

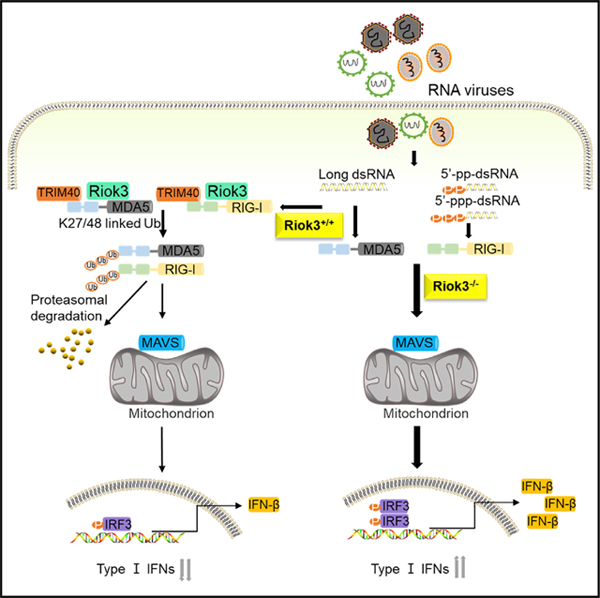
SUMMARY
The type I interferon (IFN) pathway is a key component of innate immune response upon invasion of foreign pathogens. It is also under precise control to prevent excessive upregulation and undesired inflammation cascade. In the present study, we report that Riok3, an atypical kinase, negatively regulates retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) sensing-induced type I IFN signaling. Riok3 deficiency selectively inhibits RNA viral replication in vitro, resulting from an upregulated type I IFN pathway. Mice with myeloid-specific Riok3 knockout also show a more robust induction of type I IFN upon RNA virus infection and are more resistant to RNA virus-induced pathogenesis. Mechanistically, Riok3 recruits and interacts with the E3 ubiquitin ligase TRIM40, leading to the degradation of RIG-I and melanoma differentiation-associated gene-5 (MDA5) via K48- and K27-linked ubiquitination. Collectively, our data reveal the mechanism that Riok3 employs to be a negative regulator of antiviral innate immunity.
In brief
Shen et al. show that Riok3 recruits TRIM40 to form a complex with both RIG-I and MDA5 during RNA virus infection, thus promoting TRIM40-mediated K27- and K48-linked ubiquitination and subsequent degradation of both RIG-I and MDA5 to inhibit the production of type I interferons.
Graphical Abstract
INTRODUCTION
The activation of the type I interferon (IFN) signaling pathway is the central event of an antiviral innate immune response upon recognition of pathogen-associated molecular patterns (PAMPs) by the host’s pattern recognition receptors (PRRs) (Barbalat et al., 2011; Rehwinkel and Reis e Sousa, 2010; Takeuchi and Akira, 2009).
A variety of PRRs have been identified to sense viral nucleic acids. Retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), mainly including RIG-I and melanoma differentiation-associated gene-5 (MDA5), were identified for sensing viral RNA (Loo and Gale, 2011). Both RIG-I and MDA5 contain one DExD/H box RNA helicase domain and two caspase-recruiting domains (CARDs). Upon ligand recognition, RIG-I and MDA5 activate the adaptor protein mitochondrial antiviral signaling (MAVS, also known as IPS-1, CARDIF, and VISA) through CARD domain (Kawai et al., 2005; Seth et al., 2005). MAVS further recruits the adaptor TNF receptor associated factor 3 (TRAF3) and promotes Lys63 (K63)-linked ubiquitination of TRAF3, followed by the interaction with a complex containing the kinases TANK Binding Kinase 1 (TBK1) and inhibition of IκB kinase ε (IKKε). This complex activates the phosphorylation of interferon regulatory factor 3 (IRF3) and nuclear factor κB (NF-κB), which are transcription factors of the genes encoding type I IFNs (Fitzgerald et al., 2003). Upon the production of type I IFNs, they can bind to IFN receptors to activate the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway and induce the expression of several hundreds of IFN-stimulated genes (ISGs) to restrain viral replication. Except for RLRs, much progress was achieved in characterizing other PRRs; for instance, Toll-like receptor 3 (TLR3) detects double-stranded RNA (dsRNA), while TLR7/8 detects single-stranded RNA in the endosome (Kawai and Akira, 2010). Echoing with viral RNA sensors, a group of immunostimulatory DNA sensors, such as DNA-dependent activator of IFN-regulatory factor (DAI), cyclic GMP-AMP synthase (cGAS), stimulator of interferon gene (STING), RNA III polymerase (Pol III), leucine-rich repeat flightless-interacting protein 1 (LRRFIP1), and absent in melanoma protein 2 (AIM2) were discovered (Paludan and Bowie, 2013). Similar to the cytosolic RNA-sensing pathway, recognition of cytosolic DNA also leads to the activation of TBK1 and IRF3 and the production of type I IFNs.
Precise regulation of type I IFNs is critical for efficient viral clearance and immune balance. Insufficient production of IFN leads to extensive viral replication, whereas excessive IFN is closely associated with autoimmune diseases (Kretschmer and Lee-Kirsch 2017; Snell et al., 2017). Type I IFN response is fine-tuned by opposing augmented and suppressive signals at multiple levels to shape immune responses that are appropriate for host defense and survival. Take RLRs, for example; post-translational modification (PTM) of RIG-I/MDA5 by different types of ubiquitination is a key event in the modulation of RLR activation. Several E3 ligases, such as TRIM25 (tripartite motif containing protein 25) (Gack et al., 2007), REUL (Gao et al., 2009), TRIM4 (Yan et al., 2014), MEX3C (Mex-3 RNA binding family member C) (Kuniyoshi et al., 2014), and RIPLET (Cadena et al., 2019), have been reported to promote K63-linked polyubiquitination of RIG-I to activate its downstream signals. Conversely, RNF125 (ring finger protein 125) (Arimoto et al., 2007), RNF122 (Wang et al., 2016), c-Cbl (casitas B-lineage lymphoma proto-oncogene) (Chen et al., 2013), and CHIP (carboxy-terminus of Hsc70 interacting protein) (Zhao et al., 2016) were found to facilitate covalent binding of K48-linked polyubiquitin chains to RIG-I to promote its proteasomal degradation. Functioning as E3 ligases, several TRIM (tripartite interaction motif) family members, for instance, TRIM15 (Meyer et al., 2003), TRIM26 (Ran et al., 2016; Wang et al., 2015), TRIM31 (Liu et al., 2017), and TRIM39 (Suzuki et al., 2016), were reported to play vital roles in innate immunity. In addition, TRIM13 and TRIM59 can interact with and inhibit MDA5 activity to negatively regulate IFN-β expression; however, the specific mechanism is unknown (Narayan et al., 2014). Recently, TRIM40, another TRIM family member, was reported to directly bind to both MDA5 and RIG-I, and promote their K27- and K48-linked polyubiquitination and proteasomal degradation via its E3 ligase activity, thus selectively inhibiting RLR-induced type I IFN expression (Zhao et al., 2017).
Riok3 belongs to the RIO family and is conserved in eukaryotes. It was reported to be an oncogene in pancreatic cancer, breast cancer, prostatic cancer, and glioma through a variety of regulatory mechanisms (Kalinina et al., 2010; Kimmelman et al., 2008; Singleton et al., 2015; Zhang et al., 2018). Riok3 was also found to regulate erythroblast enucleation (Zhang et al., 2011) and play as a component of pre-40S pre-ribosomal particles (Baumas et al., 2012). Riok3 was also shown to regulate the type I IFN pathway during viral infection; however, its function seems to be controversial in different systems. One group reported that Riok3 physically bridges TBK1 and IRF3 activating the downstream IFN-β pathway upon stimulation by several viruses (including Sendai virus [SeV]) and poly(inosinic-cytidylic) poly(I:C) in HEK293T cells (Feng et al., 2014), whereas another group documented that in HEK293T cells, Riok3 interacts and phosphorylates MDA5 to damage its assembly, leading to the attenuation of SeV and poly(I:C)-induced innate immune response (Takashima et al., 2015). Furthermore, a recent study revealed that during Flaviviridae viral infection in hepatocarcinoma cells, Riok3 promoted the replication of dengue virus (DENV) and Zika virus (ZIKV) but inhibited hepatitis C virus (HCV) infection (Gokhale et al., 2020). Therefore, the role and molecular mechanism of Riok3 in regulating the type I IFN pathway remains to be determined in more biologically relevant content. In the present study, by constructing myeloid-specific Riok3-deficient mice, we determined Riok3’s effect on modulating the type I IFN signaling pathway both in vivo and ex vivo. We revealed that Riok3 negatively, and selectively, regulates RLR-induced type I IFN production via recruiting E3 ligase TRIM40 for RIG-I and MDA5 degradation.
RESULTS
RNA viral replication is selectively inhibited in multiple Riok3-deficient cells
In order to investigate the role of Riok3 in host antiviral innate response, we constructed mice with myeloid-specific Riok3 knockout (KO) using the Cre/LoxP system. Western blot shows that 80%–95% of Riok3 protein was knocked out in primary peritoneal macrophages (PMs) and bone marrow-derived macrophages (BMDMs), consistent with a previous report (Clausen et al., 1999). We first examined the effect of Riok3 deficiency on viral replication using macrophages isolated from conditional KO mice. PMs were isolated from LysMCre+Riok3F/F (see as Riok3−/− below) or littermate control mice (see as Riok3+/+ below) and were challenged with RNA and DNA viruses. Viral titration assays showed that the replication of vesicular stomatitis virus (VSV), influenza A virus (IAV), and SeV were markedly restricted in Riok3−/− PMs (Figures 1A–1C and 1F). Viral gene transcripts were also significantly reduced for all tested RNA viruses (Figure 1E). However, the replication of DNA virus herpes simplex virus 1 (HSV-1) was not affected by Riok3 deficiency (Figure 1D). Consistent with PMs, we also demonstrated the reduction of RNA viral replication in BMDMs from Riok3−/− mice compared with control ones (Figure 1G). To further confirm that a lower Riok3 protein expression level restrains RNA virus replication, we tested VSV replication by knocking down Riok3 in primary mouse embryonic fibroblasts (MEFs). Using RNA interference techniques, short hairpin RNA (shRNA) targeting Riok3 was transfected into MEFs for 40 h before VSV infection. TCID50 (Median tissue culture infective dose) assay and plaque assay were followed to find that VSV replication was reduced in Riok3 knockdown primary MEFs (Figure 1H) compared with the control cells. Taken together, these data suggested that Riok3 deficiency selectively inhibited RNA viruses, but not DNA viruses, in multiple primary cell types.
Figure 1. RNA viral replication is inhibited in Riok3-deficient cells.

(A–C) Determination of viral loads in Riok3+/+ and Riok3−/− peritoneal macrophages infected with VSV (A, MOI = 1), IAV (B, MOI = 1), or SeV (C) for indicated time points. TCID50 assay was used for (A) and (B), while hemagglutination assay was used for (C).
(D) TCID50 assay for detecting viral loads in Riok3+/+ and Riok3−/− peritoneal macrophages infected with HSV-1 for indicated time points.
(E) qRT-PCR analysis of VSV-G, IAV-M, and SeV-M mRNA transcripts in Riok3+/+ and Riok3−/− peritoneal macrophages infected with VSV for 8 h and PR8 and SeV for 12 h.
(F) Plaque assay for detecting viral loads in Riok3+/+ and Riok3−/− peritoneal macrophages infected with VSV (MOI = 1), IAV (MOI = 1), or HSV-1 (MOI = 5) for indicated time points.
(G and H) Determination of viral loads by TCID50 assay in Riok3-deficient and control bone marrow-derived macrophages (F) and MEFs (G) infected with VSV for indicated time points. Before VSV infection, 2 μg/mL shRiok3 or control plasmids was transfected into MEFs for 40 h.
Data are shown as mean ± SEM. Results are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Riok3 deficiency selectively upregulates RNA virus-induced production of antiviral cytokines in primary macrophages and MEFs
We then tested whether the inhibition of viral replication in Riok3-deficient cells is due to higher expression of type I IFNs and pro-inflammatory cytokines, which are essential for antiviral defense. Through ELISA, we detected a higher induction of IFN-β, tumor necrosis factor alpha (TNF-α), and interleukin-6 (IL-6) protein in Riok3 KO PMs upon VSV, IAV, and SeV infection, whereas this increase was not detected during HSV-1 infection (Figures 2A–2C). We also knocked down Riok3 in isolated primary MEFs using shRNA, and the same phenotype was observed. MEFs with Riok3 knockdown produced higher IFN-β, TNF-α, and IL-6 with VSV challenge than those cells transfected with control shRNA (Figures 2D–2F). The upstream signaling molecules of IFNs, including TBK1, IRF3, and P65, also showed enhanced phosphorylation in Riok3−/− PMs after RNA virus infection (Figure 2G). Because type I IFNs activate the JAK/STAT pathway and induce the expression of ISGs to restrain viral replication, we analyzed several representative ISGs, including ISG15, OAS1, MX1, and MX2. After VSV infection for 4 h, higher mRNA transcripts of these ISGs were expressed in MEFs with Riok3 knockdown (Figure 2H). These results suggested that Riok3 negatively regulated RNA virus-induced type I IFNs signaling pathway.
Figure 2. Riok3 negatively regulates the production of RNA virus-induced anti-viral cytokines by promoting phosphorylation of TBK1, IRF3, and p65.

(A–C) ELISA of IFN-β (A), TNF-α (B), and IL-6 (C) in Riok3+/+ and Riok3−/− peritoneal macrophages infected for 12 h with VSV, IAV, SeV, or HSV-1, respectively.
(D–F) ELISA of IFN-β (D), TNF-α (E), and IL-6 (F) in Riok3 knockdown MEFs infected for 12 h with VSV.
(G) Immunoblot analysis of phosphorylated or total proteins in lysates of Riok3+/+ and Riok3−/− peritoneal macrophages infected for indicated hours with VSV, IAV, and SeV. Phosphorylation of TBK1 Ser172, IRF3 Ser396, and p65 Ser536 was detected.
(H) qRT-PCR analysis of anti-viral ISGs in Riok3 knockdown or control MEFs infected for 4 h with VSV.
Data are shown as mean ± SEM. Results are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Riok3 deficiency selectively strengthens the production of RLR-induced antiviral cytokines in multiple cell types
VSV, IAV, and SeV are mainly recognized by RLRs (Kato et al., 2006; Loo et al., 2008; Yoneyama and Fujita 2009), including both RIG-I and MDA5. To directly investigate if the elevated IFN response in Riok3−/− macrophages was through RLRs, we used two forms of RNA analogs poly(I:C) to mimic viral stimulation. Short-length poly(I:C) is typically sensed by RIG-I, while long-length poly(I:C) is largely recognized by MDA5 (Kato et al., 2008). In accordance with RNA viruses, the qPCR analysis showed that both short and long forms of poly(I:C) induced more IFN-β expression, as well as higher pro-inflammatory factors in Riok3−/− PMs (Figures 3A–3F). These were further confirmed by ELISA assay, with elevated protein expression levels (Figures 3G–3I). Corresponding to the raised secretion of IFN-β and pro-inflammatory cytokines, the phosphorylation of TBK1, IRF3, and p65 augmented Riok3−/− PMs with the treatment of both short-form and long-form poly(I:C) (Figure 3J). In addition, parallel experiments in Riok3 knockdown MEFs provided consistent results. Poly(I:C) targeting MDA5 induced more IFN-β, TNF-α, IL-6, and antiviral ISGs with the reduced expression of Riok3 in MEFs cells (Figures 3K–3N). Considering that Riok3 did not affect HSV-1 replication, we also validated whether Riok3 influences DNA sensors recognition-induced IFN-β production. As shown in Figures 3G–3I, PMs were transfected with poly(deoxyadenylic-deoxythymidylic) poly(dA:dT), 3′3′-cGAMP, or CpG oligonucleotides (CpG ODNs), which can activate DAI (and AIM2), STING, and TLR9, respectively. However, no significant change of IFN-β, TNF-α, or IL-6 was observed in Riok3−/− PMs compared with control. Collectively, these data indicated that Riok3 deficiency selectively enhanced the RLRs activation-induced downstream type I IFN pathway.
Figure 3. Riok3 deficiency selectively upregulates the production of RIG-I/MDA5 activation-induced anti-viral cytokines in PMs and MEFs.

(A–C) qRT-PCR analysis of IFN-β (A), TNF-α (B), and IL-6 (C) in Riok3+/+ and Riok3−/− peritoneal macrophages transfected with 5 μg/mL short-length poly(I:C) for indicated hours.
(D–F) qRT-PCR analysis of IFN-β (D), TNF-α (E), and IL-6 (F) in Riok3+/+ and Riok3−/− peritoneal macrophages transfected with 5 μg/mL long-length poly(I:C) for indicated hours.
(G–I) ELISA of IFN-β (G), TNF-α (H), and IL-6 (I) in supernatants of Riok3+/+ and Riok3−/− peritoneal macrophages transfected for 12 h with 5 μg/mL short-length poly(I:C), 5 μg/mL long-length poly(I:C), 1 μg/ml poly(dA:dT), 5 μg/mL 3′3′-cGAMP, and 5 μg/mL CpG DNA, respectively.
(J) Immunoblot analysis of phosphorylated or total proteins in lysates of Riok3+/+ and Riok3−/− peritoneal macrophages transfected for indicated times with two forms of poly(I:C). Phosphorylation of TBK1 Ser172, IRF3 Ser396, and p65 Ser536 was detected.
(K–M) ELISA of IFN-β (K), TNF-α (L), and IL-6 (M) in supernatants of Riok3-deficient and control MEFs transfected with 5 μg/mL long-length poly(I:C) for 12 h. 2 μg/mL shRiok3 or control plasmids was transfected into MEFs for 40 h to knock down Riok3 expression before poly(I:C) transfection.
(N) qRT-PCR analysis of anti-viral ISGs in Riok3 knockdown or control MEFs transfected with 5 μg/mL long-length poly(I:C) for 6 h.
Data are shown as mean ± SEM. Results are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Riok3 interacts with RIG-I/MDA5 and promotes their degradation
To further examine the mechanism underlying Riok3’s regulation of antiviral responses, we sought to determine the physical interaction between Riok3 and RLRs signaling molecules. Because the phosphorylation of two transcription factors, IRF3 and p65, was upregulated in Riok3-deficient PMs, we inferred that Riok3 may interact with key factors upstream of these two proteins. Endogenous immunoprecipitation (IP) of VSV-infected PMs was performed, which showed that Riok3 interacted with both RIG-I and MDA5, while Riok3 did not interact with MAVS, TRAF3, TBK1, or IRF3 (Figure 4A). Moreover, IP assay with anti-RIG-I and anti-MDA5 antibodies in PMs confirmed the interaction for endogenous proteins. The interaction becomes even stronger with VSV infection (Figure 4B). Considering that Riok3 may form a complex with RIG-I and MDA5, and negatively regulates RLR-induced type I IFNs, we wondered if Riok3 may inhibit the expression of RIG-I and MDA5 upon viral infection. Indeed, deficiency in Riok3 maintained a higher expression of RIG-I and MDA5 at the basal level and increased the protein abundance in macrophages after infection with VSV (Figure 4C). The cycloheximide (CHX) chase assay of macrophages showed that Riok3 deficiency increased the half-life of endogenous RIG-I and MDA5 protein during VSV infection (Figure 4D). Thus, the presence of Riok3 promotes the protein degradation of RIG-I and MDA5 upon RNA virus infection.
Figure 4. Riok3 interacts with RIG-I/MDA5 and promotes their degradation.

(A and B) Riok3 interacts with both RIG-I and MDA5. The primary macrophages were infected with VSV at MOI = 1 for indicated times, followed by immunoprecipitation using indicated antibodies.
(C) Riok3 inhibits the expression of RIG-I and MDA5 upon VSV infection. Immunoblot analysis of RIG-I and MDA5 in lysates of Riok3+/+ or Riok3−/− macrophages infected with VSV for indicated times.
(D) Riok3 promotes the degradation of RIG-I and MDA5 upon CHX treatment. Immunoblot analysis of RIG-I and MDA5 in lysates of Riok3+/+ or Riok3−/− macrophages treated with CHX (100 mg/mL) for indicated hours after infection with VSV for 2 h.
(E) Riok3 increases the polyubiquitination of RIG-I and MDA5 in 293T cells. Plasmids encoding Myc-RIG-I (or Myc-MDA5) and HA-ubiquitin were co-transfected into HEK293T cells with or without a plasmid encoding Riok3. The RIG-I and MDA5 ubiquitination was monitored by immunoprecipitation.
(F) Riok3 increases the polyubiquitination of RIG-I and MDA5 in PMs. Riok3+/+ and Riok3−/− PMs were infected with VSV for 4 h, followed by immunoprecipitation for detecting the RIG-I and MDA5 ubiquitination.
Considering the ubiquitinoylation-proteasome pathway is the major pathway for protein degradation (Liu et al., 2016), we evaluated the polyubiquitination of RIG-I and MDA5 with and without Riok3 overexpression. We observed that the level of polyubiquitination conjugated to RIG-I and MDA5 was higher both in 293T cells with ectopic Riok3 expression and in Riok3+/+ PMs (Figures 4E and 4F), which may result in the rapid turnover of proteins (Figure 4D).
Riok3 promotes TRIM40-mediated ubiquitination and degradation of RIG-I and MDA5
E3 ubiquitin ligases mediate the proteasomal degradation of their target proteins (Morreale and Walden, 2016). To examine the potential E3 ligases regulated by Riok3 and mediate the RIG-I and MDA5 ubiquitination, we performed a systematic database and literature search. For all 377 reported human E3 ligases (Medvar et al., 2016), we examined the potential interaction with both RIG-I and MDA5 through the STRING database (von Mering et al., 2003). Thirty-two E3 ligase candidates were reported to be interacting with both proteins. We first filtered the candidates without literature support. As mentioned earlier, K63-linked polyubiquitination of RLRs activates its downstream signals. Based on the result of Riok3 negatively regulating the RLR pathway, we excluded those E3 ligases performing only K-63 ubiquitin. Three E3 ligases (TRIM40, Parkin, and RNF125) were screened out (see Table S1).
Endogenous IP in PMs showed that TRIM40 is the only one that directly binds to Riok3 among the three candidates (Figure 5A). In addition, the protein expression of TRIM40 was upregulated upon VSV infection (Figure 5B), corresponding to the upregulated RIG-I and MDA5 expression (Zhao et al., 2017). Thus, we hypothesized that TRIM40 may be the E3 ligase of RIG-I and MDA5 that directly binds to and is regulated by Riok3. To further examine the role of TRIM40 in Riok3-mediated downregulation of IFN response, we first confirmed TRIM40 as the E3 ligase of both RIG-I and MDA5. The HEK293T cells were transfected with siTRIM40 or control small interfering RNA (siRNA) for 24 h, then infected with VSV for 4 h. IP assay was performed to show that TRIM40 knockdown reduced the polyubiquitination of RIG-I and MDA5, and specifically, TRIM40 knockdown reduced their K27- and K48-linked ubiquitination (Figure 5C). Then, we asked if Riok3 could affect TRIM40-mediated proteasomal degradation of RIG-I and MDA5. As shown in Figure 5D, Riok3 promotes TRIM40-mediated degradation of RIG-I and MDA5. However, this could be reversed by proteasome inhibitor ΜG132, suggesting that downregulation is through proteasomal degradation.
Figure 5. Riok3 promotes TRIM40-mediated ubiquitination and degradation of RIG-I and MDA5.

(A) TRIM40 interacts with Riok3. The primary macrophages were infected with VSV at MOI = 1 for 4 h, followed by immunoprecipitation with indicated antibodies. IgG was used as negative control.
(B) VSV infection promotes TRIM40 expression. The primary macrophages were infected with VSV at MOI = 1 for 4 h, followed by western blot (WB) with indicated antibodies.
(C) TRIM40 promotes K27- and K48-linked ubiquitination of RIG-I and MDA5. The HEK293T cells were transfected with 10 nM siTRIM40 for 24 h, then infected with VSV for 4 h. Cell lysates were immunoprecipitated with anti-RIG-I (or anti-MDA5) antibody, followed by WB with the indicated antibodies.
(D) Riok3 promotes TRIM40-mediated degradation of RIG-I and MDA5 in a dose-dependent manner. The HEK293T cells were transfected with plasmids encoding FLAG-RIG-I (or FLAG-MDA5), HA-ubiquitin, Myc-TRIM40, and varying doses of a plasmid encoding His-Riok3 (0.5 and 1 μg). Half of each cell aliquot was treated with ΜG132 (10 μM). The level of RIG-I and MDA5 proteins was examined in cells harvested 24 h later.
(E) Riok3 interacts with TRIM40. The primary macrophages were infected with VSV at MOI = 1 for indicated times, followed by immunoprecipitation using indicated antibodies.
(F) Riok3 forms a complex with TRIM40 and RIG-I (or MDA5). The primary macrophages were infected with VSV at MOI = 1 for 4 h, followed by immunoprecipitation using indicated antibodies.
(G) Riok3 is necessary for this complex formation. WT and Riok3 knockout PMs were infected with VSV at MOI = 1 for 4 and 8 h, followed by immunoprecipitation using indicated antibodies.
(H) Schematic diagram of TRIM40 and its truncate mutants.
(I) TRIM40 interacts with Riok3 via its internal CC domain. Myc-tagged TRIM40 or its mutants and FLAG-Riok3 were individually transfected into HEK293T cells. The cell lysates were immunoprecipitated with an anti-Myc antibody and then immunoblotted with the indicated antibodies.
(J) Schematic diagram of Riok3 and its truncate mutants.
(K) Riok3 interacts with TRIM40 via its RIO domain. FLAG-tagged Riok3 or its mutants and Myc-TRIM40 were individually transfected into HEK293T cells. The cell lysates were immunoprecipitated with an anti-FLAG antibody and then immunoblotted with the indicated antibodies.
(L) The deficiency of TRIM40 increases the IFN-β transcription. WT and Riok3 knockout PMs were transfected with 50 nM siTRIM40 or control siRNA to knock down TRIM40 before VSV infection for indicated time points. qRT-PCR was performed to evaluate the IFN-β mRNA expression.
B, B box; CC, coiled-coil region; R, RING domain.
To determine whether the regulation of Riok3 on TRIM40-mediated ubiquitinations of RIG-I and MDA5 is through physical interactions, we examined the interaction between Riok3 and TRIM40. IP in VSV-infected PMs showed that Riok3 interacted with TRIM40 at a basal level, which is increased during VSV infection (Figure 5E). With the same pull-down samples, RIG-I and MDA5 were also detected, indicating that Riok3 forms a protein complex containing RIG-I (or MDA5) and TRIM40 (Figure 5F). To see if Riok3 is necessary for this complex formation, we infected WT and Riok3 KO PMs with VSV, and IP was performed to find that when Riok3 was deficient, the interaction between TRIM40 and RIG-I (or MDA5) was reduced (Figure 5G).
TRIM40 contains an N-terminal RING-finger domain, B-box domain, and an internal coiled-coil (CC) domain (Tomar and Singh 2015), while Riok3 contains the RIO domain (Feng et al., 2014). To search for the domain of TRIM40 that is responsible for the interaction with Riok3, we constructed a series of Myc-tagged TRIM40-truncated mutants (Figure 5H) and FLAG-tagged Riok3-truncated mutants (Figure 5J). Riok3 was coprecipitated with TRIM40 WT, RING domain deletion mutant (ΔRING), B-box domain deletion mutant (ΔB box), and C-terminal deletion mutant (N159), but not with CC domain deletion mutant (ΔCC) (Figure 5I). Meanwhile, TRIM40 was found to be coprecipitated with Riok3 WT and all other mutants, but not with the RIO domain deletion mutant (Figure 5K). These results indicated that TRIM40 interacted with Riok3 via its internal CC domain, and Riok3 interacted with TRIM40 through its RIO domain. Finally, we validated the role of TRIM40 in regulating IFN-β expression. WT and Riok3-deficient PMs were transfected with 50 nM siTRIM40 or control siRNA to knock down TRIM40, and qRT-PCR was performed. We found that the deficiency of TRIM40 raised the IFN-β transcription, even in the presence of Riok3 KO (Figure 5L). Collectively, these results suggested that Riok3 forms a complex with TRIM40 and RIG-I/MDA5 and is required to promote TRIM40-mediated ubiquitination and degradation of RIG-I and MDA5.
Riok3 deficiency reduces viral replication in vivo after RNA virus infection
To further affirm the functional importance of Riok3 in vivo, we challenged Riok3−/− and Riok3+/+ mice with VSV and IAV. VSV was intraperitoneally injected into mice for 12 h. We examined the titer and viral copy number of VSV-G in livers and spleens. The titer and viral gene copy number of VSV-G were significantly reduced in Riok3−/− mice compared with control mice (Figures 6A and 6B). Consistent with the reduced VSV replication, we noted a higher IFN-β mRNA expression in those organs (Figure 6C) and isolated PMs (Figure 6G) of Riok3−/− mice than that in Riok3+/+ mice. The same phenotype was observed with intranasal IAV infection. Lower viral titer and higher IFN-β mRNA expression were observed at day 6 post-IAV infection in Riok3−/− mouse lungs (Figures 6D–6F). Also, immunohistochemical (IHC) staining for influenza nuclear protein (NP) demonstrated that IAV viral protein in the lungs of Riok3−/− mice was reduced compared with control mice (Figure 6H). In accordance with the limited viral replication, a lower amount of infiltrated macrophages and neutrophils was observed in the lungs of Riok3−/− mice after infection with IAV, especially at late time points (days 6 and 8 post-infection) (Figure 6I). Consistently, Riok3 deficiency also protected mice from lethal IAV challenge (Figure 6J). These data indicated that Riok3−/− mice developed a more potent IFN response against RNA viral infection, resulted in restricted viral replication, reduced pathogenesis, and improved survival.
Figure 6. Riok3-deficient mice are more resistant to RNA viral infection.

(A) Determination of VSV loads in organs by TCID50 assay after Riok3+/+ and Riok3−/− mice (n = 6 per group) were intraperitoneally injected with VSV for 12 h.
(B) qRT-PCR analysis of VSV-G gene expression in organs from Riok3+/+ and Riok3−/− mice in (A).
(C) qRT-PCR analysis of IFN-β expression in organs from Riok3+/+ and Riok3−/− mice in (A).
(D) Determination of PR8 loads in lungs by TCID50 assay after Riok3+/+ and Riok3−/− mice (n = 6 per group) were intranasally infected with PR8 for 6 days.
(E and F) qRT-PCR analysis of VSV-G (E) and IFN-β (F) expression in lungs from Riok3+/+ and Riok3−/− mice in (D).
(G) qRT-PCR analysis of VSV-G gene expression in PMs extracted from Riok3+/+ and Riok3−/− mice in (A).
(H) Immunohistochemistry result of H1N1 expression for lungs from (D). Scale bar, 100 μm.
(I) Hematoxylin and eosin staining of lung sections from mice in (D). Scale bar, 100 μm.
(J) Survival curve of Riok3+/+ and Riok3−/− mice given intranasally infection of PR8 (2,000 plaque-forming units [PFUs]/mouse) (n = 13 per group).
DISCUSSION
In the current study, we determined that Riok3 is involved in regulating the RLR-mediated antiviral innate immune response. Riok3 interacts with RIG-I, MDA5, and the E3 ligase TRIM40, leading to TRIM40-mediated K27- and K48-linked polyubiquitination of RIG-I and MDA5, which results in the downregulation of type I IFN signaling.
Recent studies have reported that Riok3 regulates multiple pathophysiological processes through interacting with cellular proteins (or relevant pathways). In pancreatic tumor models, it was discovered that Riok3 forms a complex with PAK1 (p21-activated kinase 1), a key downstream effector of Rac, to alter cytoskeletal architecture and promote the migration and invasion of pancreatic cancer cells (Kimmelman et al., 2008). In breast cancer cells, coimmunoprecipitation (coIP) studies identified that Riok3 interacted with a number of proteins, including components of the actin cytoskeleton, including actins (ACTG1, ACTA2), tropomyosin (TPM3, TPM4), and tropomodulin 3 (TMOD3) (Singleton et al., 2015). In addition, ribosomal subunits (RPS3, RPS14, RPS16, RPS18, RPS20, RPL27A, RPL30) were also identified to interact with Riok3 in breast cancer cells (Singleton et al., 2015), consistent with the reported role of Riok3 in ribosomal biogenesis in HeLa cells (Baumas et al., 2012). Riok3 was also found to regulate other tumor-related and immune response signaling pathways, such as Hedgehog pathway, AKT/mammalian target of rapamycin (AKT/mTOR) pathway, and NF-κB pathway signaling pathways (Shan et al., 2009; Tariki et al., 2013; Zhang et al., 2018). The broad cellular interactions reveal that it may perform diverse regulatory roles in different systems and under different conditions. In the context of innate immunity, different results were obtained: one group showed that Riok3 works as an adaptor protein to promote type I IFN production during both RNA and DNA viral infection in HEK293T cells through physically bridging TBK1 and IRF3 (Feng et al., 2014). However, another group declared that in HEK293T cells, Riok3 interacts with and phosphorylates MDA5 to disrupt its assembly, resulting in inhibition of type I IFN pathway upon RNA viral infection (Takashima et al., 2015). A recent study revealed that Riok3 depletion significantly reduced the production of infectious DENV and ZIKV particles but increased the production of infectious HCV particles (Gokhale et al., 2020). Importantly, through the generation of myeloid-specific Riok3 KO mice, we provided the in vivo evidence that Riok3 functions as a negative regulator of type I IFN production upon RNA viral invasion. Furthermore, our in vitro data showed that when sensing RNA viruses in primary macrophages and MEFs, Riok3 interacts with RIG-I, MDA5, and TRIM40, forming a complex to degrade RLRs and inhibit the downstream type I IFN pathway. These findings suggest that Riok3 may act as a hub protein and play multiple roles in pathophysiological processes in different cell types. Actually, protein-protein interaction (PPI) network databases, such as BioPlex Interactome, provide evidences of Riok3 interacting with other proteins, including RPS2, RPS10, KCNE3, ADAM7, RACK1, KRR1, and SLC2A13 (Huttlin et al., 2017). The role of Riok3 should be defined for each specific cell type under a defined condition.
The production of type I IFNs is one of the most important components of innate immunity to resist viral invasion. RIG-I-like receptors act as the main PRRs sensing RNA viral nucleic acids. Activation of RIG-I and MDA5 is a multi-step process consisting of viral RNA binding, conformational changes, and a series of PTMs. Ubiquitination is a key PTM for regulating RLR signaling. In this context, several E3 ligases have been implicated in promoting or inhibiting RIG-I signaling following virus infection. TRIM25, REUL, TRIM4, and MEX3C induced K63-linked polyubiquitin chains, which serve as an activation marker (Gack et al., 2007; Gao et al., 2009; Kuniyoshi et al., 2014; Yan et al., 2014). Other E3 ligases, such as RNF125, RNF122, c-Cbl, and CHIP, inhibited the RLR signaling by introducing K48-linked polyubiquitin chains for proteasomal degradation (Arimoto et al., 2007; Chen et al., 2013; Wang et al., 2016; Zhao et al., 2016). Zhao et al. (2017) illustrated that TRIM40 acts as an E3 ubiquitin ligase and directly catalyzed K27- and K48-linked polyubiquitination of RIG-I and MDA5 to increase their degradation, showing that K27-linked polyubiquitination of RLRs is also associated with their degradation through the ubiquitin-proteasome pathway. In this study, we found that Riok3 forms a complex with TRIM40, RIG-I, and MDA5 and enhances TRIM40-mediated RLRs K27- and K48-linked ubiquitination. This complex formation is required for subsequent RLR degradation and downstream antiviral signaling activation.
Multiple studies have revealed the association of RLR over-activation with autoimmune diseases, including systemic lupus erythematosus (SLE), Singleton-Merten syndrome (SMS), and type 1 diabetes (T1D) (Kato and Fujita 2014; Rutsch et al., 2015; Van Eyck et al., 2015). For example, excess activation of MDA5 by mutation G821S results in autoimmune triggering linked with the aberrant production of type I IFNs (Mino et al., 2015), whereas loss of function of MDA5 attenuates innate immune responses against viruses, such as coxsackievirus B4, which is strongly associated with triggering of T1D (Jaïdane and Hober, 2008). Interestingly, genome-wide association studies (GWASs) data showed that the SNPs of TRIM40 have an association with autoimmune diseases, such as multiple sclerosis (MS), ankylosing spondylitis (AS), rheumatoid arthritis (RA), and T1D (Sirota et al., 2009), prompting us to ask if TRIM40 may contribute to preventing autoimmune diseases through limiting the expression of RLRs at a low level. As shown here, Riok3 interacts with TRIM40, RIG-I, and MDA5 to repress the downstream pathway, indicating its potential to be a drug target for preventing autoimmune diseases. So it would be critical to clarify the role of Riok3 in the pathogenic process of autoimmunity in the context of MDA5 activation by investigating the Riok3 KO mice.
Collectively, in this study, we report that Riok3 is identified as a suppressor of RNA virus-induced production of type I IFNs by investigating both in primary macrophages in vitro and genetically modified mice. Riok3 interacts with both RIG-I and MDA5 and inhibits their expression after RNA viral infection. Mechanically, Riok3 interacts with the CC domain of the E3 ligase TRIM40 via its RIO domain, promoting TRIM40-mediated K48- and K27-linked ubiquitination and inducing subsequent proteasomal degradation of RIG-I and MDA5. Thus, Riok3 negatively regulates the RLR-activated downstream antiviral signaling pathway. These findings define one of the roles and mechanisms of Riok3 regulating the innate immune response.
Limitations of the study
This study has potential limitations. The primary limitation is only one kind of DNA virus (HSV-1) was detected in our system, and we were short of the in vivo data of HSV-1. Second, the molecular mechanism of how Riok3 recruits TRIM40 to form a complex of RIG-I and MDA5 could be performed. Also, for further in vivo study, hybrid transgenic Riok3 and TRIM40 mice (LysMCre+ Riok3F/FTRIM40F/F) may be generated.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yushen Du (lilyduyushen@zju.edu.cn).
Materials availability
The LysMCre+Riok3F/F mouse line and all plasmids generated in this study is available upon request.
Data and code availability
This study did not generate/analyze datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Myeloid-specific Riok3 knockout mice on the C57BL/6 background were generated by two steps: 1. make Riok3F/F mice with LysMCre+ mice to make heterozygous LysMCre+Riok3F/− mice; 2. make the LysMCre+Riok3F/− (male) with Riok3F/F (female) to make the homozygous LysMCre+Riok3F/F mice. The construction of the Riok3F/F transgenic mouse was designed by us and generated in the Model Animal Research Center of Nanjing University. In brief, FRT-lacZneo-FRT-loxP sequences contained vectors and loxP sequences contained vectors were microinjected into embryonic stem cells (Riok3tm1a(EUCOMM)Wtsi), which were ordered from EUCOMM. After homologous recombination, FRT-lacZneo-FRT-loxP and loxP sequences were introduced into the contiguous intron upstream of the 3rd exon and the contiguous intron downstream of the 5th exon, respectively. Antibiotic positive screening was performed, and the selected ES target cells were planted into pseudopregnant mice to generate chimeric mice, which then gave birth to the heterozygous generation with neo gene expression. By mating Flp mice with these heterozygous ones, the neo gene was eliminated, and the homozygous Riok3F/F mice (with deletion of the 3rd, 4th and 5th exons of Riok3) were selected and propagated. LysMCre+ mice were a kind gift from Prof. Ke Yuehai in Zhejiang University (Tao et al., 2014). WT pregnant mice (13–15 days old) for MEFs extraction were purchased from Shanghai SLAC Laboratory Animal Co. Ltd. All mice were bred in specific pathogen-free conditions at the Laboratory Animal Center of Zhejiang University. Female KO mice (6–8 weeks old) and their littermates were used for experiments. All animal experiments were approved by the Zhejiang University Medical Laboratory Animal Care and Use Committee. Genotypic identification was performed by PCR using toes’ genomic DNA.
Cells and reagents
Primary peritoneal macrophages (PMs) were harvested from the mice 4 days after intraperitoneally injection with 2.5ml thioglycollate broth (Sigma, USA), and cultured with RPMI1640 medium containing 10% FBS. Primary bone marrow-derived macrophages (BMDMs) were extracted from mice’ femurs and tibias, and cultured in RPMI1640 supplemented with 10ng/ml M-CSF for 7days. Murine embryonic fibroblasts (MEFs) were obtained from WT C57BL/6 mouse embryos and cultured in complete DMEM. The HEK293T, MDCK, and Vero cell lines were obtained from ATCC and grown in RPMI1640 medium or DMEM containing 10% FBS.
METHOD DETAILS
Viral infection and titration
Cells were infected with VSV (MOI = 1), IAV (MOI = 1), SeV (500HAU/ml) or HSV-1 (MOI = 5) for the indicated hours. For in vivo infection, age- and sex-matched groups of littermate mice were intraperitoneally injected with VSV (5 × 107 pfu/mouse) and intranasally infected with IAV (1000 pfu/mouse for viral titration; 2000 pfu/mouse for survival curve determination). The titration of VSV, IAV, and HSV-1 was performed by TCID50 assay and plaque assay, and hemagglutination assay was used for SeV titration.
Cell transfection
PMs were transfected with 5 μg Poly(I:C, LMW), 5 μg Poly(I:C, HMW), 1 μg Poly(dA: dT) or 5 μg 3′3’-cGAMP for indicated time points in 12-well plates, and total RNA or supernatants were collected for next experiments. MEFs were transfected with 5 μg Poly(I:C, HMW) in 12-well plates. Transfection was performed using Lipofectamine 2000 according to manufacturers’ instruction.
RNA interference and plasmid construction
MEFs were transfected with 2 μg plasmids containing short hairpin mouse Riok3 RNA(shmRiok3) or control shRNA sequences for ~40 hours in 12-well plates. The transfection was performed according to the manufacturer’s instruction of lipofectamine 2000. Riok3 cDNA and TRIM40 plasmids were obtained from DNASU plasmid repository and Vigene biosciences respectively. Full length and truncated cDNAs were PCR amplified and subcloned into pCMV-FLAG, pCMV-MYC vectors using MSCI and XhoI sites. The expression plasmids of MDA5, RIG-I, and HA-ubiquitination plasmid were kind gifts from Zhiyong Liu (Institute of Immunology, Zhejiang University). All those constructed plasmids were sequenced and aligned. For protein expression test, plasmids were transfected into HEK293T cells using Lipofectamine 2000 regentsaccording to the manufacturer’s instructions.
RT-q-PCR
Total RNA was extracted with the Ultrapure RNA Kit (CWbiotech). The first-strand cDNA was synthesized using the HiFiScript 1st Strand cDNA Synthesis Kit(CWbiotech), and q-PCR was performed with the iTaq Universal SYBR Green Supermix (Bio-Rad) on CFX96 Touch quantitative Real-Time PCR Detection System (Bio-Rad).
ELISA
Cell supernatants were collected 12 hours after a viral infection or reagent stimulation. The concentrations of IFN-β, TNF-α, and IL-6 were measured by enzyme-linked immunosorbent assay according to the manufacturer’s instructions.
Immunoblotting and immunoprecipitation
For immunoblotting, cells were directly lysed in the plate wells on ice with a mixture containing 1 × RIPA buffer, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1 × protein loading buffer. The lysates were collected and boiled at 100°C. Denatured proteins were separated by SDS-PAGE, transferred onto PVDF membrane, and blocked with milk at room temperature. After probed with the primary antibodies and the secondary antibody, the protein signal detection was performed with the FluorChem E System (Protein Simple, Santa Clara, CA). For immunoprecipitation, cells transfected with the indicated plasmids were lysed in lysis buffer (50 mM Tris-HCl [pH 7.5], 250 mM NaCl, 0.5% NP-40, 1 mM EDTA) supplemented with 0.1% protease inhibitor cocktail (Sigma-Aldrich, Oakville, Canada). Proteins were then immunoprecipitated at 4°C overnight with either anti-myc (9E10, SigmaAldrich) or anti-Flag (M2, Sigma-Aldrich) agarose beads. After three washes with lysis buffer, the immunoprecipitates were boiled and analyzed by immunoblotting.
Hematoxylin and Eosin (HE) staining and immunohistochemistry
Lungs from PBS or IAV-infected mice were dissected, fixed in 10% phosphate-buffered formalin, and sent to Servicebio Inc. (Wuhan, China). HE staining and immunohistochemical staining were performed using standard procedures. For Immunohistochemistry, the H1N1 NP antibody (Cat. No. A01506, GenScript) was used to mark IAV viral gene expression.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Results were expressed as the mean ± SEM. Statistical significance was determined using a two-tailed Student’s t test. For the mouse survival study, Kaplan-Meier survival curve was generated by GraphPad Prism 5.0.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Phospho-NFκB p65 (Ser536) | Cell Signaling Technology | Cat#3033S; RRID:AB_331284 |
| Rabbit anti-NFκB p65 | Cell Signaling Technology | Cat #8242S; RRID:AB_10859369 |
| Rabbit anti-Phospho-TBK1/NAK (Ser172) | Cell Signaling Technology | Cat#5483S; RRID:AB_10693472 |
| Rabbit anti-TBK1/NAK(D1B4) | Cell Signaling Technology | Cat#3504S; RRID:AB_2255663 |
| Rabbit anti-Phospho-IRF3(Ser396) | Cell Signaling Technology | Cat#4947S; RRID:AB_823547 |
| Rabbit anti-IRF3 | Cell Signaling Technology | Cat#4302S; RRID:AB_1904036 |
| Rabbit anti-Riok3 | Proteintech | Cat#13593-1-AP; RRID:AB_2178105 |
| Rabbit anti-RIG-1/DDX58 | Proteintech | Cat#20566-1-AP; RRID:AB_10700006 |
| Rabbit anti-IFIH1/MDA5 | Proteintech | Cat#21775-1-AP; RRID:AB_10734593 |
| Rabbit anti-TRIM40/RNF35 | Abcam | Cat#ab156583; RRID:AB_1524323 |
| Rabbit anti-β-actin | Cell Signaling Technology | Cat#4970S; RRID:AB_2223172 |
| Rabbit anti-Phospho-IκBBα (Ser32) | Cell Signaling Technology | Cat#2859S; RRID:AB_561111 |
| Mouse anti-NFκBα | Cell Signaling Technology | Cat#4814S; RRID:AB_390781 |
| Rabbit anti-Phospho-p44/42 MAPK (Thr202/Tyr204) | Cell Signaling Technology | Cat#4370S; RRID:AB_2315112 |
| Rabbit anti-p44/42 MAPK | Cell Signaling Technology | Cat#9102S; RRID:AB_330744 |
| Mouse anti-Phospho-SAPK/JNK (Thrl83/Tyrl85) | Cell Signaling Technology | Cat#9255S; RRID:AB_2307321 |
| Rabbit anti-SAPK/JNK | Cell Signaling Technology | Cat#9258S; RRID:AB_2141027 |
| Rabbit anti-Phospho-p38MAPK (Thrl80/Tyrl82) | Cell Signaling Technology | Cat#9211S; RRID:AB_331641 |
| Rabbit anti-p38 MAPK | Cell Signaling Technology | Cat#9212S; RRID:AB_330713 |
| Rabbit anti-MAVS | Proteintech | Cat#14341-1-AP; RRID:AB_10548408 |
| Mouse anti-HA-Tag | Cell Signaling Technology | Cat#2367S; RRID:AB_10691311 |
| Rabbit anti-Myc-Tag | Cell Signaling Technology | Cat#2272S; RRID:AB_10692100 |
| Rabbit anti-DYKDDDDK Tag | Cell Signaling Technology | Cat#2368S; RRID:AB_2217020 |
| K48-linkage Specific Polyubiquitin (D9D5) Rabbit mAb | Cell Signaling Technology | Cat#8081; RRID:AB_10859893 |
| Rabbit Ubiquitin Antibody | Cell Signaling Technology | Cat#3933; RRID:AB_2180538 |
| Rabbit anti-GAPDH | Proteintech | Cat#10494-1-AP; RRID:AB_2263076 |
| Rabbit Anti-parkin | Proteintech | Cat#14060-1-AP; RRID:AB_2878005 |
| Rabbit Anti-TRIM59 | Affinity | Cat#DF3343; RRID:AB_2835710 |
| Rabbit anti-RNF125 | Abcam | Cat#ab211450; RRID:AB_1524323 |
| H1N1 NP antibody | GenScript | Cat#A01506; RRID:AB_1968881 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| VSV | Chinese CDC | N/A |
| SeV | Chinese CDC | N/A |
| HSV-1 | Chinese CDC | N/A |
| IAV-H1N1-PR8(Puerto-Rico/8) strain | Chinese CDC | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Lipofectamine 2000 | ThermoFisher | Cat#11668019 |
| M-CSF | Peprotech | Cat#315-02 |
| 3′3’-cGAMP | Sigma | Cat#SML1232 |
| Poly(I:C, LMW) | InvivoGen | Cat#tlrl-picw-250 |
| Poly(I:C, HMW) | InvivoGen | Cat#tlrl-pic-5 |
| IFN-β | InvivoGen | Cat#mabg2-hifnb-3 |
| TNF-α | eBioscience | Cat#88-7324-88 |
| IL-6 | eBioscience | Cat#BMS603-2 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T cells | ATCC | ATCC CRL11268 |
| MDCK cells | ATCC | ATCC CCL-34 |
| Vero cells | ATCC | ATCC CCL-81 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| mouse: Riok3F/F transgenic C57BL/6 | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Riok3-Flox qRT-PCR primer F:ACTCAGCCATGCAAAAGAAGCACAC | Invitrogen | N/A |
| Riok3-Flox qRT-PCR primer R:AGCCAGCTTCAAACCCACAG | Invitrogen | N/A |
| mTNFa qRT-PCR primer F:CTGGGACAGTGACCTGGACT | Invitrogen | N/A |
| mTNFa qRT-PCR primer R:GCACCTCAGGGAAGAGTCTG | Invitrogen | N/A |
| mIL-6 qRT-PCR primer F:AGTTGCCTTCTTGGGACTGA | Invitrogen | N/A |
| mIL-6 qRT-PCR primer R:TCCACGATTTCCCAGAGAAC | Invitrogen | N/A |
| mIFNα4 qRT-PCR primer F:TACTCAGCAGACCTTGAACCT | Invitrogen | N/A |
| mIFNα4 qRT-PCR primer R:CAGTCTTGGCAGCAAGTTGAC | Invitrogen | N/A |
| mIFNβ qRT-PCR primer F:ATGAGTGGTGGTTGCAGGC | Invitrogen | N/A |
| mIFNβ qRT-PCR primer R:TGACCTTTCAAATGCAGTAGATTCA | Invitrogen | N/A |
| Cre qRT-PCR primer F:CCCAGAAATGCCAGATTACG | Invitrogen | N/A |
| Cre qRT-PCR primer R:CTTGGGCTGCCAGAATTTCT | Invitrogen | N/A |
| shRiok3 shRNA:GCTCAGATGCTACAGATGGAA | Invitrogen | N/A |
| shCtrl shRNAs:TTCTCCGAACGTGTCACGT | Invitrogen | N/A |
| VSV-G qRT-PCR primer F:ACGGCGTACTTCCAGATGG | Invitrogen | N/A |
| VSV-G qRT-PCR primer R:CTCGGTTCAAGATCCAGGT | Invitrogen | N/A |
| SeV-M qRT-PCR primer F:GTGATTTGGGCGGCATCT | Invitrogen | N/A |
| SeV-M qRT-PCR primer R:GATGGCCGGTTGGAACAC | Invitrogen | N/A |
| IAV-M qRT-PCR primer F:ATGAGCCTTCTAACCGAGGTCGAAACG | Invitrogen | N/A |
| IAV-M qRT-PCR primer R:TGGACAAAACGTCTACGCTGCAG | Invitrogen | N/A |
| mβ-actin qRT-PCR primer F:GTATCCTGACCCTGAAGTACC | Invitrogen | N/A |
| mβ-actin qRT-PCR primer R:TGAAGGTCTCAAACATGATCT | Invitrogen | N/A |
| mOAS1 qRT-PCR primer F:GCCTGATCCCAGAATCTATGC | Invitrogen | N/A |
| mOAS1 qRT-PCR primer R:GAGCAACTCTAGGGCGTACTG | Invitrogen | N/A |
| mISG15 qRT-PCR primer F:GGTGTCCGTGACTAACTCCAT | Invitrogen | N/A |
| mISG15 qRT-PCR primer R:TGGAAAGGGTAAGACCGTCCT | Invitrogen | N/A |
| mMX1 qRT-PCR primer F:GACCATAGGGGTCTTGACCAA | Invitrogen | N/A |
| mMX1 qRT-PCR primer R:AGACTTGCTCTTTCTGAAAAGCC | Invitrogen | N/A |
| mMX2 qRT-PCR primer F:GAGGCTCTTCAGAATGAGCAAA | Invitrogen | N/A |
| mMX2 qRT-PCR primer R:CTCTGCGGTCAGTCTCTCT | Invitrogen | N/A |
| hGapdh qRT-PCR primer F:TGCACCACCAACTGCTTAGC | Invitrogen | N/A |
| hGapdh qRT-PCR primer R:GGCATGGACTGTGGTCATGAG | Invitrogen | N/A |
| hIFNβ qRT-PCR primer F:GCTTGGATTCCTACAAAGAAGCA | Invitrogen | N/A |
| hIFNβ qRT-PCR primer R:ATAGATGGTCAATGCGGCGTC | Invitrogen | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| TRIM40 cDNA | Vigene biosciences | Cat#CH819489 |
| Riok3 cDNA | DNASU plasmid | N/A |
| pCMV-FLAG | Addgene | N/A |
| pCMV-MYC | Addgene | Cat#631604 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | ImageJ | https://imagej.net/Welcome |
| Graphpad 6.0 | Prism | https://www.graphpad.com/ |
Highlights.
Riok3 negatively and selectively inhibits RNA viruses both in vivo and in vitro
Riok3 directly interacts with TRIM40, RIG-I, and MDA5 to form a protein complex
Riok3 promotes TRIM40-mediated K27- and K48-linked ubiquitination of RIG-I and MDA5
TRIM40 induces the proteasomal degradation of both RIG-I and MDA5
ACKNOWLEDGMENTS
We appreciate Dr. Zhiyong Liu (Institute of Immunology, Zhejiang University) for his kind gifts of plasmids for RIG-I, MDA5, and hemagglutination (HA)-ubiquitination expression. This work was supported by grants from the National Natural Science Foundation of China (No. 81902046 to Y.S.) and National Institutes of Health (DE019085 and CA091791 to T.W. and R.S.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests. None of the contents of this manuscript has been previously published or is under consideration elsewhere. All the authors read and approved the final version of the manuscript before submission.
INCLUSION AND DIVERSITY
We worked to ensure diversity in experimental samples through the selection of the cell lines.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109272.
REFERENCES
- Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, and Shimotohno K. (2007). Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 104, 7500–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, and Barton GM (2011). Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol 29, 185–214. [DOI] [PubMed] [Google Scholar]
- Baumas K, Soudet J, Caizergues-Ferrer M, Faubladier M, Henry Y, and Mougin A. (2012). Human RioK3 is a novel component of cytoplasmic pre-40S pre-ribosomal particles. RNA Biol. 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena C, Ahmad S, Xavier A, Willemsen J, Park S, Park JW, Oh SW, Fujita T, Hou F, Binder M, and Hur S. (2019). Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 177, 1187–1200.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, Wang Q, Li D, Wang J, Zheng P, et al. (2013). Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152, 467–478. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, and Förster I. (1999). Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277. [DOI] [PubMed] [Google Scholar]
- Feng J, De Jesus PD, Su V, Han S, Gong D, Wu NC, Tian Y, Li X, Wu TT, Chanda SK, and Sun R. (2014). RIOK3 is an adaptor protein required for IRF3-mediated antiviral type I interferon production. J. Virol 88, 7987–7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, and Maniatis T. (2003). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol 4, 491–496. [DOI] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, and Jung JU (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. [DOI] [PubMed] [Google Scholar]
- Gao D, Yang YK, Wang RP, Zhou X, Diao FC, Li MD, Zhai ZH, Jiang ZF, and Chen DY (2009). REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS ONE 4, e5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale NS, McIntyre ABR, Mattocks MD, Holley CL, Lazear HM, Mason CE, and Horner SM (2020). Altered m6A Modification of Specific Cellular Transcripts Affects Flaviviridae Infection. Mol. Cell 77, 542–555.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaïdane H, and Hober D. (2008). Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab. 34, 537–548. [DOI] [PubMed] [Google Scholar]
- Kalinina T, Güngör C, Thieltges S, Möller-Krull M, Penas EM, Wicklein D, Streichert T, Schumacher U, Kalinin V, Simon R, et al. (2010). Establishment and characterization of a new human pancreatic adenocarcinoma cell line with high metastatic potential to the lung. BMC Cancer 10, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, and Fujita T. (2014). Autoimmunity caused by constitutive activation of cytoplasmic viral RNA sensors. Cytokine Growth Factor Rev. 25, 739–743. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. (2006). Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, and Akira S. (2008). Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med 205, 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, and Akira S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol 11, 373–384. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, and Akira S. (2005). IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol 6, 981–988. [DOI] [PubMed] [Google Scholar]
- Kimmelman AC, Hezel AF, Aguirre AJ, Zheng H, Paik JH, Ying H, Chu GC, Zhang JX, Sahin E, Yeo G, et al. (2008). Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc. Natl. Acad. Sci. USA 105, 19372–19377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer S, and Lee-Kirsch MA (2017). Type I interferon-mediated auto-inflammation and autoimmunity. Curr. Opin. Immunol 49, 96–102. [DOI] [PubMed] [Google Scholar]
- Kuniyoshi K, Takeuchi O, Pandey S, Satoh T, Iwasaki H, Akira S, and Kawai T. (2014). Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc. Natl. Acad. Sci. USA 111, 5646–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Qian C, and Cao X. (2016). Post-Translational Modification Control of Innate Immunity. Immunity 45, 15–30. [DOI] [PubMed] [Google Scholar]
- Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, Wang P, Zhao K, Hou J, Wang X, et al. (2017). The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol 18, 214–224. [DOI] [PubMed] [Google Scholar]
- Loo YM, and Gale M Jr. (2011). Immune signaling by RIG-I-like receptors. Immunity 34, 680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, García-Sastre A, Katze MG, and Gale M Jr. (2008). Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol 82, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvar B, Raghuram V, Pisitkun T, Sarkar A, and Knepper MA (2016). Comprehensive database of human E3 ubiquitin ligases: application to aquaporin-2 regulation. Physiol. Genomics 48, 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Gaudieri S, Rhodes DA, and Trowsdale J. (2003). Cluster of TRIM genes in the human MHC class I region sharing the B30.2 domain. Tissue Antigens 61, 63–71. [DOI] [PubMed] [Google Scholar]
- Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels HH, Ori D, Uehata T, Tartey S, Akira S, Suzuki Y, et al. (2015). Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell 161, 1058–1073. [DOI] [PubMed] [Google Scholar]
- Morreale FE, and Walden H. (2016). Types of Ubiquitin Ligases. Cell 165, 248–248.e1. [DOI] [PubMed] [Google Scholar]
- Narayan K, Waggoner L, Pham ST, Hendricks GL, Waggoner SN, Conlon J, Wang JP, Fitzgerald KA, and Kang J. (2014). TRIM13 is a negative regulator of MDA5-mediated type I interferon production. J. Virol 88, 10748–10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, and Bowie AG (2013). Immune sensing of DNA. Immunity 38, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran Y, Zhang J, Liu LL, Pan ZY, Nie Y, Zhang HY, and Wang YY (2016). Autoubiquitination of TRIM26 links TBK1 to NEMO in RLR-mediated innate antiviral immune response. J. Mol. Cell Biol 8, 31–43. [DOI] [PubMed] [Google Scholar]
- Rehwinkel J, and Reis e Sousa C. (2010). RIGorous detection: exposing virus through RNA sensing. Science 327, 284–286. [DOI] [PubMed] [Google Scholar]
- Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, Rice GI, Erlandsen H, Kehl HG, Thiele H, et al. (2015). A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet 96, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, and Chen ZJ (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. [DOI] [PubMed] [Google Scholar]
- Shan J, Wang P, Zhou J, Wu D, Shi H, and Huo K. (2009). RIOK3 interacts with caspase-10 and negatively regulates the NF-kappaB signaling pathway. Mol. Cell. Biochem 332, 113–120. [DOI] [PubMed] [Google Scholar]
- Singleton DC, Rouhi P, Zois CE, Haider S, Li JL, Kessler BM, Cao Y, and Harris AL (2015). Hypoxic regulation of RIOK3 is a major mechanism for cancer cell invasion and metastasis. Oncogene 34, 4713–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirota M, Schaub MA, Batzoglou S, Robinson WH, and Butte AJ (2009). Autoimmune disease classification by inverse association with SNP alleles. PLoS Genet. 5, e1000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell LM, McGaha TL, and Brooks DG (2017). Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 38, 542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Watanabe M, Nakamaru Y, Takagi D, Takahashi H, Fukuda S, and Hatakeyama S. (2016). TRIM39 negatively regulates the NFκB-mediated signaling pathway through stabilization of Cactin. Cell. Mol. Life Sci 73, 1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima K, Oshiumi H, Takaki H, Matsumoto M, and Seya T. (2015). RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 11, 192–200. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, and Akira S. (2009). Innate immunity to virus infection. Immunol. Rev 227, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao B, Jin W, Xu J, Liang Z, Yao J, Zhang Y, Wang K, Cheng H, Zhang X, and Ke Y. (2014). Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J. Immunol 193, 2801–2811. [DOI] [PubMed] [Google Scholar]
- Tariki M, Wieczorek SA, Schneider P, Bä nfer S, Veitinger S, Jacob R, Fendrich V, and Lauth M. (2013). RIO kinase 3 acts as a SUFU-dependent positive regulator of Hedgehog signaling. Cell. Signal. 25, 2668–2675. [DOI] [PubMed] [Google Scholar]
- Tomar D, and Singh R. (2015). TRIM family proteins: emerging class of RING E3 ligases as regulator of NF-κB pathway. Biol. Cell 107, 22–40. [DOI] [PubMed] [Google Scholar]
- Van Eyck L, De Somer L, Pombal D, Bornschein S, Frans G, Humblet-Baron S, Moens L, de Zegher F, Bossuyt X, Wouters C, and Liston A. (2015). Brief Report: IFIH1 Mutation Causes Systemic Lupus Erythematosus With Selective IgA Deficiency. Arthritis Rheumatol. 67, 1592–1597. [DOI] [PubMed] [Google Scholar]
- von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, and Snel B. (2003). STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zhao W, Zhao K, Zhang L, and Gao C. (2015). TRIM26 negatively regulates interferon-β production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 11, e1004726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Jiang M, Liu S, Zhang S, Liu W, Ma Y, Zhang L, Zhang J, and Cao X. (2016). RNF122 suppresses antiviral type I interferon production by targeting RIG-I CARDs to mediate RIG-I degradation. Proc. Natl. Acad. Sci. USA 113, 9581–9586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Li Q, Mao AP, Hu MM, and Shu HB (2014). TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J. Mol. Cell Biol 6, 154–163. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, and Fujita T. (2009). RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev 227, 54–65. [DOI] [PubMed] [Google Scholar]
- Zhang L, Flygare J, Wong P, Lim B, and Lodish HF (2011). miR-191 regulates mouse erythroblast enucleation by down-regulating Riok3 and Mxi1. Genes Dev. 25, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Ji D, Wang P, Liang D, Jin L, Shi H, Liu X, Meng Q, Yu R, and Gao S. (2018). The atypical protein kinase RIOK3 contributes to glioma cell proliferation/survival, migration/invasion and the AKT/mTOR signaling pathway. Cancer Lett. 415, 151–163. [DOI] [PubMed] [Google Scholar]
- Zhao K, Zhang Q, Li X, Zhao D, Liu Y, Shen Q, Yang M, Wang C, Li N, and Cao X. (2016). Cytoplasmic STAT4 Promotes Antiviral Type I IFN Production by Blocking CHIP-Mediated Degradation of RIG-I. J. Immunol 196, 1209–1217. [DOI] [PubMed] [Google Scholar]
- Zhao C, Jia M, Song H, Yu Z, Wang W, Li Q, Zhang L, Zhao W, and Cao X. (2017). The E3 Ubiquitin Ligase TRIM40 Attenuates Antiviral Immune Responses by Targeting MDA5 and RIG-I. Cell Rep. 21, 1613–1623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or code.