (RB6-8C5) – PerCP-Cy5.5 | eBioscience | Cat# 45-5931-80; RRID: AB_906247 |
| NK1.1 (PK136) – PerCP-Cy5.5 | eBioscience | Cat# 45-5941-80; RRID: AB_914359 |
| TER119 (TER-119) – PerCP-Cy5.5 | eBioscience | Cat# 45-5921-82; RRID: AB_925765 |
| TCR-β (H57-597) – PerCP-Cy5.5 | eBioscience | Cat# 45-5961-82; RRID: AB_925763 |
| ICOS (7E.17G9) – PE | eBioscience | Cat# 12-9942-82; RRID: AB_466274 |
| Thy1.2 (53-2.1) – APC | eBioscience | Cat# 17-0902-82; RRID: AB_469422 |
| ST2 (RMST2-2) – PE-Cy7 | eBioscience | Cat# 25-9335-82; RRID: AB_2637464 |
| Panx1 (D9M1C) | CST | Cat# 91137S; RRID: AB_2800167 |
| Actin-HRP (AC15) | Sigma | Cat# A3854; RRID: AB_262011 |
| SIK1 (Y-20) | Santa Cruz | Cat# sc-83754; RRID: AB_2187324 |
| FLAG (M2) | Sigma-Aldrich | Cat# A8592; RRID: AB_439702 |
| HA (F-7) | Santa Cruz | Cat# sc-7392; RRID: AB_627809 |
| GFP (B-2) | Santa Cruz | Cat# sc-9996; RRID: AB_627695 |
| p-ERK (E-4) | Santa Cruz | Cat# sc-7383; RRID: AB_627545 |
| CD3 (17A2) | Thermo Fisher | Cat# 16-0032-82; RRID: AB_468851 |
| CD28 (37.51) | Thermo Fisher | Cat# 16-0281-82; RRID: AB_468921 |
| Bacterial and virus strains | ||
| HF7C Yeast Strain | Dr. Ravichandran | Park et al., 2007 |
| Biological samples | ||
| N/A | N/A | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SgfI Restriction Enzyme | Promega | Cat# R7103 |
| Geneticin (G418) | Gibco | Cat# 10131035 |
| Cas9 Protein | PNA Bio | Cat# CP01 |
| Pfu DNA Polymerase | Agilent | Cat# 600140 |
| T4 DNA Ligase | NEB | Cat# M0202S |
| LoTox House Dust Mite | Indoor Biotech. | Cat# LTN-DPE-1 |
| Type 2 Collagenase | Worthington Bio. Corp. | Cat# LS004176 |
| 7AAD Viability Stain | Thermo Fisher | Cat# A1310 |
| Annexin V Pacific Blue | Biolegend | Cat# 640918 |
| EdU | Thermo Fisher | Cat# A10044 |
| Protease Inhibitors | Calbiochem | Cat# 539131 |
| CFSE | Invitrogen | Cat# C34554 |
| ATP | Sigma-Aldrich | Cat# FLAAS-1VL |
| Adenosine | Sigma-Aldrich | Cat# A9251-1G |
| AMP-CP | Sigma-Aldrich | Cat# M8386-5MG |
| Adenosine Deaminase | Sigma-Aldrich | Cat# 10102105001 |
| CGS-21680 | Sigma-Aldrich | Cat# C141-5MG |
| Carbenoxolone | Sigma-Aldrich | Cat# C4790-1G |
| Phenylepherine | Sigma-Aldrich | Cat# P1240000 |
| SIK Recombinant Human Protein | Thermo Fisher | Cat# PV6446 |
| GFP-Panx1 (1-355) | Dr. Yeager (University of Virginia) | N/A |
| Live Dead Yellow | Thermo Fisher | L34959 |
| Critical commercial assays | ||
| MiniElute PCR Purification Kit | Qiagen | Cat# 28004 |
| MEGAscript T7 Transcription Kit | Thermo Fisher | Cat# AMB13345 |
| MEGAClear kit | Thermo Fisher | Cat# AM1908 |
| Foxp3/Transcription Factor Staining Kit | eBioscience | Cat# 00-5523-00 |
| Click-iT Edu Cell Proliferation Kit | Thermo Fisher | Cat# C10340 |
| NucleoSpin RNA | Macherey-Nagel | Cat# 740955.50 |
| Quantitect Reverse Transcriptase kit | Qiagen | Cat# 205311 |
| Western Lightning Plus ECL Kit | Perkin-Elmer | Cat# NEL103E001EA |
| CD4+ CD25+ Regulatory T cell Isolation Kit | Miltenyi Biotec | Cat# 130-091-041 |
| CellTiter-Glo Luminescent Assay | Promega | Cat# G7570 |
| AMP-Glo Assay | Promega | Cat# V5011 |
| Adenosine Assay Kit | Abcam | Cat# ab211094 |
| Lipofectamine 2.0 | Thermo Fisher | Cat# 11668027 |
| Deposited data | ||
| N/A | N/A | N/A |
| Experimental models: Cell lines | ||
| JM8A3 | KOMP | MMRRC:050960-UCD |
| Mitomycin-treated MEF | Millipore | Cat# PMEF-CF |
| B6.SJL embryos | Dr. Xu | N/A |
| HEK293T | ATCC | Cat# CRL2316 |
| Jurkat E6.1 | ATCC | Cat# TIB-152 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Lab | Cat# 00064 |
| Panx1 fl/fl | Dr. Ravichandran | Poon et al., 2014 |
| Panx1 KO | Dr. Ravichandran | Poon et al., 2014 |
| SIK1 fl/fl | Dr. Berdeaux | Nixon et al., 2016 |
| Cd4-Cre: B6.Cg-Tg(CD4-cre)1Cwi N9 | Taconic | Model# 4196-F |
| Foxp3-Cre: B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J | The Jackson Lab | Cat# #016959 |
| Cx3cr1-Cre: B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J | The Jackson Lab | Cat# 025524 |
| Panx1 Tg | Dr. Ravichandran | This paper |
| Panx1S205A | Dr. Ravichandran | This paper |
| Oligonucleotides | ||
| 5’- CCA CTT CAA GTA CCC AAT CG-3’ sgRNA | This paper | N/A |
| 5’-GTGAAGAGAGGCTGAAGTAATAGCTCAAGTAGATACATGCCAACAGTATAACCACAAATGTCACCAGCCGGCAGCTAATGTATTTCATGATTAAATGACTCGCGTTCTTTTTTGTCTTCAAGTACTGCTCAACGATTGGGTACTTGAAGTGGCTTTCAGATATCTCCCACAGACTGAAAAACAAAGCAAATAAAAATAAT-3’ ssDNA | This paper | N/A |
| 5'-GCAGTACTTGAAGACAAAAAAGAACGCTA TCATTTAATCATGAAATACATTAGC-3' primer | This paper | N/A |
| 5'GCTAATGTATTTCATGATTAAATGACTAGCGTTCTTTTTTGTCTTCAAGTACTGC-3' primer | This paper | N/A |
| 5'-GCAGTACTTGAAGACAAAAAAGAACGATA GTCATTTAATCATGAAATACATTAGC-3' primer | This paper | N/A |
| 5'-GCTAATGTATTTCATGATTAAATGACTATC GTTCTTTTTTGT CTTCAAGTACTGC-3' primer | This paper | N/A |
| 5'-GTGCCATATGGTTATCATGTCGGAGTTCAGCGCG-3' primer | This paper | N/A |
| 5'-TCGCGGCCGCTCACTGCACCAGGACAAACGT-3' primer | This paper | N/A |
| 5'-TACAAGTCAGGAGAGCCTCTGTCCGAGTGGTGTGGGAGCCC-3' primer | This paper | N/A |
| 5’-GGGCTCCCACACCACTCGGACAGAGGCTCTCCTGACTTGTA-3’ primer | This paper | N/A |
| Mm00450899_m1 | Thermo Fisher | Cat# 4351372 |
| Mm01308054_m1 | Thermo Fisher | Cat# 4331182 |
| Mm00552586_m1 | Thermo Fisher | Cat# 4331182 |
| Mm99999981_m1 | Thermo Fisher | Cat# 4331182 |
| Recombinant DNA | ||
| Panx1Tg-STOP-eGFP-ROSA26TV | Dr. Bayliss | This paper |
| pX330 | Dr. Xu | Plasmid# 42230 |
| α1DAR-pCMV6 | OriGene | NM_013460 |
| Panx1-pEBB | Dr. Ravichandran | Chekeni et al., 2010 |
| Panx1-S205A-pEBB | Dr. Bayliss | This paper |
| Panx1-S205D-pEBB | Dr. Bayliss | This paper |
| SIK1 cDNA | Dharmacon | MHS6278-202807607 |
| SIK1-pEBB | Dr. Bayliss | This paper |
| SIK1-CA-pEBB | Dr. Bayliss | This paper |
| SIK1-pEGFP-C3 | Dr. Bayliss | This paper |
| SIK1-CA-pEGFP-C3 | Dr. Bayliss | This paper |
| pGBT10-hPanx1 C-term | Dr. Ravichandran | This paper |
| SIK2-pEGFP-C3 | Dr. Bayliss | This paper |
| SIK3-pEGFP-C3 | Dr. Bayliss | This paper |
| SIK1-HA-pcDNA3 | Dr. Bayliss | This paper |
| Software and algorithms | ||
| FlowJo 10.6.2 | FlowJo | www.flowjo.com |
| Image Lab | Bio-Rad | www.bio-rad.com |
| pCLAMP10 | Molecular Devices | moleculardevices.com |
| GraphPad Prism 7 | GraphPad Softw. | www.graphpad.com |
| Immunological Genome Project | Immgen | www.immgen.org |
| Other | ||
| RBC lysis Buffer | Sigma-Aldrich | Cat# R7757 |
| TGX-precast gels | Bio-Rad | Cat# 4569034 |
| Super-Sep Phos-tag gels | Wako Pure Chemical Corp. | Cat# 198-17981 |
Highlights:
The ATP release channel, Panx1, limits the severity of allergic airway inflammation
Panx1 on CD4+ T cells is necessary and sufficient for limiting inflammation
Treg -Teff crosstalk via Panx1 extracellular nucleotides controls effector response
Panx1 activation is dependent on phosphorylation and Salt-inducible kinase (SIK)
Acknowledgements
The authors thank members of the Ravichandran laboratory, members of the Pannexin Interest Group at UVA for numerous discussions and critical reading of the manuscript, and Dr. Wenhao Xu for help in developing transgenic and genetically targeted mice. This work is supported by grants to K.S.R. from NHLBI (P01HL120840), NIGMS R35GM122542, and the Center for Cell Clearance, University of Virginia School of Medicine, and the Odysseus Award from the FWO, Belgium, EOS Grant from the FWO (3083753-DECODE), and the NHLBI (P01HL120840) and NIAID (R21 AI139967 and R21 AI135455) to U.L. Additional support was received through the NIH T32 Pharmacology Training Grant (T32GM007055) (C.B.M). C.D.L. was supported by The Wellcome Trust (206566/Z/17/Z). Y.C. was supported by Ministry of Science and Technology Taiwan (108-2320-B-007-007-MY2). C.D.L. current affiliation is University of Edinburgh Center for Inflammation Research, Queen’s Medical Research Institute, Edinburgh BioQuarter.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in samples through the genomic datasets.
REFERENCES
- Antonioli L, Pacher P, Vizi ES, Haskó G, 2013. CD39 and CD73 in immunity and inflammation. Trends Mol Med 19, 355–367. doi: 10.1016/j.molmed.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Calderón M, Almarza G, Díaz-Vegas A, Contreras-Ferrat A, Valladares D, Casas M, Toledo H, Jaimovich E, Buvinic S, 2016. Characterization of a multiprotein complex involved in excitation-transcription coupling of skeletal muscle. Skelet Muscle 6, 15. doi: 10.1186/s13395-016-0087-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billaud M, Chiu Y-H, Lohman AW, Parpaite T, Butcher JT, Mutchler SM, DeLalio LJ, Artamonov MV, Sandilos JK, Best AK, Somlyo AV, Thompson RJ, Le TH, Ravichandran KS, Bayliss DA, Isakson BE, 2015. A molecular signature in the pannexin1 intracellular loop confers channel activation by the α1 adrenoreceptor in smooth muscle cells. Sci Signal 8, ra17–ra17. doi: 10.1126/scisignal.2005824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, Weinstein JS, Licona-Limon P, Schmid ET, Pelorosso F, Gagliani N, Craft JE, Flavell RA, Ghosh S, Rothlin CV, 2017. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 356, 1072–1076. doi: 10.1126/science.aai8132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS, 2010. Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature 467, 863–867. doi: 10.1038/nature09413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LE, Locksley RM, 2014. Allergic inflammation--innately homeostatic. Cold Spring Harb Perspect Biol 7, a016352. doi: 10.1101/cshperspect.a016352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen J-F, Enjyoji K, Linden J, Oukka M, Kuchroo VK, Strom TB, Robson SC, 2007. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265. doi: 10.1084/jem.20062512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubyak GR, 2019. Luciferase-assisted detection of extracellular ATP and ATP metabolites during immunogenic death of cancer cells. Meth. Enzymol. 629, 81–102. doi: 10.1016/bs.mie.2019.10.006 [DOI] [PubMed] [Google Scholar]
- Dwyer KM, Deaglio S, Gao W, Friedman D, Strom TB, Robson SC, 2007. CD39 and control of cellular immune responses. Purinergic Signal. 3, 171–180. doi: 10.1007/s11302-006-9050-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi VD, Davidson C, Asaduzzaman M, Nahirney D, Vliagoftis H, 2013. House dust mite interactions with airway epithelium: role in allergic airway inflammation. Curr Allergy Asthma Rep 13, 262–270. doi: 10.1007/s11882-013-0349-9 [DOI] [PubMed] [Google Scholar]
- Gold M, Marsolais D, Blanchet M-R, 2015. Mouse models of allergic asthma. Methods Mol. Biol 1220, 503–519. doi: 10.1007/978-1-4939-1568-2_31 [DOI] [PubMed] [Google Scholar]
- He N, Fan W, Henriquez B, Yu RT, Atkins AR, Liddle C, Zheng Y, Downes M, Evans RM, 2017. Metabolic control of regulatory T cell (Treg) survival and function by Lkb1. Proc. Natl. Acad. Sci. U.S.A 114, 12542–12547. doi: 10.1073/pnas.1715363114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TSP, Painter MW, Immunological Genome Project Consortium, 2008. The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094. doi: 10.1038/ni1008-1091 [DOI] [PubMed] [Google Scholar]
- Holtzman MJ, Byers DE, Alexander-Brett J, Wang X, 2014. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat. Rev. Immunol 14, 686–698. doi: 10.1038/nri3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idzko M, Ferrari D, Eltzschig HK, 2014. Nucleotide signalling during inflammation. Nature 509, 310–317. doi: 10.1038/nature13085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MAM, Muskens F, Hoogsteden HC, Luttmann W, Ferrari D, Di Virgilio F, Virchow JC, Lambrecht BN, 2007. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat. Med 13, 913–919. doi: 10.1038/nm1617 [DOI] [PubMed] [Google Scholar]
- Idzko M, K Ayata C, Müller T, Dürk T, Grimm M, Baudiß K, Vieira RP, Cicko S, Boehlke C, Zech A, Sorichter S, Pelletier J, Sévigny J, Robson SC, 2013. Attenuated allergic airway inflammation in Cd39 null mice. Allergy 68, 472–480. doi: 10.1111/all.12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski J, Perry HM, Medina CB, Huang L, Yao J, Bajwa A, Lorenz UM, Rosin DL, Ravichandran KS, Isakson BE, Okusa MD, 2018. Epithelial and Endothelial Pannexin1 Channels Mediate AKI. J. Am. Soc. Nephrol 29, 1887–1899. doi: 10.1681/ASN.2017121306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu L-F, Rudensky AY, 2012. Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol 30, 531–564. doi: 10.1146/annurev.immunol.25.022106.141623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, Ravichandran KS, 2013. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 493, 547–551. doi: 10.1038/nature11714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M, Bell T, Salek-Ardakani S, Hussell T, 2015. Macrophage adaptation in airway inflammatory resolution. Eur Respir Rev 24, 510–515. doi: 10.1183/16000617.0030-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H, 2015. The immunology of asthma. Nat. Immunol 16, 45–56. doi: 10.1038/ni.3049 [DOI] [PubMed] [Google Scholar]
- Lázár Z, Cervenak L, Orosz M, Gálffy G, Komlósi ZI, Bikov A, Losonczy G, Horváth I, 2010. Adenosine triphosphate concentration of exhaled breath condensate in asthma. Chest 138, 536–542. doi: 10.1378/chest.10-0085 [DOI] [PubMed] [Google Scholar]
- Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, Germain RN, 2015. Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528, 225–230. doi: 10.1038/nature16169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locksley RM, 2010. Asthma and allergic inflammation. Cell 140, 777–783. doi: 10.1016/j.cell.2010.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman AW, Isakson BE, 2014. Differentiating connexin hemichannels and pannexin channels in cellular ATP release. FEBS Lett. 588, 1379–1388. doi: 10.1016/j.febslet.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina CB, Mehrotra P, Arandjelovic S, Perry JSA, Guo Y, Morioka S, Barron B, Walk SF, Ghesquière B, Krupnick AS, Lorenz U, Ravichandran KS, 2020. Metabolites released from apoptotic cells act as tissue messengers. Nature 580, 130–135. doi: 10.1038/s41586-020-2121-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski K, Syrjanen JL, Henze E, Kumpf J, Furukawa H, Kawate T, 2020. The cryo-EM structure of a pannexin 1 reveals unique motifs for ion selection and inhibition. Elife 9, 213. doi: 10.7554/eLife.54670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L, Serbulea V, Onengut-Gumuscu S, Leitinger N, Kucenas S, Rathmell JC, Makowski L, Ravichandran KS, 2018. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 16, 907. doi: 10.1038/s41586-018-0735-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita H, Moro K, Koyasu S, 2016. Innate lymphoid cells in allergic and nonallergic inflammation. J. Allergy Clin. Immunol. 138, 1253–1264. doi: 10.1016/j.jaci.2016.09.011 [DOI] [PubMed] [Google Scholar]
- Nixon M, Stewart-Fitzgibbon R, Fu J, Akhmedov D, Rajendran K, Mendoza-Rodriguez MG, Rivera-Molina YA, Gibson M, Berglund ED, Justice NJ, Berdeaux R, 2016. Skeletal muscle salt inducible kinase 1 promotes insulin resistance in obesity. Mol Metab 5, 34–46. doi: 10.1016/j.molmet.2015.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Tosello-Trampont A-C, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS, 2007. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430–434. doi: 10.1038/nature06329 [DOI] [PubMed] [Google Scholar]
- Poon IKH, Lucas CD, Rossi AG, Ravichandran KS, 2014. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol 14, 166–180. doi: 10.1038/nri3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raedler D, Ballenberger N, Klucker E, Böck A, Otto R, Prazeres da Costa O, Holst O, Illig T, Buch T, Mutius, von, E., Schaub, B., 2015. Identification of novel immune phenotypes for allergic and nonallergic childhood asthma. J. Allergy Clin. Immunol 135, 81–91. doi: 10.1016/j.jaci.2014.07.046 [DOI] [PubMed] [Google Scholar]
- Ricardo-Gonzalez RR, Schneider C, Liao C, Lee J, Liang H-E, Locksley RM, 2020. Tissue-specific pathways extrude activated ILC2s to disseminate type 2 immunity. J. Exp. Med 217, 171. doi: 10.1084/jem.20191172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon ST, McKenzie ANJ, 2012. Type 2 innate lymphoid cells: new players in asthma and allergy. Curr Opin Immunol 24, 707–712. doi: 10.1016/j.coi.2012.08.009 [DOI] [PubMed] [Google Scholar]
- Sharma AK, Charles EJ, Zhao Y, Narahari AK, Baderdinni PK, Good ME, Lorenz UM, Kron IL, Bayliss DA, Ravichandran KS, Isakson BE, Laubach VE, 2018. Pannexin-1 channels on endothelial cells mediate vascular inflammation during lung ischemia-reperfusion injury. Am. J. Physiol. Lung Cell Mol. Physiol 315, L301–L312. doi: 10.1152/ajplung.00004.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P, 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet 21, 70–71. doi: 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Sun Z, Jiang Q, Li J, Guo J, 2020. The potent roles of salt-inducible kinases (SIKs) in metabolic homeostasis and tumorigenesis. Signal Transduct Target Ther 5, 150–15. doi: 10.1038/s41392-020-00265-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitsiou E, Williams AE, Moschos SA, Patel K, Rossios C, Jiang X, Adams O-D, Macedo P, Booton R, Gibeon D, Chung KF, Lindsay MA, 2012. Transcriptome analysis shows activation of circulating CD8+ T cells in patients with severe asthma. J. Allergy Clin. Immunol 129, 95–103. doi: 10.1016/j.jaci.2011.08.011 [DOI] [PubMed] [Google Scholar]
- Umetsu DT, Dekruyff RH, 2006. The regulation of allergy and asthma. Immunol. Rev 212, 238–255. doi: 10.1111/j.0105-2896.2006.00413.x [DOI] [PubMed] [Google Scholar]
- Van Dyken SJ, Nussbaum JC, Lee J, Molofsky AB, Liang H-E, Pollack JL, Gate RE, Haliburton GE, Ye CJ, Marson A, Erle DJ, Locksley RM, 2016. A tissue checkpoint regulates type 2 immunity. Nat. Immunol 17, 1381–1387. doi: 10.1038/ni.3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez S, Malik S, Lutz SE, Scemes E, Eugenin EA, 2016. Pannexin1 Channels Are Required for Chemokine-Mediated Migration of CD4+ T Lymphocytes: Role in Inflammation and Experimental Autoimmune Encephalomyelitis. J. Immunol 1502440. doi: 10.4049/jimmunol.1502440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker C, Kaegi MK, Braun P, Blaser K, 1991. Activated T cells and eosinophilia in bronchoalveolar lavages from subjects with asthma correlated with disease severity. J. Allergy Clin. Immunol 88, 935–942. [DOI] [PubMed] [Google Scholar]
- Wang Z, Takemori H, Halder SK, Nonaka Y, Okamoto M, 1999. Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett. 453, 135–139. doi: 10.1016/s0014-5793(99)00708-5 [DOI] [PubMed] [Google Scholar]
- Wei R, Wang J, Xu Y, Yin B, He F, Du Y, Peng G, Luo B, 2015. Probenecid protects against cerebral ischemia/reperfusion injury by inhibiting lysosomal and inflammatory damage in rats. Neuroscience 301, 168–177. doi: 10.1016/j.neuroscience.2015.05.070 [DOI] [PubMed] [Google Scholar]
- Xiao C, Calado DP, Galler G, Thai T-H, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K, 2007. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 131, 146–159. doi: 10.1016/j.cell.2007.07.021 [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Maruyama T, Urade Y, Nagata S, 2014. Immunosuppression via adenosine receptor activation by adenosine monophosphate released from apoptotic cells. Elife 3, e02172. doi: 10.7554/eLife.02172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F-C, Tan BC-M, Chen W-H, Lin Y-H, Huang J-Y, Chang H-Y, Sun H-Y, Hsu P-H, Liou G-G, Shen J, Chang C-J, Han C-C, Tsai M-D, Lee S-C, 2013. Reversible acetylation regulates salt-inducible kinase (SIK2) and its function in autophagy. J. Biol. Chem 288, 6227–6237. doi: 10.1074/jbc.M112.431239 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or develop any datasets or codes.