

Abstract
Endosperm is an angiosperm innovation central to their reproduction whose development, and thus seed viability, is controlled by genomic imprinting, where expression from certain genes is parent-specific. Unsuccessful imprinting has been linked to failed inter-specific and inter-ploidy hybridization. Despite their importance in plant speciation, the underlying mechanisms behind these endosperm-based barriers remain poorly understood. Here, we describe one such barrier between diploid Mimulus guttatus and tetraploid Mimulus luteus. The two parents differ in endosperm DNA methylation, expression dynamics, and imprinted genes. Hybrid seeds suffer from underdeveloped endosperm, reducing viability, or arrested endosperm and seed abortion when M. guttatus or M. luteus is seed parent, respectively, and transgressive methylation and expression patterns emerge. The two inherited M. luteus subgenomes, genetically distinct but epigenetically similar, are expressionally dominant over the M. guttatus genome in hybrid embryos and especially their endosperm, where paternal imprints are perturbed. In aborted seeds, de novo methylation is inhibited, potentially owing to incompatible paternal instructions of imbalanced dosage from M. guttatus imprints. We suggest that diverged epigenetic/regulatory landscapes between parental genomes induce epigenetic repatterning and global shifts in expression, which, in endosperm, may uniquely facilitate incompatible interactions between divergent imprinting schemes, potentially driving rapid barriers.
Diverged epigenetic/regulatory landscapes between parental genomes result in epigenetic repatterning in hybrids that drive global shifts in endosperm gene expression patterns.
Introduction
The evolution of a nutritive endosperm is thought to be closely tied to the evolutionary success of angiosperms (Friedman, 1995; Baroux et al., 2002). Uniquely, angiosperm seeds are produced by two separate fertilization events (Friedman et al., 2008): one event gives rise to the diploid zygote with the egg cell, while in the other an identical sperm cell fuses with two maternal polar nuclei (which are genetically identical to the egg) within the central cell. This secondary sexual product develops into the triploid endosperm in most angiosperm species. Nutrients and phytohormones from the maternal plant are transported to the endosperm within the seed, where they nourish and stimulate embryo development. Unlike gymnosperm seeds, where the nutritive source is maternal gametophyte tissue, resources are not wasted on unfertilized eggs in angiosperms since the endosperm typically cannot develop until fertilized (Baroux et al., 2002).

Angiosperm gametophytes are highly reduced compared with other land plant lineages, yet are a site of prominent epigenetic activity with unique patterns of chromatin state and DNA methylation (Huh and Rim, 2013). DNA methylation is an epigenetic mark that can alter the expression of DNA without altering its genetic sequence, often by physically preventing gene expression (Niederhuth et al., 2016). A methyl group is attached to cytosines in either a CG (hereon denoted as CpG), CHG, or CHH context (with H denoting any nucleotide other than guanine). CpG methylation is maintained through rounds of DNA replication by maintenance methyltransferases. CHG methylation, on the other hand, is maintained by chromomethylases that interact with methylation marks on neighboring histones, which also alter gene expression, thus producing a self-reinforcing loop. In the double-stranded DNA molecule, CHH methylation is asymmetric and must be re-established de novo after each round of DNA replication (Niederhuth and Schmitz, 2017). While CHH methylation may also be established by chromomethylases, de novo DNA methylation is largely directed by small RNAs (sRNAs) that are transcribed from noncoding DNA (Niederhuth and Schmitz, 2017). RNA-directed DNA methylation (RdDM) is highly specific and tends to target methylated heterochromatic regions such as transposon (TE) fragments near genes or coding regions of longer, autonomous TEs. RdDM establishes de novo methylation in all cytosine sequence contexts, although it is easiest to trace back patterns of CHH methylation since they cannot perpetuate through maintenance mechanisms (Niederhuth and Schmitz, 2017). Prior to fertilization, demethylation of DNA occurs in the central cell, particularly at TEs, including fragments near genes, resulting in global expression changes and TE activation (Gehring et al., 2009). Differences in DNA methylation between the central cell and the second sperm cell set the basis for genomic imprinting in the endosperm (Batista and Köhler, 2020).
After fertilization of the endosperm, genomic interactions between the maternal and paternal genomes control endosperm development and are characterized by genomic imprinting. Genomic imprinting is an epigenetic phenomenon where the alleles of certain genes are regulated differentially depending on their parent of origin, resulting in parent-specific patterns of gene expression (Matzke, 1993). RNA and protein production from an imprinted gene are largely restricted to either the maternal allele (termed a maternally expressed gene, or MEG) or the paternal allele (paternally expressed gene or PEG) (Matzke, 1993; Kinoshita, 2007). While the function or relevance of many MEGs and PEGs is unknown, several appear to have important roles in regulating development. For example, several MEGs are involved in preventing spontaneous endosperm development prior to fertilization or in metabolic and hormonal regulation after; and several PEGs are important for gene and chromosomal regulation and endosperm proliferation (Nowack et al., 2007; Figueiredo et al., 2015; Gehring and Satyaki, 2016; Roth et al., 2018b). In general, MEGs are typically predicted to restrict or regulate endosperm proliferation, while PEGs are predicted to promote it (Gehring and Satyaki, 2016). Such roles may have originated early in angiosperm evolution with the shift of the seed nutrient source from maternal tissues to the sexual endosperm (Friedman, 2001; Nowack et al., 2007), setting the foundation for advantageous parent-specific developmental cues that must reconcile into a careful balance in allelic dosage (Haig and Westoby, 1989; Haig, 2013).
Departures from the balance of the 2 maternal (m):1 paternal (p) genome dosage of triploid endosperm result in its abnormal development, apparently due to misregulation of genomic imprinting, and can act as a hybridization barrier (Haig and Westoby, 1991; Leblanc et al., 2002; Pennington et al., 2008). When the maternal progenitor is tetraploid (termed maternal excess; 4x × 2x, maternal × paternal), the endosperm has a 4m:1p ratio and thus increased maternal genomic dosage. In the reciprocal cross, when the paternal progenitor is tetraploid (paternal excess; 2x × 4x), the ratio is 2m:2p (1:1). Such crosses often result in “parent-of-origin” effects, whereby endosperm size decreases under maternal excess and increases under paternal excess (Scott et al., 1998), presumably due to altered ratios between MEGs and PEGs. Recent work has revealed global shifts in expression of both genes and TEs in endosperm with paternal excess as well as a buffer system in which the relative dosage of maternal transcripts increases in response to this paternal excess. Experimental crosses suggest that seed viability may be determined by modest changes involving only a few genes or loci that stem from paternally directed RdDM in the endosperm rather than a global departure from imprinting dosage balance (Satyaki and Gehring, 2019).
Hybridization between diverged species of the same ploidy may result in similar parent-of-origin phenotypes, which is again attributed to departures in the balance between imprinted genes. In this case, the imprinted “settings” of these genes are expected to have diverged between species (Haig and Westoby, 1991). Hypotheses describing the role of genomic imprinting as an inter-species hybridization barrier predict that the strength of imprinting differs between diverged species, thus producing “effective ploidy” differences with similar consequences to inter-ploidy hybridization (Johnston and Hanneman, 1982; Lafon-Placette et al., 2017; Roth et al., 2019). An example is the hybridization between a primarily selfing (inbreeding) species and an outcrossing species. Outcrossers are predicted to have greater selective pressures on competing parental strategies, resulting in stronger imprinting. Thus, such hybridization would lead to the same dosage imbalances as described above (Brandvain and Haig, 2005; Rebernig et al., 2015). Whether due to such pressures or not, evolutionary shifts in TE density and methylation, or other shifts affecting parent-specific dosage within regulatory networks vital to development, appear central to the imbalances that produce these inter-species endosperm-specific hybridization barriers (Lafon-Placette et al., 2018; Roth et al., 2019).
Genomic and epigenomic differences between species are known to induce genomic shock (McClintock, 1984) in their hybrids, where structural and regulatory changes generate global reprogramming and shifts in expression (Yoo et al., 2013; Lafon-Placette and Köhler, 2015). In particular, differences in DNA methylation, TE density, and sRNA production appear to be central in driving these transgressive epigenomic states, even when the parents are otherwise genetically identical (Greaves et al., 2012; Rigal et al., 2016). One common resolution to genomic, or “epigenomic”, shock and incompatibilities in hybrids is subgenome expression dominance, where the majority of genes from one “subgenome” (each subgenome being one of the two newly united parental genomes) are more highly expressed than their corresponding homologs in the other (Yoo et al., 2014). Such shifts in expression that alter parental input may be especially consequential in the endosperm where differential expression between parental alleles drives its proper development (Florez-Rueda et al., 2016). Furthermore, the disparate methylation and chromatin states, TE activity, and sRNA production between the central cell and the sperm cell present an additional layer to epigenetic differences between parental alleles in hybrid endosperm. Elucidating the consequences and mechanisms underlying endosperm-based hybridization barriers is crucial, since filial tissues are the earliest stage at which hybrid subgenomes first interact, and since endosperm development drives embryo development (Yan et al., 2014). These barriers are not only central in the study of genomic imprinting (Haig and Westoby, 1991) but are increasingly recognized to be widespread and major drivers of plant speciation (Lafon-Placette and Köhler, 2016; Lafon-Placette et al., 2017).
Here, we use a naturally occurring Mimulus (Phrymaceae) hybrid system that is both inter-ploidy and inter-species as a model to explore the underlying processes that form endosperm-based hybridization barriers. Mimulus guttatus is a diploid from North America, and M. luteus, from the Andes of South America, is an allotetraploid (formed from past hybridization of unknown origin) with two distinct subgenomes (subgenomes “A” and “B”; Figure 1) (Edger et al., 2017). Each species is capable of selfing but preferentially outcrosses (Medel et al., 2003; Lila Fishman and Willis, 2008). Both were introduced to the British Isles as ornamentals in the early 19th century (Parker, 1975; Vallejo-Marín et al., 2015), where they soon naturalized and hybridized, producing sterile triploid hybrids (named M. × robertsii; Figure 1) that can bridge to fertile allohexaploids (M. peregrinus) (Vallejo-Marín, 2012; Vallejo-Marín and Lye, 2013). Most of these hybrids, whether in nature or reproduced in the laboratory, are formed from 2x × 4x hybridization events (M. guttatus is the seed parent; i.e. paternal excess). The reciprocal cross, 4x × 2x (M. luteus is the seed parent–maternal excess) is usually nonviable (Vallejo-Marín et al., 2016; Meeus et al., 2020; Figure 1). Such asymmetry is indicative of parent-of-origin effects and offers a unique opportunity to study imprinting in a natural system.
Figure 1.
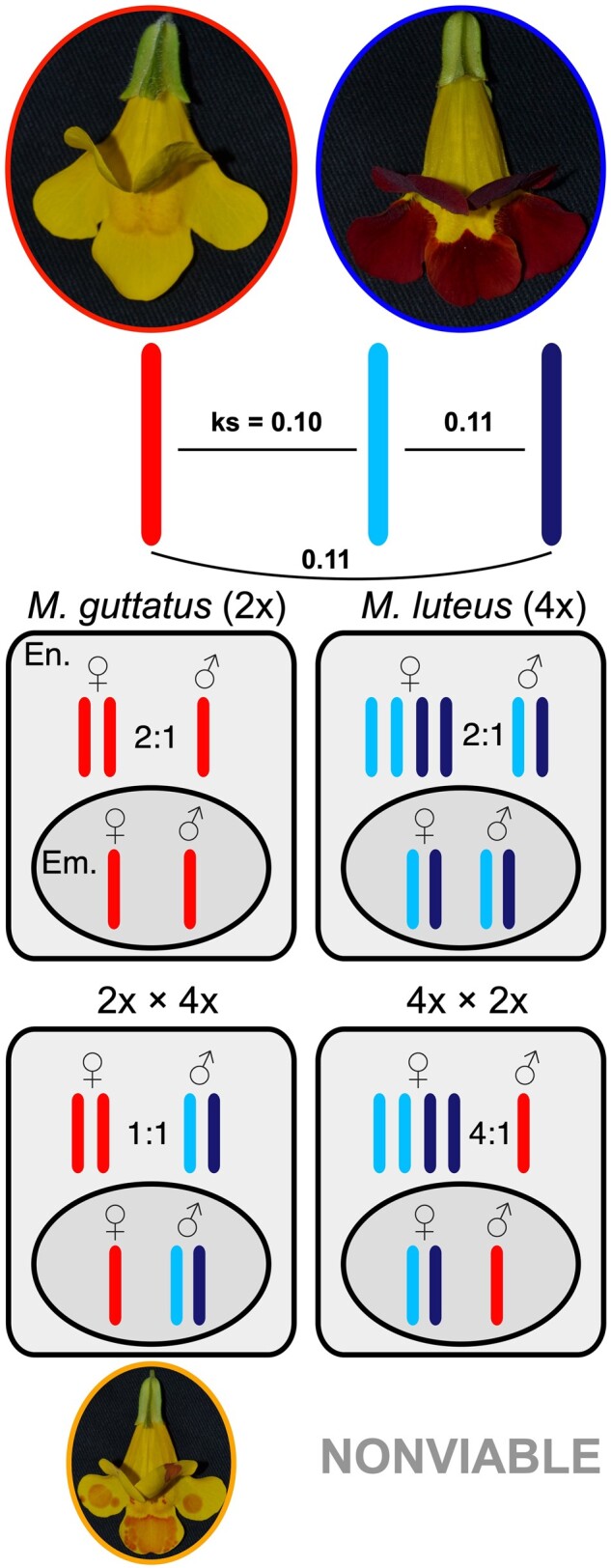
The Mimulus guttatus × M. luteus hybridization system. M. guttatus (in the red oval; left) is a diploid and M. luteus (in the blue oval; right) is a tetraploid with two distinct subgenomes (light blue and navy). Pairwise synonymous site divergences (ks) between the two M. luteus subgenomes and between each of them and M. guttatus are provided. Within seeds for each species, the genomic ratio between parental alleles is 2 maternal:1 paternal in the endosperm (En.) and 1:1 in the embryo (Em.), despite the tetraploidy of M. luteus. When hybridized, if M. guttatus is the maternal progenitor, the genomic ratio between parental alleles is 2:2 (1:1) in the endosperm, and seeds are often viable (M. × robertsii; orange). However, if M. luteus is the maternal progenitor, the ratio is 4:1 and seeds are almost always nonviable. Within the hybrids there are three distinct subgenomes (one from M. guttatus and two from M. luteus—colors are shown throughout).
Maize (Zea mays) and Arabidopsis (Arabidopsis thaliana) and relatives are invaluable models for exploring the mechanisms underlying imprinting, and now other groups are providing new opportunities for testing these mechanisms in naturally occurring systems (Florez-Rueda et al., 2016; Flores-Vergara et al., 2020). Mimulus species offer a variety of life history traits, mating systems, ongoing speciation events, and reticulate patterns (Grossenbacher and Whittall, 2011; Brandvain et al., 2014; Twyford and Friedman, 2015; Ferris et al., 2017) with which to test hypotheses on the evolution and mechanisms of genomic imprinting in seed development (Flores-Vergara et al., 2020). Another notable aspect of this system is its form of endosperm development. Most angiosperms, including maize and Arabidopsis, undergo nuclear-type endosperm development, whereby several rounds of nuclear proliferation precede cytokinesis, whereas Mimulus (and other groups within Lamiales) undergoes ab initio cellular development, during which the endosperm proliferates through typical cellular division. The timing of cellularization in nuclear-type endosperm is central to development and is under imprinted control (Ishikawa et al., 2011). Although developmental processes differ for cellular-type endosperm growth, they also appear to be under imprinted control (Roth et al., 2018b). While nuclear-type endosperm development is more common and has evolved repeatedly throughout angiosperms, cellular-type endosperm development is most likely the ancestral state (Geeta, 2003). Importantly, both M. guttatus and M. luteus have assembled and annotated genomes allowing for detailed genomic analyses (Hellsten et al., 2013; Edger et al., 2017).
In this study, we examined patterns of filial tissue development and genomic imprinting in these two Mimulus species and their hybrids to investigate the processes shaping their hybridization incompatibilities. First, we used histology, scanning electron microscopy, and germination experiments to compare seed development patterns and describe the endosperm barrier of reciprocal hybrids in detail. Second, using transcriptome deep sequencing (RNA-seq) from developing embryos and endosperm tissues, we defined the patterns of imprinting in each parental species. Third, we determined shifts in imprinting and overall expression patterns within the reciprocal hybrids, where we separated and tested relationships between the three subgenomes combined in the hybrids: the M. guttatus (sub)genome and the two M. luteus “A” and “B” subgenomes. Given that embryo and endosperm are the tissues where genomes contributed by different species first interact, we can more generally investigate subgenome interactions at the earliest stages. Finally, we utilized whole genome bisulfite sequencing of endosperm to characterize differences and changes in DNA methylation between the two species and their hybrids. In summary, we describe endosperm-based hybrid incompatibilities in the context of subgenome expression dominance and divergence between epigenetic landscapes and imprinting in the endosperm of these species.
Results
Reciprocal hybrid crosses suffer from nonreciprocal developmental abnormalities
To test for abnormalities in seed development of hybrids, we generated four sets of crosses using plants from the CS line for M. luteus and plants from the CG line for M. guttatus (Vallejo-Marín et al., 2016). Crosses are always denoted as seed parent × pollen donor. These included the two intra-specific crosses and the two hybrid crosses, which were M. guttatus × M. luteus (CG × CS, denoted as 2x × 4x) and M. luteus × M. guttatus (CS × CG, denoted as 4x × 2x). We collected developing seeds at 3, 5, 8, and 11 days after pollination (DAP) for histology, as well as mature seeds at 15–18 DAP (when fruits dehisced) to measure and visualize (by scanning electron microscopy) the entire seed. Quantitative measurements and statistics for this section are provided in Supplemental Tables S1–S5 and Supplemental Figure S1, unless otherwise noted. Qualitative descriptions for mature or developing seeds refer to Figure 2, A and B, respectively.
Figure 2.

Seed development of parental and reciprocal hybrid seeds. A, Scanning electron micrograph images of a representative seed from each cross (M. guttatus—2x × 2x [red]; 2x × 4x [orange]; M. luteus—4x × 4x [blue]; 4x × 2x [green]) is displayed. B, Histological sections were made of each cross through a developmental progression from 3, 5, 8, to 11 DAP. Crosses are displayed in columns and DAP is displayed in rows. Within the images, the seed coat is orange, embryo is green, and endosperm is blue. Scale bars, 0.1 mm
Our first goal was to characterize endosperm and embryo development in intra-specific crosses. At 5 DAP, M. guttatus and M. luteus embryos were in the globular stage. However, by 8 DAP, M. guttatus embryos were at a developmental stage between the heart and torpedo stage, while M. luteus embryos were smaller and had not yet entered the heart stage. At 11 DAP, embryos from both species had increased in size and were in the torpedo stage. While not significantly larger, M. guttatus embryos appeared slightly further developed than those of M. luteus. Similarly, the endosperm of M. luteus developed more slowly than that of M. guttatus, but in both species the endosperm was large and similar in size by 11 DAP, with M. luteus endosperm showing significant growth from 8 DAP. By maturity, the majority of M. guttatus and M. luteus seeds were relatively large, round, and plump with high germination rates (Supplemental Table S6), and seeds from both species were similar in area (Figure 3, A), although M. guttatus seeds were rounder (Supplemental Figure S2).
Figure 3.

Relationship between endosperm and seed area and effect of seed area on germination. A, Seed area in each cross (M. guttatus—2x × 2x [red]; 2x × 4x [orange]; M. luteus—4x × 4x [blue]; 4x × 2x [green]) is represented in violin plots. Dark red diamonds display group means. Letters represent Tukey–Kramer results following an ANOVA. Groups with different letters are statistically different in area. B, Regression between endosperm area and area of the entire seed. The r2 value is provided. C, Area of seeds that failed to germinate compared with successfully germinated seeds among all crosses. 4x × 2x seeds are not included to avoid biasing the comparison since none germinated, and they were the smallest of the four crosses. Dark red diamonds represent group means and the asterisk indicates they are statistically different (P < 0.05, Welch’s t test).
Next, we sought to understand developmental differences in the inter-species hybrids. Embryos of 2x × 4x (CG × CS) seeds were in the globular stage at 5 DAP and maintained the developmental progression of their maternal progenitor, M. guttatus, through 8 DAP, growing at a faster pace than M. luteus embryos. By 11 DAP, though, despite significantly increasing in size from 8 DAP, 2x × 4x embryos were smaller than M. guttatus embryos. In 2x × 4x endosperm, by contrast, we observed morphological differences from parental endosperm qualitatively as early as 3 DAP, where it appears less developed. While not significantly different in size at 8 DAP, 2x × 4x endosperm was smaller than that of both M. guttatus and M. luteus by 11 DAP and had not significantly increased in area from 8 DAP. At maturity, 2x × 4x seeds were shriveled and smaller than both M. guttatus and M. luteus seeds (Figure 3, A) and were the narrowest seeds of all the crosses (Supplemental Figure S2). The 4x × 2x (CS × CG) embryo showed no growth from 8 to 11 DAP and was the only cross type that did not reach the torpedo stage of development. The endosperm also showed little growth and development and was significantly smaller than M. guttatus and M. luteus endosperm by 11 DAP. Mature seeds were flattened and were the smallest seeds of all crosses (Figure 3, A).
Endosperm abnormalities are linked to failed or delayed germination of seeds
To test for a link between the morphological abnormalities discussed above and the viability (germination) of the seed, we aligned mature seeds (collected 15–18 DAP) from the four crosses (i.e. CG × CG [M. guttatus], CS × CS [M. luteus], CG × CS [2x × 4x], and CS × CG [4x × 2x]) along gridded filter paper within petri dishes, photographed them, measured their area in ImageJ, and then recorded their germination date. This procedure allowed us to track the germination success and timing of a given seed and correlate these to its size (area). Since these images were of the whole seed and we could therefore not measure the sizes of internal tissues, we used the histological data described above (Figure 2, B and Supplemental Figure S1) to test for relationships between whole-seed area and endosperm area. We determined that endosperm area has a close relationship with whole-seed area during development (N = 53, R2=0.70; Figure 3, B). Therefore, we consider whole-seed area at maturity to be indicative of endosperm size during development.
4x × 2x seeds were the smallest of all crosses and completely failed to germinate. Across the remaining three crosses, seeds that germinated were significantly larger than seeds that did not (Figure 3, C and Supplemental Table S7, A). When considering individual crosses, only M. luteus showed no significant difference in size between germinated and nongerminated seeds (Supplemental Table S7, C–E). Next, we compared the size of seeds that did germinate (which excludes 4x × 2x) to the timing of their germination. The earliest germinating seeds (2 days after planting) tended to be larger than those that germinated later (Supplemental Figure S3 and Supplemental Tables S8, A, S9, A). Separate ANOVAs on individual crosses revealed a significant difference among germination dates both for M. guttatus and for 2x × 4x, but subsequent pairwise Tukey’s HSD tests between dates were not significant (Supplemental Tables S8, B–D, S9, B–D).
Gene expression in the endosperm of M. luteus and of M. guttatus reveals global paternal bias
To determine patterns of imprinting and parental expression bias in M. luteus, M. guttatus, and the two hybrid crosses, we extracted total RNA from embryo and endosperm tissue of seeds at the torpedo stage (or equivalent date for 4x × 2x) for RNA-seq analysis. We identified imprinted genes by using a reciprocal crossing design to account for line-specific expression differences. To perform this reciprocal crossing design, we used two different inbred lines each for M. luteus (Mll and CS) and M. guttatus (CG and LCA). For M. guttatus, the two reciprocal crosses were CG × LCA and LCA × CG, and for M. luteus the two crosses were Mll × CS and CS × Mll. The reciprocal hybrid crosses were CG × Mll (2x × 4x) and Mll × CG (4x × 2x).
Whole genome resequencing has previously been conducted on Mll, CS, and CG (Vallejo-Marín et al., 2016; Edger et al., 2017), and RNA-seq of LCA was conducted in this study. Using these data, we identified line-specific single-nucleotide polymorphisms (SNPs) within genes, allowing RNA-seq reads generated from endosperm and embryo tissues to be uniquely mapped to specific alleles for measurement of allele expression bias (AEB). AEB is a dimensionless quantity, defined as the mean log2[RPKMpaternal/RPKMmaternal] across replicates (thus positive AEB denotes higher expression on the paternal allele). RPKM is a normalized metric of gene expression defined as the number of reads per kilobase of coding sequence per million mapped reads (Mortazavi et al., 2008). AEB measurements were standardized to dosage so that any amount of expression bias (i.e. AEB is different from 0) represents a transcriptional departure from the expected genome ratio (e.g. 2m:1p). Imprinted genes are defined here as those that show a significant shift in AEB from one reciprocal cross to the other, thus maintaining strong bias toward maternal (MEGs) or paternal (PEGs) expression.
In the endosperm of intra-specific crosses, we identified 37 candidate PEGs and 16 candidate MEGs in M. guttatus (Figure 4, A), and 270 PEGs and 6 MEGs in M. luteus (Figure 4, B). Gene ontology (GO) term enrichment analyses revealed one significantly enriched GO term among M. guttatus PEGs regarding interspecies interactions related to bacterial defense, while M. luteus PEGs were enriched for numerous GO terms related to ribosomes and translation along with GO terms for RNA binding, cytosol, proteasomes, and protein heterodimerization (Supplemental Table S10 and Supplemental Data Set S1). No significantly enriched GO terms were associated with MEGs in either species. Full lists of imprinted genes in M. guttatus and M. luteus and corresponding Basic Local Alignment Sequence Tool (BLAST) results of Arabidopsis genes and their functions as well as GO terms are provided in Supplemental Data Set S1. The distribution of the AEB scatterplot for M. luteus endosperm was largely shifted toward paternal bias, while this was not as apparent for M. guttatus endosperm (Figure 4, A and B and Supplemental Table S11). A Fisher test revealed a significantly different PEG:MEG ratio between species (P < 0.001), with a greater prevalence of PEGs in M. luteus. By contrast, the embryos of both species showed minimal parental bias, as expected. We identified only four candidate MEGs and no PEGs in M. guttatus embryos, and two PEGs and one MEG in M. luteus (Figure 4, D and E and Supplemental Table S11). Overall, we detected a few MEGs, but many PEGs in the endosperm of each species, particularly M. luteus (and with a conspicuous distribution toward paternal bias), and no clear, strong parental bias in the embryo of either species.
Figure 4.

Patterns of imprinting and parental bias. A–E, Distributions of AEB in the endosperm and embryo of M. guttatus (A and D, respectively) and M. luteus (B and E), and the endosperm of the hybrids (C). Genes in scatterplots are plotted by their AEB value in one cross on the x-axis against –AEB of the reciprocal cross on the y-axis. Due to the reciprocal crossing design, bias is divided into four quadrants. Points that fall into the bottom left or top right quadrants represent line or species-specific bias, points in the bottom right have paternal bias, and points in the top left have maternal bias. The percentage of genes in each of these quadrants is given in the corner of that quadrant. The number of PEGs and MEGs is also listed in their respective quadrants. A comparison between hybrid embryos is not included since there was insufficient tissue for Mll × CG (4x × 2x) embryo. Yellow points signify imprinted genes, as determined by a likelihood ratio test (LRT). M. luteus and M. guttatus PEGs identified within the hybrid endosperm (C) are highlighted in blue and red, respectively. The single MEG from M. guttatus is represented by a red diamond. Note these are not significantly imprinted in hybrid endosperm according to LRTs. F, AEB of M. guttatus (red) and M. luteus (blue) PEGs found in each cross, plotted onto the entire AEB distribution of that cross (shown as boxplots). AEB values of genes are averaged between reciprocal crosses for both M. guttatus (2x × 2x) and M. luteus (4x × 4x). G, Log2 fold-change of gene expression (measured in RPKM) between endosperm and embryo (positive values indicate greater endosperm expression) plotted for each allele (maternal and paternal) from M. guttatus, M. luteus, and CG × Mll (2x × 4x) crosses. RPKMs were averaged between reciprocal crosses for each tissue for both M. guttatus and M. luteus. Gray boxes indicate parental crosses (M. guttatus and M. luteus) and orange boxes indicate 2x × 4x. Red lines indicate the M. guttatus genome, and blue indicates the M. luteus genome, whether in parental crosses or inherited in 2x × 4x. The M. luteus genome is also separated into its two subgenomes: A (light blue lines) and B (navy lines). Outliers were removed for F and G
Gene expression in hybrid filial tissues is dominated by the M. luteus genome
In the hybrid, since M. luteus is tetraploid and M. guttatus is diploid, every M. guttatus gene has two M. luteus homologs. When measuring expression bias between the M. guttatus and M. luteus alleles, we counted each M. guttatus gene twice so that expression bias between it and each of its M. luteus homologs could be measured separately, thus accounting for the genomic difference in ploidy and maintaining an expected 2m:1p (1m:1p for embryo) genomic ratio. We identified no imprinted genes in the endosperm that were consistently parentally biased between the two reciprocal hybrid crosses. Furthermore, the AEB scatterplot distribution was not shifted toward paternal bias as in M. luteus endosperm. However, the distribution was strongly shifted toward expressional bias of the M. luteus genome in both reciprocal hybrid crosses (Figure 4, C and Supplemental Table S11). Due to the stunted appearance of 4x × 2x embryos, we were not able to obtain sufficient material for RNA-seq and could therefore not perform a reciprocal crossing design to distinguish between species and parental bias in embryos. Nonetheless, we did observe bias toward the paternal progenitor, M. luteus, in 2x × 4x embryos (Figure 5, D and Supplemental Data Set S4, B). Since imprinting in angiosperm embryos is rare (Batista and Köhler, 2020), as observed in the embryo of either parent species, this bias likely indicated that the M. luteus genome is expressionally dominant in hybrid tissues. In the following two sections, we focus on the endosperm.
Figure 5.

Comparison of AEB values between M. luteus subgenomes. AEB distribution of each of the two subgenomes from M. luteus (A, light blue and B, navy) in parental M. luteus (A) and reciprocal hybrid crosses (2x × 4x [CG × Mll] and 4x × 2x [Mll × CG] in D) for endosperm and embryo (other than 4x × 2x). AEB of reciprocal crosses for M. luteus are averaged. Linear regressions were performed comparing AEB values for genes on M. luteus subgenome A to AEB values of their respective homeologs on subgenome B for M. luteus (values for Mll × CS are shown here) embryo (B) and endosperm (C), and for the endosperm of 2x × 4x (E) and 4x × 2x (F). R2 values and P-values for linear regressions are provided in the top left corner of each plot. A hypothetical 1:1 relationship is shown in blue with the actual linear model in red. The gray shaded area represents the 95% confidence interval of the linear model.
Patterns of imprinting in the endosperm differ between M. luteus and M. guttatus
To begin to understand how patterns of imprinting may interact between the M. guttatus and M. luteus genomes, we sought to explore how these patterns are shared between them. First, we tested for overlap of imprinted genes between M. luteus and M. guttatus homologs, though we were highly limited by the total number of identifiable homologs. Of its 276 putative imprinted genes, M. luteus had 38 PEGs with discernable homologs in M. guttatus. Similarly, 1 out of the 16 MEGs in M. guttatus and 5 of its 37 PEGs had discernable homologs in M. luteus. Note that each of these genes in M. guttatus has two corresponding homologs in M. luteus, since M. luteus is a tetraploid. Of this reduced set of imprinted genes, one PEG overlapped between the two species (Mgu_10535 for M. guttatus and Mlu_21243 and Mlu_31004 for M. luteus—At1g51060.1 in Supplemental Data Set S1) and encodes a histone protein. Next, even though these species have little overlap in imprinted genes, we examined similarities in AEB between the set of imprinted genes from one species and their corresponding homologs in the other species to assess if at least some level of parental bias is shared. Due to sample size, we only considered PEGs. We plotted the AEB (averaged between reciprocal crosses to account for line bias) of all comparable homologs between M. guttatus and M. luteus. While M. luteus PEGs were paternally biased within the overall AEB distribution of M. luteus endosperm, their corresponding homologs in M. guttatus did not uphold clear paternal bias in M. guttatus endosperm (Figure 4, F and Supplemental Data Set S2). There was only one M. guttatus PEG for which we could confidently identify a M. luteus homolog and that had sufficient read data in the 4x × 4x (M. luteus) crosses. This gene was also a PEG in M. luteus. Overall, there appeared to be substantial differences and limited conservation in imprinting patterns between M. luteus and M. guttatus, although we are limited by our ability to compare homologs.
Within the hybrid endosperm, expression patterns of PEGs in M. luteus and M. guttatus tend to be consistent with the observed M. luteus genome expression dominance
We also examined whether the biased expression of PEGs identified in M. luteus and in M. guttatus was upheld within the hybrids by plotting the AEB of genes from each hybrid cross. Within the 2x × 4x endosperm, the distribution of expression bias was generally in the direction of the paternal progenitor, M. luteus. The specific AEB values of M. guttatus PEGs and M. luteus PEGs conformed to the overall distribution, although they were not concentrated toward its tail as they were in their respective parental AEB distributions (Figure 4, F and Supplemental Data Set S2). Within the 4x × 2x endosperm, the AEB distribution was again in the direction of M. luteus, the maternal progenitor. Mimulus luteus PEGs inherited within 4x × 2x, as in 2x × 4x, were not concentrated on the paternal tail of the overall AEB distribution. In fact, many reversed to maternal bias. However, M. guttatus PEGs maintained paternal bias within the 4x × 2x endosperm, where M. guttatus is the paternal progenitor (Figure 4, F and Supplemental Data Set S2). As evident in Figure 4, C, the imprinted genes of M. guttatus and M. luteus did not maintain strong, consistent parental bias within endosperm of the reciprocal hybrid crosses. However, M. guttatus imprints did display some consistent bias in the hybrids (e.g. six of the nine PEGs measurable within the hybrids remained in the paternal expression bias quadrant), although none of these genes reflected significant departures from expected parental expression contribution in hybrid endosperm.
This data suggest that patterns of parental bias are not obviously inherited from either parent species in the hybrids. Rather, while there may be some consistent parental bias for imprinted genes from M. guttatus, particularly for PEGs in the 4x × 2x cross where the paternal allele is inherited from M. guttatus, expression bias within hybrid endosperm appears to remain more consistent with global M. luteus expression dominance than with normal patterns of imprinting observed in parental endosperm, especially that of M. luteus.
Decreased maternal expression drives general paternal biases in parental endosperm, particularly for M. luteus, while overall expression dynamics are more similar between filial tissues in 2x × 4x
For a more specific understanding of how parental alleles are expressed in intra-specific M. luteus and M. guttatus crosses, we compared the difference in expression between embryo and endosperm, measured as log2(RPKMEndosperm/RPKMEmbryo) where positive values indicate greater endosperm expression, across all genes for each parental allele. RPKM values were averaged between reciprocal crosses to account for line bias. Genes tended to have decreased expression levels in endosperm compared with embryo tissue for each parental allele in both M. guttatus and M. luteus (Figure 4, G and Supplemental Data Set S3). Congruent with the observed prevalence of PEGs, the maternal allele was notably more downregulated than the paternal allele compared with the embryo in both species. This was especially the case for the maternal allele in M. luteus (Figure 4, G and Supplemental Data Set S3). These patterns suggested that general paternal biases are characterized by downregulation of maternal expression in Mimulus endosperm (compared with embryo), which is particularly evident in M. luteus endosperm.
We next performed the same measurement in the 2x × 4x hybrid cross. Differences in expression between embryo and endosperm tissues were less pronounced than in the parents, and they revealed unique patterns. Expression of the maternal M. guttatus allele was only slightly decreased in 2x × 4x endosperm compared with embryo (Figure 4, G and Supplemental Data Set S3). In addition, expression of the paternal M. luteus allele was somewhat increased in the endosperm compared with the embryo (Figure 4, G and Supplemental Data Set S3). Thus, expression dynamics between filial tissues differed from parental crosses in the 2x × 4x hybrid cross. We did not attempt to reproduce the equivalent measurement in 4x × 2x due to a lack of embryo tissue.
Expression patterns tend to be shared between M. luteus subgenomes
Next, we sought to determine patterns of expression and bias specific to each of the two M. luteus subgenomes (A and B subgenomes) within filial tissues from parental M. luteus and each of the reciprocal hybrid crosses. Although M. luteus, as an allotetraploid, has two distinct subgenomes, the relationships among the majority of the M. luteus genes were too ambiguous to confidently sort them into a specific subgenome; however, a subset of gene pairs did clearly sort into the A or B subgenome (Edger et al., 2017). For these gene pairs, the homolog to a gene from one M. luteus subgenome (e.g. A) on the other M. luteus subgenome (e.g. B) is referred to as a homeolog, and the pair is referred to as a homeolog pair. We again compared the difference in expression between embryo and endosperm, but here, we treated the two M. luteus subgenomes separately. The two subgenomes each retained similar patterns as when treating the M. luteus genome as a whole (discussed above), both in parental M. luteus and in 2x × 4x (Figure 4, G and Supplemental Data Set S4).
Finally, we compared levels of subgenome-specific parental expression bias (i.e. AEB) in M. luteus and the hybrid crosses. Subgenome-specific AEB measures the expression bias between maternal and paternal alleles (m and p) one subgenome at a time; here, whether in the parental M. luteus cross (i.e. Am× Ap or Bm× Bp) or the two M. luteus subgenomes and one M. guttatus (sub)genome inherited in the hybrids (e.g. M. guttatusm × Ap). Similar to treating the M. luteus genome as a whole, parental embryos had little subgenome-specific AEB, while the corresponding endosperm had similar levels of paternal AEB on each subgenome (Figure 5, A and Supplemental Data Set S4, A). However, when correlating AEB values across individual homeolog pairs (e.g. Gene1-Am× Gene1-Ap versus Gene1-Bm× Gene1-Bp, etc.), we observed no relationship between the AEB of parental alleles on one subgenome and that of their homeologs on the other in either embryos (Figure 5, B and Supplemental Tables S4, A, S12, A) or endosperm (Figure 5, C and Supplemental Tables S4, B, S12, A) in parental M. luteus. Taken together, even though the parental AEB of a specific gene on subgenome A may differ from its homeolog on subgenome B, the overall pattern of AEB across subgenome A was similar to that of subgenome B.
AEB values were more variable in the hybrids, though overall, subgenome-specific AEB patterns were similar, reflecting the overall expression bias of the M. luteus genome over the M. guttatus genome, whether paternal in 2x × 4x or maternal in 4x × 2x. Some level of M. luteus expression bias was evident even in 2x × 4x embryos—contributed by each M. luteus subgenome—though to a lesser extent than in its endosperm (Figure 5, D and Supplemental Data Set S4, B). Furthermore, there were significant relationships of AEB values between homeolog pairs within the embryo (Supplemental Tables S4, C and S12, A) and endosperm (Figure 5, E and Supplemental Table S12, A) of 2x × 4x (e.g. Gene1-M. guttatusm × Gene1-Ap versus Gene1-M.guttatusm × Gene1-Bp, etc.) and the endosperm (Figure 5, F and Supplemental Table S12, A) of 4x × 2x (e.g. Gene1-Am× Gene1-M.guttatusp versus Gene1-Bm× Gene1-M.guttatusp, etc.). This suggests that expressional biases against the M. guttatus (sub)genome were consistent between homeolog pairs of the two M. luteus subgenomes within the hybrids. Thus, expression patterns appear to be more similar between the M. luteus subgenomes, which are always inherited together, than with the M. guttatus (sub)genome within the hybrid.
M. luteus and M. guttatus endosperm differ in their DNA methylation patterns
To analyze DNA methylation patterns in M. luteus, M. guttatus, and their hybrids, we extracted DNA from endosperm tissue of seeds at the torpedo stage (or equivalent date for 4x × 2x) using the CTAB method (Porebski et al., 1997) and treated the DNA with bisulfite for Methyl-seq. The following four crosses were used: CG × LCA (for M. guttatus), Mll × CS (for M. luteus), CG × Mll (2x × 4x), and Mll × CG (4x × 2x). We measured weighted methylation levels from CpG, CHG, and CHH sequence contexts for genes and TEs in each cross. We measured the M. guttatus allele (CG) and M. luteus allele (Mll) in hybrid crosses separately, although we were not able to distinguish between maternal and paternal alleles in parental crosses. All descriptions in this and the next section refer to Figure 6.
Figure 6.

Patterns of methylation in parent and hybrid endosperm. Gene (left two columns) and transposon (right two columns) methylation of each sequence context (CpG, CHG, and CHH—1st, 2nd, and 3rd row, respectively) in the endosperm of parental crosses (red—M. guttatus, blue—M. luteus) compared with that of each allele in reciprocal hybrids (orange—2x × 4x [1st and 3rd column], green—4x × 2x [2nd and 4th column]). The inherited M. luteus allele in each of the hybrid crosses is represented by the lighter shade of their respective color, and the inherited M. guttatus allele is represented by the darker shade. Weighted methylation is on the y-axis, while the x-axis represents the gene body (TSS = transcription start site and TTS = transcription termination site) or TE body and 2 kb upstream and downstream.
We first analyzed patterns of methylation for the endosperm of parental crosses. CpG methylation was similar between M. luteus and M. guttatus upstream of genes. Downstream and, in particular, within genes, CpG methylation was higher in M. guttatus when compared with M. luteus. Conversely, non-CpG methylation (CHG and CHH) was higher in M. luteus on genes and their flanking regions. CHG methylation was especially higher on gene-flanking regions in M. luteus. Mimulus guttatus and M. luteus had similar CpG methylation levels upstream and downstream of TEs, but higher levels within TEs in M. guttatus. CHG methylation was higher in M. luteus in TE-flanking regions and lower within TEs. M. guttatus had greater CHH methylation than M. luteus on TE-flanking regions and especially within TEs. Overall, M. guttatus tended to be more heavily methylated within TEs, particularly in the CHH context, and had greater genic CpG methylation, while M. luteus tended to have more non-CpG methylation associated with genes.
Transgressive, nonreciprocal patterns of methylation emerge in hybrid endosperm
Next, we analyzed patterns of methylation for the endosperm of reciprocal hybrid crosses. In each hybrid cross, CpG methylation in genes and their flanking regions generally increased on the inherited M. luteus allele and decreased on the inherited M. guttatus allele compared with the levels in their respective parents. Within genes, the inherited M. luteus allele had higher CpG methylation than the M. guttatus allele in 2x × 4x but was marginally lower than the M. guttatus allele in 4x × 2x. 2x × 4x had considerably more non-CpG methylation than 4x × 2x, especially CHH methylation, which was very low in 4x × 2x. In addition, non-CpG methylation, and CHG in particular, was greater on the inherited M. luteus allele than the inherited M. guttatus allele in both reciprocal hybrid crosses, at least in gene-flanking regions. Each inherited allele showed decreased non-CpG methylation levels compared with their respective parental levels in 4x × 2x and increased levels in 2x × 4x on both genes and their flanking regions.
TEs had similar patterns of methylation as genes, with some notable exceptions. In both hybrids, CpG methylation increased on the inherited M. luteus allele compared with parental M. luteus, and decreased on the inherited M. guttatus allele compared with parental M. guttatus, except for within TEs in 4x × 2x. While CpG methylation was similar between hybrid crosses in flanking regions, it was greater within TEs in 4x × 2x. Overall, the inherited M. luteus allele was more CpG-methylated across TEs and their flanking regions than the M. guttatus allele in each hybrid cross. TE-flanking regions had similar patterns of non-CpG methylation as genes, with the exception that the inherited M. luteus allele in 4x × 2x and the inherited M. guttatus allele in 2x × 4x did not differ substantially from their respective parental levels for CHG and CHH contexts, respectively. The major departure from genic region patterns occurred within TEs. Each hybrid cross had CHG methylation on each allele that was more similar to their respective maternal progenitor. CHH methylation was near the mid-parental value within TEs for each allele in 2x × 4x. Otherwise, as with genes, CHH methylation was very low in 4x × 2x.
Overall, the inherited M. luteus allele generally had higher methylation levels of all types relative to the inherited M. guttatus allele in gene and TE-flanking regions, and in some cases within. CpG methylation tended to be greater within genes and TEs in 4x × 2x than in 2x × 4x, while non-CpG methylation, in general, tended to be greater in 2x × 4x than in 4x × 2x, which had very low CHH methylation. Methylation patterns typically departed from parental levels for each allele in both reciprocal hybrid crosses.
Methylation levels are similar between M. luteus subgenomes and differ from M. guttatus
Finally, we compared weighted methylation levels between the two M. luteus subgenomes (A and B) and the M. guttatus genome in parental crosses and in reciprocal hybrid crosses. We measured weighted methylation of individual coding sequences and calculated the median across each of these (sub)genomes in each cross. The two M. luteus subgenomes shared similar overall amounts of coding sequence methylation within parental M. luteus and the two reciprocal hybrids (Table 1). Only CHH sequence contexts in 4x × 2x revealed any significant differences between its two M. luteus subgenomes after applying Kolmogorov–Smirnov tests (Supplemental Table S13). Each of the two subgenomes in parental M. luteus differed significantly from parental M. guttatus. Furthermore, within each reciprocal hybrid cross, the inherited M. luteus subgenomes differed significantly from the inherited M. guttatus genome (Table 1 and Supplemental Table S13).
Table 1.
Subgenome-specific methylation levels
| Cross | Type | M. luteus A | M. luteus B | M. guttatus |
|---|---|---|---|---|
|
Parental |
CG | 0.622 | 0.625 | 0.648 |
| CHG | 0.169 | 0.171 | 0.103 | |
| CHH | 0.156 | 0.156 | 0.098 | |
|
2x × 4x |
CG | 0.610 | 0.618 | 0.556 |
| CHG | 0.203 | 0.200 | 0.181 | |
| CHH | 0.205 | 0.199 | 0.172 | |
|
4x × 2x |
CG | 0.595 | 0.599 | 0.654 |
| CHG | 0.089 | 0.090 | 0.078 | |
| CHH | 0.050 | 0.048 | 0.038 |
Median values for weighted methylation, measured per coding sequence, from each of the two subgenomes (A and B) of M. luteus and the M. guttatus genome are reported for parental crosses and the two reciprocal hybrid crosses. Note that these three (sub)genomes all co-occur in hybrid crosses.
Discussion
Inter-specific hybridization brings together two genomes that each have unique evolutionary histories. Studying how these genomes interact at the earliest stage, during embryo and endosperm development, will increase our understanding of not only basic plant processes, such as genomic imprinting and seed development, but also mechanistic barriers to hybridization. Endosperm-based hybridization incompatibilities involve divergent epigenetic and imprintome states (Lafon-Placette et al., 2018), where expression of dosage-sensitive regulatory networks between parents that control cell cycle and developmental mechanisms differs (Roth et al., 2019). Genes related to cell cycle control, gene and chromatin regulation, and regulation of auxin and other phytohormones are vital for proper endosperm development and are responsible for its failure when misregulated or incompatible (Batista et al., 2019; Roth et al., 2019; Tonosaki et al., 2020). Here, using this Mimulus hybrid system, which is both inter-specific and inter-ploidy, we demonstrate how diverged epigenetic and imprintome states in endosperm, with at least some independence from genetic divergence, can induce cross-specific outcomes that mediate the paternal cues needed for endosperm proliferation and seed viability.
Hybrid endosperm imposes an important early crossing barrier between M. luteus and M. guttatus
This hybrid system has strong crossing asymmetry in seed viability, which is linked to nonreciprocal developmental abnormalities in endosperm, leading to reduced seed size. When M. guttatus is the seed parent (2x × 4x), endosperm proliferation decreases from early stages; cells are condensed, and endosperm growth is hampered throughout development. While embryo growth follows the developmental timing of maternal M. guttatus, it decelerates, resulting in a shriveled seed with reduced viability by maturity. In contrast, when M. luteus is the seed parent (4x × 2x), endosperm growth initiates, producing large, vacuolated cells, but proliferation never properly proceeds. The embryo aborts by the early globular stage, resulting in collapsed, nonviable seeds consisting mostly of maternal seed coat. These sequences follow similar abnormalities and impaired developmental progression observed in other species with ab initio cellular endosperm (Oneal et al., 2016; Roth et al., 2018a). While developmental mechanisms differ, the overall process appears similar with nuclear-type endosperm species as well: embryos starve without successfully proliferated endosperm, reducing viability of hybrid seeds (Lafon-Placette et al., 2017).
Endosperm development requires proper parental cues of cell cycle progression, chromosome and transcription regulation, and phytohormone signaling, whether nuclear- or cellular-type (Ishikawa et al., 2011; Roth et al., 2018b; Batista et al., 2019). In ab initio cellular endosperm seeds, endosperm growth initiates first, producing large, vacuolated cells, and can become multicellular before the zygote makes its first division. As it proliferates, the maternal seed coat also expands, and its inner cells collapse, providing nutrition to peripheral endosperm cells, which become dense with cytoplasm. Endosperm cells collapse and disintegrate as the embryo enlarges and cotyledons differentiate. Thus, a clear nutritional flow follows from maternal tissue of the seed coat, to the endosperm, to the embryo (Beamish, 1955; Lee and Cooper, 1958; Oneal et al., 2016). The success of this sequence relies on parental interactions coordinating regulatory networks and cell cycle transitions in the endosperm (Roth et al., 2018b; 2019). In inter-species Solanum hybrids, abnormal endosperm with condensed cells and decreased proliferation, similar to 2x × 4x, has been linked to expedited mitotic transitions ending growth prematurely. Increased activity of phytohormone regulation and resource allocation genes, which are potentially maternally driven, is involved. Conversely, in hybrid seeds where endosperm cells remain large and vacuolar with impaired proliferation, similar to 4x × 2x, increased paternal activity of genes related to gene expression and growth may delay mitotic transitions, stunting development (Roth et al., 2018a, 2019). Proper parental cues of cell cycle and phytohormonal control are central to endosperm-based hybrid incompatibilities and likely are central in this hybrid system as well.
The reciprocal hybrid crosses between M. luteus and M. guttatus suffer from different endosperm abnormalities. 2x × 4x seeds are deficient with endosperm proliferation, a vital developmental step. Regulation of DNA replication and chromatin is critical for cell cycle transitions and secondary cell wall formation during ab initio endosperm proliferation (Oneal et al., 2016; Flores-Vergara et al., 2020). Functional genes at this step include those encoding methylases and chromomethylases (Flores-Vergara et al., 2020). Parental alleles inherited in 2x × 4x are transgressively hypermethylated in non-CpG cytosine contexts, which may involve increased chromomethylase activity that may affect DNA replication and cell cycle progression. In 4x × 2x seeds, however, the inherited alleles are non-CpG hypomethylated, with almost negligible levels of CHH methylation. The inherited M. luteus allele exhibits expression dominance over the M. guttatus allele in hybrid endosperm, consistent with observations in vegetative hybrid tissue (Edger et al., 2017). Paternal M. guttatus is presumably unable to establish the proper developmental cues and epigenetic state needed to promote endosperm proliferation when under the dominating maternal regulation of M. luteus. Interestingly, when a synthetic tetraploid M. guttatus is the seed parent to paternal M. luteus, hybrid seeds are nonviable, while the viability of the reverse cross is somewhat restored, albeit still with low germination rates (Meeus et al., 2020). The increased maternal M. guttatus dosage might have lethal levels of maternal phytohormonal regulation and cell cycle control, inhibiting endosperm proliferation, while an increased paternal dosage may provide sufficient paternal influence on replication and growth for endosperm proliferation and development to proceed.
In unique contrast from M. guttatus, M. luteus endosperm has a high prevalence of PEGs and paternal dosage, which is apparently confounded in reciprocal hybrids by its expression dominance over the inherited M. guttatus genome. Expression dominance is a common response to hybridization and appears to be affected by differential TE methylation load near genes between the inherited parental genomes, where the one with lower methylation is dominant (Alger and Edger, 2020). In hybrid vegetative tissue, dominance of the M. luteus allele appears linked to its lower methylated TE abundances near genes (Edger et al., 2017; Alger and Edger, 2020). However, in stark contrast to vegetative tissues, non-CpG methylation is generally greater around genes and TEs on the M. luteus allele, and it is transgressively CpG hypermethylated while the M. guttatus allele is CpG hypomethylated in endosperm regardless of crossing direction. Thus, M. luteus still maintains its expression dominance independently of this allele-specific transgressive methylation or demethylation that is seemingly driven by the unique epigenetic, chromosomal, and regulatory processes in the endosperm. While M. guttatus and M. luteus differ epigenetically in their vegetative tissues, their endosperm differs markedly in methylation patterns, imprinted gene set, and paternal dosage, and these differences appear to have substantial consequences in the endosperm of their hybrids. However, germinated hybrids (and fertile allohexaploids) do exhibit heterosis for several traits and increased survivability, particularly those in nature that are apparently undergoing adaptive selection (Meeus et al., 2020). While hybridization necessitates M. guttatus as the maternal progenitor here, apparently the balanced cross (with a tetraploid M. guttatus) favors M. guttatus as paternal (Meeus et al., 2020). This determinate of viability and the potential for endosperm proliferation may lie in diverged parental epigenetic and imprintome states in endosperm.
Rapid evolution and divergence of species-specific imprinting patterns is common and is likely involved in endosperm-based hybridization barriers (Lafon-Placette et al., 2018; Roth et al., 2019). Shifts in expression patterns are also common in hybrid endosperm and can directly affect the dosage of imprinted and other developmental genes (Li and Dickinson, 2010; Ishikawa et al., 2011; Burkart-Waco et al. 2015; Florez-Rueda et al., 2016). Based on our results for both morphological development and genomic patterns in seeds, along with similarities with other systems, we suspect that the endosperm represents an important barrier for hybrid seed development in this system. Below we expand on this conclusion in the context of epigenetic divergence and genomic shock.
Differing epigenetic landscapes between species can drive epigenetic reprogramming in their hybrids, which, coupled with diverged imprinting patterns, may act as an endosperm-based hybridization barrier
Differing epigenetic states between parental genomes can lead to deregulation of sRNAs, causing changes in both gene and TE methylation with downstream consequences (Shen et al., 2012; Lafon-Placette and Köhler, 2015; Rigal et al., 2016). In our system, we find clear differences between the methylation patterns of M. luteus and M. guttatus endosperm as well as transgressive methylation following hybridization. Of particular note, non-CpG methylation is greater in gene flanking regions in M. luteus endosperm than in M. guttatus, and CHG methylation is greater in TE flanking regions. The transgressive non-CpG hypermethylation of both inherited parental alleles observed in 2x × 4x may derive from a potential disjunct in sRNA production between the two alleles that results in an overproduction of sRNAs. In hybrids, transgressive methylation patterns, particularly non-CpG, often occur where parental alleles have divergent sRNA levels; transgressive sRNA production inherited from the more sRNA-rich parent methylates both alleles, including the one with lower methylation (Greaves et al., 2012; Rigal et al., 2016). Higher CHH methylation of TEs within gene-flanking regions is linked to greater PEG counts and paternal dosage (Lafon-Placette et al., 2018), characteristic of genes in M. luteus endosperm. Consistent with many of the M. luteus PEG functions, chromosomal and gene regulation are likely under paternal control in endosperm (Roth et al., 2018b), where paternal RdDM also likely plays a crucial role (Satyaki and Gehring, 2019). Thus, this divergence in epigenetic and imprintome state between M. luteus and M. guttatus may induce an influx of sRNAs from the more non-CpG methylated, PEG-rich, paternal M. luteus allele, driving transgressive non-CpG hypermethylation of both alleles. In 4x × 2x, the transgressive non-CpG hypomethylation of each parental allele, particularly the highly reduced CHH methylation, reflects diminished de novo methylation and RdDM activity (Niederhuth and Schmitz, 2017). In this case, the less non-CpG methylated, paternal M. guttatus allele of lower dosage may initiate a loss of sRNA production, resulting in reduced non-CpG methylation levels on both alleles. Thus, epigenetic differences between M. luteus and M. guttatus may drive this nonreciprocal transgressive methylation in their hybrids, where three distinct subgenomes interact (Edger et al., 2017; Alger and Edger, 2020).
The hybridization between M. luteus and M. guttatus provides a unique study system, since it brings together three subgenomes that are genetically distinct from one another (Figure 1). In contrast to the stark differences in allelic dosage and methylation between M. luteus and M. guttatus, overall expression and methylation levels are much more similar between the M. luteus “A” and “B” subgenomes, both in parental M. luteus and in hybrid seeds. The two M. luteus subgenomes each share expression dominance over the inherited M. guttatus sub(genome) in hybrid seeds despite the genetic distinctiveness among all three. Homeolog pairs (defined above) from each M. luteus subgenome tend to consistently maintain this expression bias regardless of crossing direction. While individual genes may have separate parental expression bias patterns from their corresponding homeolog when within M. luteus endosperm, overall, each subgenome displays a similar global paternal bias with greater paternal dosage than M. guttatus endosperm, apparently due to a larger, shared global downregulation of each of their maternal alleles. Thus, regulation of parental expression bias in endosperm appears to act consistently across the two M. luteus subgenomes. It is possible that, even though M. luteus “A” and “B” subgenomes are no less genetically diverged from each other than they are from the M. guttatus genome (Edger et al., 2017), due to their long coexistence, their epigenetic and regulatory patterns are largely shared and remain linked in the hybrids. The inherited M. luteus subgenomes tend to be similarly hypermethylated compared with the M. guttatus allele in hybrid endosperm, so, presumably, cis-regulatory expression bias specific to these M. luteus subgenomes (Stupar and Springer, 2006; Alger and Edger, 2020) maintains the expression dominance observed in other tissues (Edger et al., 2017). The role of DNA methylation on gene expression is complicated in the endosperm, as chromosomal regulation is central to imprinted expression, particularly paternally (Batista and Köhler, 2020). The outcome of these subgenome interactions appears to have greater consequences amidst the epigenetic and regulatory activity of hybrid endosperm compared with other tissues: they are generally more pronounced than in vegetative tissues (Edger et al. 2017), and, in 2x × 4x, the paternal M. luteus allele is somewhat upregulated in endosperm compared with embryo, increasing both paternal and M. luteus expression bias. Such consequences in endosperm can affect dosage-sensitive imprinted regulation.
A lack of overlap and dosage imbalances in imprinted genes may result in the absence or mismatch of an interacting component(s) of an imprinted gene (Comai et al., 2003; Josefsson et al., 2006). Such incompatible genomic interactions between parents often manifest cross-specific outcomes in their hybrids (Rebernig et al., 2015). While CpG methylation remains high, 4x × 2x undergoes transgressive non-CpG hypomethylation and disrupted RdDM, suggesting a deficit in chromomethylase activity and sRNA production, which are interrelated with chromatin state (Stroud et al., 2014). While the effects of DNA methylation on gene expression can be indirect or even not apparent, DNA methylation tends to more directly affect TE activity (Rigal et al., 2016). TEs in M. guttatus endosperm are generally more methylated, particularly in CHH contexts, than in M. luteus endosperm. Thus, these regions are likely more silenced and heterochromatic in M. guttatus endosperm (Stroud et al., 2014). Therefore, a paternal M. guttatus may induce a derepressed TE state in 4x × 2x, perhaps especially those on its own allele, which tend to be less methylated. Such a state may contribute or be related to lethality in 4x × 2x seeds, and nevertheless, highlights the nonreciprocal epigenetic consequences in hybrid endosperm. Non-CpG methylation is greater on TEs in 2x × 4x, often transgressively. TEs on the M. luteus allele are also consistently CpG-hypermethylated in the hybrids. Thus, TE activity, along with sRNAs and chromomethylase activity, may be starkly different on the M. luteus subgenomes and in the viable 2x × 4x hybrid endosperm. Mimulus luteus endosperm has high paternal dosage with many putative PEGs, which are involved in various aspects of gene expression and protein production. However, while M. guttatus PEGs tended to retain some paternal bias in the hybrids (though nonsignificant), M. luteus PEGs generally reflected the expression dominance of the M. luteus subgenomes; whether paternal in 2x × 4x or maternal in 4x × 2x. Ultimately, dissimilarities in the epigenetic and imprintome states of the endosperm between M. luteus and M. guttatus likely cause cross-specific epigenomic shock, while similarities between the M. luteus “A” and “B” subgenomes retain the processes that uphold their expression dominance. The progression of endosperm proliferation thus may rely on the success of a paternal role in regulating balanced or proper expression of key genes.
Conclusions
Based on the above ideas, we suggest that the following processes may contribute to hybrid barriers in M. luteus × M. guttatus crosses: (1) diverged imprinting patterns between parents generate functional incongruence in hybrid endosperm; (2) disparity of epigenetic characteristics between the parent species underlies an epigenomic shock in the hybrids; (3) transgressive methylation and other epigenetic reprogramming following hybridization may alter regulatory networks between the parental genomes, however, these processes likely act similarly on the two M. luteus subgenomes due to their shared epigenetic and regulatory states; (4) compared with other tissues, the diverged epigenetic and imprintome states specific to endosperm tissue yields unique and consequential epigenomic outcomes that are dependent on crossing direction, and (5) altered regulatory networks and diverged parental interactions, potentially involving sRNA production and methylation cues, between the two M. luteus subgenomes and the M. guttatus (sub)genome impact paternal function involved in endosperm proliferation resulting in successful, albeit decreased, endosperm development when the tetraploid M. luteus is the paternal progenitor and aborted endosperm when diploid M. guttatus is paternal. Given this logic and observations from other systems (Josefsson et al., 2006; Greaves et al., 2012; Satyaki and Gehring, 2019), we suggest that epigenetic repatterning accompanied with global shifts in expression patterns may result from diverged epigenetic states and regulatory networks of parental genomes; this phenomenon can be particularly prominent and critical in endosperm where parental interactions and instructions coordinate its development and may also diverge. Importantly, even if imprinting patterns are mostly conserved, epigenetic differences that shift global networks and dosage may affect parental interactions with nonreciprocal outcomes. Such processes may underlie genomic imprinting in endosperm-based hybrid incompatibilities. Since such incompatibilities can likely precede genetic incompatibilities in the embryo (Lafon-Placette et al., 2018), these processes could be important components of hybridization barriers in general.
Further investigation into allele-specific patterns of sRNA production and TE activation; links between DNA (and histone) methylation, heterochromatin, and expression; and imprinting function and loss of function, are necessary for further elucidating imprinting patterns in these Mimulus species and their role in hybrid incompatibility. Additional studies in this system and the many other intriguing Mimulus hybrid and speciation systems (Oneal et al., 2014; Garner et al., 2016; Kooyers et al., 2017; Flores-Vergara et al., 2020), along with its genomic and genetic resources (Ding and Yuan, 2016; Vallejo-Marín et al., 2016; Edger et al., 2017), will make Mimulus a valuable model for elucidating the mechanisms and evolutionary drivers of genomic imprinting.
Materials and methods
Crossing design
Four accessions were used: CS and Mll for M. luteus and CG and LCA for M. guttatus. LCA is from Lake Alamanor, California, and was inbred for 11 generations, and Mll is from Embalse el Yeso, Chile, and was inbred for 13 generations. For more information on CS (CS-4-JP1) and CG (CG-1-1-JP1), refer to Table 2 in Vallejo-Marín et al. (2016). For histology, seed area, and germination experiments, first- or second-generation CS and CG plants were used. Flowers were emasculated before blooming, and then pollinated by a flower from a different plant the next day. There were four unique crosses: 4x × 4x (M. luteus; CS pollinated by CS), 2x × 2x (M. guttatus; CG pollinated by CG), 2x × 4x (CG pollinated by CS), and 4x × 2x (CS pollinated by CG). For RNA-seq, Mll, CS, LCA, and CG were used to produce six unique crosses. There were two reciprocal crosses for M. luteus, Mll × CS and CS × Mll, two for M. guttatus, CG × LCA and CG × LCA, and the two hybrid reciprocal crosses, CG × Mll (2x × 4x) and Mll × CG (4x × 2x). There were four crosses used for Methyl-seq: Mll × CS, CG × LCA, CG × Mll, and Mll × CG. Independently fertilized ovaries for each cross from each experiment came from different maternal and paternal progenitors (i.e. no two ovaries came from or were fertilized by the same plant). The number of replicated ovaries collected for each experiment is given in the following sections. Ovaries were always collected at the same time of day for consistency. All plants were grown in the College of William and Mary greenhouse with a photoperiod of 16-h light/8-h dark to mimic the natural growing season of these plants.
Seed area and histology
Four mature ovaries with 100–200 seeds each were collected for each cross (refer to the “Crossing design” section) at 15–18 DAP. All measurements and experiments related to mature seed size used seeds from these four ovaries. Images of a subset of seeds were taken (used below in the “Seed germination” section) under a dissecting microscope with the same magnification for all images, and area and aspect ratio were measured in ImageJ with a custom script to automate measurements. Each imaged seed across all harvested ovaries was used as an independent data point (replicate) for each cross. ANOVAs with post hoc Tukey–Kramer tests were used to compare crosses in R.
To investigate seeds throughout their development, one to three ovaries were collected at 3, 5, 8, and 11 DAP for each cross. Ovaries were immediately placed into 4% paraformaldehyde and vacuum infiltrated for 15–20 min and kept at 4°C for 48 h. They were then washed with phosphate-buffered saline (PBS) (pH 7.4) three times for 30 min each and left in fresh PBS at 4°C overnight. Ovaries were then dehydrated using the following dehydration series: 10%, 30%, 50%, and 70% ethanol for 15 min each, 95% for 20 min, and two times in 100% for 30 min, all on a shaker. Ovaries were next infiltrated through the following infiltration series: 100% propylene oxide three times for 20 min each, 2:1 propylene oxide to Spurr’s resin mixture (EMS Catalog #14300) for 1 h, and 1:1 propylene oxide to Spurr’s for 1 h, all on a shaker. All samples were embedded into full resin, changed after 2 h, and left overnight in fresh resin at room temperature on a shaker. The next day, resin was changed twice, each after 2 h. Ovaries were then placed into individual molds with resin and left in an oven at 6°C for 48 h. Using an ultramicrotome, 0.5 μm sections were produced from the resin molds. Sections were adhered to slides in water at 50°C on a heat block. Slides were next stained with Azure II for 5 min at 50°C on a heat block. Coverslips were mounted onto slides using acrylamide, and images were taken on a compound microscope using SPOT Imaging software. We scanned through serial sections of individual seeds and selected the section closest to the longitudinal median of the embryo (for consistency among seeds) within the series. Such sections were selected from each cross for 8 DAP and 11 DAP. Due to the difficulty of producing such sections, some ovaries contained multiple seeds that were used in the following analysis and others contained none. However, most conditions (cross and DAP) had seeds that were used from at least two independent ovaries. Each individual seed from each condition was treated as a biological replicate. ImageJ was used to measure the area of the endosperm, embryo, and whole seed by manually tracing these tissues. Measurements were adjusted based on the magnification of the image. ANOVAs with post hoc Tukey–Kramer tests were used in R to compare endosperm and embryo area among crosses at 8 and 11 DAP each. Welch’s t tests were used to identify shifts in area from 8 to 11 DAP for endosperm and embryo for each cross. Images from all histological sections, with those selected for measurement denoted, and quantitative data from this and the “Seed germination” section below are available on Dryad (https://doi.org/10.5061/dryad.x0k6djhj4).
Scanning electron microscopy
Ten mature seeds (see above) from each of the four crosses were fixed in 2% glutaraldehyde with PBS for 2 h. After fixation, seeds were washed with PBS and put through a dehydration series at concentrations of 50%, 70%, 85%, 95%, and 100% ethanol: PBS for 1 h at each step. Seeds were left in 100% ethanol overnight. One hour before critical point drying, 100% ethanol was renewed. After critical point drying with a Samdri PVT-3B Critical Point Drier (Tousimis Research Corporation), seeds were sputter-coated with gold-paladium using a Hummer sputtering system from Anatech Ltd. and mounted on pin stubs for scanning electron microscopy (SEM). All images were acquired using Phenom Tabletop SEM.
Seed germination
To link the observed seed morphologies to germination rates, a germination experiment was performed in which morphology was measured. Mature ovaries were collected at 15–18 DAP within a week of the experiment (these were the same mature ovaries and seeds used for measurement of seed area above). Seeds were soaked in 3% calcium hypochlorite for 10 min and rinsed in PBS three times for 5 min each time, all on a shaker. Surface sterilized seeds were then placed on 60 mm petri dishes with gridded filter paper atop additional filter paper to retain moisture. The petri dishes and filter paper had been sterilized with UV for 30 min first. Images of seeds on the plates were taken with SPOT Imaging software, area was measured in ImageJ using a custom script to automate measurements, and seed germination was tracked specifically for each seed based off its position on the grid. Five plates were used per cross, with 16–32 seeds per plate. Seeds were placed under growth lights for 8 days with a photoperiod of 16-h light/8-h dark. The final data sheet contained a list of seeds, their size, which cross they belonged to, whether or not they germinated over the 8-day period, and if so, on which day they germinated. Welch’s t tests were used in R to compare the area of germinated seeds to nongerminated seeds for seeds from all crosses, seeds from all crosses excluding 4x × 2x, and seeds from each individual cross. Next, ANOVAs were used in R to compare the area of seeds at different germination dates (2, 3, 4, 5, 6, and 7 days after the start of the experiment) for all crosses and for each individual cross. Finally, since we could not measure endosperm area for this germination experiment, a linear regression was performed in R between endosperm and seed area using the histology data above for 8 and 11 DAP in order to link whole seed area to endosperm area.
RNA-seq data collection
Three or four ovaries were collected at 11 DAP for each cross (or 10 DAP for M11 × CS, since little endosperm was left at 11 DAP). After collection, ovaries were stored in RNAlater (Ambion) at 4°C and dissected on the same day. Each individual ovary was treated as a biological replicate. In order to separate endosperm tissue from the rest of the seeds, a modified protocol was used from Gehring et al. (2009). Endosperm and embryo were dissected from seeds in a 0.3-M sorbitol, 5-mM MES (2-[N-morpholino] ethanesulfonic acid) solution (Gehring et al., 2009) on a glass slide under a dissecting microscope using 30-gauge hypodermic needles. Endosperm and embryo from 20 to 40 seeds were pooled (separately) per biological replicate (ovary) and rinsed with the same solution 5–10 times. Pooled endosperm and embryo were then placed into the Lysis solution of RNAqueous-Micro Total RNA Isolation Kit (Ambion) and disrupted with a pestle (rotating 50–60 times). The remainder of the RNA extraction protocol from the kit was performed. RNA was converted into cDNA and libraries were constructed using KAPA Stranded mRNA-Seq Kit. During library construction, sequence-specific Illumina TruSeq adapters were added to distinguish each library. Using an Agilent 2100 Bioanalyzer, average fragment lengths were determined to be between 250 and 300 bp. Libraries were then pooled and sequenced by the Duke Center for Genomic and Computational Biology on an Illumina HiSeq 2500 instrument. All raw sequencing data are available at SRA PRJNA715438.
Analysis of RNA-seq data
The overview of our RNA-seq analysis is as follows. Previously (Edger et al., 2017; Puzey et al., 2017) we have shown that M. guttatus and the two M. luteus subgenomes (A and B) are distinct from each other. The two M. luteus subgenomes are 11% diverged at synonymous sites, while subgenome A is 10% and B is 11% diverged from the M. guttatus genome (Edger et al., 2017). This level of divergence between species allows unique mapping of RNA-seq reads to specific parental alleles of homologs in inter-specific crosses. In addition, we used diverged inbred lines of M. luteus (CS and Mll) and M. guttatus (CG and LCA) for unique mapping to homologs in intra-specific crosses. We define homologs generally as the two parental alleles (i.e. maternal and paternal) of a given gene in a given cross (whether in an intra-specific or inter-specific cross). Due to their sequence divergence, in crosses involving M. luteus, reads could either be mapped to the M. luteus genome as a whole or separately to its two subgenomes, allowing subgenome-specific comparisons as well. Homologous genes between M. luteus subgenomes A and B from the same parental allele are defined as homeologs. Using homolog- (and/or homeolog)-specific mappings, we proceeded with a traditional HT-seq-based approach to count the number of actual reads aligning to individual homologs. We then used a tool developed by Smith et al. (2019) to test for allele-specific expression bias in the embryo and endosperm of each cross (refer to the “Crossing design” section above), with which downstream analyses were performed (in R). Specific details follow.
Regarding alignment, parental genomes were SNP-corrected by first mapping whole genome (CG, CS, Mll) and transcriptome (LCA) data to previously assembled reference genomes (Hellsten et al., 2013; Edger et al., 2017) using bowtie2 in very-sensitive-local mode (Langmead and Salzberg, 2012). Picardtools were used to fix mate information for paired-end reads (“FixMateInformation”), remove PCR duplicates (“MarkDuplicates”), and add read groups (“AddOrReplaceReadGroups”) (https://broadinstitute.github.io/picard). Sequence variants were then called using the GATK UnifiedGenotyper (McKenna et al., 2010) with parameters: “-R genome -T UnifiedGenotyper -rf MaxInsertSize –maxInsertSize 10000 -rf DuplicateRead -rf BadMate -rf BadCigar –minbasequalityscore 25 -rf MappingQuality –minmappingqualityscore 25 -ploidy 2 –genotypelikelihoodsmodel BOTH –outputmode EMITALLSITES –maxalternatealleles 2 $inputBams -o $outputVCF.” The GATK FastaAlternateReferenceMaker (McKenna et al., 2010) was used to generate the new SNP-corrected fasta files. For each cross (and tissue), the SNP-corrected coding regions of the parental genomes (or transcriptome) were combined into a single fasta file, and the fastq reads were aligned to these references using bowtie2. For aligning RNA-seq reads to the combined reference, the following settings were used: “–local -5 10 -D 25 -R 4 -N 0 -L 10 -i S,1,0.5 –mp 46,42.” Counts were generated using HTSeq-count with default options (Anders et al., 2015). Homologs with no sequence differences were excluded from further analysis. Final alignments and RPKM values can be accessed in via Dryad (https://doi.org/10.5061/dryad.x0k6djhj4).
Next, parental allelic expression biases (AEBs) and shifts in AEB (AEBS) were calculated and compared between crosses using the methods described in Smith et al. (2019). Maternal bias has a negative AEB, paternal bias has a positive AEB, and an AEB of 0 indicates no bias. Differences in genomic dosage between the two alleles, due to the 2m:1p parental genome ratio in the endosperm, were accounted for by adjusting the gene length accordingly in AEB measurements (i.e. for a 2m:1p ratio, the gene length for the maternal allele is multiplied by 2 to represent the doubling of genetic material when calculating RPKM). In hybrid crosses, we managed the difference in ploidy between parental species (2x in M. guttatus versus 4x in M. luteus) by testing each M. guttatus gene twice; they were compared with each of their two M. luteus homologs as separate homolog pairs, thus maintaining an expected 2m:1p (or 1m:1p for embryo) genomic ratio for every comparison. For every homolog pair within each cross, a likelihood ratio test was used to test whether, after normalizing for gene length and sequencing depth differences, the mean expression level of the two homologs was the same or different, assuming the mean expression level follows a negative binomial distribution. False discovery rates for all tests were controlled using the R package “fdrtool” (Strimmer, 2008), and AEB or AEBS values with a q-value less than 0.05 were called significant. The inspection of unadjusted P-value distributions revealed one case, the embryo of the CG × LCA (M. guttatus) cross, with a highly conservative (theoretically impossible) skew. This might be indicative of unknown covariates in the data, or an otherwise mis-specified null model. To correct for this, we ran DESeq2 (Love et al., 2014) for each gene (as maternal versus paternal) and extracted the shrunken log2 fold-changes as well as their estimated standard errors, then performed a Z-test between each homolog pair. This method also produced P-values with a highly conservative skew; however, fdrtool was able to correct the Z-scores using its empirical null modeling approach. We did not consider genes that had less than 10 RPKM in both the paternal and maternal allele or had no mapped reads in at least one of the biological replicates. For AEBS tests, the same filter was extended to apply to both reciprocal crosses (e.g. CG × LCA and LCA × CG). Due to the reciprocal crossing design used (e.g. CG × LCA and LCA × CG), genes can either be consistently biased to the maternal or the paternal allele, or consistently biased to either one of the inbred lines (or have no bias in at least one cross). Imprinted genes were those that consistently had significant (after controlling FDR < 0.05) maternal or paternal bias between the reciprocal crosses (MEG or PEG, respectively). AEB values of genes in one cross were plotted against the AEB values of the same genes in the reciprocal cross (the direction of parental bias is reversed for visualization) for M. guttatus (i.e. CG × LCA and LCA × CG), M. luteus (i.e. CS × Mll and Mll × CS), and the hybrid (i.e. CG × Mll and Mll × CG). Finally, a Fisher’s test was used to determine differences in the PEG:MEG ratio between M. luteus and M. guttatus.
This section applies only to endosperm. We first quantified the amount of overlap in imprinted genes between M. guttatus and M. luteus. The list of genes for the 2x × 4x and 4x × 2x crosses represents all the known clear homologs between M. guttatus and M. luteus (Edger et al., 2017). Therefore, to assess overlap between M. guttatus and M. luteus imprinted genes, we counted only those imprinted genes within this list of homologs. Of its 276 putative imprinted genes, M. luteus had 38 PEGs with clear homologs in M. guttatus, and 1 out of the 16 MEGs in M. guttatus and 5 of its 37 PEGs had homologs in M. luteus (note that each gene in M. guttatus has two corresponding homologs in M. luteus since it is a tetraploid). Next, we wanted to understand how the parental expression bias (i.e. AEB) of the set of imprinted genes in one species differs or is consistent in the other crosses. We focused only on PEGs. To visualize these differences, we plotted the AEB distributions of each cross using the subset of genes from the M. guttatus and M. luteus homolog list (Edger et al., 2017). For each gene, AEB values were averaged between reciprocal crosses for M. guttatus and for M. luteus (e.g. CG × LCA and LCA × CG) to avoid line-specific bias. We plotted the averaged AEB values of the five M. guttatus and 38 M. luteus PEGs represented in this homolog list on top of the 2x × 2x and 4x × 4x distributions, respectively. We also plotted the averaged AEB of the homologs of M. luteus PEGs in M. guttatus on top of the 2x × 2x distribution and the averaged AEB of the homologs of M. guttatus PEGs in M. luteus on top of the 4x × 4x distribution. Finally, we plotted the AEB values of the M. guttatus and M. luteus PEGs from the 2x × 4x and 4x × 2x crosses on top of each of their respective distributions. Only genes filtered for AEBS tests (above) were used for M. guttatus and M. luteus and genes filtered for AEB tests (above) for 2x × 4x and 4x × 2x crosses.
We next compared expression differences between filial tissues in M. guttatus, M. luteus, and 2x × 4x. For both embryo and endosperm, RPKM values were averaged between reciprocal crosses for M. guttatus and for M. luteus (e.g. CG × LCA and LCA × CG) to avoid line-specific bias. The log2 fold-change between the two tissues (log2(RPKMEndosperm/RPKMEmbryo)) was then calculated for each gene in each species. Infinite (as a result of zeroes) or not available values were removed. The same measurements were made for 2x × 4x, except we did not average RPKM values with its reciprocal cross, 4x × 2x. We could not produce this measurement for 4x × 2x since it lacked sufficient embryo tissue.
For crosses with the M. luteus genome (CS × Mll, Mll × CS, CG × Mll, Mll × CG), alignments were prepared for those homeolog pairs that could unambiguously be sorted into the specific M. luteus subgenomes (A and B). The same endosperm to embryo comparison of expression as described previously was reproduced for each subgenome for M. luteus (4x × 4x) and 2x × 4x. Next, we performed AEB measurements, as discussed above, between homeologous pairs from parental alleles for each subgenome from each cross (e.g. Amaternal versus Apaternal and Bmaternal versus Bpaternal in CS × Mll, or M. guttatusmaternal versus Apaternal and M. guttatusmaternal versus Bpaternal in CG × Mll). Genes were filtered according to AEBS filtrations (above) and averaged between the M. luteus reciprocal crosses (CS × Mll and Mll × CS) for each subgenome. For the hybrid crosses (CG × Mll and Mll × CG), genes were filtered according to AEB filtrations (above). To compare the relationship of AEB between homeolog pairs in M. luteus, a linear regression was performed between AEB values calculated from comparing the maternal and paternal alleles of the A subgenome and the corresponding AEB values calculated from comparing their homeologs on maternal and paternal alleles of the B subgenome. That is, for each homeolog pair, parental expression bias between A subgenome alleles was compared with parental expression bias between B subgenome alleles. Homeolog pairs were filtered out if the number of reads in any replicate was 0 for either homeolog. We did not filter for low RPKM, because sample sizes were low for the linear regressions, though even after filtering out genes that had less than 10 RPKM on both the maternal and paternal allele (for each subgenome), results of linear regressions were very similar (Supplemental Figure S5, A–D and Supplemental Table S12, B). Linear regressions were performed for each reciprocal cross (i.e. Mll × CS and CS × Mll) in the embryo and the endosperm. Similar methods and filtration were used for the hybrid crosses (i.e. 2x × 4x and 4x × 2x) except linear regressions were performed between AEB values calculated from comparing the M. guttatus (maternal for 2x × 4x and paternal for 4x × 2x) and A subgenome alleles (paternal for 2x × 4x and maternal for 4x × 2x) and the corresponding AEB values calculated from comparing the M. guttatus and B subgenome alleles. Linear regressions were performed in the embryo and endosperm of 2x × 4x and the endosperm of 4x × 2x. Again, genes were not filtered for low RPKM, but even if filtered, results were also similar (Supplemental Figure S5, E–F and Supplemental Table S12, B).
BLAST of imprinted genes
Using our lists of putative M. luteus and M. guttatus imprinted genes, we took the gene name and found the sequence from a specific reference genome (either M. luteus or M. guttatus). This reference sequence was then BLASTed against a database of A. thaliana genes (TAIR). We used a BLAST NT query and set outputs to include Araport 11 transcripts (DNA). Gene descriptions were pulled from A. thaliana genes with e-values below 0.01.
GO enrichment
We used coding sequences derived from the CS (for M. luteus) and CG (for M. guttatus) genomes for functional annotation. For each genome, we translated DNA sequences with Trinotate (version 3.0.1) and identified protein domains with HMMER (version 3.2.1), signal peptides with SignalP (version 5.0b), and transmembrane regions with tmHMM (version 2.0c) using the Pfam database. We also captured BLAST homologies with BLASTx and BLASTp from the UniProt protein database (The UniProt Consortium 2018). We loaded these annotation results into a Trinotate SQLite Database and gathered GO assignments from Pfam and UniProt. Using all annotated coding sequences as the background gene set, we performed a GO enrichment analysis for both MEGs and PEGs using the GOseq pipeline from Trinity for both M. luteus and M. guttatus (using their respective genomes). We consider significant GO terms with a false discovery rate adjusted P-value below 0.05.
Collection and analysis of Methyl-seq data
Endosperm was collected and pooled using the same methods as above (see RNA-seq data collection section for details), and DNA was extracted using a CTAB-based protocol. There were three replicates (ovaries) for each of the four crosses used (refer to the “Crossing design” section above). Bisulfite conversion was performed using the EZ DNA Methylation Kit (Zymo) and libraries were immediately constructed using the TruSeq DNA Methylation Kit (Illumina) with adapters from TruSeq DNA Methylation Index PCR Primers (Illumina). DNA was sequenced on an Illumina HiSeq 2500, generating 50-bp reads. All raw sequencing data are available at SRA PRJNA715438.
Methylation analyses were conducted using the methylpy pipeline (Schultz et al., 2015). Methylpy was specifically designed to analyze high-throughput bisulfite sequencing data. Reads from each replicate for a given cross were pooled and analyzed together through this pipeline. Pooled reads were aligned to a combined reference file using the following parameters: “num_procs = 20, illumina adapter sequence, quality_version=“1.8”, bowtie_options=[“-S”,“-k 1”,“-m 1”,“–chunkmbs 3072”,“–best”,“–strata”,“-o 4”,“-e 80”,“-l 20”,“-n 0”], max_adapter_removal=None, overlap_length=None, zero_cap=None, error_rate=None, min_qual_score = 10, min_read_len = 30, sig_cutoff = 0.05, min_cov = 3, binom_test=True, num_reads=-1.” The combined reference file contained both M. guttatus (CG) and M. luteus (Mll) genomes as well as the mitochondrial genome (CG) (NCBI accession NC_018041). Following alignment, methylpy calls methylated bases based on SNPs relative to the reference. The output of methylpy includes, for each site, the number of methylated bases sequenced, the total number of bases sequenced, and the sequence context (CpG, CHG, CHH). With these data, we calculated the weighted methylation level per site for each sequence context in all crosses. This data in combination with the genome annotation files was used to analyze the data by gene, TE, their flanking regions, and sequence context. Nonconversion rates were calculated by comparing the number of methylated sites (as called by the methylpy pipeline) against unmethylated sites for the mitochondrial genome for sites with at least 10 reads. These rates were 0.0554, 0.0369, 0.0322, and 0.0278 for CG × Mll, Mll × CG, CG × LCA, and Mll × C5, respectively. Raw methylation outputs from the Methylpy pipeline are available on Dryad (https://doi.org/10.5061/dryad.x0k6djhj4).
The impact of differences in nonconversion rates across samples may bias comparisons between samples: samples with lower nonconversion rates would appear to have slightly higher weighted methylation. This impact would be most extreme for samples with low methylation. Our primary goal here was to compare global methylation patterns within a sample, and not methylation levels of individual genes. With this in mind, when comparing two distributions, with the prior knowledge that each distribution has a unique correction factor (i.e. nonconversion rate), it is possible to shift the distribution accordingly and then compare the rescaled distributions. Thus, we corrected for nonconversion rate for each of our samples (crosses) by multiplying the per-site weighted methylation value (Schultz et al., 2012) by (1 – nonconversion rate). Preceding this correction, we filtered out any sites with 20 or fewer mapped reads. We produced metaplots by binning upstream and downstream regions every 20 bp over a length of 2 kb in each direction. We binned genes and TE bodies every 0.1% of their regions. Weighted methylation levels were calculated for each of these bins as described above. For hybrid crosses (CG × Mll and Mll × CG), we were able to produce these measurements for each allele (genome: CG and Mll) separately. These data are accessible via Dryad (https://doi.org/10.5061/dryad.x0k6djhj4).
Accession numbers
The raw RNA-seq and bisulfite sequencing reads are deposited on SRA (PRJNA715438). The processed data files used in analysis are available on Dryad (https://doi.org/10.5061/dryad.x0k6djhj4).
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Quantitative analysis for seed development of parental and reciprocal hybrids seeds.
Supplemental Figure S2. Aspect ratio (roundness) of parental and reciprocal hybrid seeds.
Supplemental Figure S3. Effect of seed area on germination timing.
Supplemental Figure S4. Additional comparisons of AEB values between M. luteus subgenomes.
Supplemental Figure S5. Comparisons of AEB values between M. luteus subgenomes using stricter filtration.
Supplemental Table S1. Means of seed area and AR for each cross.
Supplemental Table S2. Tukey’s HSD results for seed area and AR between crosses.
Supplemental Table S3. Endosperm and embryo size at different developmental stages for each cross.
Supplemental Table S4. Tukey’s HSD results for endosperm and embryo size at different developmental stages between crosses.
Supplemental Table S5. Welch’s t test results for changes in size of seed tissues over time.
Supplemental Table S6. Germination rates and area for each cross.
Supplemental Table S7. Area of germinated and nongerminated seeds.
Supplemental Table S8. Area of seeds at different germination dates.
Supplemental Table S9. Tukey’s HSD comparisons for seed area between different germination dates.
Supplemental Table S10. Significant results from GO enrichment analysis.
Supplemental Table S11. Sample size (N) for AEB plots in Figure 4.
Supplemental Table S12. Sample sizes (N) for AEB subgenome regressions.
Supplemental Table S13. Weighted methylation of coding sequences across genomes.
Supplemental Table S14. Raw read counts for methylome sequencing and nonconversion rates.
Supplemental Data Set S1. Imprinted genes in M. luteus, M. guttatus, and significantly enriched GO categories for M. luteus and M. guttatus PEGs.
Supplemental Data Set S2. Subgenome-specific AEB values in M. luteus and of the M. luteus subgenomes inherited in the hybrids.
Supplemental File S1. Reduced dataset of genes (M. luteus and M. guttatus homologs) that could be compared among all crosses.
Supplemental File S2. Log fold-change of endosperm compared with embryo tissue.
Supplementary Material
Acknowledgments
The authors would like to thank Dr Diane Shakes and Dr Harmony Dalgleish for their support and constructive critique during the development and implementation of this project. Dr Abigail Reft provided support and training for seed sectioning and SEM. Colleen Flynn performed SEM. Hunter Call wrote scripts for automated measurements of seed morphology in ImageJ. Sara Meier, Nora Flynn, and Scott Teresi assisted in laboratory work. They would also like to thank Dr Mary Gehring for providing insight into seed dissection methods for RNA and DNA extraction.
Funding
This work was supported by The College of William and Mary Research Award to JRP, NSF 1754080 to J.R.P., and NSF 1754075 to A.M.C.
Conflict of interest statement. None declared.
T.J.K. and J.R.P. conceived of and designed all aspects of this research. A.M.C and M.V.-M. contributed initial ideas for research development, edited this article and its conclusions, and provided seeds. T.J.K. performed all crosses for histology, germination experiments, and sequencing; performed seed sectioning, staining, histological measurements, germination experiments, and all associated data analyses; collected samples and performed RNA sequencing and bisulfite treatment/methylation sequencing. R.D.S. performed transcriptome assembly and all homolog expression analyses and likelihood ratio tests with input from J.R.P. and T.J.K. R.D.S. and G.D.C.S. modified these analyses for this system and the specific questions addressed in this study. Furthermore, R.D.S. provided general guidance regarding statistical analyses. A.H.L. performed methylation analyses with input from J.R.P and T.J.K. T.J.K. performed downstream analyses of homolog expression and methylation data with input from J.R.P. T.J.K. wrote this article with contributions from R.D.S., J.R.P., and A.H.L.
The author(s) responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: Joshua R. Puzey (jrpuzey@wm.edu).
References
- Alger EI, Edger PP (2020) One subgenome to rule them all: underlying mechanisms of subgenome dominance. Curr Opin Plant Biol 54: 108–113 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. (2015). HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroux C, Spillane C, Grossniklaus U. (2002). Evolutionary origins of the endosperm in flowering plants. Genome Biol 3: 1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista RA, Figueiredo DD, Santos-González J, Köhler C (2019) Auxin regulates endosperm cellularization in Arabidopsis. Genes Dev 33: 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista RA, Köhler C (2020) Genomic imprinting in plants—revisiting existing models. Genes Dev 34: 24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beamish KI (1955) Seed failure following hybridization between the hexaploid Solanum demissum and four diploid Solanum species. Am J Bot 42: 297–304 [Google Scholar]
- Brandvain Y, Haig D (2005) Divergent mating systems and parental conflict as a barrier to hybridization in flowering plants. Am Nat 166: 330–338 [DOI] [PubMed] [Google Scholar]
- Brandvain Y, Kenney AM, Flagel L, Coop G, Sweigart AL (2014) Speciation and introgression between Mimulus nasutus and Mimulus guttatus. PLoS Genet 10: e1004410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkart-Waco D, Ngo K, Lieberman M, Comai L (2015) Perturbation of parentally biased gene expression during interspecific hybridization. PLoS ONE 10: e0117293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L, Madlung A, Josefsson C, Tyagi A (2003) Do the different parental ‘heteromes’ cause genomic shock in newly formed allopolyploids? Phil Trans R Soc Lond B 358: 1149–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B, Yuan Y-W (2016) Testing the utility of fluorescent proteins in Mimulus lewisii by an Agrobacterium-mediated transient assay. Plant Cell Rep 35: 771–777 [DOI] [PubMed] [Google Scholar]
- Edger PP, Smith R, McKain MR, Cooley AM, Vallejo-Marín M, Yuan Y, Bewick AJ, Ji L, Platts AE, Bowman MJ, et al. (2017) Subgenome dominance in an interspecific hybrid, synthetic allopolyploid, and a 140-year-old naturally established neo-allopolyploid monkeyflower. Plant Cell 29: 2150–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris KG, Barnett LL, Blackman BK, Willis JH (2017) The genetic architecture of local adaptation and reproductive isolation in sympatry within the Mimulus guttatus species complex. Mol Ecol 26: 208–224 [DOI] [PubMed] [Google Scholar]
- Figueiredo DD, Batista RA, Roszak PJ, Köhler C (2015) Auxin production couples endosperm development to fertilization. Nat Plants 1: 15184. [DOI] [PubMed] [Google Scholar]
- Fishman L, Willis JH(2008) Pollen limitation and natural selection on floral characters in the yellow monkeyflower, Mimulus guttatus. New Phytol 177: 802–810 [DOI] [PubMed] [Google Scholar]
- Flores-Vergara MA, Oneal E, Costa M, Villarino G, Roberts C, De Luis Balaguer MA, Coimbra S, Willis J, Franks RG (2020) Developmental analysis of Mimulus seed transcriptomes reveals functional gene expression clusters and four imprinted, endosperm-expressed genes. Front Plant Sci 11: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florez-Rueda AM, Paris M, Schmidt A, Widmer A, Grossniklaus U, Städler T (2016) Genomic imprinting in the endosperm is systematically perturbed in abortive hybrid tomato seeds. Mol Biol Evol 33: 2935–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman WE (1995) Organismal duplication, inclusive fitness theory, and altruism: understanding the evolution of endosperm and the angiosperm reproductive syndrome. Proc Natl Acad Sci USA 92: 3913–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman WE (2001) Developmental and evolutionary hypotheses for the origin of double fertilization and endosperm. Comptes Rendus Acad Sci 324: 559–567 [DOI] [PubMed] [Google Scholar]
- Friedman WE, Madrid EN, Williams JH (2008) Origin of the fittest and survival of the fittest: relating female gametophyte development to endosperm genetics. Int J Plant Sci 169: 79–92 [Google Scholar]
- Garner AG, Kenney AM, Fishman L, Sweigart AL (2016) Genetic loci with parent-of-origin effects cause hybrid seed lethality in crosses between Mimulus species. New Phytol 211: 319–331 [DOI] [PubMed] [Google Scholar]
- Geeta R (2003) The origin and maintenance of nuclear endosperms: viewing development through a phylogenetic lens. Proc Biol Sci R Soc 270: 29–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M, Bubb KL, Henikoff S (2009) Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 324: 1447–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M, Satyaki PR (2016) Endosperm and imprinting, inextricably linked. Plant Physiol 173: 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves IK, Groszmann M, Ying H, Taylor JM, Peacock WJ, Dennis ES (2012) Trans chromosomal methylation in Arabidopsis hybrids. Proc Natl Acad Sci USA 109: 3570–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossenbacher DL, Whittall JB (2011) Increased floral divergence in sympatric monkeyflowers. Evolution 65: 2712–2718 [DOI] [PubMed] [Google Scholar]
- Haig D ((2013) Kin conflict in seed development: an interdependent but fractious collective. Annu Rev Cell Dev Biol 29: 189–211 [DOI] [PubMed] [Google Scholar]
- Haig D, Westoby M (1989) Parent-specific gene expression and the triploid endosperm. Am Nat 134: 147–155 [Google Scholar]
- Haig D, Westoby M (1991) Genomic imprinting in endosperm: its effect on seed development in crosses between species, and between different ploidies of the same species, and its implications for the evolution of apomixis. Phil Trans R Soc Lond B 333: 1–13 [Google Scholar]
- Hellsten U, Wright KM, Jenkins J, Shu S, Yuan Y, Wessler SR, Schmutz J, Willis JH, Rokhsar DS (2013) Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc Natl Acad Sci USA 110: 19478–19482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JH, Rim HJ (2013) DNA demethylation and gene imprinting in flowering plants. In Gideon Grafi, Nir Ohad (eds) Epigenetic Memory and Control in Plants. Springer, Berlin, Heidelberg, pp 201–232 [Google Scholar]
- Ishikawa R, Ohnishi T, Kinoshita Y, Eiguchi M, Kurata N, Kinoshita T (2011) Rice interspecies hybrids show precocious or delayed developmental transitions in the endosperm without change to the rate of syncytial nuclear division. Plant J 65: 798–806 [DOI] [PubMed] [Google Scholar]
- Johnston SA, Hanneman RE Jr (1982) Manipulations of endosperm balance number overcome crossing barriers between diploid Solanum species. Science 217: 446–448 [DOI] [PubMed] [Google Scholar]
- Josefsson C, Dilkes B, Comai L (2006) Parent-dependent loss of gene silencing during interspecies hybridization. Curr Biol 16: 1322–1328 [DOI] [PubMed] [Google Scholar]
- Kinoshita T ((2007) Reproductive barrier and genomic imprinting in the endosperm of flowering plants. Genes Genet Syst 82: 177–186 [DOI] [PubMed] [Google Scholar]
- Kooyers NJ, James B, Blackman BK (2017) Competition drives trait evolution and character displacement between Mimulus species along an environmental gradient. Evolution 71: 1205–1221 [DOI] [PubMed] [Google Scholar]
- Lafon-Placette C, Johannessen IM, Hornslien KS, Ali MF, Bjerkan KN, Bramsiepe J, Glöckle BM, Rebernig CA, Brysting AK, Grini PE, et al. (2017) Endosperm-based hybridization barriers explain the pattern of gene flow between Arabidopsis lyrata and Arabidopsis arenosa in Central Europe. Proc Natl Acad Sci USA 114: E1027–E1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Placette C, Köhler C (2015) Epigenetic mechanisms of postzygotic reproductive isolation in plants. Curr Opin Plant Biol 23: 39–44 [DOI] [PubMed] [Google Scholar]
- Lafon-Placette C, Köhler C (2016) Endosperm-based postzygotic hybridization barriers: developmental mechanisms and evolutionary drivers. Mol Ecol 25: 2620–2629 [DOI] [PubMed] [Google Scholar]
- Lafon-Placette C, Hatorangan MR, Steige KA, Cornille A, Lascoux M, Slotte T, Köhler C (2018) Paternally expressed imprinted genes associate with hybridization barriers in Capsella. Nat Plants 4: 352–357 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc O, Pointe C, Hernandez M (2002) Cell cycle progression during endosperm development in Zea mays depends on parental dosage effects. Plant J 32: 1057–1066 [DOI] [PubMed] [Google Scholar]
- Lee JH, Cooper DC (1958) Seed development following hybridization between diploid Solanum species from Mexico, Central and South America. Am J Bot 45: 104–110. [Google Scholar]
- Li N, Dickinson HG (2010) Balance between maternal and paternal alleles sets the timing of resource accumulation in the maize endosperm. Proc Biol Sci R Soc 277: 3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke M (1993) Genomic imprinting in plants: parental effects and trans-inactivation phenomena. Annu Rev Plant Physiol Plant Mol Biol 44: 53–76 [Google Scholar]
- McClintock B (1984) The significance of responses of the genome to challenge. Science 226: 792–801 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medel R, Botto-Mahan C, Kalin-Arroyo M (2003) Pollinator-mediated selection on the nectar guide phenotype in the Andean Monkey flower, Mimulus luteus. Ecology 84: 1721–1732 [Google Scholar]
- Meeus S, Šemberová K, De Storme N, Geelen D, Vallejo-Marín M (2020) Effect of whole-genome duplication on the evolutionary rescue of sterile hybrid monkeyflowers. Plant Commun [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Niederhuth CE, Bewick AJ, Ji L, Alabady MS, Do Kim K, Li Q, Rohr NA, Rambani A, Burke JM, Udall JA, et al. (2016) Widespread natural variation of DNA methylation within angiosperms. Genome Biol 17: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederhuth CE, Schmitz RJ (2017) Putting DNA methylation in context: from genomes to gene expression in plants. Biochim Biophys Acta 1860: 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowack MK, Shirzadi R, Dissmeyer N, Dolf A, Endl E, Grini PE, Schnittger A (2007) Bypassing genomic imprinting allows seed development. Nature 447: 312. [DOI] [PubMed] [Google Scholar]
- Oneal E, Lowry DB, Wright KM, Zhu Z, Willis JH (2014) Divergent population structure and climate associations of a chromosomal inversion polymorphism across the Mimulus guttatus species complex. Mol Ecol 23: 2844–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oneal E, Willis JH, Franks RG (2016) Disruption of endosperm development is a major cause of hybrid seed inviability between Mimulus guttatus and Mimulus nudatus. New Phytol 210: 1107–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker PF (1975) Mimulus in Great Britain-a cytotaxonomic note. New Phytol 74: 155–160 [Google Scholar]
- Porebski S, Grant Bailey L, Baum BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep 15: 8–15 [Google Scholar]
- Pennington PD, Costa LM, Gutierrez-Marcos JF, Greenland AJ, Dickinson HG (2008) When genomes collide: Aberrant seed development following maize interploidy crosses. Ann. Botany 101: 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzey JR, Willis JH, Kelly JK (2017) Population structure and local selection yield high genomic variation in Mimulus guttatus. Mol Ecol 26: 519–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebernig CA, Lafon-Placette C, Hatorangan MR, Slotte T, Köhler C (2015) Non-reciprocal interspecies hybridization barriers in the Capsella genus are established in the endosperm. PLoS Genet 11: e1005295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigal M, Becker C, Pélissier T, Pogorelcnik R, Devos J, Ikeda Y, Weigel D, Mathieu O (2016) Epigenome confrontation triggers immediate reprogramming of DNA methylation and transposon silencing in Arabidopsis thaliana F1 epihybrids. Proc Natl Acad Sci USA 113: E2083–E2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M, Florez-Rueda AM, Griesser S, Paris M, Städler T (2018a) Incidence and developmental timing of endosperm failure in post-zygotic isolation between wild tomato lineages. Ann Bot 121: 107–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M, Florez‐Rueda AM, Paris M, Städler T. (2018b) Wild tomato endosperm transcriptomes reveal common roles of genomic imprinting in both nuclear and cellular endosperm. Plant J 95: 1084–1101 [DOI] [PubMed] [Google Scholar]
- Roth M, Florez-Rueda AM, Städler T (2019) Differences in effective ploidy drive genome-wide endosperm expression polarization and seed failure in wild tomato hybrids. Genetics 212: 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyaki PR, Gehring M (2019) Paternally acting canonical RNA-directed DNA methylation pathway genes sensitize Arabidopsis endosperm to paternal genome dosage. Plant Cell 31: 1563–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MD, Schmitz RJ, Ecker JR (2012) ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet 28: 583–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, et al. (2015) Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523: 212–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RJ, Spielman M, Bailey J, Dickinson HG (1998) Parent-of-origin effects on seed development in Arabidopsis thaliana. Development 125: 3329–3341 [DOI] [PubMed] [Google Scholar]
- Shen H, He H, Li J, Chen W, Wang X, Guo L, Peng Z, He G, Zhong S, Qi Y, et al. (2012) Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 24: 875–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RD, Kinser TJ, Smith GD, Puzey JR (2019) A likelihood ratio test for changes in homeolog expression bias. BMC Bioinform 20: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE (2014) Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol 21: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K (2008) fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24: 1461–1462 [DOI] [PubMed] [Google Scholar]
- Stupar RM, Springer NM (2006) Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 173: 2199–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonosaki K, Ono A, Kunisada M, Nishino M, Nagata H, Sakamoto S, Kijima ST, Furuumi H, Nonomura KI, Sato Y, et al. (2020) Mutation of the imprinted gene OsEMF2a induces autonomous endosperm development and delayed cellularization in rice. Plant Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyford AD, Friedman J (2015) Adaptive divergence in the monkey flower Mimulus guttatus is maintained by a chromosomal inversion. Evolution 69: 1476–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium (2018) UniProt: the universal protein knowledgebase. Nucleic Acids Res 46: 2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Marín M (2012) Mimulus peregrinus (Phrymaceae): a new British allopolyploid species. PhytoKeys 14: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Marín M, Lye GC (2013) Hybridisation and genetic diversity in introduced Mimulus (Phrymaceae). Heredity 110: 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Marín M, Buggs RJA, Cooley AM, Puzey JR. (2015). Speciation by genome duplication: repeated origins and genomic composition of the recently formed allopolyploid species Mimulus peregrinus. Evolution 69: 1487–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Marín M, Cooley AM, Lee MY, Folmer M, McKain MR, Puzey JR. (2016). Strongly asymmetric hybridization barriers shape the origin of a new polyploid species and its hybrid ancestor. Am J Bot 103: 1272–1288 [DOI] [PubMed] [Google Scholar]
- Yan D, Duermeyer L, Leoveanu C, Nambara E (2014) The functions of the endosperm during seed germination. Plant Cell Physiol 55: 1521–1533 [DOI] [PubMed] [Google Scholar]
- Yoo M-J, Liu X, Chris Pires J, Soltis PS, Soltis DE. (2014). Nonadditive gene expression in polyploids. Annu Rev Genet 48: 485–517 [DOI] [PubMed] [Google Scholar]
- Yoo M-J, Szadkowski E, Wendel JF (2013) Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110: 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.