

Abstract
The advent of personalized medicine has driven the development of novel approaches for obtaining detailed cellular and molecular information from clinical tissue samples. Tissue cytometry is a promising new technique that can be used to enumerate and characterize each cell in a tissue and, unlike flow cytometry and other single-cell techniques, does so in the context of the intact tissue, preserving spatial information that is frequently crucial to understanding a cell’s physiology, function and behavior. However, the wide-scale adoption of tissue cytometry as a research tool has been limited by the fact that published examples utilize specialized techniques that are beyond the capabilities of most laboratories. Here we describe a complete and accessible pipeline, including methods of sample preparation, microscopy, image analysis and data analysis for large-scale three-dimensional tissue cytometry of human kidney tissues. In this workflow, multiphoton microscopy of unlabeled tissue is first conducted to collect autofluorescence and second harmonic images. The tissue is then labeled with 8 fluorescent probes, and imaged using spectral confocal microscopy. The raw 16 channel images are spectrally deconvolved into 8 channel images, and analyzed using the VTEA (Volumetric Tissue Exploration and Analysis) software developed by our group. We applied this workflow to analyze millimeter-scale tissue samples obtained from human nephrectomies and from renal biopsies from individuals diagnosed with diabetic nephropathy, generating a quantitative census of tens of thousands of cells in each. Such analyses can provide useful insights that can be linked to the biology or pathology of kidney disease. The approach utilizes common laboratory techniques, is compatible with most commercially-available confocal microscope systems and all image and data analysis is conducted using the VTEA image analysis software, which is available as a plug-in for ImageJ.
For nearly 70 years, histological analysis of biopsy samples has represented the “gold standard” of renal pathology, with clinical diagnoses largely based upon structural changes detected using morphological stains. However, as we enter the era of personalized medicine, researchers increasingly appreciate the need to obtain and quantify additional molecular and genetic information from clinical tissue samples, information that can be used to develop mechanism-based diagnoses and individualized therapies.
Advances in molecular biology have made it possible to collect a rich molecular phenotype of individual samples, even to characterize the genome and transcriptome of individual cells. In the realm of renal research, single cell “omics” techniques are poised to provide new insights into the mechanisms underlying kidney development, disease and treatment (1, 2). However, a general shortcoming of these approaches is that in the process of homogenizing a tissue for cellular analysis one loses all spatial context, discarding crucial information about the organization of structures and cells in the tissue and the relationships that inform the in situ behaviors of each cell. Spatial context is particularly important to understanding cell function in the kidney, whose structure reflects multiple layers of cellular organization. Endothelia and epithelia are organized into sheets that are organized into capillaries and tubules, which are in turn are organized into capillary networks and nephrons whose function is mediated by their organization within the cortex and medulla. This spatial complexity is compounded in the context of disease in which the structure and cellular relationships are disrupted in a time-dependent way, with the additional complication of the local infiltration and activation of immune cells.
An alternative approach is to use multiplexed fluorescence microscopy to characterize the protein or RNA phenotype of cells in fixed tissue sections. Confocal microscopy extends the spatial context of fluorescence studies into three dimensions (3D), which is critical for capturing the structure and inter-relationship of large multicellular structures such as glomeruli and tubules. Whereas light microscopy has traditionally been used to sample representative regions of a tissue sample, automated systems can now be used to collect multiplexed images of entire millimeter-scale samples with subcellular resolution. The analysis of image volumes of this size and complexity is well beyond the capabilities of visual inspection and depends upon advanced methods of digital image analysis that are used to identify and quantitatively characterize each cell, essentially providing a census of every cell in the specimen. The resulting data, which may consist of hundreds of features measured for each of hundreds of thousands of cells, are then analyzed using methods of multi-dimensional data analysis to test hypotheses and to identify unforeseen patterns and relationships.
This approach, which we have termed “tissue cytometry” has been used to obtain unique insights into cellular function in lymphatic tissues (3–9), tumors (10, 11), bone marrow (12) and by our group in human and mouse kidney (13–18). Although these studies demonstrate its power, the wide-scale adoption of tissue cytometry as a tool in biomedical research has been limited by the fact that published examples utilize specialized techniques that are beyond the capabilities of most laboratories. Here we describe a complete and accessible pipeline, including methods of sample preparation, microscopy, image analysis and data analysis for large-scale tissue cytometry of human kidney tissues.
Materials and methods
Reference kidney samples (from tumor-free regions of nephrectomies or deceased donors) were obtained from the KPMP (Kidney Precision Medicine Project, https://kpmp.org) and diabetic nephropathy biopsy specimens were obtained from the Biopsy Biobank Cohort of Indiana (BBCI). This study was approved by the Institutional Review Board (IRB) of Indiana University. The diabetic nephropathy biopsies were indicated because of rapidly progressing kidney disease and the patients were at stage 2 or 3 CKD at the time of biopsy with nephrotic range proteinuria (Supplementary Table 1).
Sample sectioning for multiphoton and confocal microscopy
Fresh-frozen human kidney samples are placed in cold OCT (Optimal Cutting Temperature) compound for 3 minutes and then transferred to a cryomold with partially frozen OCT in the bottom on a block of dry ice. Once the OCT is completely frozen, the tissue block is wrapped in parafilm and stored at 80°Celsius. Frozen tissues are sectioned to a thickness of 50 μm using a cryostat and then immediately fixed in 4% fresh paraformaldehyde (PFA) for 24 hrs, and subsequently stored at 4°C in 0.25% PFA.
Multiphoton microscopy
The imaging strategy described here consists of two steps. Multiphoton microscopy is first used to collect autofluorescence and second harmonic images of the unlabeled tissue. The tissue is then labeled with fluorescent compounds and antibodies and confocal microscopy is used to collect multiplexed images of the fluorescent labels. Since fluorescence labeling was accomplished with the tissue suspended in a solution, this sequential approach required that multiphoton microscopy be conducted on unlabeled tissue mounted in a temporary mount from which the tissue could be easily removed. Accordingly, multiphoton microscopy was conducted on tissues that were rinsed in PBS and temporarily mounted in a non-hardening mounting medium on low-charge slides and sealed via removable rubber cement sealant.
Both multiphoton and confocal microscopy were conducted using a Leica SP8 confocal scan-head mounted to an upright DM6000 microscope with computer-controlled motorized stage. In order to collect images of entire millimeter-scale tissues at submicron resolution, the Leica Tile Scan function was used to collect a mosaic of smaller image volumes using a high-power, high-numerical aperture objective. The Leica LASX software was then used to stitch these component volumes into a single image volume of the entire sample. The scanner zoom and focus motor control were set to provide voxel dimensions of 0.5 by 0.5 microns laterally and 1 micron axially.
Multiphoton excitation microscopy was conducted using a 20× 0.75 NA Leica multi-immersion objective (adjusted for water immersion), with excitation provided by a MaiTai DeepSee tunable titanium-sapphire laser (Spectra Physics, Santa Clara, CA) adjusted to 910 nm. The descanned pathway was configured for multiphoton imaging by fully opening the confocal pinhole and adjusting photomultiplier detectors to collect emissions from 439 to 465 nm (for second harmonic imaging, or SHG) and from 473 to 497 nm (for autofluorescence).
Fluorescence labeling for confocal microscopy
Following multiphoton microscopy, the temporary mount is dismantled, and the unlabeled tissue is carefully retrieved for fluorescence labeling (fluorescence reagents are listed in Supplementary Table 2). The tissue is washed in PBS (phosphate-buffered saline) for ten minutes and placed in blocking buffer (PBS with 0.1% Triton X-100 (MP Biomedical) and 10% Normal Donkey Serum (Jackson Immuno Research)). The tissue is gently rocked for 4–8 hrs at room temperature after which it is placed in 200 μl of a solution containing antibodies to be used for indirect immunofluorescence (AQP1, MPO, CD68 and SIGLEC-8) and rocked for 8–16 hrs at room temperature. The tissue is then washed in two changes of 2 mls of PBST (PBS with 0.1% Triton X-100) over 6 hrs, and placed in blocking buffer. After 4–8 hrs of gentle rocking at room temperature, the tissue is then incubated in the panel of secondary antibodies. Following an 8–16 hr incubation the tissue is washed in two changes of 2 mls of PBST over 6 hrs, and placed in blocking buffer for 4–8 hrs. The tissue is then incubated at room temperature in 200 μl of a solution containing the directly-labeled antibodies (THP, CD3) as well as DAPI and OregonGreen phalloidin. After 8–16 hrs in this solution, the tissue is washed in two changes of PBST over 6 hrs and mounted in Prolong Glass (Thermo Fisher) on acid-cleaned slides.
Confocal microscopy of labeled tissue
Confocal microscopy was conducted using a Leica 20× 0.75 NA multi-immersion objective (adjusted for oil immersion), with excitation provided by a solid state laser launch with laser lines at 405 nm, 488 nm, 552 nm and 635 nm. A total of 16 images is collected for each focal plane of each panel of the 3D mosaic. The sample is first illuminated at 405nm, and the four confocal detectors (two PMT, two HyD) adjusted to collect 410–430nm, 430–450nm, 450–470nm and 470–490 nm emissions. The sample is then illuminated at 488nm and the confocal detectors adjusted to collect 500–509nm, 510–519nm, 520–530nm and 530nm-540nm emissions. The sample is then illuminated at 552nm and the confocal detectors adjusted to collect 570–590nm, 590–610nm, 610–630nm and 631–651nm emissions. Finally, the sample is illuminated at 633nm and the confocal detectors adjusted to collect 643–664nm, 664–685nm, 685–706nm and 706–726nm emissions. The resulting 16-channel image is then spectrally deconvolved (via linear unmixing, as described below) to discriminate the 8 fluorescent probes in the sample.
Spectral deconvolution
The excitation and emission spectra of many of the fluorescent probes used in these studies overlap with one another. In order to discriminate each of the 8 fluorescent probes, the 16 channel images were spectrally deconvolved using the Leica LASX linear unmixing software. The unmixing matrix is derived from 16 channel reference images of singly-labeled samples (tissue labeled with DAPI or OregonGreen alone, or samples containing microspheres (ThermoFisher, cat# C37253) labeled with one of the fluorophores used for immunofluorescence.) Spectral deconvolution results were validated by collecting 16 channel images of three samples containing 6 different fluorescently-labeled microspheres in different known proportions, and then comparing the numbers of microspheres identified by linear unmixing with the numbers expected based upon their known proportions. Regression of the pooled results indicated an excellent agreement between the observed and expected counts (Supplementary Figure 1, R2 = .981) and a G-test of independence detected no significant difference (P=0.98). The effectiveness of the linear unmixing process is also illustrated in Supplementary Figure 2, which shows a multiple channel image before and after linear unmixing.
Quantitative image analysis - tissue cytometry
All tissue cytometry was conducted using the Volumetric Tissue Exploration and Analysis (VTEA) software. VTEA is unique 3D image processing workspace that was developed by our group as a plug-in for ImageJ (14). Compatible with essentially any personal computer system, VTEA can be obtained using the ImageJ updater system as described at https://imagej.net/VTEA. The details of VTEA’s operation and utility are described in more detail in the Results section. Validation of the gates drawn on the analytical scatterplots was performed by visual mapping of the gated cells over the image volume and kept constant between specimens in the particular study. Cell density isomaps (e.g. Figure 4B) were generated in R using ggplot. Tissue volumes were quantified as the product of the lateral dimension of the tissue (quantified in FIJI) multiplied by the mean depth of the tissue, as measured by the distance between the first and last sections containing at least one segmented nucleus.
Figure 4. 3D confocal fluorescence microscopy and cytometry of structural and immune cell markers.

Panel A - Maximum projection of combined fluorescence images of DAPI (grey), phalloidin (green) and antibodies to THP (cyan), AQP1 (magenta), myeloperoxidase (MPO, red), CD68 (yellow) and CD3 (white). Arrows indicate two regions of immune cell infiltrates. (A video of a volume rendering of this volume is presented in Supplementary Video 1). Panel B - Projection of 3D image volume of binary map of nuclei following image segmentation using VTEA. Each segmented nucleus is presented in an arbitrary color. Inset is an isomap of cell density with darker colors depicting higher cell densities. Panel C - Distribution of different cell types following scatterplot gating. Green – phalloidin, Cyan – thick ascending limb, Magenta – proximal tubules. Red – neutrophils, Yellow – macrophages, White – T-cells. Panels D and E show corresponding 4X magnification images of the regions indicated in the two boxes shown in panel A. Panels F and G show scatterplots of the fluorescence intensity of THP vs. AQP1 and MPO vs. CD68, respectively. In each scatterplot, the color of each point represents the fluorescence intensity of a third probe (phalloidin and AQP1, respectively were chosen for these examples). Panels H and I show the locations of the different cell types in the regions shown in panels F and G, respectively, as determined by scatterplot gating. Scale bar in panel A represents 1 mm and 250 microns in panel D.
Image processing for figures
As described above, mosaic image volumes were assembled using the Leica Suite X software. Quantitative analysis was conducted using raw image data, but micrograph images presented in figures were contrast enhanced in a way that preserved the visibility of both the dim and bright structures of the original images. Images in figures were processed, assembled and annotated using Adobe Photoshop. The video of the volume rendering of the data shown in Figure 4A (Supplementary Video 1) was generated using Imaris Software (Version 8.1.2, Oxford Instruments, Concord, MA).
Statistics
When applicable, average values are reported ± standard errors. Two-tailed Student’s t-tests were used as tests of significance.
Results
Overview of the approach
The defining goal of our approach is to maximize the information content of images collected from human kidney samples. Each additional parameter improves our ability to unravel the complexity of the kidney, to test hypotheses and to reveal unforeseen relationships and processes. The need to extract maximum information from each sample is particularly acute for human tissue samples. First, these samples are frequently rare, particularly for specific disease states. Second, human tissue samples incur an ethical responsibility to the donor/patient to justify their sacrifice by extracting maximum information from each precious sample. Thus, we developed a tissue preparation, imaging and image analysis workflow that provides a quantitative characterization of every cell in an entire 50 micron thick tissue sample with respect to 8 different fluorescent probes, tissue autofluorescence and second harmonic generation. Described in detail in subsequent sections, the entire process is summarized below and schematized in Figure 1.
Figure 1. Overview of the tissue cytometry workflow.
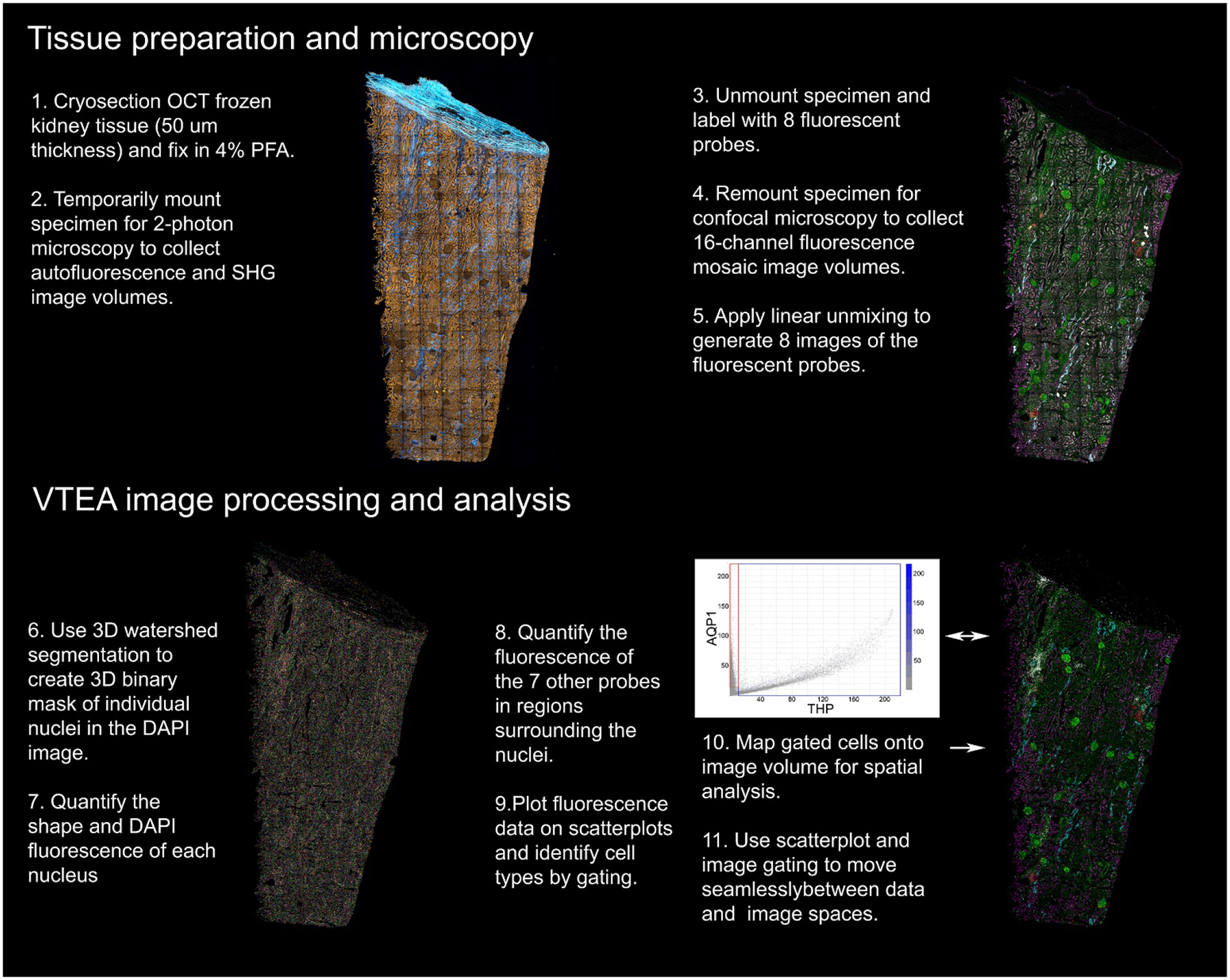
Major steps involved in the tissue imaging and analysis pipeline.
Cryosectioned samples are first placed in a temporary mount and imaged using multiphoton microscopy to collect high-resolution autofluorescence and second-harmonic generation (SHG) images of the entire sample. The section is then removed from its temporary mount and processed for fluorescence labeling with six antibodies, fluorescent phalloidin and DAPI. The tissue is then mounted permanently on a slide, where it is imaged in its entirety using spectral confocal microscopy. The resulting 16 channel image volume is spectrally deconvolved to discriminate the fluorescence of the eight fluorescent markers. VTEA software is then used to segment the resulting DAPI volume into the individual nuclei of each cell and to quantify the fluorescence in each of the 8 channels. VTEA provides scatterplots of the fluorescence associated with each nucleus that can be gated to identify specific cell types, that are then mapped back onto the original image volume. The combination of image maps and scatterplots are then used to interactively explore the image volume. In the following sections, we present an expanded demonstration of this workflow and how it can be used to extract and explore the cellular and molecular constitution of human tissue samples.
Multiphoton microscopy of unlabeled human kidney tissue
The endogenous fluorescence of tissues is frequently considered an objectionable impediment to immunofluorescence microscopy. However autofluorescence, arising from a variety of different molecular sources (e.g., NADH, flavins, porphyrins, collagen, elastin, lipofuscin) has also been used as a “label-free” approach for characterizing tissue structure and detecting pathologies (19, 20). The fact that tissue autofluorescence can be efficiently stimulated by two-photon processes means that multiphoton microscopy can be used simultaneously image tissue autofluorescence alongside signals arising from SHG, another label-free imaging modality. The predominant source of SHG signals in biological tissues is fibrillary collagen. Accordingly, SHG has been previously used to detect fibrosis and other pathological alterations of the extracellular matrix (21, 22).
Figure 2 shows the results of a study in which multiphoton microscopy was used to collect a 3D volume of autofluorescence and SHG signals from a 4 mm by 9 mm, 50 micron thick section of fixed human nephrectomy tissue. Since tissue cytometry requires images collected at a spatial resolution sufficient to distinguish individual nuclei, we employed an approach in which the image volume of the tissue is constructed as a mosaic of smaller image volumes (each ~400 microns across), collected using a 20x objective, with a numerical aperture large enough (0.75) to provide subcellular resolution. For each panel of the mosaic, the tissue was illuminated at 910 nm, and two-channel images (SHG and autofluorescence) were collected at one micron intervals through the entire 50 micron thickness of the tissue. The process was then repeated across the entire sample, and the resulting volumes stitched together into a single mosaic.
Figure 2. 3D multiphoton microscopy of unlabeled nephrectomy.
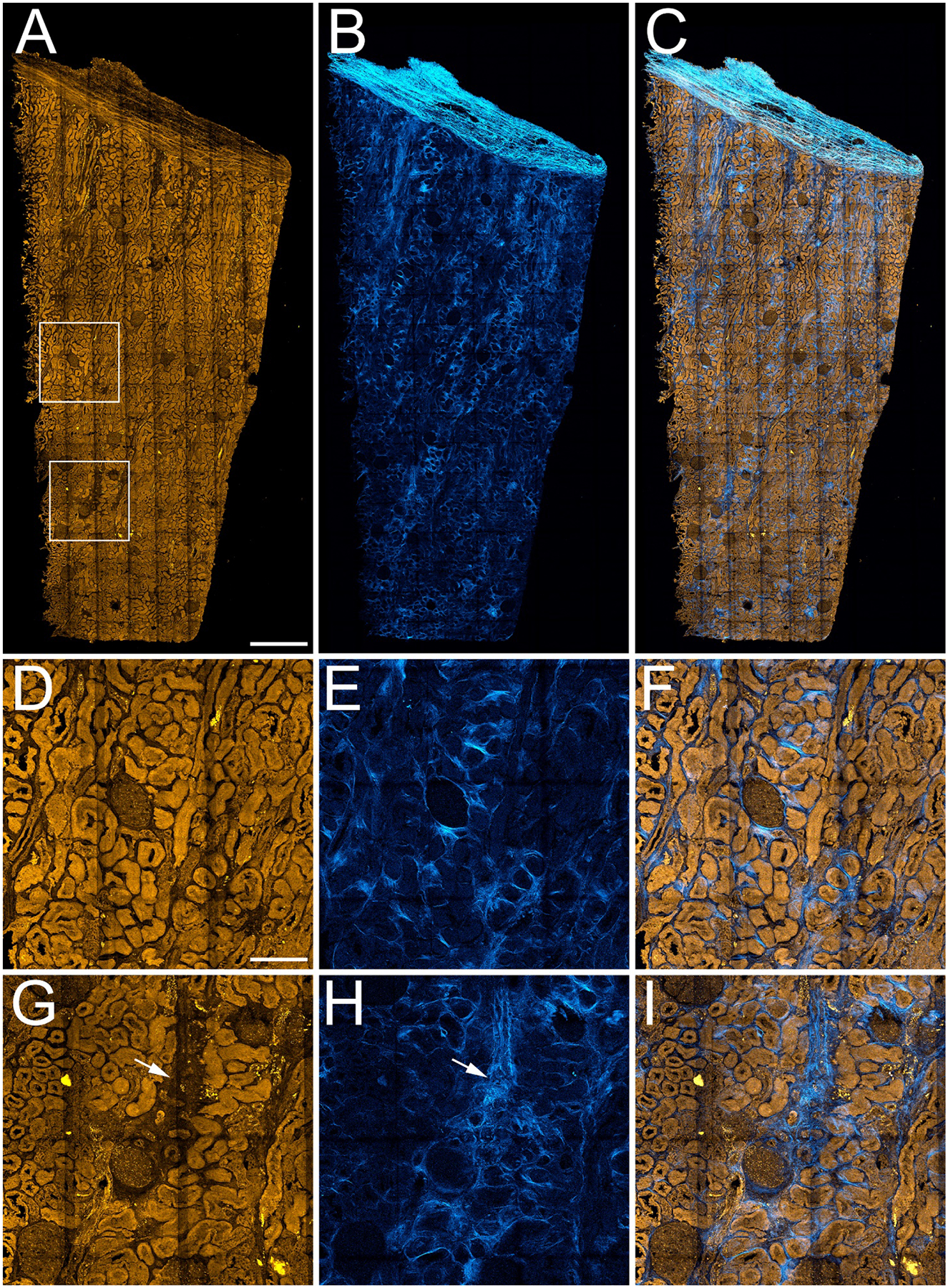
Mosaic of high-resolution image volumes collected from a 4 mm by 9 mm, 50 micron thick section of paraformaldehyde-fixed human nephrectomy tissue. Panel A – Maximum projection image of 3D volume of tissue autofluorescence. Panel B- Maximum projection of 3D volume of second harmonic generation (SHG) images. Panel C – Overlay of autofluorescence and SHG. Panels D-F show corresponding 4X magnification images of the region indicated in the upper box in panel A, and panels G-I show corresponding 4X magnification images of the region indicated in the lower box in panel A. Arrows in panels G and H indicate regions of apparent tubular dropout and fibrosis. Scale bar in panel A represents 1 mm. Scale bar in panel D represents 250 microns in D-I.
The images of tissue autofluorescence (panels A, D, G) demonstrate that tissue autofluorescence provides structural information sufficient to identify glomeruli and tubules, but also regions of apparent tubular drop-out (arrows in panel G). The images of SHG, that largely reflect fibrillary collagen, provide a complementary characterization of the structure of the extracellular matrix, including regions of collagen deposition likely to reflect fibrosis. Interestingly, SHG signals are frequently strong in regions lacking autofluorescence, consistent with the expectation of fibrosis in regions of tubular dropout. Table 1 shows the results of quantitative analyses of the autofluorescence SHG images of this and four other nephrectomies, which were used to determine the number of glomeruli and the percentage of the tissue section collected from the cortex (based upon autofluorescence) and the percentage of the tissue area that is fibrotic (based upon SHG). Autofluorescence and SHG images of the additional nephrectomies are shown in Supplementary Figure 3.
Table 1 –
Results of label free and VTEA analysis of 5 nephrectomy samples
| Sample | % Cortex | Tissue volume mm3 |
SHG+ area % total |
Total cell | Cell density cells/mm3 |
THP+ cells/mm3 (%) |
AQP1+ cells/mm3 (%) |
MPO+ cells/mm3 (%) |
CD68+ cells/mm3 (%) |
CD3+ cells/mm3 (%) |
Siglec8+ cells/mm3 (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 Figs 3, 4 |
90.5 | 1.73 | 11.7 | 248,050 | 143,506 | 4495 (3.1) |
21807 (15.2) |
2152 (1.5) |
551 (0.3) |
5481 (3.8) |
15 (0.0001) |
| 2 Supp2A |
89.9 | 0.62 | 10.9 | 154,471 | 249,772 | 14166 (5.7) |
21281 (8.5) |
864 (0.3) |
11 (0.0) |
298 (0.1) |
0 (0) |
| 3 Supp2B |
60.4 | 1.03 | 12.3 | 214,769 | 207,947 | 38840 (18.7) |
40221 (19.4) |
5852 (2.8) |
435 (0.2) |
4131 (2.0) |
0 (0) |
| 4 Supp2C |
100 | 0.45 | 10.4 | 112,360 | 250,786 | 23826 (9.5) |
31533 (12.6) |
11595 (4.6) |
1129 (0.5) |
16452 (16.6) |
0 (0) |
| 5 Supp2D |
44.7 | 0.72 | 7.7 | 234,685 | 326,625 | 99141 (30.4) |
63357 (19.4) |
3592 (1.1) |
1063 (0.3) |
8706 (2.7) |
0 (0) |
Spectral confocal microscopy of human kidney labeled with 8 fluorescent probes
Although the autofluorescence image provides structural information, and the SHG indicates the distribution of fibrillary collagen, a characterization of the cellular constitution of the sample requires immunofluorescence labeling of cell-specific proteins. Thus, after multiphoton microscopy, the tissue was removed from its temporary mount and labeled with eight fluorescent probes. As described in Methods, the tissue was processed for immunofluorescence labeling, using antibodies to aquaporin-1 (AQP1, to label proximal tubule cells), Tamm-Horsfall protein (THP, to label thick ascending limbs), and CD68, CD3, SIGLEC8 and myeloperoxidase (MPO) (to label macrophages, T-cells, eosinophils and neutrophils, respectively). Following immunofluorescence labeling, the tissue was incubated with fluorescent phalloidin (to label filamentous actin) and DAPI (to label cell nuclei). The tissue was then mounted in a permanent mount and imaged using spectral confocal microscopy and the same 3D mosaic imaging approach described above.
Although the excitation and emission spectra of the fluorescent probes are offset from one another, they nonetheless overlap with one another to varying degrees. In order to selectively distinguish the fluorescence of each probe, we employed an approach in which spectral information obtained separately for each probe was used to “unmix” the images of the combined fluorescence (see Methods). In order to capture as much spectral information as possible, the sample was illuminated sequentially at 405 nm, 488 nm, 552 nm and 635 nm, and 4 channels of fluorescence were collected for each illumination. This procedure was repeated for each focal plane for each panel of the mosaic, resulting in a rich 16-channel image volume of the tissue that was then spectrally deconvolved, using linear unmixing, to distinguish the 8 different fluorescent probes.
Figure 3 shows how the addition of probes to detect filamentous actin, THP and AQP1 augments the information provided by autofluorescence and SHG. As expected, fluorescent phalloidin strongly labeled the filamentous actin of the vasculature, glomerular capillaries, basement membranes and tubule brush borders. The magnified images shown in panels D-I demonstrate that AQP1 immunofluorescence correlates well with the phalloidin fluorescence of the brush borders of proximal tubule cells. In contrast, immunolabeling for THP identifies a distinct set of tubules of the thick ascending limb, but also strongly labels structures in tubule lumens that appear to be tubular casts. Interestingly, regions showing strong SHG fluorescence and amorphous phalloidin labeling correlate with regions lacking immunofluorescence (indicated with arrows in Figures 3G, H and I), consistent with the loss of tubular markers in fibrotic regions.
Figure 3. 3D confocal immunofluorescence of structural markers.
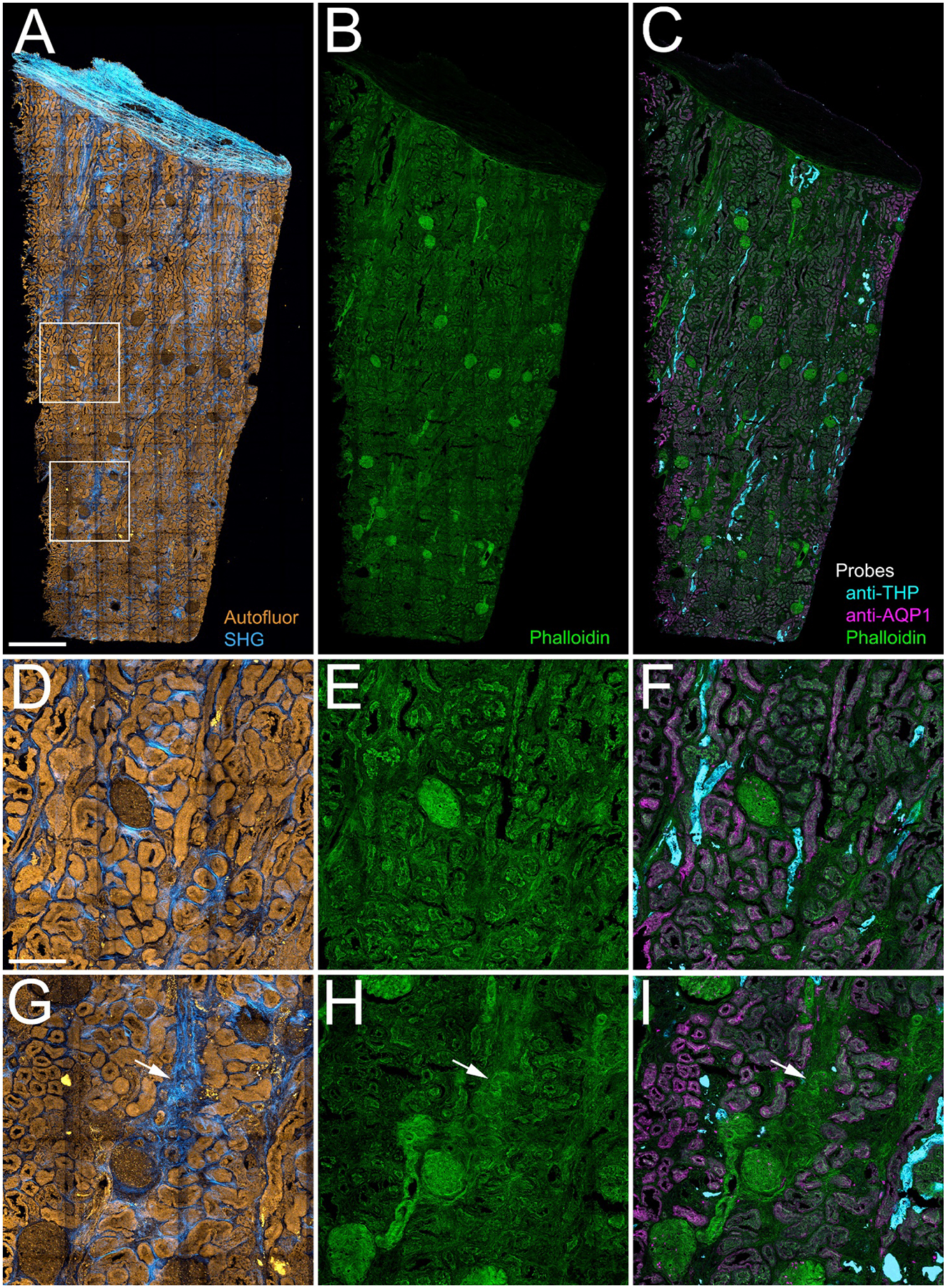
Panel A - Maximum projection of combined autofluorescence and SHG images. Panel B - Maximum projection of mosaic of confocal fluorescence image volumes of Oregon-Green phalloidin. Panel C - Overlay of maximum projection confocal fluorescence images of phalloidin (green) and antibodies to Tamm-Horsfall Protein (THP, cyan) and aquaporin-1 (AQP1, magenta). Panels D-F show corresponding 4X magnification images of the region indicated in the upper box in panel A, and panels G-I show corresponding 4X magnification images of the region indicated in the lower box in panel A. Arrows in panels G-I indicate regions of fibrosis and apparent tubular dropout. Scale bar in panel A represents 1 mm. Scale bar in panel D represents 250 microns.
3D tissue cytometry of human kidney
Figure 4A shows a projection of the same tissue volume shown in Figure 3C, with the addition of the nuclear fluorescence of DAPI and the immunofluorescence detected for CD68, CD3, and MPO (labeling macrophages, T-cells and neutrophils, respectively). In order to provide a sense of the 3D structure of this tissue, a video of a volume rendering is also presented in Supplementary Video 1. Although several interesting patterns are apparent in the figure and in the video, it is clear that the richness of the data in this 9800 × 19,800 × 50 voxel, 8-probe image volume exceeds our ability to evaluate it visually. In order to fully explore and discover meaningful patterns in an image of this scale and complexity, the image must be converted into quantitative data.
Quantification of large-scale three dimensional images of tissues has been conducted using an approach that has been variously termed histo-cytometry (9, 23), 3D confocal cytometry (12), 3D imaging cytometry (10) and tissue cytometry (13–15). Conceptually similar to flow cytometry, tissue cytometry is based upon the detection and attribution of immunofluorescence signal levels to individual cells, providing a multi-parameter analysis of a cell population. However, as it is based upon fluorescence measurements detected as a laser scans over a tissue, tissue cytometry provides this information along with the spatial location of each cell, as demarcated by methods of digital image analysis (segmentation). The combination of large-scale microscopy with automated methods of digital image analysis transforms microscopy from a tool for characterizing sample regions into one providing a census of every cell in a tissue sample.
While a variety of software tools have been developed to support quantitative analysis of two-dimensional images, few have been developed for the analysis of large-scale 3D image volumes. Moreover, none integrate all of the necessary tools of image processing, segmentation, quantification and data analysis into a single package. We developed VTEA in response to the need for an accessible, yet powerful solution to 3D tissue cytometry. Developed as a plug-in for the widely-used ImageJ freeware, VTEA organizes each of the steps of tissue cytometry into a bi-directional workflow that facilitates interactive exploration and analysis of complex, 3D image volumes.
VTEA tissue cytometry is based upon a strategy in which each individual cell is identified by its fluorescently-labeled nucleus and then characterized by quantification of the immunofluorescence of the cell markers in the region immediately surrounding the nucleus. A 3D watershed segmentation of the nuclei in the image shown in Figure 4A identified 248,050 nuclei from the 1.7 mm3 volume of the tissue, each of which is depicted in a different color in Figure 4B. Although difficult to discern in this image, the tissue includes several high-density regions, which are shown in a density plot in the inset. Some of these regions are associated with regions of immune cell infiltrates that are apparent in the original image (indicated with arrows in Figure 4A).
The nuclear segmentation is used to define regions that are used to quantify nuclear fluorescence and, in the region immediately surrounding the segmented nucleus, the fluorescence of each of the 8 fluorescent probes following linear unmixing. VTEA presents these data in csv files, as well as in interactive scatterplots that can be used to visualize the distributions of values any two measurement parameters along the x and y axes (Figures 4F and 4G). In order to extend the dimensionality of these scatterplots, each point can be rendered in a color representing the value of a third measurement parameter of the user’s choice.
The VTEA scatterplots can be used to identify specific cell types in fluorescence images in an approach similar to that used by FlowJo to identify cells in flow cytometry analyses of suspended cells. Like FlowJo, VTEA provides the user with a tool to draw “gates” on the scatterplots to identify specific cell types based upon the levels of fluorescence of specific cell markers. Figure 4F shows examples of gates drawn onto a scatterplot of AQP1 and THP fluorescence to define cells of the proximal tubule and thick ascending limb, respectively. This gating procedure is repeated for each of the cell markers to identify each cell type and the resulting gated data is used by VTEA to generate a new version of image in which each the identity of each cell is mapped onto its nucleus in a unique color. Figure 4C shows the results of scatterplot mapping of different cell types based upon gating the fluorescence levels obtained from the image shown in Figure 4A. Figures 4H and 4I show the results of gating the magnified sub-regions shown in Figure 4D and E, respectively. In these examples, the fluorescence of antibodies against THP, AQP1, MPO, CD68 and CD3 was used to identify cells of the thick ascending limb, proximal tubule, neutrophils, macrophages and T cells, respectively. The fluorescence of phalloidin was used to detect cells rich in F-actin, primarily cells of the glomeruli and peritubular capillaries. As this mapping is immediately updated as gates are adjusted, it provides an effective method for visually validating the gating strategy. As the gated cells are rendered in a single, bright color, the mapped image also serves to more clearly delineate the distribution of dimly-labeled cells (e.g., CD3+ cells).
VTEA’s scatterplot functions are crucial to accurate identification of specific cell populations in highly multiplexed image volumes. The quantitative analysis and gating procedures described above were conducted on fluorescence data following linear unmixing which, as described above, was used to distinguish the fluorescence of each individual probe from the combined spectrum of the 8 fluorescent probes. Although linear unmixing is effective for distinguishing probes whose fluorescence levels are similar, it frequently fails to completely eliminate crosstalk of strong signals into the spectral channels used to detect weaker signals. In the samples analyzed here, the fluorescence of the THP probe was much stronger than the other probes. However, VTEA scatterplot tools can be used to selectively identify cells whose target probe signals are contaminated by fluorescence crosstalk by the intense THP signal. For example, the crosstalk from THP cells with high signal spilling into the AQP1 channel can be parsed out by plotting AQP1 vs. THP (Figure 4F). VTEA’s ability to represent a third dimension of data in 2D scatterplots using color can also be used to isolate the effect of cross-talk. In Figure 4G, color is used to represent AQP1 fluorescence. When viewed in this way, the cells whose CD68 signal derives from AQP1 crosstalk are immediately apparent, and easily excluded from the “true” CD68+ gate.
The data derived from VTEA analysis of this volume are summarized in Table 1, along with those from four other nephrectomy tissue samples, whose images are shown in Supplementary Figure 3. The degree of fibrosis varied between a minimum of 7.7 to a maximum of 12.3% of total areas. As expected, the distribution and abundance of tubular cells varies depending on various renal areas sampled (cortex vs medulla). Interestingly, there was a consistent immune cell infiltration in all the specimens imaged, distributed in a focal pattern within each tissue. The average number of immune cells (reported as % of total cells) were: 2.1± 0.9, 0.3± 0.1 and 5.0± 2.2, for neutrophils, inflammatory macrophages and T Cells, respectively.
Multiplexed analysis of tissues at this scale offers unique benefits. First, the extended volume of sampling increases the likelihood of detecting rare events. The scale of the image is sufficient to detect multiple highly-localized foci of inflammation, which may be missed when surveying a small area of tissue. More significantly, the ability to conduct a multiparametric analysis of multiple events provides the opportunity to identify meaningful correlations that might otherwise be missed. For example, four regions of inflammation are indicated in Figure 5A, each with a different constitution of T cells (white) and neutrophils (red) (Figure 5B–E). While the underlying basis of these differences is unknown, an evaluation of the corresponding immunofluorescence, autofluorescence and SHG images suggests that they may reflect regions at different stages of inflammation, associated with various degrees of fibrosis (increased SGH density) and tubular drop-out (loss of autofluorescence, AQP1 and THP immunofluorescence). While this interpretation may be speculative, it demonstrates how an image of an entire tissue might provide a snapshot in time of multiple regions of injury, each at a different stage, and how a multiparameter analysis might be used to understand the progression of injury.
Figure 5. 3D confocal fluorescence and multiphoton autofluorescence/SHG microscopy of regions of apparent injury.
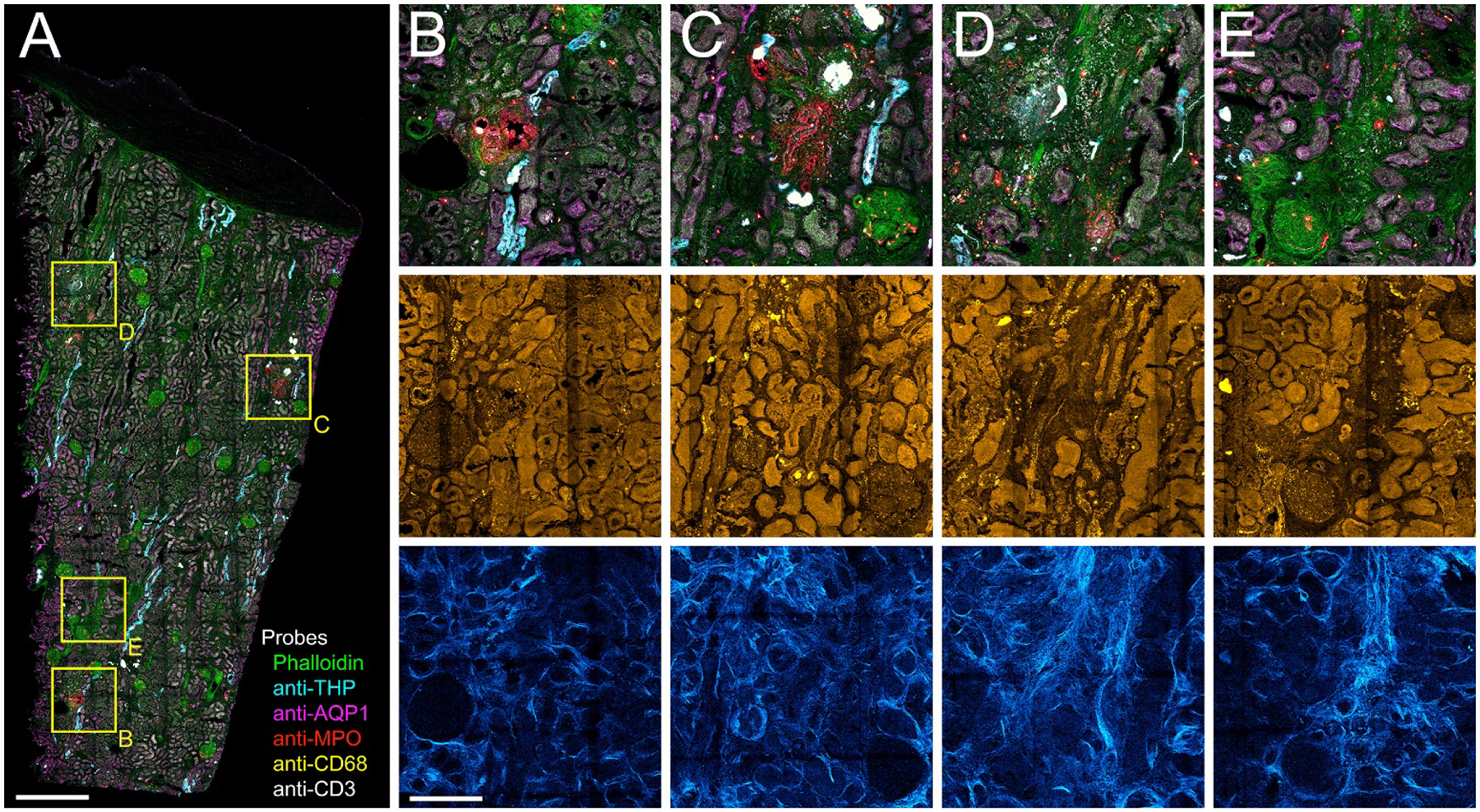
Panel A - Maximum projection of combined 3D fluorescence image volume of phalloidin (green) and antibodies to THP (cyan), AQP1 (magenta), myeloperoxidase (MPO, red), CD68 (yellow) and CD3 (white). Panels B-E - 4X magnifications of regions indicated in boxes in panel A. Top row – confocal fluorescence images. Middle row – corresponding autofluorescence images. Bottom row – corresponding SHG images. Scale bar in panel A represents 1 mm and in panel B 250 um for B-E.
Application of large scale 3D tissue cytometry to kidney biopsies
Figure 6 shows an example of one of the most exciting applications of tissue cytometry - the analysis of clinical biopsy samples, in this case kidney core biopsies obtained from patients diagnosed with diabetic nephropathy (details listed in Supplementary Table 1). Label-free imaging uncovers the structural make-up of the biopsies (glomeruli, tubules, renal capsule, etc.) and the associated area of collagen deposition through SHG. Spectral confocal imaging and results of tissue cytometry with VTEA are shown in Figure 6 and Table 2, respectively. In the example shown, we can infer an advanced degree of fibrosis, which corresponds to the advanced clinical disease of the patients (CKD stages 3 and 4). Although it may be premature to derive meaningful conclusions from this small sample size, the analysis already underscores the heterogeneity of diabetic kidney disease and the presence of active foci of inflammation with neutrophil and macrophage infiltration even in advanced disease. Other observations of interest are the change in structural markers, especially THP, which is significantly decreased compared to reference tissue with comparable amount of cortical tissue (samples 1,2 and 4, Table 1): THP+ cells (%): 0.2 ± 0.2 vs. 6.1±1.9, respectively (p= 0.03). The absence of T cells is also noted (P<0.05 compared to reference tissue) and needs to be validated in a larger series.
Figure 6. 3D Multiphoton autofluorescence/SHG and confocal immunofluorescence microscopy of diabetic renal biopsies.
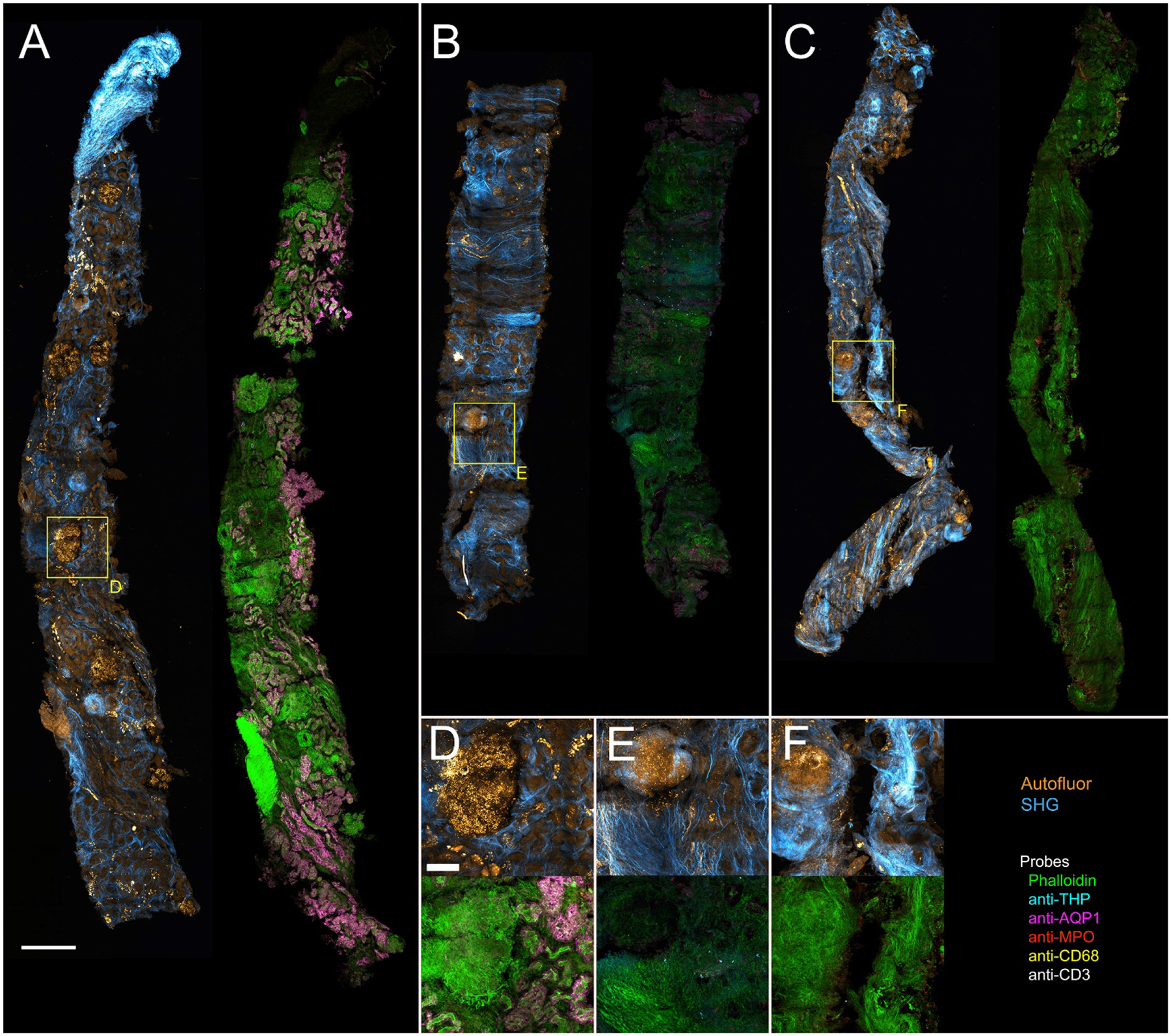
Gallery of 50 um sections of diabetic biopsies. Panels A–C - Maximum projections of combined 3D label-free autofluorescence and SHG images (Autofluor/SHG) (left0 and immunofluorescence confocal images (right). Phalloidin (green), THP (cyan), AQP1 (magenta), myeloperoxidase (MPO, red), CD68 (yellow) and CD3 (white). Panels D-F - insets from biopsies representing highlighted regions in panels A through C, respectively. The top row shows maximum projections of combined autofluorescence and SHG images and the bottom row shows corresponding immunofluorescence images. Scale bar in panel A represents 2 mm and in panel D represents 100 microns (D-F)
Table 2 –
Results of label free and VTEA analysis of 3 diabetic biopsies
| Sample | % Cortex | Tissue volume mm3 |
SHG+ area % total |
Total cell | Cell density cells/mm3 |
THP+ cells/mm3 (%) |
AQP1+ cells/mm3 (%) |
MPO+ cells/mm3 (%) |
CD68+ cells/mm3 (%) |
CD3+ cells/mm3 (%) |
Siglec8+ cells/mm3 (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DN1 Fig 6A |
90.8 | 0.1281183 | 9.4 | 59,852 | 467,163 | 343 (0.07) |
52131 (11.2) |
991 (0.2) |
4355 (0.9) |
0 (0) |
47 (0.001) |
| DN2 Fig 6B |
100 | 0.1018575 | 12.6 | 48,827 | 479,365 | 2,926 (0.6) |
88 (0.02) |
2258 (0.5) |
216 (0.05) |
0 (0) |
30 (0.006) |
| DN3 Fig 6C |
100 | 0.0729754 | 27.3 | 41,536 | 569,178 | 260 (0.05) |
96 (0.02) |
2480 (0.3) |
12443 (2.2) |
0 (0) |
41 (0.007) |
Figure 7 shows a specific example of how tissue cytometry can be used to study the cellularity of glomeruli, a key site of pathology in diabetic kidney disease. Compared to reference tissue, where the range of glomerular cellular density is consistent, diabetes induces an increase in glomerular cellular density in many but not all the studied biopsy specimens (Figure 7A). Interestingly, cellularity remains homogeneous within each specimen, suggesting that glomerular pathology induced by diabetes is global and not focal in nature. Furthermore, diabetes does not increase immune cell infiltration of glomeruli, as shown in Figure 7B.
Figure 7. Scatterplots of glomerular nuclear density and immune cell density.
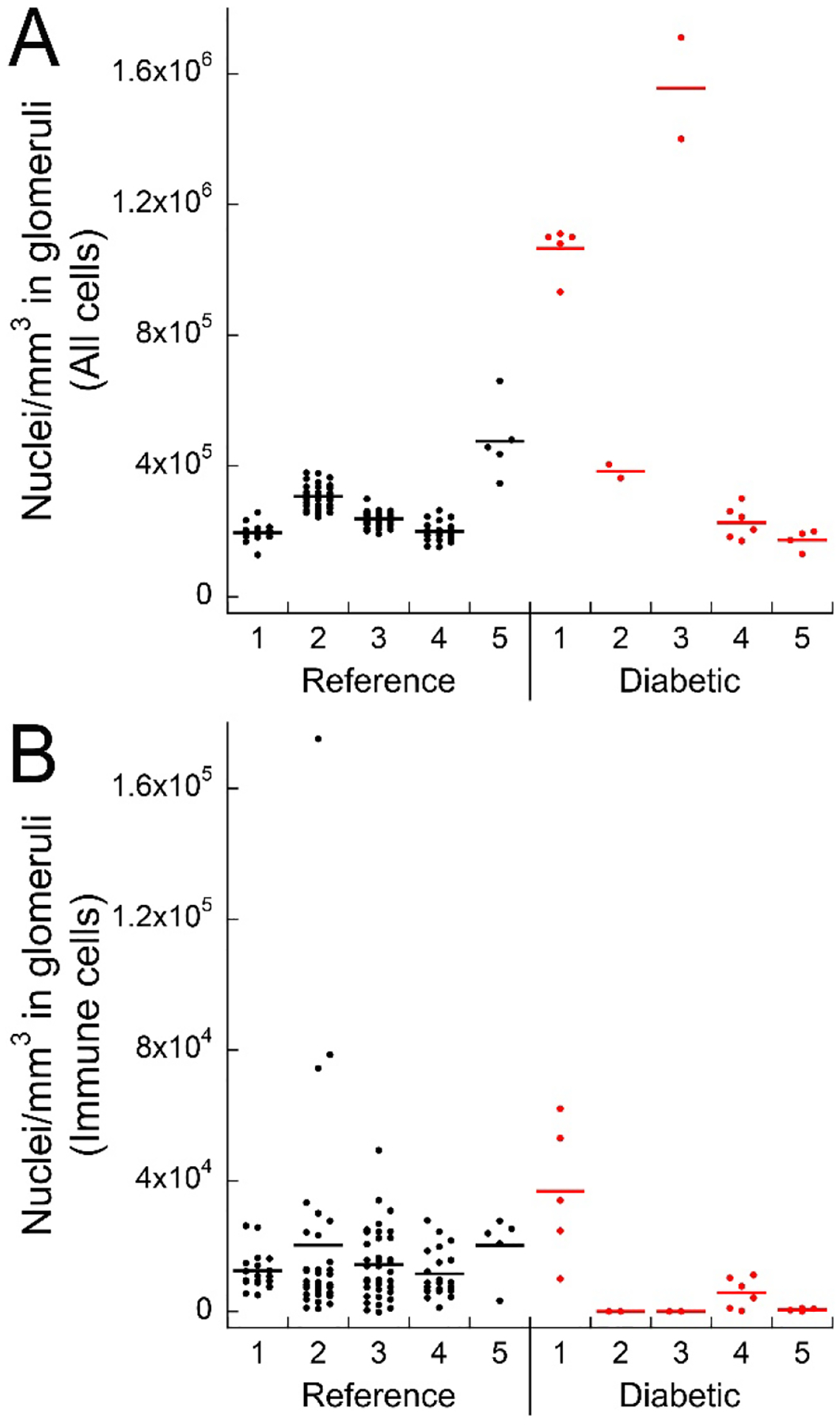
Panel A - Total cell densities for 5 reference (black dots) and 5 diabetic (red dots) cases. Panel B - Density of all probed immune cells (myeloperoxidase (MPO), CD68, CD3 and SIGLEC8 positive cells) from the same samples. Each dot represents a single glomerulus.
Discussion
Here we have demonstrated an accessible approach to tissue cytometry, a technique that is increasingly used to analyze and understand the complex biology of human and animal tissues. As compared with traditional methods of histology, fluorescence-based tissue cytometry offers several advantages. First, the fluorescence modality is compatible with confocal or multiphoton microscopy, which can be used to collect 3D image volumes, providing unique structural information, more accurate representations of spatial relationships and a more accurate and extensive census of cells in a tissue. Second, fluorescently-labeled antibodies, lectins and toxins can be used to provide molecular specificity, which supports enumeration of specific cell types and structures. Third, fluorescence supports multiplexing, which can be used to detect multiple cell types, and thus their interactions, in the same tissue. Finally, quantitative methods of digital image analysis are more fully developed and implemented for the analysis of fluorescence images, facilitating automated analysis of large and complex image volumes.
The nascent field of tissue cytometry developed naturally (and inevitably) from the development of methods for large-scale, high-content, high-resolution imaging. The automation of confocal microscope systems gave researchers the capability to collect fluorescence images of centimeter-scale samples at submicron resolution. The development and implementation of methods of tissue clearing extended the depth of image volumes from tens of microns into the range of millimeters. Whereas once confocal image volumes might consist of the fluorescence of two or three probes, new laser systems and methods of spectral deconvolution now enable researchers to simultaneously resolve 7 or more probes in the same sample.
The scale and complexity of these image volumes presents both an opportunity and a challenge. Capable of providing rich molecular data for entire tissue biopsies, tissue cytometry can transform fluorescence microscopy from a tool used to qualitatively characterize “representative fields” into a tool for generating a quantitative, multi-parameter census of every cell in a tissue. However, tissue cytometry is still a developing field – effectively extracting information from such large and complex image volumes depends upon developing methods of microscopy optimized for quantitative analysis, methods of image analysis suited to quantitatively characterizing individual cells in a tissue and methods of data analysis that can be used to explore and, ideally, discover unforeseen patterns in the data. Thus, tissue cytometry remains a tool that is utilized by relatively small number of laboratories with particular skill in quantitative microscopy. Here we have presented a straightforward workflow for tissue cytometry based upon commonly used methods of tissue processing, microscopy techniques that can be implemented by nearly all commercially-available confocal microscope systems and an image and data analysis workflow based upon the easy-to-use VTEA freeware that we developed as a plug-in for ImageJ. We specifically show how this approach can be used to study human kidney tissue both in nephrectomy reference tissue and from kidney biopsies from patients with diabetes.
Although we have described a workflow that is specific in its details, tissue cytometry is a highly flexible research tool that is compatible with a variety of different methods of tissue preparation and fluorescence microscopy. Our approach is based upon analyses of OCT-frozen tissue cryosections. While frozen tissues are common to research laboratories and tissue banks, a rich repository of clinical samples are embedded in paraffin. Such samples are compatible with immunofluorescence once “deparaffinized” by treatment with organic solvents. This approach was used in recent studies in which quantitative multiplexed immunofluorescence was used to analyze human tumor biopsies (10, 11). These studies demonstrate the exciting potential of tissue cytometry as a tool for extracting new data from archived clinical samples. Cellular and molecular data obtained from tissue cytometry could be combined with corresponding clinical data to provide unique insights into disease and therapy. However, the potential large-scale studies of this kind will critically depend upon the quality and consistency of the clinical samples.
For our studies, tissue samples were cut to a nominal thickness of 50 microns, a thickness that could be rapidly labeled and imaged, and yielded substantial amounts of high-quality data. However, spatial scale of tissue cytometry can be significantly increased through the use of “tissue clearing” techniques that extend the depth of fluorescence microscopy by reducing light scatter. Tissue cytometry has been applied to cleared tissues to characterize cell populations in tissues samples that are hundreds of microns thick (10, 12, 23). While tissue clearing significantly extends the scope of fluorescence imaging, it does so at the cost of time. First the process of tissue clearing typically requires days in itself. Second, extending the thickness of tissues extends the time required for all incubations as well as the time required to collect image volumes.
The process of collecting multiplexed fluorescence images required 24–36 hrs per sample, largely due to the inherently slow process of single-point scanning confocal microscopy. Alternative modes of microscopy could be used to accelerate image capture. For example, microscope systems such as spinning-disk confocal and light-sheet microscopes speed the process of image capture by collecting images of entire fields in a matter of milliseconds. A light-sheet microscope system, modified to accommodate the geometry of tissue sections on slides, was demonstrated to be capable of collecting a 1 mm3 volume in less than one minute (24). Another alternative is the “ribbon-scanning” confocal microscope system, which uses a high-speed resonant scanner to continuously scan a sample as the stage translates though the sample. A commercially-available system was recently demonstrated that could scan a two-channel image of an entire 1 mm2 field in less than 3 minutes (25). The impressive speed advantage of the light-sheet and ribbon scanning systems is reduced somewhat for studies involving more than one or two probes, since collection of additional channels requires repeated, time-consuming filter changes (for the light-sheet system) or additional stage scans (for the ribbon scanning confocal).
In order to glean as much information as possible from each sample, we first used multiphoton microscopy to acquire autofluorescence and second harmonic images of unlabeled tissue, and then used confocal microscopy to acquire 8-channel fluorescence images following labeling with fluorescent probes. Multiphoton-excited autofluorescence and SHG have previously been used to analyze mouse experimental mouse models of kidney injury (26, 27) and to characterize unlabeled human bladder biopsies (28), human prostate (29) and human colorectal cancer (30). However, we are unaware of studies in which multiphoton microscopy of unlabeled tissue was followed by confocal microscopy of the same tissue following fluorescent labeling. Imaging the same tissue before and after labeling is complicated by the need to manipulate the sample between the two image collections. In our protocol, the unlabeled tissue was removed from the temporary mount for fluorescent labeling, subsequent confocal imaging was conducted after the tissue was mounted a second time. The process of tissue incubation and remounting introduced small changes in the overall shape of the tissue, so that the multiphoton and confocal images were only approximately registered with one another, compromising our ability to include autofluorescence and SHG data in the VTEA cytometry analysis. We have since developed a new technique whereby tissues are fluorescently labeled while still mounted on the slide, eliminating the misregistration of multiphoton and confocal fluorescence images. Doing so will increase the data-space of each cell in the tissue so that it includes not only the 8 fluorescent probes, but also the autofluorescence and SHG associated with each cell.
Multiplexed fluorescence microscopy is complicated by the extensive overlap in the excitation and emission spectrum of fluorescent probes, which limits the discrimination of any single fluorophore in a multiply-labeled sample. Imaging up to 3 or 4 probes can be accomplished by judicious choice of optical filters. However, distinguishing additional fluorescent probes typically requires spectral deconvolution of fluorescence images, particularly in the presence of tissue autofluorescence. In our studies, we used linear unmixing to discriminate 8 fluorescent probes. We found that linear unmixing was frequently only partially successful in removing the strong fluorescence of the probes used to detect AQP1 and THP from that of probes used to identify immune cells. Whereas residual signal contamination would ordinarily be difficult to detect and correct, we describe how VTEA’s scatterplot tools simplify the process of discriminating cell populations in the presence of signal crosstalk. The VTEA scatterplots can also be used to identify signal saturation, an issue that is frequently encountered due to between-sample variability, and compromises the signal linearity that is prerequisite to linear unmixing.
The problem of spectral crosstalk in multiplexed imaging can be avoided in two ways. First, a number of techniques have been developed whereby highly multiplexed fluorescence images are assembled from a sequence of images of tissues labeled with between one and three probes at a time. In this approach, following imaging, the first set of probes are removed (31–34) or their fluorescence is quenched (35–38), the sample is labeled with a new set of probes, and the tissue is imaged again. As this process is repeated, highly-specific images of 50 or more different antigens can be collected from the same tissue sample. The second approach to avoiding spectral crosstalk is to abandon fluorescence as the source of image contrast. Imaging mass cytometry uses heavy metal isotopes to label antibodies, which are then used like fluorescently-labeled antibodies to label tissues. The isotopes are detected using mass spectrometer following ablation of the tissue with either a laser (39) or ion beam (40). Using isotopes with appropriately distinct mass-to-charge ratios, images of more than 30 probes can be collected with subcellular resolution (39). The major drawback of sequential fluorescence and imaging mass cytometry is that neither is compatible with tissues more than a few microns in thickness, limiting their ability to characterize tissues in three dimensions.
Perhaps the most challenging aspects of tissue cytometry are those involved with extracting and exploring the complex data obtained from large multiplexed 3D image volumes. In most published examples of 3D tissue cytometry (3–10, 41), image analysis is first conducted using combinations of custom-designed and commercial software, and the resulting data are then exported to Excel and/or FlowJo for data analysis. The Schroeder laboratory developed an alternative approach, in which images are first segmented using Imaris, and then analyzed using the custom-designed XiT software which provides additional capabilities for image quantification and data exploration (42). We developed VTEA in response to the need for powerful, yet accessible software that incorporates the entire image and data analysis into a single platform. This design provides a seamless bidirectional conduit between the image and data space, facilitating refinement of image analysis, interactive exploration of image data and discovery of unique patterns and associations. Insofar as VTEA is an evolving platform that is continuously being refined and extended, the version demonstrated here represents only a fraction of the capabilities that we will have incorporated into the version available at the time of publication. In addition to the features demonstrated here, the new version will provide tools for (1) analysis of structural features, (2) analysis of spatial relationships and (3) unsupervised analysis of multiplex data.
In conclusion, we provide a complete workflow to perform large scale 3D imaging and tissue cytometry on human kidney tissue, which could be used to extract “big data” type spatial information from kidney biopsies from patients with kidney disease. We showed examples of how such analyses can provide useful insights that can be linked to the biology or pathology of kidney disease. We anticipate that this imaging and analytical approach will complement other Omics type outputs and could help spatially map pathological changes that occur with kidney disease.
Supplementary Material
Acknowledgements
This work was supported by grants from the NIDDK: UH3DK114923 and DiaComp. We thank the KPMP consortium members for their significant input and support
Footnotes
Disclosures/Conflicts of Interest
The authors declare that they have no conflicts of interest
Supplementary information is available at Laboratory Investigation’s website.
References
- 1.Malone AF, Wu H, Humphreys BD. Bringing Renal Biopsy Interpretation Into the Molecular Age With Single-Cell RNA Sequencing. Semin Nephrol. 2018;38:31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park J, Liu CL, Kim J, Susztak K. Understanding the kidney one cell at a time. Kidney Int. 2019; 96:862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrovas C, Ferrando-Martinez S, Gerner MY, Casazza JP, Pegu A, Deleage C, et al. Follicular CD8 T cells accumulate in HIV infection and can kill infected cells in vitro via bispecific antibodies. Sci Transl Med. 2017;9:eaag2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Gerner MY, Van Panhuys N, Levine AG, Rudensky AY, Germain RN. Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature. 2015;528:225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fonseca DM, Hand TW, Han SJ, Gerner MY, Glatman Zaretsky A, Byrd AL, et al. Microbiota-Dependent Sequelae of Acute Infection Compromise Tissue-Specific Immunity. Cell. 2015;163:354–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radtke AJ, Kastenmuller W, Espinosa DA, Gerner MY, Tse SW, Sinnis P, et al. Lymph-node resident CD8alpha+ dendritic cells capture antigens from migratory malaria sporozoites and induce CD8+ T cell responses. PLoS Pathog. 2015;11:e1004637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerner MY, Torabi-Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity. 2015;42:172–85. [DOI] [PubMed] [Google Scholar]
- 9.Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 2012;37:364–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SS, Bindokas VP, Lingen MW, Kron SJ. Nondestructive, multiplex three-dimensional mapping of immune infiltrates in core needle biopsy. Lab Invest. 2019;99:1400–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halse H, Colebatch AJ, Petrone P, Henderson MA, Mills JK, Snow H, et al. Multiplex immunohistochemistry accurately defines the immune context of metastatic melanoma. Sci Rep. 2018;8:11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coutu DL, Kokkaliaris KD, Kunz L, Schroeder T. Three-dimensional map of nonhematopoietic bone and bone-marrow cells and molecules. Nat Biotechnol. 2017;35:1202–10. [DOI] [PubMed] [Google Scholar]
- 13.Winfree S, Ferkowicz MJ, Dagher PC, Kelly KJ, Eadon MT, Sutton TA, et al. Large-scale 3-dimensional quantitative imaging of tissues: state-of-the-art and translational implications. Transl Res. 2017;189:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winfree S, Khan S, Micanovic R, Eadon MT, Kelly KJ, Sutton TA, et al. Quantitative Three-Dimensional Tissue Cytometry to Study Kidney Tissue and Resident Immune Cells. J Am Soc Nephrol. 2017;28:2108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winfree S, Dagher PC, Dunn KW, Eadon MT, Ferkowicz M, Barwinska D, et al. Quantitative Large-Scale Three-Dimensional Imaging of Human Kidney Biopsies: A Bridge to Precision Medicine in Kidney Disease. Nephron. 2018;140:134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Micanovic R, Khan S, Janosevic D, Lee ME, Hato T, Srour EF, et al. Tamm-Horsfall Protein Regulates Mononuclear Phagocytes in the Kidney. J Am Soc Nephrol. 2018;29:841–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LaFavers KA, Macedo E, Garimella PS, Lima C, Khan S, Myslinski J, et al. Circulating uromodulin inhibits systemic oxidative stress by inactivating the TRPM2 channel. Sci Transl Med. 2019;11:eaaw3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makki MS, Winfree S, Lingeman JE, Witzmann FA, Worcester EM, Krambeck AE, et al. A Precision Medicine Approach Uncovers a Unique Signature of Neutrophils in Patients With Brushite Kidney Stones. Kidney Int Rep. 2020;5:663–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croce AC, Bottiroli G. Autofluorescence spectroscopy and imaging: a tool for biomedical research and diagnosis. Eur J Histochem. 2014;58:2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Croce AC, Ferrigno A, Bottiroli G, Vairetti M. Autofluorescence-based optical biopsy: An effective diagnostic tool in hepatology. Liver Int. 2018;38:1160–74. [DOI] [PubMed] [Google Scholar]
- 21.Dong CY, Campagnola PJ. Optical diagnostics of tissue pathology by multiphoton microscopy. Expert Opin Med Diagn. 2010;4:519–29. [DOI] [PubMed] [Google Scholar]
- 22.Ranjit S, Dobrinskikh E, Montford J, Dvornikov A, Lehman A, Orlicky DJ, et al. Label-free fluorescence lifetime and second harmonic generation imaging microscopy improves quantification of experimental renal fibrosis. Kidney International. 2016;90:1123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Germain RN, Gerner MY. Multiplex, quantitative cellular analysis in large tissue volumes with clearing-enhanced 3D microscopy (Ce3D). Proc Natl Acad Sci U S A. 2017;114:E7321–E7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glaser AK, Reder NP, Chen Y, Yin C, Wei L, Kang S, et al. Multi-immersion open-top light-sheet microscope for high-throughput imaging of cleared tissues. Nat Commun. 2019;10:2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson AM, Rose AH, Gibson GA, Gardner CL, Sun C, Reed DS, et al. Ribbon scanning confocal for high-speed high-resolution volume imaging of brain. PLoS One. 2017;12:e0180486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ranjit S, Dobrinskikh E, Montford J, Dvornikov A, Lehman A, Orlicky DJ, et al. Label-free fluorescence lifetime and second harmonic generation imaging microscopy improves quantification of experimental renal fibrosis. Kidney Int. 2016;90:1123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strupler M, Hernest M, Fligny C, Martin JL, Tharaux PL, Schanne-Klein MC. Second harmonic microscopy to quantify renal interstitial fibrosis and arterial remodeling. J Biomed Opt. 2008;13:054041. [DOI] [PubMed] [Google Scholar]
- 28.Jain M, Robinson BD, Scherr DS, Sterling J, Lee MM, Wysock J, et al. Multiphoton microscopy in the evaluation of human bladder biopsies. Arch Pathol Lab Med. 2012;136:517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tewari AK, Shevchuk MM, Sterling J, Grover S, Herman M, Yadav R, et al. Multiphoton microscopy for structure identification in human prostate and periprostatic tissue: implications in prostate cancer surgery. BJU Int. 2011;108:1421–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsui T, Mizuno H, Sudo T, Kikuta J, Haraguchi N, Ikeda JI, et al. Non-labeling multiphoton excitation microscopy as a novel diagnostic tool for discriminating normal tissue and colorectal cancer lesions. Sci Rep. 2017;7:6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, et al. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell. 2018;174:968–81 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agasti SS, Wang Y, Schueder F, Sukumar A, Jungmann R, Yin P. DNA-barcoded labeling probes for highly multiplexed Exchange-PAINT imaging. Chem Sci. 2017;8:3080–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Woehrstein JB, Donoghue N, Dai M, Avendano MS, Schackmann RCJ, et al. Rapid Sequential in Situ Multiplexing with DNA Exchange Imaging in Neuronal Cells and Tissues. Nano Lett. 2017;17:6131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wahlby C, Erlandsson F, Bengtsson E, Zetterberg A. Sequential immunofluorescence staining and image analysis for detection of large numbers of antigens in individual cell nuclei. Cytometry. 2002;47:32–41. [PubMed] [Google Scholar]
- 35.Hillert R, Gieseler A, Krusche A, Humme D, Rowert-Huber HJ, Sterry W, et al. Large molecular systems landscape uncovers T cell trapping in human skin cancer. Sci Rep. 2016;6:19012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schubert W, Bonnekoh B, Pommer AJ, Philipsen L, Bockelmann R, Malykh Y, et al. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol. 2006;24:1270–8. [DOI] [PubMed] [Google Scholar]
- 37.Lin JR, Fallahi-Sichani M, Sorger PK. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat Commun. 2015;6:8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A. 2013;110:11982–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417–22. [DOI] [PubMed] [Google Scholar]
- 40.Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. 2014;20:436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moreau HD, Lemaitre F, Terriac E, Azar G, Piel M, Lennon-Dumenil AM, et al. Dynamic in situ cytometry uncovers T cell receptor signaling during immunological synapses and kinapses in vivo. Immunity. 2012;37:351–63. [DOI] [PubMed] [Google Scholar]
- 42.Coutu DL, Kokkaliaris KD, Kunz L, Schroeder T. Multicolor quantitative confocal imaging cytometry. Nat Methods. 2018;15:39–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.