

Abstract
The developing nervous system is sensitive to environmental and physiological perturbations in part due to its protracted period of prenatal and postnatal development. Epidemiological and experimental studies link developmental exposures to persistent organic pollutants (POPs) including polychlorinated biphenyls, polychlorinated dibenzo-p-dioxins, polybrominated diphenyl ethers, and benzo(a)pyrene to increased risk for neurodevelopmental disorders in children. Mechanistic studies reveal that many of the complex cellular processes that occur during sensitive periods of rapid brain development are cellular targets for developmental neurotoxicants. One area of research interest has focused on synapse formation and plasticity, processes that involve the growth and retraction of dendrites and dendritic spines. For each chemical discussed in this review, we summarize the morphological and electrophysiological data that provide evidence that developmental POP exposure produces long-lasting effects on dendritic morphology, spine formation, glutamatergic and GABAergic signaling systems, and synaptic transmission. We also discuss shared intracellular mechanisms, with a focus on calcium and thyroid hormone homeostasis, by which these chemicals act to modify synapses. We conclude our review highlighting research gaps that merit consideration when characterizing synaptic pathology elicited by chemical exposure. These gaps include low-dose and non-monotonic dose-response effects, revealing the temporal relationship between dendritic growth, spine formation, and synaptic activity, excitation-inhibition balance, hormonal effects, and the critical need for more studies in females to identify sex differences. By identifying converging pathological mechanisms elicited by POP exposure at the synapse, we can define future research directions that will advance our understanding of these chemicals on synapse structure and function.
Keywords: Neurodevelopment, dendrites, dendritic spines, synaptic plasticity, low-dose effects
1. Introduction:
Nervous system development occurs over a protracted period, beginning in early gestation and continuing through the neonatal period and adolescence, and in some brain regions into adulthood (Semple et al., 2013; Stiles and Jernigan, 2010; Tau and Peterson, 2010). In utero, the human nervous system begins to form following the closure of the neural tube (gestation week 4), when brain structures undergo temporally distinct periods of rapid growth (Dobbing and Sands, 1973, 1979) characterized by increased dendritic complexity, synapse formation, axon targeting, and synaptic pruning. While the peak formation of synaptic contacts occurs between the third trimester and the first two years of life in the human neocortex (Bianchi et al., 2013; Huttenlocher and Dabholkar, 1997; Rakic et al., 1986), the cerebellum exhibits two waves of rapid brain growth, corresponding to the proliferation of Purkinje cells (soon after neural tube closure) and later granule cells (third trimester and early postnatal life) (Sathyanesan et al., 2019). In the hippocampus, rapid expansion of granule cells begins early during prenatal development and continues postnatally and these neurons continue to proliferate and form synaptic contacts well into adulthood, albeit at slower rates (Li et al., 2009). These distinct temporal and regional periods of rapid brain development confer selective vulnerability of the nervous system to injury and, if perturbed during critical developmental periods, can predispose the developing fetus or child to neurodevelopmental disorders (Rice and Barone, 2000; Rodier, 1994; Selevan et al., 2000).
1.1. Environmental exposures and neurodevelopment
Exposure to man-made environmental chemicals during sensitive developmental periods have been postulated to result in lifelong structural and functional changes in the brain (Bondy and Campbell, 2005; Grandjean, 2008; Landrigan et al., 2005; Rice and Barone, 2000; Rodier, 1994). For example, early exposures to the fungicide maneb (Meco et al., 1994) and the insecticide dieldrin (Weisskopf et al., 2010) have been implicated in the development of Parkinson’s disease. Epidemiologic evidence has also associated early exposure to heavy metals with psychiatric disorders (Bouchard et al., 2009), schizophrenia (Opler et al., 2008), and cognitive decline (Weisskopf et al., 2007) later in life. As a result, the prenatal and early postnatal periods are important time-windows for understanding how and to what degree exposure to environmental chemicals can lead to permanent and life-long impacts on the brain and behavior (Koger et al., 2005; Maffini and Neltner, 2015; Rauh and Margolis, 2016; Tyler et al., 2008).
While more than 80,000 chemicals are registered for commercial use with the U.S. Environmental Protection Agency (CDC, 2019; EPA, 2020; Muir and Howard, 2006), only a small number have been studied in humans during developmentally sensitive time-windows (Grandjean and Landrigan, 2006, 2014). Fewer have been rigorously identified to be developmentally neurotoxic, and none have been systematically assessed for their effects on brain development before commencing commercial use (Grandjean and Landrigan, 2006, 2014). This is alarming as developmental exposures to environmental toxicants are common as shown in a 2013 National Health and Nutrition Examination Survey which detected these chemicals in biological samples provided by a representative subset of the US population (Thompson and Boekelheide, 2013). More recently, the Centers for Disease Control (CDC, 2019) and the Global Burden of Disease study, supervised by the Institute for Health Metrics and Evaluation (Shaffer et al., 2019), also urgently recommended additional studies on environmental health risk factors of disease including developmental neurotoxicants. These recommendations are supported by studies demonstrating negative effects on brain development and increased risk for neurodevelopmental disorders after environmental exposure to lead (Lanphear et al., 2005), mercury (Axelrad et al., 2007; Oken et al., 2008), air pollution (Chiu et al., 2013; Siddique et al., 2011; Suglia et al., 2008), organophosphorus pesticides (Bouchard et al., 2011; Engel et al., 2011), brominated flame retardants (Lam et al., 2017), polychlorinated biphenyls (PCBs; Pessah et al., 2019), polybrominated diphenyl ethers (PBDEs; Gibson et al., 2018), and dioxin (Ames et al., 2019; Nishijo et al., 2012; Pham et al., 2019; Tai et al., 2013). Additionally, the relative contributions of environmental exposures to the prevalence of neurodevelopmental disorders have been reported to be more substantial than nonchemical risk factors such as preterm birth, type 1 diabetes, and congenital heart disease as well as socioeconomic, nutritional, and psychosocial factors (Bellinger, 2012).
The EPA defines neurotoxicity as an adverse change in the structure and/or function of the central and/or peripheral nervous system measured at the neurochemical, behavioral, neurophysiological, or anatomical levels (Tilson, 2000; Tilson et al., 1995). Given the multifaceted process of neurodevelopment (Stiles and Jernigan, 2010; Tau and Peterson, 2010), it is no surprise that there is a wealth of published literature that reviews the effects of various environmental contaminants on biological processes such as cell proliferation, differentiation, and apoptosis (Heyer and Meredith, 2017; Klocke and Lein, 2020; Rock and Patisaul, 2018; Tamm and Ceccatelli, 2017). However, less is known about how chemical exposure effects synaptic pathology (Klocke et al., 2020; Stamou et al., 2013; Vester and Caudle, 2016). This is a critical area of research because synaptogenesis is a protracted process, beginning in utero and lasting into the early postnatal years and, in neurogenic areas, also into adulthood. Synaptogenesis is followed by a period of significant synapse elimination and circuit remodeling to configure mature functional neural networks (Bianchi et al., 2013; Huttenlocher and Dabholkar, 1997; Rakic et al., 1986). The period of greatest synaptic density represents a sensitive time-window of significant structural and functional synaptic plasticity when there is a rapid forming and reforming of synaptic structures (Sala and Segal, 2014), processes that involve the growth and retraction of dendrites and dendritic spines. Deficits in these processes are thought to contribute to neurodevelopmental disorders such as autism (Herbert, 2011), Rett syndrome (Belichenko et al., 1994; Zoghbi, 2003), and fragile X syndrome (Kaufmann and Moser, 2000; Portera-Cailliau, 2012). Thus, the idea that neurotoxic exposures to man-made chemicals modify synapses and circuits, leading to synaptic dysfunction and neurotoxicity, merits careful investigation (Stamou et al., 2013; Vester and Caudle, 2016).
1.2. Developmental neurotoxicity of persistent organic pollutants (POPs)
One group of chemicals that the EPA has identified as high risk as part of the Toxic Substances Control Act and in need of further risk evaluation are persistent organic pollutants (POPs). POPs are carbon-containing industrial compounds that, to a varying degree, resist photochemical, biological, and chemical degradation. Because of their persistence and high lipid solubility, POPs are not readily destroyed and persist in ecosystems, bioaccumulate in food chains, and ultimately accumulate in body tissue. Human exposure occurs through several routes including inhalation, ingestion, and dermal contact with contaminated air, food, water, house dust, and soil. Both the developing fetus and children tend to be exposed to higher levels of these chemicals than adults for several reasons. First, many toxicants can cross the placenta and the fetal blood-brain barrier (BBB) to reach the developing brain (Grandjean and Landrigan, 2006). Fat-soluble toxicants such as POPs can also accumulate in maternal adipose tissue and breast milk and be passed on to the developing newborn (Grandjean and Landrigan, 2006). Second, the BBB and the function of enzymes that detoxify and metabolize exogenous chemicals are not fully developed until well after birth (Choudhary et al., 2003; Risau and Wolburg, 1990; Rodier, 1994). Third, because of their rapid development, children have greater energy demands than adults. When factoring in their body weight, children exhibit higher rates of inhalation and food consumption, leading to potentially higher chemical exposure (Koger et al., 2005). Lastly, infants and children engage in behaviors that increase their contact with contaminants such as crawling and frequent hand-to-mouth behaviors (Koger et al., 2005). Given the developmentally sensitive time-windows and the increased level of exposure in the developing brain, it is crucial to understand how and what chemicals can impact the developing brain in order to mitigate the risk of long-lasting neurological effects (Bellinger, 2012; Maffini and Neltner, 2015).
For the purpose of this review, we focus on POPs that have documented experimental evidence for modifying synaptic morphology and function during critical periods of neurodevelopment. These chemicals include polychlorinated biphenyls (PCBs; Figure 1A), polychlorinated dibenzodioxins (PCDDs; Figure 1B) including the most toxic PCDD congener, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD; Figure 1C), polybrominated diphenyl ethers (PBDEs; Figure 1D), and the five-ring polycyclic aromatic hydrocarbon benzo(a)pyrene ((B(a)P; Figure 1E). Although human body burdens for some of these chemicals including dioxin-like PCBs and TCDD have been steadily declining since the 1970s due to environmental regulations (Aylward and Hays, 2002; Pessah et al., 2019), these chemicals remain detectable in humans (Thompson and Boekelheide, 2013). Before discussing the experimental evidence for synaptic pathologies for each of these chemicals, we first provide a brief overview of epidemiologic studies that have documented abnormal neuropsychological function in children exposed to these chemicals.
Figure 1.
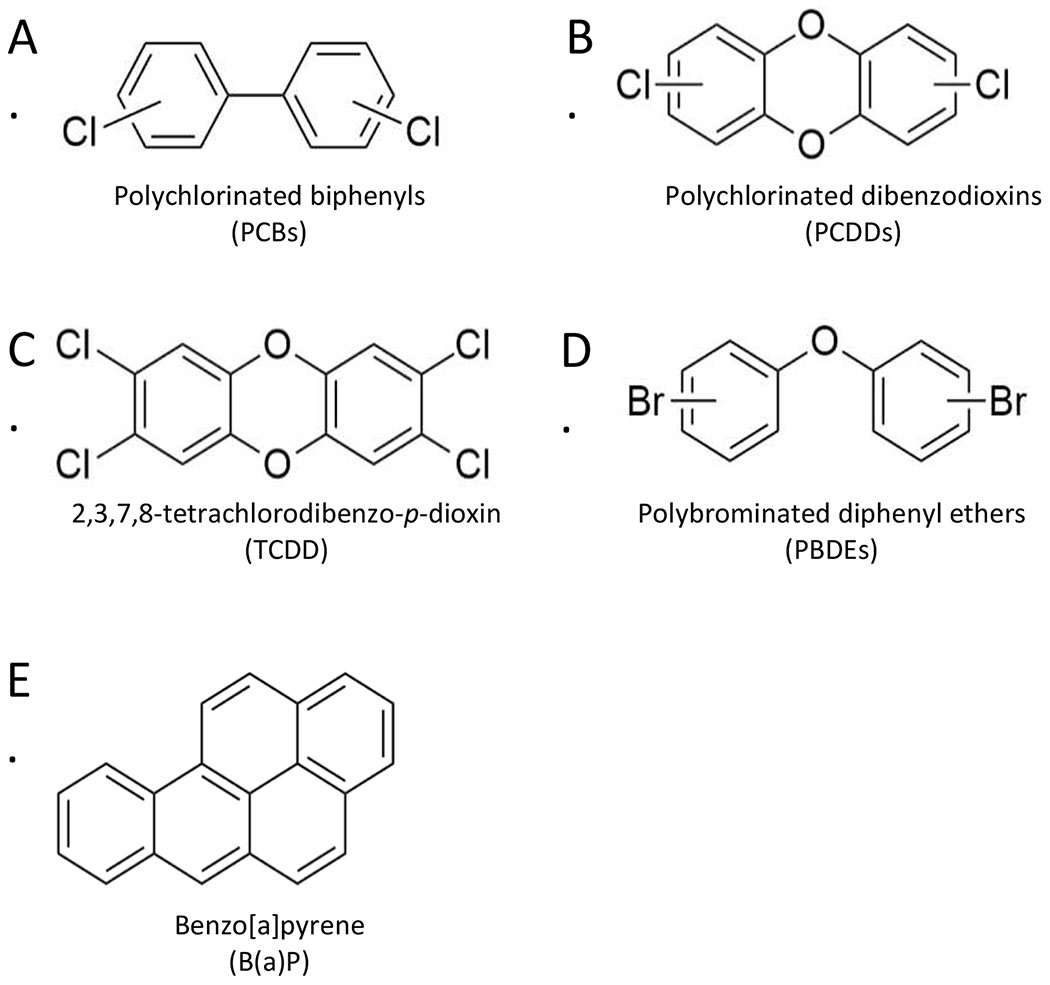
Chemical structures of common persistent organic pollutants (POPs). Many POPs including (A) polychlorinated biphenyls (PCBs), (B) polychlorinated dibenzodioxins (PCDDs) including (C) the most toxic PCDD congener 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), and (D) polybrominated diphenyl ethers (PBDEs) are carbon-based compounds with high-molecular weight, high lipid solubility, and varying degrees of halogenation. These chemical properties confer resistance to chemical and biological degradation, allowing for bioaccumulation of these pollutants in lipid-rich body tissues, including the brain, even at low doses. (E) Benzo(a)pyrene lacks the typical halogen substitutions but is equally lipid soluble and resistant to biodegradation. Figure was created with ChemDraw software.
1.2.1. Polychlorinated biphenyls (PCBs)
PCBs represent a large class of 209 structurally related chemicals comprised of a biphenyl with a variable number of chlorine substitutions in varying ortho, meta, and para positions on the benzene rings (Fig 1A; Klocke et al., 2020). The 209 related chemicals, or congeners, are generally classified as dioxin-like or non-dioxin-like based on their chemical structure and affinity for the aryl hydrocarbon receptor (AhR). Dioxin-like congeners typically have chlorine substitutions at the meta and/or para positions, with no chlorines at the ortho positions. These congeners take on a coplanar structure and bind to the AhR with high affinity. In contrast, non-dioxin-like congeners typically have multiple chlorine substitutions at the ortho position, adopt a non-coplanar structure, and do not bind to the AhR (Avilla et al., 2020; Klocke et al., 2020; Pessah et al., 2010). Before their initial ban in the United States in 1977 followed by a more global ban in 2001, PCBs were synthesized as commercial mixtures and contained heavily chlorinated dioxin-like and non-dioxin-like congeners. Despite regulatory efforts, both dioxin-like and non-dioxin-like PCBs persist at high concentrations in the environment and in animal and human tissues (Consonni et al., 2012; Hopf et al., 2009; Koh et al., 2015). Background levels of PCBs in various environmental sources are reported to range from 1 to 100 pg/m3 in air and 100 to 1,000 pg/g dry weight in soil (Hornbuckle and Robertson, 2010). Of concern for developmental neurotoxicity, levels of lightly chlorinated PCB congeners have been detected in the plasma of pregnant women at levels ranging from 0.005 ng/mL to 1.7 ng/mL (Sethi et al., 2017a) and continue to increase in the environment and in human tissues (Hornbuckle and Robertson, 2010).
Both dioxin-like and non-dioxin-like congeners can negatively impact neuropsychological function in infancy and childhood (Boucher et al., 2009; Carpenter, 2006; Korrick and Sagiv, 2008; Pessah et al., 2019; Schantz et al., 2003; Winneke, 2011). In utero and lactational PCB exposures have been correlated with decreased intelligence, decreased learning and memory, psychomotor impairments, and attention deficits (Korrick and Sagiv, 2008; Schantz et al., 2003; Winneke, 2011) and are postulated to increase the risk for neurodevelopmental disorders (Grandjean and Landrigan, 2014). More recently, lightly chlorinated congeners, which were not synthesized prior to the ban on PCB production, have emerged as contemporary concern for developmental neurotoxicity (Koh et al., 2015; Sethi et al., 2017a). Of special concern, the lightly chlorinated congener, PCB 11, was detected in all 241 pregnant women who were at increased risk for having a child with a neurodevelopmental disorder (Granillo et al., 2019; Sethi et al., 2017a). Collectively, despite the ban on production of PCBs, the continued presence of PCB congeners in the general population underscores the need for a clear understanding of how this chemical class may lead to impaired synaptogenesis and adverse neurodevelopmental outcomes.
1.2.2. Polychlorinated dibenzodioxins (PCDDs)
Polychlorinated dibenzodioxins (PCDDs; Figure 1B) represent a class of 75 structurally related chemicals that contain two phenyl rings joined together with a central dioxin ring containing two oxygen atoms. The toxicity of PCDDs depends on the number and positions of the chlorine atoms that are attached on either phenyl ring. With four chlorine substitutions at the meta and para positions, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, “dioxin”; Figure 1C) is considered to be the most potent of all PCDDs. PCDDs are mainly by-products of industrial processes where organic compounds are burned in the presence of chlorine, including metal smelting, chlorine bleaching, and the manufacturing of some herbicides and pesticides. PCDDs have similar lipophilic properties as PCBs and are also extremely resistant to biodegradation with an estimated half-life of 7 to 11 years in humans. While all humans in industrialized countries are presumed to carry a body burden of dioxins primarily from the environment and food intake, the mean lipid levels of TCDD has steadily declined over the last several decades due to regulatory efforts (Aylward and Hays, 2002). Populations with known exposure to TCDD have body burdens ranging from 96 to 7,000 ng/kg body weight, while the general population is estimated to have an average background TCDD concentration of approximately 58 ng/kg serum lipid, corresponding to a body burden of 13.5 ng/kg body weight (DeVito et al., 1995; Hojo et al., 2002). While there is evidence in humans correlating high-level, acute exposures with deficits in various metrics of neurodevelopment (Ames et al., 2019; Nishijo et al., 2012; Pham et al., 2019; Tai et al., 2013), the effects of lower, environmentally relevant levels of TCDD exposure are less well understood. Experimental studies in rodents, however, have linked gestational exposure to environmentally relevant levels of TCDD with impaired spatial learning and memory (Markowski et al., 2002), altered executive functions (Endo et al., 2012; Haijima et al., 2010; Kakeyama et al., 2014), and abnormal development of multiple brain areas including the cortex (Mitsuhashi et al., 2010) and the hippocampus (Latchney et al., 2011; Nguyen et al., 2013). These indicate that synaptogenesis may also be vulnerable to TCDD exposure in humans.
1.2.3. Polybrominated diphenyl ethers (PBDEs)
Structurally akin to PCBs, polybrominated diphenyl ethers (PBDEs) represent a class of 209 structurally related brominated compounds consisting of two phenyl rings connected by an ether link (Figure 1D). Commercial PBDEs exist as pentabrominated BDE, octabrominated BDE, and decabrominated BDE and are mainly used as flame retardants to decrease the flammability of a variety of consumer products including electronics, furniture, plastics, and textiles. Being highly lipophilic, these chemicals are also extremely resistant to environmental and biological degradation, with a half-life of 2 to 12 years in humans, and their widespread use has resulted in global contamination (Klincic et al., 2020). The general population is estimated to have an average background PBDE concentration of approximately 5 ng/g lipid in Europe and Asia but are 10 to 100 times greater in North America (Klincic et al., 2020). Of concern are infants and toddlers having the highest body burden of PBDE exposure via breast milk (Toms et al., 2009). Epidemiologic studies have demonstrated possible neurotoxic effects of prenatal and childhood exposure to PBDEs (Gibson et al., 2018), including deficits in fine motor coordination (Eskenazi et al., 2013; Herbstman et al., 2010; Roze et al., 2009), attention (Eskenazi et al., 2013; Gascon et al., 2011; Roze et al., 2009), impulsivity (Hoffman et al., 2012), and cognition (Eskenazi et al., 2013).
1.2.4. Benzo(a)pyrene (B(a)P)
While B(a)P does not possess the halogen substitutions of PCBs, PCDDs, and PBDEs, it is a five-ring polycyclic aromatic hydrocarbon formed by the fusion of a benzene ring with a pyrene (Figure 1F). The five-ringed structure imparts a high molecular weight, lipophilic properties, and resistance to biodegradation. It is most well-known for its carcinogenic properties as B(a)P metabolites react and bind to DNA, resulting in genetic mutations and cancer. Given its chemical properties, B(a)P can also easily cross the placenta and the blood brain barrier during pregnancy and directly access the developing brain (Balansky et al., 2007; Das et al., 1985). Environmental levels of B(a)P have been steadily increasing in industrialized countries due to the incomplete combustion of organic compounds (Van Metre and Mahler, 2005), exposing humans via ingestion of contaminated foods, water, and inhalation of polluted air (Waldman et al., 1991). B(a)P exposures ranges from 2.2 μg/day average daily intake by the general population (Edwards et al., 2010) to 20 to 800 ng/kg in individuals living near contaminated hazardous waste sites (Lioy et al., 1988). While B(a)P initially received significant scientific attention for its role in chemical carcinogenesis, the current literature suggests that B(a)P-induced neurotoxicity may occur at lower doses and at earlier times than B(a)P-induced carcinogenicity (Chepelev et al., 2015), making B(a)P relevant to human health risk assessment (Landrigan et al., 2004; Perera et al., 2003; Perera et al., 2006; Perera et al., 2005). Supporting this, human studies have shown that prenatal exposure to B(a)P adversely affects fetal development, resulting in reduced head circumference and neurobehavioral deficits in infants and children (Chepelev et al., 2015). Increased levels of B(a)P-DNA adducts in umbilical cord or blood of neonates have been correlated with maternal exposure to B(a)P in air (Perera et al., 2008; Perera et al., 1999; Perera et al., 2007). Neuropsychological manifestations of prenatal exposure include delayed mental development (Perera et al., 2007), decreased intelligence (Edwards et al., 2010; Perera et al., 2009), and impaired attention (Perera et al., 2012). Experimental evidence in rodents also demonstrates that developmental B(a)P exposure increases the risk of adverse neuropsychological function, impacting locomotor activity, spatial memory (Chen et al., 2012) and neuromuscular, autonomic, sensorimotor, and physiological functions (Saunders et al., 2001).
1.3. Synaptic effects of POPs may underlie cognitive and neurological impairments
In this review, we summarize evidence supporting a hypothesis that abnormalities in synapse structure, function, and plasticity as a result of POP exposure during developmentally sensitive time-windows contribute to cognitive and behavioral dysfunction (Stamou et al., 2013; Vester and Caudle, 2016). Due to similar chemical structure and properties, it is plausible that POPs share common mechanisms of neurotoxicity. To this end, we review the evidence that the synapse is a common vulnerable target for PCBs, PCDDs, PBDEs, and B(a)P, which alter the morphology of dendrites and dendritic spines, as well as synaptic functions. We also briefly summarize shared toxicological mechanisms that may underlie the structural and functional changes within the synapse, focusing on calcium and thyroid hormone dependent mechanisms. By recognizing converging pathological mechanisms among similar POPs at the synapse, we can more clearly define future research that will advance our understanding of POPs, and possibly other environmental pollutants on synapse structure, function, and plasticity.
2. Dendritic morphology and spines:
A neuron’s function and its ability to integrate into neuronal circuits is largely determined by its connectivity to neighboring neurons, which in turn is tightly linked to the structure of its dendrites (Kennedy, 2000; Miller and Jacobs, 1984). Dendrites serve as the primary site for signal reception and processing and their growth, shape, and branching complexity can determine synaptic input (Miller and Jacobs, 1984; Segal, 2010). Neighboring or distant neurons send axons to form synapses with dendrites, and excitatory synapses are largely formed on tiny dendritic protrusions called dendritic spines. Inhibitory synapses can be formed onto the soma, dendritic shaft, but also on the dendritic spine, including on the small neck that connects the spine to the dendrite. During development, changes in dendritic morphology and spine number facilitate the formation, maintenance, and elimination of excitatory synapses, allowing the establishment and remodeling of neuronal connectivity (Dailey and Smith, 1996; Dunaevsky et al., 1999; Fiala et al., 1998). At the cellular level, dendritic and spine structural plasticity is tightly coordinated with synaptic function and plasticity (Sala and Segal, 2014; Segal, 2010). For example, spine enlargement parallels long-term potentiation (LTP) (Matsuzaki et al., 2004), whereas long-term depression (LTD) is associated with spine shrinkage (Zhou et al., 2004). These subtle changes in dendritic spine structure may have striking effects on synaptic function, plasticity, and patterns of connectivity in neuronal circuits. Multiple lines of evidence implicate excitatory and inhibitory synapses as key cellular substrates of human neurodevelopmental disorders including autism and schizophrenia, with alterations in dendritic structure and spine formation being a hallmark of these disorders (Copf, 2016; Penzes et al., 2011; Supekar et al., 2013).
2.1. Developmental POP exposure has complex effects on dendritic growth
An analysis of the literature revealed 21 studies to date reporting the effects of POPs on the morphology of dendrites (Table 1). The majority of these studies used Golgi-Cox staining to visualize the dendritic branching pattern after exposure to polychlorinated biphenyls (PCBs) and commercial mixtures of PCBs (Figure 1A) known as Aroclor mixtures (also reviewed in Klocke and Lein, 2020). Pregnant rat dams were exposed to low (1 mg/kg/d) or high (6 mg/kg/d) doses of Aroclor 1254 before and during gestation and during lactation. This resulted in total PCB levels of 0.5 to 3.0 ng/g wet weight in exposed pups at weaning (Yang et al., 2009), well within the range of total PCB levels measured in human postmortem brain (Dewailly et al., 1999; Mitchell et al., 2012). Golgi staining of cerebellar Purkinje cells and pyramidal neurons in the neocortex and CA1 subfield of the hippocampus revealed several effects of PCBs on dendritic morphology. First, adult offspring developmentally exposed to Aroclor 1254 exhibited a significant increase in dendritic complexity (Lein et al., 2007; Yang et al., 2009) but decreased experience-dependent dendritic growth (Yang et al., 2009). Interestingly, the growth promoting dendritic effects were more pronounced in low dose compared to the high dose exposure. The low, but not high, dose was also associated with decreased spatial learning and memory in the Morris water maze (Yang et al., 2009). A third study reported no changes in the dendritic arbor for primary dendrite length or branching area of Purkinje cells in offspring exposed to the 6 mg/kg/d dose of Aroclor 1254 (Roegge et al., 2006). While it is unclear why adverse effects were observed preferentially at the low dose, this observation has been well-documented for other organic pollutants, including endocrine disrupting chemicals such as bisphenol A (Vandenberg et al., 2012). It also remains unknown why the high dose increased dendritic length in CA1 pyramidal neurons (Lein et al., 2007) and neocortical pyramidal cells (Yang et al., 2009), but not in cerebellar Purkinje cells (Roegge et al., 2006; Yang et al., 2009). Some of these discrepancies may be due to differences in the age of the animals at which dendritic morphology was examined, the dosing regimen, and whether or not PCB exposure began before gestation (Klocke and Lein, 2020; Lein et al., 2007). For example, in the Lein et al. (2007) study, dendritic growth decreased in pups exposed to Aroclor 1254 when analyzed at weaning age (postnatal day (PND) 22). However, when analyzed at PND 60, dendritic growth increased compared to pups born to dam-exposed to vehicle (Lein et al., 2007). In both the Roegge et al. (2006) and Yang et al. (2009) studies – where the 6 mg/kg/d dose did not produce any observed changes in dendritic growth – dendritic length was measured at PND 21 (Roegge et al., 2006) and 31 (Yang et al., 2009), similar to the age in which Aroclor 1254 did not have any effects in the Lein et al. (2007) study. These discrepancies demonstrate that PCB mixtures have age-dependent effects on the growth of dendrites at different stages of synaptogenesis.
Table 1.
Summary of effects of developmental POP exposure on dendritic morphology and spine formation.
| Toxicant | Dose | Route of Exposure | Species | Sex | Brain region | Exposure Period | Effect on dendrite growth and spine formation | Reference |
|---|---|---|---|---|---|---|---|---|
| Aroclor 1254 | 6 mg/kg/day | Gavage | Rat | Males vs. Females | Cerebellum | Before, during, after gestation | No change in dendrite growth | Roegge et al., 2006 |
| 6 mg/kg/day | Gavage | Rat | Males only | Hippocampus, Cortex | During and after gestation | Increased dendrite growth | Lein et al., 2007 | |
| 1, 6 mg/kg/day | Diet | Rat | Males only | Cerebellum, Cortex | Before, during, after gestation | Increased dendrite growth; Decreased experience-dependent dendrite growth | Yang et al., 2009 | |
| PCB 95 | 0.1, 1, 6 mg/kg/day | Diet; in vitro | Rat | Not specified | Hippocampus | Before, during, after gestation; primary cell cultures | Increased dendrite growth | Wayman et al., 2012 |
| 200 nM | In vitro | Rat | Male and Females Combined | Hippocampus | Primary cell cultures | Increased spine formation | Lesiak et al., 2014 | |
| 2, 20, 200 nM | In vitro | Rat | Not specified | Hippocampus | Primary cell cultures | Increased dendrite growth | Wayman et al., 2012 | |
| 1 nM | In vitro | Rat | Male and Females Combined | Hippocampus | Primary cell cultures | Increased dendrite growth | Keil et al., 2018 | |
| 100 fM to 1 μM | In vitro | Mouse | Male vs. Females | Hippocampus, Cortex | Primary cell cultures | Increased dendrite growth | Keil et al., 2019 | |
| PCB 136 | 0.1 to 1000 nM | In vitro | Rat | Not specified | Hippocampus | Primary cell cultures | Increased dendrite growth | Yang et al., 2014 |
| PCB 11 | 1 aM to 1 μM | In vitro | Rat | Male and Females Combined | Hippocampus | Primary cell cultures | Increased dendrite growth | Sethi et al., 2017 |
| 1 fM to 1 nM | In vitro | Rat; Mouse | Male vs. Females | Hippocampus, Cortex | Primary cell cultures | Increased dendrite growth (species, sex, and region specific) | Sethi et al., 2017 | |
| 1 fM to 1 nM | In vitro | Rat | Not specified | Cortex | Primary cell cultures | Increased dendrite growth | Sethi et al., 2018 | |
| PCB 106, 112, 121, 159, 165, | 0.5 nM, 5 nM, 50 nM | In vitro | Mouse | Not specified | Cerebellum | Primary cell cultures | PCBs 112, 165, 187 increased dendritic growth; PCBs 106, 121, and 159 decreasd dendritic growth | Kimura-Kuroda et al., 2007 |
| PCB 121, 159 | 0.5 pM, 50 pM, 50 nM | In vitro | Mouse | Not specified | Cerebellum | Primary cell cultures | Decreased thyroid-induced dendrite growth | Kimura-Kuroda et al., 2005 |
| TCDD | 0.6, 3.0 μg/kg | Gavage | Mouse | Males only | Hippocampus, Amygdala | GD 12.5 | Decreased dendrite growth and spine density | Kimura et al., 2015 |
| B(a)P | 0.1, 1 μM/d | Intraperitoneal | Rat | Males only | Hippocampus | Early adolescence | Decreased dendrite growth and spine density | Das et al., 2019 |
| PBDE 209 | 10^-10 M | In vitro | Rat | Not specified | Cerebellum | Primary cell cultures | Inhibited TH-induced dendrite growth | Ibhazehiebo et al., 2011 |
| 10^-10 M | In vitro | Rat | Not specified | Cerebellum | Primary cell cultures | Inhibited TH-induced dendrite growth | Xiong et al., 2012 | |
| 10^-10 M | In vitro | Rat | Not specified | Cerebellum | Primary cell cultures | Inhibited TH-induced dendrite growth | Ibhazehiebo et al., 2012 | |
| PBDE 47, 49 | 20 pM to 2 μM | In vitro | Rat | Male and Females Combined | Hippocampus | Primary cell cultures | Delayed neurite growth (high concentrations) | Chen et al., 2017 |
| BP-6 | 10^-7 to 10^-12 M | In vitro | Rat | Not specified | Cerebellum | Primary cell cultures | Inhibited TH-induced dendrite growth | Ibhazehiebo et al., 2011 |
Abbreviations: GD = gestational day; PND = postnatal day; TH = thyroid hormone
In addition to PCB mixtures, individual PCB compounds have been shown to influence dendritic arborization during development. In five studies to date, the non-dioxin-like PCB congener PCB 95 in particular, produced similar dendritic effects as Aroclor 1254. In these studies, Golgi staining revealed a significant increase in the dendritic complexity of CA1 hippocampal pyramidal neurons in adult offspring developmentally exposed to low doses (0.1 or 1 mg/kg/d) of PCB 95, but not the high dose (6 mg/kg/d) (Wayman et al., 2012b), reproducing the low-dose effects observed in Aroclor 1254 exposed offspring (Yang et al., 2009). Because Aroclor 1254 is composed predominantly of non-dioxin-like PCB congeners and PCB 95 alone produced similar dendritic promoting effects, it is likely that it is the non-dioxin-like PCBs that produce dendrite-promoting effects (Wayman et al., 2012b). Substantiating these in vivo studies, in vitro studies using primary rat hippocampal and cortical neurons also demonstrated that PCB 95 promoted dendritic growth at picomolar to nanomolar concentrations (Keil et al., 2018; Keil et al., 2019; Lesiak et al., 2014; Wayman et al., 2012a;Wayman et al., 2012b), but these dendritic promoting effects were lost at higher concentrations in the micromolar range (Wayman et al., 2012b; Yang et al., 2014). Similar dendrite-promoting effects in vitro have also been observed at nanomolar concentrations for additional non-dioxin-like PCBs including PCB 11 (Sethi et al., 2017a; 2018) and PCB 136 (Yang et al., 2014). Moreover, from the studies to date, there seems to be no apparent additional dendrite-promoting effects from dioxin-like PCBs in the Aroclor mixtures, suggesting a common convergent mechanism that non-dioxin-like PCBs may exert on dendritogenesis (Klocke and Lein, 2020).
In addition to the parent compound, the oxidation of PCBs by hepatic cytochrome P (CYP)-450 enzymes produces neurotoxic metabolites that also influence dendritic growth, but with varying effects. Nanomolar concentrations of the 4-hydroxy metabolites of the dioxin-like PCBs 112, 165, and 187 enhanced dendritic arborization in primary mouse cerebellar Purkinje cells (Kimura-Kuroda et al., 2007). In contrast, metabolites of the non-dioxin-like PCBs 106, 121, and 159 did not increase dendritic arborization but did inhibit dendritic growth that is typically stimulated by thyroid hormone (Kimura-Kuroda et al., 2005, 2007). These results suggest that different CYP enzyme superfamilies may produce PCB metabolites that trigger different effects on dendritic growth. The dioxin-like PCB congeners are typically metabolized by CYP1A enzymes, while non-dioxin-like PCBs are metabolized by CYP2B enzymes (Lu et al., 2013; Lu and Wong, 2011; McGraw and Waller, 2006). Interestingly, however, it was noted that PCB 66, a congener similar to the non-dioxin-like PCB 95 but with only one ortho-chlorine substitution, exhibited no effect on dendritic arborization in primary rat hippocampal neurons at similar concentrations of PCB 95 that increases dendritic growth (Wayman et al., 2012a; Wayman et al., 2012b). While beyond the scope of this review, these data suggest that there may be multiple detoxification pathways that can make PCB metabolites more or less toxic than the parent compound, contributing to differing effects on dendritic growth and morphogenesis (Lu et al., 2013; Lu and Wong, 2011; McGraw and Waller, 2006).
In light of these original studies on developmental PCB exposure, recent work has begun to tease out the dendritic effects of additional POPs, including PCDDs, with TCDD being the most toxic. A collective review of the TCDD neurotoxicity literature suggests that, in vivo, 1 μg/kg is the cut-off for “low” vs. “high” doses (Hood et al., 2006). Effects on synaptic plasticity and behavior have been observed at “low” TCDD doses between 500 and 700 ng/kg (Hood et al., 2006; Markowski et al., 2002; Wormley et al., 2004a), suggesting that synapses and dendrites are vulnerable to low level TCDD exposure. Similar to Aroclor 1254 and PCB 95, TCDD exposure was found to alter dendritic arborization in a dose-dependent manner in studies that used Golgi-Cox staining and GFP labeling of Thy1-positive neurons. Pregnant rat dams were exposed to a single low (0.6 μg/kg) or high (3 μg/kg) dose of TCDD on gestational day (GD) 12.5 and dendritic length and branching of hippocampal CA1 pyramidal neurons were quantified in the offspring (Kimura et al., 2015). At PND 14, the growth of second-order branches was arrested in offspring exposed to the high dose, but the length of the third-order branches was increased in those exposed to the low dose (Kimura et al., 2015). While these dendritic effects did not persist into adulthood, the transient increase in dendritic growth may have long lasting effects on hippocampal circuits (Klocke and Lein, 2020), as a previous study by the same group demonstrated that mice born to dams exposed to the low (but not high) TCDD dose showed abnormal behavioral flexibility and social interaction compared to the control group (Endo et al., 2012). These findings are consistent with the non-monotonic dose-response effects seen in developmentally exposed PCB animals, warranting additional dose-response studies investigating the relationship between POP exposure and dendritogenesis (Vandenberg et al., 2012).
Studies investigating PBDEs and B(a)P exposure on dendritic morphology are noticeably limited. To date, only in vitro studies have investigated dendritic effects of PBDE exposure. A series of in vitro studies from the same research group showed that the dendritic complexity of cultured cerebellar neurons immunoreactive for calbindin decreased following exposure to 10−10 M of PBDE 209 (Ibhazehiebo et al., 2011a; Xiong et al., 2012). Similar results were obtained in an in vitro studying using a PBDE mixture (Ibhazehiebo et al., 2011b) and the effects were time-dependent (Ibhazehiebo and Koibuchi, 2012). More recently, however, in vitro exposure of primary hippocampal neurons to concentrations ranging from 200 pM to 2 μM of PBDE 47 or 49 did not alter dendritic arborization (Chen et al., 2017), suggesting that PBDE effects on dendritic growth may be cell-type and congener specific. To date, no studies have investigated the dendritic effects of perinatal B(a)P exposure. Rats exposed to 0.1 or 1 μM of B(a)P daily for 14 consecutive days via an intraperitoneal injection during early adolescence (PND 30-44), however, developed fewer and shorter dendrites, and this correlated with learning and memory impairments in the T-maze (Das et al., 2019). Given the similar chemical properties of B(a)P with other POPs, it is likely that developmental exposure to B(a)P may also lead to changes in neuronal morphology, effects that would also be highly dependent on the age, dose, and duration of B(a)P exposure.
2.2. Developmental POP exposure has variable effects on dendritic spine density
While much more remains to be done to explore dendritic impairments in response to developmental POP exposure, the effects of environmentally relevant doses of POPs on dendritic spines have only just begun to be elucidated. There are only three studies to date that have investigated the effects of these chemicals on dendritic spines, with each study reporting varying effects. In vitro exposure to nanomolar concentrations of PCB 95 in primary dissociated rat hippocampal cultures and organotypic hippocampal slice cultures increased spine density to 50% above that observed in vehicle control cultures (Lesiak et al., 2014), reflecting an increase in both mushroom-shaped and stubby spines, which are thought to represent mature synapses. In contrast, mice born to dams administered a low (0.6 μg/kg) or high (3 μg/kg) dose of TCDD on GD 12.5 exhibited significantly reduced dendritic spine densities in hippocampal pyramidal CA1 neurons and in the amygdala at 16 months of age (Kimura et al., 2015), a time point at which dendritic arbor size was similar to that in non-exposed animals. Spine densities were also reduced in concert with a reduction in dendritic size in hippocampal pyramidal CA1 neurons in mice exposed to 0.1 μM and 1.0 μM B(a)P, doses that are within the range of B(a)P exposures in the general population (Edwards et al., 2010), during early adolescence (Das et al., 2019).
While it is not clear why non-dioxin-like PCB congeners and TCDD produce opposite effects on spine formation, it is possible that the different metabolism of these congeners may produce distinct metabolites that subsequently exert different synaptic outcomes (Lu et al., 2013; Lu and Wong, 2011; McGraw and Waller, 2006). It is also known that TCDD and dioxin-like PCBs bind with relatively high affinity to the aryl hydrocarbon (AhR), a receptor that regulates the expression of many dioxin-responsive genes (Avilla et al., 2020) and is implicated in neurodevelopment (Latchney et al., 2011; Williamson et al., 2005). In contrast, non-dioxin-like PCBs such as PCB 95 have low binding affinity for the AhR. Interestingly, while environmentally relevant exposures to TCDD and dioxin-like PCBs have been correlated with immune, liver, and skin pathologies (Bock, 2019), specific evidence for TCDD-induced developmental neurotoxicity in humans is less established. On the other hand, there is accumulating evidence that associates non-dioxin-like PCB exposure with developmental neurotoxicity (Pessah et al., 2019), suggesting that AhR-dependent mechanisms may, in part, contribute to different effects of these chemicals on spine formation.
3. Synaptic transmission:
Neuronal activity modulates the formation and refinement of synaptic connections during neurodevelopment, partly through its effects on dendrite morphology, spine morphogenesis, and synaptic transmission (Dailey and Smith, 1996; Dunaevsky et al., 1999; Fiala et al., 1998; Matsuzaki et al., 2004; Zhou et al., 2004). While many different neurotransmitters and neuromodulators control neuronal transmission in the developing cortex and hippocampus, glutamatergic and gamma-aminobutyric acid (GABAergic) signaling have long been recognized as important targets of the developmental actions of neurotoxicants and can bidirectionally alter synaptic strength, altering synaptic structure and shaping neural circuit function long-term (Segal, 2010).
3.1. Effects of developmental POP exposure on neuronal activity
There is a broad consensus that developmental exposure to POPs in rodents dampens neuronal activity in the hippocampus and cortex through post-synaptic mechanisms (Hong et al., 1998; Hood et al., 2006; McCallister et al., 2008; Xing et al., 2010). One common exposure paradigm used to study the effects of TCDD on neuronal activity in rats administers a single, low dose (< 1 μg/kg) of TCDD orally to pregnant dams on GD 14 or 15, a time period where excitatory synapses are being formed for the first time. In these studies, TCDD exposed offspring had reduced spontaneous and evoked activity in the whisker barrel column neurons of the somatosensory cortex (Hood et al., 2006), an indicator of decreased synaptic function. This effect of TCDD on synaptic transmission may be very rapid as acute exposure to TCDD decreased excitatory transmission in hippocampal slices within 5 minutes of application (Hong et al., 1998). In vitro studies suggest that acute TCDD exposure (20 nM) increases calcium influx through voltage sensitive calcium channels as well as synaptic NMDA receptors, and synergizes with network activity to effect long term changes through alterations of gene expression (Lin et al., 2009; Lin et al., 2008). An oral subacute exposure regimen to 25 and 150 μg/kg B(a)P between GD 14 to 17, a similar time period used for the oral TCDD studies, also decreased neuronal activity by decreasing the amplitude of inward currents in the hippocampus (Brown et al., 2007) and the barrel cortex (McCallister et al., 2008). Similarly, acute in vitro exposure of primary cultured rat hippocampal neurons to micromolar concentrations (0.05 to 2.0 μM) of PBDE 209 irreversibly decreased voltage-gated sodium channel currents in a concentration-dependent manner and shifted the activation and inactivation of the inward sodium current in the hyperpolarizing direction, slowing down the recovery of sodium channels from their inactivation (Xing et al., 2010). Thus, it appears that many different POPs can rapidly and persistently reduce neuronal activity, dampening the firing of individual neurons and reducing synaptic activity. It is interesting to note, that this downregulation of network activity is a result of exposure at a time when excitatory synapses are being formed for the first time in the developing nervous system.
These changes in the activity of nascent neuronal networks may be the result of changes in the ability of synapses to strengthen in an activity-dependent manner. Many of the exposures described above also resulted in a lack of LTP induction (Wormley et al., 2004b). For instance, rats exposed to an aerosol concentration of 100 μg/m3 B(a)P for four hours a day between GD 11 and 21 exhibited decreased LTP in synapses of the hippocampal perforant path (Wormley et al., 2004a). This inhalational exposure of B(a)P is consistent with the legally enforceable limit of 100 μg/m3 for polycyclic aromatic hydrocarbons, including B(a)P, by the United States Occupational Safety and Health Administration, and represents 50% less than the “permissible exposure limit” for coal tar products of 200 μg/m3 as recommended by the US National Institute for Occupational Safety and Health. Mice developmentally exposed orally to various PBDE congeners including PBDE 209 (20 μM; Xing et al., 2009) and PBDE 47 (6.8 mg/kg; Dingemans et al., 2007) also demonstrated reduced hippocampal LTP. This suggests that even very low levels of exposure to different POPs can inhibit the ability of nascent synapses to strengthen.
3.2. Developmental POP exposure differentially affects glutamatergic and GABA-ergic synaptic activity
Experimental evidence demonstrates that developmental exposure to POPs leads to reductions in synaptic activity through a dual effect of inhibiting glutamate receptor-mediated currents while potentiating those mediated by GABA (Table 2). In one study, the effects of maternal exposure to TCDD during gestation on cortical glutamate transmission was investigated in PND 14 and PND 60 rat offspring. A single dose (0.7 μg/kg) of TCDD orally administered to dams on GD 18 reduced basal and potassium-evoked extracellular glutamate levels in PND 14 offspring, an effect which persisted to adulthood, although the reduced viability of glutamatergic neurons after TCDD administration complicated the interpretation of this result (Tomasini et al., 2012). Additionally, the authors observed reduced glutamate uptake following TCDD exposure, which could also be explained by the presence of fewer functioning neurons or could be a compensatory measure counteracting the decreased glutamate release. The decreased uptake of glutamate was postulated to be a result of lower glutamate availability, although this will need to be directly tested. Likewise, and as discussed in section 3.1, reduced glutamatergic signaling was also seen after oral exposure to 1 mg/kg per day throughout gestation and lactation (GD 7 to PND 21) to the non-dioxin-like PCB congeners 138 and 180 (Boix et al., 2010).
Table 2.
Summary of effects of developmental POP exposure on glutamatergic and GABAergic transmission.
| Toxicant | Dose | Route of Exposure | Species | Sex | Brain region | Exposure | Effects on synaptic plasticity | Reference |
|---|---|---|---|---|---|---|---|---|
| TCDD | 200, 800 ng/kg | Gavage | Rat | Females only | Hippocampus, Cortex | GD 15 | Decreased NR2B NMDAR; Increased NR2A NMDAR | Kakeyama et al., 2001 |
| 700 ng/kg | Gavage | Rat | Males only | Hippocampus | GD 15 | Decreased NR1 NMDAR | Nayyar et al., 2003 | |
| 700 ng/kg | Gavage | Rat | Males only | Hippocampus | GD 14 | Decreased LTP; Decreased NR2B NMDAR; Incresed NR2A NMDAR | Wormley et al., 2004 | |
| 100, 700 ng/kg | Gavage | Rat | Males and Females Combined | Barrel cortex | GD 15 | Decreased activity; Decreased NR2B NMDAR; Decreased GluR1 AMPAR | Hood et al., 2006 | |
| 700 ng/kg | Gavage | Rat | Males only | Cortex | GD 18 | Decreased glutamate transmission | Tomasini et al., 2012 | |
| 100 nM | In vitro | Rat | Males only | Hippocampus | PND 26-47 | Decreased EPSPs | Hong et al., 1998 | |
| 20 nmol/L | In vitro | Rat | Not specified | Cortex | GD 17 primary cortical neurons | Decreased activity | Lin et al., 2008 | |
| 20 nM | In vitro | Rat | Not specified | Cortex | GD 17 primary cortical neurons | Increased NR2A NMDAR | Lin et al., 2009 | |
| 1 μg/kg | Diet | Rat | Males vs. Females | Preoptic area | GD 15 | Decreased GAD 67 expression | Hays et al., 2002 | |
| B(a)P | 25, 150 μg/kg/d | Gavage | Rat | Not specified | Hippocampus, Cortex | GD 14-17 | Decreased NR2A and NR2B NMDARs; Decreased GluR1 AMPAR; Decreased inward currents | Brown et al., 2007 |
| 300 μg/kg/d | Gavage | Rat | Males only | Cortex | GD 14-17 | Decreased NR2B NMDAR and activity | McCallister et al., 2008 | |
| 100 μg/m3;4 h/day | Inhalation | Rat | Males only | Hippocampus | GD 11-21 | Decreased NR1 NMDAR; Decreased LTP | Wormley et al., 2004 | |
| 100 μg/m3; 4 h/day | Inhalation | Rat | Males only | Hippocampus | GD 14 | Decreased LTP; Decreased NR2B NMDAR; Incresed NR2A NMDAR | Wormley et al., 2004 | |
| PCB 95 | 200 nM | In vitro | Rat | Males and Females Combined | Hippocampus | Primary cell cultlures | Increased mEPSCs | Lesiak et al., 2014 |
| 6 mg/kg/d | Diet | Rat | Male and Females Combined | Auditory Cortex | GD 5-PND21 | Disrupted excitation/inhibition balance | Kenet et al., 2007 | |
| PCB Mixture | 6 mg/kg/d | Diet | Rat | Male and Females Combined | Auditory Cortex | Gestation and Lactation | Increased spontaneous and miniature IPSCs; EPSCs not affected; Disrupted excitation/inhibition balance | Lee et al., 2021 |
| PCB 52, 138, 180 | 1 mg/kg/d | Gavage | Rat | Males vs. Females | Cerebellum | GD 7-PND21 | PCB 138 and 180 decreased NR1 NMDAR; Increased extracellular GABA; Decreased glutamate signaling | Boix et al., 2010 |
| 22 PCB congeners | 0.3 - 10 μM | In vitro | Xenopus laevis oocytes | Females | N/A | N/A | GABA(A) receptor activation | Fernandes et al., 2010 |
| PCB 28, 52, 101, 138, 153, 180 | 0.3 - 10 μM | In vitro | Xenopus laevis oocytes | Females | N/A | N/A | GABA(A) receptor potentiation | Fernandes et al., 2010 |
| PCB 47, PBDE 47, 6-OH-PBDE 47 | 0.3 - 10 μM | In vitro | Xenopus laevis oocytes | Females | N/A | N/A | GABA(A) receptor potentiation | Hendriks et al., 2010 |
| PBDE 47 | 6.8 mg/kg | Gavage | Mouse | Males only | Hippocampus | PND 10 | Decreased LTP; Decreased NR2B NMDAR; Decreased GluR1 AMPAR | Dingemans et al., 2007 |
| PBDE 209 | 20 μM | Gavage | Rat | Males and Females Combined | Hippocampus | Before and after gesation | Decreased input/output functions; Decreased paired-pulse ratio; Decreased LTP | Xing et al., 2009 |
| PBDE 209 | 0.05 - 2.0 μM | In vitro | Rat | Not specified | Hippocampus | PND 0 primary hippocampal neurons | Decreased inward currents | Xing et al., 2010 |
Accompanying reduced glutamate activity, POPs have also been shown to potentiate GABAA receptors, increase presynaptic GABA release, and increase postsynaptic GABA responses in the cerebellum (Boix et al., 2010), hippocampus (Fernandes et al., 2010a; Fernandes et al., 2010b; Hendriks et al., 2010), and preoptic area (Hays et al., 2002). Notably, the strongest evidence for GABAA potentiation comes not from neuronal cultures, but from in vitro experiments using X. laevis oocytes. Lightly chlorinated PCBs such as PCB 28 and PCB 52 interact with the GABAA receptor to potentiate GABA-induced ion current in a concentration-dependent manner, ranging from 0.3 to 10 μM (Fernandes et al., 2010a). This observation was not found in heavily chlorinated PCBs such as PCBs 101, 138, 153, and 180. Moreover, in the same X. laevis culture system, micromolar concentrations (0.3 to 10 μM) of the lightly chlorinated PCBs 19, 47, and 51 directly activated the GABAA receptor in the absence of GABA, with modulation of ion current being dependent on the chlorination pattern of the congeners (Fernandes et al., 2010b; Hendriks et al., 2010). A palpable limitation to these X. laevis studies, however, is the physiological relevance of using oocytes to quantify GABAA potentiation and extending experimental evidence from oocytes to neurons.
Additional evidence suggests that inhibitory signaling is affected by POP exposure. In vivo studies demonstrated that gestational exposure to PCB 52 in rats, but to not the more heavily chlorinated PCBs 138 or 180, increased extracellular GABA levels in the cerebellum and impaired motor coordination (Boix et al., 2010). The effects of POP exposure on GABAergic neurons and synapses may be complex, however, as levels of glutamate decarboxylase 67 (GAD 67), one of the enzymes that synthesizes GABA from glutamate, were reduced in the preoptic area of rats exposed to TCDD on GD 15 (Hays et al., 2002), although this effect was not found in other brain areas. One possibility is that decreased GAD 67 expression could be a compensatory response to elevated levels of GABA, a hypothesis that remains to be tested.
While the evidence documented to date largely suggests that POPs decrease excitatory signaling and increase inhibitory signaling, there are contradictory reports that suggest that the effects of POPs on neurotransmission may be complex. For instance, primary rat hippocampal neurons exposed to 200 nM of PCB 95, a non-dioxin-like congener, for five days in vitro had significantly increased spine density and frequency of miniature EPSCs (Lesiak et al., 2014). This is in contrast to the in vivo exposures where male and female rats developmentally exposed to the non-dioxin-like PCBs 138 and 180 demonstrated reduced function of the NMDA-dependent glutamate-nitric oxide-cGMP pathway in the cerebellum (Boix et al., 2010). Most recently, developmental exposure to a high (6 mg/kg/d) dose of an Aroclor mixture increased spontaneous and miniature IPSCs in layer 2/3 of the auditory cortex with no changes in spontaneous EPSCs (Lee et al., 2021). While the exact reasons for these discrepancies remain unknown, it is conceivable that cell type specificity, and the differences between the in vivo and in vitro environment contribute. Additionally, individual congeners may target different signaling systems as in vivo exposure to the more heavily chlorinated PCB 138 and 180 significantly modulated glutamate signaling whereas the lightly chlorinated PCB 52 significantly modulated GABA signaling (Boix et al., 2010). Corroborating this are additional studies in Xenopus laevis (X. laevis) oocytes showing that micromolar concentrations (0.3 to 10 μM) of the lightly chlorinated PCBs 19, 28, 47, 51 and 52, but not the heavily chlorinated PCBs 101, 138, 153, and 180, potentiate GABA currents (Fernandes et al., 2010a; Fernandes et al., 2010b; Hendriks et al., 2010). Therefore, the degree of chlorine substitution may distinctively influence how individual POPs interact with neurotransmitter systems to promote or inhibit synaptic transmission.
3.3. Effects of POPs on glutamatergic receptors
The effects of POPs on glutamatergic transmission are likely mediated through their actions on receptors expressed at excitatory synapses. POP exposure during critical periods of rapid brain development has been shown to influence the subunit composition of the glutamate N-methyl-D-aspartate (NMDA) receptor, by decreasing the expression and activity of its specific subunits. The majority of NMDA receptors consist of two NR1 and two NR2A or two NR2B subunits, and changes in the expression of all three of these subunits after developmental exposures have been reported. To quantify receptor expression, most studies used quantitative reverse transcription polymerase chain reaction (RT-PCR) and immunoblotting to quantify the mRNA and protein level, respectively, of various glutamatergic receptors. In studies in which pregnant dams were exposed to a low dose (1 mg/kg/d) of non-dioxin-like PCBs during gestation and lactation, the NR1 subunit of NMDA receptors was significantly reduced in rat offspring exposed to PCBs 138 or 180, but not by PCB 52 (Boix et al., 2010). As a functional consequence, rats exposed to PCBs 138 or 180, but not PCB 52, had reduced function of the glutamate-nitric oxide-cGMP pathway in the cerebellum. Quantitative analysis of mRNA transcript expression also revealed a downregulation of the NR1 subunit during the first postnatal month in the hippocampus of rat offspring orally exposed to TCDD on GD 15 (Nayyar et al., 2003) and at PND 60 and 65 in rats exposed to B(a)P via inhalation between GD 11 and 21 (Wormley et al., 2004a).
The NR2A and NR2B subunits are also vulnerable synaptic targets during the period of rapid brain growth. Immunoblotting of proteins in the postsynaptic density revealed that neonatal (PND 10) exposure to a single dose (6.8 mg/kg) of PBDE 47 decreased the expression of the NR2B subunit in the mouse hippocampus (Dingemans et al., 2007). Dams exposed to a single, low dose of TCDD (< 1 μg/kg) on GD 15 also had decreased NR2B mRNA and protein levels in the barrel cortex (Hood et al., 2006), hippocampus (Wormley et al., 2004b), and cortex (Kakeyama et al., 2001) but increased expression of the NR2A subunit (Kakeyama et al., 2001; Lin et al., 2009; Wormley et al., 2004b). Changes in the ratio of NR2A to NR2B expression have functional consequences as NMDA receptors composed of NR2A subunits gate smaller Ca2+ currents, have a lower affinity for glutamate, and desensitize faster than those with NR2B subunits (Kutsuwada et al., 1992). Therefore, an increased NR2A/NR2B ratio as a result of expression changes in individual subunits is likely to result in a higher threshold for LTP induction, which could partially explain the reduction in neuronal activity and synaptic plasticity. This interpretation is supported by in vitro studies of cultured cortical neurons from GD 17 rats in which activity-dependent synapse toxicity from acute TCDD exposure (20 μM) was attenuated with NMDA receptor blockers (Lin et al., 2008). However, it should be noted that not all POPs trigger this specific change in subunit composition. In one study, gestational exposure to B(a)P between GD 14 to 17 downregulated both NR2A and NR2B NMDA receptors as early as PND 2 in the hippocampus (Brown et al., 2007; McCallister et al., 2008), but still yielded decreased NMDA-dependent neuronal activity and synaptic transmission.
In addition to NMDA receptor subunits, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are also vulnerable targets. In addition to decreasing NR2B expression, acute neonatal PBDE 47 exposure (6.8 mg/kg) on PND10 also decreased the expression of the AMPA GluR1subunit (Dingemans et al., 2007). Gestational exposure to a single, low dose of TCDD (0.1 μg/kg) on GD 15 also decreased AMPA mRNA and protein levels in the rat whisker barrel (Hood el al., 2006). Similar effects were seen following oral exposure to B(a)P (25 and 150 μg/kg/d) between GD 14 to 17 in the rat hippocampus (Brown et al., 2007). AMPA receptors mediate increases in synaptic strength that implement LTP. Thus, decreasing the signaling of both NMDA and AMPA receptors could have significant implications for synaptic plasticity, decreasing the ability of neuronal networks throughout the brain to strengthen and remodel synapses.
What remains to be clarified is the precise time course in which changes in receptor expression occur. In some studies, changes in receptor expression following gestational exposure were detected as early as PND 2 and continued throughout the first month of postnatal life (Brown et al., 2007; Hood et al., 2006), whereas in others, changes were not detectable until later in life (Kakeyama et al., 2001; Wormley et al., 2004a). The reason for the discrepancy is unknown and requires additional studies by independent laboratories to systematically tease out the developmental expression profiles of individual receptor subunits and the time course in which they may be affected by developmental POP exposure.
4. Changes in intracellular Ca2+ signaling may underlie the effects of POPs on dendrites and synapses.
The precise control of intracellular calcium is critical for neuronal development (Berridge, 2006), and the formation of dendritic arbors and synapses (Konur and Ghosh, 2005; Lohmann and Wong, 2005). One converging mechanism by which POPs interfere with dendritic and spine plasticity is the modulation of calcium signaling within developing neurons (Klocke et al., 2020; Pessah et al., 2010; Stamou et al., 2013). Current studies have focused on PCBs and suggest that non-dioxin-like PCBs, but not dioxin-like PCBs, potently increase intracellular Ca2+ levels (Klocke et al., 2020; Pessah et al., 2010), by altering signaling through receptors in both the plasma membrane (ionotropic glutamate receptors (Gafni et al., 2004) and L-type voltage-gated Ca2+ channels (Inglefield and Shafer, 2000)), and those in the endoplasmic reticulum (ER) membrane (inositol 1,4,5-triphosphate receptors (IP3; Inglefield et al., 2001; Mundy et al., 1999) and ryanodine receptors (RyR; Pessah et al., 2010)).
The most sensitive receptor to non-dioxin-like PCBs appears to be the RyR. RyRs are ion channels that regulate the release of Ca2+ from the ER and modulate the cell’s response to ion channels in the plasma membrane, including NMDA receptors (Pessah et al., 2010). Picomolar to nanomolar concentrations of PCB 95 potently activate RyRs in primary rat hippocampal (Wayman et al., 2008; Wayman et al., 2006; Wong et al., 1997a; Wong and Pessah, 1997) and cortical (Yang et al., 2009) neurons and subsequently increase Ca2+ release from intracellular stores (Pessah et al., 2010), thereby stimulating dendritic growth (Keil et al., 2018; Wayman et al., 2012a; Wayman et al., 2012b; Yang et al., 2014). In cultured rat hippocampal neurons, PCB 95 increases the frequency and amplitude of Ca2+ oscillations (Wayman et al., 2012b) and alters the plasticity of hippocampal neurons (Wong et al., 1997b). Supporting this, blocking RyR activity using either pharmacological approaches or siRNA knockdown of RyR prevents the PCB 95-induced increase in Ca2+ oscillations (Wayman et al., 2012a) and dendritic growth (Wayman et al., 2012b). In addition to the parent compound, hydroxylated PCB metabolites also increase intracellular Ca2+ oscillations at high micromolar concentrations, and pharmacological blockade of the RyR prevents this effect (Londono et al., 2010). From a neurotoxicity perspective, these Ca2+-dependent, PCB-induced effects closely resemble neurodevelopmental disorders that are characterized by increased dendritic arborization (Copf, 2016; Penzes et al., 2011; Stamou et al., 2013; Supekar et al., 2013), further underscoring the vulnerability of synapses to POP exposure during critical periods of synaptogenesis.
PCB 95-mediated modulation of RyR results in the activation of the cAMP response element-binding protein (CREB) which changes downstream signaling of several Ca2+-dependent intracellular signaling pathways to influence dendritic arborization and synapse formation (Keil et al., 2018; Wayman et al., 2008; Wayman et al., 2006). RyR sensitization through PCB 95 exposure can activate CaMKK, MEK/ERK, and CREB to increase the expression of Wnt2 to stimulate dendritic growth in rat hippocampal neurons (Wayman et al., 2012a). PCB 95 can also increase synaptogenesis by activating CREB and upregulating the expression of the microRNA miR132 (Lesiak et al., 2014). In both cases, pharmacological inhibition or siRNA knockdown of CREB prevented the PCB 95-induced dendritic arborization (Lesiak et al., 2014; Wayman et al., 2012a; Wayman et al., 2012b). Similar dendrite promoting effects have been observed in primary rat cortical neurons exposed to femtomolar to nanomolar concentrations of PCB 11 (Sethi et al., 2017a) where pharmacologic blockade or shRNA knockdown of CREB also significantly decreased dendritic growth (Sethi et al., 2018). Collectively, these data highlight CREB signaling as a molecular convergence point for multiple PCBs (Klocke et al., 2020). This has functional implications in the context of developmental neurotoxicity as the untimely activation of CREB from PCB exposure could predispose the developing fetus or child to neurodevelopmental disorders (Bu et al., 2017).
Dendritic growth is also modulated by Ca2+-dependent translational mechanisms (Tsokas et al., 2005; Vickers et al., 2005), which are sensitive to POP exposures. Recently, Keil et al. (2018) demonstrated that nanomolar concentrations of PCB 95 promoted dendritic growth in primary hippocampal neurons by activating the Ca2+-dependent translational mechanisms involving mechanistic target of rapamycin (mTOR). Pharmacological inhibition and siRNA knockdown of mTOR or the mTOR complex binding proteins blocked the PCB 95-induced dendritic growth. This recent study also adds to the growing list of Ca2+-dependent mechanisms underlying PCB neurotoxicity.
In line with their similar effects on dendritic growth, TCDD and PCBs both elicit changes in Ca2+ signaling, accentuating the converging role of Ca2+-dependent processes in POP-induced synaptic toxicity. In vitro exposure to nanomolar concentrations of TCDD can activate ionotropic glutamate receptors (Hong et al., 1998; Lin et al., 2009; Lin et al., 2008), leading to a rapid increase in Ca2+ levels in hippocampal (Hanneman et al., 1996) and cerebellar granule cells (Kim and Yang, 2005). These rapid increases in Ca2+ subsequently activate multiple Ca2+-dependent players including ERK-1/2 (Kim et al., 2007; Kim and Yang, 2005; Lee et al., 2007), protein kinase C (PKC; Kim et al., 2007; Kim and Yang, 2005; Lee et al., 2007) and its adaptor protein, receptor for activated C kinase-1 (RACK-1; Lee et al., 2007), and calcium/calmodulin-dependent kinase II (CaMK; Lin et al., 2008). Ultimately, and similar to PCB 95, these pathways converge onto CREB (Lin et al., 2009; Lin et al., 2008), which may explain the similarities in dendritic growth following exposures to each of these POPs. However, distinct from PCB 95, TCDD-induced increases in Ca2+ signaling required the AhR as blocking the AhR with the inhibitor alpha-naphthoflavone or with an antisense oligonucleotide prevented the TCDD-induced calcium activity (Kim and Yang, 2005; Lee et al., 2007; Lin et al., 2009; Lin et al., 2008).
Taken together, the PCB and TCDD studies suggest that regardless of the receptor being activated – whether it is ionotropic glutamate or L-type voltage-gated Ca2+ receptors in the plasma membrane or IP3 and RyRs in the ER membrane – intracellular Ca2+ homeostasis and Ca2+-dependent transcriptional and translational events are a common platform on which POPs act to modify the growth and morphology of dendrites and spines, thereby influencing synaptogenesis. Moreover, these Ca2+-dependent mechanisms may operate upstream of and/or couple to the activation of glutamatergic and GABA-ergic signaling systems that are critical for synaptogenesis (Pessah et al., 2010). In vivo studies will be necessary to verify the findings on calcium signaling in a physiological setting, as the majority of the studies linking Ca2+ deregulation with POP exposure have been carried out in reduced preparations which lack the complex environment of the intact brain. Additionally, while a few studies have begun to elucidate the involvement of RyRs (Chen et al., 2017; Kim et al., 2011) and Ca2+-dependent pathways with other POPs such as PBDEs (Gassmann et al., 2014; Viberg, 2009; Viberg et al., 2008), more detailed mechanistic studies are needed to generalize the involvement of these signaling pathways to other POP-mediated synaptic toxicity.
6. Perspectives for Future Research
6.1. The temporal relationship between structural and functional changes elicited at excitatory synapses by developmental POP exposure.
Collectively, the studies reviewed above provide evidence that developmental exposure to PCBs, TCDD, B(a)P, and PBDEs all lead to changes in the subunit composition of glutamatergic receptors, reduced excitatory synaptic activity, an imbalance in excitation and inhibition, and reduced LTP. This is accompanied by an exuberant growth of the dendritic arbor and, in some cases, a reduction in the density of dendritic spines. It is thus plausible that there exists a common synaptic phenotype in which effects of POPs on synaptic receptors either directly or through dysregulation of intracellular Ca2+ signaling cause a change in the development of both excitatory and inhibitory connectivity. The increased dendritic growth and branching complexity observed following most developmental POP exposures may serve to stabilize synapses in order to compensate for reduced neuronal activity (Figure 2), although this hypothesis remains to be directly tested. In this model, a neuron may be programmed to expect a certain level of activity, and when this level is not met (due to POP-mediated reductions in excitatory signaling), increased dendritic growth could be a response that provides more opportunity for the neuron to form synapses with nearby axons. More detailed time-course analyses to detect whether the reduced glutamatergic transmission and subsequent weakening of synaptic activity occurs before the growth of dendrites would help address this relationship, as it is equally possible that reduced spine densities are the result of excessive dendritic growth, which elicits a “spreading” of existing connections to a larger dendritic surface area. Chronic in vivo imaging approaches, such as those recently used to address synaptic changes in neurodevelopmental disorders (Nakai et al., 2018), could provide answers to these questions. They could also illuminate the time course of dendritic growth between exposure and adulthood to determine whether dynamic dendritic remodeling includes phases of growth and retraction, processes that have been tied to plasticity in inhibitory neurons (Chen et al., 2011). Another under-explored area is the effects of POPs on different types of dendritic spines. Spines can take on various shapes and sizes, that are thought to serve different functions in synaptic transmission (Nimchinsky et al., 2002). While developmental exposures can selectively alter specific spine shapes and sizes (Lesiak et al., 2014), more detailed and comprehensive studies are needed to characterize the dynamic effects of developmental exposures on the morphology of the spine neck and spine head. In the one study where spine types were quantified, it appeared that developmental exposure increased the density of mature mushroom spines without altering filopodia (Lesiak et al., 2014), suggesting that POPs may stabilize some excitatory connections without altering the dynamics of new filopodial growth. Moreover, spines are remarkably dynamic, changing their shape and continually being formed and eliminated. These dynamic structural changes are thought to be critical to synaptic plasticity (Moyer and Zuo, 2018). In vivo imaging could also address the dynamics of spine formation, enlargement, shrinkage, and retraction during and following chemical exposure, as has recently been done for developmental exposure to Bisphenol A (Kelly et al., 2014) to further illuminate the short and long-term effects of POPs on synapses.
Figure 2.
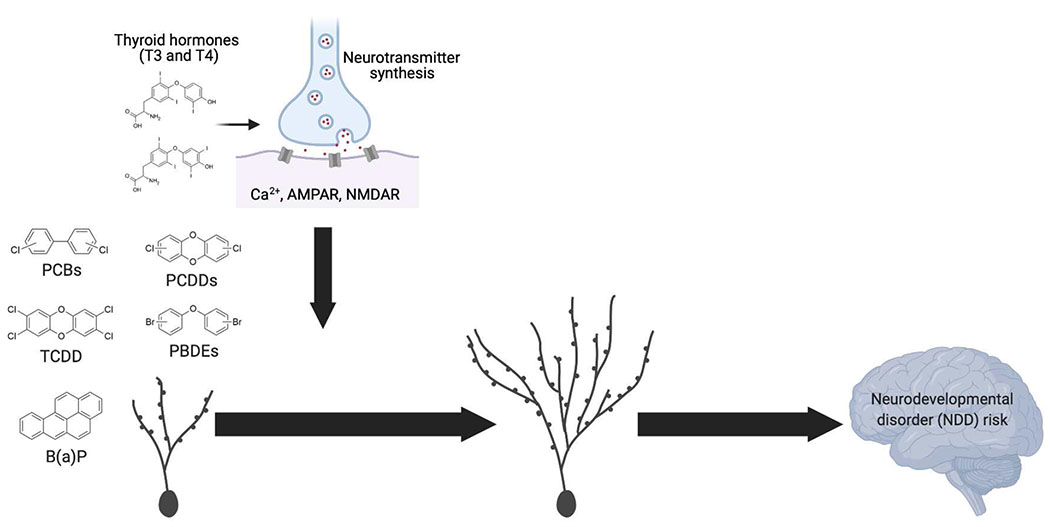
Proposed model of the effects of developmental POP exposure on synapse remodeling. Morphological and electrophysiological data suggest that developmental exposure to persistent organic pollutants (POPs) impairs several parameters of synapse formation, including AMPA and NMDA receptor expression, Ca2+ signaling, thyroid hormone, excitation/inhibition balance, and synaptic activity. While not yet directly tested, these alterations may decrease neuronal activity and destabilize synapses. Over time, neurons may compensate for this by increasing dendritic growth and spine formation in an attempt to restore synaptic stability and function. As a result, altered dendritic and spine structures following developmental POP exposure may predispose children to neurodevelopmental disorders.
6.2. POPs target network processes that impact synaptic plasticity.
While it is not yet clear how the different POPs elicit their effects on synaptic and circuit activity, the mechanisms that translate POP exposure to synaptic deficits are likely to center on molecular and cellular processes that modulate activity-dependent plasticity (Stamou et al., 2013). The level of neuronal excitability is largely determined by the balance between excitation and inhibition, and a proper balance between the two signaling systems is essential for cortical and hippocampal function (Takesian and Hensch, 2013). An excessive shift towards excitation or inhibition could lead to a wide range of neuropsychological deficits (Froemke, 2015), and the proper balance between excitatory and inhibitory signaling is thought to be critical for opening and closing critical periods for experience-dependent plasticity (Takesian and Hensch, 2013). The experimental data reviewed here identify both glutamatergic and GABAergic signaling as targets for developmental exposure to POPs and underscore the importance of appropriate excitation-inhibition balance in the developing brain. An elegant example is provided by a study where perinatal exposure (GD 5 to PND 21) to a high dose (6 mg/kg/d) of PCB 95 in rats disrupted excitation/inhibition balance, and reduced responsiveness to tonal stimuli during the critical period of sound-exposure-driven auditory plasticity resulting in a reversed topographic organization of the auditory cortex (Kenet et al., 2007). Mechanistically, a more recent study determined that developmental exposure to 6 mg/kg/d of an Aroclor mixture induced long-lasting changes in both inhibitory and excitatory neurotransmission in the auditory cortex (Lee et al., 2021). As an increase in the excitation-inhibition ratio can stabilize synapses and circuits (Antoine et al., 2019), it is possible that the decreased excitatory and increased inhibitory signaling seen in most developmental POP exposure studies can destabilize synapses by preventing plastic changes that stabilize and strengthen connections. The deregulation of the excitatory/inhibitory ratio may also synergize with other changes such as those in the NR2A:NR2B ratio and the handling of calcium postsynaptically, which is also known to modulate activity-dependent plasticity and the timing of critical periods for circuit remodeling (Smith et al., 2009).
6.3. Thyroid hormone-dependent neurotoxicity mechanisms
Many POPs are known endocrine disrupting chemicals (EDCs). EDCs perturb the synthesis, secretion, transport, metabolism, binding, and/or elimination of endogenous hormones, such as thyroid hormone (TH). TH is required throughout fetal life and early childhood for proper brain development and severe deficiencies in TH are associated with irreversible damage to the nervous system (Rovet, 2014). In the normal developing brain, increased TH levels increase dendritic arborization of cerebellar Purkinje cells (Hatsukano et al., 2017; Ibhazehiebo and Koibuchi, 2012; Kimura-Kuroda et al., 2002) while decreased TH levels decrease dendritic arborization (Kapfhammer, 2004; Salazar et al., 2019), suggesting that TH dysregulation may be a mechanism in which POP may modify synapses (Zoeller, 2010). Although epidemiologic studies provide conflicting and inconsistent data, the prevailing view is that many POPs, including PCBs (Hagmar, 2003; Itoh et al., 2018; Li et al., 2018; Osius et al., 1999; Sala et al., 2001), PCDDs (Baccarelli et al., 2008; Goodman et al., 2010; Warner et al., 2020), and PBDEs (Herbstman et al., 2008; Mazdai et al., 2003) may interfere with the TH system by reducing circulating levels of TH, thereby limiting the availability of the hormone that is needed for synaptogenesis.
Similarly, in vivo studies in rats attempting to link developmental PCB exposure with reduced TH levels and abnormal dendrite development have yielded conflicting results (Roegge et al., 2006; Sethi et al., 2018; Yang et al., 2009). In one study, a high dose of Aroclor 1254 (6 mg/kg/d) had no significant effects on the morphology of cerebellar neurons dendrites, but significantly decreased serum T4 and T3 concentrations in exposed PND 21 pups (Roegge et al., 2006). In another study, low-dose (1 mg/kg/d) developmental Aroclor 1254 exposure increased the dendritic growth of cerebellar Purkinje cells and cortical pyramidal neurons, and both the low and high dose of Aroclor 1254 (1 mg and 6 mg/kg/d) significantly decreased serum concentrations of TH levels at PND 21. By PND 31, serum levels had recovered to control levels in the 1 mg/kg/d group, but continued to be significantly reduced in the high dose group (Yang et al., 2009). The authors concluded that the increased dendrite branching in the Aroclor exposed group was unlikely due to decreased TH levels because it has previously been shown that decreased TH levels decrease dendritic growth (Ruiz-Marcos et al., 1994; Yang et al., 2009). Furthermore, individual PCBs, such as PCB 11, were found to have no agonistic activity for the TH receptor and TH receptor antagonism did not block the dendritic growth promoting effects of PCB 11 (Sethi et al., 2018). Therefore, these studies indicate that PCBs may not target the TH system in the developing brain. However, one in vitro study demonstrated that hydroxylated PCB metabolites inhibit TH-induced dendritic growth in isolated neocortical cells (Kimura-Kuroda et al., 2005), suggesting that further studies are warranted to investigate additional PCB congeners (Martin and Klaassen, 2010) and the effects of PCB metabolism on the TH system.
When considering other chemicals, the TH-targeting hypothesis may be more convincing for POPs that have been shown to decrease dendritic growth, resembling a hypothyroidism phenotype. For example, PBDE-exposed experimental animals have been repeatedly found to have decreased serum thyroid hormone levels (Hallgren and Darnerud, 2002; Hallgren et al., 2001; Szabo et al., 2009), providing evidence for thyroid disruption. In a series of studies by the same laboratory, in vitro exposures to low doses of the flame retardant PBDE 209 (10−10 M) inhibited the dendritic growth of cerebellar Purkinje neurons cultured with and without TH (Ibhazehiebo et al., 2011a; Ibhazehiebo et al., 2011b) in a time-dependent manner (Ibhazehiebo and Koibuchi, 2012), resembling a hypothyroidism phenotype. These results were also replicated in an in vitro study with cerebellar Purkinje neurons that used a PBDE mixture model (Xiong et al., 2012). However, this growth arresting phenotype was not observed in primary hippocampal neurons exposed to PBDE 47 or 49 (Chen et al., 2017). Regardless of these inconsistencies, it is intriguing to note that the structure of PBDEs – with two halogenated phenyl rings attached by an ether link – is considerably more similar to TH than that of PCBs, making it more likely that PBDE congeners may target the TH system. More recently, additional POPs that structurally resemble TCDD have been shown to reduce TH levels and dendritic growth, also producing a hypothyroidism phenotype. For example, rats developmentally exposed to 3,3′,4,4′-tetrachloroazobenzene (TCAB; 0.1 to 10 mg/kg/d) showed a dose-dependent reduction in serum T4 levels (Harry et al., 2014). Golgi staining of Purkinje neurons in rats receiving the high TCAB dose (10 mg/kg/d) showed a diminished dendritic arbor at PND 21 and PND 150 (Harry et al., 2014). While the evidence suggesting developmental exposure to POPs may target the TH system is inconsistent, it may be that only certain compounds (those that reduce dendritic growth) elicit their effects through the TH system. More studies are warranted to test this hypothesis and to determine the precise TH-dependent intracellular mechanisms of developmental POP toxicity at the synapse.
6.4. Non-monotonic responses to POP developmental exposure
As discussed in the last section, not all POPs and exposure paradigms have the same effects on the structure and function of excitatory synapses, suggesting that the relationship between exposure and synapse pathology is complex. Some of this may be due to the differential effects of low vs. high exposure doses. For decades, high doses typically seen following accidental or occupational exposures and intentional poisoning to POPs including PCBs (Chu et al., 2019; Lai et al., 2001) and TCDD (Eskenazi et al., 2018; Marnane, 2012; Pham et al., 2019) have shown striking effects on neurodevelopment, including altered functional connectivity in humans exposed to high levels of PCBs (Chu et al., 2019). However, accumulating evidence suggests that low doses of these chemicals may also be neurotoxic and, in some cases, may even pose greater toxicity than high doses, challenging the long-standing dogma in toxicology that “the dose makes the poison.” Low-dose effects, as defined by the National Toxicology Program, are doses that occur in the range of human exposures or doses below those used in traditional toxicological studies. This “low-dose” hypothesis, also known as non-monotonic dose-response effects, postulates that low doses of long-lasting chemicals including pesticides, flame retardants, and plasticizers may have neurotoxic effects that may not necessarily be predicted from their effects at high doses. Related to synaptic pathologies, it is unknown why a non-monotonic dose-response relationship exists whereby higher doses (of PCB for example), do not affect dendritic morphology (Wayman et al., 2012b; Yang et al., 2014), although several mechanisms have been proposed (Vandenberg et al., 2012; Yang et al., 2009). These synaptic effects are not thought to be mediated by increased cell death, whereby stunted cells may be expected to die leaving only those cells that were relatively unaffected by exposure, and appears to be driven instead by a resistance to PCB at higher doses (Keil et al., 2019; Wayman et al., 2012b; Yang et al., 2014).
While there are a limited number of studies examining low doses and non-monotonic dose-response relationships in the context of dendritic and spine plasticity, there are published reports of low-dose effects on neurobehavioral endpoints in rodents, most notably with TCDD exposure where more pronounced neurobehavioral impairments are seen with low doses. For example, male offspring born to rat dams exposed to 0.1 μg TCDD/kg demonstrated decreased working memory, whereas those exposed to a higher dose of TCDD (0.2 μg/kg) were unaffected (Seo et al., 2000). In another study, male and female rat offspring prenatally exposed to 0.18 μg TCDD/kg were significantly impaired in operant behavior testing paradigms compared with the control and high dose (0.54 μg/kg) groups (Markowski et al., 2002). In addition, U-shaped dose-response behaviors in male and female offspring born to TCDD-exposed rats have also been reported (Hojo et al., 2008; Hojo et al., 2002; Kakeyama et al., 2014). Rats exposed to 200 ng/kg had increased behavioral performance compared to the control (0 ng/kg), low dose (50 ng/kg), or the high dose (800 ng/kg) (Hojo et al., 2008). Gestational exposure to 200 ng/kg, but not 800 ng/kg, TCDD also impaired learning in a paired-associate learning task (Kakeyama et al., 2014). These low-dose effects are not limited to rats as this phenomenon has also been observed in mice born to dams exposed to 0, 0.6, or 3.0 μg TCDD/kg (Endo et al., 2012). Only the low-dose group (0.6 μg/kg) demonstrated significant impairments in behavioral flexibility parameters. Moreover, low-dose effects have also been documented for developmental exposures to heavy metals including methyl mercury (Weston et al., 2014) and lead (Rooney et al., 2018; Zhao et al., 2018) and other lipophilic organic compounds including Bisphenol A (Vandenberg et al., 2012), PCBs (Yang et al., 2009), and pesticides (Cassidy et al., 1994; Chapin et al., 1997; Palanza et al., 1999; Timofeeva et al., 2008). Therefore, it is important that future studies focus on the effects of doses that exemplify common human exposure (Vandenberg et al., 2012) as these low exposures may be particularly detrimental to circuit function.
6.5. Sex-specific effects of POP exposures.
There are a scarce number of studies investigating the sex-specific effects of POP developmental exposures on dendritic and spine morphology. Only two studies to date have methodically investigated sex-dependent effects of PCBs on dendritic arborization (Keil et al., 2019; Sethi et al., 2017b). In mouse neuron-glia co-cultures, the non-dioxin-like PCB 11 increased dendritic complexity in female, but not male, hippocampal neurons and in male, but not female, cortical neurons (Sethi et al., 2017b). In rat cultures, PCB 11 increased dendritic arborization in both male and female hippocampal and cortical neurons (Sethi et al., 2017b), demonstrating that the effects of PCBs on dendritic phenotypes is influenced by neuronal cell type, sex, and species. In the second study, in vitro exposure to PCB 95 increased dendritic arborization of cultured hippocampal and cortical neurons isolated from female but not male mice (Keil et al., 2019). Sex-specific effects were also observed in inhibitory circuits after TCDD exposure (Hays et al., 2002). Additionally, many lines of evidence suggest that sex and sex hormones affect dendritic spine remodeling and synaptic plasticity (Hyer et al., 2018). Although the effect of sex hormones is most pronounced in females, where spine density can oscillate with the hormone cycle (Shors et al., 2001; Woolley et al., 1990), sex hormones can also affect spine density and morphology in males (Skucas et al., 2013) and some studies have demonstrated male sensitivity to developmental exposure to low doses of POP chemicals (Sobolewski et al., 2014). Additionally, many other pathways that impact synaptogenesis and synapse remodeling, such as immune activation (which may be particularly sensitive to neurotoxic exposures), also show sex-specific developmental trajectories (VanRyzin et al., 2020). Developmental POP exposure has also been shown to alter sexual differentiation of the brain in animal models, impacting cortical dominance, cognition, and behavior (Bell et al., 2016; Bircsak et al., 2020; Hojo et al., 2002; Hojo et al., 2006; Ikeda et al., 2002; Ikeda et al., 2005; Lenters et al., 2019; Tian et al., 2011; Weiss, 2002), making it paramount that future studies examine the importance of sex in neuronal responses related to dendritic and spine morphology to environmental POPs.
7. Concluding remarks:
The goal of this review was to evaluate studies investigating the developmental neurotoxicity of a subset of POPs including PCBs, PCDDs, PBDEs, and B(a)P. While definitive conclusions on the effects of developmental exposure to many of these POPs on dendritic morphology, spine formation, and the development of synaptic circuits are still lacking, lessons learned from current studies can be applied to a new list of recently prioritized chemicals that are believed to be developmentally neurotoxic (Pellizzari et al., 2019; Shaffer et al., 2019). This new list of prioritized chemicals includes alternative flame retardants, alternative plasticizers, aromatic amines, environmental phenols, organophosphorus-based flame retardants, perfluoroalkyl substances, and pesticides, all of which share similar chemical properties of POPs making it likely that dendritic and synaptic plasticity may also be vulnerable to these newly prioritized chemicals (Stamou et al., 2013; Vester and Caudle, 2016). Consideration of the temporal relationship between dendritic growth, spine formation, and synaptic activity, the balance between excitation and inhibition, converging intracellular signaling systems, low-dose and non-monotonic dose-response effects, and sex-specific effects should be taken into consideration for future studies investigating the neurotoxic effects of developmental toxicants.
Footnotes
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the VersionofRecord. Please cite this article as doi: 10.1002/DNEU.22825
Conflict of interest statement: The authors declare they have no competing financial interests.
References:
- Ames J, Warner M, Siracusa C, Signorini S, Brambilla P, Mocarelli P, & Eskenazi B (2019). Prenatal dioxin exposure and neuropsychological functioning in the Seveso Second Generation Health Study. Int J Hyg Environ Health, 222(3), 425–433. doi: 10.1016/j.ijheh.2018.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine MW, Langberg T, Schnepel P, & Feldman DE (2019). Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron, 101(4), 648–661 e644. doi: 10.1016/j.neuron.2018.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilla MN, Malecki KMC, Hahn ME, Wilson RH, & Bradfield CA (2020). The Ah Receptor: Adaptive Metabolism, Ligand Diversity, and the Xenokine Model. Chem Res Toxicol, 22(4), 860–879. doi: 10.1021/acs.chemrestox.9b00476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrad DA, Bellinger DC, Ryan LM, & Woodruff TJ (2007). Dose-response relationship of prenatal mercury exposure and IQ: an integrative analysis of epidemiologic data. Environ Health Perspect, 115(4), 609–615. doi: 10.1289/ehp.9303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward LL, & Hays SM (2002). Temporal trends in human TCDD body burden: decreases over three decades and implications for exposure levels. J Expo Anal Environ Epidemiol, 12(5), 319–328. doi: 10.1038/sj.jea.7500233 [DOI] [PubMed] [Google Scholar]
- Baccarelli A, Giacomini SM, Corbetta C, Landi MT, Bonzini M, Consonni D, Grillo P, Patterson DG, Pesatori AC, & Bertazzi PA (2008). Neonatal thyroid function in Seveso 25 years after maternal exposure to dioxin. PLoS Med, 5(7), e161. doi: 10.1371/journal.pmed.0050161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balansky R, Ganchev G, Iltcheva M, Steele VE, D’Agostini F, & De Flora S (2007). Potent carcinogenicity of cigarette smoke in mice exposed early in life. Carcinogenesis, 28(10), 2236–2243. doi: 10.1093/carcin/bgm122 [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Oldfors A, Hagberg B, & Dahlstrom A (1994). Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport, 5(12), 1509–1513. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/7948850 [PubMed] [Google Scholar]
- Bell MR, Hart BG, & Gore AC (2016). Two-hit exposure to polychlorinated biphenyls at gestational and juvenile life stages: 2. Sex-specific neuromolecular effects in the brain. Mol Cell Endocrinol, 420, 125–137. doi: 10.1016/j.mce.2015.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger DC (2012). A strategy for comparing the contributions of environmental chemicals and other risk factors to neurodevelopment of children. Environ Health Perspect, 120(4), 501–507. doi: 10.1289/ehp.1104170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (2006). Calcium microdomains: organization and function. Cell Calcium, 40(5-6), 405–412. doi: 10.1016/j.ceca.2006.09.002 [DOI] [PubMed] [Google Scholar]
- Bianchi S, Stimpson CD, Duka T, Larsen MD, Janssen WG, Collins Z, Bauernfeind AL, Schapiro SJ, Baze WB, McArthur MJ, Hopkins WD, Wildman DE, Lipovich L, Kuzawa CW, Jacobs B, Hof PR, & Sherwood CC (2013). Synaptogenesis and development of pyramidal neuron dendritic morphology in the chimpanzee neocortex resembles humans. Proc Natl Acad Sci U S A, 110 Suppl 2, 10395–10401. doi: 10.1073/pnas.1301224110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bircsak KM, Copes LT, King S, Prantner AM, Hwang WT, & Gerton GL (2020). The aryl hydrocarbon receptor mediates sex ratio distortion in the embryos sired by TCDD-exposed male mice. Reprod Toxicol, 94, 75–83. doi: 10.1016/j.reprotox.2020.04.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock KW (2019). Aryl hydrocarbon receptor (AHR): From selected human target genes and crosstalk with transcription factors to multiple AHR functions. Biochem Pharmacol, 168, 65–70. doi: 10.1016/j.bcp.2019.06.015 [DOI] [PubMed] [Google Scholar]
- Boix J, Cauli O, & Felipo V (2010). Developmental exposure to polychlorinated biphenyls 52, 138 or 180 affects differentially learning or motor coordination in adult rats. Mechanisms involved. Neuroscience, 167(4), 994–1003. doi: 10.1016/j.neuroscience.2010.02.068 [DOI] [PubMed] [Google Scholar]
- Bondy SC, & Campbell A (2005). Developmental neurotoxicology. J Neurosci Res, 81(5), 605–612. doi: 10.1002/jnr.20589 [DOI] [PubMed] [Google Scholar]
- Bouchard MF, Bellinger DC, Weuve J, Matthews-Bellinger J, Gilman SE, Wright RO, Schwartz J, & Weisskopf MG (2009). Blood lead levels and major depressive disorder, panic disorder, and generalized anxiety disorder in US young adults. Arch Gen Psychiatry, 66(12), 1313–1319. doi: 10.1001/archgenpsychiatry.2009.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard MF, Chevrier J, Harley KG, Kogut K, Vedar M, Calderon N, Trujillo C, Johnson C, Bradman A, Barr DB, & Eskenazi B (2011). Prenatal exposure to organophosphate pesticides and IQ in 7-year-old children. Environ Health Perspect, 119(8), 1189–1195. doi: 10.1289/ehp.1003185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher O, Muckle G, & Bastien CH (2009). Prenatal exposure to polychlorinated biphenyls: a neuropsychologic analysis. Environ Health Perspect, 117(1), 7–16. doi: 10.1289/ehp.11294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LA, Khousbouei H, Goodwin JS, Irvin-Wilson CV, Ramesh A, Sheng L, McCallister MM, Jiang GC, Aschner M, & Hood DB (2007). Down-regulation of early ionotrophic glutamate receptor subunit developmental expression as a mechanism for observed plasticity deficits following gestational exposure to benzo(a)pyrene. Neurotoxicology, 28(5), 965–978. doi: 10.1016/j.neuro.2007.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu Q, Wang A, Hamzah H, Waldman A, Jiang K, Dong Q, Li R, Kim J, Turner D, & Chang Q (2017). CREB Signaling Is Involved in Rett Syndrome Pathogenesis. J Neurosci, 37(13), 3671–3685. doi: 10.1523/JNEUROSCI.3735-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter DO (2006). Polychlorinated biphenyls (PCBs): routes of exposure and effects on human health. Rev Environ Health, 21(1), 1–23. doi: 10.1515/reveh.2006.21.1.1 [DOI] [PubMed] [Google Scholar]
- Cassidy RA, Vorhees CV, Minnema DJ, & Hastings L (1994). The effects of chlordane exposure during pre- and postnatal periods at environmentally relevant levels on sex steroid-mediated behaviors and functions in the rat. Toxicol Appl Pharmacol, 126(2), 326–337. doi: 10.1006/taap.1994.1123 [DOI] [PubMed] [Google Scholar]
- CDC. (2019). Centers for Disease Control and Prevention. Fourth Report on Human Exposure to Environmental Chemicals, Updated Tables, January2019. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention. https://www.cdc.gov/exposurereport/index.html [last accessed 7 December 2020]. [Google Scholar]
- Chapin RE, Harris MW, Davis BJ, Ward SM, Wilson RE, Mauney MA, Lockhart AC, Smialowicz RJ, Moser VC, Burka LT, & Collins BJ (1997). The effects of perinatal/juvenile methoxychlor exposure on adult rat nervous, immune, and reproductive system function. Fundam Appl Toxicol, 40(1), 138–157. doi: 10.1006/faat.1997.2381 [DOI] [PubMed] [Google Scholar]
- Chen C, Tang Y, Jiang X, Qi Y, Cheng S, Qiu C, Peng B, & Tu B (2012). Early postnatal benzo(a)pyrene exposure in Sprague-Dawley rats causes persistent neurobehavioral impairments that emerge postnatally and continue into adolescence and adulthood. Toxicol Sci, 125(1), 248–261. doi: 10.1093/toxsci/kfr265 [DOI] [PubMed] [Google Scholar]
- Chen H, Streifel KM, Singh V, Yang D, Mangini L, Wulff H, & Lein PJ (2017). From the Cover: BDE-47 and BDE-49 Inhibit Axonal Growth in Primary Rat Hippocampal Neuron-Glia Co-Cultures via Ryanodine Receptor-Dependent Mechanisms. Toxicol Sci, 156(2), 375–386. doi: 10.1093/toxsci/kfw259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Lin WC, Cha JW, So PT, Kubota Y, & Nedivi E (2011). Structural basis for the role of inhibition in facilitating adult brain plasticity. Nat Neurosci, 14(5), 587–594. doi: 10.1038/nn.2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepelev NL, Moffat ID, Bowers WJ, & Yauk CL (2015). Neurotoxicity may be an overlooked consequence of benzo[a]pyrene exposure that is relevant to human health risk assessment. Mutat Res Rev Mutat Res, 764, 64–89. doi: 10.1016/j.mrrev.2015.03.001 [DOI] [PubMed] [Google Scholar]
- Chiu YH, Bellinger DC, Coull BA, Anderson S, Barber R, Wright RO, & Wright RJ (2013). Associations between traffic-related black carbon exposure and attention in a prospective birth cohort of urban children. Environ Health Perspect, 121(7), 859–864. doi: 10.1289/ehp.1205940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary D, Jansson I, Schenkman JB, Sarfarazi M, & Stoilov I (2003). Comparative expression profiling of 40 mouse cytochrome P450 genes in embryonic and adult tissues. Arch Biochem Biophys, 414(1), 91–100. doi: 10.1016/s0003-9861(03)00174-7 [DOI] [PubMed] [Google Scholar]
- Chu CP, Wu SW, Huang YJ, Chiang MC, Hsieh ST, & Guo YL (2019). Neuroimaging signatures of brain plasticity in adults with prenatal exposure to polychlorinated biphenyls: Altered functional connectivity on functional MRI. Environ Pollut, 250, 960–968. doi: 10.1016/j.envpol.2019.04.105 [DOI] [PubMed] [Google Scholar]
- Consonni D, Sindaco R, & Bertazzi PA (2012). Blood levels of dioxins, furans, dioxin-like PCBs, and TEQs in general populations: a review, 1989-2010. Environ Int, 44, 151–162. doi: 10.1016/j.envint.2012.01.004 [DOI] [PubMed] [Google Scholar]
- Copf T (2016). Impairments in dendrite morphogenesis as etiology for neurodevelopmental disorders and implications for therapeutic treatments. Neurosci Biobehav Rev, 68, 946–978. doi: 10.1016/j.neubiorev.2016.04.008 [DOI] [PubMed] [Google Scholar]
- Dailey ME, & Smith SJ (1996). The dynamics of dendritic structure in developing hippocampal slices. J Neurosci, 16(9), 2983–2994. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/8622128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das L, Patel B, & Patri M (2019). Adolescence benzo[a]pyrene treatment induces learning and memory impairment and anxiolytic like behavioral response altering neuronal morphology of hippocampus in adult male Wistar rats. Toxicol Rep, 6, 1104–1113. doi: 10.1016/j.toxrep.2019.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M, Seth PK, & Mukhtar H (1985). Distribution of benzo(a)pyrene in discrete regions of rat brain. Bull Environ Contam Toxicol, 35(4), 500–504. doi: 10.1007/BF01636545 [DOI] [PubMed] [Google Scholar]
- DeVito MJ, Birnbaum LS, Farland WH, & Gasiewicz TA (1995). Comparisons of estimated human body burdens of dioxinlike chemicals and TCDD body burdens in experimentally exposed animals. Environ Health Perspect, 103(9), 820–831. doi: 10.1289/ehp.95103820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewailly E, Mulvad G, Pedersen HS, Ayotte P, Demers A, Weber JP, & Hansen JC (1999). Concentration of organochlorines in human brain, liver, and adipose tissue autopsy samples from Greenland. Environ Health Perspect, 107(10), 823–828. doi: 10.1289/ehp.99107823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans MM, Ramakers GM, Gardoni F, van Kleef RG, Bergman A, Di Luca M, van den Berg M, Westerink RH, & Vijverberg HP (2007). Neonatal exposure to brominated flame retardant BDE-47 reduces long-term potentiation and postsynaptic protein levels in mouse hippocampus. Environ Health Perspect, 115(6), 865–870. doi: 10.1289/ehp.9860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, & Sands J (1973). Quantitative growth and development of human brain. Arch Dis Child, 48(10), 757–767. doi: 10.1136/adc.48.10.757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, & Sands J (1979). Comparative aspects of the brain growth spurt. Early Hum Dev, 3(1), 79–83. doi: 10.1016/0378-3782(79)90022-7 [DOI] [PubMed] [Google Scholar]
- Dunaevsky A, Tashiro A, Majewska A, Mason C, & Yuste R (1999). Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci U S A, 96(23), 13438–13443. doi: 10.1073/pnas.96.23.13438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SC, Jedrychowski W, Butscher M, Camann D, Kieltyka A, Mroz E, Flak E, Li Z, Wang S, Rauh V, & Perera F (2010). Prenatal exposure to airborne polycyclic aromatic hydrocarbons and children’s intelligence at 5 years of age in a prospective cohort study in Poland. Environ Health Perspect, 118(9), 1326–1331. doi: 10.1289/ehp.0901070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo T, Kakeyama M, Uemura Y, Haijima A, Okuno H, Bito H, & Tohyama C (2012). Executive function deficits and social-behavioral abnormality in mice exposed to a low dose of dioxin in utero and via lactation. PLoS One, 7(12), e50741. doi: 10.1371/journal.pone.0050741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel SM, Wetmur J, Chen J, Zhu C, Barr DB, Canfield RL, & Wolff MS (2011). Prenatal exposure to organophosphates, paraoxonase 1, and cognitive development in childhood. Environ Health Perspect, 119(8), 1182–1188. doi: 10.1289/ehp.1003183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA. (2020). US Environmental Protection Agency. Toxic Substances Control Act Chemical Substance Inventory. Availalbe online: https://www.epa.gov/tsca-inventory.Last accessed on 7 December 2020.
- Eskenazi B, Chevrier J, Rauch SA, Kogut K, Harley KG, Johnson C, Trujillo C, Sjodin A, & Bradman A (2013). In utero and childhood polybrominated diphenyl ether (PBDE) exposures and neurodevelopment in the CHAMACOS study. Environ Health Perspect, 121(2), 257–262. doi: 10.1289/ehp.1205597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskenazi B, Warner M, Brambilla P, Signorini S, Ames J, & Mocarelli P (2018). The Seveso accident: A look at 40years of health research and beyond. Environ Int, 121(Pt 1), 71–84. doi: 10.1016/j.envint.2018.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes EC, Hendriks HS, van Kleef RG, Reniers A, Andersson PL, van den Berg M, & Westerink RH (2010a). Activation and potentiation of human GABAA receptors by non-dioxin-like PCBs depends on chlorination pattern. Toxicol Sci, 118(1), 183–190. doi: 10.1093/toxsci/kfq257 [DOI] [PubMed] [Google Scholar]
- Fernandes EC, Hendriks HS, van Kleef RG, van den Berg M, & Westerink RH (2010b). Potentiation of the human GABA(A) receptor as a novel mode of action of lower-chlorinated non-dioxin-like PCBs. Environ Sci Technol, 44(8), 2864–2869. doi: 10.1021/es902321a [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, & Harris KM (1998). Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci, 18(21), 8900–8911. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9786995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC (2015). Plasticity of cortical excitatory-inhibitory balance. Annu Rev Neurosci, 38, 195–219. doi: 10.1146/annurev-neuro-071714-034002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Wong PW, & Pessah IN (2004). Non-coplanar 2,2′,3,5′,6-pentachlorobiphenyl (PCB 95) amplifies ionotropic glutamate receptor signaling in embryonic cerebellar granule neurons by a mechanism involving ryanodine receptors. Toxicol Sci, 77(1), 72–82. doi: 10.1093/toxsci/kfh004 [DOI] [PubMed] [Google Scholar]
- Gascon M, Vrijheid M, Martinez D, Forns J, Grimalt JO, Torrent M, & Sunyer J (2011). Effects of pre and postnatal exposure to low levels of polybromodiphenyl ethers on neurodevelopment and thyroid hormone levels at 4 years of age. Environ Int, 37(3), 605–611. doi: 10.1016/j.envint.2010.12.005 [DOI] [PubMed] [Google Scholar]
- Gassmann K, Schreiber T, Dingemans MM, Krause G, Roderigo C, Giersiefer S, Schuwald J, Moors M, Unfried K, Bergman A, Westerink RH, Rose CR, & Fritsche E (2014). BDE-47 and 6-OH-BDE-47 modulate calcium homeostasis in primary fetal human neural progenitor cells via ryanodine receptor-independent mechanisms. Arch Toxicol, 88(8), 1537–1548. doi: 10.1007/s00204-014-1217-7 [DOI] [PubMed] [Google Scholar]
- Gibson EA, Siegel EL, Eniola F, Herbstman JB, & Factor-Litvak P (2018). Effects of Polybrominated Diphenyl Ethers on Child Cognitive, Behavioral, and Motor Development. Int J Environ Res Public Health, 15(8). doi: 10.3390/ijerph15081636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman JE, Kerper LE, Boyce CP, Prueitt RL, & Rhomberg LR (2010). Weight-of-evidence analysis of human exposures to dioxins and dioxin-like compounds and associations with thyroid hormone levels during early development. Regul Toxicol Pharmacol, 58(1), 79–99. doi: 10.1016/j.yrtph.2010.04.008 [DOI] [PubMed] [Google Scholar]
- Grandjean P (2008). Late insights into early origins of disease. Basic Clin Pharmacol Toxicol, 102(2), 94–99. doi: 10.1111/j.1742-7843.2007.00167.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean P, & Landrigan PJ (2006). Developmental neurotoxicity of industrial chemicals. Lancet, 368(9553), 2167–2178. doi: 10.1016/S0140-6736(06)69665-7 [DOI] [PubMed] [Google Scholar]
- Grandjean P, & Landrigan PJ (2014). Neurobehavioural effects of developmental toxicity. Lancet Neurol, 13(3), 330–338. doi: 10.1016/S1474-4422(13)70278-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granillo L, Sethi S, Keil KP, Lin Y, Ozonoff S, Iosif AM, Puschner B, & Schmidt RJ (2019). Polychlorinated biphenyls influence on autism spectrum disorder risk in the MARBLES cohort. Environ Res, 171, 177–184. doi: 10.1016/j.envres.2018.12.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagmar L (2003). Polychlorinated biphenyls and thyroid status in humans: a review. Thyroid, 13(11), 1021–1028. doi: 10.1089/105072503770867192 [DOI] [PubMed] [Google Scholar]
- Haijima A, Endo T, Zhang Y, Miyazaki W, Kakeyama M, & Tohyama C (2010). In utero and lactational exposure to low doses of chlorinated and brominated dioxins induces deficits in the fear memory of male mice. Neurotoxicology, 31(4), 385–390. doi: 10.1016/j.neuro.2010.04.004 [DOI] [PubMed] [Google Scholar]
- Hallgren S, & Darnerud PO (2002). Polybrominated diphenyl ethers (PBDEs), polychlorinated biphenyls (PCBs) and chlorinated paraffins (CPs) in rats-testing interactions and mechanisms for thyroid hormone effects. Toxicology, 177(2-3), 227–243. doi: 10.1016/s0300-483x(02)00222-6 [DOI] [PubMed] [Google Scholar]
- Hallgren S, Sinjari T, Hakansson H, & Darnerud PO (2001). Effects of polybrominated diphenyl ethers (PBDEs) and polychlorinated biphenyls (PCBs) on thyroid hormone and vitamin A levels in rats and mice. Arch Toxicol, 75(4), 200–208. doi: 10.1007/s002040000208 [DOI] [PubMed] [Google Scholar]
- Hanneman WH, Legare ME, Barhoumi R, Burghardt RC, Safe S, & Tiffany-Castiglioni E (1996). Stimulation of calcium uptake in cultured rat hippocampal neurons by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology, 112(1), 19–28. doi: 10.1016/0300-483x(96)03346-x [DOI] [PubMed] [Google Scholar]
- Harry GJ, Hooth MJ, Vallant M, Behl M, Travlos GS, Howard JL, Price CJ, McBride S, Mervis R, & Mouton PR (2014). Developmental neurotoxicity of 3,3′,4,4′-tetrachloroazobenzene with thyroxine deficit: Sensitivity of glia and dentate granule neurons in the absence of behavioral changes. Toxics, 2(3), 496–532. doi: 10.3390/toxics2030496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsukano T, Kurisu J, Fukumitsu K, Fujishima K, & Kengaku M (2017). Thyroid Hormone Induces PGC-1alpha during Dendritic Outgrowth in Mouse Cerebellar Purkinje Cells. Front Cell Neurosci, 11, 133. doi: 10.3389/fncel.2017.00133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays LE, Carpenter CD, & Petersen SL (2002). Evidence that GABAergic neurons in the preoptic area of the rat brain are targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin during development. Environ Health Perspect, 110 Suppl 3, 369–376. doi: 10.1289/ehp.02110s3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks HS, Antunes Fernandes EC, Bergman A, van den Berg M, & Westerink RH (2010). PCB-47, PBDE-47, and 6-OH-PBDE-47 differentially modulate human GABAA and alpha4beta2 nicotinic acetylcholine receptors. Toxicol Sci, 118(2), 635–642. doi: 10.1093/toxsci/kfq284 [DOI] [PubMed] [Google Scholar]
- Herbert MR (2011). SHANK3, the synapse, and autism. N Engl J Med, 365(2), 173–175. doi: 10.1056/NEJMcibr1104261 [DOI] [PubMed] [Google Scholar]
- Herbstman JB, Sjodin A, Apelberg BJ, Witter FR, Halden RU, Patterson DG, Panny SR, Needham LL, & Goldman LR (2008). Birth delivery mode modifies the associations between prenatal polychlorinated biphenyl (PCB) and polybrominated diphenyl ether (PBDE) and neonatal thyroid hormone levels. Environ Health Perspect, 116(10), 1376–1382. doi: 10.1289/ehp.11379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbstman JB, Sjodin A, Kurzon M, Lederman SA, Jones RS, Rauh V, Needham LL, Tang D, Niedzwiecki M, Wang RY, & Perera F (2010). Prenatal exposure to PBDEs and neurodevelopment. Environ Health Perspect, 118(5), 712–719. doi: 10.1289/ehp.0901340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer DB, & Meredith RM (2017). Environmental toxicology: Sensitive periods of development and neurodevelopmental disorders. Neurotoxicology, 58, 23–41. doi: 10.1016/j.neuro.2016.10.017 [DOI] [PubMed] [Google Scholar]
- Hoffman K, Adgent M, Goldman BD, Sjodin A, & Daniels JL (2012). Lactational exposure to polybrominated diphenyl ethers and its relation to social and emotional development among toddlers. Environ Health Perspect, 120(10), 1438–1442. doi: 10.1289/ehp.1205100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojo R, Kakeyama M, Kurokawa Y, Aoki Y, Yonemoto J, & Tohyama C (2008). Learning behavior in rat offspring after in utero and lactational exposure to either TCDD or PCB126. Environ Health Prev Med, 13(3), 169–180. doi: 10.1007/s12199-008-0026-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojo R, Stern S, Zareba G, Markowski VP, Cox C, Kost JT, & Weiss B (2002). Sexually dimorphic behavioral responses to prenatal dioxin exposure. Environ Health Perspect, 110(3), 247–254. doi: 10.1289/ehp.02110247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojo R, Zareba G, Kai JW, Baggs RB, & Weiss B (2006). Sex-specific alterations of cerebral cortical cell size in rats exposed prenatally to dioxin. J Appl Toxicol, 26(1), 25–34. doi: 10.1002/jat.1101 [DOI] [PubMed] [Google Scholar]
- Hong SJ, Grover CA, Safe SH, Tiffany-Castiglioni E, & Frye GD (1998). Halogenated aromatic hydrocarbons suppress CA1 field excitatory postsynaptic potentials in rat hippocampal slices. Toxicol Appl Pharmacol, 148(1), 7–13. doi: 10.1006/taap.1997.8317 [DOI] [PubMed] [Google Scholar]
- Hood DB, Woods L, Brown L, Johnson S, & Ebner FF (2006). Gestational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure effects on sensory cortex function. Neurotoxicology, 27(6), 1032–1042. doi: 10.1016/j.neuro.2006.05.022 [DOI] [PubMed] [Google Scholar]
- Hopf NB, Ruder AM, & Succop P (2009). Background levels of polychlorinated biphenyls in the U.S. population. Sci Total Environ, 407(24), 6109–6119. doi: 10.1016/j.scitotenv.2009.08.035 [DOI] [PubMed] [Google Scholar]
- Hornbuckle K, & Robertson L (2010). Polychlorinated biphenyls (PCBs): sources, exposures, toxicities. Environ Sci Technol, 44(8), 2749–2751. doi: 10.1021/es100801f [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR, & Dabholkar AS (1997). Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol, 387(2), 167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z [DOI] [PubMed] [Google Scholar]
- Hyer MM, Phillips LL, & Neigh GN (2018). Sex Differences in Synaptic Plasticity: Hormones and Beyond. Front Mol Neurosci, 11, 266. doi: 10.3389/fnmol.2018.00266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibhazehiebo K, Iwasaki T, Kimura-Kuroda J, Miyazaki W, Shimokawa N, & Koibuchi N (2011a). Disruption of thyroid hormone receptor-mediated transcription and thyroid hormone-induced Purkinje cell dendrite arborization by polybrominated diphenyl ethers. Environ Health Perspect, 119(2), 168–175. doi: 10.1289/ehp.1002065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibhazehiebo K, Iwasaki T, Okano-Uchida T, Shimokawa N, Ishizaki Y, & Koibuchi N (2011b). Suppression of thyroid hormone receptor-mediated transcription and disruption of thyroid hormone-induced cerebellar morphogenesis by the polybrominated biphenyl mixture, BP-6. Neurotoxicology, 32(4), 400–409. doi: 10.1016/j.neuro.2011.02.008 [DOI] [PubMed] [Google Scholar]
- Ibhazehiebo K, & Koibuchi N (2012). Temporal effects of thyroid hormone (TH) and decabrominated diphenyl ether (BDE209) on Purkinje cell dendrite arborization. Niger J Physiol Sci, 27(1), 11–17. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/23235302 [PubMed] [Google Scholar]
- Ikeda M, Inukai N, Mitsui T, Sone H, Yonemoto J, Tohyama C, & Tomita T (2002). Changes in fetal brain aromatase activity following in utero 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure in rats. Environ Toxicol Pharmacol, 11(1), 1–7. doi: 10.1016/s1382-6689(01)00094-1 [DOI] [PubMed] [Google Scholar]
- Ikeda M, Mitsui T, Setani K, Tamura M, Kakeyama M, Sone H, Tohyama C, & Tomita T (2005). In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats disrupts brain sexual differentiation. Toxicol Appl Pharmacol, 205(1), 98–105. doi: 10.1016/j.taap.2004.09.010 [DOI] [PubMed] [Google Scholar]
- Inglefield JR, Mundy WR, & Shafer TJ (2001). Inositol 1,4,5-triphosphate receptor-sensitive Ca(2+) release, store-operated Ca(2+) entry, and cAMP responsive element binding protein phosphorylation in developing cortical cells following exposure to polychlorinated biphenyls. J Pharmacol Exp Ther, 297(2), 762–773. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/11303068https://jpet.aspetjournals.org/content/297/2/762.long [PubMed] [Google Scholar]
- Inglefield JR, & Shafer TJ (2000). Polychlorinated biphenyl-stimulation of Ca(2+) oscillations in developing neocortical cells: a role for excitatory transmitters and L-type voltage-sensitive Ca(2+) channels. J Pharmacol Exp Ther, 295(1), 105–113. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/10991967 [PubMed] [Google Scholar]
- Itoh S, Baba T, Yuasa M, Miyashita C, Kobayashi S, Araki A, Sasaki S, Kajiwara J, Hori T, Todaka T, Fujikura K, Nakajima S, Kato S, & Kishi R (2018). Association of maternal serum concentration of hydroxylated polychlorinated biphenyls with maternal and neonatal thyroid hormones: The Hokkaido birth cohort study. Environ Res, 167, 583–590. doi: 10.1016/j.envres.2018.08.027 [DOI] [PubMed] [Google Scholar]
- Kakeyama M, Endo T, Zhang Y, Miyazaki W, & Tohyama C (2014). Disruption of paired-associate learning in rat offspring perinatally exposed to dioxins. Arch Toxicol, 88(3), 789–798. doi: 10.1007/s00204-013-1161-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakeyama M, Sone H, & Tohyama C (2001). Changes in expression of NMDA receptor subunit mRNA by perinatal exposure to dioxin. Neuroreport, 12(18), 4009–4012. doi: 10.1097/00001756-200112210-00031 [DOI] [PubMed] [Google Scholar]
- Kapfhammer JP (2004). Cellular and molecular control of dendritic growth and development of cerebellar Purkinje cells. Prog Histochem Cytochem, 39(3), 131–182. doi: 10.1016/j.proghi.2004.07.002 [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, & Moser HW (2000). Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex, 10(10), 981–991. doi: 10.1093/cercor/10.10.981 [DOI] [PubMed] [Google Scholar]
- Keil KP, Miller GW, Chen H, Sethi S, Schmuck MR, Dhakal K, Kim JW, & Lein PJ (2018). PCB 95 promotes dendritic growth in primary rat hippocampal neurons via mTOR-dependent mechanisms. Arch Toxicol, 92(10), 3163–3173. doi: 10.1007/s00204-018-2285-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil KP, Sethi S, & Lein PJ (2019). Sex-Dependent Effects of 2,2′,3,5′,6-Pentachlorobiphenyl on Dendritic Arborization of Primary Mouse Neurons. Toxicol Sci, 168(1), 95–109. doi: 10.1093/toxsci/kfy277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly EA, Opanashuk LA, & Majewska AK (2014). The effects of postnatal exposure to low-dose bisphenol-A on activity-dependent plasticity in the mouse sensory cortex. Front Neuroanat, 8, 117. doi: 10.3389/fnana.2014.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenet T, Froemke RC, Schreiner CE, Pessah IN, & Merzenich MM (2007). Perinatal exposure to a noncoplanar polychlorinated biphenyl alters tonotopy, receptive fields, and plasticity in rat primary auditory cortex. Proc Natl Acad Sci U S A, 104(18), 7646–7651. doi: 10.1073/pnas.0701944104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB (2000). Signal-processing machines at the postsynaptic density. Science, 290(5492), 750–754. doi: 10.1126/science.290.5492.750 [DOI] [PubMed] [Google Scholar]
- Kim KH, Bose DD, Ghogha A, Riehl J, Zhang R, Barnhart CD, Lein PJ, & Pessah IN (2011). Para- and ortho-substitutions are key determinants of polybrominated diphenyl ether activity toward ryanodine receptors and neurotoxicity. Environ Health Perspect, 119(4), 519–526. doi: 10.1289/ehp.1002728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Lee HG, Choi EJ, Park KY, & Yang JH (2007). TCDD alters PKC signaling pathways in developing neuronal cells in culture. Chemosphere, 67(9), S421–427. doi: 10.1016/j.chemosphere.2006.05.138 [DOI] [PubMed] [Google Scholar]
- Kim SY, & Yang JH (2005). Neurotoxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in cerebellar granule cells. Exp Mol Med, 37(1), 58–64. doi: 10.1038/emm.2005.8 [DOI] [PubMed] [Google Scholar]
- Kimura E, Kubo K, Matsuyoshi C, Benner S, Hosokawa M, Endo T, Ling W, Kohda M, Yokoyama K, Nakajima K, Kakeyama M, & Tohyama C (2015). Developmental origin of abnormal dendritic growth in the mouse brain induced by in utero disruption of aryl hydrocarbon receptor signaling. Neurotoxicol Teratol, 52(Pt A), 42–50. doi: 10.1016/j.ntt.2015.10.005 [DOI] [PubMed] [Google Scholar]
- Kimura-Kuroda J, Nagata I, & Kuroda Y (2005). Hydroxylated metabolites of polychlorinated biphenyls inhibit thyroid-hormone-dependent extension of cerebellar Purkinje cell dendrites. Brain Res Dev Brain Res, 154(2), 259–263. doi: 10.1016/j.devbrainres.2004.1L004 [DOI] [PubMed] [Google Scholar]
- Kimura-Kuroda J, Nagata I, & Kuroda Y (2007). Disrupting effects of hydroxy-polychlorinated biphenyl (PCB) congeners on neuronal development of cerebellar Purkinje cells: a possible causal factor for developmental brain disorders? Chemosphere, 67(9), S412–420. doi: 10.1016/j.chemosphere.2006.05.137 [DOI] [PubMed] [Google Scholar]
- Kimura-Kuroda J, Nagata I, Negishi-Kato M, & Kuroda Y (2002). Thyroid hormone-dependent development of mouse cerebellar Purkinje cells in vitro. Brain Res Dev Brain Res, 137(1), 55–65. doi: 10.1016/s0165-3806(02)00408-x [DOI] [PubMed] [Google Scholar]
- Klincic D, Dvorscak M, Jagic K, Mendas G, & Herceg Romanic S (2020). Levels and distribution of polybrominated diphenyl ethers in humans and environmental compartments: a comprehensive review of the last five years of research. Environ Sci Pollut Res Int, 27(6), 5744–5758. doi: 10.1007/s11356-020-07598-7 [DOI] [PubMed] [Google Scholar]
- Klocke C, & Lein PJ (2020). Evidence Implicating Non-Dioxin-Like Congeners as the Key Mediators of Polychlorinated Biphenyl (PCB) Developmental Neurotoxicity. Int J Mol Sci, 21(3). doi: 10.3390/ijms21031013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klocke C, Sethi S, & Lein PJ (2020). The developmental neurotoxicity of legacy vs. contemporary polychlorinated biphenyls (PCBs): similarities and differences. Environ Sci Pollut Res Int, 27(9), 8885–8896. doi: 10.1007/s11356-019-06723-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koger SM, Schettler T, & Weiss B (2005). Environmental toxicants and developmental disabilities: a challenge for psychologists. Am Psychol, 60(3), 243–255. doi: 10.1037/0003-066X.60.3.243 [DOI] [PubMed] [Google Scholar]
- Koh WX, Hornbuckle KC, & Thorne PS (2015). Human Serum from Urban and Rural Adolescents and Their Mothers Shows Exposure to Polychlorinated Biphenyls Not Found in Commercial Mixtures. Environ Sci Technol, 49(13), 8105–8112. doi: 10.1021/acs.est.5b01854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konur S, & Ghosh A (2005). Calcium signaling and the control of dendritic development. Neuron, 46(3), 401–405. doi: 10.1016/j.neuron.2005.04.022 [DOI] [PubMed] [Google Scholar]
- Korrick SA, & Sagiv SK (2008). Polychlorinated biphenyls, organochlorine pesticides and neurodevelopment. Curr Opin Pediatr, 20(2), 198–204. doi: 10.1097/MOP.0b013e3282f6a4e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M, & et al. (1992). Molecular diversity of the NMDA receptor channel. Nature, 358(6381), 36–41. doi: 10.1038/358036a0 [DOI] [PubMed] [Google Scholar]
- Lai TJ, Guo YL, Guo NW, & Hsu CC (2001). Effect of prenatal exposure to polychlorinated biphenyls on cognitive development in children: a longitudinal study in Taiwan. Br J Psychiatry Suppl, 40, s49–52. doi: 10.1192/bjp.178.40.s49 [DOI] [PubMed] [Google Scholar]
- Lam J, Lanphear BP, Bellinger D, Axelrad DA, McPartland J, Sutton P, Davidson L, Daniels N, Sen S, & Woodruff TJ (2017). Developmental PBDE Exposure and IQ/ADHD in Childhood: A Systematic Review and Meta-analysis. Environ Health Perspect, 125(8), 086001. doi: 10.1289/EHP1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrigan PJ, Lioy PJ, Thurston G, Berkowitz G, Chen LC, Chillrud SN, Gavett SH, Georgopoulos PG, Geyh AS, Levin S, Perera F, Rappaport SM, Small C, & Group NWTCW (2004). Health and environmental consequences of the world trade center disaster. Environ Health Perspect, 112(6), 731–739. doi: 10.1289/ehp.6702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrigan PJ, Sonawane B, Butler RN, Trasande L, Callan R, & Droller D (2005). Early environmental origins of neurodegenerative disease in later life. Environ Health Perspect, 113(9), 1230–1233. doi: 10.1289/ehp.7571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanphear BP, Hornung R, Khoury J, Yolton K, Baghurst P, Bellinger DC, Canfield RL, Dietrich KN, Bornschein R, Greene T, Rothenberg SJ, Needleman HL, Schnaas L, Wasserman G, Graziano J, & Roberts R (2005). Low-level environmental lead exposure and children’s intellectual function: an international pooled analysis. Environ Health Perspect, 113(7), 894–899. doi: 10.1289/ehp.7688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchney SE, Lioy DT, Henry EC, Gasiewicz TA, Strathmann FG, Mayer-Proschel M, & Opanashuk LA (2011). Neural precursor cell proliferation is disrupted through activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Stem Cells Dev, 20(2), 313–326. doi: 10.1089/scd.2009.0529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Sadowski RN, Schantz SL, & Llano DA (2021). Developmental PCB Exposure Disrupts Synaptic Transmission and Connectivity in the Rat Auditory Cortex, Independent of Its Effects on Peripheral Hearing Threshold. eNeuro, 8(1). doi: 10.1523/ENEURO.0321-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HG, Kim SY, Choi EJ, Park KY, & Yang JH (2007). Translocation of PKC-betaII is mediated via RACK-1 in the neuronal cells following dioxin exposure. Neurotoxicology, 28(2), 408–414. doi: 10.1016/j.neuro.2006.04.007 [DOI] [PubMed] [Google Scholar]
- Lein PJ, Yang D, Bachstetter AD, Tilson HA, Harry GJ, Mervis RF, & Kodavanti PR (2007). Ontogenetic alterations in molecular and structural correlates of dendritic growth after developmental exposure to polychlorinated biphenyls. Environ Health Perspect, 115(4), 556–563. doi: 10.1289/ehp.9773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenters V, Iszatt N, Forns J, Cechova E, Kocan A, Legler J, Leonards P, Stigum H, & Eggesbo M (2019). Early-life exposure to persistent organic pollutants (OCPs, PBDEs, PCBs, PFASs) and attention-deficit/hyperactivity disorder: A multi-pollutant analysis of a Norwegian birth cohort. Environ Int, 125, 33–42. doi: 10.1016/j.envint.2019.01.020 [DOI] [PubMed] [Google Scholar]
- Lesiak A, Zhu M, Chen H, Appleyard SM, Impey S, Lein PJ, & Wayman GA (2014). The environmental neurotoxicant PCB 95 promotes synaptogenesis via ryanodine receptor-dependent miR132 upregulation. J Neurosci, 34(3), 717–725. doi: 10.1523/JNEUROSCI.2884-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Mu Y, & Gage FH (2009). Development of neural circuits in the adult hippocampus. Curr Top Dev Biol, 87, 149–174. doi: 10.1016/S0070-2153(09)01205-8 [DOI] [PubMed] [Google Scholar]
- Li ZM, Hernandez-Moreno D, Main KM, Skakkebaek NE, Kiviranta H, Toppari J, Feldt-Rasmussen U, Shen H, Schramm KW, & De Angelis M (2018). Association of In Utero Persistent Organic Pollutant Exposure With Placental Thyroid Hormones. Endocrinology, 159(10), 3473–3481. doi: 10.1210/en.2018-00542 [DOI] [PubMed] [Google Scholar]
- Lin CH, Chen CC, Chou CM, Wang CY, Hung CC, Chen JY, Chang HW, Chen YC, Yeh GC, & Lee YH (2009). Knockdown of the aryl hydrocarbon receptor attenuates excitotoxicity and enhances NMDA-induced BDNF expression in cortical neurons. J Neurochem, 111(3), 777–789. doi: 10.1111/j.1471-4159.2009.06364.x [DOI] [PubMed] [Google Scholar]
- Lin CH, Juan SH, Wang CY, Sun YY, Chou CM, Chang SF, Hu SY, Lee WS, & Lee YH (2008). Neuronal activity enhances aryl hydrocarbon receptor-mediated gene expression and dioxin neurotoxicity in cortical neurons. J Neurochem, 104(5), 1415–1429. doi: 10.1111/j.1471-4159.2007.05098.x [DOI] [PubMed] [Google Scholar]
- Lioy PL, Waldman JM, Greenberg A, Harkov R, & Pietarinen C (1988). The Total Human Environmental Exposure Study (THEES) to benzo(a)pyrene: comparison of the inhalation and food pathways. Arch Environ Health, 43(4), 304–312. doi: 10.1080/00039896.1988.10545954 [DOI] [PubMed] [Google Scholar]
- Lohmann C, & Wong RO (2005). Regulation of dendritic growth and plasticity by local and global calcium dynamics. Cell Calcium, 37(5), 403–409. doi: 10.1016/j.ceca.2005.01.008 [DOI] [PubMed] [Google Scholar]
- Londono M, Shimokawa N, Miyazaki W, Iwasaki T, & Koibuchi N (2010). Hydroxylated PCB induces Ca2+ oscillations and alterations of membrane potential in cultured cortical cells. J Appl Toxicol, 30(4), 334–342. doi: 10.1002/jat.1501 [DOI] [PubMed] [Google Scholar]
- Lu Z, Kania-Korwel I, Lehmler HJ, & Wong CS (2013). Stereoselective formation of mono-and dihydroxylated polychlorinated biphenyls by rat cytochrome P450 2B1. Environ Sci Technol, 47(21), 12184–12192. doi: 10.1021/es402838f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, & Wong CS (2011). Factors affecting phase I stereoselective biotransformation of chiral polychlorinated biphenyls by rat cytochrome P-450 2B1 isozyme. Environ Sci Technol, 45(19), 8298–8305. doi: 10.1021/es200673q [DOI] [PubMed] [Google Scholar]
- Maffini MV, & Neltner TG (2015). Brain drain: the cost of neglected responsibilities in evaluating cumulative effects of environmental chemicals. J Epidemiol Community Health, 69(5), 496–499. doi: 10.1136/jech-2014-203980 [DOI] [PubMed] [Google Scholar]
- Markowski VP, Cox C, Preston R, & Weiss B (2002). Impaired cued delayed alternation behavior in adult rat offspring following exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on gestation day 15. Neurotoxicol Teratol, 24(2), 209–218. doi: 10.1016/s0892-0362(02)00186-1 [DOI] [PubMed] [Google Scholar]
- Marnane I (2012). Comprehensive environmental review following the pork PCB/dioxin contamination incident in Ireland. J Environ Monit, 14(10), 2551–2556. doi: 10.1039/c2em30374d [DOI] [PubMed] [Google Scholar]
- Martin L, & Klaassen CD (2010). Differential effects of polychlorinated biphenyl congeners on serum thyroid hormone levels in rats. Toxicol Sci, 117(1), 36–44. doi: 10.1093/toxsci/kfq187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, & Kasai H (2004). Structural basis of long-term potentiation in single dendritic spines. Nature, 429(6993), 761–766. doi: 10.1038/nature02617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazdai A, Dodder NG, Abernathy MP, Hites RA, & Bigsby RM (2003). Polybrominated diphenyl ethers in maternal and fetal blood samples. Environ Health Perspect, 111(9), 1249–1252. doi: 10.1289/ehp.6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallister MM, Maguire M, Ramesh A, Aimin Q, Liu S, Khoshbouei H, Aschner M, Ebner FF, & Hood DB (2008). Prenatal exposure to benzo(a)pyrene impairs later-life cortical neuronal function. Neurotoxicology, 29(5), 846–854. doi: 10.1016/j.neuro.2008.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw JE Sr., & Waller DP (2006). Specific human CYP 450 isoform metabolism of a pentachlorobiphenyl (PCB-IUPAC# 101). Biochem Biophys Res Commun, 344(1), 129–133. doi: 10.1016/j.bbrc.2006.03.122 [DOI] [PubMed] [Google Scholar]
- Meco G, Bonifati V, Vanacore N, & Fabrizio E (1994). Parkinsonism after chronic exposure to the fungicide maneb (manganese ethylene-bis-dithiocarbamate). Scand J Work Environ Health, 20(4), 301–305. doi: 10.5271/sjweh.1394 [DOI] [PubMed] [Google Scholar]
- Miller JP, & Jacobs GA (1984). Relationships between neuronal structure and function. J Exp Biol, 112, 129–145. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/6392465 [DOI] [PubMed] [Google Scholar]
- Mitchell MM, Woods R, Chi LH, Schmidt RJ, Pessah IN, Kostyniak PJ, & LaSalle JM (2012). Levels of select PCB and PBDE congeners in human postmortem brain reveal possible environmental involvement in 15q11-q13 duplication autism spectrum disorder. Environ Mol Mutagen, 53(8), 589–598. doi: 10.1002/em.21722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuhashi T, Yonemoto J, Sone H, Kosuge Y, Kosaki K, & Takahashi T (2010). In utero exposure to dioxin causes neocortical dysgenesis through the actions of p27Kip1. Proc Natl Acad Sci USA, 107(37), 16331–16335. doi: 10.1073/pnas.1002960107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer CE, & Zuo Y (2018). Cortical dendritic spine development and plasticity: insights from in vivo imaging. Curr Opin Neurobiol, 53, 76–82. doi: 10.1016/j.conb.2018.06.002 [DOI] [PubMed] [Google Scholar]
- Muir DC, & Howard PH (2006). Are there other persistent organic pollutants? A challenge for environmental chemists. Environ Sci Technol, 40(23), 7157–7166. doi: 10.1021/es061677a [DOI] [PubMed] [Google Scholar]
- Mundy WR, Shafer TJ, Tilson HA, & Kodavanti PR (1999). Extracellular calcium is required for the polychlorinated biphenyl-induced increase of intracellular free calcium levels in cerebellar granule cell culture. Toxicology, 136(1), 27–39. doi: 10.1016/s0300-483x(99)00052-9 [DOI] [PubMed] [Google Scholar]
- Nakai N, Takumi T, Nakai J, & Sato M (2018). Common Defects of Spine Dynamics and Circuit Function in Neurodevelopmental Disorders: A Systematic Review of Findings From in Vivo Optical Imaging of Mouse Models. Front Neurosci, 12, 412. doi: 10.3389/fnins.2018.00412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayyar T, Wu J, & Hood DB (2003). Downregulation of hippocampal NMDA receptor expression by prenatal exposure to dioxin. Cell Mol Biol (Noisy-le-grand), 49(8), 1357–1362. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/14984010 [PubMed] [Google Scholar]
- Nguyen MN, Nishijo M, Nguyen AT, Bor A, Nakamura T, Hori E, Nakagawa H, Ono T, & Nishijo H (2013). Effects of maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on parvalbumin- and calbindin-immunoreactive neurons in the limbic system and superior colliculus in rat offspring. Toxicology, 314(1), 125–134. doi: 10.1016/j.tox.2013.09.005 [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, & Svoboda K (2002). Structure and function of dendritic spines. Annu Rev Physiol, 64, 313–353. doi: 10.1146/annurev.physiol.64.081501.160008 [DOI] [PubMed] [Google Scholar]
- Nishijo M, Tai PT, Nakagawa H, Maruzeni S, Anh NT, Luong HV, Anh TH, Honda R, Morikawa Y, Kido T, & Nishijo H (2012). Impact of perinatal dioxin exposure on infant growth: a cross-sectional and longitudinal studies in dioxin-contaminated areas in Vietnam. PLoS One, 7(7), e40273. doi: 10.1371/journal.pone.0040273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oken E, Radesky JS, Wright RO, Bellinger DC, Amarasiriwardena CJ, Kleinman KP, Hu H, & Gillman MW (2008). Maternal fish intake during pregnancy, blood mercury levels, and child cognition at age 3 years in a US cohort. Am J Epidemiol, 167(10), 1171–1181. doi: 10.1093/aje/kwn034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opler MG, Buka SL, Groeger J, McKeague I, Wei C, Factor-Litvak P, Bresnahan M, Graziano J, Goldstein JM, Seidman LJ, Brown AS, & Susser ES (2008). Prenatal exposure to lead, delta-aminolevulinic acid, and schizophrenia: further evidence. Environ Health Perspect, 116(11), 1586–1590. doi: 10.1289/ehp.10464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osius N, Karmaus W, Kruse H, & Witten J (1999). Exposure to polychlorinated biphenyls and levels of thyroid hormones in children. Environ Health Perspect, 107(10), 843–849. doi: 10.1289/ehp.99107843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanza P, Parmigiani S, Liu H, & vom Saal FS (1999). Prenatal exposure to low doses of the estrogenic chemicals diethylstilbestrol and o,p′-DDT alters aggressive behavior of male and female house mice. Pharmacol Biochem Behav, 64(4), 665–672. doi: 10.1016/s0091-3057(99)00151-3 [DOI] [PubMed] [Google Scholar]
- Pellizzari ED, Woodruff TJ, Boyles RR, Kannan K, Beamer PI, Buckley JP, Wang A, Zhu Y, & Bennett DH (2019). Identifying and Prioritizing Chemicals with Uncertain Burden of Exposure: Opportunities for Biomonitoring and Health-Related Research. Environ Health Perspect, 127(12), 126001. doi: 10.1289/EHP5133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, & Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci, 14(3), 285–293. doi: 10.1038/nn.2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera F, Li TY, Zhou ZJ, Yuan T, Chen YH, Qu L, Rauh VA, Zhang Y, & Tang D (2008). Benefits of reducing prenatal exposure to coal-burning pollutants to children’s neurodevelopment in China. Environ Health Perspect, 116(10), 1396–1400. doi: 10.1289/ehp.11480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera FP, Jedrychowski W, Rauh V, & Whyatt RM (1999). Molecular epidemiologic research on the effects of environmental pollutants on the fetus. Environ Health Perspect, 107 Suppl 3, 451–460. doi: 10.1289/ehp.99107s3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera FP, Li Z, Whyatt R, Hoepner L, Wang S, Camann D, & Rauh V (2009). Prenatal airborne polycyclic aromatic hydrocarbon exposure and child IQ at age 5 years. Pediatrics, 124(2), e195–202. doi: 10.1542/peds.2008-3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera FP, Rauh V, Tsai WY, Kinney P, Camann D, Barr D, Bernert T, Garfinkel R, Tu YH, Diaz D, Dietrich J, & Whyatt RM (2003). Effects of transplacental exposure to environmental pollutants on birth outcomes in a multiethnic population. Environ Health Perspect, 111(2), 201–205. doi: 10.1289/ehp.5742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera FP, Rauh V, Whyatt RM, Tsai WY, Tang D, Diaz D, Hoepner L, Barr D, Tu YH, Camann D, & Kinney P (2006). Effect of prenatal exposure to airborne polycyclic aromatic hydrocarbons on neurodevelopment in the first 3 years of life among inner-city children. Environ Health Perspect, 114(8), 1287–1292. doi: 10.1289/ehp.9084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera FP, Tang D, Rauh V, Lester K, Tsai WY, Tu YH, Weiss L, Hoepner L, King J, Del Priore G, & Lederman SA (2005). Relationships among polycyclic aromatic hydrocarbon-DNA adducts, proximity to the World Trade Center, and effects on fetal growth. Environ Health Perspect, 113(8), 1062–1067. doi: 10.1289/ehp.7908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera FP, Tang D, Rauh V, Tu YH, Tsai WY, Becker M, Stein JL, King J, Del Priore G, & Lederman SA (2007). Relationship between polycyclic aromatic hydrocarbon-DNA adducts, environmental tobacco smoke, and child development in the World Trade Center cohort. Environ Health Perspect, 115(10), 1497–1502. doi: 10.1289/ehp.10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera FP, Tang D, Wang S, Vishnevetsky J, Zhang B, Diaz D, Camann D, & Rauh V (2012). Prenatal polycyclic aromatic hydrocarbon (PAH) exposure and child behavior at age 6-7 years. Environ Health Perspect, 120(6), 921–926. doi: 10.1289/ehp.1104315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessah IN, Cherednichenko G, & Lein PJ (2010). Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity. Pharmacol Ther, 125(2), 260–285. doi: 10.1016/j.pharmthera.2009.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessah IN, Lein PJ, Seegal RF, & Sagiv SK (2019). Neurotoxicity of polychlorinated biphenyls and related organohalogens. Acta Neuropathol, 138(3), 363–387. doi: 10.1007/s00401-019-01978-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham NT, Nishijo M, Pham TT, Tran NN, Le VQ, Tran HA, Phan HAV, Nishino Y, & Nishijo H (2019). Perinatal dioxin exposure and neurodevelopment of 2-year-old Vietnamese children in the most contaminated area from Agent Orange in Vietnam. Sci Total Environ, 678, 217–226. doi: 10.1016/j.scitotenv.2019.04.425 [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C (2012). Which comes first in fragile X syndrome, dendritic spine dysgenesis or defects in circuit plasticity? Neuroscientist, 18(1), 28–44. doi: 10.1177/1073858410395322 [DOI] [PubMed] [Google Scholar]
- Rakic P, Bourgeois JP, Eckenhoff MF, Zecevic N, & Goldman-Rakic PS (1986). Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science, 232(4747), 232–235. doi: 10.1126/science.3952506 [DOI] [PubMed] [Google Scholar]
- Rauh VA, & Margolis AE (2016). Research Review: Environmental exposures, neurodevelopment, and child mental health - new paradigms for the study of brain and behavioral effects. J Child Psychol Psychiatry, 57(7), 775–793. doi: 10.1111/jcpp.12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice D, & Barone S Jr. (2000). Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect, 108 Suppl 3, 511–533. doi: 10.1289/ehp.00108s3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risau W, & Wolburg H (1990). Development of the blood-brain barrier. Trends Neurosci, 13(5), 174–178. doi: 10.1016/0166-2236(90)90043-a [DOI] [PubMed] [Google Scholar]
- Rock KD, & Patisaul HB (2018). Environmental Mechanisms of Neurodevelopmental Toxicity. Curr Environ Health Rep, 5(1), 145–157. doi: 10.1007/s40572-018-0185-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier PM (1994). Vulnerable periods and processes during central nervous system development. Environ Health Perspect, 102 Suppl 2, 121–124. doi: 10.1289/ehp.94102121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roegge CS, Morris JR, Villareal S, Wang VC, Powers BE, Klintsova AY, Greenough WT, Pessah IN, & Schantz SL (2006). Purkinje cell and cerebellar effects following developmental exposure to PCBs and/or MeHg. Neurotoxicol Teratol, 28(1), 74–85. doi: 10.1016/j.ntt.2005.10.001 [DOI] [PubMed] [Google Scholar]
- Rooney JPK, Woods NF, Martin MD, & Woods JS (2018). Genetic polymorphisms of GRIN2A and GRIN2B modify the neurobehavioral effects of low-level lead exposure in children. Environ Res, 165, 1–10. doi: 10.1016/j.envres.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovet JF (2014). The role of thyroid hormones for brain development and cognitive function. Endocr Dev, 26, 26–43. doi: 10.1159/000363153 [DOI] [PubMed] [Google Scholar]
- Roze E, Meijer L, Bakker A, Van Braeckel KN, Sauer PJ, & Bos AF (2009). Prenatal exposure to organohalogens, including brominated flame retardants, influences motor, cognitive, and behavioral performance at school age. Environ Health Perspect, 117(12), 1953–1958. doi: 10.1289/ehp.0901015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Marcos A, Cartagena-Abella P, Martinez-Galan JR, Calvo R, Morreale de Escobar G, & Escobar del Rey F (1994). Thyroxine treatment and the recovery of pyramidal cells of the cerebral cortex from changes induced by juvenile-onset hypothyroidism. J Neurobiol, 25(7), 808–818. doi: 10.1002/neu.480250706 [DOI] [PubMed] [Google Scholar]
- Sala C, & Segal M (2014). Dendritic spines: the locus of structural and functional plasticity. Physiol Rev, 94(1), 141–188. doi: 10.1152/physrev.00012.2013 [DOI] [PubMed] [Google Scholar]
- Sala M, Sunyer J, Herrero C, To-Figueras J, & Grimalt J (2001). Association between serum concentrations of hexachlorobenzene and polychlorobiphenyls with thyroid hormone and liver enzymes in a sample of the general population. Occup Environ Med, 58(3), 172–177. doi: 10.1136/oem.58.3.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar P, Cisternas P, Martinez M, & Inestrosa NC (2019). Hypothyroidism and Cognitive Disorders during Development and Adulthood: Implications in the Central Nervous System. Mol Neurobiol, 56(4), 2952–2963. doi: 10.1007/s12035-018-1270-y [DOI] [PubMed] [Google Scholar]
- Sathyanesan A, Zhou J, Scafidi J, Heck DH, Sillitoe RV, & Gallo V (2019). Emerging connections between cerebellar development, behaviour and complex brain disorders. Nat Rev Neurosci, 20(5), 298–313. doi: 10.1038/s41583-019-0152-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CR, Shockley DC, & Knuckles ME (2001). Behavioral effects induced by acute exposure to benzo(a)pyrene in F-344 rats. Neurotox Res, 5(6), 557–579. doi: 10.1007/BF03033211 [DOI] [PubMed] [Google Scholar]
- Schantz SL, Widholm JJ, & Rice DC (2003). Effects of PCB exposure on neuropsychological function in children. Environ Health Perspect, 111(3), 357–576. doi: 10.1289/ehp.5461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M (2010). Dendritic spines, synaptic plasticity and neuronal survival: activity shapes dendritic spines to enhance neuronal viability. Eur J Neurosci, 31(12), 2178–2184. doi: 10.1111/j.1460-9568.2010.07270.x [DOI] [PubMed] [Google Scholar]
- Selevan SG, Kimmel CA, & Mendola P (2000). Identifying critical windows of exposure for children’s health. Environ Health Perspect, 108 Suppl 5, 451–455. doi: 10.1289/ehp.00108s3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, & Noble-Haeusslein LJ (2013). Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol, 106-107, 1–16. doi: 10.1016/j.pneurobio.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo BW, Powers BE, Widholm JJ, & Schantz SL (2000). Radial arm maze performance in rats following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Neurotoxicol Teratol, 22(4), 511–519. doi: 10.1016/s0892-0362(00)00070-2 [DOI] [PubMed] [Google Scholar]
- Sethi S, Keil KP, Chen H, Hayakawa K, Li X, Lin Y, Lehmler HJ, Puschner B, & Lein PJ (2017a). Detection of 3,3′-Dichlorobiphenyl in Human Maternal Plasma and Its Effects on Axonal and Dendritic Growth in Primary Rat Neurons. Toxicol Sci, 158(2), 401–411. doi: 10.1093/toxsci/kfx100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi S, Keil KP, & Lein PJ (2017b). Species and Sex Differences in the Morphogenic Response of Primary Rodent Neurons to 3,3′-Dichlorobiphenyl (PCB 11). Toxics, 6(1). doi: 10.3390/toxics6010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi S, Keil KP, & Lein PJ (2018). 3,3′-Dichlorobiphenyl (PCB 11) promotes dendritic arborization in primary rat cortical neurons via a CREB-dependent mechanism. Arch Toxicol, 92(11), 3337–3345. doi: 10.1007/s00204-018-2307-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer RM, Sellers SP, Baker MG, de Buen Kalman R, Frostad J, Suter MK, Anenberg SC, Balbus J, Basu N, Bellinger DC, Birnbaum L, Brauer M, Cohen A, Ebi KL, Fuller R, Grandjean P, Hess JJ, Kogevinas M, Kumar P, Landrigan PJ, Lanphear B, London SJ, Rooney AA, Stanaway JD, Trasande L, Walker K, & Hu H (2019). Improving and Expanding Estimates of the Global Burden of Disease Due to Environmental Health Risk Factors. Environ Health Perspect, 127(10), 105001. doi: 10.1289/EHP5496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, Chua C, & Falduto J (2001). Sex differences and opposite effects of stress on dendritic spine density in the male versus female hippocampus. J Neurosci, 21(16), 6292–6297. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/11487652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddique S, Banerjee M, Ray MR, & Lahiri T (2011). Attention-deficit hyperactivity disorder in children chronically exposed to high level of vehicular pollution. Eur J Pediatr, 170(7), 923–929. doi: 10.1007/s00431-010-1379-0 [DOI] [PubMed] [Google Scholar]
- Skucas VA, Duffy AM, Harte-Hargrove LC, Magagna-Poveda A, Radman T, Chakraborty G, Schroeder CE, MacLusky NJ, & Scharfman HE (2013). Testosterone depletion in adult male rats increases mossy fiber transmission, LTP, and sprouting in area CA3 of hippocampus. J Neurosci, 33(6), 2338–2355. doi: 10.1523/JNEUROSCI.3857-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GB, Heynen AJ, & Bear MF (2009). Bidirectional synaptic mechanisms of ocular dominance plasticity in visual cortex. Philos Trans R Soc Lond B Biol Sci, 364(1515), 357–367. doi: 10.1098/rstb.2008.0198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolewski M, Conrad K, Allen JL, Weston H, Martin K, Lawrence BP, & Cory-Slechta DA (2014). Sex-specific enhanced behavioral toxicity induced by maternal exposure to a mixture of low dose endocrine-disrupting chemicals. Neurotoxicology, 45, 121–130. doi: 10.1016/j.neuro.2014.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamou M, Streifel KM, Goines PE, & Lein PJ (2013). Neuronal connectivity as a convergent target of gene x environment interactions that confer risk for Autism Spectrum Disorders. Neurotoxicol Teratol, 36, 3–16. doi: 10.1016/j.ntt.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles J, & Jernigan TL (2010). The basics of brain development. Neuropsychol Rev, 20(4), 327–348. doi: 10.1007/s11065-010-9148-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suglia SF, Gryparis A, Wright RO, Schwartz J, & Wright RJ (2008). Association of black carbon with cognition among children in a prospective birth cohort study. Am J Epidemiol, 167(3), 280–286. doi: 10.1093/aje/kwm308 [DOI] [PubMed] [Google Scholar]
- Supekar K, Uddin LQ, Khouzam A, Phillips J, Gaillard WD, Kenworthy LE, Yerys BE, Vaidya CJ, & Menon V (2013). Brain hyperconnectivity in children with autism and its links to social deficits. Cell Rep, 5(3), 738–747. doi: 10.1016/j.celrep.2013.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo DT, Richardson VM, Ross DG, Diliberto JJ, Kodavanti PR, & Birnbaum LS (2009). Effects of perinatal PBDE exposure on hepatic phase I, phase II, phase III, and deiodinase 1 gene expression involved in thyroid hormone metabolism in male rat pups. Toxicol Sci, 107(1), 27–39. doi: 10.1093/toxsci/kfn230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai PT, Nishijo M, Anh NT, Maruzeni S, Nakagawa H, Van Luong H, Anh TH, Honda R, Kido T, & Nishijo H (2013). Dioxin exposure in breast milk and infant neurodevelopment in Vietnam. Occup Environ Med, 70(9), 656–662. doi: 10.1136/oemed-2012-101021 [DOI] [PubMed] [Google Scholar]
- Takesian AE, & Hensch TK (2013). Balancing plasticity/stability across brain development. Prog Brain Res, 207, 3–34. doi: 10.1016/B978-0-444-63327-9.00001-1 [DOI] [PubMed] [Google Scholar]
- Tamm C, & Ceccatelli S (2017). Mechanistic insight into neurotoxicity induced by developmental insults. Biochem Biophys Res Commun, 482(3), 408–418. doi: 10.1016/j.bbrc.2016.10.087 [DOI] [PubMed] [Google Scholar]
- Tau GZ, & Peterson BS (2010). Normal development of brain circuits. Neuropsychopharmacology, 35(1), 147–168. doi: 10.1038/npp.2009.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MR, & Boekelheide K (2013). Multiple environmental chemical exposures to lead, mercury and polychlorinated biphenyls among childbearing-aged women (NHANES 1999-2004): Body burden and risk factors. Environ Res, 121, 23–30. doi: 10.1016/j.envres.2012.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian YH, Hwan Kim S, Lee SY, & Jang CG (2011). Lactational and postnatal exposure to polychlorinated biphenyls induces sex-specific anxiolytic behavior and cognitive deficit in mice offspring. Synapse, 65(10), 1032–1041. doi: 10.1002/syn.20934 [DOI] [PubMed] [Google Scholar]
- Tilson HA (2000). Neurotoxicology risk assessment guidelines: developmental neurotoxicology. Neurotoxicology, 21(1-2), 189–194. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/10794399 [PubMed] [Google Scholar]
- Tilson HA, MacPhail RC, & Crofton KM (1995). Defining neurotoxicity in a decision-making context. Neurotoxicology, 16(2), 363–375. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/7566695 [PubMed] [Google Scholar]
- Timofeeva OA, Sanders D, Seemann K, Yang L, Hermanson D, Regenbogen S, Agoos S, Kallepalli A, Rastogi A, Braddy D, Wells C, Perraut C, Seidler FJ, Slotkin TA, & Levin ED (2008). Persistent behavioral alterations in rats neonatally exposed to low doses of the organophosphate pesticide, parathion. Brain Res Bull, 77(6), 404–411. doi: 10.1016/j.brainresbull.2008.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini MC, Beggiato S, Ferraro L, Tanganelli S, Marani L, Lorenzini L, & Antonelli T (2012). Prenatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin produces alterations in cortical neuron development and a long-term dysfunction of glutamate transmission in rat cerebral cortex. Neurochem Int, 61(5), 759–766. doi: 10.1016/j.neuint.2012.07.004 [DOI] [PubMed] [Google Scholar]
- Toms LM, Sjodin A, Harden F, Hobson P, Jones R, Edenfield E, & Mueller JF (2009). Serum polybrominated diphenyl ether (PBDE) levels are higher in children (2-5 years of age) than in infants and adults. Environ Health Perspect, 117(9), 1461–1465. doi: 10.1289/ehp.0900596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R, Landau EM, & Blitzer RD (2005). Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci, 25(24), 5833–5843. doi: 10.1523/JNEUROSCI.0599-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler CV Jr., White-Scott S, Ekvall SM, & Abulafia L (2008). Environmental health and developmental disabilities: a life span approach. Fam Community Health, 31(4), 287–304. doi: 10.1097/01.FCH.0000336092.39066.a0 [DOI] [PubMed] [Google Scholar]
- Van Metre PC, & Mahler BJ (2005). Trends in hydrophobic organic contaminants in urban and reference lake sediments across the United States, 1970-2001. Environ Sci Technol, 39(15), 5567–5574. doi: 10.1021/es0503175 [DOI] [PubMed] [Google Scholar]
- Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr., Lee DH, Shioda T, Soto AM, vom Saal FS, Welshons WV, Zoeller RT, & Myers JP (2012). Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev, 33(3), 378–455. doi: 10.1210/er.2011-1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRyzin JW, Marquardt AE, Pickett LA, & McCarthy MM (2020). Microglia and sexual differentiation of the developing brain: A focus on extrinsic factors. Glia, 68(6), 1100–1113. doi: 10.1002/glia.23740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester A, & Caudle WM (2016). The Synapse as a Central Target for Neurodevelopmental Susceptibility to Pesticides. Toxics, 4(3). doi: 10.3390/toxics4030018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viberg H (2009). Neonatal ontogeny and neurotoxic effect of decabrominated diphenyl ether (PBDE 209) on levels of synaptophysin and tau. Int J Dev Neurosci, 27(5), 423–429. doi: 10.1016/j.ijdevneu.2009.05.007 [DOI] [PubMed] [Google Scholar]
- Viberg H, Mundy W, & Eriksson P (2008). Neonatal exposure to decabrominated diphenyl ether (PBDE 209) results in changes in BDNF, CaMKII and GAP-43, biochemical substrates of neuronal survival, growth, and synaptogenesis. Neurotoxicology, 29(1), 152–159. doi: 10.1016/j.neuro.2007.10.007 [DOI] [PubMed] [Google Scholar]
- Vickers CA, Dickson KS, & Wyllie DJ (2005). Induction and maintenance of late-phase long-term potentiation in isolated dendrites of rat hippocampal CA1 pyramidal neurones. J Physiol, 568(Pt 3), 803–813. doi: 10.1113/jphysiol.2005.092924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman JM, Lioy PJ, Greenberg A, & Butler JP (1991). Analysis of human exposure to benzo(a)pyrene via inhalation and food ingestion in the Total Human Environmental Exposure Study (THEES). J Expo Anal Environ Epidemiol, 1(2), 193–225. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/1824316 [PubMed] [Google Scholar]
- Warner M, Rauch S, Ames J, Mocarelli P, Brambilla P, Signorini S, & Eskenazi B (2020). Prenatal dioxin exposure and thyroid hormone levels in the Seveso second generation study. Environ Res, 183, 109280. doi: 10.1016/j.envres.2020.109280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Bose DD, Yang D, Lesiak A, Bruun D, Impey S, Ledoux V, Pessah IN, & Lein PJ (2012a). PCB-95 modulates the calcium-dependent signaling pathway responsible for activity-dependent dendritic growth. Environ Health Perspect, 120(7), 1003–1009. doi: 10.1289/ehp.1104833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Davare M, Ando H, Fortin D, Varlamova O, Cheng HY, Marks D, Obrietan K, Soderling TR, Goodman RH, & Impey S (2008). An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc Natl Acad Sci U S A, 105(26), 9093–9098. doi: 10.1073/pnas.0803072105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, & Soderling TR (2006). Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron, 50(6), 897–909. doi: 10.1016/j.neuron.2006.05.008 [DOI] [PubMed] [Google Scholar]
- Wayman GA, Yang D, Bose DD, Lesiak A, Ledoux V, Bruun D, Pessah IN, & Lein PJ (2012b). PCB-95 promotes dendritic growth via ryanodine receptor-dependent mechanisms. Environ Health Perspect, 120(7), 997–1002. doi: 10.1289/ehp.1104832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B (2002). Sexually dimorphic nonreproductive behaviors as indicators of endocrine disruption. Environ Health Perspect, 110 Suppl 3, 387–391. doi: 10.1289/ehp.02110s3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Knekt P, O’Reilly EJ, Lyytinen J, Reunanen A, Laden F, Altshul L, & Ascherio A (2010). Persistent organochlorine pesticides in serum and risk of Parkinson disease. Neurology, 74(13), 1055–1061. doi: 10.1212/WNL.0b013e3181d76a93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Proctor SP, Wright RO, Schwartz J, Spiro A 3rd, Sparrow D, Nie H, & Hu H (2007). Cumulative lead exposure and cognitive performance among elderly men. Epidemiology, 18(1), 59–66. doi: 10.1097/01.ede.0000248237.35363.29 [DOI] [PubMed] [Google Scholar]
- Weston HI, Sobolewski ME, Allen JL, Weston D, Conrad K, Pelkowski S, Watson GE, Zareba G, & Cory-Slechta DA (2014). Sex-dependent and non-monotonic enhancement and unmasking of methylmercury neurotoxicity by prenatal stress. Neurotoxicology, 41, 123–140. doi: 10.1016/j.neuro.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson MA, Gasiewicz TA, & Opanashuk LA (2005). Aryl hydrocarbon receptor expression and activity in cerebellar granule neuroblasts: implications for development and dioxin neurotoxicity. Toxicol Sci, 83(2), 340–348. doi: 10.1093/toxsci/kfi031 [DOI] [PubMed] [Google Scholar]
- Winneke G (2011). Developmental aspects of environmental neurotoxicology: lessons from lead and polychlorinated biphenyls. J Neurol Sci, 308(1-2), 9–15. doi: 10.1016/j.jns.2011.05.020 [DOI] [PubMed] [Google Scholar]
- Wong PW, Brackney WR, & Pessah IN (1997a). Ortho-substituted polychlorinated biphenyls alter microsomal calcium transport by direct interaction with ryanodine receptors of mammalian brain. J Biol Chem, 272(24), 15145–15153. doi: 10.1074/jbc.272.24.15145 [DOI] [PubMed] [Google Scholar]
- Wong PW, Joy RM, Albertson TE, Schantz SL, & Pessah IN (1997b). Ortho-substituted 2,2′,3,5′,6-pentachlorobiphenyl (PCB 95) alters rat hippocampal ryanodine receptors and neuroplasticity in vitro: evidence for altered hippocampal function. Neurotoxicology, 18(2), 443–456. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9291493 [PubMed] [Google Scholar]
- Wong PW, & Pessah IN (1997). Noncoplanar PCB 95 alters microsomal calcium transport by an immunophilin FKBP12-dependent mechanism. Mol Pharmacol, 51(5), 693–702. doi: 10.1124/mol.51.5.693 [DOI] [PubMed] [Google Scholar]
- Woolley CS, Gould E, Frankfurt M, & McEwen BS (1990). Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci, 10(12), 4035–4039. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/2269895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormley DD, Chirwa S, Nayyar T, Wu J, Johnson S, Brown LA, Harris E, & Hood DB (2004a). Inhaled benzo(a)pyrene impairs long-term potentiation in the F1 generation rat dentate gyrus. Cell Mol Biol (Noisy-le-grand), 50(6), 715–721. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/15641162 [PubMed] [Google Scholar]
- Wormley DD, Ramesh A, & Hood DB (2004b). Environmental contaminant-mixture effects on CNS development, plasticity, and behavior. Toxicol Appl Pharmacol, 197(1), 49–65. doi: 10.1016/j.taap.2004.01.016 [DOI] [PubMed] [Google Scholar]
- Xing T, Chen L, Tao Y, Wang M, Chen J, & Ruan DY (2009). Effects of decabrominated diphenyl ether (PBDE 209) exposure at different developmental periods on synaptic plasticity in the dentate gyrus of adult rats In vivo. Toxicol Sci, 110(2), 401–410. doi: 10.1093/toxsci/kfp114 [DOI] [PubMed] [Google Scholar]
- Xing TR, Yong W, Chen L, Tang ML, Wang M, Chen JT, & Ruan DY (2010). Effects of decabrominated diphenyl ether (PBDE 209) on voltage-gated sodium channels in primary cultured rat hippocampal neurons. Environ Toxicol, 25(4), 400–408. doi: 10.1002/tox.20511 [DOI] [PubMed] [Google Scholar]
- Xiong Y, Ibhazehiebo K, Iwasaki T, & Koibuchi N (2012). An in vitro method to study the effects of thyroid hormone-disrupting chemicals on neuronal development. Neurotoxicology, 33(4), 753–757. doi: 10.1016/j.neuro.2012.04.021 [DOI] [PubMed] [Google Scholar]
- Yang D, Kania-Korwel I, Ghogha A, Chen H, Stamou M, Bose DD, Pessah IN, Lehmler HJ, & Lein PJ (2014). PCB 136 atropselectively alters morphometric and functional parameters of neuronal connectivity in cultured rat hippocampal neurons via ryanodine receptor-dependent mechanisms. Toxicol Sci, 138(2), 379–392. doi: 10.1093/toxsci/kft334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Kim KH, Phimister A, Bachstetter AD, Ward TR, Stackman RW, Mervis RF, Wisniewski AB, Klein SL, Kodavanti PR, Anderson KA, Wayman G, Pessah IN, & Lein PJ (2009). Developmental exposure to polychlorinated biphenyls interferes with experience-dependent dendritic plasticity and ryanodine receptor expression in weanling rats. Environ Health Perspect, 117(3), 426–435. doi: 10.1289/ehp.11771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZH, Zheng G, Wang T, Du KJ, Han X, Luo WJ, Shen XF, & Chen JY (2018). Low-level Gestational Lead Exposure Alters Dendritic Spine Plasticity in the Hippocampus and Reduces Learning and Memory in Rats. Sci Rep, 8(1), 3533. doi: 10.1038/s41598-018-21521-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, & Poo MM (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron, 44(5), 749–757. doi: 10.1016/j.neuron.2004.11.011 [DOI] [PubMed] [Google Scholar]
- Zoeller TR (2010). Environmental chemicals targeting thyroid. Hormones (Athens), 9(1), 28–40. doi: 10.14310/horm.2002.1250 [DOI] [PubMed] [Google Scholar]
- Zoghbi HY (2003). Postnatal neurodevelopmental disorders: meeting at the synapse? Science, 302(5646), 826–830. doi: 10.1126/science.1089071 [DOI] [PubMed] [Google Scholar]