

Abstract
Benzene is a known genotoxic carcinogen linked to many hematological abnormalities. S-phenylmercapturic acid (PHMA, N-acetyl-S-(phenyl)-L-cysteine, CAS# 4775-80-8) is a urinary metabolite of benzene and is used as a biomarker to assess benzene exposure. Pre-S-phenylmercapturic acid (pre-PHMA) is a PHMA precursor that dehydrates to PHMA at acidic pH. Published analytical methods that measure urinary PHMA adjust urine samples to a wide range of pH values using several types of acid, potentially leading to highly variable results depending on the concentration of pre-PHMA in a sample. Information is lacking on the variation in sample preparation among laboratories regularly measuring PHMA and the effect of those differences on PHMA quantitation in human urine samples. To investigate the differences in PHMA quantitation, we conducted an inter-laboratory comparison that included the analysis of 50 anonymous human urine samples (25 self-identified smokers and 25 self-identified non-smokers), quality control samples and commercially available reference samples in five laboratories using different analytical methods. Observed urinary PHMA concentrations were proportionally higher at lower pH, and results for anonymous urine samples varied widely among the methods. The method with the neutral preparation pH yielded results about 60% lower than the method using the most acidic conditions. Samples spiked with PHMA showed little variation, suggesting that the variability in results in human urine samples across methods is driven by the acid-mediated conversion of pre-PHMA to PHMA.
Introduction
Benzene is a known genotoxic carcinogen linked to many hematological abnormalities including acute myeloid and acute non-lymphocytic leukemia (1–4). It is a starting material for a variety of synthetic resins, plastics and fibers. Sources of occupational exposure to benzene include manufacturing processes such as paint, tire and rubber production and the petrochemical industry. Other environmental sources of benzene include traveling in or proximity to combustion vehicles and pumping gasoline (1, 5–7). However, tobacco smoke is the greatest single source of benzene exposure in the population (5, 8) as would be expected because of the microgram quantities of benzene found in the mainstream smoke of a single cigarette (9). Previous studies showed that S-phenylmercapturic acid (PHMA), a urinary biomarker of benzene exposure, is elevated in cigarette smokers (10–12). PHMA levels have been quantified in the general population in studies such as the US National Health and Nutrition Examination Survey (NHANES) (13), the Canadian Health Measures Survey (CHMS) (14), the Population Assessment of Tobacco and Health study (15) and in workers to monitor occupational exposure (6, 16, 17). There remains a compelling need for robust PHMA analysis to assess human exposure to benzene.
A variety of urine sample preparation approaches (17–20) are described in the literature. Discrepancies in quantitative urinary PHMA values due to differences in sample preparation are well documented (21, 22). Pre-PHMA is a benzene oxide metabolite in the glutathione metabolic pathway that undergoes acid dehydration to form PHMA (6, 23, 24) (Figure 1). The presence of pre-S-phenylmercapturic acid (pre-PHMA) in urine can influence PHMA quantitation. The ratio of pre-PHMA to PHMA varies from person to person, and the conversion from pre-PHMA to PHMA is sensitive to pH. Therefore, conversion of pre-PHMA to the more stable PHMA is a typical analytical approach.
Figure 1.
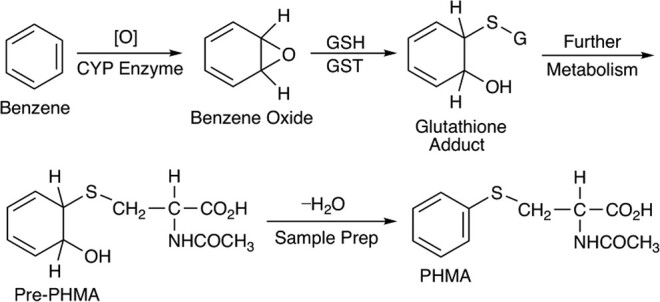
Metabolism of benzene to pre-PHMA followed by dehydration to PHMA during sample prep. CYP, Cytochrome P-450; GSH, Glutathione; GST, Glutathione S-transferase.
Estimates of the percentage of pre-PHMA as a portion of total PHMA in urine vary from a low of 75% to a high of 99% in exposed workers and smokers (21, 22). Paci et al. (22) measured urinary PHMA by treating urine samples with a strong mineral acid (9M H2SO4, presumed full conversion of pre-PHMA to PHMA), an organic acid (glacial acetic acid, presumed partial conversion) and without pH adjustment (free PHMA). They found that total urinary PHMA concentration increased with decreasing pH, with pH 2 capturing, on average, 44% of the total PHMA. Sterz et al. (25) measured PHMA in samples from occupationally exposed individuals using four different pH conditions: untreated, formic acid (pH 2), HCl (pH ∼1) and sulfuric acid (pH ∼0.6). They also observed that PHMA increased as pH decreased. However, Sterz et al. suggested that the type of acid used to achieve the pre-PHMA to PHMA conversion may also influence quantitation. They found that while the presumed pre-PHMA mass spectral peak (m/z 256→109) disappeared at pH ∼1 after successive additions of 37% HCl, samples prepared in concentrated H2SO4 (pH ∼0.6) had significantly higher PHMA concentration than samples prepared with 37% HCl (pH 0.5–1.0). They suggested unidentified side reactions and/or other sources of PHMA as the cause of the increase and advised adjusting the pH of urine samples to 0.5–1.0 by adding HCl to ensure complete conversion of pre-PHMA to PHMA.
While several groups have explored the effects of sample preparation pH on PHMA quantitation (22, 25), a systematic, multi-laboratory study to evaluate these differences using a single set of samples has not been published. A small, two-laboratory comparison found agreement between two methods analyzing the same set of 149 urine samples from petrochemical workers (26). The authors concluded that their results assured the comparability of their previous studies that used these two methods. However, the difference in sample preparation pH between the two methods was small relative to the difference in pH observed in other published methods. An existing global effort to support laboratories that quantitate urinary PHMA is the German External Quality Assessment Scheme (G-EQUAS), a program maintained by the University of Erlangen-Nuremberg. Unfortunately, this program does not collect information from participating laboratories regarding the method used to prepare samples for PHMA quantitation. It is not clear whether G-EQUAS samples contain levels of pre-PHMA high enough to mimic an exposed population. These drawbacks limit its usefulness in understanding how differences in samples preparation affect PHMA results.
Given that sample pH has a large, documented impact on urinary PHMA concentration and the possibility that other factors may also influence quantitation [e.g., type of acid used to achieve conversion, unidentified sources of PHMA (25)], we asked five laboratories using different liquid chromatography–tandem mass spectrometry (LC–MS-MS) methods to analyze the same set of samples. We invited laboratories that currently and/or regularly analyze human urine samples for PHMA for benzene exposure studies. The methods included an assay used by the Centers for Disease Control and Prevention (CDC) to measure urinary PHMA for NHANES cycles 2005–2006 and 2011–2012 through 2015–2016 (method A) (18), a recently developed benzene metabolite assay by CDC (method B) (27), the assay used in the CHMS cycles 2–4 (method C) (28) and two other assays used by laboratories that regularly investigate benzene exposure in the population, method D (29) (modified to include an initial acid treatment step) and method E (20). We supplied each laboratory with 50 anonymous human urine samples (25 self-identified smokers and 25 self-identified non-smokers), spiked quality control (QC) materials and a commercially available reference material. We compared accuracy and precision across the methods using the QC pools and reference material and assessed agreement of urinary PHMA results across methods using the anonymous human urine samples.
Methods
Preparation of human urine samples
Fifty unique, spot urine samples (10 mL) from 25 self-identified non-smokers and 25 self-identified smokers (Tennessee Blood Services, Memphis, TN, USA) were thawed to room temperature and placed on a rotating mixer for 15 minutes. Samples were then aliquoted into 5- or 2-mL cryogenic vials (P/N 1050027 and P/N 1050026, ThermoFisher Scientific, Waltham, MA, USA) using a Microlab STAR automated liquid handling system (Hamilton, Reno, NV, USA).
Preparation of QC pools
QC pools consisted of spiked human urine anonymously collected at CDC (IRB Approved Protocol #3994). Urine samples were collected and measured for urinary PHMA using Method B before pooling and spiking. All samples were found to have PHMA levels below the limit of detection (LOD), and the spiked urines were within 9% of the targeted concentrations. Selected urine samples were pooled (∼400 mL each) into a low (QCL) and high portion (QCH) and shipped to an external ISO 17034 and ISO/IEC 17025 certified lab (o2si, Charleston, SC, USA) for spiking and aliquoting. Urine pools were spiked by o2si with a PHMA intermediate stock solution prepared in methanol to bring the pool concentrations within the lower and upper reportable ranges of participating laboratories. The PHMA intermediate stock solution was prepared by o2si from neat PHMA (CAS 4775-80-8, catalogue #P33560, lot #4-TKN-68-1, Toronto Research Chemicals, Toronto, ON, Canada).
We characterized the QC pools by inspecting the distribution of results from both high and low pools across 20 characterization runs. Subsequent analysis of the slope of the QC pool concentration measurements ordered by analysis date yielded a 95% confidence interval (CI) of -0.0102 to 0.0654 for the QCH slope estimate and 0.00141–0.0132 for the QCL slope estimate. The slope estimate for QCH pool included zero and the slope estimate for QCL pool was negligible; therefore, we concluded that the pool was homogeneous for the purpose of this study.
Reference samples
We purchased ClinChek Parameters for Occupational Medicine Level I and II (product #8922, lot #1316, RECIPE Chemicals + Instruments, Munich, Germany) from Iris Technologies International (Olathe, KS, USA). We requested that participating laboratories reconstitute and store the lyophilized reference samples according to the manufacturer’s instructions. Mean values and control ranges for each level were based on the material lot documentation. These values were established using results from independent reference laboratories using chromatographic techniques following the Guideline of the German Medical Association on Quality Assurance (30).
Sample analysis and reporting
Laboratories received the following samples frozen on dry ice: 50 anonymous human urine samples (laboratories were blind to self-identified smoking status), two QC pools and a lyophilized reference material. We asked laboratories to measure PHMA in the 50 urine samples and report one result for each sample using their typical analytical, QC and reporting practices. We requested that laboratories analyze each QC pool three times, each from an unopened vial, over at least 5 days and ClinChek Level I and II samples at least three and no more than five times in different analytical runs, from the same ampoule. Results were compiled by the CDC laboratory and combined into a single file for statistical analysis.
Statistical analysis
Statistical analyses were performed using RStudio 1.1.423 (R Foundation for Statistical Computing, Vienna, Austria). We checked for the presence of outliers in both QC pools using the Grubbs test. Two outliers were identified in the QCL pool for method A and were removed before subsequent analysis. We evaluated precision using the coefficient of variance (%CV) (31) calculated from two QC pools run in triplicate across five analytical batches. Intra-run %CV was calculated for QCL and QCH based on the intra-run mean of the three sample results. Inter-run %CV was calculated by averaging intra-run means across the five batches. For unknown samples, results less than the LOD were imputed (32) using each method’s reported LOD/ . One exceedingly high sample (R503543 in Supplemental Table 1) was removed from the dataset due to a large influence on regression estimates.
. One exceedingly high sample (R503543 in Supplemental Table 1) was removed from the dataset due to a large influence on regression estimates.
We compared result means for each method using a Student’s two-sided t-test with Tukey post-hoc P-value adjustment (P < 0.05). We then performed a second analysis on a scatter plot of paired results using a linear weighted (1/x2) Deming regression (33) with the mcreg function in R’s mcr package. The Deming regression accounts for imprecision present in both axes using a variance ratio (34). We averaged the %CV values from QCL and QCH for each method in the pairwise comparison and ratioed them to obtain the variance ratio. Similarity between methods was assessed by comparing slope and intercept estimates and 95% CI and Pearson correlation values from the Deming regression.
Results
We analyzed results from the five participating laboratories, each of which used different LC–MS-MS methods (identified as methods A–E) to quantify PHMA in identical urine aliquots. We summarized each method’s sample preparation parameters (Table I) and analytical parameters (Table II). In addition, we calculated the precision and accuracy of each method, and we used the human urine samples to ascertain differences and trends in calculated PHMA values between them.
Table I.
Selected Sample Preparation Parameters of Urinary PHMA Methods
| Method | |||||
|---|---|---|---|---|---|
| Parameter | A | B | C | D | E |
| Sample preparation | Dilute and shoot | Dilute and shoot | SPE | SPE | Liquid/liquid extraction |
| Sample treatment pHa | 6.8 | 2.9 | 1.4 | ∼0.5 | ∼4.5 |
| Treatment time (min) | – | – | No treatment time | 10 | 10 |
| Acid used for sample pretreatment | – | – | 4% H3PO4 | 36% HCl | KH2PO4 |
| Sample diluent | 15 mM NH4OAc | 5 mM NH4HCO2, 0.15% HCOOH | – | – | – |
| Reconstitution solvent | – | – | 0.05% CH3COOH | 30% methanol in 15 mM NH4OAc | Methanol |
Used diluent pH for dilute-and-shoot methods. “–” is not applicable.
Table II.
Selected Analytical Parameters of Urinary PHMA Methods
| Method | A | B | C | D | E |
|---|---|---|---|---|---|
| Sample volume (mL) | 0.050 | 0.050 | 0.400 | 0.200 | 0.300 |
| Internal standard | PHMA-13C6 | PHMA-13C6 | PHMA-13C6 | PHMA-d5 (phenyl) | PHMA-d3 (acetyl) |
| PHMA standard manufacturer (lot#) | Toronto Research Chemicals (7-ECGW-117-1) | Toronto Research Chemicals (4-TKN-68-1) | Cambridge Isotope Laboratories (PR-15201) | Toronto Research Chemicals (7-ECGW-115-2) | Tokyo Chemical Industry Co., Ltd (not provided) |
| Mobile phase A | 15 mM NH4OAc | 5 mM NH4HCO2 | 0.05% CH3COOH | 15 mM NH4OAc | Water |
| Mobile phase B | Acetonitrile | Methanol | Acetonitrile | Methanol | Methanol |
| Column | Waters HSS T3, 100Å, 1.8 µm, 2.1 mm × 150 mm |
Waters HSS PFP, 100Å, 1.8 µm, 2.1 mm × 100 mm |
Waters HSS T3, 100Å 1.8 µm, 2.1 mm × 50 mm |
Phenomenex Synergi Max-RP, C12, 100Å 2.5 µm, 3.0 mm × 50 mm | Phenomenex Synergi Polar RP, 80Å, 4 µm, 4.6 mm × 150 mm |
| Source/mode | ESI/– | ESI/– | ESI/– | APCI/– | EC-APCI/– |
Laboratory method parameters
The participating laboratories used a wide range of sample preparation techniques (Table I). Two laboratories (A–B) used direct injection after dilution (dilute-and-shoot) methods, two used solid-phase extraction (SPE, C–D) and one used liquid/liquid extraction and derivatization (E). Methods used a wide pH range in the acid-treatment step or in the sample diluent, ranging from ∼0.5 to 6.8. Method A used a sample diluent with the highest pH (6.8) and method B used a sample diluent with the median pH (2.9). Two methods, C (pH 1.4) and D (pH ∼0.5), used a strong acid-treatment step but used different acids, H3PO4 and HCl, respectively. Method E’s extraction process used KH2PO4 during extraction at pH ∼4.5. In the dilute-and-shoot methods, samples were prepared (1:9) in sample diluent and held at the diluent pH prior to analysis. In contrast, samples in methods using SPE were acid-treated for a specific time and then pH-adjusted before injection onto the instrument.
All laboratories used LC–MS-MS in negative-ion mode to measure PHMA using the same quantitative transition, m/z 238 → 109. Selected analytical parameters of laboratory methods are summarized in Table II. Methods A, B and C used electrospray ionization as the ionization source and PHMA-13C6 as the internal standard. Methods D and E used atmospheric pressure chemical ionization (APCI) and electron capture APCI, respectively, and two different deuterated internal standards.
Precision
We evaluated method precision using the two spiked QC pools and the ClinChek reference material. One of the five methods did not report one of the individual QCH results for Day 2. All methods reported QC results greater than their LOD. We assessed precision using two QC pools run in triplicate across 5 days. Intra-run precision was calculated from QC pool replicate results in the same run (Supplemental Tables 2 and 3). Inter-run precision is summarized in Table III. The range of CVs for QCL and QCH were 3.08–15.7% and 1.83–11.9%, respectively. Methods with the highest CV for QCL and QCH were A (14.5% and 11.9%, respectively) and E (15.7% and 8.27%, respectively).
Table III.
Inter-day Variation for PHMA Results (µg/L) by Method and QC Levela
| Pool | Method | Mean (µg/L) | SD | CV (%) |
|---|---|---|---|---|
| A | 1.41 | 0.204 | 14.5 | |
| B | 1.03 | 0.060 | 5.78 | |
| QCL | C | 0.934 | 0.029 | 3.08 |
| D | 1.05 | 0.050 | 4.77 | |
| E | 1.12 | 0.176 | 15.7 | |
| Overall | 1.11 | 0.180 | 16.2 | |
| A | 11.6 | 1.38 | 11.9 | |
| B | 13.1 | 0.255 | 1.94 | |
| QCH | C | 11.2 | 0.482 | 4.31 |
| D | 14.0 | 0.256 | 1.83 | |
| E | 12.7 | 1.05 | 8.27 | |
| Overall | 12.5 | 1.14 | 9.08 |
Inter-day variation was calculated based on intra-run means (average of triplicate sample results) averaged across 5 days.
Precision from replicate analysis of ClinChek samples showed that Method A had CV >15% for both ClinChek levels, whereas CVs for all other methods were <10% (Table IV).
Table IV.
PHMA ClinChek Results (µg/L) by Reference Material Level and Method
| Ref. Mat. | Observed mean | SD | CV (%) | N | % Error | Established mean value | Control range |
|---|---|---|---|---|---|---|---|
| Method | |||||||
| ClinChek Level I | |||||||
| A | 4.41 | 0.728 | 16.5 | 5 | –8.7 | 4.83 | 3.86–5.79 |
| B | 5.37 | 0.438 | 8.17 | 3 | 11.2 | ||
| C | 4.25 | 0.256 | 6.01 | 5 | –12.0 | ||
| D | 5.11 | 0.272 | 5.32 | 5 | 5.8 | ||
| E | 4.44 | 0.399 | 8.99 | 5 | –8.1 | ||
| ClinChek Level II | |||||||
| A | 38.5 | 5.87 | 15.3 | 5 | –12.5 | 44.0 | 35.2–52.8 |
| B | 48.5 | 3.58 | 7.37 | 3 | 10.2 | ||
| C | 38.0 | 1.53 | 4.03 | 5 | –13.6 | ||
| D | 46.7 | 3.53 | 7.56 | 5 | 6.1 | ||
| E | 41.7 | 2.94 | 7.05 | 5 | –5.2 |
% Error was calculated as [(observed mean − established mean value)/established mean value] × 100.
Accuracy
Method accuracy was evaluated by comparing each method’s ClinChek results (Supplemental Table 4) to the manufacturer’s published mean value and control range. All ClinChek I and ClinChek II PHMA results (Figure 2) were within the control range, except for results from method A, where two of the five results from each level were below the bottom of the control range. The mean of results submitted by each laboratory were all within the upper and lower control ranges and within less than a 15% error of ClinChek’s established mean value (Table IV).
Figure 2.
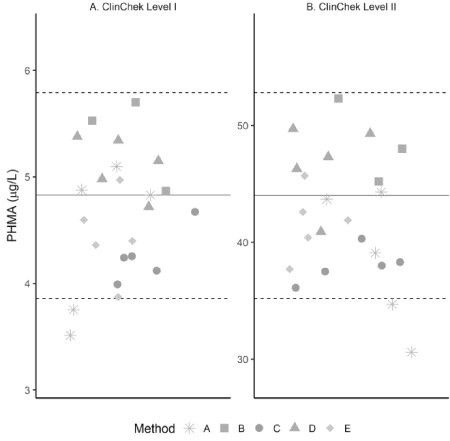
Scatterplot depicting ClinChek Level I (A) and ClinChek Level II (B) urinary PHMA results for each method. Solid lines represent manufacturer’s established mean values, and dotted lines represent upper and lower control range.
Unknown urine samples
All laboratories reported a result for each of the 50 samples (Supplemental Table 1). The number of unknown samples with results greater than LOD and the range of results are summarized in Table V. Method LODs ranged from a low value of 0.024 µg/L (method D) to a high value of 0.600 µg/L (method A). Four of the five participating laboratories reported numeric values for results less than LOD, while one lab reported their LOD. Sample results were below all methods’ upper limit of quantitation, defined as the highest concentration still within the calibration range. Sample-result ranges varied widely across methods. Method D had the largest range (µg/L) of 0.035–29.7, while method A had the smallest range (µg/L) of 0.607–9.03.
Table V.
Unknown Urine Sample Results (µg/L) by Method (n = 50)
| Method | LOD (µg/L) |
ULOQ (µg/L) |
n > LOD | Measured PHMA (µg/L) | |
|---|---|---|---|---|---|
| Minimum | Maximum | ||||
| A | 0.600 | 100 | 40 | 0.607 | 9.03 |
| B | 0.150 | 100 | 35 | 0.202 | 18.7 |
| C | 0.080 | 120 | 42 | 0.094 | 26.2 |
| D | 0.024 | 100 | 47 | 0.035 | 29.7 |
| Ea | 0.200 | 50.0 | 45 | 0.201 | 16.6 |
Laboratory E uses a limit of quantitation (LOQ) rather than an LOD. For the purposes of this study, we used their LOQ as the method LOD. ULOQ, upper limit of quantitation.
We performed linear regression analyses on the average of all PHMA unknown results against each laboratory’s individual results. Quantitative results correlated well between methods that used some form of sample acidification (methods B–E), with R values ranging from 0.770 to 0.983 (Figure 3). The correlations improved with decreasing pH. Method A had the highest pH of all participating laboratories and the lowest correlation (R = 0.403) with the mean of all method results. In general, the observed concentration of PHMA in human urine samples was higher when prepared at lower pH, which is consistent with previous reports.
Figure 3.
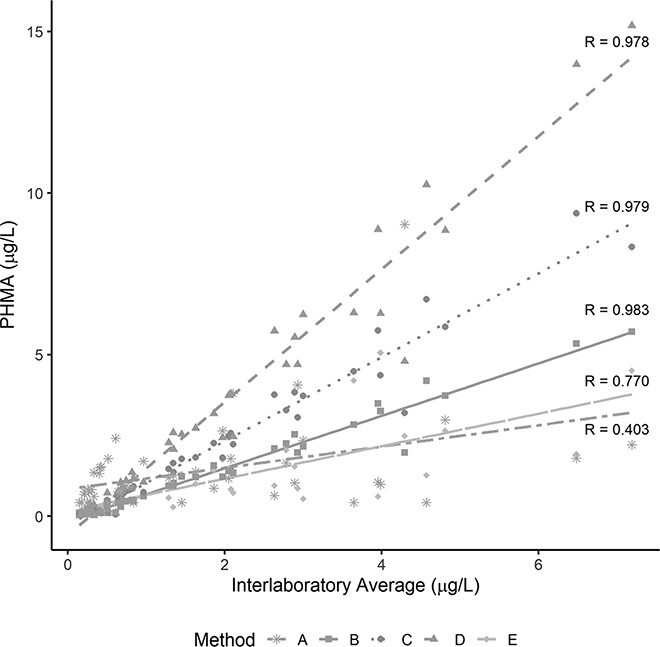
PHMA results (µg/L) from unknown samples (n = 49) plotted against the corresponding averaged results across the five methods.
A pairwise comparison found a significant difference between the means of methods A and D (P = 0.008), B and D (P = 0.002) and E and D (P = 0.0002). Method C, the only other method using strong acidic conditions aside from method D, was the only method mean not statistically different from method D’s mean. Results of the follow-up evaluation using a Deming regression of a scatterplot of unknown results (Table VI) show that the methods in this study do not provide statistically similar urinary PHMA results compared to the precision of the methods, despite pairwise means comparison testing suggesting otherwise.
Table VI.
Deming Regression of PHMA Results (Urine Samples) Arranged in Order of Increasing Pearson Correlation
| Method X | Method Y |
Pearson correlation | Slope (95% CI) |
Intercept (95% CI) |
Variance ratio |
|---|---|---|---|---|---|
| D | C | 0.994 | 0.632 (0.600, 0.675) | 0.037 (0.017, 0.043) | 0.893 |
| C | B | 0.994 | 0.580 (0.548, 0.611) | 0.064 (0.053, 0.071) | 0.957 |
| D | B | 0.992 | 0.368 (0.338, 0.399) | 0.087 (0.075, 0.093) | 0.855 |
| E | B | 0.744 | 1.28 (1.04, 1.63) | –0.176 (–0.298, –0.103) | 3.10 |
| E | C | 0.695 | 1.84 (1.51, 2.36) | –0.292 (–0.473, –0.206) | 3.24 |
| E | D | 0.691 | 2.32 (1.88, 2.98) | –0.348 (–0.532, –0.273) | 3.63 |
| A | E | 0.250 | 0.346 (–13.6, 17.8) | 0.083 (–13.6, 11.3) | 1.10 |
| A | C | 0.248 | 1.53 (–7.02, 15.7) | –0.936 (–11.5, 7.65) | 3.57 |
| A | B | 0.244 | 1.07 (0.483, 11.3) | –0.559 (–7.84, –0.148) | 3.42 |
| A | D | 0.241 | 2.04 (–8.07, 18.7) | –1.38 (–18.1, 9.52) | 4.00 |
A slope estimate of 1.0 (or a slope estimate CI that contains 1.0) and an intercept estimate of 0 (or an intercept estimate CI that contains 0) indicate that the two methods provide results that are statistically equal. None of the slopes and intercepts from the pairwise Deming regressions meet this threshold, even when the Pearson correlations are >0.990. The slope estimates from a pairwise comparison of method A and all other methods contained 1.0, but method A correlated poorly with all other methods (0.241–0.250) and had large CIs around the slope and intercept estimates. Methods with at least some form of acid-treatment step correlated much better with one another and had smaller CIs around the slope and intercept estimates. However, the slope estimates from the pairwise comparisons show systematic biases among these methods.
Discussion
This inter-laboratory comparison is the first, to our knowledge, to obtain quantitative urinary PHMA values from multiple LC–MS-MS methods that include different sample preparation techniques and pH. Estimating slopes and intercepts in linear space allowed us to directly assess the magnitude of differences in quantitation between methods (Table VI). The three method pairs with the best Pearson correlation were B and C, B and D and C and D. However, these comparisons uncovered substantial biases between methods. Method B reported results that were 58% of the values reported by method C and 37% of the values reported by method D. Results for method D, which had the lowest sample preparation pH, matched method C results most closely. However, with a slope (CI) estimate of 0.632 (0.600, 0.675), observed PHMA results from method C are about 63% of those observed using method D. The intercept estimate is 0.037 (0.017, 0.043) and the CI does not contain 0, but since the intercept is the same order of magnitude as the LODs (method D: 0.024 µg/L, method C: 0.08 µg/L), it does not have a large impact on quantitation when the observed concentration is above the LOD.
The results from Deming regressions of these split-sample results reveal large, systematic biases among all methods, even methods with strong acidic sample preparation conditions (i.e., method C and method D). Method D is the only method to use strong acidic sample preparation parameters like those recommended by Sterz (pH of urine adjusted to 0.5–1.0 using HCl) to ensure complete pre-PHMA to PHMA conversion. Laboratories may choose more alkaline sample preparation conditions depending on the need to quantify additional analytes in the same method (18). The authors of method A acknowledge measuring only “free” PHMA (18), noting that acidic conditions negatively affected the stability of other analytes in a method measuring 28 volatile organic compound metabolites. While measuring only “free” PHMA should yield lower observed PHMA concentrations in unknowns, observed results with method A were highest for 17 of the 40 unknowns greater than LOD (0.600 µg/L), suggesting that method A may also not be selective for PHMA in some urine samples. PHMA does not have a reliable confirmation transition because it does not produce any other fragments of reasonable intensity in negative ion mode. Less acidic sample preparation conditions (pH >2) may be preferred for analyses without SPE to prevent corrosion of LC–MS-MS components and/or acid hydrolysis of stationary phase functional groups. While SPE can remove this limitation, it may be cost-prohibitive for large studies. The compromises and limitations of multianalyte methods highlight the importance of evaluating and comparing the effects of assay conditions on quantitation of individual analytes.
We used QC pools and reference materials spiked with PHMA to determine the precision and accuracy of each method. Sample preparation parameters (Table I) covered a wide pH range of <1.0 to 6.8 using three different preparation techniques (dilute-and-shoot, SPE and liquid/liquid extraction). Analytical parameters also varied (Table II), including the use of different stable isotope internal standards. While not investigated in this study, deuterated internal standards can undergo deuterium exchange (though highly unlikely for PHMA-d5 [phenyl]), resulting in a reduction in the concentration of the internal standard and leading to an artificial increase in quantitation (35). Deuterium-labelled internal standards may not accurately capture ion suppression due to retention-time offset from the native analyte (36). Yet, despite differences in analytical parameters (such as different internal standards) and sample preparation parameters, results for ClinChek levels I and II were within range (Table IV) for all methods, with the exception of two measurements from method A. The QCL and QCH observed mean concentrations were similar across the five methods (Table III), except for method A, where QCL mean is about 40% higher than the other methods. This difference may be due, in part, to the actual concentration of QCL being only slightly above two times the LOD of method A (0.600 µg/L). The relationship between sample preparation pH and PHMA values in human urine samples in this study, but not in spiked samples, suggests that differences in analytical parameters have a negligible impact on quantitation compared to differences in sample preparation pH.
Laboratories typically assess their accuracy through an external quality-assurance program. For example, CDC has participated in G-EQUAS since 2017 and has passed the PHMA proficiency tests using method A. Other laboratories in this study have also successfully participated in G-EQUAS. However, this study shows that spiked reference materials are not a reliable indicator of method accuracy in urine samples from exposed individuals due to the absence of a significant amount of pre-PHMA. Pre-PHMA is lacking in the QC pools and ClinChek samples used in this study but is presumably present in the smokers’ urine samples. Moreover, non-spiked reference materials such as NIST SRM 3672, a material containing only smoker’s urine and therefore likely to contain pre-PHMA, may also be of limited use since the PHMA value assigned to the material may be dependent on the sample preparation conditions and analytical method used to characterize it. The above variations should be considered when evaluating test results for urinary PHMA from different analytical methods.
An alternative to non-spiked urine samples from exposed donors would be samples enriched with a known concentration of pre-PHMA. However, pre-PHMA is not commercially available, possibly due to the instability of the product (37). Therefore, we cannot assess the relationship between factors such as pre-PHMA concentration, time, pH and type of acid on the extent of conversion of pre-PHMA to PHMA in the absence of the urine matrix. A pre-PHMA solution could be used to systematically evaluate the effect of different pre-PHMA dehydration conditions to determine whether discrepancies in PHMA measurements are solely due to pH or whether other sources of PHMA in human urine samples and/or side reactions also play a role. In the absence of a solution of known pre-PHMA concentration, a urine sample with a large concentration of pre-PHMA could be used to investigate the relationship between PHMA concentration, time and pH. Repeat analysis of such a sample over time would show whether a sample treated at higher pH (e.g., method C, where samples are treated for a very short time at pH 1.4) can achieve results comparable to lower pH methods (e.g., method D, where samples are treated for 10 minutes at pH ∼0.5) given an extended treatment time.
Conclusion
Among methods with results that correlated well with the average of all other results (R > 0.9), urinary PHMA results for individual samples from the method with the highest pH (method B, pH 2.9) were only 37% of the results from the method using the lowest pH (method D, pH ∼0.5). This result is consistent with previous observations which showed that PHMA quantitation is pH dependent. Two methods in this study (C and D) used strong acidic sample preparation conditions (pH 1.4 and ∼0.5, respectively) but generated dissimilar results (method C results were only %58 of method D) using two different acids (H3PO4 vs HCl, respectively). These discrepancies underscore the need for a comprehensive understanding of the relationship between pH, incubation time and acid type on pre-PHMA conversion to PHMA. Four of the five methods in this study provided PHMA results within the target range for a commercially available reference material for all five measurements. While the results for PHMA-spiked QC pools and ClinChek were similar across the five methods, the results for “real” urine samples were not. PHMA-spiked urine samples like those used in proficiency testing programs and reference materials like ClinChek do not provide information on the accuracy of the method when measuring “real” urine samples that contain significant quantities of pre-PHMA. Though the pH dependence of PHMA quantitation is well known, multi-analyte methods may not be optimized for PHMA analysis. Methods may not be extensively validated using non-spiked samples containing large quantities of pre-PHMA to determine the level of conversion to PHMA achieved using the method’s sample preparation conditions relative to complete conversion. These compromises lead to large differences in PHMA quantitation across analytical methods. The results of this method comparison suggest that care must be taken when drawing comparisons across studies using different analytical methods to measure urinary PHMA, as variations in sample preparation parameters like pH lead to large differences in the quantitation of urine samples that contain pre-PHMA.
Supplementary Material
Acknowledgements
Laboratory resources for analytical chemistry at the University of California, San Francisco, were supported by the National Institutes of Health, P30 DA012393. Mass spectrometry carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, was supported in part by Cancer Center Support Grant CA-077598, and this study was supported by grant CA-203851 from the US National Cancer Institute and the Food and Drug Administration Center for Tobacco Products.
Contributor Information
Denise S Tevis, Tobacco and Volatiles Branch, Division of Laboratory Sciences, National Center for Environmental Health, Centers for Disease Control and Prevention, Atlanta, GA, 30341, USA.
Andrew Willmore, Oak Ridge Institute for Science and Education, Knoxville, TN, 37831, USA.
Deepak Bhandari, Tobacco and Volatiles Branch, Division of Laboratory Sciences, National Center for Environmental Health, Centers for Disease Control and Prevention, Atlanta, GA, 30341, USA.
Brett Bowman, Oak Ridge Institute for Science and Education, Knoxville, TN, 37831, USA.
Chloe Biren, Oak Ridge Institute for Science and Education, Knoxville, TN, 37831, USA.
Brandon M Kenwood, Tobacco and Volatiles Branch, Division of Laboratory Sciences, National Center for Environmental Health, Centers for Disease Control and Prevention, Atlanta, GA, 30341, USA.
Peyton Jacob, Clinical Pharmacology Program, Division of Cardiology, Department of Medicine, University of California San Francisco, San Francisco, CA, 94143, USA.
Jia Liu, Clinical Pharmacology Program, Division of Cardiology, Department of Medicine, University of California San Francisco, San Francisco, CA, 94143, USA.
Kristina Bello, Clinical Pharmacology Program, Division of Cardiology, Department of Medicine, University of California San Francisco, San Francisco, CA, 94143, USA.
Stephen S Hecht, Masonic Cancer Center, University of Minnesota, Minneapolis, MN, 55455, USA.
Steven G Carmella, Masonic Cancer Center, University of Minnesota, Minneapolis, MN, 55455, USA.
Menglan Chen, Masonic Cancer Center, University of Minnesota, Minneapolis, MN, 55455, USA.
Eric Gaudreau, Centre de Toxicologie du Québec, Unité Laboratoire de Toxicologie, Institut National de Santé Publique du Québec, Direction de la santé environnementale et de la toxicologie, Québec, G1V 5B3, Canada.
Jean-François Bienvenu, Centre de Toxicologie du Québec, Unité Laboratoire de Toxicologie, Institut National de Santé Publique du Québec, Direction de la santé environnementale et de la toxicologie, Québec, G1V 5B3, Canada.
Benjamin C Blount, Tobacco and Volatiles Branch, Division of Laboratory Sciences, National Center for Environmental Health, Centers for Disease Control and Prevention, Atlanta, GA, 30341, USA.
Víctor R De Jesús, Tobacco and Volatiles Branch, Division of Laboratory Sciences, National Center for Environmental Health, Centers for Disease Control and Prevention, Atlanta, GA, 30341, USA.
Disclaimer
The views and opinions expressed in this report are those of the authors and do not necessarily represent the views, official policy or position of the US Department of Health and Human Services or any of its affiliated institutions or agencies. Use of trade names is for identification purposes and does not imply endorsement by the Centers for Disease Control and Prevention, the Public Health Service or the US Department of Health and Human Services. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Food and Drug Administration.
Supplementary data
Supplementary data is available at Journal of Analytical Toxicology online.
References
- 1.Toxicological profile for Benzene. Agency for Toxic Substances and Disease Registry (ATSDR). https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=40&tid=14#bookmark02 [PubMed]
- 2.Smith M.T. (2010) Advances in understanding benzene health effects and susceptibility. Annual Review of Public Health, 31, 133–148. doi: 10.1146/annurev.publhealth.012809.103646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fiebelkorn S., Meredith C. (2018) Estimation of the leukemia risk in human populations exposed to benzene from tobacco smoke using epidemiological data. Risk Analysis, 38, 1490–1501. doi: 10.1111/risa.12956 [DOI] [PubMed] [Google Scholar]
- 4.Korte J.E., Hertz-Picciotto I., Schulz M.R., Ball L.M., Duell E.J. (2000) The contribution of benzene to smoking-induced leukemia. Environmental Health Perspectives, 108, 333–339. doi: 10.1289/ehp.00108333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wallace L.A. (1989) Major sources of benzene exposure. Environmental Health Perspectives, 82, 165–169. doi: 10.1289/ehp.8982165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson E.S., Langård S., Lin Y.-S. (2007) A critique of benzene exposure in the general population. Science of the Total Environment, 374, 183–198. doi: 10.1016/j.scitotenv.2006.11.045 [DOI] [PubMed] [Google Scholar]
- 7.Javelaud B., Vian L., Molle R., Allain P., Allemand B., Andre B., et al. (1998) Benzene exposure in car mechanics and road tanker drivers. International Archives of Occupational and Environmental Health, 71, 277–283. doi: 10.1007/s004200050281 [DOI] [PubMed] [Google Scholar]
- 8.Protano C., Andreoli R., Manini P., Guidotti M., Vitali M. (2012) A tobacco-related carcinogen: assessing the impact of smoking behaviours of cohabitants on benzene exposure in children. Tobacco Control, 21, 325–329. doi: 10.1136/tc.2010.039255 [DOI] [PubMed] [Google Scholar]
- 9.Pazo D.Y., Moliere F., Sampson M.M., Reese C.M., Agnew-Heard K.A., Walters M.J., et al. (2016) Mainstream smoke levels of volatile organic compounds in 50 U.S. domestic cigarette brands smoked with the ISO and Canadian intense protocols. Nicotine Tobacco Research, 18, 1886–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng S., Roethig H.J., Liang Q., Kinser R., Jin Y., Scherer G., et al. (2006) Evaluation of urinary 1-hydroxypyrene, S-phenylmercap-turic acid, trans,trans-muconic acid, 3-methyladenine, 3-ethyladenine, 8-hydroxy-2ʹ-deoxyguanosine and thioethers as biomarkers of expo-sure to cigarette smoke. Biomarkers, 11, 28–52. doi: 10.1080/13547500500399730 [DOI] [PubMed] [Google Scholar]
- 11.Campagna M., Satta G., Campo L., Flore V., Ibba A., Meloni M., et al. (2012) Biological monitoring of low-level exposure to benzene. La Medicina Del Lavoro, 103, 338–346. [PubMed] [Google Scholar]
- 12.Carmella S.G., Chen M., Han S., Briggs A., Jensen J., Hatsukami D.K., et al. (2009) Effects of smoking cessation on eight urinary tobacco carcinogen and toxicant biomarkers. Chemical Research in Toxicology, 22, 734–741. doi: 10.1021/tx800479s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.National Health and Nutrition Examination Survey (NHANES)Centers for Disease Control and Prevention. https://wwwn.cdc.gov/nchs/nhanes/Search/DataPage.aspx?Component=Laboratory
- 14.Second, Third, and Fourth report on Human Biomonitoring of Environmental Chemicals in Canada. Canadian Health Measures Survey (CHMS). https://www.canada.ca/en/health-canada/services/environmental-workplace-health/environmental-contaminants/human-biomonitoring-environmental-chemicals/canadian-health-measures-survey.html
- 15.Population Assessment of Tobacco and Health (PATH) Study [United States]. United States Department of Health and Human Services, National Institutes of Health, National Institute on Drug Abuse, Food and Drug Administration Center for Tobacco Products. 10.3886/ICPSR36498.v10 (accessed Mar 16, 2020) [DOI]
- 16.European Petroleum Refiners Association. https://www.concawe.eu/wp-content/uploads/BenzeneMonitoring-_DEF.pdf
- 17.National Institute for Occupational Safety and Health (NIOSH). NIOSH Manual of Analytical Methods, 2017.
- 18.Alwis K.U., Blount B.C., Britt A.S., Patel D., Ashley D.L. (2012) Simultaneous analysis of 28 urinary VOC metabolites using ultra high performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry (UPLC-ESI/MSMS). Analytica Chimica Acta, 750, 152–160. doi: 10.1016/j.aca.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Y.S., Blount B.C., Valentin-Blasini L., Applewhite H.S., Xia Y., Watson C.H., et al. (2009) Simultaneous determination of six mer-capturic acid metabolites of volatile organic compounds in human urine. Chemical Research in Toxicology, 22, 1018–1025. doi: 10.1021/tx800468w [DOI] [PubMed] [Google Scholar]
- 20.Jacob P. 3rd, Abu Raddaha A.H., Dempsey D., Havel C., Peng M., Yu L., et al. (2013) Comparison of nicotine and carcinogen exposure with water pipe and cigarette smoking. Cancer Epidemiology, Biomarkers & Prevention : A Publication of the American Association for Cancer Research, Cosponsored by the American Society of Preventive Oncology, 22, 765–772. doi: 10.1158/1055-9965.EPI-12-1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inoue O., Kanno E., Kakizaki M., Watanabe T., Higashikawa K., Ikeda M. (2000) Urinary phenylmercapturic acid as a marker of occupational exposure to benzene. Industrial Health, 38, 195–204. doi: 10.2486/indhealth.38.195 [DOI] [PubMed] [Google Scholar]
- 22.Paci E., Pigini D., Cialdella A.M., Faranda P., Tranfo G. (2007) Determination of free and total S-phenylmercapturic acid by HPLC/MS/MS in the biological monitoring of benzene exposure. Biomarkers, 12, 111–122. doi: 10.1080/13547500601007943 [DOI] [PubMed] [Google Scholar]
- 23.Sabourin P.J., Bechtold W.E., Henderson R.F. (1988) A high pressure liquid chromatographic method for the separation and quantitation of water-soluble radiolabeled benzene metabolites. Analytical Biochemistry, 170, 316–327. doi: 10.1016/0003-2697(88)90637-9 [DOI] [PubMed] [Google Scholar]
- 24.Henderson A.P., Barnes M.L., Bleasdale C., Cameron R., Clegg W., Heath S.L., et al. (2005) Reactions of benzene oxide with thiols including glutathione. Chemical Research in Toxicology, 18, 265–270. doi: 10.1021/tx049781y [DOI] [PubMed] [Google Scholar]
- 25.Sterz K., Köhler D., Schettgen T., Scherer G. (2010) Enrichment and properties of urinary pre-S-phenylmercapturic acid (pre-SPMA). Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 878, 2502–2505. doi: 10.1016/j.jchromb.2009.08.043 [DOI] [PubMed] [Google Scholar]
- 26.Fustinoni S., Campo L., Mercadante R., Manini P. (2010) Methodological issues in the biological monitoring of urinary benzene and S-phenylmercapturic acid at low exposure levels. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 878, 2534–2540. doi: 10.1016/j.jchromb.2009.11.045 [DOI] [PubMed] [Google Scholar]
- 27.Bhandari D., McCarthy D., Biren C., Movassaghi C., Blount B.C., De Jesus V.R. (2019) Development of a UPLC-ESI-MS/MS method to measure urinary metabolites of selected VOCs: benzene, cyanide, furfural, furfuryl alcohol, 5-hydroxymethylfurfural, and N-methyl-2-pyrrolidone. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 1126–1127, 121746. doi: 10.1016/j.jchromb.2019.121746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Analytical method for the determination of benzene metabolites in urine by UPLC-MS-MS (E-460), condensed version for CHMS. Institut National de Santé Publique du Québec (INSPQ). https://www.canada.ca/en/health-canada/services/environmental-workplace-health/reports-publications/environmental-contaminants/second-report-hum-an-biomonitoring-environmental-chemicals-canada-health-canada-2013.html (accessed Mar 16, 2020)
- 29.Carmella S.G., Chen M., Zarth A., Hecht S.S. (2013) High throughput liquid chromatography-tandem mass spectrometry assay for mercapturic acids of acrolein and crotonaldehyde in cigarette smokers’ urine. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 935, 36–40. doi: 10.1016/j.jchromb.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Occupational Medicine in Urine. RECIPE. https://recipe.de/products/occupational-medicine-urine-2/
- 31.Brown C.E.Coefficient of variation. Applied Multivariate Statistics in Geohydrology and Related Sciences. Springer Berlin Heidelberg: Berlin, Heidelberg, 1998; 155–157. [Google Scholar]
- 32.Hornung R.W., Reed L.D. (1990) Estimation of average concentration in the presence of nondetectable values. Applied Occupational and Environmental Hygiene, 5, 46–51. doi: 10.1080/1047322X.1990.10389587 [DOI] [Google Scholar]
- 33.CLSI . Measurement Procedure Comparison and Bias Estimation Using Patient Samples. 3rd ed. CLSI guideline EP09c. Wayne, PA: Clinical and Laboratory Standards Institute; 2018. [Google Scholar]
- 34.Linnet K. (2019) Evaluation of regression procedures for methods comparison studies. Clinical Chemistry, 39, 424–432. doi: 10.1093/clinchem/39.3.424 [DOI] [PubMed] [Google Scholar]
- 35.Davies N.W., Smith J.A., Molesworth P.P., Ross J.J. (2010) Hydrogen/deuterium exchange on aromatic rings during atmospheric pressure chemical ionization mass spectrometry. Rapid Communications in Mass Spectrometry : RCM, 24, 1105–1110. doi: 10.1002/rcm.4488 [DOI] [PubMed] [Google Scholar]
- 36.Wang S., Cyronak M., Yang E. (2007) Does a stable isotopically labeled internal standard always correct analyte response? A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. Journal of Pharmaceutical and Biomedical Analysis, 43, 701–707. doi: 10.1016/j.jpba.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 37.Micova K., Linhart I. (2012) Reactions of benzene oxide, a reactive metabolite of benzene, with model nucleophiles and DNA. Xenobiotica, 42, 1028–1037. doi: 10.3109/00498254.2012.669872 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.